

Abstract
The emerging N501Y mutation in severe acute respiratory syndrome coronavirus 2, which becomes prevalent in the UK rapidly, is one of the major challenges of COVID-19 control. To explore the transmission advantage, we estimate that the N501Y substitution increases the infectivity by 52% (95% confidence interval: 46, 58) in terms of the reproduction number.
Keywords: COVID-19, mutation, transmission advantage, reproduction number, statistical modelling, B.1.1.7 lineage
Main text
Exploring the relationship between mutation activities and disease transmissibility is important to manage the epidemiological outcomes at population scale. In September 2020, genetic variants carrying the N501Y substitution on the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike (S) protein began to spread first in Wales and then otherwhere in the UK1 and reached dominance among SARS-CoV-2 variants in other lineages by the end of 2020. The rapid spread of 501Y variant indicates a possible transmission advantage over the 501N variant. In this work, we adopted a statistical framework to infer the transmission advantage associated with N501Y substitution using the surveillance and sequencing data in the UK.
We reconstructed the variant-specified instantaneous reproduction number by using the commonly adopted renewable equation.2 The transmission advantage of 501Y variant against 501N variant is defined as the ratio, η, between variant-specified reproduction numbers, see Supplementary Information S1. In other words, given the reproduction number of cases infected by 501N variant as Rt, the reproduction number of cases infected by the 501Y variant is η·Rt. Thus, if η > 1, the mutated variant may be more infectious than the original genetic variant and vice versa.
We obtained the SARS-CoV-2 sequencing data and COVID-19 surveillance data (Figure 1A) collected from 1 August to 31 December 2020, via the public databases released by GISAID and World Health Organization, respectively, see Supplementary Information S2. With the likelihood-based inference framework, we reconstruct the variant-specified instantaneous reproduction number in Figure 1B. The reproduction number of 501Y variant appears higher than that of 501N variant. Although Rt of the 501N variant fluctuated around 1, the η·Rt of 501Y variant appears generally above 1 during the same period, which implies a higher potential for causing larger epidemics, and more intensive intervention strategies are thus desired. Figure 1C shows that the estimated proportion of 501Y variant fits the observed proportion well. We estimated that the 501Y variant is 52% (95% confidence interval: 46, 58) more transmissible than the 501N variant, which indicates a transmission advantage of N501Y substitution. To check the sensitivity of estimates, we repeat the estimating process of η with alternative shorter or longer mean generation time. We find that the η estimates are consistently and significantly larger than 1 in similar scales (data not shown), which validates our main results.
Figure 1.
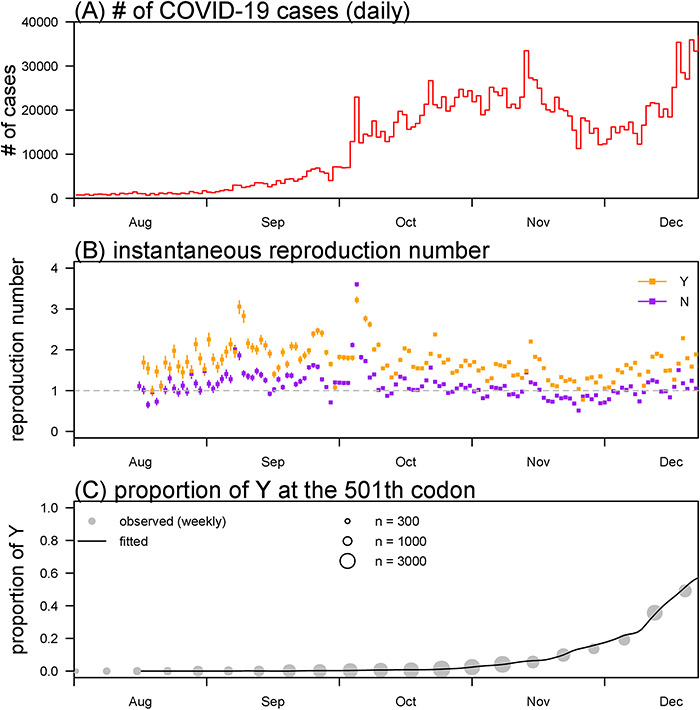
The daily number of COVID-19 cases (panel A), the reconstructed reproduction number (Rt, panel B) and proportion of the Y on the 501th codon of the S protein (panel C). Panel A shows the daily number of COVID-19 time series in the UK. Panel B shows the estimated reproduction numbers of Y (in orange) and N (in purple). The dots are the estimates, and bars are the 95% confidence intervals. Panel C shows the observed (dots) and fitted (curve) proportion of the Y on the 501th codon of the S protein
The increase in transmissibility associated with N501Y substitution appears consistent with the statistical evidence and structural biological insights in literature.3,4 Similarly, we learn from the recent D614G substitution of SARS-CoV-2, as well as the seasonal influenza virus, that single but key AA substitution may alter the conformation of protein trimer and binding-competent profiles.5
Provided the rapidly growing prevalence and spread of 501Y variant, several concerns of public health importance are raised, which need further exploration. The transmission advantage of the mutation implies increase in both herd immunity threshold and intrinsic growth rate of epidemic, and thus, the non-pharmaceutical interventions for COVID-19 control may require enforcement to flatten the epidemic curve. Clinical severity remains largely unassessed for the 501Y variant, which needs relatively a long-term observation, and unexpected clinical outcomes may warrant adjustments in the treatment strategies. Given the challenges in the control of spatial spreading of COVID-19, we highlight the potential risks of SARS-CoV-2 with 501Y when it invades places having groups of population with vulnerability. There is a limited investigation for the roles of other mutations co-circulating with N501Y, which might be of importance in compensating or enhancing the viral fitness or functionality. In addition, the matching level of vaccine candidates in development or in production is also unknown, which requires further analysis from both biological and epidemiological sides. Some limitations about the analytical procedures, mainly due to lack of data or information, are discussed in Supplementary Information S3.
Ethics approval and consent to participate
The COVID-19 number of cases and sequencing data are collected via public domains, and thus, neither ethical approval nor individual consent is applicable.
Availability of materials
All data used in this work are publicly available.
Consent for publication
Not applicable.
Authors’ contributions
S.Z. conceived the study and drafted the first manuscript. S.Z. and J.L. collected and processed the data and carried out the analysis. S.Z., L.C. and M.H.W. discussed the results. All authors critically read and revised the manuscript and gave final approval for publication.
Supplementary Material
Acknowledgements
This study was conducted using the resources of Alibaba Cloud Intelligence High Performance Cluster computing facilities, which is made free for COVID-19 research.
Contributor Information
Shi Zhao, JC School of Public Health and Primary Care, Chinese University of Hong Kong, Hong Kong; CUHK Shenzhen Research Institute, Shenzhen, China.
Jingzhi Lou, JC School of Public Health and Primary Care, Chinese University of Hong Kong, Hong Kong.
Lirong Cao, JC School of Public Health and Primary Care, Chinese University of Hong Kong, Hong Kong.
Hong Zheng, JC School of Public Health and Primary Care, Chinese University of Hong Kong, Hong Kong.
Marc K C Chong, JC School of Public Health and Primary Care, Chinese University of Hong Kong, Hong Kong; CUHK Shenzhen Research Institute, Shenzhen, China.
Zigui Chen, Department of Microbiology, Chinese University of Hong Kong, Hong Kong.
Renee W Y Chan, Department of Paediatrics, Chinese University of Hong Kong, Hong Kong.
Benny C Y Zee, JC School of Public Health and Primary Care, Chinese University of Hong Kong, Hong Kong; CUHK Shenzhen Research Institute, Shenzhen, China.
Paul K S Chan, Department of Microbiology, Chinese University of Hong Kong, Hong Kong.
Maggie H Wang, JC School of Public Health and Primary Care, Chinese University of Hong Kong, Hong Kong; CUHK Shenzhen Research Institute, Shenzhen, China.
Funding
This work is supported by CUHK grant [PIEF/Ph2/COVID/06, 4054456], the Health and Medical Research Fund (HMRF) Commissioned Research [COVID190103, INF-CUHK-1] of Hong Kong, China and partially supported by the National Natural Science Foundation of China (NSFC) [31871340, 71974165].
Disclaimer
The funding agencies had no role in the design and conduct of the study; collection, management, analysis and interpretation of the data; preparation, review or approval of the manuscript or decision to submit the manuscript for publication.
Conflict of interests
M.H.W. is a shareholder of Beth Bioinformatics Co., Ltd. B.C.Y.Z. is a shareholder of Beth Bioinformatics Co., Ltd and Health View Bioanalytics Ltd. Other authors declared no competing interests.
References
- 1. Tang JW, Tambyah PA, Hui DSC. Emergence of a new sars-cov-2 variant in the UK. J Infect. doi: 10.1016/j.jinf.2020.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cori A, Ferguson NM, Fraser C, Cauchemez S. A new framework and software to estimate time-varying reproduction numbers during epidemics. Am J Epidemiol 2013; 178:1505–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Leung K, Shum MH, Leung GM et al. Early transmissibility assessment of the n501y mutant strains of sars-cov-2 in the United Kingdom, October to November 2020. Eurosurveillance 2021; 26:2002106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Teruel N, Maihot O, Najmanovich RJ. Modelling conformational state dynamics and its role on infection for sars-cov-2 spike protein variants. bioRxiv 2020; 2020.12.16.423118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yurkovetskiy L, Wang X, Pascal KE et al. Structural and functional analysis of the d614g sars-cov-2 spike protein variant. Cell 2020; 183:739–51.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.