

Abstract
Introduction
Patient-derived skin fibroblasts offer a unique translational model to study molecular mechanisms of multiple human diseases. Metabolomics profiling allows to track changes in a broad range of metabolites and interconnected metabolic pathways that could inform on molecular mechanisms involved in disease development and progression, and on the efficacy of therapeutic interventions. Therefore, it is important to establish standardized protocols for metabolomics analysis in human skin fibroblasts for rigorous and reliable metabolic assessment.
Objectives
We aimed to develop an optimized protocol for concurrent measure of the concentration of amino acids, acylcarnitines, and components of the tricarboxylic acid (TCA) cycle in human skin fibroblasts using gas (GC) and liquid chromatography (LC) coupled with mass spectrometry (MS).
Methods
The suitability of four different methods of cell harvesting on the recovery of amino acids, acylcarnitines, and TCA cycle metabolites was established using GC/MS and LC/MS analytical platforms. For each method, metabolite stability was determined after 48 hours, two weeks and one month of storage at −80°C.
Results
Harvesting cells in 80% methanol solution allowed the best recovery and preservation of metabolites. Storage of samples in 80% methanol up to one month at −80°C did not significantly impact metabolite concentrations.
Conclusion
We developed a robust workflow for metabolomics analysis in human skin fibroblasts suitable for a high-throughput multiplatform analysis. This method allows a direct side-by-side comparison of metabolic changes in samples collected at different time that could be used for studies in large patient cohorts.
Keywords: Skin fibroblasts, Cell harvesting, Metabolomics, Amino acids, Acylcarnitines, TCA cycle
1. Introduction
Metabolomics is a systems biology approach that helps to understand mechanisms of complex diseases associated with dysregulated metabolic processes including cancer, diabetes, and neurodegenerative disorders (DeBerardinis and Thompson, 2012). Normal mitochondrial dynamics and function are crucial for proper metabolic and energy homeostasis (Nunnari and Suomalainen, 2012). Mitochondrial dysfunction is implicated in mechanisms of multiple human diseases affecting cell metabolism (Sorrentino et al., 2018, Nunnari and Suomalainen, 2012). The tricarboxylic acid (TCA) cycle is linked to the oxidative phosphorylation (OXPHOS) machinery and fatty acid oxidation, two major metabolic pathways important for the generation of ATP and precursors for multiple amino acids. Fatty acids, transported into mitochondria as acylcarnitines, are oxidized to generate acetyl-CoA, which is further oxidized in the TCA cycle to power OXPHOS and ATP production. Under normal conditions, glucose is the primary source catabolized via glycolysis to produce acetyl-CoA. Under disease conditions, such as Alzheimer’s disease (AD), glucose utilization could be decreased, and the TCA cycle could be powered by auxiliary fuels including fatty acids and amino acids (Wilkins and Trushina, 2017). Therefore, monitoring changes in salient energy metabolites could provide mechanistic insight into the disease development, identify potential targets for therapeutic interventions, and serve as biomarkers for disease progression and therapeutic efficacy.
Several recent studies reported changes in energy metabolites indicative of mitochondrial abnormalities using metabolomics profiling in biofluids from patients with cancer, diabetes, and neurodegenerative diseases (Guasch-Ferre et al., 2016, Lecuyer et al., 2018, Wu et al., 2016, Trushina et al., 2013). In our earlier study, application of untargeted metabolomics detected changes in metabolic pathways related to mitochondrial function and energy metabolism in cerebrospinal fluid (CSF) and plasma from patients with mild cognitive impairment (MCI), a prodromal stage of AD (Trushina et al. 2013). These changes became more pronounced in CSF and plasma from AD patients supporting the hypothesis that alterations in mitochondrial function underlie early disease mechanisms. Affected metabolic pathways in MCI and AD patients included the TCA cycle, amino acid metabolism, and lipid metabolism (Trushina et al., 2013) where later studies also identified changes in acylcarnitines (Toledo et al., 2017). While easily accessible biofluids such as plasma represent an excellent source of biomarkers for recurrent measures, they have limited use for mechanistic studies and the development of experimental therapeutics. A complementary approach could include the use of patient-derived skin fibroblasts where individualized changes associated with disease conditions could be evaluated using multiple techniques including metabolomics. Fibroblasts derived from patients with sporadic diseases (e.g. late onset AD) could be particularly useful as they may recapitulate underlying genetic, epigenetic, and metabolic changes that would be impossible to reproduce in animal models (Perez et al., 2017). In addition, metabolomics in fibroblasts offers allow us to study disease specific changes that are not influenced by variable factors inherent to human biological fluids such as diet, medications, and circadian cycle among others.
Despite major advancements in the field of metabolomics, standardized protocols suitable for the assessment of metabolic changes in cultured cells are lacking. Importantly, methods utilized for cell culture, harvest, and storage prior to the metabolomics analysis could significantly affect metabolite concentration and stability. For example, several harvesting techniques including trypsinization, mechanical detachment, and direct fixation of adherent cells were described in protocols used for metabolomics analysis making a direct comparison of results difficult (Dettmer et al., 2011, Kapoore et al., 2017, Muschet et al., 2016). Here, we compared four methods that differ in cell harvesting techniques and time of sample storage to develop an optimal method for the detection of amino acids, acylcarnitines, and metabolites of the TCA cycle in cultured human skin fibroblasts for high-throughput analysis using multiplatform targeted metabolomics.
2. Materials and methods
2.1. Skin fibroblasts and cell culture
Experiments with human fibroblasts were approved by the Mayo Clinic Internal Review Board. All experiments were conducted using the same fibroblast cell line derived from the skin biopsy of a healthy Caucasian male 21 years of age. Cells were received from the Mayo Clinic Biorepository (Cat. # 10268) and were passage 10 at the time of analysis. Fibroblasts were grown on 6 cm dishes for amino acid analysis and on 10 cm dishes for the TCA and acylcarnitine analysis. Cells were maintained in Minimum Essential Media (MEM; Corning Cat. # 10-010-CV) supplemented with 15% fetal bovine serum (FBS; Sigma Cat. # F2442), 1X non-essential amino acids (Corning Cat. # 25-025-CI), 2 mM L-glutamine (Gibco Cat. # 25030081), and 1X penicillin-streptomycin (Sigma Cat. # P0781). Cells were maintained at 37°C and 5 % CO2/95 % air. Cells were cultured for 48 hours prior to harvest.
2.2. Cell harvest
Skin fibroblasts were grown to 95% confluency. For cells grown on 6 cm dishes, half the volume of all buffers and solutions listed in the following methods was used. Four methods (Methods I - IV, Fig. 1) were utilized for cell collection. Prior to harvest, media was removed and cells were rinsed with PBS. In Method I, cells were dissociated by incubation with 0.05% trypsin at 37°C for 5 minutes. Dissociated cells were suspended in MEM (containing 15% FBS) to neutralize trypsin activity. Cells were centrifuged at 1000 x g for 5 min at 4°C. Pelleted cells were suspended in PBS and centrifuged at 1000 x g for 5 min at 4°C. The supernatant was aspirated, cell pellets were flash frozen in liquid nitrogen, and frozen pellets were stored at −80°C. In Method II, cells were detached in 1 mL PBS using a rubber scraper. Detached cells were centrifuged at 1000 x g for 5 min at 4°C, and PBS was aspirated. Cell pellets were flash frozen in liquid nitrogen and stored at −80°C. In Method III, 1 mL 80% methanol (pre-chilled on dry ice) was added to cells. Tissue culture plates were rocked by hand for about 20 seconds to evenly distribute methanol solution. Cells were detached in 80% methanol using a rubber scraper. Suspensions were transferred to an Eppendorf tube and stored at −80°C. In Method IV, cells were harvested in 1 mL 80% methanol, and the supernatant was evaporated using a Speed Vacuum at room temperature. Dried samples were stored at −80°C. Frozen samples collected using Methods I - IV were stored at −80°C for 48 hours, two weeks, and four weeks prior to metabolomics analysis.
Fig. 1.
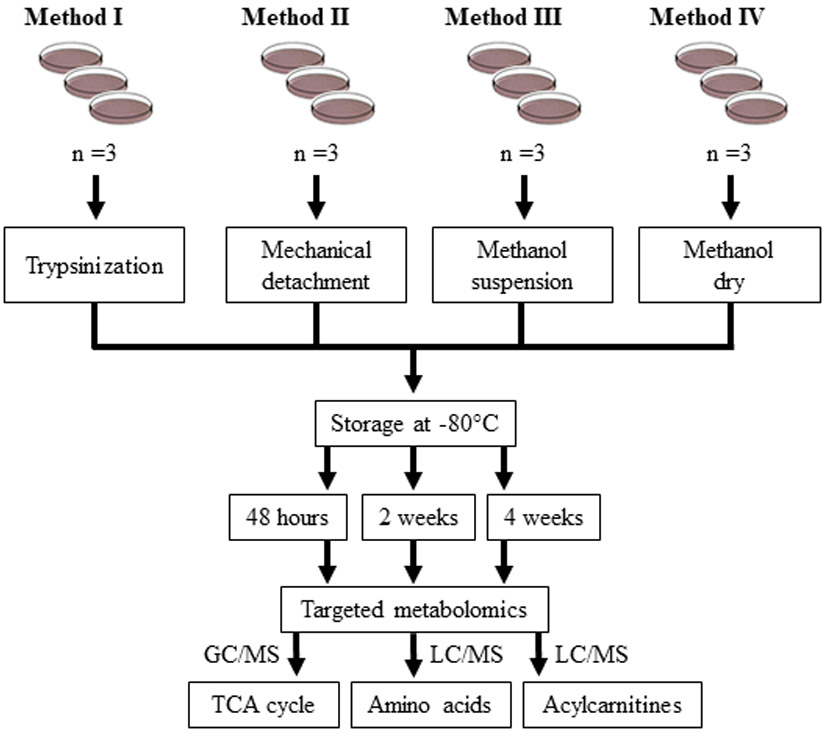
Workflow for the harvesting, storage, and targeted metabolomics analysis in skin fibroblasts. Cultured skin fibroblasts (n = 3 per group) were collected using four different methods (Methods I - IV) followed by storage at −80°C for 48 hours, two weeks, or four weeks. Targeted metabolomics analysis of the TCA cycle metabolites was carried out using gas chromatography/mass spectrometry (GC/MS). Amino acids and acylcarnitines were profiled using liquid chromatography/mass spectrometry (LC/MS).
2.3. Targeted metabolomics
Targeted metabolomics analysis of amino acids, acylcarnitines, and metabolites of the TCA cycle was conducted at the Mayo Clinic Metabolomics Core using previously established analytical techniques (Koek et al., 2006, Dutta et al., 2016, Lanza et al., 2010, Chace et al., 2001). The metabolites were classified using the Metabolomics Standards Initiative (MSI) guidelines (Salek et al., 2013). Supplementary Table 1 lists the Human Metabolome Database IDs and MSI classification level. Prior to the analyses, samples harvested in methanol (Method III) were dried using a Speed Vacuum. Cell pellets were sonicated in 100 μL PBS. Each sample was spiked with 15 - 25 μL of the respective internal standards (Supplementary Table 1). Samples were incubated in an ice bath for 15 minutes. Proteins were removed by adding 450 μL of cold 1:1 methanol/acetonitrile solution with subsequent centrifugation for 15 minutes (18,000 x g at 4°C). Supernatants were transferred to a 1 mL dram and dried under a nitrogen stream for approximately 30 minutes. Sodium hydroxide solution (400 μL of 0.3N NaOH) was added to the protein pellets and incubated overnight at 4°C prior to measuring protein concentration using BCA protein assay kit (Thermo Fisher Scientific, Cat. # 23235) (Kruger, 1994). Prior to detection, TCA cycle metabolites were derivatized using ethoxyamine following with MtBSTFA + 1% tBDMCS (N-Methyl-N-(t-Butyldimethylsilyl)-Trifluoroacetamide + 1% t-Butyldimethylchlorosilane). Amino acids were derivatized with 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate using the Waters AccQ-Fluor Reagent Kit (Cat. # WATO52880). Acylcarnitines were reconstituted in buffer containing 99% MeOH, 1% H2O, 1 mM ammonium formate, and 0.1% formic acid.
Metabolites of the TCA cycle (Supplementary Table 1) were detected with an Agilent 5977A gas chromatography/mass spectrometry (GC/MS) under electron impact and single ion monitoring conditions in positive mode (Koek et al., 2006). Analytes were separated on an Agilent DB-5MS column (30 m x 0.25 mm x 0.25 μm). Sample injection volume was 1 μL performed in splitless mode. The inlet temperature was maintained at 250°C. The carrier gas was helium set at a flow rate of 0.9 ml/min. The initial oven temperature was 120°C set with the following ramp rates: Ramp to 180°C at 25°C/min; Ramp to 270°C at 6°C/min; Ramp to 325°C at 30°C/min. The transfer line temperature was 280°C. Concentrations of lactic acid, fumaric acid, succinic acid, oxaloacetic acid, alpha-ketoglutaric acid, malic acid, 2-hydroxyglutaric acid, cis-aconitic acid, citric acid, and isocitric acid were measured against a calibration curve (Koek et al., 2006, Dutta et al., 2016).
Amino acids (Supplementary Table 1) were analyzed using Thermo TSQ Quantum Ultra mass spectrometer (West Palm Beach, FL) coupled with a Waters ACQUITY ultra performance liquid chromatography (UPLC) system. Analytes were separated on a Waters BEH C18 column (2.1 mm x 150 mm x. 1.7 μm) prior to entering the mass spectrometer. Data acquisition was performed using selected reaction monitoring (SRM) and positive electrospray ionization (ESI). Injection volume was 2 μL. The column flow rate was 400 μL/min with an isothermal set at 43°C. Mobile phase A was 1% acetonitrile in 0.1% formic acid, and mobile phase B was 100% acetonitrile. Elution of analytes was achieved by the following gradient: 0-1.0 minute 0.1% B, 2.0 minutes 1.5% B, 5.5 minutes 1.9% B, 6.5 minutes 2.0% B, 10.0 2.4% B, 12.0 minutes 4% B, 20 minutes 12% B, 27 minutes 13.5% B, 30 minutes 20% B, 31 minutes 98% B, isocratic for 3 minutes, 34.5 minutes 0.1% B, hold for 4 minutes. The mass spectrometer was operated with 4000 capillary voltage, 50 sheath gas, 20 auxiliary gas, and 15 sweep gas. The capillary temperature was 270°C. Collision gas was 1.5 Torr and collision energy was 25 V. The tube lens was kept at 90 V. The concentration of amino acids (Supplementary Table 1) was calculated against a calibration curve (Lanza et al., 2010).
Acylcarnitines (Supplementary Table 1) were analyzed using a Waters ACQUITY UPLC system (Milford, MA) coupled with a Thermo TSQ Quantiva tandem mass spectrometer (West Palm Beach, FL) in SRM and positive ESI mode. Two microliters of sample was injected for analysis. Analytes were separated on a Waters BEH C8 column (2.1 mm x 150 mm x. 1.7 μm) with an isothermal temperature of 43°C using the following mobile phases and gradient, A: 1% methanol in 2 mM ammonium formate and 0.1% formic acid, B: 99% methanol in 1 mM ammonium formate and 0.1% formic acid, starting at 1% B, increasing to 80% B at 6.5 minutes, then to 99% B at 10 minutes, hold for 2 minutes at 99% B then decreasing to starting conditions at 13 minutes for three minutes. The mass spectrometer capillary voltage was set to 4000 with a sheath gas 30, auxiliary gas 5, and sweep gas 2. The ion transfer tube was maintained at 300°C with the vaporizer at 40°C, collision gas at 1.5 Torr, and collision energy at 12 V. Concentrations of carnitine, acetylcarnitine, propionylcarnitine, butyrylcarnitine, isovalerylcarnitine, octanoylcarnitine, lauroylcarnitine, myristoylcarnitine, palmitoylcarnitine, oleoylcarnitine, and stearoylcarnitine were measured against a calibration curve (Chace et al., 2001).
Data sets collected by GC/MS were analyzed using Mass Hunter GC/MS Quantitation software version B.07 (Agilent). Analysis of LC/MS data sets was performed using Xcalibur Quant browser (Thermo Scientific).
2.4. Statistical analysis
Metabolite concentrations in each sample were normalized to protein content measured from the protein pellet after the deproteinization step (see materials and methods section 2.3). Data analysis was performed in GraphPad Prism 7 using ANOVA with post-hoc Tukey test. The heat map was generated using Partek software after normalizing data so each column mean equals zero with a standard deviation of one. Transformed data was used to ensure each metabolite had an equal weight in the heat map.
3. Results and discussion
Our goal was to establish a protocol for the detection and quantitation of salient metabolites involved in mitochondrial energy metabolism in primary human skin fibroblasts. We focused on standardization and optimization of pre-analytical steps of the protocol that are usually carried out in the investigator’s laboratory (e.g., cell harvest and sample storage) that could influence data integrity rather than on procedures established within an analytical facility (e.g., drying conditions, derivatization, etc.) that have already undergone rigorous optimization as part of standard operating procedures (SOP) established at the Mayo Metabolomics Core. Untargeted metabolomics profiling permits the detection of thousands of metabolites using one analytical platform. Although such an agnostic approach is a tremendous discovery tool, this method does not allow precise quantitation of metabolites of interest. Targeted metabolomics provides the measurement of metabolite concentrations. However, the complexity of this analysis is associated with the utilization of multiple analytical platforms where metabolites of the TCA cycle are detected using GC/MS and amino acids and acylcarnitines using LC/MS/MS. Moreover, the requirements of current clinical research necessitate large patient cohorts to ensure reproducibility, scientific rigor, and adequate power to evaluate sex-specific differences. Since culturing fibroblasts demands a substantial amount of time, we also determined to what extent the duration of sample storage affects metabolite stability in order to provide a protocol where multiple samples collected at different time could be subjected to simultaneous metabolite measurements as a single batch. We compared four methods of cell harvesting and the effect of storage at −80°C on metabolite concentration using the same cell line (Fig. 1). Our results provide a method for optimum metabolite recovery that allows storing samples without significant metabolite degradation for up to one month.
3.1. Effect of cell harvesting techniques on metabolite concentration
We compared four different methods previously reported for cell harvesting for metabolomics analysis on metabolite recovery (Fig. 1) (Dettmer et al., 2011, Kapoore et al., 2017, Muschet et al., 2016). Cells were collected using trypsinization (Method I), mechanical detachment (Method II), and methanol fixation (Methods III and IV) (Fig. 1). Principal component analysis demonstrates that each method has consistent recovery profiles within three replicates but significantly differs in the concentrations of metabolites detected using different techniques (Fig. 2). In Method I, cells were detached using standard trypsinization protocol. Dissociation by trypsin is considered a gentle form of cell detachment that should preserve intact cells. However, Method I appeared to be time consuming, as it required several minutes of incubation with trypsin and multiple centrifugation steps to ensure cells were thoroughly rinsed and free of residual media and trypsin prior to storage. While Method I had the greatest recovery of acylcarnitines, the overall recovery of the TCA cycle metabolites and amino acids was lower compared to Methods III and IV (Fig. 3 and Supplementary Table 2). In Method II, fibroblasts were detached in PBS using a rubber scraper. The total time required for cell freezing using Method II was less than ten minutes. Unexpectedly, Method II had the lowest recovery of all metabolites compared to any other Methods (Fig. 3 and Supplementary Table 2). Reduced recovery of metabolites in Method II was most likely associated with the damage to cell membranes and the leakage of metabolites during scraping. In Methods III and IV, cells were directly quenched with ice-cold methanol. Method III demonstrated the best recovery of all metabolites despite the fact that cells were collected after quenching using a rubber scraper as in Method II. In Method III, we also did not remove methanol prior to placing samples in −80°C, which significantly reduced the time required for sample collection (about 5 min). Method IV differed from Method III in one additional step where methanol was evaporated prior to storage at −80°C. In Method IV, the recovery of five metabolites including alpha-ketoglutaric acid, isocitric acid, glutamic acid, gamma-aminobutyric acid, and aminoadipic acid was reduced compared to Method III (Fig. 3 and Supplementary Table 2). Thus, Method III was deemed as the most efficient allowing preservation of most metabolites (Fig. 3 and Supplementary Table 2).
Fig. 2.
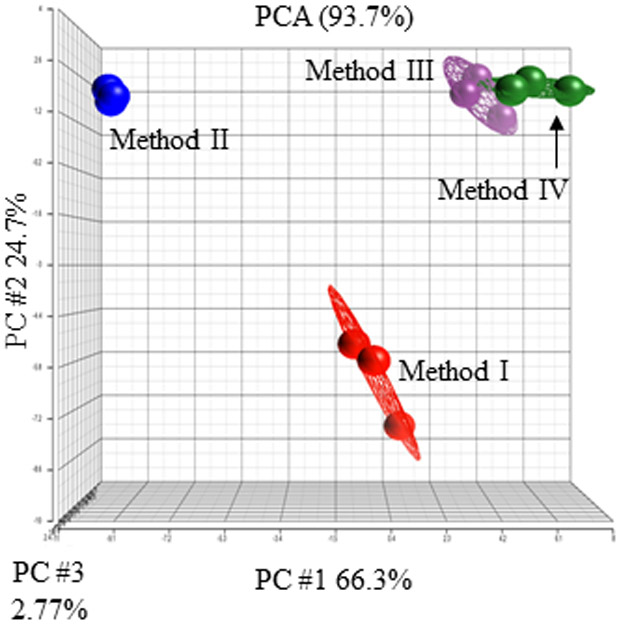
Cell culture harvesting methods influence metabolite profiles in skin fibroblasts. Principal component analysis comparing metabolic profiles in skin fibroblast harvested using Methods I - IV. Results are shown for three replicates per each Method. Each circle represents an individual sample.
Fig. 3.

Hierarchical clustering showing relative concentrations of metabolites in skin fibroblasts collected by Methods I – IV. The average metabolite value obtained for each Method was used for the analysis. Each row represents a different collection Method and columns signify a metabolite. The green and black colors indicate higher and lower metabolite abundances, respectively.
3.2. Effect of storage on metabolite concentration
In many cases, projects require prolonged storage of samples prior to metabolomics analyses. To ensure that such analysis is feasible without compromising data quality, we determined the composition of samples collected using Methods I - IV after 48 hours, two weeks, and one month of storage at −80°C (Supplementary Table 2). After 4 weeks of storage, samples collected by Methods I and II had a significant reduction in the concentration of 3 and 19 metabolites, respectively (Supplementary Table 2). Methods III and IV had significant change in two (alpha-ketoglutaric acid and glutamic acid) and one (glutamine) metabolites, respectively (Supplementary Table 2). Some of the metabolites including alpha-ketoglutaric acid and oxaloacetic acid are known to be unstable and be present at low abundance (Han et al., 2013, Mamer et al., 2013). Indeed, we had difficulty detecting oxaloacetic acid in all of our samples. As metabolites have a wide range of physicochemical properties (e.g. charge, volatility, hydrophilicity, etc.) some variation in metabolite detection after storage is expected warranting additional consideration particularly when measuring unstable analytes of interest. Overall, these results demonstrate that storage up to one month minimally impacted metabolite concentration when collected using Methods III and IV.
Application of Method III provided the best outcomes representing the most efficient technique for metabolomics analysis of energy metabolites in human fibroblasts. An optimized workflow for the collection of skin fibroblasts for targeted analysis of amino acids, acylcarnitines, and the TCA cycle is presented in Figure 4. This method resulted in the greatest enrichment of energy metabolites, which was minimally impacted during storage. In our study, targeted metabolomics was carried out at the Mayo Clinic Metabolomics Core, which is one of six NIH Regional Comprehensive Metabolomics Resource Cores (RCMRCs) supported by the NIH (https://commonfund.nih.gov/metabolomics). Services at these RCMRCs are available for interested researchers nationwide. Simultaneous daily assessment of samples for targeted metabolomics at the Mayo Clinic RCMRC includes approximately 30 samples for the TCA cycle, 40 for amino acids, and 60 for acylcarnitines (Fig. 4). Harvesting 60 samples using the optimized workflow (Fig. 4) would take an individual approximately five hours and could be completed in a single day. Inclusion of additional samples would likely require multiple days for metabolomics analysis. Therefore, it is important to randomize samples for analysis to account for potential variability due to batch-to-batch variation. Randomization during sample preparation, analysis, and peak-picking is also essential to reduce bias. Together, the standardized protocols for sample preparation, robust quality control procedures, and validated SOPs established in the RCMRCs ensure consistency between analyses performed on different days.
Fig. 4.
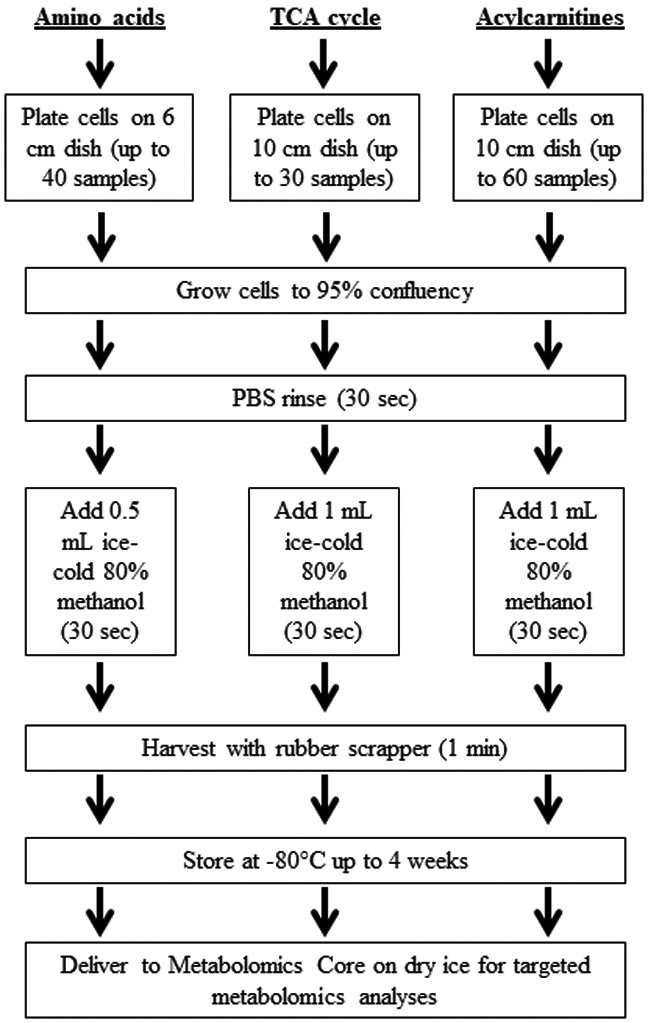
Optimized workflow for targeted analysis of amino acids, acylcarnitines, and TCA cycle metabolites in human skin fibroblasts. Fibroblasts are grown to 95% confluency on 6 cm or 10 cm dishes for amino acid or TCA cycle and acylcarnitine analysis, respectively. Approximately 30 samples for the TCA cycle, 40 samples for amino acids, and 60 samples for acylcarnitines can be measured in a single run. Cells are rinsed with PBS to remove residual media. Ice-cold 80% methanol is added to cells and harvested using a rubber scraper. All cells and methanol are transferred to an Eppendorf tube; samples could be stably stored up to one month at −80 °C.
4. Additional considerations for metabolomics analysis in cells
Measurements of metabolites with diverse chemical properties and concentration range in biological samples such as human fibroblasts require the use of multiple analytical platforms emphasizing the importance of the standardized procedures so that data generated in different laboratories could be easily compared. We found that metabolites could be significantly influenced by common techniques utilized for cell harvest. In our hands, quick quenching of cells with cold methanol and storing as a slurry or dried pellet resulted in the greatest enrichment and stability of most metabolites after one month of storage. Neither scraping nor trypsinization of cells produced optimum results most likely due to the damage to cellular membranes and changes in metabolic activity during prolonged procedures. While most of metabolites evaluated in this study were concentrated and well presetved in methanol samples, some metabolites including few amino acids and acylcarnitines were particularly enriched when cells were harvested using trypsin (Method I). In comparison, carnitine was most abundant when cells were harvested in methanol (Methods III and IV). Similar, glutamine, ethanolamine, phosphoethanolamine, glycine, 5-hydroxylysine and cystathionine were enriched in samples collected with Method I but not III or IV. This observation emphasizes the requirement to know the behavior (stability, reactivity, solubility, etc.) of metabolites of interest prior to the analysis since changes in carnitine concentrations, for example, could be very important informing on the mechanism of multiple metabolic diseases (Khan et al., 2014, Shibbani et al., 2014, Lodeiro et al., 2014).
Additional factors including cell culture medium composition, culture dishes (e.g., glass vs. plastic), and cell characteristics such as sex, age, the race of the donor, and passages in culture could impact the outcomes and need to be carefully documented. It is especially important in case of cell passages since with age in culture, fibroblasts acquire senescence phenotype that could significantly change cell metabolism. Rapid quenching of cellular activity is essential to obtain an accurate snapshot of the metabolome, especially in samples with high metabolic activity (Martano et al., 2015). Several methods have been utilized to quench cellular metabolism and facilitate metabolite extraction including the use of methanol, sample freezing or heating, and adjusting the pH (Martano et al., 2015, Puchalska et al., 2019, Fiehn et al., 2000, Shukla et al., 2015, Ser et al., 2015, Chrysanthopoulos et al., 2010). Multiple extraction solvents have also been proposed including hot/cold methanol, ethanol, chloroform/methanol, acid/basic solutions, and acetonitrile (Hernandez Bort et al., 2014). Depending on the nature of the sample and metabolites of interest, it might be necessary to test different extraction protocols to achieve the best recovery of metabolites. Furthermore, given that samples are likely to be frozen prior to metabolomics analysis, it is important to ensure that cell pellets are sufficiently disrupted in order to maximize the extraction and precipitation of proteins after defrosting. Incomplete disruption could reduce the total concentration of protein, which is often used to normalize metabolite concentrations. Techniques including ultrasonication, bead beating, freeze-thaw cycle, and homogenization are effective methods for cell disruption prior to metabolite extraction (Luo and Li, 2017, Danielsson et al., 2010, Cao et al., 2011). In our case, sonication allowed to achieve reproducible results. After the extraction, solvents are often dried in order to further prepare samples GC/MS or LC/MS analysis (as detailed in the Materials and Methods) and (Lanza et al., 2010, Koek et al., 2006, Dutta et al., 2016, Chace et al., 2001). However, volatile compounds (e.g., acetone and acetic acid) can be lost during drying steps when desiccated under a vacuum and/or nitrogen stream requiring careful planning if such metabolites are of interest. In contrast, other metabolites including those of the TCA cycle and amino acids require a derivatization to render them more volatile prior to the analysis (Koek et al., 2006, Lanza et al., 2010). Therefore, with metabolomics profiling becoming increasingly adapted for research and clinical applications, the development of standardized protocols specific for particular model and sets of metabolites becomes the key to success. However, each protocol has its limitations, so it is advisable that the investigators carefully consider the suitability of a particular method to achieve their objectives.
5. Concluding remarks
There is an increasing need for models to study mechanisms of human diseases. Transgenic animal models commonly used for translational research have limitations that could affect successful translation of clinically relevant findings. Patient-derived skin fibroblasts provide a promising model that may retain underlying genetic, epigenetic, and metabolic changes associated with complex diseases. Additionally, skin fibroblasts could be used to generate specialized cells including neurons that normally cannot be obtained from living individuals. This versatility of skin fibroblasts offers opportunities for individualized medicine. The metabolome can reflect the status of an entire organism in health and disease that is influenced by the genome, proteome, and environmental exposures. Thus, metabolomics analysis of human skin fibroblasts could provide a snapshot that is characteristic of an individual’s physical traits. Generating standardized methods to study metabolic mechanisms in patient-derived cells is important for establishing a human translational model for diagnosis, prognosis, and testing of therapeutic efficacy.
Supplementary Material
Acknowledgements
This study was supported by the National Institutes of Health NIEHS, NIA, NINDS (grant numbers R01ES020715, RF1AG 55549, R01NS107265) and Mayo Clinic Metabolomics Core (NIDDK grant number U24DK100469); Mayo Clinic Clinomics; and Mayo Clinic Center for Multiple Sclerosis and Autoimmune Neurology
Footnotes
Data availability All data generated or analyzed during this study are included in this publication. All raw data collected in this study was uploaded to the Metabolomics Workbench: NIH Data Repository (website: https://www.metabolomicsworkbench.org/).
Software availability No software was developed in this study.
Conflict of interest All authors declare no conflict of interest.
Compliance with ethical standards All ethical guidelines and procedures established by the Mayo Clinic Institutional Review Board for the use of human skin fibroblasts were followed in this study.
References
- CAO B, AA J, WANG G, WU X, LIU L, LI M, SHI J, WANG X, ZHAO C, ZHENG T, GUO S & DUAN J 2011. GC-TOFMS analysis of metabolites in adherent MDCK cells and a novel strategy for identifying intracellular metabolic markers for use as cell amount indicators in data normalization. Anal Bioanal Chem, 400, 2983–93. [DOI] [PubMed] [Google Scholar]
- CHACE DH, DIPERNA JC, MITCHELL BL, SGROI B, HOFMAN LF & NAYLOR EW 2001. Electrospray tandem mass spectrometry for analysis of acylcarnitines in dried postmortem blood specimens collected at autopsy from infants with unexplained cause of death. Clin Chem, 47, 1166–82. [PubMed] [Google Scholar]
- CHRYSANTHOPOULOS PK, GOUDAR CT & KLAPA MI 2010. Metabolomics for high-resolution monitoring of the cellular physiological state in cell culture engineering. Metab Eng, 12, 212–22. [DOI] [PubMed] [Google Scholar]
- DANIELSSON AP, MORITZ T, MULDER H & SPEGEL P 2010. Development and optimization of a metabolomic method for analysis of adherent cell cultures. Anal Biochem, 404, 30–9. [DOI] [PubMed] [Google Scholar]
- DEBERARDINIS RJ & THOMPSON CB 2012. Cellular metabolism and disease: what do metabolic outliers teach us? Cell, 148, 1132–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DETTMER K, NURNBERGER N, KASPAR H, GRUBER MA, ALMSTETTER MF & OEFNER PJ 2011. Metabolite extraction from adherently growing mammalian cells for metabolomics studies: optimization of harvesting and extraction protocols. Anal Bioanal Chem, 399, 1127–39. [DOI] [PubMed] [Google Scholar]
- DUTTA T, KUDVA YC, PERSSON XM, SCHENCK LA, FORD GC, SINGH RJ, CARTER R & NAIR KS 2016. Impact of Long-Term Poor and Good Glycemic Control on Metabolomics Alterations in Type 1 Diabetic People. J Clin Endocrinol Metab, 101, 1023–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FIEHN O, KOPKA J, TRETHEWEY RN & WILLMITZER L 2000. Identification of uncommon plant metabolites based on calculation of elemental compositions using gas chromatography and quadrupole mass spectrometry. Anal Chem, 72, 3573–80. [DOI] [PubMed] [Google Scholar]
- GUASCH-FERRE M, HRUBY A, TOLEDO E, CLISH CB, MARTINEZ-GONZALEZ MA, SALAS-SALVADO J & HU FB 2016. Metabolomics in Prediabetes and Diabetes: A Systematic Review and Meta-analysis. Diabetes Care, 39, 833–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAN J, GAGNON S, ECKLE T & BORCHERS CH 2013. Metabolomic analysis of key central carbon metabolism carboxylic acids as their 3-nitrophenylhydrazones by UPLC/ESI-MS. Electrophoresis, 34, 2891–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HERNANDEZ BORT JA, SHANMUKAM V, PABST M, WINDWARDER M, NEUMANN L, ALCHALABI A, KREBIEHL G, KOELLENSPERGER G, HANN S, SONNTAG D, ALTMANN F, HEEL C & BORTH N 2014. Reduced quenching and extraction time for mammalian cells using filtration and syringe extraction. J Biotechnol, 182-183, 97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAPOORE RV, COYLE R, STATON CA, BROWN NJ & VAIDYANATHAN S 2017. Influence of washing and quenching in profiling the metabolome of adherent mammalian cells: a case study with the metastatic breast cancer cell line MDA-MB-231. Analyst, 142, 2038–2049. [DOI] [PubMed] [Google Scholar]
- KHAN H,S ALHOMIDA A, HABIB S, ALI KHAN A, OLA M,J SIDDIQUI N, SOBKI S & AL MADANI H. 2014. Blood carnitine as a biomarker for acute myocardial infarction.
- KOEK MM, MUILWIJK B, VAN DER WERF MJ & HANKEMEIER T 2006. Microbial metabolomics with gas chromatography/mass spectrometry. Anal Chem, 78, 1272–81. [DOI] [PubMed] [Google Scholar]
- KRUGER NJ 1994. The Bradford method for protein quantitation. Methods Mol Biol, 32, 9–15. [DOI] [PubMed] [Google Scholar]
- LANZA IR, ZHANG S, WARD LE, KARAKELIDES H, RAFTERY D & NAIR KS 2010. Quantitative metabolomics by H-NMR and LC-MS/MS confirms altered metabolic pathways in diabetes. PLoS One, 5, e10538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LECUYER L, VICTOR BALA A, DESCHASAUX M, BOUCHEMAL N, NAWFAL TRIBA M, VASSON MP, ROSSARY A, DEMIDEM A, GALAN P, HERCBERG S, PARTULA V, LE MOYEC L, SROUR B, FIOLET T, LATINO-MARTEL P, KESSE-GUYOT E, SAVARIN P & TOUVIER M 2018. NMR metabolomic signatures reveal predictive plasma metabolites associated with long-term risk of developing breast cancer. Int J Epidemiol, 47, 484–494. [DOI] [PubMed] [Google Scholar]
- LODEIRO M, IBANEZ C, CIFUENTES A, SIMO C & CEDAZO-MINGUEZ A 2014. Decreased cerebrospinal fluid levels of L-carnitine in non-apolipoprotein E4 carriers at early stages of Alzheimer’s disease. J Alzheimers Dis, 41, 223–32. [DOI] [PubMed] [Google Scholar]
- LUO X & LI L 2017. Metabolomics of Small Numbers of Cells: Metabolomic Profiling of 100, 1000, and 10000 Human Breast Cancer Cells. Anal Chem, 89, 11664–11671. [DOI] [PubMed] [Google Scholar]
- MAMER O, GRAVEL SP, CHOINIERE L, CHENARD V, ST-PIERRE J & AVIZONIS D 2013. The complete targeted profile of the organic acid intermediates of the citric acid cycle using a single stable isotope dilution analysis, sodium borodeuteride reduction and selected ion monitoring GC/MS. Metabolomics, 9, 1019–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARTANO G, DELMOTTE N, KIEFER P, CHRISTEN P, KENTNER D, BUMANN D & VORHOLT JA 2015. Fast sampling method for mammalian cell metabolic analyses using liquid chromatography-mass spectrometry. Nat Protoc, 10, 1–11. [DOI] [PubMed] [Google Scholar]
- MUSCHET C, MOLLER G, PREHN C, DE ANGELIS MH, ADAMSKI J & TOKARZ J 2016. Removing the bottlenecks of cell culture metabolomics: fast normalization procedure, correlation of metabolites to cell number, and impact of the cell harvesting method. Metabolomics, 12, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NUNNARI J & SUOMALAINEN A 2012. Mitochondria: in sickness and in health. Cell, 148, 1145–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PEREZ MJ, PONCE DP, OSORIO-FUENTEALBA C, BEHRENS MI & QUINTANILLA RA 2017. Mitochondrial Bioenergetics Is Altered in Fibroblasts from Patients with Sporadic Alzheimer’s Disease. Front Neurosci, 11, 553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PUCHALSKA P, MARTIN SE, HUANG X, LENGFELD JE, DANIEL B, GRAHAM MJ, HAN X, NAGY L, PATTI GJ & CRAWFORD PA 2019. Hepatocyte-Macrophage Acetoacetate Shuttle Protects against Tissue Fibrosis. Cell Metab, 29, 383–398 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SALEK RM, STEINBECK C, VIANT MR, GOODACRE R & DUNN WB 2013. The role of reporting standards for metabolite annotation and identification in metabolomic studies. Gigascience, 2, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SER Z, LIU X, TANG NN & LOCASALE JW 2015. Extraction parameters for metabolomics from cultured cells. Anal Biochem, 475, 22–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIBBANI K, FAHED AC, AL-SHAAR L, ARABI M, NEMER G, BITAR F & MAJDALANI M 2014. Primary carnitine deficiency: novel mutations and insights into the cardiac phenotype. Clin Genet, 85, 127–37. [DOI] [PubMed] [Google Scholar]
- SHUKLA SK, GUNDA V, ABREGO J, HARIDAS D, MISHRA A, SOUCHEK J, CHAIKA NV, YU F, SASSON AR, LAZENBY AJ, BATRA SK & SINGH PK 2015. MUC16-mediated activation of mTOR and c-Myc reprograms pancreatic cancer metabolism. Oncotarget, 6, 19118–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SORRENTINO V, MENZIES KJ & AUWERX J 2018. Repairing Mitochondrial Dysfunction in Disease. Annu Rev Pharmacol Toxicol, 58, 353–389. [DOI] [PubMed] [Google Scholar]
- TOLEDO JB, ARNOLD M, KASTENMULLER G, CHANG R, BAILLIE RA, HAN X, THAMBISETTY M, TENENBAUM JD, SUHRE K, THOMPSON JW, JOHN-WILLIAMS LS, MAHMOUDIANDEHKORDI S, ROTROFF DM, JACK JR, MOTSINGER-REIF A, RISACHER SL, BLACH C, LUCAS JE, MASSARO T, LOUIE G, ZHU H, DALLMANN G, KLAVINS K, KOAL T, KIM S, NHO K, SHEN L, CASANOVA R, VARMA S, LEGIDO-QUIGLEY C, MOSELEY MA, ZHU K, HENRION MY, VAN DER LEE SJ, HARMS AC, DEMIRKAN A, HANKEMEIER T, VAN DUIJN CM, TROJANOWSKI JQ, SHAW LM, SAYKIN AJ, WEINER MW, DORAISWAMY PM, KADDURAH-DAOUK R, ALZHEIMER’S DISEASE NEUROIMAGING, I. & THE ALZHEIMER DISEASE METABOLOMICS, C. 2017. Metabolic network failures in Alzheimer’s disease-A biochemical road map. Alzheimers Dement. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TRUSHINA E, DUTTA T, PERSSON XM, MIELKE MM & PETERSEN RC 2013. Identification of altered metabolic pathways in plasma and CSF in mild cognitive impairment and Alzheimer’s disease using metabolomics. PLoS One, 8, e63644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILKINS JM & TRUSHINA E 2017. Application of Metabolomics in Alzheimer’s Disease. Front Neurol, 8, 719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WU J, WUOLIKAINEN A, TRUPP M, JONSSON P, MARKLUND SL, ANDERSEN PM, FORSGREN L & ÖHMAN A 2016. NMR analysis of the CSF and plasma metabolome of rigorously matched amyotrophic lateral sclerosis, Parkinson’s disease and control subjects. Metabolomics, 12, 101. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.