

ABSTRACT
Intestinal epithelial cell endoplasmic reticulum (ER) stress has been implicated in intestinal inflammation. It remains unclear whether ER stress is an initiator of or a response to inflammation. Winnie mice, carrying a Muc2 gene mutation resulting in intestinal goblet cell ER stress, develop spontaneous colitis with a depleted mucus barrier and increased bacterial translocation. This study aims to determine whether the microbiota was required for the development of Winnie colitis, and whether protein misfolding itself can initiate inflammation directly in absence of the microbiota. To assess the role of microbiota in driving Winnie colitis, WT and Winnie mice on the same background were rederived into the germ-free facility and housed in the Trexler-type soft-sided isolators. The colitis phenotype of these mice was assessed and compared to WT and Winnie mice housed within a specific pathogen-free facility. We found that Winnie colitis was substantially reduced but not abolished under germ-free conditions. Expression of inflammatory cytokine genes was reduced but several chemokines remained elevated in absence of microbiota. Concomitantly, ER stress was also diminished, although mucin misfolding persisted. RNA-Seq revealed that Winnie differentiated colon organoids have decreased expression of the negative regulators of the inflammatory response compared to WT. This data along with the increase in Mip2a chemokine expression, suggests that the epithelial cells in the Winnie mice are more responsive to stimuli. Moreover, the data demonstrate that intestinal epithelial intrinsic protein misfolding can prime an inflammatory response without initiating the unfolded protein response in the absence of the microbiota. However, the microbiota is necessary for the amplification of colitis in Winnie mice. Genetic predisposition to mucin misfolding in secretory cells initiates mild inflammatory signals. However, the inflammatory signal sets a forward-feeding cycle establishing progressive inflammation in the presence of microbiota.
Abbreviations: Endoplasmic Reticulum: ER; Mucin-2: Muc-2; GF: Germ-Free; Inflammatory Bowel Disease: IBD
KEYWORDS: Microbiota, epithelial cells, Germ free, Inflammation, mucin
Introduction
Inflammatory bowel diseases (IBD), including Crohn’s disease (CD) and ulcerative colitis (UC), are chronic inflammatory disorders with multiple contributing factors. Accumulating evidence demonstrates that dysregulation or altered balance between composition of the intestinal microbiota, intestinal epithelial integrity, and mucosal immune responses can trigger IBD. Among all three compartments, epithelial integrity is particularly important as it is a primary target of inflammation and also contributes to the initiation of inflammation.1 Studies in mouse models of UC highlight the importance of the protective mucus barrier and the role of mucin-producing goblet cells in regulating barrier function. Defects in mucin glycosylation and the major intestinal mucin Muc2 result in increased experimental colitis severity.2–5 Winnie mice with single missense mutation in the Muc2 mucin gene have depleted goblet cells and a diminished mucus layer with increased epithelial permeability, and intestinal inflammation akin to ulcerative colitis.6 Winnie colitis is characterized by a TH17 immune phenotype involving both innate and adaptive immunity.7 This mouse model provides important evidence that protein misfolding in secretory cells and associated endoplasmic reticulum (ER) stress and can be a primary trigger of colitis. ER stress and the unfolded protein response (UPR) that alleviates protein misfolding and restores ER homeostasis, can also drive intestinal inflammation.8–10 This potentially sets up a forward feeding cycle of ER stress and inflammation because the UPR leads to immune activation via multiple pathways and, in turn, inflammatory cytokines, and reactive oxygen species (ROS) produced by immune cells exacerbate ER stress (these relationships are reviewed in11). Importantly, Winnie mice respond to anti-inflammatory drugs that are used clinically to treat patients with UC, including glucocorticoids12 and thiopurines.13 In both cases, suppression of Winnie colitis is accompanied by reduced ER stress and UPR activation, less Muc2 misfolding, restoration of mature mucin production, and amelioration of goblet cell depletion. While glucocorticoids have a co-contributing direct effect on goblet cell oxidative and ER stress, thiopurines had no direct effects on secretory cell ER stress. These data are consistent with inflammation exacerbating Muc2 misfolding and ER stress.
Intestinal microbiota play an essential role in regulating epithelial function and maintaining intestinal homeostasis, however in most settings microbes are required for intestinal inflammation. Mice carrying genetic defects predisposing to colitis that are born and raised in a germ-free (GF) environment without exposure to any live microbes generally fail to develop intestinal inflammation.14 Studies in GF mice have also shown that microbiota-dependent signals regulate mucus production by goblet cells and epithelial integrity.1,15 Consistent with the requirement for microbes to trigger inflammation, upon DSS challenge, GF mice only had minimal inflammatory response, but somewhat paradoxically these mice suffered enhanced epithelial injury and mortality as a consequence of a weakened intestinal barrier and direct effects of DSS on the epithelium.16
Although both secretory cell ER stress and gut microbiota dysregulation can initiate inflammation, the relative contribution of these two parameters in driving colitis progression remains unknown. It is also unclear whether protein misfolding associated ER stress itself can directly trigger inflammation in the absence of microbiota, and whether inflammation influences the degree of ER stress experienced by epithelial cells, particularly in the cells that undertake substantial biosynthesis of proteins in the secretory pathway. In this study, we have used the Winnie spontaneous colitis model to address these questions demonstrating that in the absence of the microbiota, epithelial intrinsic protein misfolding initiates inflammation via downregulation of the anti-inflammatory regulators. Gut microbiota is required to fully activate and exacerbate inflammation however, protein misfolding still persists in the absence of microbes.
Results
Reduced histological colitis in Winnie mice raised under germ-free (GF) conditions
To determine the role of the microbiota in the development of intestinal inflammation, we re-derived male Winnie mice into a GF facility and compared them to male Winnie mice housed in a conventional specific-pathogen-free facility. Age-matched male GF C57BL/6 (WT) mice from the same facilities were used as controls. 6–8 week old GF Winnie mice had lower body weight than conventional Winnie mice or GF WT or conventional WT mice (Figure 1a). Colon weight and diarrhea scores of GF Winnie were reduced compared to Winnie mice, suggesting Winnie spontaneous colitis is substantially reduced in the absence of the microbiota (Figure 1b,c). No major changes were observed with microbiota depletion in the histological appearance of the small intestine and proximal colon of Winnie mice where the inflammation is mild (Figure 1e-f). However, while the absence of microbiota did not alter the colonic histology of WT mice, it substantially reduced histological colitis in the mid and distal colonic regions of Winnie mice where colitis is most severe (Figure 1e-f). Reduced crypt length in all regions of colon (Figure 1d), improved crypt structure and decreased inflammatory cell infiltration (Figure 1e) contributed to reduced histological scores in GF Winnie compared to Winnie mice (Figure 1f).
Figure 1.
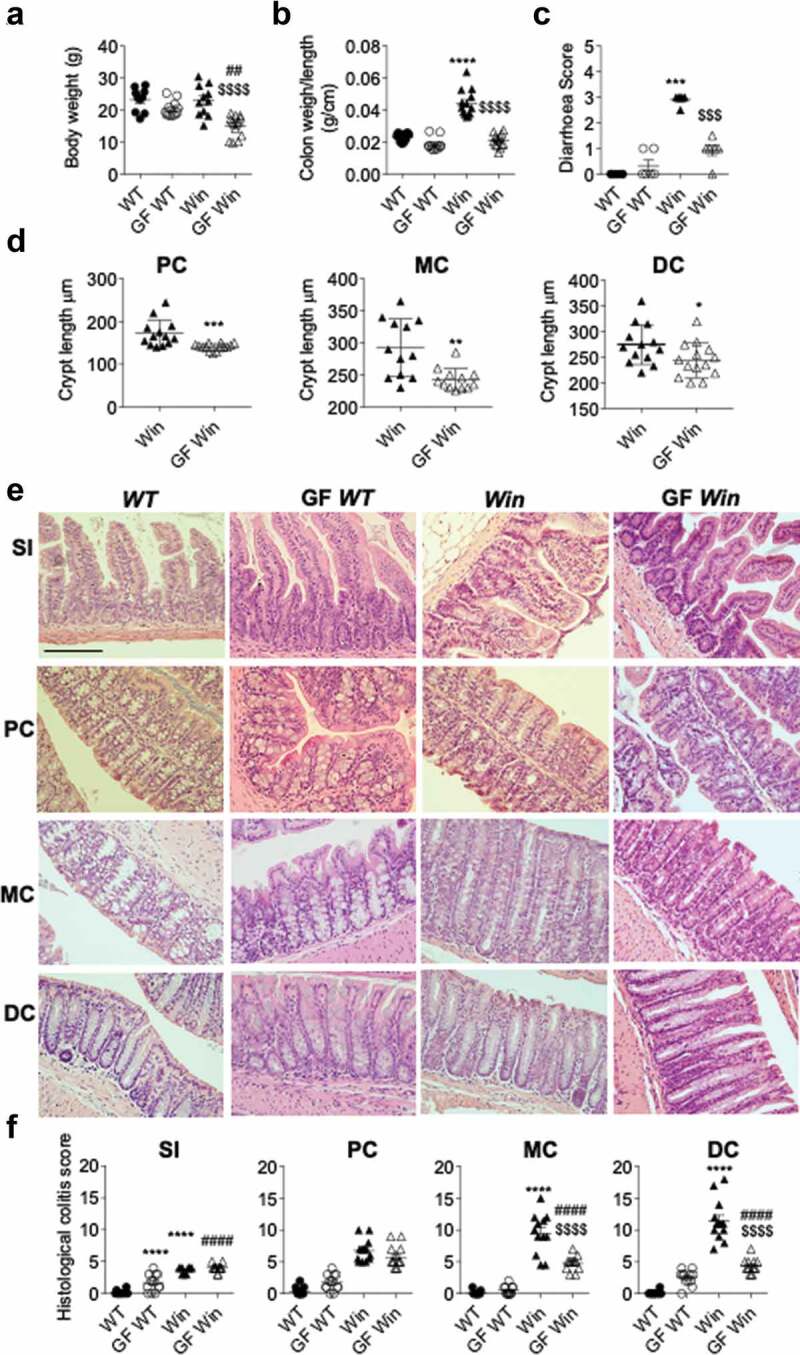
Absence of microbiota alleviated colitis in Winnie mice. WT, GF WT, Winnie and GF Winnie mice were sacrificed at 6–8 weeks of age. (a), Body weight. (b), Colon weight to length ratio. (c), Diarrhea score. (d), Crypt length in all mice was measured microscopically and presented as average crypt length per mouse in proximal (PC), middle (MC) and distal colon (DC) regions. (e), Representative H&E-stained small intestine and colon sections. (f), Blind assessment of histological colitis of mice in abased on H&E stained sections (maximum score = 20). Values are expressed as mean ± SEM and individual data points, n = 10 to 14. a, b, cand f, One-way ANOVA with Bonferroni’s multiple comparison test; *compared to WT control, #compared to GF WT, and $compared to Win (**P < .01; ***P < .001; ****P < .0001). Scale bar = 200 µm
Reduced colonic inflammation and ER stress in Winnie mice in the absence of the microbiota
The intestinal inflammation of Winnie mice progresses with prominent distal colitis featuring elevated expression of inflammatory cytokines and chemokines as well as increased ER stress.6,7 mRNA expression of multiple inflammatory effector cytokines was decreased in the distal colon of GF Winnie compared to Winnie mice (Figure 2a). Ifng, Il1b, Il17a and Il23a mRNA levels decreased to the levels comparable to WT mice. In WT mice cytokine gene expression was unaffected by microbiota status. In contrast, variation was seen in the response of chemokine genes to GF conditions in Winnie mice. While the chemokines, Cxcl2 (encodes Mip2α) and Cxcl9 were downregulated in GF Winnie compared to Winnie mice, in contrast, Ccl1 and Ccl3 (encodes Mip1α) were up-regulated in GF Winnie mice compared to Winnie mice.
Figure 2.
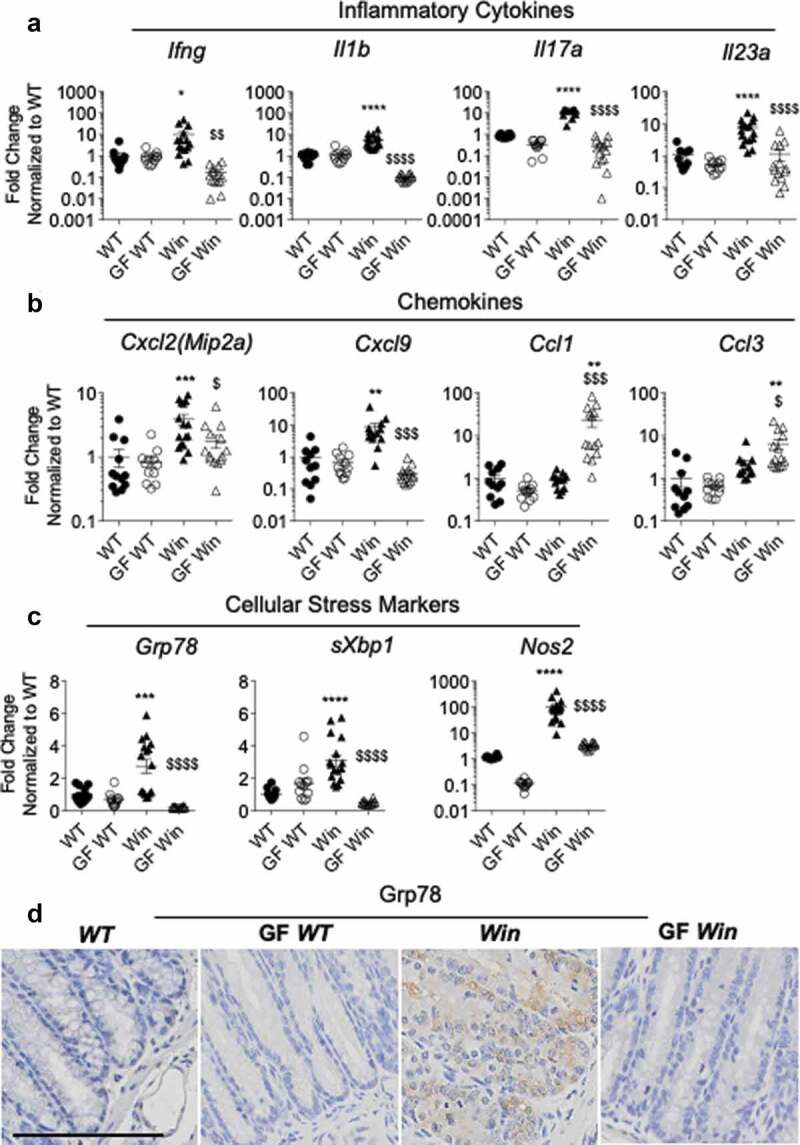
Inflammation and ER stress were reduced in the distal colon of GF Winnie. (a-c): Distal colonic mRNA expression of genes encoding inflammatory cytokines (a), intestinal epithelial-specific chemokines (b), and cellular stress markers (c) in mice described in Figure 1. (d), Representative immunohistochemistry with Grp78 antibody reflective of ER stress in distal colon sections. (a-c): Data presented as fold change corrected to b actin and normalized to WT mice. Values are expressed as mean ± SEM and individual data points, n = 10 to 14. One-way ANOVA with Bonferroni’s multiple comparison test; *compared to WT control, #compared to GF WT, and $compared to Win (*P < .05; **P < .01; ***P < .001; ****P < .0001)
Next, we sought to determine whether ER stress was alleviated because of the altered intestinal inflammation in GF Winnie mice. Using qRT-PCR and immunohistochemistry, we found lower levels of ER stress and oxidative stress markers in the absence of microbiota. Expression of Grp78, sXbp1, and Nos2 was increased in distal colon of Winnie mice compared to WT mice, and lower in the absence of microbiota to levels seen in WT mice (Figure 2c). Confirming the mRNA data, immunohistochemistry showed the chaperone Grp78, that is elevated during protein misfolding and ER stress, accumulated in the ER region of colonic goblet cells of Winnie mice, but was not seen in GF Winnie mice (Figure 2d). These data show that reduced ER stress in GF Winnie mice occurs concomitantly with reduced inflammation, suggesting that in conventional conditions inflammation exacerbates protein misfolding and ER stress.
Reduced Intestinal Epithelial Proliferation and Restored Paneth and Goblet Cell Secretion in Winnie Mice in the Germ-Free Environment
Epithelial hyperproliferation is a key feature of colitis in Winnie mice. To determine the proliferation rate of crypt epithelial cells, we performed immunohistochemistry staining for Ki67. The number of Ki67-positive proliferating cells were lower in small intestine of GF WT compared to WT, and also in the distal colon of the GF Winnie mice compared to Winnie mice (Supplemental Figure 1a-d), consistent with previously reported decreased epithelial turnover in the absence of luminal microbes.17 Winnie mice had increased proliferation in the distal colon (Supplemental Figure 1d) consistent with an inflammation-driven proliferative response associated with crypt elongation, as previously described,6 and under GF conditions proliferation reduced to near levels seen in conventional WT mice though greater than in GF WT mice (Figure 2b).
Reduced mucin volume in the theca of goblet cells and reduced Paneth cell granule formation are also key features of Winnie colitis. The expression of Paneth cell-derived anti-microbial peptides has been shown to be modulated and dependent on, the presence of signals from the microbiota.18 Lysozyme immunohistochemistry revealed that in conventional conditions Paneth cells in Winnie mice possess fewer secretory granules compared to WT mice due to chronic ER stress in these Muc2-producing cells (Supplemental Figure 1e). In addition, both WT and Winnie mice housed in GF conditions had reduced number of secretory granules in Paneth cells compared to their counterparts raised in the conventional environment, consistent with previous findings that the microbiota stimulates anti-microbial peptide production (Supplemental Figure 1f). mRNA expression of genes encoding Paneth cell signature anti-microbial peptides Reg3b and Reg3g (Supplemental Figure 1e) mirrored the decrease in lysozyme granule immunohistochemistry in GF conditions in both WT and Winnie mice (Supplemental Figure 1f-g).
Microbiota colonization and TLR/NOD ligands have been shown to regulate mucin gene expression and promote mucin secretion from goblet cells.19 To further elucidate the complex relationship between ER stress, inflammation, and mucin production in intestinal epithelial cells, we examined colonic goblet cell morphology and function in GF WT and Winnie mice. Under GF conditions Muc2 mRNA levels decreased ~10-fold in both WT and Winnie mice, consistent with the previous finding17 that diminished secretion of Muc2 (and therefore a decreased requirement for biosynthesis) is potentially as a result of lacking stimulation from bacterial products including TLR/NOD ligands (Figure 3c-f). In WT mice, GF conditions were accompanied by little change (if anything a slight increase) in the size of the goblet cell theca representing stored mucin granules for secretion (Figure 3a-d). As previously published,6 Winnie mice showed the characteristic small goblet cell theca in distal (Figure 3a-c) as well as proximal colon and small intestine (Supplemental Figure 2). The Winnie mice goblet cell theca size slightly increased under GF conditions (Figure 3a-d). Co-staining with the DBA lectin which binds O-glycans added in the Golgi apparatus and a Muc2 peptide antibody demonstrated the accumulation of the unglycosylated misfolded Muc2 precursor in Winnie goblet cells (red staining in Figure 3c). Under GF conditions the accumulation of Muc2 precursor was reduced (Figure 3e). Moreover, mRNA expression of the secretory cell lineage-determining transcription factor Atoh1 was not altered while enterocyte lineage transcription factor Hes1 expression was lower in both WT and Winnie under GF conditions (Figure 3f). Expression of transcription factor Spdef which drives goblet cell secretory function was lower in Winnie mice probably reflecting premature death of stressed goblet cells, however its target gene Agr2 was strongly induced in Winnie, which may reflect upregulation of this ER protein disulfide isomerase by the UPR. Spdef expression decreased substantially in WT and Winnie mice under GF conditions, and consequently, the genes it drives, Muc2 and Agr2, decreased similarly (Figure 3f), showing that the restored goblet cell theca could be a result of increased translation or reduced secretion, rather than a direct consequence of elevated Muc2 transcription. Overall, this result indicates that intestinal bacterial colonization promotes intestinal epithelial cell proliferation, goblet cell mucin production, and Paneth cell secretory function. In Winnie mice carrying a Muc2 misfolding mutation, the increased mucin production driven by bacterial colonization results in greater Muc2 misfolding, ER stress, and goblet cell depletion.
Figure 3.
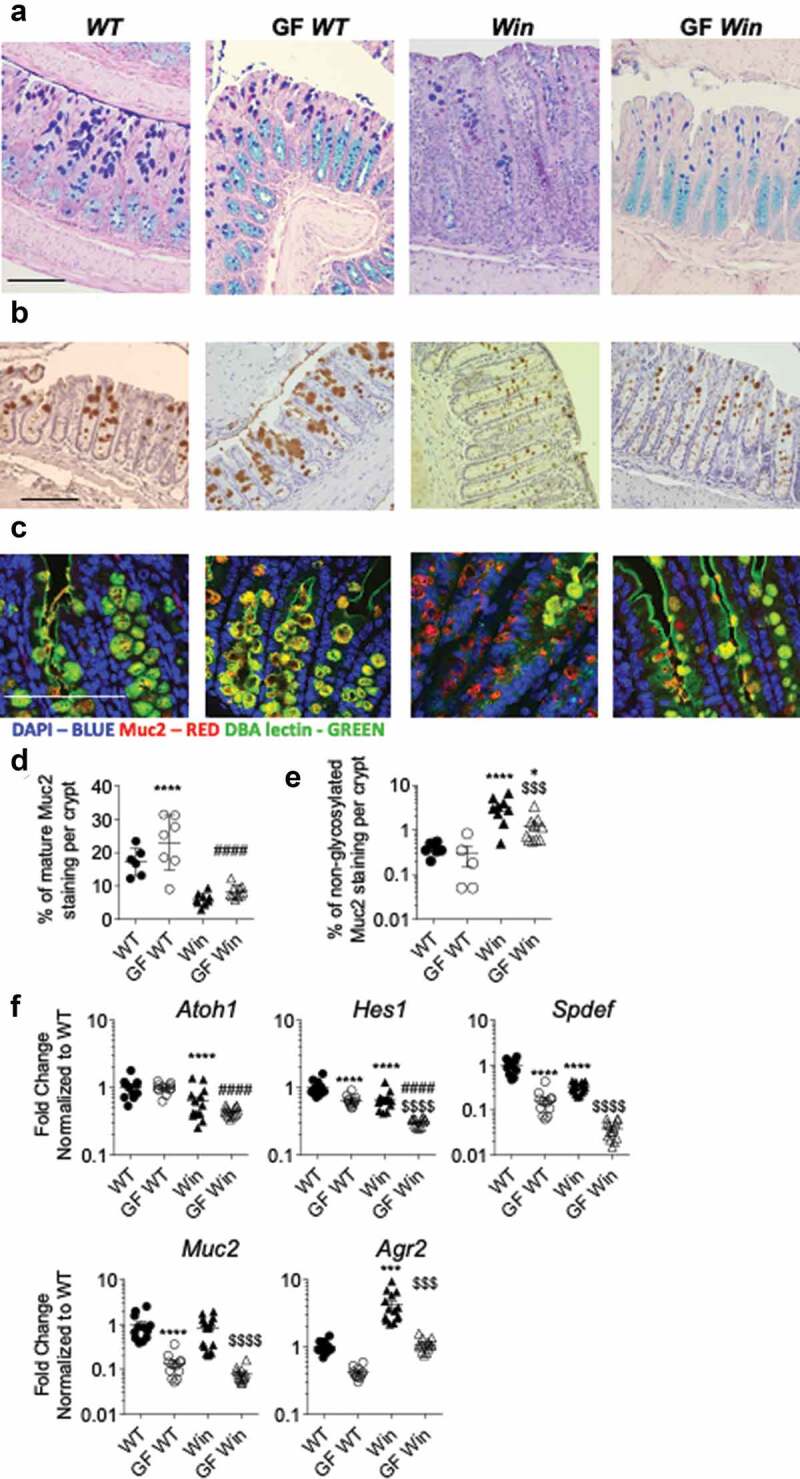
Muc2 misfolding and goblet cell mucin storage were partially improved in the distal colon of GF Winnie mice. (a), Representative sections of Alcian blue and periodic-acid Schiff stain, (b) immunohistochemistry with mature Muc2 antibody showing goblet cell morphology and (c) immunofluorescent staining of Muc2 core peptide (Red) and DBA lectin O-glycan staining (Green) showing accumulation of non-glycosylated misfolded Muc2 precursor in the distal part of the colon of mice described in Figure 1. D, Quantification of total Muc2 staining in the distal colon. (e), Quantification of non-glycosylated Muc2 staining (misfolded Muc2 in Red) in the distal colon. (f), mRNA expression of Atoh1(Math1), Hes1, Spdef, Muc2 and Agr2 in the distal colon of mice in Figure 1. Values are expressed as mean ± SEM and individual data points, n = 10 to 14, mixed gender. One-way ANOVA with Bonferroni’s multiple comparison test; *compared to WT control, #compared to GF WT, and $compared to Win (***P < .001; ****P < .0001). Scale bar = 200 µm
Muc2 misfolding initiates inflammation directly via downregulation of the endogenous anti-inflammatory regulators in epithelial cells
In GF Winnie mice, expression of ER stress and UPR markers were downregulated to a similar level as WT GF mice (Figure 2c,d). Intestinal inflammation was significantly reduced but not abolished in GF Winnie mice, mucin misfolding was reduced, and molecular markers of ER stress and activation of the UPR returned to levels seen in WT mice. Together, this suggests that Muc2 misfolding can initiate inflammatory signals directly, thus initiating inflammatory pathology when microbes are present. To test this hypothesis, colonic crypts were isolated from both WT and Winnie mice and cultured in vitro to remove the epithelium from the influence of microbial products and non-epithelial immune factors. Morphologically, organoids from Winnie mice were larger with slightly less budding observed compare to those from WT mice (Supplemental Figure 3a). Colonic organoids were next cultured and differentiated in presence of a Notch γ-secretase inhibitor (DAPT). This condition primarily promotes goblet cell differentiation and mucin biosynthesis, making it a preferred method to assess Muc2 protein misfolding associated functional changes. The differentiation was confirmed by the upregulation of Muc2 mRNA expression (Supplemental Figure 3b). Using RNA-Seq the differences in transcriptome between differentiated WT and Winnie epithelial cells were investigated 48 h after differentiation. We found that amongst all genes that are differentially regulated between WT and Winnie, most genes were downregulated in Winnie compare to WT organoids (Supplemental Figure 4a). An RNA-Seq signature comprising of 119 genes was generated based on greater than two-fold enriched with FDR of <0.01 in WT organoids compared to Winnie organoids. Pathway analysis using DAVID Bioinformatics was performed and the most significant keywords associated with the 119 gene signature were found including “negative regulation of inflammatory response” and “immune response” (Figure 4a and Supplemental Figure 4b). A number of genes known to negatively regulate inflammatory responses including Slpi, Nov, Calcrl, Cma1, Cxcl11, Tnfrsf8, Nt5e and Enpp3 were downregulated in differentiated Winnie compared to WT organoids (Figure 4b). We validated this result using quantitative RT-PCR in a separate set of differentiated WT and Winnie organoids for all genes (Figure 4c), except for Tnfrsf8 whose expression trended downward but was not significant (Supplemental Figure 4c). Concurrently, chemokine receptor Ccr4 and the trefoil factor (TFF) family member, spasmolytic peptide (SP)/Tff2 were upregulated, although these genes could not be confirmed in a second experiment using qRT-PCR validation (Supplemental Figure 4c). The downregulation of genes associated with negative regulation of inflammatory responses in Winnie compared to WT organoids suggests the ability of Winnie epithelial cells to prime a highly regulated tolerogenic immune response under sterile conditions is impaired.
Figure 4.
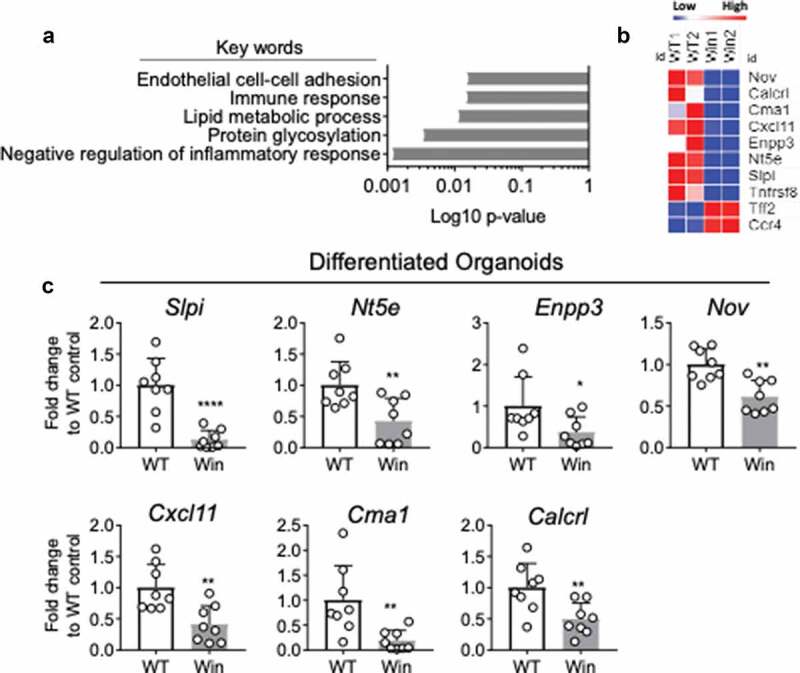
RNA-sequencing reveals lower expression of negative regulators of inflammation in differentiated Winnie compare to WT organoids. (a), DAVID gene ontology analysis reveals top five pathways/keywords associated with the 119 mostly differentially expressed genes between differentiated Winnie and WT organoids. (b), Heatmap showing expression levels of genes in “Negative regulation of inflammatory response”. (c), mRNA expression of Slpi, Nt5e, Enpp3, Nov, Cxcl11, Cma1 and Calcrl in a separate set of colon organoids isolated from WT and Winnie mice and differentiated with DAPT for 48 h. n = 8 in C per group, each dot represents organoid isolated from one mouse. Mann-Whitney test (*P < .05)
Winnie spontaneous colitis originates from Muc2 misfolding associated ER stress and UPR activation, which further leads to impaired barrier integrity with increased bacteria translocation. To assess whether Muc2 misfolding itself is driving sterile inflammation via the induction of ER stress and UPR activation in Winnie epithelial cells, WT and Winnie colonic organoids were differentiated with DAPT for 48 h (promotes Muc2 biosynthesis and folding). DAPT increased Muc2 protein synthesis in both WT and Winnie colonic organoids, however Muc2 was not appropriately glycosylated (less co-localization of DBA lectin with Muc2) in Winnie compared to WT organoids (Figure 5a, lower magnification in Supplemental Figure 5a). This data suggests that Muc2 mutation-induced protein misfolding occurs in Winnie organoids in vitro. Based on this, it was hypothesized that Muc2 misfolding could induce ER stress and initiate the UPR. Interestingly, gene expression of ER stress and UPR markers were similar or lower in differentiated Winnie organoids compared to WT organoids (Figure 5d), suggesting that Muc2 misfolding is not sufficient to drive UPR activation in vitro in absence of microbial and inflammatory factors in the environment. To address whether Muc2 misfolding itself triggered inflammatory signals, the same chemokines altered in vivo (Figure 2b) were measured by qPCR in organoids and by ELISA in culture supernatants (Figure 5b). Expression of Ccl1 was increased with differentiation, while others including Ccl3, Cxcl9 were unchanged (Figure 5b). Chemokines at protein level were undetectable in both differentiated WT and Winnie organoids culture supernatants, which may be due to the in vitro culture environment. To demonstrate the epithelial-specific chemokine involvement in Winnie colitis in vivo, colonic epithelial cells were isolated from WT and Winnie mice using an established protocol.20 Then, Mip2a protein was measured by ELISA in ex vivo isolated crypt lysates, then normalized to total protein quantity. Colonic epithelial cells in Winnie mice, primed by the in vivo microenvironment, in the presence of microbiota and immune cells, contained higher levels of Mip2a compared to WT mice (Figure 5c). Overall, our data show that mucin production can trigger subtle inflammatory signals directly at transcriptional level. In absence of a comprehensive anti-inflammatory network in Winnie epithelial cells, these signals become amplified in vivo in the presence of microbiota and inflammation.
Figure 5.
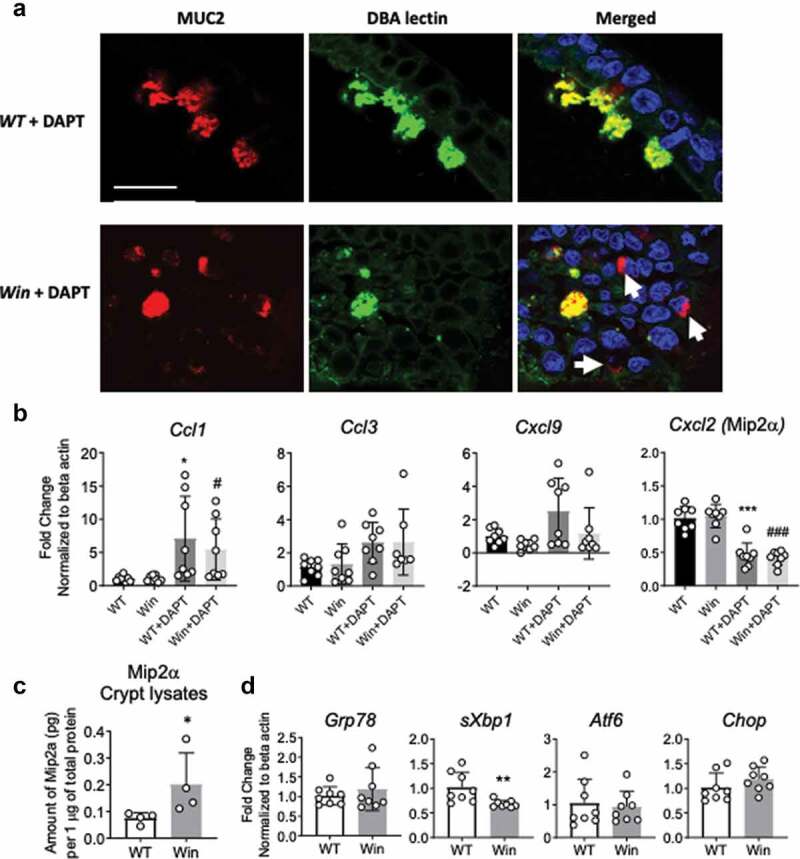
Muc2 misfolding persists in in vitro differentiated colonic organoids resulting in altered chemokine expression in epithelial cells. (a), Immunofluorescent staining of Muc2 core peptide (Red) and DBA lectin O-glycan staining (Green) showing Muc2 precursor accumulating in the non-glycosylated form in the DAPT differentiated Winnie organoids compared to WT. (b), mRNA expression of chemokines Ccl1, Ccl3,Cxcl9 and Mip2a in the organoids in (a). (c), Levels of Mip2a protein in colonic crypt isolated from whole colon of 8 weeks old WT and Winnie mice raised in conventional condition. (d), mRNA expression of ER stress and UPR markers in organoids in (a). For b and d, each dot indicates organoid isolated from one mouse, n = 8 per group, values are expressed as mean ± SD and individual data points: For C, each dot indicates one mouse, n = 4 per group. B, One-way ANOVA with Bonferroni’s multiple comparison test; *compared to WT control, #compared to Winnie (*P < .05; ** P < .01; ***P < .001). C and D, Mann-Whitney test, ns – not significant. Scale bar in A = 20 µm
Discussion
In this study, we have determined the effects of the intestinal microbiota on spontaneous colitis development in a mouse model with an intrinsic goblet cell protein misfolding defect. By comparing mice in conventional and germ-free conditions, and by in vitro organoid culture we demonstrate that mucin misfolding in goblet cells is sufficient to condition epithelial cells to become more responsive to stimuli, but that the microbiota is required for the full-scale development of spontaneous colitis in Winnie mice. The reduced severity of colitis in GF conditions was characterized by reduced inflammatory cell infiltration and effector cytokine expression in the mucosa. Therefore, our results suggest that while protein misfolding and ER stress prime intestinal inflammation, the microbiota exacerbates inflammation and ER stress in this model and is required for the full disease phenotype, including diarrhea. Our findings are consistent with previous reports where mice housed under GF environment failed to develop intestinal inflammation in multiple experimental colitis models, including genetic deficiencies in IL-10 and IL-2, and the T cell transfer and chemically induced colitis models.21–23 The major reason for the reduced inflammation is likely due to the absence of microbiota driving activation of innate and adaptive immunity24–26 (Figure 6). One limitation of the current study is that WTand Winnie mice were not littermates and therefore could harbor different gut microbiota. However, we have compared Winnie mice to littermate control WT mice, and Winnie mice to WT mice ordered from a different animal facility, and the same colitis phenotype was consistently observed in Winnie mice.6,20 In addition, Winnie mice housed either under SPF or clean conventional conditions showed the same colitis phenotype in both housing conditions.6 A recent paper used 16S rRNA metagenetic analysis to study the gut microbiota composition of age-matched littermate WT and Winnie mice. It showed that dysbiosis in Winnie mice was already established at 4 weeks of age, before substantial histologic evidence of gut inflammatory changes, and these microbial communities diverged from that derived from their heterozygous mothers.27 Results from this study demonstrated that Muc2 genetic mutation is involved in the complex regulation of intestinal homeostasis, such as promoting early gut dysbiosis that is independent from maternal microbial transfer.27 These results are consistent with our current findings that the Muc2 mutation drives the colitis phenotype, while microbiota is the amplifier.
Figure 6.
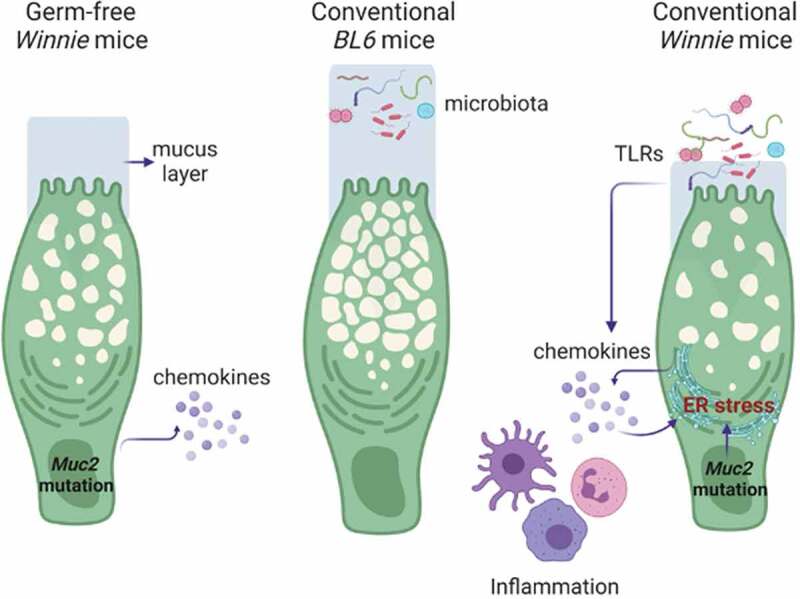
Complex relationship of epithelium defect, microbiome translocation and mucosal inflammation. Under a germ-free environment, Muc2 misfolding in epithelial cells initiate inflammatory signals directly, but the signal is only significantly amplified by the presence of gut microbiota and immune cells in a conventional environment. In turn, the inflammation and microbiota translocation (or TLR leakage) can exacerbate Muc2 misfolding and ER stress resulting in mucus layer depletion and increased bacteria translocation. This sets a feed-forward cycle for progressive intestinal inflammation in Winnie mice. Figure created using BioRender
Muc2 protein misfolding and ER stress in intestinal secretory cells of Winnie mice can be exacerbated by inflammation.6,7 This is evident with anti-inflammatory drug treatment12 and depletion of T and B cells7,28 in the Winnie mice which alleviate both ER stress and colitis. It is important to note that anti-inflammatory drugs or biologics we have used including glucocorticoids,12 thioguanine13, and IL-23p19 antibody,29 not only suppressed inflammation, but also alleviated ER stress in Winnie mice providing direct evidence that inflammation exacerbates ER stress in this model. Although a mucin misfolding mutation is the primary driver of Winnie colitis, it is also recognized that secretory cells such as goblet and Paneth cells are targeted by inflammation.28,30 Accompanying reduced inflammation and ER stress, in the absence of microbiota, there was an increase in stored mucins in the goblet cells of GF Winnie when compared to Winnie mice. Goblet cell differentiation marker Math1 (Atoh1) is unchanged in GF compare to conventional conditions, but goblet cell markers including Spdef and Muc2 gene expression were markedly reduced in GF mice compared to relative conventional raised mice, suggesting that microbiota regulates intestinal epithelial secretory function, which has been reported in multiple studies.1,31 Paneth cell production of anti-microbial molecules was also diminished under GF conditions compared to conventionally raised mice, confirming that microbiota regulates intestinal secretory function in both major intestinal secretory cell types.32
Although ER stress and UPR activation are involved in IBD pathogenesis,33 Muc2 misfolding did not induce measurable ER stress and UPR activation in Winnie mice housed under GF conditions. Expression of inflammatory chemokines were largely similar in differentiated WT and Winnie organoids. There was an increase in chemokine expression in the Winnie mice in vivo, suggesting that these changes are either lost due to the removal of inflammatory milieu in culture or the main source of chemokines seen in vivo are leukocytes. Additionally, Mip2a expression in organoids is decreased with DAPT differentiation, suggesting enterocytes, not goblet cells, are the major cellular source. These transcriptional changes of chemokines were too subtle to be detected at the protein level in organoid culture supernatants. When we look at the global transcriptome landscape, Winnie epithelial cells carrying Muc2 mutation present a less immunosuppressive phenotype with lower expression of negative regulators of inflammation compared to WT epithelial cells. Among all immune suppressors, the most inhibited genes in Winnie compared to WT organoids was Slpi (secretory leukocyte protease inhibitor) – a well-known peptide protecting mucosal tissue against the detrimental consequences of inflammation.34 Another well-known negative regulator of inflammation is the adenosine pathway involving Nt5e (CD73) and Enpp3 (ecto-pyrophosphatase/phosphodiesterase-3) which are ectoenzymes that control extracellular nucleotide and nucleoside levels. Both enzymes catalyze ATP to AMP, therefore driving a shift from an ATP-driven proinflammatory environment to an anti-inflammatory milieu induced by adenosine.35,36 Significantly decreased CD73 expression in Winnie epithelial cells compared to WT could result in high ATP, therefore, sustained inflammatory signals. This is intriguing, since ATP has been shown to be one of the damage-associated molecular patterns (DAMPs) in cancer cells causing immunogenic cell death.37 Although detailed mechanism of Muc2 mutation driven downregulation of purinergic pathways is unknown, unfolded Muc2 in the ER could modify the cellular redox state and initiate inflammatory signals. Other negative regulators of inflammation include Nov also known as CCN3 – a matricellular protein that negatively regulates nuclear factor kappaB (NF-kappaB) activity;38 and Cxcl11 expression drives CD4+ T cell polarization into IL-10-producing type 1 regulatory T cells.39,40 Genes like Cma1 and Calcrl have not been reported to be expressed in epithelial cells, but their immune-modulatory roles are known in other settings. Cma1 expression in immune cells (Mast cells) plays a role in maintaining intestinal barrier function and to expel parasitic worms.41,42 Calcrl encoding calcitonin receptor-like receptor is important in maintaining lymphatic tissue homeostasis. Loss of Calcrl expression in lymphatic tissue exacerbates intestinal pathology following mucosal injury demonstrating the importance of lymphatic function in promoting recovery from intestinal inflammation.43 Taken together, our in vitro data in primary organoids highlights the important function of mucosal epithelial cells in priming a more inflammatory response to the luminal microbiota.
Defects in intestinal epithelial barrier function are a characteristic feature of human IBD. Although Muc2 gene polymorphisms have not been associated with increased risk of IBD in GWAS, epithelial integrity is linked with IBD pathogenesis. Mucosal barrier perturbations can affect different levels of the protective mechanisms including alterations of PRRs, a defective mucus layer, alterations in the process of autophagy, or an increased epithelial barrier permeability, and thereby result in inadequate protection against microbial adherence and invasion. Like the Winnie mice, bacterial translocation due to impaired barrier function in humans results in chronic intestinal inflammation in IBD. Since IBD affects predominantly the most heavily colonized gut segments – terminal ileum and colon, an involvement of the microbiota in disease progression is obvious and supported by success of fecal microbiota transplants. Post-operative recurrence of CD is also triggered by luminal contents.44 Because the microbiota plays important roles in driving and amplifying chronic inflammation, it is reasonable to target the microbiota in order to dampen inflammation. Meta-analysis revealed that fecal transplantation could be a promising treatment of IBD patients.45,46 This approach can be an exciting opportunity for patients carrying genetic mutations predisposing them to increased risk of having IBD.
In the current study, we have shown that in absence of microbiota, intestinal inflammation, and epithelial ER stress are significantly alleviated in a Muc2 misfolding induced spontaneous colitis mouse model. Data from this study provide valuable information on how epithelial cell intrinsic defects, inflammation, and bacteria translocation inter-regulate each other in the complex intestinal mucosal system. This work explains how genetic predispositions to protein misfolding could combine with the microbiota to drive progressive barrier depletion and inflammation.
Materials and methods
Animals
Conventionally raised Winnie mice on a C57BL/6 background (bred in-house) and C57BL/6 mice (purchased from ARC) were housed within a specific pathogen-free Biological Research Facility (BRF) at the Translational Research Institute (TRI, free of Norovirus and Helicobacter) and fed autoclaved food. C57BL/6 mice from ARC were acclimatized for 2 weeks in the TRI-BRF before used for experiments. C57BL/6 and Winnie mice were rederived into the GF facility at Water and Eliza Hall Institute in Melbourne and housed in the trexler-type soft-sided isolators. For assessing the colitis phenotype of these mice, age and gender-matched (only male animals were used in this study). GF C57BL/6 and Winnie mice were shipped to the TRI BRF at Brisbane in microisolator cages. All animal research and experiment protocols were approved by the Animal Ethics Committee of the University of Queensland and conducted according to National Health and Medical Research Council guidelines.
Mouse tissue end-point collection
Upon arrival, diarrhea scoring was performed on all mice. The diarrhea scoring system can be interpreted as follows: 0 – no diarrhea: solid stool; 1 – very mild diarrhea: formed stools that appear moist on the outside; 2 – mild diarrhea: formed stools that easily submit to pressure applied with forceps; 3 – diarrhea: no fully formed stools with a mucous-like appearance; 4 – severe, watery diarrhea: mostly clear or mucous-like liquid stool with very minimal solid present. Then, GF C57BL/6 and Winnie mice were sacrificed on the same day and colonic tissues were harvested for downstream analysis. The whole colon, the cecum and ileum (distal 1/3rd part of the small intestine) were removed. Each segment of the intestine was opened longitudinally and the fecal content was gently removed. Colon weight was measured and then the colon was dissected longitudinally into two sections. One of the sections was rolled (Swiss rolled) from the rectum along with cecum and ileum and fixed in 10% neutral buffered formalin for histological examination. The other section of the colon was dissected in equal halves, designated as ‘proximal’ and ‘distal’ colon and snap frozen in dry ice for later mRNA expression analysis.
Quantitative reverse-transcription polymerase chain reaction
Tissue samples (snap frozen and homogenized) were extracted into TRIzol before RNA purification using ISOLATE II RNA Mini Kit (Bioline, London, UK), followed by cDNA synthesis using SensiFAST™ cDNA Synthesis Kit (Bioline, London, UK) containing oligo (dT) and random hexamer primers. SensiFAST™ SYBR® Lo-ROX Kit (Bioline, London, UK) was used for quantitative polymerase chain reaction using a ViiA 7 (Life Technologies, Carlsbad, CA); passive reference dye ROX was used. Results were analyzed using ViiA 7 RUO software v1.2 (Applied Biosystems, Life Technology). Primer efficiencies were determined using complementary DNA and primer dilutions for each gene of interest. Primers used to determine gene expressions are summarized in Supplemental Table 1. All cytokine gene expression data were normalized to the housekeeping gene beta actin as a fold change compared with the average of the control group. Samples having non-detectable cytokine cDNA amplification were given an arbitrary Ct value of 37 representing the limit of detection.
Hematoxylin & eosin staining
Formalin-fixed tissue samples were processed into paraffin blocks and sectioned by the TRI Histology Core Service. After de-waxing in xylene and re-hydrating through ethanol gradients (100%, 95%, 90% 70%, and dH2O), paraffin sections were placed in Hematoxylin reagent (Gill’s formula, No.2, Sigma-Aldrich) for 30 s and then washed under running tap water for 5 min. Slides were then placed in Eosin Y reagent (Sigma-Aldrich) for 30 s. After Eosin staining, slides were dehydrated in 90%, 100% ethanol and xylene, followed by mounting with Pertex® mounting medium (Medite, Burgdorf, Germany) and examination under a bright field microscope.
Histological scoring of colitis
H&E stained mouse intestinal sections were scored microscopically in a blinded manner by RW including assessments of crypt architecture (0-normal to 5-large ulcers), crypt abscesses (0-none to 3->10 crypt abscesses), tissue damage (0-none to 3-extensive mucosal damage), goblet cell depletion (0-normal to 3->50% depletion), inflammatory cell infiltration (0-occasional infiltration to 3-transmural infiltration) and local neutrophil counts (0-<5 to 3->20 in one field under 40X lenses). Crypt length (0–4) was measured using CellSens software (Olympus). This scoring system determines a combined score of histological colitis (maximum of 25) via the assessment of the above-mentioned parameters which reflect tissue damage and colitis severity.6,28,29
Immunohistochemistry and immunofluorescence staining
Different antigen retrieval methods were used for different antigens. For Muc2, Grp78, and Ki67 staining, slides were boiled in citric acid buffer (10 mM citric acid and 0.05% Tween-20, pH 6.0) for 20 min in a microwave oven. For lysozyme staining, slides were boiled in Tris-EDTA buffer (10 mM Tris Base; 1 mM EDTA; 0.05% Tween-20, pH 9.0) for 20 min in a microwave oven. After antigen retrieval steps, the endogenous hydrogen peroxidase activity was quenched by incubating slides with 3% hydrogen peroxidase in PBS for 10 min. Sections were then blocked with 10% KPL blocking solution (KPL Inc, USA) in PBS for 1 h at room temperature (RT). Each primary antibody was titrated first to define the optimal staining concentration. For mature Muc2, 1 in 500 diluted rabbit anti-mouse Muc2 antibody (Muc2.3, in-house, against peptide NGLKPVRVPDADNC, previously described in6) was incubated with slides overnight at 4°C. For Grp78 staining, rabbit anti-mouse Grp78 (clone: GL-19, Sigma) antibody was diluted 1 in 100 in PBS with 5% KPL and incubated with sections overnight at 4°C. For lysozyme, rabbit anti-lysozyme antibody (Dako, USA) was diluted 1 in 500 in PBS with 5% FCS and applied with sections overnight at 4°C. For Ki-67, 1 in 200 diluted rabbit anti-human/mouse Ki-67 antibody (SP6, Thermo Fisher, MA5-14520) was diluted in 5% KPL in PBS and applied with sections for 2 h at RT. For detection of Muc2 maturation, 20 µg/ml FITC-conjugated Dolichos biflorus agglutinin (DBA) lectin (Sigma) was applied with sections for 30 min at RT after Muc2 primary antibody staining. After incubation with primary antibody, slides were washed 3 times in PBS. HRP-conjugated secondary antibodies against the species primary antibody raised in were incubated with sections for 2 h at room temperature. Unbound antibodies were washed with PBS and signal was detected using diaminobenzidine solution (Dako, CA, US). After counter-staining with hematoxylin, sections were dehydrated in an ethanol gradient and cleared in xylene before mounting with Pertex® mounting medium (Medite, Belgium). For Muc2 staining in differentiated organoids, BME-embedded organoids were dislodged with a spatula and solubilized in 5 mM EDTA in PBS for 1 h at 4°C on a rotating platform. Organoids were gently pelleted at 100 g for 5 min before fixed in 2% paraformaldehyde (PFA) in 1X PBS for 30 minutes at room temperature. Fixed organoids were washed extensively in PBS for 3 times at 100 g for 5 min before resuspended in 50 µl of 2% agarose. Once solidified, the agarose plug containing organoids are gently removed from the tube and then paraffin-embedded and sectioned at 10 µm thickness on super frost plus slides for staining. Organoids are permeabilized in PBS with 3% bovine serum albumin, 1% Triton X-100, and 1% Saponin for 30 mins. Mucin was reduced with 50 mM DTT in 0.1 M Tris-HCl (pH8) for 30 min, followed by alkylation using 25 mM iodoacetamide in 0.1 M Tris-HCl (pH8) in the dark at RT for 30 min. 1 in 500 diluted rabbit anti-mouse Muc2 antibody (Muc2.3) was incubated with organoids overnight at 4°C. Then, organoids were stained with anti-rabbit Alexa Flour-texas red secondary antibody (Invitrogen) and 20 µg/ml FITC-conjugated Dolichos biflorus lectin (Sigma). Organoids were stained with DAPI before imaged using the Olympus FV1200 Confocal Microscope.
Alcian blue and periodic acid Schiff’s (PAS) staining
Alcian blue and PAS staining was used to assess the goblet cell mucin storage. After de-waxing in xylene and re-hydrating through ethanol gradients, paraffin sections were placed in Alcian blue solution (1% Alcian blue in 3% acetic acid, pH 2.5) for 10 min and washed under running tap water. Then, slides were treated with freshly prepared 1% periodic acid for 5 min and washed with dH2O before staining in Schiff’s reagent (Sigma) for 10 min. Slides were washed under running tap water for 5 min and then counterstained with hematoxylin (Gill’s Formula No.2, Sigma) for 30 s, dehydrated, cleared, and mounted with Pertex® mounting medium (Medite, Belgium).
Morphometric assessments
To quantify positive Muc2 staining in tissue sections after immunohistochemistry staining, multiple pictures from areas with longitudinally sectioned crypts were taken randomly using an Olympus bright field microscope with a 40× lens to keep sampling process unbiased. Similarly, to quantify non-glycosylated Muc2 staining in tissue sections after immunofluorescent staining, multiple pictures from areas with longitudinally sectioned crypts were taken randomly using an Olympus FV1200 confocal microscopy with a 40× lens. In each image, positive staining was defined by RGB pixel picking tool in NIS-Elements software v.3.0 (Nikon, Tokyo, Japan). The percentage of the longitudinally sectioned crypt area occupied by positive staining was calculated for multiple crypts within each specimen and the average for that specimen determined and used for statistical analysis. For Ki-67 positive cell quantification, the number of positive staining nuclei per every 10 longitudinally cut crypts was counted.
Colonic organoid culture and treatments
For WT and Winnie (n = 4) colonic organoid culture, whole colon was dissected and opened longitudinally in cold PBS, cut into 10 mm pieces, and incubated in EDTA solution for 1 h at room temperature, then digested with collagenase type I (2 mg/mL) supplemented with gentamicin (50 µg/mL) for 15–20 min at 37°C for 15 min. Then, colonic epithelia containing crypts were released from the tissue after vigorous pipetting. The isolated crypts were washed with DMEM/F12 medium and centrifuged at 50 x g for 5 min at 4°C and cells were embedded in Basement Membrane Extract (Invitrogen) in a 1:1 ratio. Then, 20 µL of the mixture was plated in a 24 well tissue culture plate and cultured in 50% L-WRN conditioned medium (a kind gift from Stappenbeck Lab) together with Y27632 at 10 µM (Rho-associated protein kinase inhibitor) and SB431542 at 10 µM (the transforming growth factor (TGF)-β type I receptor inhibitor). The crypts were expanded by serial culture until sufficient numbers were obtained for experimentation. To stimulate mucin production, organoids were differentiated in 5% L-WRN conditioned medium +95% of advanced DMEM supplemented with 20% FBS, 1% L-glutamine and 1% Penicillin/Streptomycin and SB431542 at 10 µM in presence of DAPT at 5 µM for 48 h. Organoids were harvested in TRIzol for RNA isolation and qPCR to look at gene expressions.
Epithelial isolation and protein extraction for ELISA
Conventionally raised WT and Winnie (n = 4) were used for colonic epithelial (crypts) isolation. The same protocol was used as above for crypt isolation. The isolated crypts were lysed in 50 µL of cold RIPA buffer (50 mM Tris-HCI, pH 7.5, 150 mM NaCI, 1.0% Nonidet P-40, 0.1% sodium deoxycholate supplemented with Complete protease inhibitors and phosphate stop cocktail) for 15 min on ice and vortexed every 5 min. The lysates were centrifuged at 16,000 g for 20 min at 4°C to remove cell debris and stored at −80°C for further analysis. BCA assay was conducted to determine the protein concentration. 50 µL of the epithelial cell lysate was used for ELISA to measure Mip2a concentration. Final concentration was assessed by ELISA and then normalized to the total amount of protein determined by BCA assay.
RNA-sequencing
To determine the transcriptome changes in differentiated Winnie compared to WT organoids. Organoids from 2 WT and 2 Winnie mice were established as above. After 48 h differentiation with DAPT at 20 µM, organoids were harvested and total mRNA was extracted using the TRizol/Chloroform method in combination with RNA column (RNA mini column, Bio-line). The RNA library preparation and pair-end sequencing was done by Australian Genome Research Facility (AGRF) using a HiSeq 2500 instrument (Illumina, San Diego, CA) with a maximal read length of 100 bp. The differential gene expression was analyzed through edgeR (version 3.22.3) using R (The R Foundation; version 3.5.0). Raw gene counts per million were used for transcriptional comparison. The gene ontology analyses were done using David Pathway online tool to identify the pathways.
Supplementary Material
Acknowledgments
We would like to acknowledge the support of Translational Research Institute (TRI) Core facilities, in particular, the Microscopy Core and Histology Core for helping with the sample preparation, the Biological Resource Facility at TRI for support with animal husbandry.
Funding Statement
This work is supported by Mater Foundation and the National Health and Medical Research Council, Australia. RW is supported by the Bushell Postdoctoral Fellowship from the Gastroenterology Society of Australia and Mater Foundation. SZH is supported by Mater Foundation and National Health and Medical Research Council.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author contributions
RW, KY, PW, HT and SZH performed all in vivo and ex vivo experiments, analyzed results and drafted the manuscript. MM performed the RNA-Seq experiments. MM, KY, PW, AH, RG and JC conducted organoids culture, and were involved in RNA-Seq genes validation. JB provided intellectual input to the project including manuscript editing. MAM and SZH conceived and provided all support needed for the project and edited the paper.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Kim YS, Ho SB.. Intestinal goblet cells and mucins in health and disease: recent insights and progress. Curr Gastroenterol Rep. 2010;12(5):319–18. doi: 10.1007/s11894-010-0131-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Velcich A, Yang W, Heyer J, Fragale A, Nicholas C, Viani S, Kucherlapati R, Lipkin M, Yang K, Augenlicht L. Colorectal cancer in mice genetically deficient in the mucin Muc2. Science. 2002;295(5560):1726–1729. doi: 10.1126/science.1069094. [DOI] [PubMed] [Google Scholar]
- 3.Van Der Sluis M, De Koning BAE, De Bruijn ACJM, Velcich A, Meijerink JPP, Van Goudoever JB, Büller HA, Dekker J, Van Seuningen I, Renes IB, et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology. 2006;131(1):117–129. doi: 10.1053/j.gastro.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 4.An G, Wei B, Xia B, McDaniel JM, Ju T, Cummings RD, Braun J, Xia L. Increased susceptibility to colitis and colorectal tumors in mice lacking core 3-derived O-glycans. J Exp Med. 2007;204(6):1417–1429. doi: 10.1084/jem.20061929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tobisawa Y, Imai Y, Fukuda M, Kawashima H. Sulfation of colonic mucins by N-acetylglucosamine 6-O-sulfotransferase-2 and its protective function in experimental colitis in mice. J Biol Chem. 2010;285(9):6750–6760. doi: 10.1074/jbc.M109.067082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heazlewood CK, Cook MC, Eri R, Price GR, Tauro SB, Taupin D, Thornton DJ, Png CW, Crockford TL, Cornall RJ. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med. 2008;5(3):e54. doi: 10.1371/journal.pmed.0050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eri RD, Adams RJ, Tran TV, Tong H, Das I, Roche DK, Oancea I, Png CW, Jeffery PL, Radford-Smith GL, et al. An intestinal epithelial defect conferring ER stress results in inflammation involving both innate and adaptive immunity. Mucosal Immunol. 2011;4(3):354–364. doi: 10.1038/mi.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park SW, Zhen G, Verhaeghe C, Nakagami Y, Nguyenvu LT, Barczak AJ, Killeen N, Erle DJ. The protein disulfide isomerase AGR2 is essential for production of intestinal mucus. Proc Natl Acad Sci U S A. 2009;106(17):6950–6955. doi: 10.1073/pnas.0808722106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsuru A, Fujimoto N, Takahashi S, Saito M, Nakamura D, Iwano M, Iwawaki T, Kadokura H, Ron D, Kohno K, et al. Negative feedback by IRE1beta optimizes mucin production in goblet cells. Proc Natl Acad Sci U S A. 2013;110(8):2864–2869. doi: 10.1073/pnas.1212484110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107(7):881–891. doi: 10.1016/S0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 11.Hasnain SZ, Lourie R, Das I, Chen ACH, McGuckin MA. The interplay between endoplasmic reticulum stress and inflammation. Immunol Cell Biol. 2012;90(3):260–270. doi: 10.1038/icb.2011.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Das I, Png CW, Oancea I, Hasnain SZ, Lourie R, Proctor M, Eri RD, Sheng Y, Crane DI, Florin TH, et al. Glucocorticoids alleviate intestinal ER stress by enhancing protein folding and degradation of misfolded proteins. J Exp Med. 2013;210(6):1201–1216. doi: 10.1084/jem.20121268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oancea I, Movva R, Das I, Aguirre De Cárcer D, Schreiber V, Yang Y, Purdon A, Harrington B, Proctor M, Wang R, et al. Colonic microbiota can promote rapid local improvement of murine colitis by thioguanine independently of T lymphocytes and host metabolism. Gut. 2017;66(1):59–69. doi: 10.1136/gutjnl-2015-310874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Min YW, Rhee PL. The role of microbiota on the gut immunology. Clin Ther. 2015;37(5):968–975. doi: 10.1016/j.clinthera.2015.03.009. [DOI] [PubMed] [Google Scholar]
- 15.Hormann N, Brandão I, Jäckel S, Ens N, Lillich M, Walter U, Reinhardt C. Gut microbial colonization orchestrates TLR2 expression, signaling and epithelial proliferation in the small intestinal mucosa. PLoS One. 2014;9(11):e113080. doi: 10.1371/journal.pone.0113080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hernandez-Chirlaque C, Aranda CJ, Ocón B, Capitán-Cañadas F, Ortega-González M, Carrero JJ, Suárez MD, Zarzuelo A, Sánchez De Medina F, Martínez-Augustin O, et al. Germ-free and antibiotic-treated mice are highly susceptible to epithelial injury in DSS colitis. J Crohns Colitis. 2016;10(11):1324–1335. doi: 10.1093/ecco-jcc/jjw096. [DOI] [PubMed] [Google Scholar]
- 17.Yu LC, Wang JT, Wei SC, Ni YH. Host-microbial interactions and regulation of intestinal epithelial barrier function: from physiology to pathology. World J Gastrointest Pathophysiol. 2012;3(1):27–43. doi: 10.4291/wjgp.v3.i1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, Ley R, Wakeland EK, Hooper LV. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science. 2011;334(6053):255–258. doi: 10.1126/science.1209791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Birchenough GM, Nystrom EEL, Johansson MEV, Hansson GC. A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science. 2016;352(6293):1535–1542. doi: 10.1126/science.aaf7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gulhane M, Murray L, Lourie R, Tong H, Sheng YH, Wang R, Kang A, Schreiber V, Wong KY, Magor G, et al. High fat diets induce colonic epithelial cell stress and inflammation that is reversed by IL-22. Sci Rep. 2016;6(1):28990. doi: 10.1038/srep28990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, Rennick DM, Sartor RB. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun. 1998;66(11):5224–5231. doi: 10.1128/IAI.66.11.5224-5231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schultz M, Tonkonogy SL, Sellon RK, Veltkamp C, Godfrey VL, Kwon J, Grenther WB, Balish E, Horak I, Sartor RB, et al. IL-2-deficient mice raised under germfree conditions develop delayed mild focal intestinal inflammation. Am J Physiol. 1999;276(6 Pt 1):G1461–72. doi: 10.1152/ajpgi.1999.276.6.G1461. [DOI] [PubMed] [Google Scholar]
- 23.Stepankova R, Powrie F, Kofronova O, Kozakova H, Hudcovic T, Hrncir T, Uhlig H, Read S, Rehakova Z, Benada O, et al. Segmented filamentous bacteria in a defined bacterial cocktail induce intestinal inflammation in SCID mice reconstituted with CD45RBhigh CD4+ T cells. Inflamm Bowel Dis. 2007;13(10):1202–1211. doi: 10.1002/ibd.20221. [DOI] [PubMed] [Google Scholar]
- 24.Chiba T, Seno H. Indigenous clostridium species regulate systemic immune responses by induction of colonic regulatory T cells. Gastroenterology. 2011;141(3):1114–1116. doi: 10.1053/j.gastro.2011.07.013. [DOI] [PubMed] [Google Scholar]
- 25.Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 2011;331(6015):337–341. doi: 10.1126/science.1198469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hirota K, Turner J-E, Villa M, Duarte JH, Demengeot J, Steinmetz OM, Stockinger B. Plasticity of Th17 cells in Peyer’s patches is responsible for the induction of T cell-dependent IgA responses. Nat Immunol. 2013;14(4):372–379. doi: 10.1038/ni.2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liso M, De Santis S, Verna G, Dicarlo M, Calasso M, Santino A, Gigante I, Eri R, Raveenthiraraj S, Sobolewski A. A specific mutation in Muc2 determines early dysbiosis in colitis-prone Winnie mice. Inflamm Bowel Dis. 2019; 26(4): p. 546-556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hasnain SZ, Tauro S, Das I, Tong H, Chen AC–, Jeffery PL, McDonald V, Florin TH, McGuckin MA. IL-10 promotes production of intestinal mucus by suppressing protein misfolding and endoplasmic reticulum stress in goblet cells. Gastroenterology. 2013;144(2):357–368. e9. doi: 10.1053/j.gastro.2012.10.043. [DOI] [PubMed] [Google Scholar]
- 29.Wang R, Hasnain SZ, Tong H, Das I, Che-Hao Chen A, Oancea I, Proctor M, Florin TH, Eri RD, McGuckin MA, et al. Neutralizing IL-23 is superior to blocking IL-17 in suppressing intestinal inflammation in a spontaneous murine colitis model. Inflamm Bowel Dis. 2015;21(5):973–984. doi: 10.1097/MIB.0000000000000353. [DOI] [PubMed] [Google Scholar]
- 30.Hasnain SZ, Borg DJ, Harcourt BE, Tong H, Sheng YH, Ng CP, Das I, Wang R, Chen ACH, Loudovaris T, et al. Glycemic control in diabetes is restored by therapeutic manipulation of cytokines that regulate beta cell stress. Nat Med. 2014;20(12):1417–1426. doi: 10.1038/nm.3705. [DOI] [PubMed] [Google Scholar]
- 31.Becker S, Oelschlaeger TA, Wullaert A, Pasparakis M, Wehkamp J, Stange EF, Gersemann M. Bacteria regulate intestinal epithelial cell differentiation factors both in vitro and in vivo. PLoS One. 2013;8(2):e55620. doi: 10.1371/journal.pone.0055620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bevins CL, Salzman NH. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol. 2011;9(5):356–368. doi: 10.1038/nrmicro2546. [DOI] [PubMed] [Google Scholar]
- 33.Kaser A, Martinez-Naves E, Blumberg RS. Endoplasmic reticulum stress: implications for inflammatory bowel disease pathogenesis. Curr Opin Gastroenterol. 2010;26(4):318–326. doi: 10.1097/MOG.0b013e32833a9ff1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Doumas S, Kolokotronis A, Stefanopoulos P. Anti-inflammatory and antimicrobial roles of secretory leukocyte protease inhibitor. Infect Immun. 2005;73(3):1271–1274. doi: 10.1128/IAI.73.3.1271-1274.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsai SH, Kinoshita M, Kusu T, Kayama H, Okumura R, Ikeda K, Shimada Y, Takeda A, Yoshikawa S, Obata-Ninomiya K, et al. The ectoenzyme E-NPP3 negatively regulates ATP-dependent chronic allergic responses by basophils and mast cells. Immunity. 2015;42(2):279–293. doi: 10.1016/j.immuni.2015.01.015. [DOI] [PubMed] [Google Scholar]
- 36.Antonioli L, Pacher P, Vizi ES, Haskó G. CD39 and CD73 in immunity and inflammation. Trends Mol Med. 2013;19(6):355–367. doi: 10.1016/j.molmed.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T, Rubio N, Firczuk M, Mathieu C, Roebroek AJM, et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. Embo J. 2012;31(5):1062–1079. doi: 10.1038/emboj.2011.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin Z, Natesan V, Shi H, Hamik A, Kawanami D, Hao C, Mahabaleshwar GH, Wang W, Jin Z-G, Atkins GB, et al. A novel role of CCN3 in regulating endothelial inflammation. J Cell Commun Signal. 2010;4(3):141–153. doi: 10.1007/s12079-010-0095-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karin N, Wildbaum G. The role of chemokines in shaping the balance between CD4(+) T cell subsets and its therapeutic implications in autoimmune and cancer diseases. Front Immunol. 2015;6:609. doi: 10.3389/fimmu.2015.00609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zohar Y, Wildbaum G, Novak R, Salzman AL, Thelen M, Alon R, Barsheshet Y, Karp CL, Karin N. CXCL11-dependent induction of FOXP3-negative regulatory T cells suppresses autoimmune encephalomyelitis. J Clin Invest. 2018;128(3):1200–1201. doi: 10.1172/JCI120358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Knight PA, Wright SH, Lawrence CE, Paterson YYW, Miller HRP. Delayed expulsion of the nematode Trichinella spiralis in mice lacking the mucosal mast cell-specific granule chymase, mouse mast cell protease-1. J Exp Med. 2000;192(12):1849–1856. doi: 10.1084/jem.192.12.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caughey GH. Mast cell proteases as protective and inflammatory mediators. Mast Cell Biol. 2011;716:212–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Davis RB, Kechele DO, Blakeney ES, Pawlak JB, Caron KM. Lymphatic deletion of calcitonin receptor-like receptor exacerbates intestinal inflammation. Jci Insight. 2017;2(6). doi: 10.1172/jci.insight.92465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.D’Haens GR, Geboes K, Peeters M, Baert F, Penninckx F, Rutgeerts P. Early lesions of recurrent Crohn’s disease caused by infusion of intestinal contents in excluded ileum. Gastroenterology. 1998;114(2):262–267. doi: 10.1016/S0016-5085(98)70476-7. [DOI] [PubMed] [Google Scholar]
- 45.Imdad A, Nicholson MR, Tanner-Smith EE, Zackular JP, Gomez-Duarte OG, Beaulieu DB, Acra S. Fecal transplantation for treatment of inflammatory bowel disease. Cochrane Database Syst Rev. 2018;11:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Levy AN, Allegretti JR. Insights into the role of fecal microbiota transplantation for the treatment of inflammatory bowel disease. Therap Adv Gastroenterol. 2019;12:175628481983689. doi: 10.1177/1756284819836893. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.