

Abstract
Discovering efficient drugs and identifying target proteins are still an unmet but urgent need for curing coronavirus disease 2019 (COVID-19). Protein structure-based docking is a widely applied approach for discovering active compounds against drug targets and for predicting potential targets of active compounds. However, this approach has its inherent deficiency caused by e.g. various different conformations with largely varied binding pockets adopted by proteins, or the lack of true target proteins in the database. This deficiency may result in false negative results. As a complementary approach to the protein structure-based platform for COVID-19, termed as D3Docking in our previous work, we developed in this study a ligand-based method, named D3Similarity, which is based on the molecular similarity evaluation between the submitted molecule(s) and those in an active compound database. The database is constituted by all the reported bioactive molecules against the coronaviruses, viz., severe acute respiratory syndrome coronavirus (SARS), Middle East respiratory syndrome coronavirus (MERS), severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), human betacoronavirus 2c EMC/2012 (HCoV-EMC), human CoV 229E (HCoV-229E) and feline infectious peritonitis virus (FIPV), some of which have target or mechanism information but some do not. Based on the two-dimensional (2D) and three-dimensional (3D) similarity evaluation of molecular structures, virtual screening and target prediction could be performed according to similarity ranking results. With two examples, we demonstrated the reliability and efficiency of D3Similarity by using 2D × 3D value as score for drug discovery and target prediction against COVID-19. The database, which will be updated regularly, is available free of charge at https://www.d3pharma.com/D3Targets-2019-nCoV/D3Similarity/index.php.
Keywords: D3Similarity, COVID-19, target prediction, database, virtual screening
INTRODUCTION
The coronavirus disease 2019 (COVID-19) is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [1–5] that is highly transmissible and pathogenic [6, 7]. As of 31 August 2020, COVID-19 has caused nearly 800 000 deaths with rapidly increasing number of infected people [8]. However, there is still no cure for COVID-19 treatment despite that numerous efforts have been made to satisfy the unmet clinical need.
Previously, we embedded a structure-based module named D3Docking in the D3Targets-2019-nCoV web server [9, 10], which utilized molecular docking to explore the potential protein-ligand binding energies. Though we tried to consider the influence of conformations and pockets in the D3Docking module, it is very difficult to comprehensively and precisely predict all the possible conformations and druggable pockets in a structural-based approach using molecular docking, and thus may lead to false negative prediction. Moreover, a few other docking platforms and computational tools have been developed and reported for the treatment of COVID-19. For example, COVID-19 Docking Server predicts the binding modes between COVID-19 targets and the ligands [11]. Shennong carries out structure-based virtual screening for COVID-19 using FDA approved drugs and drugs that are currently undergoing phase 3 clinical trials as the library [12]. MolAICal supplies a way for generating 3D drugs in the 3D pocket of SARS-CoV-2 main protease [13]. Virus-CKB applies pharmacology-target mapping to rapidly predict the FDA-approved drugs [14] and a network-based approach has been developed for drug repurposing [15]. However, there is no platform to predict target, perform virtual screening and discover drug candidates for COVID-19 with a ligand-based approach. Therefore, developing another parallel approach for target identification and virtual screening is necessary as an alternative to the structure-based scheme. Active compounds against coronavirus could serve as the starting points for a ligand-based scheme.
Quite a number of compounds including natural products have been reported to be active against various coronavirus at different levels. For example, glycyrrhizin was found to have bioactivity in inhibiting the replication, absorption and penetration of SARS-CoV [16]. Tanshinones, which are a series of natural products derived from Salvia miltiorrhiza, were reported as inhibitors against the 3C-like and papain-like proteases of SARS-CoV [17], with one of the compounds in this series exhibiting nanomolar level activity (IC50 = 0.8 ± 0.2 μM) against the papain-like protease of SARS-CoV. Several FDA approved drugs, including chloroquine, chlorpromazine, loperamide and lopinavir [18], showed in vitro activity in the inhibition of MERS-CoV replication at low-micromolar level (3–8 μM). For the novel coronavirus SARS-CoV-2, compounds represented by remdesivir and chloroquine [19] have demonstrated promising based on their in vitro or in vivo bioactivity. Dexamethasone, a type of corticosteroid medication, has been found to reduce the 28-day mortality of patients with SARS-CoV-2 [20]. Although the action mechanism of some of the reported bioactive molecules have been explored, there may still be a large proportion of compounds, of which the corresponding target protein and the mechanism behind the bioactivity remain uncovered.
Here we presented a ligand-based approach, named D3Similarity, to predict active compounds against SARS-CoV-2, and to identify the potential target proteins for molecules with potential bioactivity in a scheme that is irrelevant to the reliability and availability of protein 3D structures. This was realized by evaluating the molecular similarity between the input molecules and the active compounds in the D3Similarity database. We hope D3Similarity would provide another efficient way for target identification and virtual screening to meet the need for curing COVID-19.
MATERIALS AND METHODS
Preparation of the ligand-based database
A total of 604 molecules with potential bioactivity in the treatment of different types of coronavirus infection, which are downloadable in sdf format from the D3Similarity webpage, were collected and used to construct the ligand-based database, involving targets of both viral (including SARS, MERS, SARS-CoV-2, HCoV-EMC, HCoV-229E, FIPV) and human proteins. The ligand-based database of D3Similarity will be continuously updated in our future work.
Preprocessing of small molecules
All the small molecule files, including that inputted by the user and those already existing in the ligand-based database, would be preprocessed under the identical workflow. Generally, the small molecule file would be first transformed to the mol format with Open Babel [21]; following optimization under the MMFF94 force field with the RDKit package [22], the mol file outputted by RDKit with optimized structures would then be transformed to the mol2 format again with Open Babel to be prepared for the molecular similarity evaluation task.
Notably, we found that small molecules involving the nitro group (-NO2) could not be well handled by RDKit if essential information besides atomic coordinates and bonds is missing in the structure file. This essential information involves additional atomic charge definition of the nitro group, termed as the ‘unity atom attributes’ in a qualified mol2 format file, which put one positive charge unit on the nitrogen atom while one negative charge unit on one of the oxygen atoms. Thus, we recommend that molecules containing nitro groups be preprocessed with a SMILES-based workflow. The input molecule will first be transformed to the SMILES string if our program determines that the molecule is substituted with the nitro group. Subsequently, the SMILES of this small molecule would be revised (to include the ‘unity atom attributes’ of the nitro group) and used as the input, following optimization using RDKit with the MMFF94 force field to get the three-dimensional structure, and finally be transformed to the mol2 format with Open Babel.
Evaluating the 2D molecular similarity
The 2D molecular similarity was evaluated based on the Tanimoto coefficient (Tc) values between the SMILES of the input structure and the sdf file containing all molecules in the database, which was obtained using Open Babel based on the mol2 files generated in part 2.2. The Tc values were calculated with Open Babel using the default FP2 fingerprint.
Evaluating the 3D molecular similarity
The 3D molecular similarly between the input molecule and those in the ligand-based database was evaluated based on the mol2 files generated in part 2.2 using MolShaCS (Molecular Shape and Charge Similarity) [23], which is a computational tool to assess the molecular shape and charge similarity between two molecules. Parameters used in the evaluation task (Table 1) were set to the recommended values as mentioned in the MolShaCS manual.
Table 1.
Parameters used in the molecular similarity evaluation task in MolShaCS
| Parameter name | Value |
|---|---|
| minimizer | nlopt_mma |
| align_molecules | yes |
| timeout | 60 |
| write_coordinates | yes |
| mol2_aa | no |
| box_size | 30.0 |
| step | 1.0E-5 |
| tol | 1.0E-4 |
| delta | 1.0E-5 |
Ligand-based virtual screening
The ligand-based virtual screening would be conducted based either on target protein-related compounds or on the active compounds without any target information. Molecular similarity would be evaluated between the molecules in the input sdf or mol2 file and those in a subset of the ligand-based database. The output result would simply be ranked by 2D and 3D molecular similarity for all involved pairs of input molecule and database ligands, and thus offer suggestions in choosing promising molecules for further experimental exploration.
The workflow
All anti-coronavirus active compounds were collated and used as the backend database. Open Babel is used to convert various molecular formats and perform molecular 2D structural similarity evaluation based on molecular fingerprints. RDKit is used to generate 3D conformations from 2D molecular structures. MolShaCS is used to evaluate the similarity of 3D structures between the user submitted molecules and the ligands collected in D3Similarity. Molecular similarity evaluation was performed by 2D, 3D and 2D × 3D scores. Upon job completion, the predicted target or screened ligands, similarity scores and related information will be reported on the website, which are downloadable. The workflow of D3Similarity server is illustrated in Figure 1.
Figure 1.
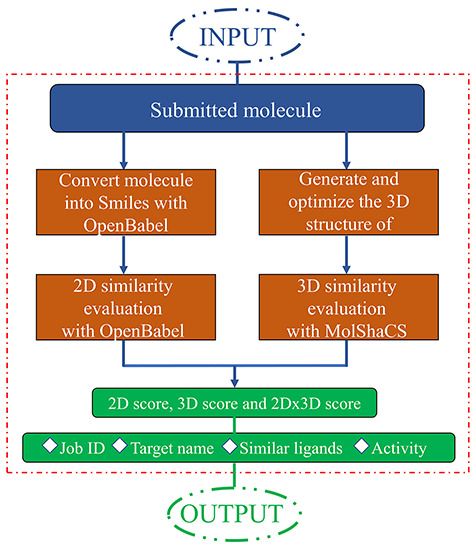
The workflow of the D3Similarity server for target prediction and for ligand-based virtual screening.
RESULTS AND DISCUSSION
Overview of molecules and potential target proteins included in the database
After carefully searching in SciFinder and Pubmed about coronavirus active compounds, the compounds in the literature were collected one by one, including molecular structure, activity, target, coronavirus type and crystal structure. Currently, 32 targets were identified for 430 compounds of the 604 potential bioactive molecules that are contained in our current ligand-based database. Inhibitors for the 3C-like (44.7%) and papain-like (17%) proteases account for the two largest proportions among all involved molecules (Figure 2). Notably, molecules with multiple targets were also counted for multiple times in the pie chart plotted in Figure 2. Details of ligand structures and the associated information for the target(s) are provided on the webpage (see the CoViLigands module, https://www.d3pharma.com/D3Targets-2019-nCoV/CoViLigands/2019-nCoV.php).
Figure 2.
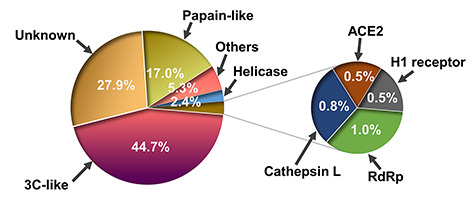
Pie chart for the percentage of associated targets or types for small molecules composing the ligand-based database.
Representative active compounds in the database
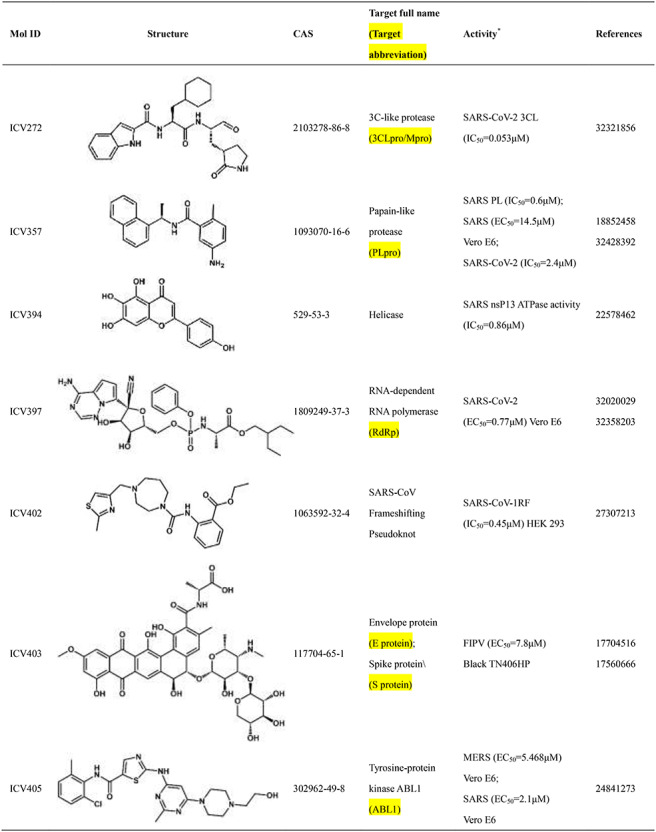
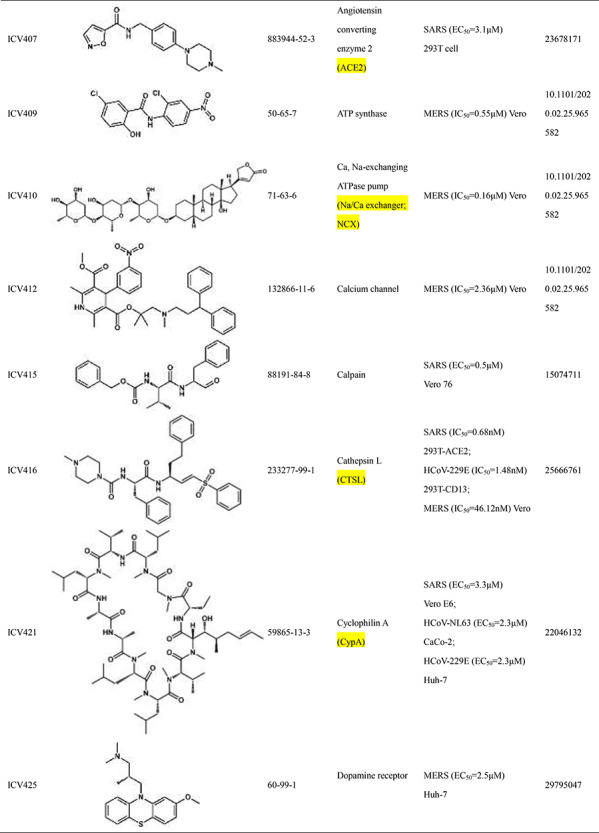
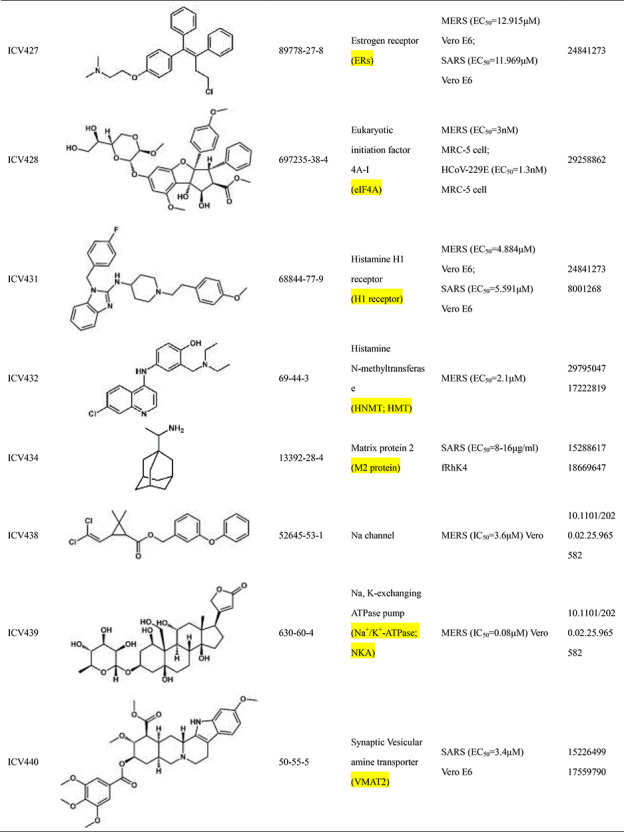
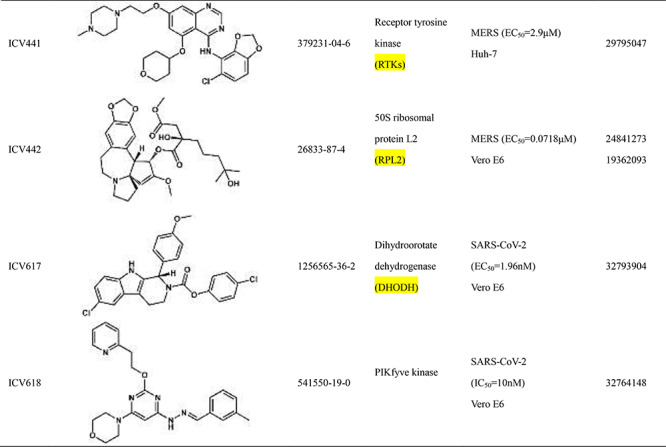
Scientists are committed to the research and development of drugs against SARS-CoV-2 since the outbreak of COVID-19. All currently reported active compounds against coronavirus and their potential targets are included in the D3Similarity database in order to discover more effective compounds against COVID-19. Representative active compounds and targets in the database are shown in Table 2. ICV272 [24] was designed and synthesized targeting main protease that plays a pivotal role in mediating viral replication and transcription [25]. ICV272 showed good pharmacokinetic properties in vivo and low toxicity, suggesting that it is a promising drug candidate. The requisite roles of papain-like protease suggest that papain-like protease is an attractive target for antiviral drugs [26, 27]. ICV357 [28, 29] acts as a noncovalent competitive inhibitor of papain-like protease by inducing a conformational change. Natural compound ICV394 [30] potently inhibits the SARS-CoV helicase protein in vitro by affecting the ATPase activity. Remdesivir (ICV397) has been recognized as a promising antiviral drug against a wide array of RNA viruses including SARS-CoV-2 by inhibiting viral RNA-dependent RNA polymerase (RdRp) [19, 31]. The above compounds exert antiviral effects by acting on the protein targets of the coronavirus. In addition, host targets also play a very important role in the invasion of coronaviruses into organisms. In addition to its importance in regulation of hypertension, angiotensin-converting enzyme 2 (ACE2) has been demonstrated to be a target for the coronavirus [32]. ICV407 [33] inhibits SARS-CoV by blocking early interactions of SARS-S with ACE2. As the COVID-19 continues, active compounds against SARS-CoV-2 have been continuously reported. For example, Dihydroorotate dehydrogenase (DHODH) [34] was considered as drug target for SARS-CoV-2 treatment [35]. The DHODH Inhibitor ICV617 is a highly potent inhibitor of SARS-CoV-2 replication (EC50 = 1.96 nM) [36]. ICV618 can prevent infection by SARS-CoV-2 as an inhibitor of PIKfyve kinase (IC50 = 10 nM) [37]. Besides, ICV402 [38], ICV416 [39] and ICV421 [40] also have potential bioactivity against coronavirus. Active compounds against coronavirus in D3Similarity provide useful leads for the discovery of antivirals that could prevent SARS-CoV-2 and SARS-related infections and may also become a useful tool for studying fundamental mechanisms of compound.
Table 2.
Introduction of representative active compounds of coronavirus in the database
|
|
|
|
*Semicolons is used to separate activity data for the same compound. There are two types of activity data including cell activity and protein activity. For example, SARS-CoV-2 (IC50 = 10 nM) Vero E6 means the IC50 of the compound against SARS-CoV-2 measured in Vero E6 cells is 10 nM. SARS-CoV-2 3CL (IC50 = 0.053 μM) means IC50 of the compound against 3C-like protease of SARS-CoV-2 is 0.053 μM.
Input and output
D3Similarity is provided free of charge for registered users of the D3Targets-2019-nCoV web server (https://www.d3pharma.com/D3Targets-2019-nCoV/D3Similarity/index.php). For target prediction, a graphical interface for user is shown in Figure 3. We recommend that the users submit the input structure in common file formats such as mol2 or sdf to ensure the input file could be well handled by D3Similarity. Usually the evaluation of molecular similarity between the submitted molecule and those in the database would last for several minutes after the beginning of the calculation before the output result is returned, in which the information of the molecular structures and associated targets for top-ranking ligands will be provided on the webpage. The targets for top-ranking ligands are predicted potential targets for the submitted molecule. For example, human histamine N-methyltransferase, 3C-like protease and papain-like protease were predicted to be the top three potential targets of chloroquine, among which 3C-like protease was reported recently based on in vitro results [41, 42].
Figure 3.
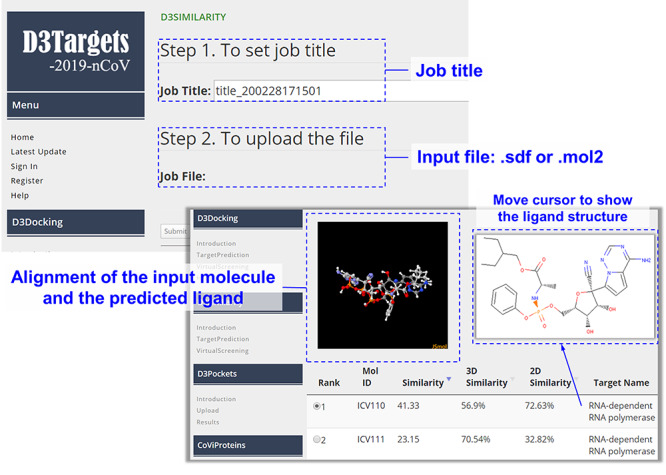
Graphical interface for input and output of the target identification module of D3Similarity.
For virtual screening, a similar graphical interface as shown in Figure 4 is also provided. In this module, the users must provide the input database file in sdf or mol2 format to guarantee the program could correctly split different molecules out of the input database. Usually the calculation would last for several minutes for each molecule in the input database, and thus, the total running time would depend on the size of the input database. In the output result, molecules in the input database would be labeled as ‘MOL_1’, ‘MOL_2’, … based on the order in which they appear in the input file. By default, the results would be ranked by molecular similarity between the input compounds and the database ligands.
Figure 4.
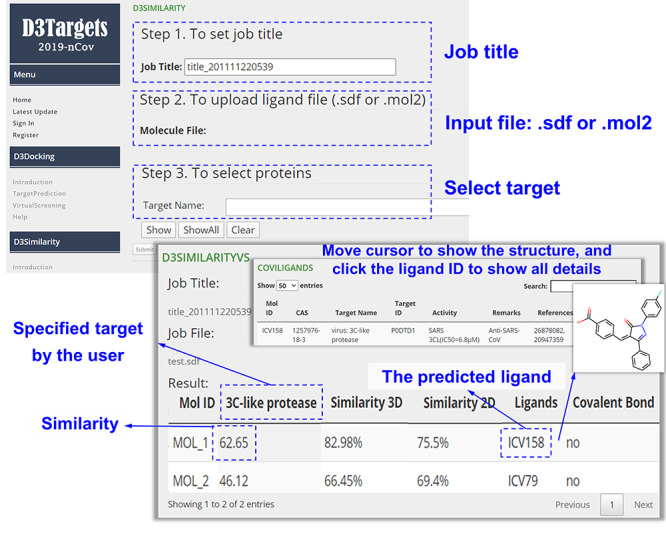
Graphical interface for input and output of the virtual screening module of D3Similarity.
Evaluation of different ranking methods
As mentioned above, both 2D and 3D similarity evaluations were conducted in our ligand-based module. However, considering that the SMILES-based 2D scheme may lose sight of the molecular geometry while the 3D scheme would be slightly affected by the difference in molecular conformations, here, we additionally included the evaluation results ranked by the product of 2D similarity score and 3D similarity score (2D × 3D). Case studies were presented to explore the efficiency of the three ranking methods, and thus, that of D3Similarity in identifying potential targets.
Inhibitors of the 3C-like protease and papain-like protease were selected as two typical examples for the evaluation, considering these two subsets account for the largest proportions of the ligand-based database. Molecules in the rest of the database excluding inhibitors of 3C-like protease or papain-like protease were also selected as negative control subsets to explore the potential influence of database components. The evaluations were then conducted between the input ligands in the subsets and in the database excluding the input ligand itself.
As shown in the pie charts of Figure 5, in the case study of 3C-like protease inhibitors, we presented the average percentage composition of 3C-like protease, papain-like protease, unknown and other targets that correspond to the molecules in the top 10 similarity rankings (hereinafter referred to as ‘top 10’ targets and ‘top 10’ molecules) using 3C-like protease inhibitors in the database as input structures. As demonstrated in the pie charts, in the evaluation results ranked by 2D similarity, 3D similarity and the 2D × 3D scheme, 3C-like protease accounts for a significantly larger percentage among the ‘top 10’ targets for 3C-like protease inhibitors (Figure 5a–c) than that for molecules in the negative control subset (Figure 5d–f). This observation suggested that the large proportion of 3C-like protease in the ‘top 10’ targets for 3C-like protease inhibitors results not only from the database component itself, but also from the successful prediction of our ligand-based approach.
Figure 5.
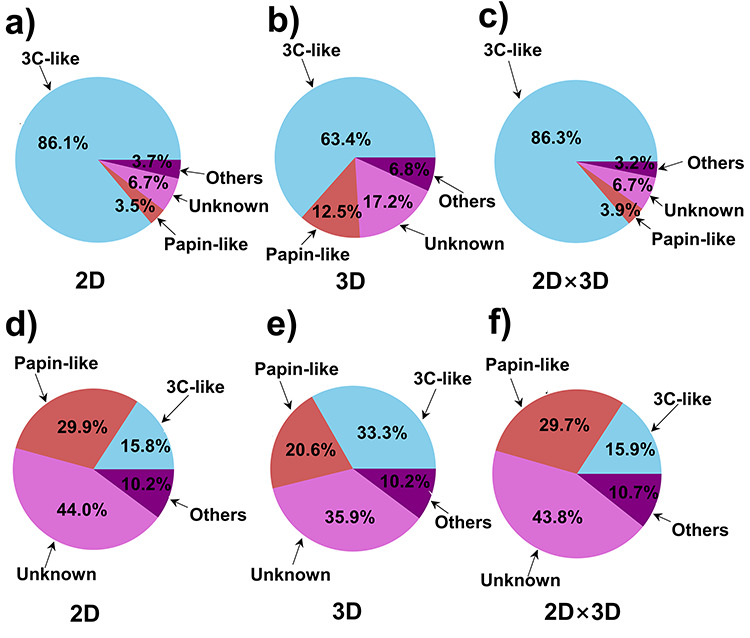
Case study of the 3C-like protease inhibitors using D3Similarity. Plotted pie charts are for average percentage composition of 3C-like protease, papain-like protease, unknown and other targets that correspond to the molecules in the top 10 similarity rankings using 3C-like protease inhibitors in the database as input structures ranked by (a) 2D similarity, (b) 3D similarity, (c) the product of 2D similarity score and 3D similarity score, and using molecules in the rest of the database as input structures ranked by (d) 2D similarity, (e) 3D similarity, (f) the product of 2D similarity score and 3D similarity score.
The observations of the case of papain-like protease inhibitors are similar to that of the case of 3C-like protease inhibitors. In the ‘top 10’ targets of the similarity evaluation result, papain-like protease also accounts for a larger proportion for its reported inhibitors (Figure 6a–c) compared with that for other molecules (Figure 6d–f). What’s more, in general, in both the case studies, the usage of the ‘2D × 3D’ scheme to rank the molecular similarity yielded better results than using either 2D similarity score or 3D similarity score alone, suggesting that the 2D and 3D score may complement each other after the multiplication. Thus, we recommend the users to employ the ‘2D × 3D’ scheme as the default ranking scheme for molecular similarity.
Figure 6.
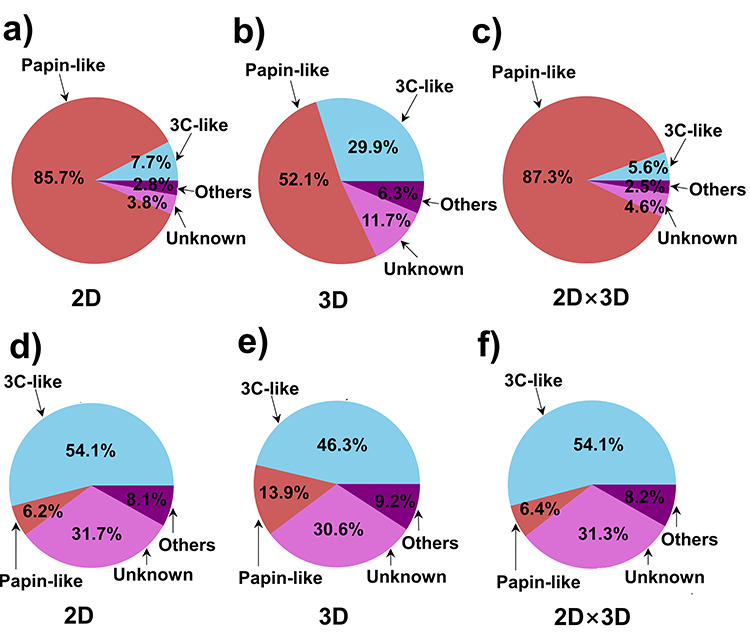
Case study of the papain-like protease inhibitors using D3Similarity. Plotted pie charts are for average percentage composition of 3C-like protease, papain-like protease, unknown and other targets that correspond to the molecules in the top 10 similarity rankings using papain-like protease inhibitors in the database as input structures ranked by (a) 2D similarity, (b) 3D similarity, (c) the product of 2D similarity score and 3D similarity score, and using molecules in the rest of the database as input structures ranked by (d) 2D similarity, (e) 3D similarity, (f) the product of 2D similarity score and 3D similarity score.
Predicting covalent binding by D3Similarity
Covalent binding plays an important role in SARS-CoV-2 drug research [24, 43]. By checking molecular structures, we found that 24 ligands in D3Similarity are potential covalent binders to SARS-CoV-2 related targets. If a submitted molecule is similar to the covalent binders in D3Similarity with at least one of the warheads (e.g. Michael acceptor, trifluoromethyl ketone, aldehyde group, and aldehyde bisulfite), the submitted molecule would be classified as a potential covalent binder.
D3Similarity works together with D3Docking
D3Similarity is a ligand-based approach which does not rely on target structures, while D3Docking relies on the targets’3D structures. There are 32 targets in D3Similarity and 46 targets in D3Docking, while only 12 targets are identical in the two target datasets, suggesting that these two approaches are complementary well with each other. D3Similarity is much faster for target prediction than D3Docking, i.e. 5–15 min for a job by D3Similarity versus 1–2 h for a job by D3Docking. If the predicted targets by D3Similarity are found in D3Docking (https://www.d3pharma.com/D3Targets-2019-nCoV/CoViProteins/2019-nCoV.php), the potential binding modes could be obtained by running D3Docking.
Overall, we believe that D3Similarity should be a complementary approach to docking-based methods for ligand-based target prediction and virtual screening.
CONCLUSIONS
The SARS-CoV-2 infection has led to nearly 800 000 deaths in more than 200 countries worldwide as of 31 August 2020, while no approved drug is available for clinical treatment. Virtual screening is a highly efficient approach to find potential antivirals, while identifying the potential targets is of great importance for understanding the bioactivity mechanism of both now-existing and to-be-developed molecules against the coronavirus infection. On the basis of the previously reported D3Targets-2019-nCoV web server, which has already been embedded with a structure-based module named D3Docking, we released in this study the ligand-based module, termed as D3Similarity, which utilizes the molecular similarity evaluation with bioactive molecules with known targets or/and well-explored mechanism. 604 molecules were included in the ligand-based database and will be updated regularly. In the evaluation of different ranking methods, when applying the product of 2D similarity score and 3D similarity score (2D × 3D) to rank the results, D3Similarity correctly predicted the target of the inhibitors of 3C-like and papain proteases and outperformed the ranking results using either 2D similarity score or 3D similarity score alone. These observations demonstrated D3Similarity should be a complementary approach to docking-based methods for virtual screening and target identification of potential coronavirus antivirals. We hope this ligand-based module would be helpful to the drug development against SARS-CoV-2 and other coronaviruses. The module is available free of charge at https://www.d3pharma.com/D3Targets-2019-nCoV/D3Similarity/index.php.
Key Points
The COVID-19 pandemic caused by the virus SARS-CoV-2 has become a humanitarian crisis, so we have established a database constituted by all the reported bioactive molecules against coronaviruses.
We developed the ligand-based method, named D3Similarity, which is based on the molecular similarity evaluation between the submitted molecule(s) and those in our active compound database.
D3Similarity can be used to predict target proteins for active compounds observed from experimental studies, and to perform virtual screen via 2D and 3D similarity evaluation.
The similarity results can be sorted according to 2D similarity, 3D similarity and 2D × 3D, respectively. The case studies were provided for evaluating these three sorting methods.
D3Similarity is provided free of charge for registered users of the D3Targets-2019-nCoV web server (https://www.d3pharma.com/D3Targets-2019-nCoV/D3Similarity/index.php)
Yanqing Yang is a postgraduate at Shanghai Institute of Materia Medica. His research interests are molecular docking, virtual screening and molecular dynamics. His affiliation is with CAS Key Laboratory of Receptor Research; Drug Discovery and Design Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203, China; School of Pharmacy, University of Chinese Academy of Sciences, No.19A Yuquan Road, Beijing, 100049, China.
Zhengdan Zhu got his Ph.D. degree at Shanghai Institute of Materia Medica in 2020. His research interest is halogen bond interaction. His affiliation is with CAS Key Laboratory of Receptor Research; Drug Discovery and Design Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203, China; School of Pharmacy, University of Chinese Academy of Sciences, No.19A Yuquan Road, Beijing, 100049, China.
Xiaoyu Wang got her master’s degree in 2020 at Shanghai University of Electric Power. Her research interest is database construction. Her affiliation is with College of Mathematics and Physics, Shanghai University of Electric Power, Shanghai, 200090, China.
Xinben Zhang got his master’s degree at East China University of Science and Technology. His research interest is software development. His affiliation is with CAS Key Laboratory of Receptor Research; Drug Discovery and Design Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203, China.
Kaijie Mu is a postgraduate at Nano Science and Technology Institute, University of Science and Technology of China. Her research interest is QM/MM calculations and molecular modeling. Her affiliation is with Nano Science and Technology Institute, University of Science and Technology of China, Suzhou, Jiangsu, 215123, China.
Yulong Shi is a Ph.D. student at Shanghai Institute of Materia Medica. His research interest is molecular docking method development. His affiliation is with CAS Key Laboratory of Receptor Research; Drug Discovery and Design Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203, China; School of Pharmacy, University of Chinese Academy of Sciences, No.19A Yuquan Road, Beijing, 100049, China.
Cheng Peng is a Ph.D. student at Shanghai Institute of Materia Medica. His research interest is molecular dynamics. His affiliation is with CAS Key Laboratory of Receptor Research; Drug Discovery and Design Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203, China; School of Pharmacy, University of Chinese Academy of Sciences, No.19A Yuquan Road, Beijing, 100049, China.
Zhijian Xu got his Ph.D. degree at Shanghai Institute of Materia Medica in 2012. His research interests include drug-target noncovalent interaction, virtual screening and target prediction.
Weiliang Zhu received his Ph.D. degree from Shanghai Institute of Materia Medica in 1998. His main research fields are computer-aided drug design, computational biology, computational chemistry and pharmaceutical chemistry, with a special focus on the theoretical research and method development of drug design.
Contributor Information
Yanqing Yang, Shanghai Institute of Materia Medica; CAS Key Laboratory of Receptor Research; Drug Discovery and Design Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203, China; School of Pharmacy, University of Chinese Academy of Sciences, No.19A Yuquan Road, Beijing, 100049, China.
Zhengdan Zhu, Shanghai Institute of Materia Medica in 2020. His research interest is halogen bond interaction. His affiliation is with CAS Key Laboratory of Receptor Research; Drug Discovery and Design Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203, China; School of Pharmacy, University of Chinese Academy of Sciences, No.19A Yuquan Road, Beijing, 100049, China.
Xiaoyu Wang, Shanghai University of Electric Power. Her research interest is database construction. Her affiliation is with College of Mathematics and Physics, Shanghai University of Electric Power, Shanghai, 200090, China.
Xinben Zhang, East China University of Science and Technology. His research interest is software development. His affiliation is with CAS Key Laboratory of Receptor Research; Drug Discovery and Design Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203, China.
Kaijie Mu, Nano Science and Technology Institute, University of Science and Technology of China. Her research interest is QM/MM calculations and molecular modeling. Her affiliation is with Nano Science and Technology Institute, University of Science and Technology of China, Suzhou, Jiangsu, 215123, China.
Yulong Shi, Shanghai Institute of Materia Medica. His research interest is molecular docking method development. His affiliation is with CAS Key Laboratory of Receptor Research; Drug Discovery and Design Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203, China; School of Pharmacy, University of Chinese Academy of Sciences, No.19A Yuquan Road, Beijing, 100049, China.
Cheng Peng, Shanghai Institute of Materia Medica. His research interest is molecular dynamics. His affiliation is with CAS Key Laboratory of Receptor Research; Drug Discovery and Design Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203, China; School of Pharmacy, University of Chinese Academy of Sciences, No.19A Yuquan Road, Beijing, 100049, China.
Zhijian Xu, Shanghai Institute of Materia Medica in 2012.
Weiliang Zhu, Shanghai Institute of Materia Medica in 1998.
Data Availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
Funding
This work was supported by the National Key Research and Development Program of China [2016YFA0502800, 2016YFA0502301].
Conflict of interest
There are no conflicts to declare.
References
- 1. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature 2020;579:265–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 2020;382:727–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shan C, Yao YF, Yang XL, et al. Infection with novel coronavirus (SARS-CoV-2) causes pneumonia in rhesus macaques. Cell Res 2020;30:670–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Latinne A, Hu B, Olival KJ, et al. Origin and cross-species transmission of bat coronaviruses in China. Nat Commun 2020;11:4235. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 5. Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020;579:270–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chan JF-W, Yuan S, Kok K-H, et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. The Lancet 2020;395:514–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. The Lancet 2020;395:565–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. World Health Organization . Coronavirus disease (COVID-2019) situation reports. https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200831-weekly-epi-update-3.pdf?sfvrsn=d7032a2a_4 (31 August 2020, date last accessed).
- 9. Shi Y, Zhang X, Mu K, et al. D3Targets-2019-nCoV: a webserver for predicting drug targets and for multi-target and multi-site based virtual screening against COVID-19. Acta Pharm Sin B 2020;10:1239–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen Z, Zhang X, Peng C, et al. D3Pockets: a method and web server for systematic analysis of protein pocket dynamics. J Chem Inf Model 2019;59:3353–8. [DOI] [PubMed] [Google Scholar]
- 11. Kong R, Yang G, Xue R, et al. COVID-19 docking server: a meta server for docking small molecules, peptides and antibodies against potential targets of COVID-19. Bioinformatics 2020. doi: 10.1093/bioinformatics/btaa645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu C, Ke Z, Liu C, et al. Systemic in silico screening in drug discovery for coronavirus disease (COVID-19) with an online interactive web server. J Chem Inf Model 2020. doi: 10.1021/acs.jcim.0c00821. [DOI] [PubMed] [Google Scholar]
- 13. Bai Q, Tan S, Xu T, et al. MolAICal: a soft tool for 3D drug design of protein targets by artificial intelligence and classical algorithm. Brief Bioinform 2020. doi: 10.1093/bib/bbaa161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Feng Z, Chen M, Liang T, et al. Virus-CKB: an integrated bioinformatics platform and analysis resource for COVID-19 research. Brief Bioinform 2020. doi: 10.1093/bib/bbaa155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhou Y, Hou Y, Shen J, et al. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov 2020;6:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cinatl J, Morgenstern B, Bauer G, et al. Glycyrrhizin, an active component of liquorice roots, and replication of SARS-associated coronavirus. Lancet 2003;361:2045–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Park JY, Kim JH, Kim YM, et al. Tanshinones as selective and slow-binding inhibitors for SARS-CoV cysteine proteases. Bioorg Med Chem 2012;20:5928–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. de Wilde AH, Jochmans D, Posthuma CC, et al. Screening of an FDA-approved compound library identifies four small-molecule inhibitors of Middle East respiratory syndrome coronavirus replication in cell culture. Antimicrob Agents Chemother 2014;58:4875–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang M, Cao R, Zhang L, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res 2020;30:269–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Group RC, Horby P, Lim WS, et al. Dexamethasone in hospitalized patients with Covid-19- preliminary report. N Engl J Med . doi: 10.1056/NEJMoa2021436.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. O'Boyle NM, Banck M, James CA, et al. Open babel: an open chemical toolbox. J Chem 2011;3:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Landrum G. A. RDKit: open-source cheminformatics software. version 2020.03.1.
- 23. Vaz de Lima LA, Nascimento AS. MolShaCS: a free and open source tool for ligand similarity identification based on Gaussian descriptors. Eur J Med Chem 2013;59:296–303. [DOI] [PubMed] [Google Scholar]
- 24. Dai W, Zhang B, Jiang XM, et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020;368:1331–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hsu MF, Kuo CJ, Chang KT, et al. Mechanism of the maturation process of SARS-CoV 3CL protease. J Biol Chem 2005;280:31257–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Clementz MA, Chen Z, Banach BS, et al. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J Virol 2010;84:4619–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Frieman M, Ratia K, Johnston RE, et al. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-kappaB signaling. J Virol 2009;83:6689–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Freitas BT, Durie IA, Murray J, et al. Characterization and noncovalent inhibition of the Deubiquitinase and deISGylase activity of SARS-CoV-2 papain-like protease. ACS Infect Dis 2020;6:2099–109. [DOI] [PubMed] [Google Scholar]
- 29. Ratia K, Pegan S, Takayama J, et al. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc Natl Acad Sci U S A 2008;105:16119–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yu MS, Lee J, Lee JM, et al. Identification of myricetin and scutellarein as novel chemical inhibitors of the SARS coronavirus helicase, nsP13. Bioorg Med Chem Lett 2012;22:4049–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yin W, Mao C, Luan X, et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 2020;368:1499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li W, Moore MJ, Vasilieva N, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003;426:450–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Adedeji AO, Severson W, Jonsson C, et al. Novel inhibitors of severe acute respiratory syndrome coronavirus entry that act by three distinct mechanisms. J Virol 2013;87:8017–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Munier-Lehmann H, Vidalain PO, Tangy F, et al. On dihydroorotate dehydrogenases and their inhibitors and uses. J Med Chem 2013;56:3148–67. [DOI] [PubMed] [Google Scholar]
- 35. Zheng J, Zhang Y, Liu Y, et al. Multi-omics study revealing tissue-dependent putative mechanisms of SARS-CoV-2 drug targets on viral infections and complex diseases. medRxiv 2020. doi: 10.1101/2020.05.07.20093286. [DOI]
- 36. Luban J, Sattler R, Muhlberger E, et al. The DHODH inhibitor PTC299 arrests SARS-CoV-2 replication and suppresses induction of inflammatory cytokines. bioRxiv 2020. doi: 10.1101/2020.08.05.238394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kang YL, Chou YY, Rothlauf PW, et al. Inhibition of PIKfyve kinase prevents infection by Zaire ebolavirus and SARS-CoV-2. Proc Natl Acad Sci U S A 2020;117:20803–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hermann T. Small molecules targeting viral RNA. Wiley Interdiscip Rev RNA 2016;7:726–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhou Y, Vedantham P, Lu K, et al. Protease inhibitors targeting coronavirus and filovirus entry. Antiviral Res 2015;116:76–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pfefferle S, Schopf J, Kogl M, et al. The SARS-coronavirus-host interactome: identification of cyclophilins as target for pan-coronavirus inhibitors. PLoS Pathog 2011;7:e1002331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li Z, Li X, Huang YY, et al. Identify potent SARS-CoV-2 main protease inhibitors via accelerated free energy perturbation-based virtual screening of existing drugs. Proc Natl Acad Sci U S A 2020;117:27381–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tripathi PK, Upadhyay S, Singh M, et al. Screening and evaluation of approved drugs as inhibitors of main protease of SARS-CoV-2. Int J Biol Macromol 2020;164:2622–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vuong W, Khan MB, Fischer C, et al. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat Commun 2020;11:4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request.