

Abstract
Brain injury resulting from stroke or trauma can be exacerbated by the release of pro-inflammatory cytokines, proteases, and reactive oxygen species by activated microglia. The microglial activation resulting from brain injury is mediated in part by alarmins, which are signaling molecules released from damaged cells. The nuclear enzyme poly(ADP-ribose) polymerase-1 (PARP-1) has been shown to regulate microglial activation after brain injury, and here we show that signaling effects of the alarmin S100B are regulated by PARP-1. S100B is a protein localized predominantly to astrocytes. Exogenous S100B added to primary microglial cultures induced a rapid change in microglial morphology, upregulation of IL-1β, TNFα and iNOS gene expression, and release of matrix metalloproteinase 9 and nitric oxide. Most, though not all of these effects were attenuated in PARP-1−/− microglia and in wild-type microglia treated with the PARP inhibitor, veliparib. Microglial activation and gene expression changes induced by S100B injected directly into brain were likewise attenuated by PARP-1 inhibition. The anti-inflammatory effects of PARP-1 inhibitors in acutely injured brain may thus be mediated in part through effects on S100B signaling pathways.
Keywords: stroke, trauma, astrocyte, PARP-1, nitric oxide, MMP9, veliparib
Graphical Abstract

INTRODUCTION
Microglia, the primary immune cells in brain, normally exist in a “resting” state with highly branched processes. When activated, they retract these processes and release pro-inflammatory cytokines, reactive oxygen species and proteases (Banati et al. 1993; Ransohoff and Perry 2009). This evolutionarily conserved response is likely a first line of defense against the microbial infections that commonly accompany tissue injury. However, activated microglia can also kill neurons and other neighboring cells (Chao et al. 1992; Kauppinen and Swanson 2005; Lehnardt 2010; Zhang et al. 2003), such that acute microglial activation can have a net deleterious effect in conditions such as closed head trauma and stroke.
Microglial activation in response to several stimuli is regulated by the nuclear enzyme poly(ADP-ribose) polymerase-1 (PARP-1). When activated by DNA damage, cytokine signaling, or other factors, PARP-1 forms branched ADP-ribose chains on target proteins (Gibson and Kraus 2012). These polymers affect interactions between the target proteins and other proteins, and form a scaffold for other protein – protein interactions. PARP inhibitors have potent anti-inflammatory effects (Weltin et al. 1995) and these anti-inflammatory effects can improve outcomes after experimental stroke and brain trauma (d’Avila et al. 2012; Kauppinen et al. 2009; Stoica et al. 2014). The PARP inhibitors include minocycline (Alano et al. 2006), which is now widely used to suppress microglial activation (Chen et al. 2014; Zhang et al. 2003). The mechanism by which PARP-1 inhibitors suppresses inflammatory responses has not been fully resolved, but likely involves regulation of the activity of pro-inflammatory transcription factors (Bai and Virag 2012; Gibson and Kraus 2012; Ha et al. 2002; Hassa and Hottiger 1999; Kauppinen et al. 2013; Kauppinen and Swanson 2005; Martinez-Zamudio and Ha 2014). In addition to PARP-1, mammalian cells also express other PARP isoforms. Though less abundant than PARP-1, these other PARPs may also influence cellular inflammatory responses (Kamboj et al. 2013; Phulwani and Kielian 2008).
Most of what is now known about microglial activation comes from studies in which the bacterial cell wall constituent lipopolysaccharide (LPS) was used as a stimulus; however, LPS usually has no role in the initial brain response to brain injury. Instead, the rapid microglial activation induced by stroke or trauma is thought to result from the actions of alarmins, which are signaling molecules released from cells by tissue damage (Bianchi 2007). Microglial detection and response to alarmins differs in many respects from microglial detection and response to LPS (Adami et al. 2001; Bianchi 2007; Lehnardt 2010). Two alarmins known to be released in brain injury are the high mobility group box 1 protein (HMGB1) and the calcium-binding protein B (S100B). HMGB1 is a non-histone DNA binding protein that is ubiquitously expressed by eukaryotic cells. S100B is expressed most abundantly by astrocytes and is implicated in regulation of astrocyte shape and cytosolic calcium concentration (Hayakata et al. 2004). Both HMBG1 and S100B can induce aspects of microglial activation (Bianchi et al. 2010; Kabadi et al. 2015; Lee et al. 2014).
Given the robust effect of PARP-1 inhibitors on microglial activation in conditions such as stroke and head trauma, where bacterial infection is rarely a factor, the present study aimed to identify alarmins whose effects may be influenced by PARP-1. Our findings show that S100B has potent effects on microglia in vitro and in situ, and that these effects are attenuated by PARP-1 inhibition.
MATERIALS AND METHODS
Animals.
Studies were approved by the San Francisco Veterans Affairs Medical Center animal studies committee and follow the NIH guidelines for humane care of animals. PARP-1−/− mice were initially obtained from the Jackson Laboratory (Bar Harbor, ME) and subsequently bred for more than 10 generations onto the C57BL/6 background. Wild-type C57BL/6 mice were obtained from Simonsen Labs (Gilroy, CA). Studies using adult mice employed males, age 3–4 months. Animals were housed on a 12h light/dark cycle with free access to food and water.
Reagents.
Human recombinant high-mobility group box 1 (HMGB1) and IFN-γ were acquired from Biolegend (San Diego, CA). Mouse recombinant S100 calcium-binding protein B (S100B) was obtained from ProSpec (East Brunswick, NJ). Cell culture reagents were obtained from Invitrogen (Carlsbad, CA) and Corning (Corning, NY). High pure RNA isolation and Transcriptor First Strand cDNA Synthesis kits were acquired from Roche (Basel, Switzerland), and SYBR Green PCR Master Mix from Life technologies (Carlsbad, CA). The antibodies used in this study were as follows: anti Iba-1, Abcam #ab107159 (Cambridge, MA); anti β-actin, Cell Signaling #8H10D10 (Danvers, MA); anti-MMP9 for western blots, EMD Millipore #AB19016 (Hayward, CA); anti MMP9 for immunofluorescence, Cloud-Clone #PAA553Mu02 (Houston, TX); goat anti-rabbit IgG and horse anti-mouse IgG, Vector Laboratories #PI-1000 and #PI-2000 (Burlingame, CA); rabbit anti-goat IgG and donkey anti-rabbit IgG, Thermo Fisher Scientific #A-11080 and #A-21206 (Waltham, MA). Protein concentrators and Griess reagent were procured from Thermo Fisher Scientific (Waltham, MA). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO).
Primary microglia and astrocyte cultures.
Mixed astrocyte-microglia cultures were prepared from whole brains of 1-day old mouse pups as previously described (Kauppinen et al. 2008). After 2–3 weeks, the flasks were gently shaken by hand and the floating microglia were re-plated in MEM supplemented with 2.5% FBS. The microglia cultures were maintained at 37°C in an incubator with a physiological gas mixture (5% CO2, 6% O2 and 89% N2) and used for experiments at days 2–3 after re-plating. To generate astrocyte cultures without microglia, the cultures were treated with 25 μM cytosine β-D-arabinofuranoside for 48 hours when the astrocytes reached confluency.
mRNA quantification.
Total RNA was extracted from cultured cells or mice brain tissue using the High Pure RNA Isolation kit (Roche) and immediately reverse transcribed to cDNA with a cDNA Synthesis Kit (Thermo Fisher). Samples were analyzed in triplicate. The primers were designed according to Pubmed GenBank and synthesized by Eurofins Genomics. The primer sequences were as follows: GAPDH, f: GGGTGTGAACCACGAGAAAT; r: CCTTCCA-CAATGCCAAAGTT; IL-1b, f: CGACAAAATACCTGTGGCCT; r: TTCTTTGGGTATTGCTTGGG; TNF-a, f: TCGTAGCAAACCACCAAGTG; r: TTGTCTTTGAGATCCATGCC; iNOS, f: GTTCTCA -GCCCAACAATACAAGA; r: GTGGACGGGTCGATGTGTCAC. Quantitative real time PCR analysis was performed with an Mx3000P system (San Diego, CA), using SYBR Green to measure double strand DNA content. A dissociation step was added at the end of the PCR to confirm the amplifcation of a single product. The transcript level of each gene was normalized to the GAPDH mRNA in the same sample using the 2 –ΔΔCT method (Livak and Schmittgen 2001).
Microglial activation scoring.
Three randomly selected fields were photographed in each culture well by an observer blinded to the experimental conditions, and the soma diameter was measured in each cell by a second observer. Experiments were performed using triplicate wells in each of 3 independent culture preparations per condition.
Matrix metalloproteinase 9 (MMP9) release.
Culture medium was collected after 24 hours incubation under the designated conditions and concentrated from 6 ml to 50 μl by centrifugation in protein concentrators (Thermo Fisher). The cell layer was lysed with radioimmunoprecipitation assay buffer containing protease inhibitors. The cell lyates and concentrated medium were mixed with loading buffer, incubated at 100°C for 10 min, electrophoresed on 10% SDS-PAGE gels, and transferred onto polyvinylidene difluoride (PVDF) membranes. After incubation in 5% skim milk in Tris-buffered saline with 0.5% Tween 20 (TBST) at room temperature for 2 h, membranes were incubated over night at 4°C with MMP9 antibody at 1:2000 dilution and with β-actin antibody at 1:5000 dilution. The membranes were then washed and incubated in secondary antibodies for 2 hours at room temperature. Bands were visualized using enhanced chemiluminescence imaging.
Nitric oxide production.
Culture medium was collected after 24 hours incubations under the designated conditions and centrifuged to remove cell debris. Samples were combined with Griess reagent to complex nitrite, and OD was measured at 540 nm. Values were calibrated to nitrite standards prepared in MEM.
Needle track injury and S100B intracerebral injection.
S100B was prepared in saline vehicle and injected into the striatum using a stereotaxic injector (Stoelting, Wood Dale, IL) with the animals under isoflurane / nitrous oxide anesthesia. Injection coordinates (relative to the bregma) were 1.0 mm anterior, 1.9 mm lateral and 3.5 mm deep. Needle track injuries were induced with vehicle injections alone.
Brain tissue preparation.
For immunostaining studies, the animals were euthanized 1 or 3 days after injections and perfusion fixed with 4% buffered formaldehyde. Brains were removed, post-fixed in 4% buffered formaldehyde, cyroprotected in 20% sucrose, and stored at −80°C before use. For mRNA analysis, the animals were euthanized 4 hours or 24 hours after S100B injection and perfused with ice-cold saline. The brains were removed, coronally bisected 3 mm posterior to the injection site. The striata were dissected free on ice and stored at −80°C.
Immunostaining.
Coronal brain sections, 40μ thick, were collected from 2 mm anterior to 2 mm posterior to the injection site. Sections were immunostained in parallel and photographed with a confocal microscope under uniform conditions. For the quantification of Iba-1 intensity, measurements were made on 6 evenly spaced sections spanning the injection sites. Photographs were taken 200 μm from the needle track in studies of needle track injury, and at the lateral edge of the striatum (> 2 mm from the needle track) for studies using S100B injection. Iba-1 expression level in each photographed area was calculated by multiplying the area of Iba-1 staining by the intensity of Iba-1 staining, using the NIH ImageJ program as described previously (d’Avila et al. 2012).
Statistical analysis.
Values are presented as means ± s.e.m. The “n” values denote the number of animals or, for cell culture studies, the number of independent experiments. Each independent experiment used cells from different mice and was repeated in triplicate culture wells. Statistical analyses were performed with ANOVA followed by the Tukey–Kramer test where multiple groups were compared to one another, or Dunnett’s test where multiple groups are compared to a common treatment group.
RESULTS
Consistent with prior reports, we found that brain trauma (here produced by needle penetration) induced microglial activation in the surrounding brain tissue and that this response could be suppressed by a PARP-1 inhibitor (Fig. 1). As a first step in evaluating the role of alarmins in this process, we measured pro-inflammatory gene expression changes induced by HMGB1, S100B, or LPS (as a positive control) in cultured microglia. LPS produced a concentration-dependent increase in each of the three mRNA species evaluated; interleukin-1 ß (IL-1b), tumor necrosis factor α (TFNα), and inducible nitric oxide synthase (iNOS) (Fig. 2A). Both HMGB1 and S100B also increased expression of these mRNA species, with S100B being the more potent and effective agent (Fig. 2B,C). The effects of S100B at 200 ng/ml were comparable to those induced by 1 ng/ml LPS, and the effects of 2000 ng/ml S100B were in all cases greater than the maximal effects achieved with LPS (Fig. 2C). This result confirms that the effects of S100B are not caused by LPS contamination of the recombinant protein. As the effects of HMGB1 were much less robust, we focused on S100B for the remainder of the studies and used the 200 ng/ml (~ 10 nM) concentration. S100B in brain is localized primarily to astrocytes (Adami et al. 2001; Donato et al. 2012); consistent with this, microglia exposed to lysed cultured astrocytes responded with changes in gene expression similar to those induced by recombinant S100B (Fig. 2D).
Figure 1.
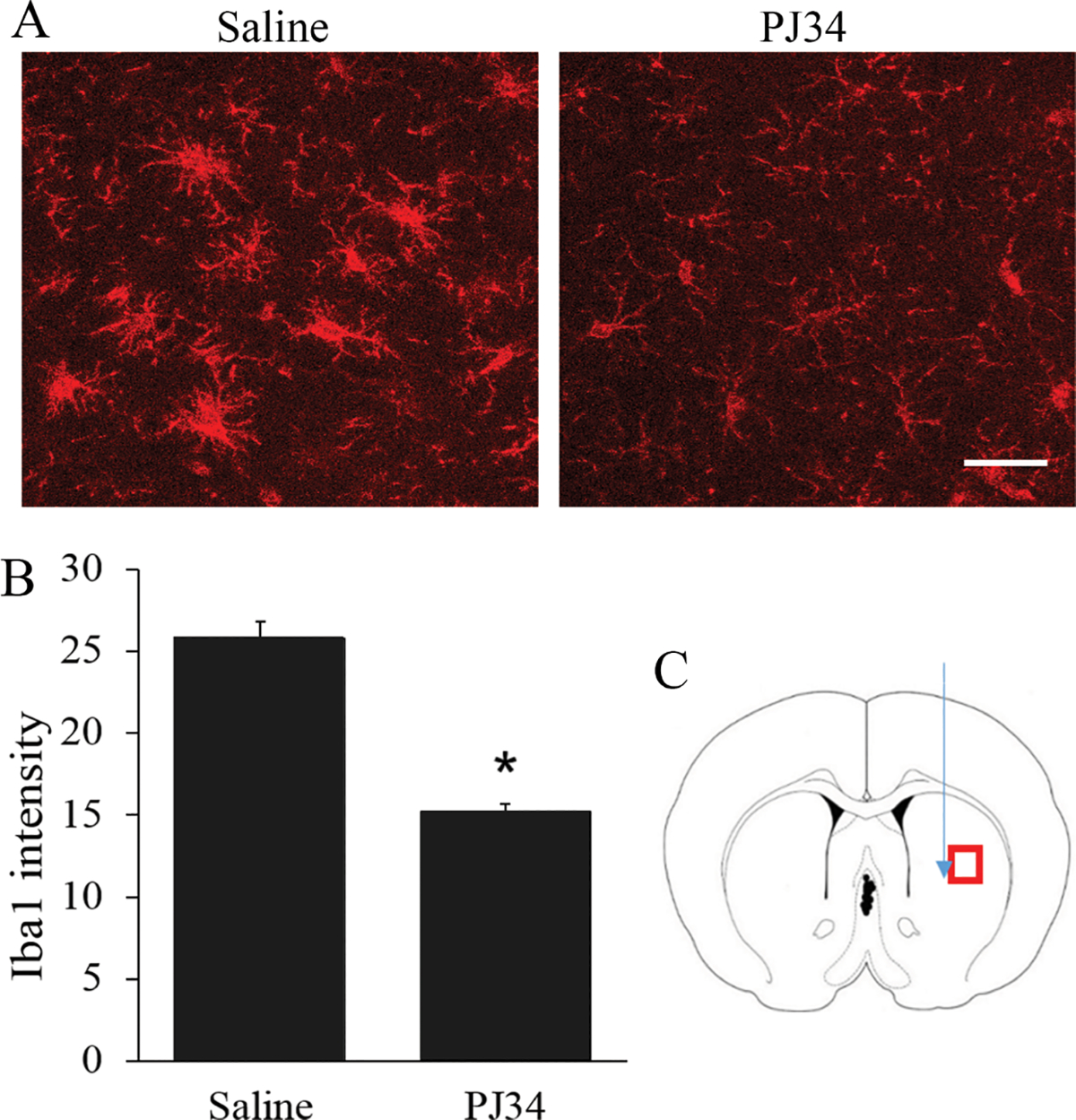
The PARP-1 inhibitor PJ34 suppresses microglial activation induced by trauma. A. Representative photomicrographs of Iba-1 staining adjacent to needle tracks. The microglia show enlarged soma, shortened processes, and increased Iba-1 expression. B. Quantification of Iba-1 immunostaining intensity, n = 3; * P < 0.05. C. Diagram shows the locations of photographed areas (red box) and needle track (green arrow). Scale bar = 50 μm
Figure 2.
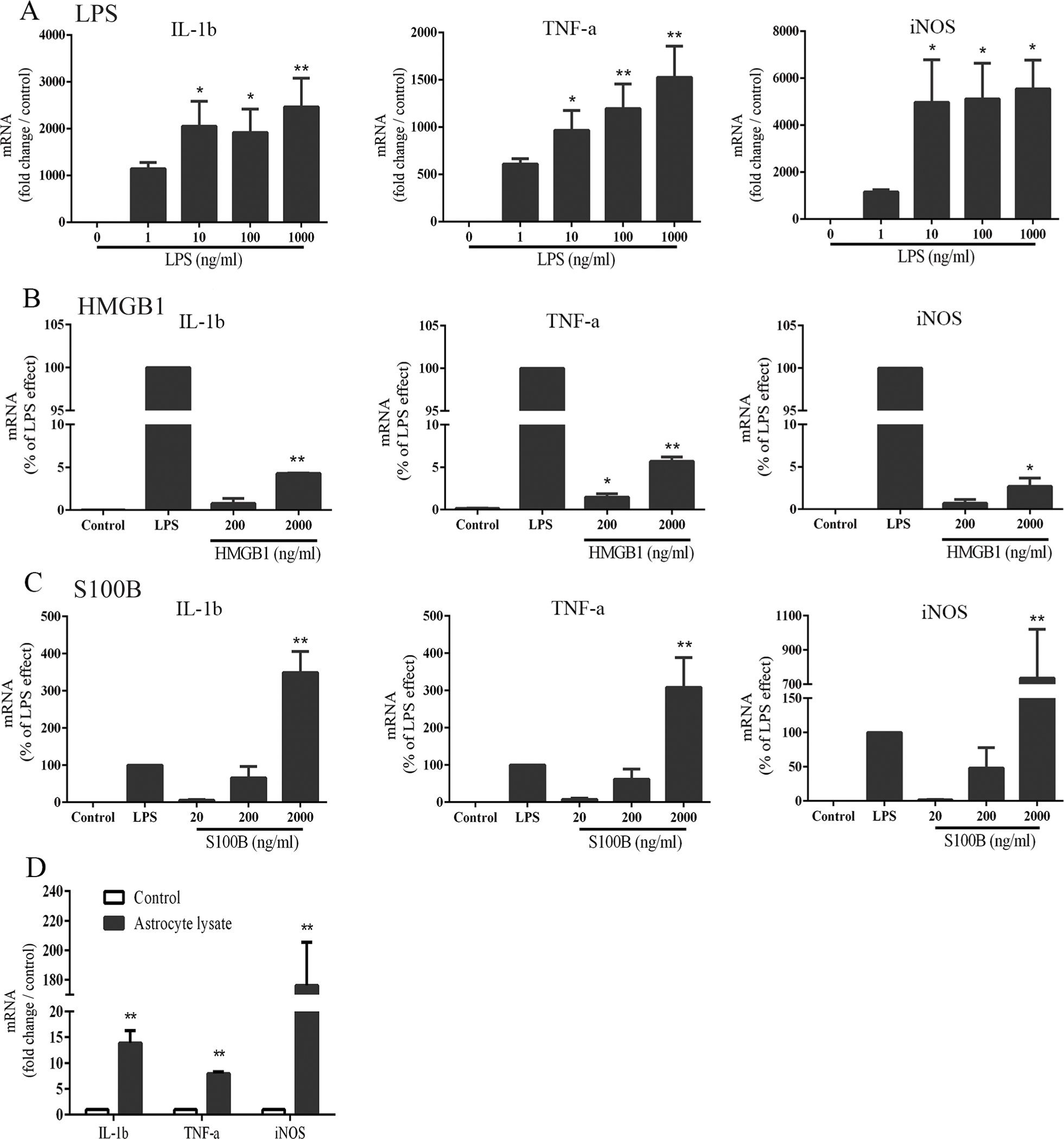
Alarmins up-regulate pro-inflammatory gene expression in cultured microglia. mRNA levels were measured after 4 hour incubations with the designated stimulus. A. Effects of LPS on gene expression. B,C. Effects of HMGB1 and S100B on gene expression, expressed relative to that induced by 1 ng/ml LPS. n ≥ 3; * P < 0.05, ** P < 0.01 vs. control. D. Lysates of astrocyte cultures mimic the effect of S100B. n = 3; ** P < 0.01
PARP inhibition attenuates the gene expression and morphology changes induced by S100B
To evaluate the role of PARP-1 in the gene expression changes induced by S100B, we used both the PARP inhibitor veliparib and microglia from PARP-1−/− mice. Veliparib reduced, but did not eliminate, the effect of S100B on IL-1β, TNFα, and iNOS gene expression (Fig 3A). A very similar pattern was observed in PARP-1−/− microglia (Fig. 3B). Microglia exposed to S100B also underwent process retraction and soma enlargement, characteristic of an activated morphology, in a manner and rate similar to that induced by LPS (Fig. 4A,B). These changes, like the gene expression changes, were attenuated but not eliminated by both veliparib and in PARP-1−/− microglia (Fig. 4C,D)
Figure 3.
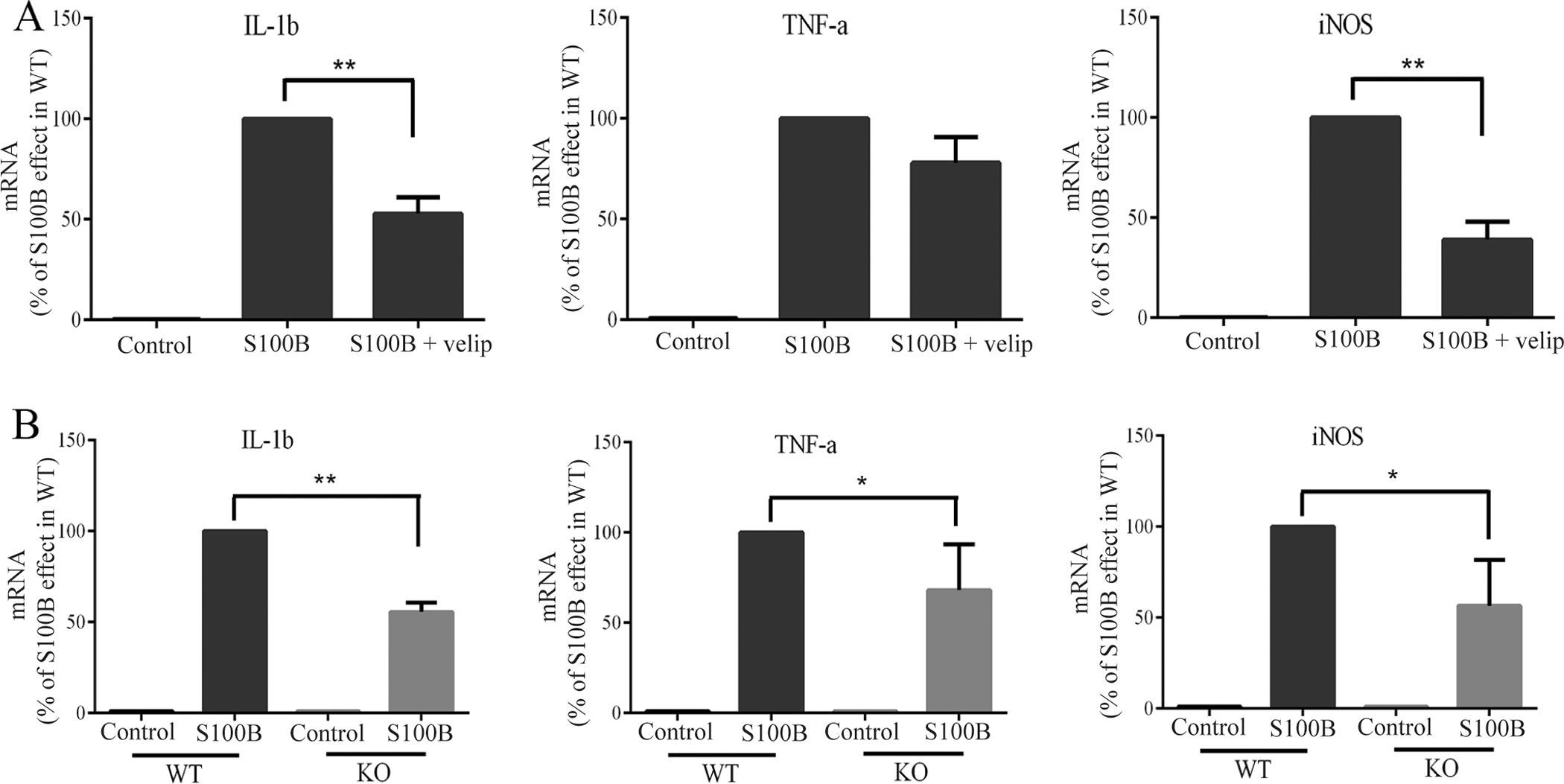
PARP-1 inhibition attenuates the pro-inflammatory gene expression changes induced by S100B. A. Effects of the PARP inhibitor veliparib (5 μM) on gene expression changes induced by 200 ng/ml S100B. B. Effects of PARP-1−/− genotype on gene expression changes induced by 200 ng/ml S100B. n = 3, *P < 0.05, **P < 0.01.
Figure 4.
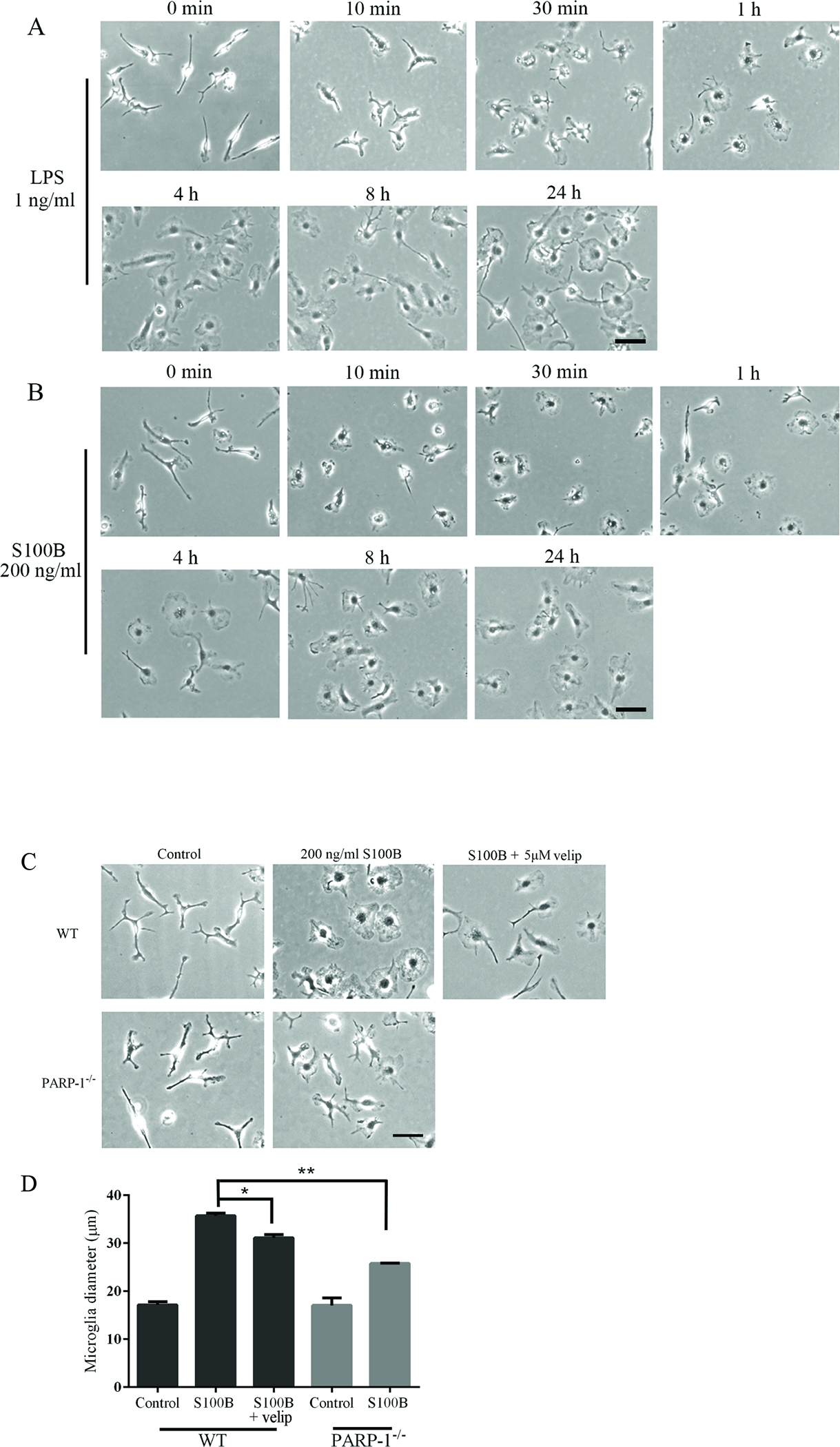
PARP-1 inhibition attenuates the microglial morphology changes induced by S100B. Phase contrast micrographs show microglia after incubation with LPS (A) or S100B (B). Representative of 3 experiments; scale bar = 50 μm. The effects of S100B were attenuated by the PARP inhibitor veliparib and in PARP-1−/− microglia (C,D). n = 3, *P < 0.05, **P < 0.01.
Effects of PARP-1 inhibition on MMP9 and nitric oxide release
Microglial release of matrix metalloproteinase-9 (MMP9) and nitric oxide release can have cytotoxic effects may thus directly contribute to secondary injury after brain trauma. Microglia were incubated with S100B for 24 hours exhibited a roughly 2-fold increase in MMP9 release and 10-fold increase in nitric oxide release (as assessed by nitrite accumulation). Both of these increases were strikingly attenuated in PARP-1−/− cells (Fig. 5B,D), though not significantly reduced by veliparib (Fig. 5A,C). Note that for the studies of nitric oxide release, S100B was co-incubated with 20 ng/ml IFNγ. S100B alone and IFNγ alone had no measurable effect (not shown). LPS likewise required co-incubation with IFNγ to induce NO release. This is consistent with prior reports (Bianchi et al. 2010; Kabadi et al. 2015), and may reflect a “priming” effect of factors present in situ but not present in the cell culture system.
Figure 5.
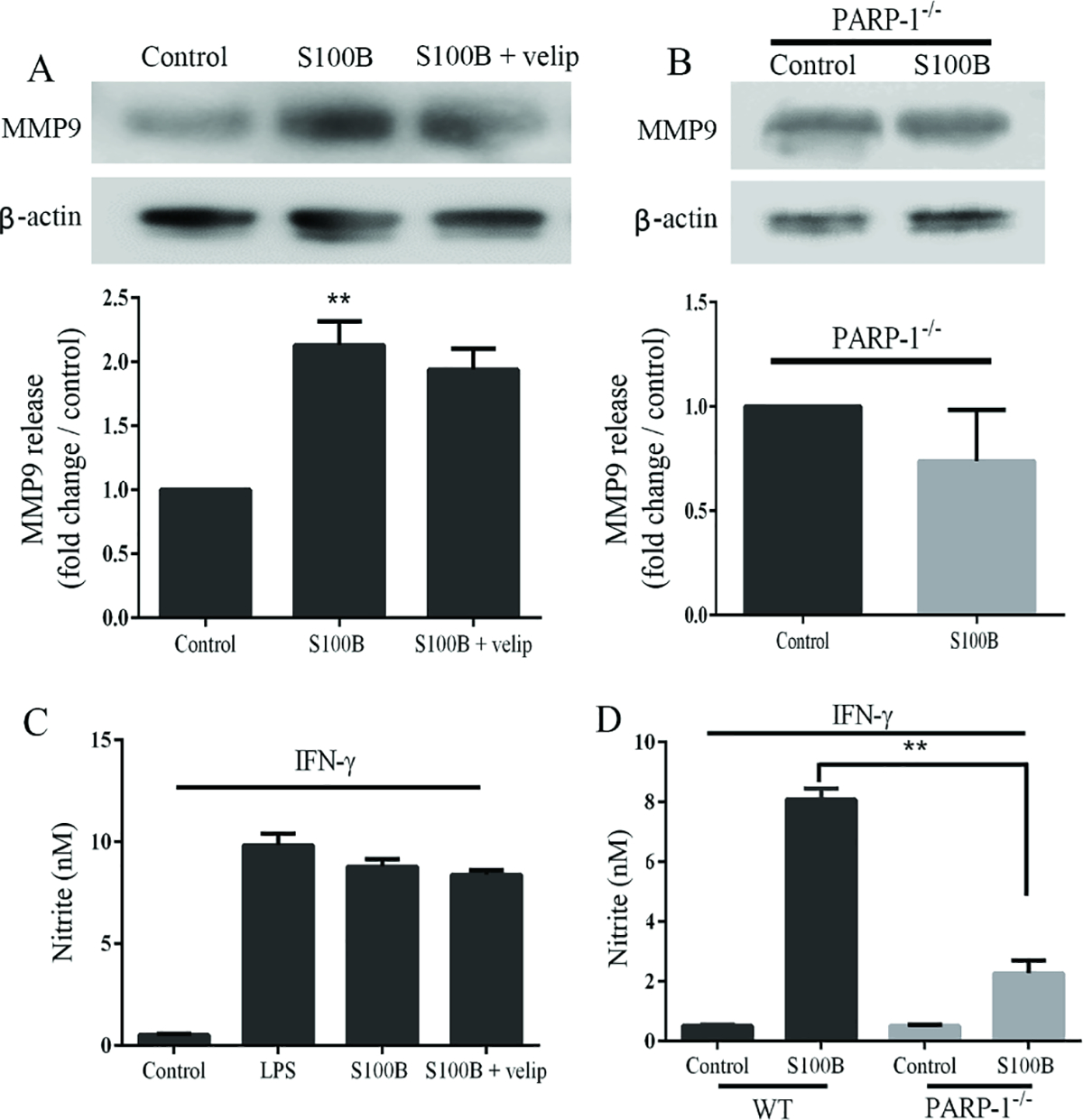
Effects of PARP-1 inhibition on S100B-induced nitric oxide (NO) and matrix metalloproteinase-9 (MMP9) release. A. Representative MMP9 immunoblots prepared from the medium of microglial cultures treated for 24 hours with 200 ng/ml S100B, ± 5 μM veliparib. B. MMP9 immunoblots prepared from the medium of PARP-1−/− microglial cultures treated for 24 hours with 200 ng/ml S100B. C. Nitrite concentrations in medium of microglial cultures treated for 24 hours with 200 ng/ml S100B or 1 ng / ml LPS ± 5 μM veliparib. D. Nitrite concentrations in medium of PARP-1−/− microglial cultures treated for 24 hours with 200 ng/ml S100B. Cultures in (C) and (D) were also treated with 20 ng/ml IFN-γ. For all panels n = 3, ** P < 0.01.
PARP-1 inhibition attenuates S100B-induced microglial activation in vivo
S100B injected directly into brain (striatum) induced a robust change in microglial morphology and increase in Iba-1 expression throughout the striatum (Fig. 6). These microglial changes were attenuated both in PARP-1−/− mice, and in wild-type mice treated with veliparib. S100B injections also produced a robust increase in MMP9 immunoreactivity in brain, which was reduced in PARP-1−/− mice (Fig. 6F). The MMP9 signal was not exclusively co-localized with Iba-1, likely reflecting the fact that this is a secreted protein. We also measured pro-inflammatory mRNA levels in wild-type and PARP-1−/− mice after S100B injection. The increases (relative to saline injections) were relatively small at 4 hours after S100B injections, but much larger at 24 hours (Fig. 7). The increases of all three mRNA species examined were attenuated in the PARP-1−/− mice.
Figure 6.
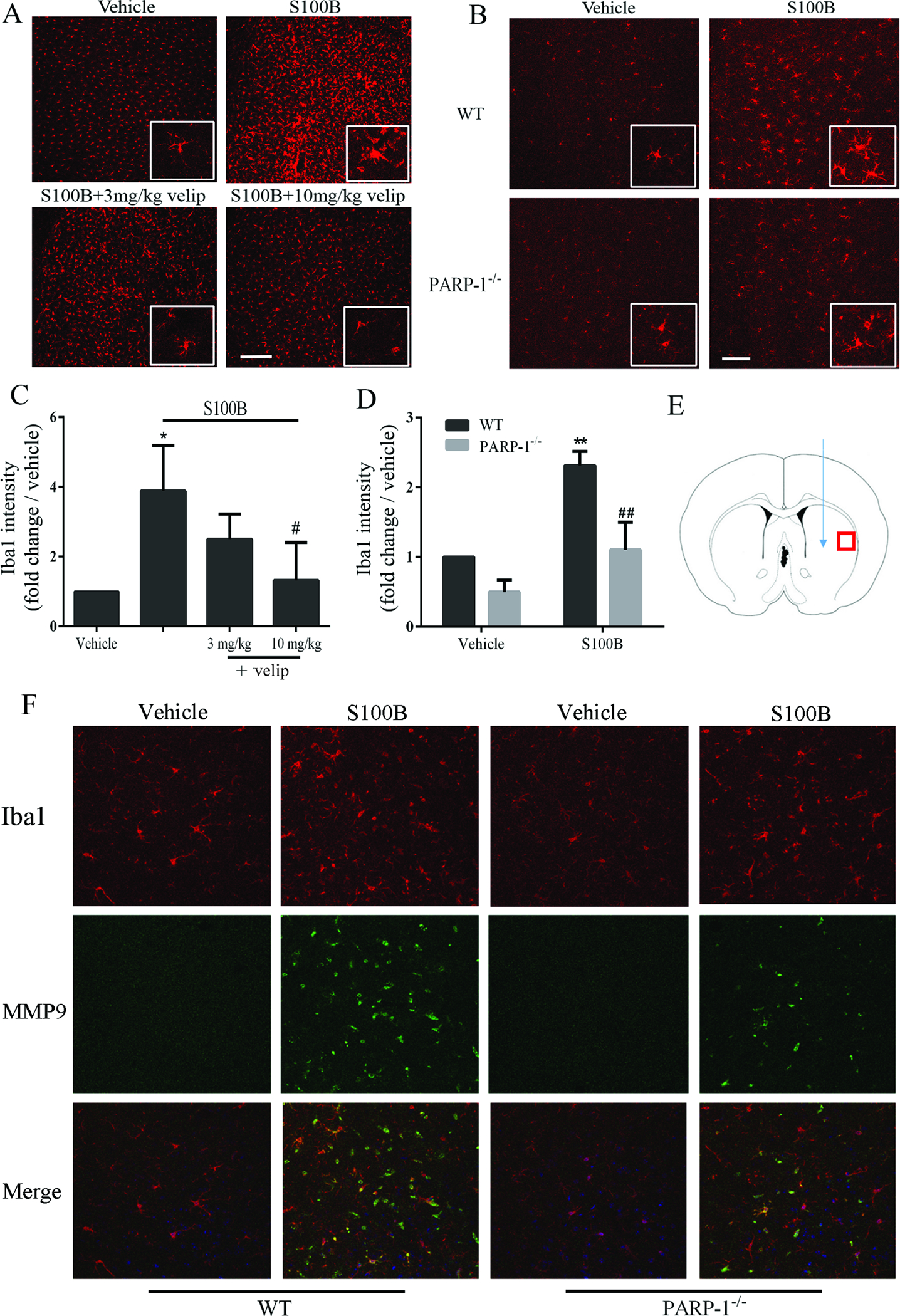
Microglial activation and MMP9 production induced by S100B in situ is blocked by PARP-1 inhibition. A. Iba-1 immunostaining in striatum 3 days after injection with 0.5 μg S100B or saline vehicle and subsequent daily i.p. injection with veliparib or saline vehicle by. Scale bar = 200 μm. Insert boxes are magnified 4-fold to show cell morphology. B. Iba-1 immunostaining in brain sections from wild-type and PARP-1−/− mice. The mice received 0.5 μg/ml intrastriatal injections of S100B, and brain sections were prepared 24 hours later. Scale bar = 100 μm. C, D. Quantification of Iba-1 immunostaining intensity. n = 3, *P < 0.05 vs. vehicle, # P < 0.05 vs. S100B. E. Diagram shows the relative locations of S100B injection (green arrow) and photographs (red box). Note that the distance between photographic field and injection site iss much greater here than in the study using needle track injury alone as the stimulus (Fig. 1C). F. Confocal micrographs show immunostaining for MMP9 (green) show that the increase observed 24 hours after injection of S100B into striatum was attenuated in PARP-1−/− mice. Scale bar = 20 μm; representative of n = 3.
Figure 7.
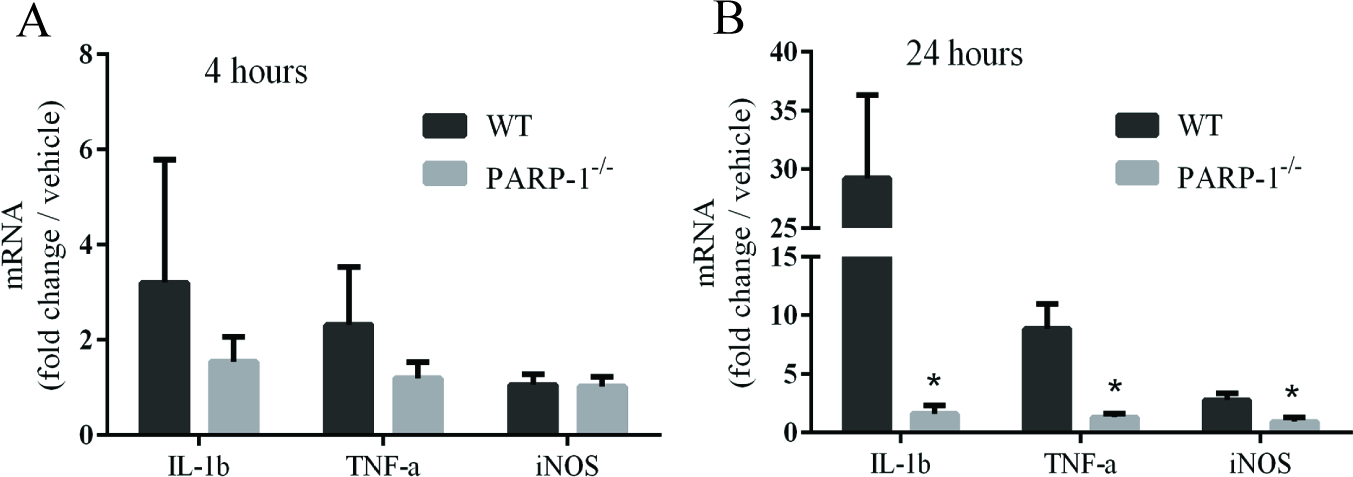
Relative changes in mRNA levels in mouse striatum harvested 4 hours (A) and 24 hours (B) after S100B injections with 0.5 μg S100B. Note differences in Y-axis scales. Data are expressed relative to vehicle-injected brains. n = 3–4, *P < 0.05.
DISCUSSION
Our results show that S100B is a potent and effective stimulus for microglial activation. In primary microglial cultures, S100B induced a rapid change in microglial morphology, upregulation of IL-1β, TNFα and iNOS gene expression, and stimulated release of MMP9 and nitric oxide. Most, though not all of these effects were markedly attenuated in both PARP-1−/− microglia and in wild-type microglia treated with the PARP inhibitor veliparib. S100B injected directly into brain similarly induced microglial morphology changes and upregulated pro-inflammatory gene expression, and these effects were likewise attenuated in PARP-1−/− mice.
Recombinant S100B added to microglial cultures increased pro-inflammatory gene expression in a dose-dependent manner. LPS, used as a positive control, also increased pro-inflammatory gene expression and did so more potently (at lower concentrations) than S100B; however, the maximal effect of S100B on the expression of all three pro-inflammatory genes far exceeded the maximal effect achieved by LPS. These observations confirmed that the effects of S100B were not simply due to LPS contamination of the recombinant S100B protein, and showed that the capacity for S100B to induce microglial reaction is greater than that of LPS. For most cell culture studies we used an S100B concentration that produced roughly 50% of the maximal effect on these gene expression changes (200 ng/ml; ~ 10 nM). The actual extracellular concentrations of S100B after brain injury are unknown, but measured S100B concentrations in human CSF after severe brain trauma are well above this range (Brandner et al. 2013; Hayakata et al. 2004).
MMP9 and nitric oxide can both be cytotoxic to neighboring cells, and may thereby contribute to secondary injury after brain trauma and stroke. Our findings also showed that S100B induces MMP9 and nitric oxide release from primary microglia, and that this effect is blocked in PARP-1−/− cells. Moreover, direct injection of S100B into wild-type mouse brains activated microglial morphology, upregulated of pro-inflammatory gene expression, and increased MMP9 release, all of which were attenuated in PARP-1−/− mice.
Bianchi and colleagues (Bianchi et al. 2010) previously showed that S100B stimulates cell migration and upregulates chemokine expression in the BV2 cell line. Our results are consistent with these findings, but were obtained with substantially lower S100B concentrations. This may because our studies used primary microglia rather than BV2 cells, or because we took other measures, including culture at a physiological (6%) oxygen tension, to limit the baseline level of cell activation.
Cells from PARP-1−/− mice have no known deficit in other PARP family member expression, but could have other developmental changes in response to PARP-1 deficiency. PARP inhibitors and PARP-1 gene deletion may also have differing effects on cell physiology (Shen et al. 2015). The use of a PARP inhibitor and PARP-1−/− cells in the present studies thus provide complementary outcome measures. The PARP inhibitor veliparib is currently being used in phase III clinical trials as a cancer therapeutic, in part because it has excellent brain penetration and long functional half-life (Li et al. 2011; Wagner 2015). Veliparib, does, however, also inhibit of other PARP isoforms (Passeri et al. 2015). The gene expression changes induced by S100B in our studies were attenuated similarly by both veliparib and PARP-1−/− gene deficiency, but the effect of veliparib on nitric oxide release and MMP9 release was much less than observed with PARP-1−/− cells. Though our findings do not identify the reason for this difference, the long (24 hour) incubations required for these experiments may affect veliparib stability or permit development of secondary autocrine influences.
The effects of PARP-1 and S100B on inflammatory responses are likely both mediated through effects on pro-inflammatory transcription factors. PARP-1 may promote transcription factor activity, either by direct poly(ADP-ribosyl)ation or by indirect effects mediated by changes in chromatin structure (Martinez-Zamudio and Ha 2014), poly(ADP-ribose) polymer signaling in the extracellular space (Krukenberg et al. 2015), or release of HMGB1 into the extracellular space (Ditsworth et al. 2007). S100B signaling similarly promotes the activity of pro-inflammatory transcription factors, and evidence suggests that this occurs through activation of Receptors for Advanced Glycation End-products (RAGE) (Bianchi et al. 2010; Donato et al. 2012, but see also Kabadi et al. 2015). More indirect S100B pro-inflammatory effects are also possible, e.g. by S100B induction of chemokine release from microglia or astrocytes and subsequent triggering of pro-inflammatory cascades (Bianchi et al. 2010; Villarreal et al. 2014). Extracellular receptor kinase (ERK) is activated by RAGE ligands (Ott et al. 2014), and is also a regulator of PARP-1 activity (Kauppinen et al. 2006; Vuong et al. 2016), thus providing one potential site of direct crosstalk between the PARP-1 and S100B signaling pathways.
Though much of what is known about microglial activation comes from studies using LPS as a stimulus, this is rarely a significant factor in stroke or brain trauma. The present work identifies several microglial responses to the endogenous alarmin S100B, including release of the cytotoxic protease MMP9, and shows that these effects are modulated by PARP-1. The findings thus identify cytotoxic S100B-induced microglial responses as a target amenable to PARP inhibition.
Main points.
The astrocyte-derived alarmin, S100B, is a potent and effective stimulus for microglial activation.
This effect of S100B is mediated in part by poly(ADP-ribose) polymerase-1 (PARP-1)
ACKNOWLEDGEMENTS
This work was supported by the U.S. Department of Veterans Affairs (RAS), the National Institutes of Health (NS041421; RAS) and the China Scholarship Council (JX). We thank Colleen Hefner for excellent technical assistance.
REFERENCES
- Adami C, Sorci G, Blasi E, Agneletti AL, Bistoni F, Donato R. 2001. S100B expression in and effects on microglia. Glia 33(2):131–42. [PubMed] [Google Scholar]
- Alano CC, Kauppinen TM, Valls AV, Swanson RA. 2006. Minocycline inhibits poly(ADP-ribose) polymerase-1 at nanomolar concentrations. Proc Natl Acad Sci U S A 103(25):9685–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai P, Virag L. 2012. Role of poly(ADP-ribose) polymerases in the regulation of inflammatory processes. FEBS Lett 586(21):3771–7. [DOI] [PubMed] [Google Scholar]
- Banati RB, Gehrmann J, Schubert P, Kreutzberg GW. 1993. Cytotoxicity of microglia. Glia 7(1):111–8. [DOI] [PubMed] [Google Scholar]
- Bianchi ME. 2007. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol 81(1):1–5. [DOI] [PubMed] [Google Scholar]
- Bianchi R, Giambanco I, Donato R. 2010. S100B/RAGE-dependent activation of microglia via NF-kappaB and AP-1 Co-regulation of COX-2 expression by S100B, IL-1beta and TNF-alpha. Neurobiol Aging 31(4):665–77. [DOI] [PubMed] [Google Scholar]
- Brandner S, Thaler C, Buchfelder M, Kleindienst A. 2013. Brain-derived protein concentrations in the cerebrospinal fluid: contribution of trauma resulting from ventricular drain insertion. J Neurotrauma 30(13):1205–10. [DOI] [PubMed] [Google Scholar]
- Chao CC, Hu S, Molitor TW, Shaskan EG, Peterson PK. 1992. Activated microglia mediate neuronal cell injury via a nitric oxide mechanism. J Immunol 149(8):2736–41. [PubMed] [Google Scholar]
- Chen Y, Won SJ, Xu Y, Swanson RA. 2014. Targeting Microglial Activation in Stroke Therapy: Pharmacological Tools and Gender Effects. Curr Med Chem 21(19):2146–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d’Avila JC, Lam TI, Bingham D, Shi J, Won SJ, Kauppinen TM, Massa S, Liu J, Swanson RA. 2012. Microglial activation induced by brain trauma is suppressed by post-injury treatment with a PARP inhibitor. J Neuroinflammation 9:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditsworth D, Zong WX, Thompson CB. 2007. Activation of poly(ADP)-ribose polymerase (PARP-1) induces release of the pro-inflammatory mediator HMGB1 from the nucleus. J Biol Chem 282(24):17845–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato R, Cannon BR, Sorci G, Riuzzi F, Hsu K, Weber DJ, Geczy CL. 2012. Functions of S100 proteins. Curr Mol Med 13(1):24–57. [PMC free article] [PubMed] [Google Scholar]
- Gibson BA, Kraus WL. 2012. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol 13(7):411–24. [DOI] [PubMed] [Google Scholar]
- Ha HC, Hester LD, Snyder SH. 2002. Poly(ADP-ribose) polymerase-1 dependence of stress-induced transcription factors and associated gene expression in glia. Proc Natl Acad Sci U S A 99(5):3270–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassa PO, Hottiger MO. 1999. A role of poly (ADP-ribose) polymerase in NF-kappaB transcriptional activation. Biol Chem 380(7–8):953–9. [DOI] [PubMed] [Google Scholar]
- Hayakata T, Shiozaki T, Tasaki O, Ikegawa H, Inoue Y, Toshiyuki F, Hosotubo H, Kieko F, Yamashita T, Tanaka H and others. 2004. Changes in CSF S100B and cytokine concentrations in early-phase severe traumatic brain injury. Shock 22(2):102–7. [DOI] [PubMed] [Google Scholar]
- Kabadi SV, Stoica BA, Zimmer DB, Afanador L, Duffy KB, Loane DJ, Faden AI. 2015. S100B inhibition reduces behavioral and pathologic changes in experimental traumatic brain injury. J Cereb Blood Flow Metab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamboj A, Lu P, Cossoy MB, Stobart JL, Dolhun BA, Kauppinen TM, de Murcia G, Anderson CM. 2013. Poly(ADP-ribose) polymerase 2 contributes to neuroinflammation and neurological dysfunction in mouse experimental autoimmune encephalomyelitis. J Neuroinflammation 10:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppinen TM, Chan WY, Suh SW, Wiggins AK, Huang EJ, Swanson RA. 2006. Direct phosphorylation and regulation of poly(ADP-ribose) polymerase-1 by extracellular signal-regulated kinases 1/2. Proc Natl Acad Sci U S A 103(18):7136–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppinen TM, Gan L, Swanson RA. 2013. Poly(ADP-ribose) polymerase-1-induced NAD depletion promotes nuclear factor-kappaB transcriptional activity by preventing p65 de-acetylation. Biochim Biophys Acta 1833(8):1985–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppinen TM, Higashi Y, Suh SW, Escartin C, Nagasawa K, Swanson RA. 2008. Zinc triggers microglial activation. J Neurosci 28(22):5827–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppinen TM, Suh SW, Berman AE, Hamby AM, Swanson RA. 2009. Inhibition of poly(ADP-ribose) polymerase suppresses inflammation and promotes recovery after ischemic injury. J Cereb Blood Flow Metab 29(4):820–9. [DOI] [PubMed] [Google Scholar]
- Kauppinen TM, Swanson RA. 2005. Poly(ADP-ribose) polymerase-1 promotes microglial activation, proliferation, and matrix metalloproteinase-9-mediated neuron death. J Immunol 174(4):2288–96. [DOI] [PubMed] [Google Scholar]
- Krukenberg KA, Kim S, Tan ES, Maliga Z, Mitchison TJ. 2015. Extracellular poly(ADP-ribose) is a pro-inflammatory signal for macrophages. Chem Biol 22(4):446–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Nam Y, Koo JY, Lim D, Park J, Ock J, Kim J, Suk K, Park SB. 2014. A small molecule binding HMGB1 and HMGB2 inhibits microglia-mediated neuroinflammation. Nat Chem Biol 10(12):1055–60. [DOI] [PubMed] [Google Scholar]
- Lehnardt S 2010. Innate immunity and neuroinflammation in the CNS: the role of microglia in Toll-like receptor-mediated neuronal injury. Glia 58(3):253–63. [DOI] [PubMed] [Google Scholar]
- Li X, Delzer J, Voorman R, de Morais SM, Lao Y. 2011. Disposition and drug-drug interaction potential of veliparib (ABT-888), a novel and potent inhibitor of poly(ADP-ribose) polymerase. Drug Metab Dispos 39(7):1161–9. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25(4):402–8. [DOI] [PubMed] [Google Scholar]
- Martinez-Zamudio RI, Ha HC. 2014. PARP1 enhances inflammatory cytokine expression by alteration of promoter chromatin structure in microglia. Brain Behav 4(4):552–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott C, Jacobs K, Haucke E, Navarrete Santos A, Grune T, Simm A. 2014. Role of advanced glycation end products in cellular signaling. Redox Biol 2:411–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passeri D, Camaioni E, Liscio P, Sabbatini P, Ferri M, Carotti A, Giacche N, Pellicciari R, Gioiello A, Macchiarulo A. 2015. Concepts and Molecular Aspects in the Polypharmacology of PARP-1 Inhibitors. ChemMedChem. [DOI] [PubMed] [Google Scholar]
- Phulwani NK, Kielian T. 2008. Poly (ADP-ribose) polymerases (PARPs) 1–3 regulate astrocyte activation. J Neurochem 106(2):578–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM, Perry VH. 2009. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol 27:119–45. [DOI] [PubMed] [Google Scholar]
- Shen Y, Aoyagi-Scharber M, Wang B. 2015. Trapping Poly(ADP-Ribose) Polymerase. J Pharmacol Exp Ther 353(3):446–57. [DOI] [PubMed] [Google Scholar]
- Stoica BA, Loane DJ, Zhao Z, Kabadi SV, Hanscom M, Byrnes KR, Faden AI. 2014. PARP-1 inhibition attenuates neuronal loss, microglia activation and neurological deficits after traumatic brain injury. J Neurotrauma 31(8):758–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarreal A, Seoane R, Gonzalez Torres A, Rosciszewski G, Angelo MF, Rossi A, Barker PA, Ramos AJ. 2014. S100B protein activates a RAGE-dependent autocrine loop in astrocytes: implications for its role in the propagation of reactive gliosis. J Neurochem 131(2):190–205. [DOI] [PubMed] [Google Scholar]
- Vuong B, Hogan-Cann ADJ, Alano CC, Stevenson M, Chan WY, Anderson CM, Swanson RA, Kauppinen TM. 2016. NF-κB transcriptional activation by TNFα requires phospholipase C, extracellular signal-regulated kinase 2 and poly(ADP-ribose) polymerase-1. Journal of Neuroinflammation 12:229–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner LM. 2015. Profile of veliparib and its potential in the treatment of solid tumors. Onco Targets Ther 8:1931–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weltin D, Picard V, Aupeix K, Varin M, Oth D, Marchal J, Dufour P, Bischoff P. 1995. Immunosuppressive activities of 6(5H)-phenanthridinone, a new poly(ADP-ribose)polymerase inhibitor. Int J Immunopharmacol 17(4):265–71. [DOI] [PubMed] [Google Scholar]
- Zhang SC, Goetz BD, Duncan ID. 2003. Suppression of activated microglia promotes survival and function of transplanted oligodendroglial progenitors. Glia 41(2):191–8. [DOI] [PubMed] [Google Scholar]