

Streptococcus pneumoniae is a major causative bacterium of community-acquired pneumonia. Dendritic cell-associated C-type lectin-2 (dectin-2), one of the C-type lectin receptors (CLRs), was previously reported to play a pivotal role in host defense against pneumococcal infection through regulating phagocytosis by neutrophils while not being involved in neutrophil accumulation. In the present study, to elucidate the possible contribution of other CLRs to neutrophil accumulation, we examined the role of caspase recruitment domain-containing protein 9 (CARD9), a common adaptor molecule for signal transduction triggered by CLRs, in neutrophilic inflammatory response against pneumococcal infection.
KEYWORDS: CARD9, CLRs, host defense, neutrophils, pneumococcal infection
ABSTRACT
Streptococcus pneumoniae is a major causative bacterium of community-acquired pneumonia. Dendritic cell-associated C-type lectin-2 (dectin-2), one of the C-type lectin receptors (CLRs), was previously reported to play a pivotal role in host defense against pneumococcal infection through regulating phagocytosis by neutrophils while not being involved in neutrophil accumulation. In the present study, to elucidate the possible contribution of other CLRs to neutrophil accumulation, we examined the role of caspase recruitment domain-containing protein 9 (CARD9), a common adaptor molecule for signal transduction triggered by CLRs, in neutrophilic inflammatory response against pneumococcal infection. Wild-type (WT), CARD9 knockout (KO), and dectin-2 KO mice were infected intratracheally with pneumococcus, and the infected lungs were histopathologically analyzed to assess neutrophil accumulation at 24 h postinfection. Bronchoalveolar lavage fluids (BALFs) were collected at the same time point to count the neutrophils and assess the production of inflammatory cytokines and chemokines. Neutrophil accumulation was significantly decreased in CARD9 KO mice, but not in dectin-2 KO mice. Tumor necrosis factor alpha (TNF-α), keratinocyte-derived chemokine (KC), and macrophage inflammatory protein-2 (MIP-2) production in BALFs were also attenuated in CARD9 KO mice, but not in dectin-2 KO mice. Production of TNF-α and KC by alveolar macrophages stimulated with pneumococcal culture supernatants was significantly attenuated in CARD9 KO mice, but not in dectin-2 KO mice, compared to that in each group’s respective control mice. In addition, pneumococcus-infected CARD9 KO mice showed larger bacterial burdens in the lungs than did WT mice. These data indicate that CARD9 is required for neutrophil migration after pneumococcal infection, as well as inflammatory cytokine and chemokine production by alveolar macrophages, and suggest that a CLR distinct from dectin-2 may be involved in this response.
INTRODUCTION
Pneumonia remains a major cause of death worldwide. Streptococcus pneumoniae (pneumococcus) is the most frequently isolated causative bacterium of community-acquired pneumonia (1, 2). This bacterium causes invasive infections such as bacteremia and meningitis, especially in infants and the elderly (3, 4). Therefore, it is important to clarify the immunological mechanisms underlying host defense against pneumococcal infection.
The early phase of the immune response is critical for elimination of this bacterium. Upon infection of the lungs with pneumococcus, the initial immune response is initiated by activation of alveolar macrophages triggered via recognition of the bacterial elements, such as pathogen-associated molecular patterns (PAMPs) including capsular polysaccharides, teichoic acid, and pneumolysin (5). Subsequently, neutrophils accumulate in the infected alveolar spaces and eradicate the infection via phagocytic killing, which is promoted by opsonization of the bacterium with IgG antibody against pneumococcal capsular polysaccharides (6).
Macrophages and dendritic cells (DCs) are activated via recognition of the PAMPs from microbial pathogens by their pattern recognition receptors (PRRs) (7). In previous studies, Toll-like receptor 2 (TLR2), TLR4, and TLR9 have been reported to play important roles in recognition of pneumococcal infection (8–10). C-type lectin receptors (CLRs) are also PRRs and possess carbohydrate recognition domains, through which they mainly recognize polysaccharides (11). Interaction of CLRs with their own ligands triggers signaling via intramolecular immunoreceptor tyrosine-based activation motif (ITAM) within their intracellular domain or by associating with ITAM-bearing adaptor molecules such as Fc receptor γ (FcRγ) or DAP12 (12–16). Subsequently, the activation signals are delivered via phosphorylation of the tyrosine kinase Syk and activation of caspase recruitment domain-containing protein 9 (CARD9), a downstream adaptor molecule common to most CLRs (17, 18). These processes trigger the production of inflammatory cytokines and chemokines, leading to initiation of the inflammatory responses necessary for eradication of this bacterium (19–21).
The CLR dendritic cell-associated C-type lectin-2 (dectin-2) contributes to recognition of Candida albicans and plays a protective role by eliciting neutrophilic inflammatory responses (22). Previous studies by McGreal and coworkers have demonstrated that dectin-2 binds directly to pneumococcal polysaccharides (23). In our previous studies, dectin-2-deficient (knockout [KO]) mice showed shorter survival time and failed to eliminate bacteria from their lungs after pulmonary infection with S. pneumoniae (24). The resulting exacerbated infection in dectin-2 KO mice was associated with decreased production of anti-pneumococcal polysaccharide type-3 (PPS3) antibody (Ab) and attenuated phagocytosis of this bacterium by neutrophils, but not with impaired accumulation of these phagocytic cells (24). These findings indicate the important role of dectin-2 in the host defense against pneumococcal infection, a role that may consist of promoting phagocytosis of this bacterium by neutrophils. Other than dectin-2, however, it remains unclear which CLRs contribute to the accumulation of neutrophils in the infected lungs and which do not.
Because CARD9 is a common adaptor molecule for signaling triggered by CLRs, the present study focused on the effect of CARD9 deficiency on the accumulation of these cells after pneumococcal infection. We found that CARD9 KO mice were prone to this infection in combination with the impaired accumulation of neutrophils in the infected lungs, suggesting the involvement of a CLR other than dectin-2 in this neutrophilic response.
RESULTS
CARD9-dependent neutrophil accumulation in the lungs after pneumococcal infection.
To clarify whether CARD9 deficiency affected neutrophil accumulation in the lungs, CARD9 KO and wild-type (WT) mice were infected with pneumococcus, and histopathological analysis was conducted at 24 h after infection. As shown in Fig. 1A, infiltration of neutrophils into the alveolar spaces was drastically reduced in CARD9 KO mice compared to that in WT mice. In a striking contrast, both dectin-2 KO and WT mice showed equivalent levels of neutrophil infiltration into the lungs after pneumococcal infection (Fig. 1B). In addition, to compare the numbers of neutrophils infiltrating into the lungs among the genotypes, bronchoalveolar lavage fluids (BALFs) were collected from pneumococcus-infected mice, and the numbers of neutrophils in the BALFs were compared between CARD9 KO or dectin-2 KO mice and the respective control group for each genotype. As Fig. 2A shows, most of the infiltrating cells in the BALFs of WT mice were morphologically identifiable as neutrophils, whereas macrophages were predominant among the infiltrating cells in the BALFs of CARD9 KO mice. The BALFs of dectin-2 KO mice, in contrast, had neutrophil counts equivalent to those in WT mice, according to our morphological analysis (data not shown). In keeping with these results, the proportion and number of neutrophils in BALFs were markedly reduced in CARD9 KO mice compared to those in WT mice, whereas no such differences were observed between WT and dectin-2 KO mice (Fig. 2B). The number of neutrophils, but not that of macrophages, in BALFs was nearly 100 times lower in CARD9 KO mice than in WT mice, whereas no such difference was observed in either neutrophils or macrophages between WT and dectin-2 KO mice (Fig. 2C).
FIG 1.
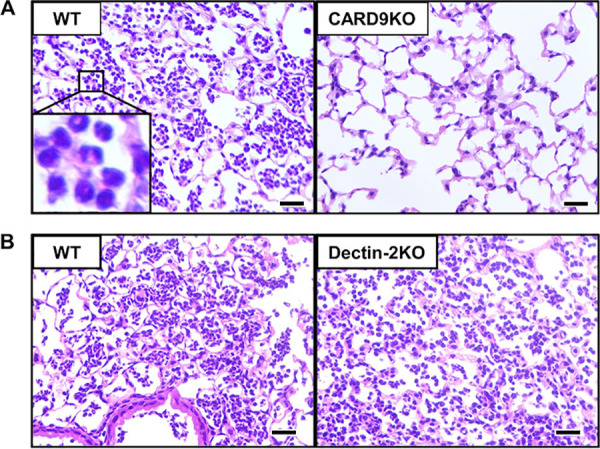
Effect of CARD9 deficiency on neutrophil accumulation in the lungs after pneumococcal infection. Mice were infected intratracheally with pneumococcus. Lung sections obtained at 24 h after infection were stained with hematoxylin-eosin (H&E) and observed under optical microscopy. Representative pictures of three or four mice are shown. Original magnification, ×400. Neutrophil accumulation was significantly diminished in CARD9 KO mice (A), but not in dectin-2 KO mice (B), compared to that in each genotype’s respective control mice. Bars, 25 μm.
FIG 2.
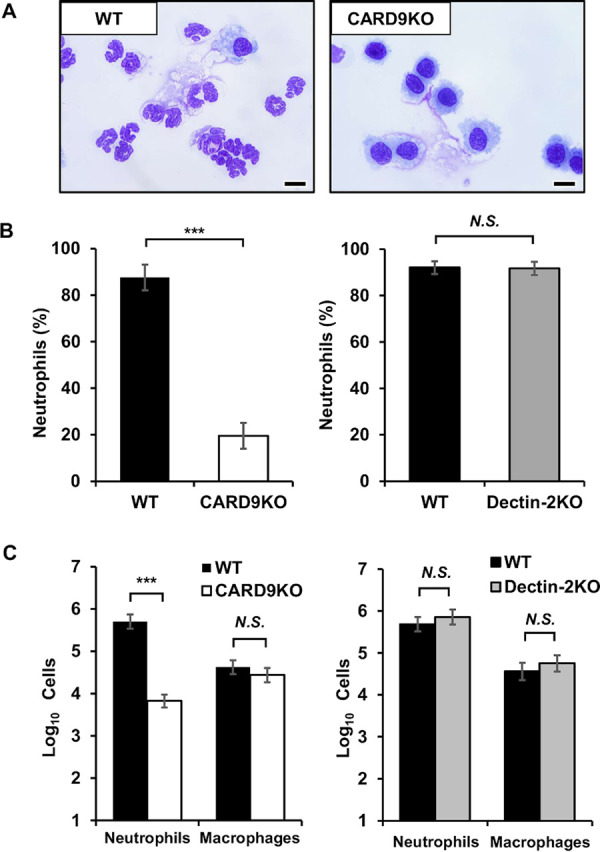
Effect of CARD9 deficiency on the number of neutrophils in BALFs after pneumococcal infection. BALFs obtained from infected mice at 24 h after pneumococcal infection were centrifuged, and then the sedimented cells stained with Diff-Quik were analyzed. (A) Representative pictures of five to eight mice are shown. Original magnification, ×1,000. Bars, 10 μm. The proportion (B) and the absolute number (C) of neutrophils in BALFs were shown. Each column represents the mean ± standard deviation (SD) of five to eight mice. Neutrophils were significantly lower in CARD9 KO mice, but not in dectin-2 KO mice, than they were in each genotype’s respective control mice. ***, P < 0.005; N.S., not significant.
Effect of CARD9 deficiency on the production of cytokines and chemokines in BALFs after pneumococcal infection.
BALFs were collected from CARD9 KO or dectin-2 KO mice and their respective control groups at 24 h after pneumococcal infection, and the production of inflammatory cytokines and neutrophil-attracting chemokines by each genotype was measured. Tumor necrosis factor alpha (TNF-α) increases the expression of adhesion molecules, enabling the adhesion of neutrophils to vascular endothelial cells to promote their migration from the vessels to the infected sites (25–27). In addition, keratinocyte-derived chemokine (KC) and macrophage inflammatory protein-2 (MIP-2) are known to play critical roles in the recruitment of neutrophils (28). As Fig. 3A and B show, production levels of TNF-α, KC, and MIP-2 in BALFs were significantly lower in CARD9 KO mice than in WT mice. In contrast, TNF-α production was almost equivalent between WT and dectin-2 KO mice, and production levels of KC and MIP-2 were significantly greater in dectin-2 KO mice than in WT mice.
FIG 3.
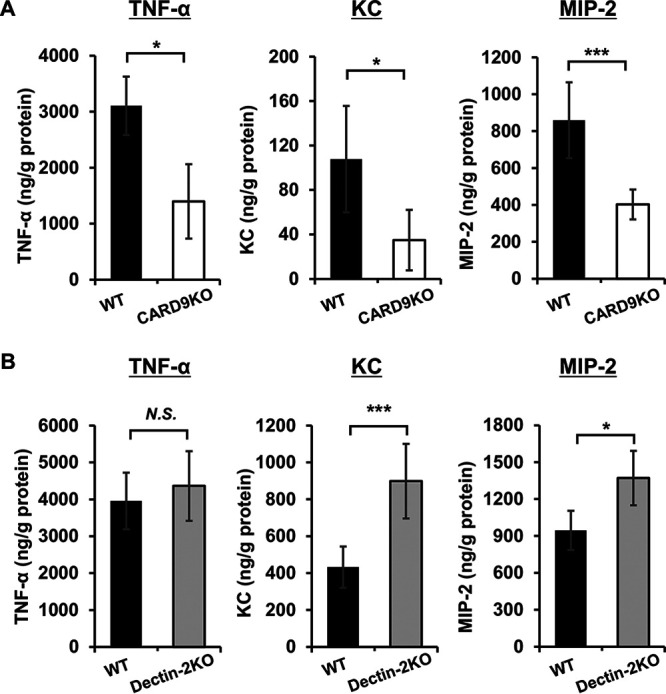
Effect of CARD9 deficiency on the production of cytokines and chemokines in BALFs after pneumococcal infection. BALFs were collected from infected mice at 24 h after pneumococcal infection and the cytokines and chemokines in these BALFs were measured by enzyme-linked immunosorbent assay (ELISA). Each column represents the mean ± SD of five to eight mice. Tumor necrosis factor alpha (TNF-α), keratinocyte-derived chemokine (KC), and macrophage inflammatory protein-2 (MIP-2) production in BALFs were attenuated in CARD9 KO mice (A), but not in dectin-2 KO mice (B), compared to that in each genotype’s respective control mice. *, P < 0.05; ***, P < 0.005; NS, not significant.
Effect of CARD9 deficiency on the production of cytokines and chemokines by alveolar macrophages.
In lung infections, alveolar macrophages are often the first cells to encounter the pathogens (5). When alveolar macrophages were stimulated with whole bacteria and their lysates, we did not observe any influence of CARD9 deficiency on cytokine production by alveolar macrophages (data not shown), perhaps because whole bacteria contain various PAMPs, including TLR and CLR ligands, as well as other ligands, on their surfaces and within their cells. Therefore, alveolar macrophages collected from uninfected mice were instead stimulated with the supernatants of pneumococcal cultures or with the culture broth as negative controls. As Fig. 4A shows, macrophages from CARD9 KO mice exhibited significantly decreased but not completely eradicated production of both TNF-α and KC when stimulated with pneumococcal culture supernatants at any concentration; in macrophages from WT mice, in contrast, production of TNF-α and KC was completely abrogated upon stimulation with trehalose 6,6′-dimycolate (TDM), a Mincle ligand. Macrophages from dectin-2 KO mice, on the other hand, produced significantly more TNF-α and KC than those from WT mice did, although this production was significantly reduced when macrophages from dectin-2 KO mice were stimulated with furfurman, a dectin-2-specific agonist (Fig. 4B).
FIG 4.
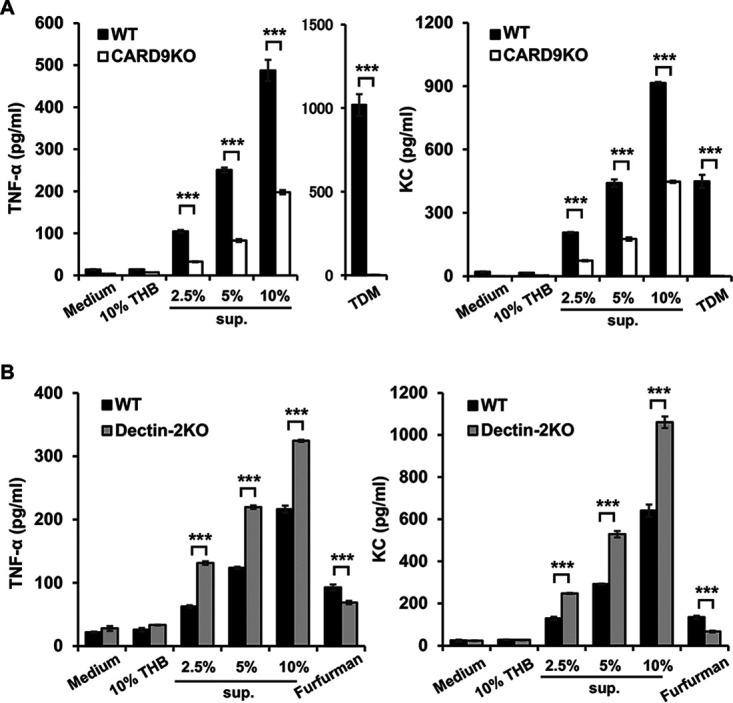
Effect of CARD9 deficiency on the production of cytokines and chemokines by alveolar macrophages. Alveolar macrophages from CARD9 KO, dectin-2 KO, and their respective control mice were stimulated with pneumococcus culture supernatants for 24 h, and production of TNF-α and KC was then measured. Each column represents the mean ± SD of triplicate cultures. TNF-α and KC production were significantly attenuated in CARD9 KO mice (A), but not in dectin-2 KO mice (B), compared to that in each genotype’s respective control mice. ***, P < 0.005, THB, Todd-Hewitt broth; sup., pneumococcus culture supernatants; TDM, trehalose 6,6'-dimycolate.
Effect of CARD9 deficiency on the phagocytosis of pneumococcus by neutrophils.
Our previous study showed that neutrophils from dectin-2 KO mice had a reduced capacity to phagocytize pneumococcus during in vivo infection (24). Therefore, to elucidate the effect of CARD9 deficiency on the capacity of neutrophils to phagocytize this bacterium, BALFs were collected from WT or CARD9 KO mice at 24 h after pneumococcal infection, and the phagocytic rate was calculated as the proportion of neutrophils engulfing bacteria relative to total neutrophils, as seen in Gram-stained samples (Fig. 5A). As Fig. 5B shows, the phagocytic rate was significantly lower in CARD9 KO mice than in WT mice.
FIG 5.
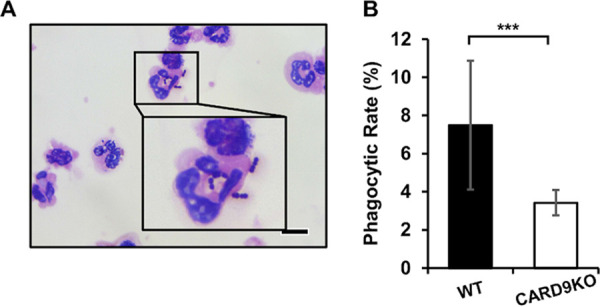
Effect of CARD9 deficiency on the phagocytosis of pneumococcus by neutrophils. BALFs from infected mice obtained at 24 h after pneumococcal infection were centrifuged, and the sedimented cells were stained by the Gram method and analyzed under optical microscopy. (A) A picture representative of 8 WT mice is shown. Original magnification, ×1,000. Bars, 10 μm. (B) The proportion of all neutrophils that were engulfing pneumococcus cells was calculated as the phagocytic rate. Each column represents the mean ± SD of 8 to 10 mice. This rate was significantly lower in CARD9 KO mice (B) than in WT mice. ***, P < 0.005.
Effect of CARD9 deficiency on IgG3 production against pneumococcal capsular polysaccharides in lungs.
Pneumococcus is known to be resistant to complement-mediated opsonization that leads to phagocytic killing by neutrophils (29). Therefore, anti-capsular polysaccharide Ab plays a critical role as an alternative opsonin (30). In our previous study (24), neutrophils from dectin-2 KO mice exhibited lower phagocytic efficiency, which may result from decreased production of anti-PPS3 IgG3, a major IgG subclass that is produced in response to polysaccharides in mice and that contributes to host defense against septic pneumococcal infection (30, 31). To explore this possibility, we measured the BALF levels of anti-PPS3 IgG3 at 24 h after pneumococcal infection. As Fig. 6A shows, production of anti-PPS3 IgG3 was significantly lower in CARD9 KO mice than in WT mice. Previously, Snapper and coworkers reported that interferon (IFN)-γ plays a major role in class switching from IgM to IgG3 in mice (32). To assess the effect of CARD9 deficiency on this phenomenon, BALFs were collected from WT or CARD9 KO mice at 24 h after pneumococcal infection, and levels of IFN-γ in these samples were measured. As Fig. 6B shows, IFN-γ production was significantly reduced in the lungs of CARD9 KO mice compared to that in those of WT mice.
FIG 6.
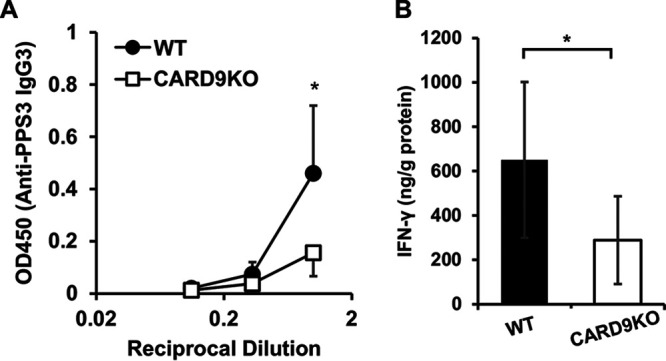
Effect of CARD9 deficiency on the production of anti-PPS3 IgG3. (A) BALFs were collected from infected mice at 24 h after pneumococcal infection, and anti-PPS3 IgG3 in these BALFs was measured. Each column represents the mean ± SD of six to eight mice. Anti-PPS3 IgG3 production in BALFs was attenuated in CARD9 KO mice compared to WT mice. *, P < 0.05. (B) BALFs were collected from infected mice at 24 h after pneumococcal infection, and IFN-γ in BALFs were measured. Each column represents the mean ± SD of six to eight mice. IFN-γ production in BALFs was attenuated in CARD9 KO mice compared to that in WT mice. *, P < 0.05 (B).
Effect of CARD9 deficiency on IL-12 production by BM-DCs.
Our previous study reported that natural killer T (NKT) cell-derived IFN-γ is involved in the class switching of IgG3 and that interleukin 12 (IL-12) is essential for activating NKT cells for the production of IFN-γ (33). Therefore, bone marrow-derived dendritic cells (BM-DCs) prepared from CARD9 KO, dectin-2 KO, or their respective control mice were stimulated with pneumococcal culture supernatants to elucidate the effects of these gene deficiencies on the production of IL-12p40, which we chose because IL-12p70 production was not detected in BM-DCs (data not shown). As Fig. 7 shows, BM-DCs from both CARD9 KO mice and dectin-2 KO mice produced significantly lower levels of IL-12p40 than those from each genotype’s respective control group; this pattern is similar to what we had previously observed in the production of this cytokine upon stimulation with heat-killed Candida albicans (34). When BM-DCs were stimulated with the TLR9 ligand CpG1826, in contrast, neither CARD9 nor dectin-2 deficiency appeared to have any effect on the resulting production of IL-12p40. On the other hand, IL-12p40 was not produced by alveolar macrophages stimulated with pneumococcal culture supernatants (data not shown).
FIG 7.
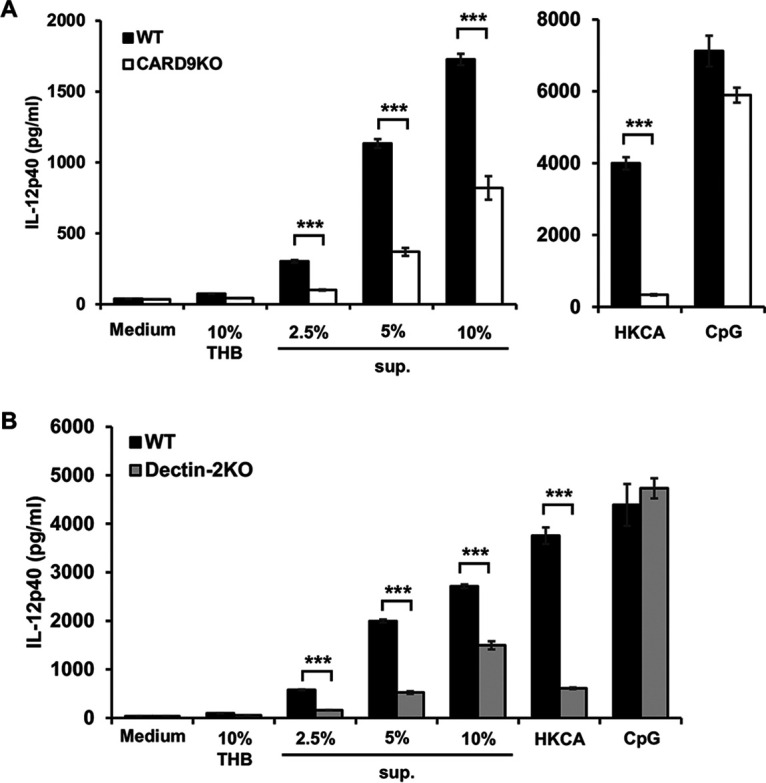
Effect of CARD9 deficiency on the production of IL-12p40 by BM-DCs. BM-DCs from CARD9 KO, dectin-2 KO, and their respective control mice were stimulated with pneumococcus culture supernatants for 24 h, after which IL-12p40 production was measured. Each column represents the mean ± SD of triplicate cultures. IL-12p40 production was significantly reduced in both CARD9 KO mice (A) and dectin-2 KO mice (B) compared to that in each genotype’s respective control mice. ***, P < 0.005, THB, Todd-Hewitt broth; sup., pneumococcus culture supernatants; HKCA, heat-killed C. albicans; CpG, CpG-motif containing oligonucleotides.
Effect of CARD9 deficiency on the elimination of pneumococcus in lungs.
The attenuating effects of CARD9 deficiency on neutrophil-mediated inflammatory responses may lead to the impairment of host defense against pneumococcal infection in the lungs. Therefore, we assessed whether CARD9 deficiency affected the elimination of pneumococcal bacteria from the lungs. As Fig. 8 shows, although the different genotypes of control mice had different distributions and amounts of CFU in the lungs due to their different backgrounds, the number of viable pneumococcus cells was significantly greater in both CARD9 KO mice and dectin-2 KO mice compared with that in each genotype’s respective control group.
FIG 8.
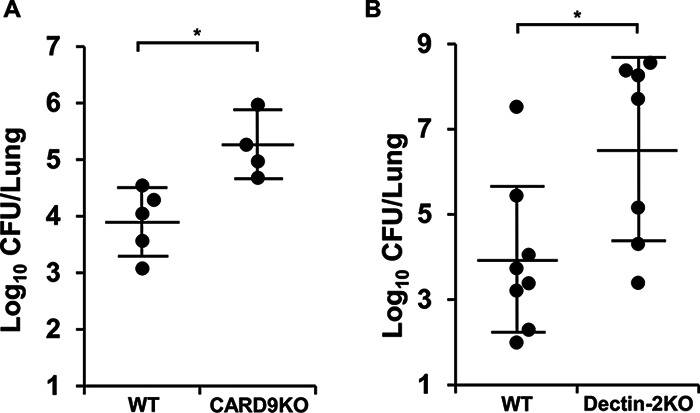
Effect of CARD9 deficiency on bacterial burden in the lungs after pneumococcal infection. CARD9 KO, dectin-2 KO, and their respective control mice were infected with pneumococcus. Lungs were collected on day 3 after infection, and live colonies were counted. Each symbol represents a mouse, and bars indicate the mean ± SD of four to eight mice. Bacterial burdens were significantly greater in CARD9 KO and in dectin-2 KO mice compared to those in each genotype’s respective control mouse population (A and B). *, P < 0.05.
DISCUSSION
Our earlier study revealed the critical role played by dectin-2 in the phagocytosis of bacteria by neutrophils while showing that it does not play a role in the accumulation of these cells in the lungs after infection with S. pneumoniae (24). It remains to be clarified how the detection of this bacterium by immune cells contributes to neutrophilic inflammatory responses after pneumococcal infection. In the present study, therefore, we sought to explore the possibility that a certain CLR may play a role in the lung accumulation of neutrophils following pneumococcal infection by clarifying how these responses are influenced by deficiency of CARD9, a common adapter molecule involved in the downstream signal transduction triggered by CLRs (17, 18).
Both our pathological and BALF analysis confirmed that neutrophil accumulation in the lungs after pneumococcal infection is markedly lower in CARD9 KO mice than that in WT mice. In contrast, as in our previous study (24), equivalent quantities of neutrophils infiltrated into the infected lungs of dectin-2 KO and WT mice. From these results, we conclude that CARD9-mediated signaling plays a critical role in the accumulation of neutrophils, as well as in the elimination of bacteria in the lungs after infection with S. pneumoniae. In keeping with this conclusion, previous studies using a mouse model of systemic infection with C. albicans have demonstrated that neutrophil infiltration into the kidneys is not impaired but is rather accelerated in dectin-2 KO mice compared to WT mice, although dectin-2 KO mice develop markedly worse infections (22). In addition, not only the elimination of C. albicans but also the infiltration of neutrophils is reported to be impaired in CARD9 KO mice compared to WT mice (35, 36). Considering our present findings alongside these previous observations, we propose that an unidentified CLR other than dectin-2 may be involved in the recognition of pneumococcus leading to neutrophil accumulation at pneumococcal infection sites.
In the second portion of our experiments, we measured BALF levels of TNF-α, KC, and MIP-2, each of which plays a critical role in the accumulation of neutrophils (25, 26), at 24 h after infection with S. pneumoniae. In line with the present results indicating attenuated neutrophil infiltration in CARD9 KO mice, the production of TNF-α, KC, and MIP-2 was significantly reduced in these mice compared to that in WT mice. No such influence, however, was observed in dectin-2 KO mice. These results suggest that pneumococcal infection may stimulate the activation signals that induce the production of cytokines and chemokines, leading to neutrophil infiltration, and that these signals may be triggered via an unidentified CLR other than dectin-2 that is delivered through a process involving CARD9 (e.g., Mincle) (14). Alveolar macrophages contribute to early host defense in the alveolar spaces not only by engulfing microorganisms but also by producing various cytokines and chemokines (37, 38). In vitro production of TNF-α and KC by alveolar macrophages in response to stimulation with S. pneumoniae culture supernatants was significantly lower in CARD9 KO mice than that in WT mice, as we had observed in BALFs in our in vivo experiments, suggesting that some CLR ligands are not only expressed on the capsules but also secreted from these bacteria and that these ligands may stimulate these cells in a CARD9-dependent manner. In dectin-2 KO mice, in contrast, TNF-α and KC production was not lower than that in WT mice but rather was higher, suggesting that decreased phagocytosis by neutrophils may trigger a feedback increase in the production of these chemokines that leads to more accumulation of these cells and/or that dectin-2 may be involved in the anti-inflammatory response, although the precise explanation for our findings is not clear. Although it is not known whether the higher TNF-α and KC production by alveolar macrophages in dectin-2-deficient mice is significant in any way, these results are consistent with the current findings regarding the respective contributions of CARD9 and dectin-2 to neutrophil infiltration. Interestingly, TNF-α and KC production was reduced by 50 to 60% in CARD9 KO mice, suggesting the possible involvement of an unidentified CARD9-independent signaling mechanism such as a TLRs-MyD88-mediated pathway. Despite this reduction in TNF-α and KC production, the number of accumulated neutrophils was strikingly lower in CARD9 KO mice than that in WT mice. To interpret this apparent inconsistency, we speculate that the significant difference may result from the distinct effects of TNF-α and KC/MIP-2 on promoting neutrophil infiltration. TNF-α increases the expression of Mac1, an adhesion molecule that is expressed on neutrophils, and promotes the neutrophil adhesion to vascular endothelial cells by increasing the expression of intercellular adhesion molecule 1 (ICAM1, also known as CD54), a ligand for Mac1; this in turn accelerates the extravasation and migration of neutrophils into the infected alveolar spaces (25–27). Chemokines like KC and MIP-2, meanwhile, directly facilitate the migration of neutrophils to the primary infected sites (28). During infection with S. pneumoniae under CARD9-deficient conditions, both functions are thought to be attenuated, which may result in a synergistic impairment of the migration of neutrophils to the infected alveolar spaces.
In addition to the infiltration of neutrophils, their phagocytosis of pneumococcus is also hampered in CARD9 KO mice. In our previous study, neutrophils in dectin-2 KO mice likewise exhibited a reduced capacity for phagocytosis of this bacterium, although neutrophil infiltration into the lungs was almost equivalent to that in WT mice (24). Anti-capsular polysaccharide Ab plays a critical role as an opsonin enabling the effective phagocytosis of pneumococcus by neutrophils (30), especially given that the activation of complement, an alternative opsonin, is known to be inhibited by this bacterium (29). IgG3, a major subclass of Ab against pneumococcal polysaccharides, relies for its production on the production of IL-12 by dendritic cells or macrophages. IL-12 signaling, as well as antigen presentation, contributes to the activation of NKT cells (39, 40), which, when activated, produce abundant IFN-γ, which facilitates class switching of anti-pneumococcal polysaccharide Ab to IgG3 Ab (32). IL-12p40 production by BM-DCs upon stimulation with pneumococcus culture supernatants was significantly lower in CARD9 KO mice than in WT mice. Similarly, anti-PPS3 IgG3 production was decreased in dectin-2 KO mice (24). Importantly, IFN-γ production in BALFs was also reduced in both CARD9 KO and dectin-2 KO mice. Considering all these findings together, we propose that the activation of CARD9-mediated signaling may lead to IFN-γ production through the recognition of pneumococcus via dectin-2, which may contribute to the capacity of neutrophils to phagocytose this bacterium by promoting the production of IgG3 anti-pneumococcal polysaccharide Ab. However, Yamamoto and coworkers have reported that IgG3 antibodies do not mediate early protection against pneumococcal pneumonia, as shown by their results indicating that, in a pneumococcal pneumonia model, there was no difference in survival rates or lung burdens of this bacterium between WT and IgG3 KO mice under a BALB/c background (41). The fact that different studies have used different strains of mice (C57BL/6 versus BALB/c) could explain the conflicting interpretations of the possible role of IgG3 antibodies. Another possibility is that some compensatory mechanism might be operating to minimize the undesired effect of IgG3 deficiency, which would account for the failure of IgG3 deficiency to affect the host’s protection against this infection. Additionally, in our study, the precise role of IgG3 remains to be substantiated. Further investigation is necessary to address this issue.
In conclusion, the current study demonstrated that CARD9-mediated signaling plays a critical role in host defense against pneumococcal infection through both enhancing neutrophil infiltration and promoting neutrophil phagocytosis of this bacterium, and it suggested that the two functions of neutrophils may be separately regulated by distinct pattern recognition receptors, namely, dectin-2 and an unidentified CLR or other type of receptor, both of which deliver their signals via CARD9 (Fig. 9). Phagocytosis of pneumococcus by neutrophils is thought to be regulated by signaling through a dectin-2-CARD9-dependent mechanism. In contrast, it remains to be clarified how neutrophil infiltration is regulated via the CARD9-mediated signaling pathway after pneumococcal infection. Recently, glucosyl-diacylglycerol, a lipid component of pneumococcus, has been reported as a ligand recognized by Mincle, another CLR (42). In our unpublished experiments, however, neutrophil infiltration into the lungs was only marginally affected by Mincle deficiency. Thus, further investigation is still necessary to define the precise mechanism for neutrophil-mediated host defense against pneumococcal infection.
FIG 9.
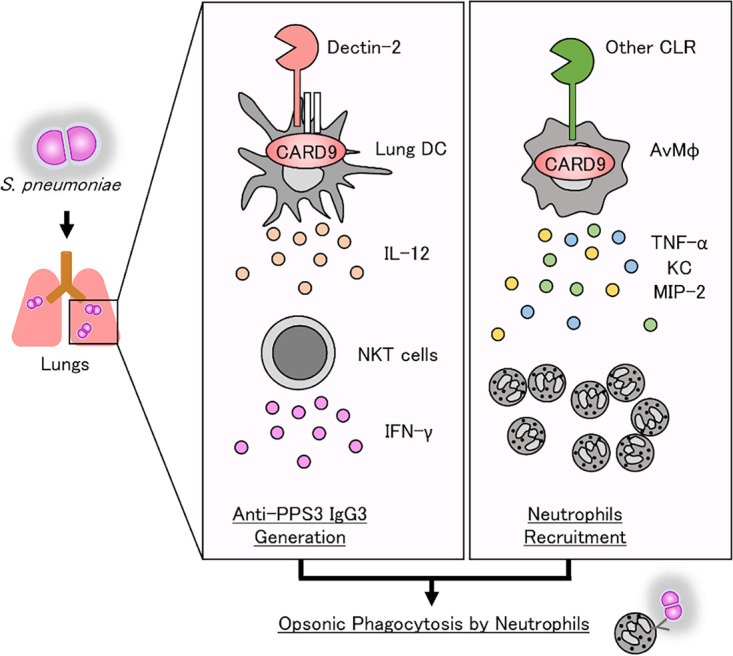
Schematic illustration of neutrophil-mediated host defense against pulmonary infection with S. pneumoniae. CARD9-mediated signaling plays a critical role in host defense against pneumococcal infection through both enhancing neutrophil recruitment and promoting neutrophil phagocytosis. Phagocytosis of pneumococcus by neutrophils is regulated by the production of serotype-specific IgG through IFN-γ synthesis by NKT cells induced by IL-12 produced from dendritic cells via the recognition of pneumococcal polysaccharides by dectin-2 (33). In contrast, neutrophil infiltration may be regulated via the other CLR-CARD9 axis pathway after pneumococcal infection. DC, dendritic cell(s); AvMφ, alveolar macrophage(s), PPS3: pneumococcal polysaccharide type-3.
MATERIALS AND METHODS
Mice.
CARD9 KO mice, generated and established as described previously (18) and backcrossed to C57BL/6 mice for more than eight generations, were kindly provided by Hiromitsu Hara (Kagoshima University, Kagoshima, Japan). C57BL/6 mice, purchased from CLEA Japan (Tokyo, Japan), were used as WT control mice. Dectin-2 KO mice, generated by homologous recombination of the Clec4n gene as described previously (22) and backcrossed to C57BL/6 mice for seven generations, were kindly provided by Yoichiro Iwakura (Tokyo University of Science, Noda, Chiba, Japan). Because the dectin-2 KO mice had been backcrossed to C57BL/6 for fewer than eight generations, WT littermate mice of the dectin-2 KO mice were used as their control group, whereas a different control group was used for comparison with CARD9 KO mice. Male or female mice at 6 to 8 weeks of age were used for the experiments. The mice were bred under specific pathogen-free conditions at the Animal Facility, Tohoku University Graduate School of Medicine (Sendai, Japan). All experimental procedures involving animals followed the Regulations for Animal Experiments and Related Activities at Tohoku University and were approved by the ethics committees of Tohoku University.
Bacteria.
A serotype 3 clinical strain of S. pneumoniae, designated URF918, was isolated from a patient with pneumococcal pneumonia. The bacteria were cultured in Todd-Hewitt broth (BD Biosciences, East Rutherford, NJ) at 37°C in a 5% CO2 incubator, harvested at the mid-log phase of growth, and then washed twice in phosphate-buffered saline (PBS). The inoculum was stored at −80°C until use.
S. pneumoniae infection.
CARD9 KO and dectin-2 KO mice and the mice from their respective control groups were anaesthetized by an intramuscular injection of 0.3 mg/kg midazolam (Teva Takeda Pharma, Ltd., Nagoya, Japan) and 0.02 mg/kg medetomidine (Nippon Zenyaku Kogyo Co., Ltd., Fukushima, Japan) and by an intraperitoneal injection of 25 mg/kg pentobarbital (Abbott Laboratories, North Chicago, IL). Live S. pneumoniae bacteria (0.75 × 105 to 10 × 105 CFU) at 50 μl per mouse were inoculated by insertion of a 24-gauge intravenous catheter (Terumo, Tokyo, Japan) into the trachea. Colony counts were performed to confirm the accuracy of the inoculum CFU count as a factor used to calculate CFU/ml for S. pneumoniae using a 5% sheep blood tryptic soy agar plate (BD Biosciences).
Preparation of S. pneumoniae culture supernatants.
S. pneumoniae culture was diluted with Todd-Hewitt broth until the turbidity as measured with a spectrophotometer reached an optical density at 600 nm (OD600) of 0.5, then inoculated into 19 volumes of Todd-Hewitt broth and incubated on an orbital shaker (150 rpm) at 37°C in a 5% CO2 incubator for 24 h. The culture supernatants were centrifuged, passed through a 0.2-μm membrane filter (Sartorius, Göttingen, Germany) and stored at −80°C until use. Todd-Hewitt broth incubated without S. pneumoniae was used as a control.
Preparation and culture of dendritic cells.
CARD9 KO and dectin-2 KO mice and the mice from their respective control groups were anaesthetized by isoflurane. Bone marrow cells obtained from these mice were cultured at 2 × 105 cells/ml in 10 ml RPMI 1640 medium (Sigma-Aldrich, St. Louis, MO) supplemented with 10% fetal calf serum (FCS; Biowest, Nuaillé, France), 100 U/ml penicillin G, 100 μg/ml streptomycin (Sigma-Aldrich), and 50 μM 2-mercaptoethanol (Sigma-Aldrich) containing 20 ng/ml murine granulocyte-macrophage colony-stimulating factor (GM-CSF; Wako Pure Chemical Industries, Ltd., Osaka, Japan). On day 3, 10 ml of the same medium was added; on day 6, half of the medium was replaced with GM-CSF-containing culture medium. On day 8, nonadherent cells were collected and used as bone-marrow derived dendritic cells (BM-DCs).
Preparation of alveolar macrophages.
CARD9 KO and dectin-2 KO mice and the mice from their respective control groups were anaesthetized by isoflurane. Each specimen’s chest was opened and the trachea was cannulated with the outer sheath of a 22-gauge intravenous catheter/needle unit (Terumo, Tokyo, Japan). Lung lavage was performed three times with 1 ml of cold PBS. The BALFs were centrifuged, and the sedimented cells were collected and used as alveolar macrophages.
In vitro stimulation.
BM-DCs and alveolar macrophages (1 × 105 cells/ml) were cultured with S. pneumoniae culture supernatants, Todd-Hewitt broth as the vehicle control, trehalose 6,6′-dimycolate (TDM; Sigma-Aldrich), furfurman (mannan derived from Malassezia furfur; InvivoGen, San Diego, CA), heat-killed C. albicans (HKCA), and CpG1826 (Hokkaido System Science, Sapporo, Japan) at 37°C in a 5% CO2 incubator for 24 h. The collected culture supernatants were stored at −80°C until use.
Preparation and analysis of BALFs.
Bronchoalveolar lavage fluids (BALFs) were collected as described above using a 22-gauge intravenous catheter/needle unit (Terumo). Lung lavage was performed three times with 1 ml of cold PBS. The first 1-ml and the subsequent 2-ml aliquots were pooled separately (tube 1 and tube 2, respectively). Both tubes were centrifuged, and the supernatants from tube 1 were collected and stored at −80°C until use. The number of sedimented cells in both tubes was counted, then 1 × 105 cells were centrifuged onto a glass slide using a StatSpin CytoFuge 2 (Iris Sample Processing, Westwood, MA), and stained with Diff-Quik (Sysmex, Kobe, Japan) or Gram stain using a Favor-G kit (Nissui Pharmaceutical Co., Ltd., Tokyo, Japan). To obtain the differential leukocyte counts and the numbers of neutrophils that were engulfing S. pneumoniae, at least 500 cells were examined under optical microscopy.
Measurement of cytokine and chemokine concentrations.
Collected BALFs were tested using enzyme-linked immunosorbent assay (ELISA) kits to measure tumor necrosis factor alpha (TNF-α), interferon (IFN)-γ (BioLegend, San Diego, CA), keratinocyte-derived chemokine (KC, also known asCXCL1) and macrophage inflammatory protein-2 (MIP-2, also known asCXCL2) (R&D Systems, Minneapolis, MN). Supernatants of the in vitro cell stimulation were assessed using ELISA kits for TNF-α, KC, and IL-12p40 (BioLegend).
Measurement of total protein in BALFs.
To calibrate the concentrations of cytokines and chemokines in the BALFs, the total protein was quantified by bicinchoninic acid (BCA) assay (Life Technologies, Carlsbad, CA).
Lung histopathology.
Lungs isolated from CARD9 KO and dectin-2 KO mice and the mice from their respective control groups at 24 h after pneumococcal infection were fixed in 10% buffered formalin, dehydrated, and embedded in paraffin. Sections were cut and stained with hematoxylin-eosin (H&E) at the Biomedical Research Core, Animal Pathology Platform of Tohoku University Graduate School of Medicine (Sendai, Japan).
Counting viable S. pneumoniae colonies.
CARD9 KO and dectin-2 KO mice and the mice from their respective control groups were sacrificed on day 3 after pneumococcal infection, and their lungs were carefully dissected and excised. The lungs were then homogenized in 5 ml of sterile half saline by teasing through a stainless mesh at room temperature. The homogenates (100 μl) were diluted in a 10-fold series using half saline and inoculated onto a 5% sheep blood tryptic soy agar plate (BD Biosciences). The homogenates were then cultured overnight at 37°C in 5% CO2, and the number of live colonies was counted.
Statistical analysis.
Statistical analysis was conducted using Excel 2016 (Microsoft Corporation, Redmond, WA). Data are presented as the mean ± standard deviation (SD). Differences between the two groups were tested using a two-tailed analysis in an unpaired Student’s t test. A P value of less than 0.05 was considered significant.
ACKNOWLEDGMENTS
We thank Hiromitsu Hara (Kagoshima University, Kagoshima, Japan) and Yoichiro Iwakura (Tokyo University of Science, Noda, Chiba, Japan) for kindly providing CARD9 KO mice and dectin-2 KO mice, respectively.
This work was supported in part by a Grant-in-Aid for Scientific Research (C) (grant 17K10013) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
We have no financial conflict of interest to declare.
S.I., R.Y., R.S., H.Y., K.T., and T.K. performed the experiments and analyzed the data; S.I., T.M., J.K., E.K., H.T., K.I., and K.K. conceived and designed the experiments; and S.I., K.S., and K.K. wrote the manuscript. All authors read and approved the final manuscript.
REFERENCES
- 1.Shindo Y, Ito R, Kobayashi D, Ando M, Ichikawa M, Shiraki A, Goto Y, Fukui Y, Iwaki M, Okumura J, Yamaguchi I, Yagi T, Tanikawa Y, Sugino Y, Shindoh J, Ogasawara T, Nomura F, Saka H, Yamamoto M, Taniguchi H, Suzuki R, Saito H, Kawamura T, Hasegawa Y, Shindo Y, Okumura J, Central Japan Lung Study Group . 2013. Risk factors for drug-resistant pathogens in community acquired and healthcare-associated pneumonia. Am J Respir Crit Care Med 188:985–995. doi: 10.1164/rccm.201301-0079OC. [DOI] [PubMed] [Google Scholar]
- 2.Ishiguro T, Takayanagi N, Yamaguchi S, Yamakawa H, Nakamoto K, Takaku Y, Miyahara Y, Kagiyama N, Kurashima K, Yanagisawa T, Sugita Y, Ishiguro T, Sugita Y. 2013. Etiology and factors contributing to the severity and mortality of community-acquired pneumonia. Intern Med 52:317–324. doi: 10.2169/internalmedicine.52.8830. [DOI] [PubMed] [Google Scholar]
- 2.O'Brien KL, Wolfson LJ, Watt JP, Henkle E, Deloria-Knoll M, McCall N, Lee E, Mulholland K, Levine OS, Cherian T, Hib and Pneumococcal Global Burden of Disease Study Team . 2009. Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: global estimates. Lancet 374:893–902. doi: 10.1016/S0140-6736(09)61204-6. [DOI] [PubMed] [Google Scholar]
- 4.Morimoto K, Suzuki M, Ishifuji T, Yaegashi M, Asoh N, Hamashige N, Abe M, Aoshima M, Ariyoshi K, Adult Pneumonia Study Group-Japan (APSG-J) . 2015. The burden and etiology of community-onset pneumonia in the aging Japanese popula-tion: a multicenter prospective study. PLoS One 10:e0122247. doi: 10.1371/journal.pone.0122247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quinton LJ, Walkey AJ, Mizgerd JP. 2018. Integrative physiology of pneumonia. Physiol Rev 98:1417–1464. doi: 10.1152/physrev.00032.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Musher DM, Phan HM, Watson DA, Baughn RE. 2000. Antibody to capsular polysaccharide of Streptococcus pneumoniae at the time of hospital admission for pneumococcal pneumonia. J Infect Dis 182:158–167. doi: 10.1086/315697. [DOI] [PubMed] [Google Scholar]
- 7.Bieber K, Autenrieth SE. 2020. Dendritic cell development in infection. Mol Immunol 121:111–117. doi: 10.1016/j.molimm.2020.02.015. [DOI] [PubMed] [Google Scholar]
- 8.Echchannaoui H, Frei K, Schnell C, Leib SL, Zimmerli W, Landmann R. 2002. Toll-like receptor 2-deficient mice are highly susceptible to Streptococcus pneumoniae meningitis because of reduced bacterial clearing and enhanced inflammation. J Infect Dis 186:798–806. doi: 10.1086/342845. [DOI] [PubMed] [Google Scholar]
- 9.Malley R, Henneke P, Morse SC, Cieslewicz MJ, Lipsitch M, Thompson CM, Kurt-Jones E, Paton JC, Wessels MR, Golenbock DT. 2003. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc Natl Acad Sci U S A 100:1966–1971. doi: 10.1073/pnas.0435928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luxameechanporn T, Kirtsreesakul V, Klemens J, Khoury P, Thompson K, Naclerio RM. 2005. Evaluation of importance of Toll-like receptor 4 in acute Streptococcus pneumoniae sinusitis in mice. Arch Otolaryngol Head Neck Surg 131:1001–1006. doi: 10.1001/archotol.131.11.1001. [DOI] [PubMed] [Google Scholar]
- 11.Hou H, Guo Y, Chang Q, Luo T, Wu X, Zhao X. 2017. C-type lectin receptor: old friend and new player. Med Chem 13:536–543. doi: 10.2174/1573406413666170510103030. [DOI] [PubMed] [Google Scholar]
- 12.Ariizumi K, Shen GL, Shikano S, Xu S, Ritter R, 3rd, Kumamoto T, Edelbaum D, Morita A, Bergstresser PR, Takashima A. 2000. Identification of a novel, dendritic cell- associated molecule, dectin-1, by subtractive cDNA cloning. J Biol Chem 275:20157–20167. doi: 10.1074/jbc.M909512199. [DOI] [PubMed] [Google Scholar]
- 13.Sato K, Yang XL, Yudate T, Chung JS, Wu J, Luby-Phelps K, Kimberly RP, Underhill D, Cruz PD, Jr, Ariizumi K. 2006. Dectin-2 is a pattern recognition receptor for fungi that couples with the Fc receptor gamma chain to induce innate immune responses. J Biol Chem 281:38854–38866. doi: 10.1074/jbc.M606542200. [DOI] [PubMed] [Google Scholar]
- 14.Yamasaki S, Ishikawa E, Sakuma M, Hara H, Ogata K, Saito T. 2008. Mincle is an ITAM-coupled activating receptor that senses damaged cells. Nat Immunol 9:1179–1188. doi: 10.1038/ni.1651. [DOI] [PubMed] [Google Scholar]
- 15.Miyake Y, Toyonaga K, Mori D, Kakuta S, Hoshino Y, Oyamada A, Yamada H, Ono K, Suyama M, Iwakura Y, Yoshikai Y, Yamasaki S. 2013. C-type lectin MCL is an FcRγ-coupled receptor that mediates the adjuvanticity of mycobacterial cord factor. Immunity 38:1050–1062. doi: 10.1016/j.immuni.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 16.Bakker ABH, Baker E, Sutherland GR, Phillips JH, Lanier LL. 1999. Myeloid DAP12-associateing lectin (MDL)-1 is a cell surface receptor involved in the activation of myeloid cells. Proc Natl Acad Sci U S A 96:9792–9796. doi: 10.1073/pnas.96.17.9792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gross O, Gewies A, Finger K, Schäfer M, Sparwasser T, Peschel C, Förster I, Ruland J. 2006. Card9 controls a non-TLR signaling pathway for innate anti-fungal immunity. Nature 442:651–656. doi: 10.1038/nature04926. [DOI] [PubMed] [Google Scholar]
- 18.Hara H, Ishihara C, Takeuchi A, Imanishi T, Xue L, Morris SW, Inui M, Takai T, Shibuya A, Saijo S, Iwakura Y, Ohno N, Koseki H, Yoshida H, Penninger JM, Saito T. 2007. The adaptor protein CARD9 is essential for the activation of myeloid cells through ITAM-associated and Toll-like receptors. Nat Immunol 8:619–629. doi: 10.1038/ni1466. [DOI] [PubMed] [Google Scholar]
- 19.Robinson MJ, Osorio F, Rosas M, Freitas RP, Schweighoffer E, Gross O, Verbeek JS, Ruland J, Tybulewicz V, Brown GD, Moita LF, Taylor PR, Reis e Sousa C. 2009. Dectin-2 is a Syk-coupled pattern recognition receptor crucial for Th17 responses to fungal infection. J Exp Med 206:2037–2051. doi: 10.1084/jem.20082818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bertin J, Guo Y, Wang L, Srinivasula SM, Jacobson MD, Poyet JL, Merriam S, Du MQ, Dyer MJ, Robison KE, DiStefano PS, Alnemri ES. 2000. CARD9 is a novel caspase recruitment domain-containing protein that interacts with BCL10/CLAP and activates NF-kappa B. J Biol Chem 275:41082–44186. doi: 10.1074/jbc.C000726200. [DOI] [PubMed] [Google Scholar]
- 21.Zhao XQ, Zhu LL, Chang Q, Jiang C, You Y, Luo T, Jia XM, Lin X. 2014. C-type lectin receptor dectin-3 mediates trehalose 6,6′-dimycolate (TDM)-induced Mincle expression through CARD9/Bcl10/MALT1-dependent nuclear factor (NF)-κB activation. J Biol Chem 289:30052–30062. doi: 10.1074/jbc.M114.588574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saijo S, Ikeda S, Yamabe K, Kakuta S, Ishigame H, Akitsu A, Fujikado N, Kusaka T, Kubo S, Chung SH, Komatsu R, Miura N, Adachi Y, Ohno N, Shibuya K, Yamamoto N, Kawakami K, Yamasaki S, Saito T, Akira S, Iwakura Y. 2010. Dectin-2 recognition of α-mannans and induction of Th17 cell differentiation is essential for host defense against Candida albicans. Immunity 32:681–691. doi: 10.1016/j.immuni.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 23.McGreal EP, Rosas M, Brown GD, Zamze S, Wong SY, Gordon S, Martinez-Pomares L, Taylor PR. 2006. The carbohydrate-recognition domain of dectin-2 is a C-type lectin with specificity for high mannose. Glycobiology 16:422–430. doi: 10.1093/glycob/cwj077. [DOI] [PubMed] [Google Scholar]
- 24.Akahori Y, Miyasaka T, Toyama M, Matsumoto I, Miyahara A, Zong T, Ishii K, Kinjo Y, Miyazaki Y, Saijo S, Iwakura Y, Kawakami K. 2016. Dectin-2-dependent host defense in mice infected with serotype 3 Streptococcus pneumoniae. BMC Immunol 17:1. doi: 10.1186/s12865-015-0139-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mackay F, Loetscher H, Stueber D, Gehr G, Lesslauer W. 1993. Tumor necrosis factor alpha (TNF-alpha)-induced cell adhesion to human endothelial cells is under dominant control of one TNF receptor type, TNF-R55. J Exp Med 177:1277–1286. doi: 10.1084/jem.177.5.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. 2007. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol 7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 27.Dunne JL, Ballantyne CM, Beaudet AL, Ley K. 2002. Control of leukocyte rolling velocity in TNF-α–induced inflammation by LFA-1 and Mac-1. Blood 99:336–341. doi: 10.1182/blood.V99.1.336. [DOI] [PubMed] [Google Scholar]
- 28.Sokol CL, Luster AD. 2015. The chemokine system in innate immunity. Cold Spring Harb Perspect Biol 7:a016303. doi: 10.1101/cshperspect.a016303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feldman C, Anderson R. 2014. Review: current and new generation pneumococcal vaccines. J Infect 69:309–325. doi: 10.1016/j.jinf.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 30.Briles DE, Claflin JL, Schroer K, Forman C. 1981. Mouse IgG3 antibodies are highly protective against infection with Streptococcus pneumoniae. Nature 294:88–90. doi: 10.1038/294088a0. [DOI] [PubMed] [Google Scholar]
- 31.Perlmutter RM, Hansburg D, Briles DE, Nicolotti RA, Davie JM. 1978. Subclass restriction of murine anti-carbohydrate antibodies. J Immunol 121:566–572. [PubMed] [Google Scholar]
- 32.Snapper CM, Rosas F, Moorman MA, Jin L, Shanebeck K, Klinman DM, Kehry MR, Mond JJ, Maliszewski CR. 1996. IFN-gamma is a potent inducer of Ig secretion by sort-purified murine B cells activated through the mIg, but not the CD40, signaling pathway. Int Immunol 8:877–885. doi: 10.1093/intimm/8.6.877. [DOI] [PubMed] [Google Scholar]
- 33.Miyasaka T, Akahori Y, Toyama M, Miyamura N, Ishii K, Saijo S, Iwakura Y, Kinjo Y, Miyazaki Y, Oishi K, Kawakami K. 2013. Dectin-2-dependent NKT cell activation and serotype-specific antibody production in mice immunized with pneumococcal polysaccharide vaccine. PLoS One 8:e78611. doi: 10.1371/journal.pone.0078611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanno D, Yokoyama R, Kawamura K, Kitai K, Yuan X, Ishii K, De Jesus M, Yamamoto H, Sato K, Miyasaka T, Shimura H, Shibata N, Adachi Y, Ohno N, Yamasaki S, Kawakami K. 2019. Dectin-2 mediated signaling triggered by the cell wall polysaccharide of Cryptococcus neoformans. Microbiol Immunol 63:500–512. doi: 10.1111/1348-0421.12746. [DOI] [PubMed] [Google Scholar]
- 35.Drummond RA, Collar AL, Muthulekha S, Rodriguez CA, Lim JK, Mendez LM, Fink DL, Hsu AP, Zhai B, Karauzum H, Mikelis CM, Rose SR, Ferre EMN, Yockey L, Lemberg K, Kuehn HS, Rosenzweig SD, Lin X, Chittiboina P, Datta SK, Belhorn TH, Weimer ET, Hernandez ML, Hohl TM, Kuhns DB, Lionakis MS. 2015. CARD9-dependent neutrophil recruitment protects against fungal invasion of the central nervous system. PLoS Pathog 11:e1005293. doi: 10.1371/journal.ppat.1005293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Drummond RA, Swamydas M, Oikonomou V, Zhai B, Dambuza IM, Schaefer BC, Bohrer AC, Mayer-Barber KD, Lira SA, Iwakura Y, Filler SG, Brown GD, Hube B, Naglik JR, Hohl TM, Lionakis MS. 2019. CARD9+ microglia promote antifungal immunity via IL-1β- and CXCL1-mediated neutrophil recruitment. Nat Immunol 20:559–570. doi: 10.1038/s41590-019-0377-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sibille Y, Reynolds HY. 1990. Macrophages and polymorphonuclear neutrophils in lung defense and injury. Am Rev Respir Dis 141:471–501. doi: 10.1164/ajrccm/141.2.471. [DOI] [PubMed] [Google Scholar]
- 38.Haslett C. 1999. Granulocyte apoptosis and its role in the resolution and control of lung inflammation. Am J Respir Crit Care Med 160:S5–S11. doi: 10.1164/ajrccm.160.supplement_1.4. [DOI] [PubMed] [Google Scholar]
- 39.Tyznik AJ, Tupin E, Nagarajan NA, Her MJ, Benedict CA, Kronenberg M. 2008. Cutting edge: the mechanism of invariant NKT cell responses to viral danger signals. J Immunol 181:4452–4456. doi: 10.4049/jimmunol.181.7.4452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nagarajan NA, Kronenberg M. 2007. Invariant NKT cells amplify the innate immune response to lipopolysaccharide. J Immunol 178:2706–2713. doi: 10.4049/jimmunol.178.5.2706. [DOI] [PubMed] [Google Scholar]
- 41.Yamamoto N, Kerfoot SM, Hutchinson AT, Dela Cruz CS, Nakazawa N, Szczepanik M, Majewska-Szczepanik M, Nazimek K, Ohana N, Bryniarski K, Mori T, Muramatsu M, Kanemitsu K, Askenase PW. 2016. Expression of activation-induced cytidine deaminase enhances the clearance of pneumococcal pneumonia: evidence of a subpopulation of protective anti-pneumococcal B1a cells. Immunology 147:97–113. doi: 10.1111/imm.12544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Behler F, Takano T, Maus R, Stolper J, Jonigk D, Tort Tarrés M, Fuehner T, Prasse A, Welte T, Timmer MS, Stocker BL, Nakanishi Y, Miyamoto T, Yamasaki S, Maus UA. 2016. C-type lectin Mincle recognizes glucosyl-diacylglycerol of Streptococcus pneumoniae and plays a protective role in pneumococcal pneumonia. PLoS Pathog 12:e1006038. doi: 10.1371/journal.ppat.1006038. [DOI] [PMC free article] [PubMed] [Google Scholar]