

Abstract
Human coronaviruses (CoVs) can cause respiratory infection epidemics that sometimes expand into globally relevant pandemics. All human CoVs have sister strains isolated from animal hosts and seem to have an animal origin, yet the process of host jumping is largely unknown. RNA interference (RNAi) is an ancient mechanism in many eukaryotes to defend against viral infections through the hybridization of host endogenous small RNAs (miRNAs) with target sites in invading RNAs. Here, we developed a method to identify potential RNAi-sensitive sites in the viral genome and discovered that human-adapted coronavirus strains had deleted some of their sites targeted by miRNAs in human lungs when compared to their close zoonic relatives. We further confirmed using a phylogenetic analysis that the loss of RNAi-sensitive target sites could be a major driver of the host-jumping process, and adaptive mutations that lead to the loss-of-target might be as simple as point mutation. Up-to-date genomic data of severe acute respiratory syndrome coronavirus 2 and Middle-East respiratory syndromes-CoV strains demonstrate that the stress from host miRNA milieus sustained even after their epidemics in humans. Thus, this study illustrates a new mechanism about coronavirus to explain its host-jumping process and provides a novel avenue for pathogenesis research, epidemiological modeling, and development of drugs and vaccines against coronavirus, taking into consideration these findings.
Keywords: coronavirus, adaptive mutation, host tropisms, RNAi, miRNA
Introduction
Severe acute respiratory syndrome coronavirus 2 [SARS-CoV-2 (or nCov-19)], the causal viral pathogen of COVID-19 [1], has caused worldwide pandemics and claimed millions of lives in the past year [2]. SARS-CoV-2 is the 7th coronavirus species that is known to have humans as hosts. The other six include SARS-CoV and MERS-CoV, which can cause severe acute respiratory symptoms, and the remaining four (OC43, 229E, NL-63 and HKU-1) usually lead to mild respiratory infections similar to a common cold [3]. Besides humans, coronavirus species can be hosted by a wide range of mammals, and five of the human CoVs have sister strains hosted by bats, which are proposed to be the source of these human CoVs [4, 5]. Although the transfer of coronaviruses among different hosts (host jumping) is at this moment in 2020 one of the most widely discussed topics in life science [6], much remains unknown about the mechanisms through which this process occurs, and how adaptive mutations in coronavirus genomes may enable host jumping into humans.
To control the inherited capability of viruses in propagation and invasion, hosts often equip with several defense apparatuses to hamper the transmission of viruses. RNA interference (RNAi) is one of such ancient defensive mechanisms utilizing the spontaneous hybridization against viruses or mobile elements that has been extensively documented as a major source of immunity in plants and invertebrates [7, 8]. In this mechanism, small RNAs of 20–30 nt (nucleotide) in length with various sequences and abundance are capable of recognizing exogenous long RNA molecules through the formation of double-stranded RNA duplex with a target sequence on them via hybridization. Such duplexes subsequently induce a set of complex RNases-centric reactions to degrade the recognized RNA molecules [9]. To overcome the barrier of RNAi defense, viruses have evolved many counteracting mechanisms, including mutations in their target sites, to escape binding by host small RNA sequences [10].
In mammals, the RNAi apparatus has long been known to function in regulating endogenous gene expression via a molecular mechanism similar to that for viral control [11]. Recently, the role of endogenous small RNA (miRNA) in defending against viruses in mammals has gradually established, supported by growing evidence [12–18]. In these studies, particularly in human cell lines and animal models, both endogenous and externally introduced microRNAs (including miRNAs and siRNAs) can strongly inhibit the proliferation and transmission of viruses, provided that a given microRNA sequence matches a target on the viral genome [19–23]. Other supportive evidence includes the discovery of anti-RNAi mechanisms in human viruses [10, 24] and the necessity of some components in the mammalian RNAi apparatus in viral resistance [25]. Therefore, endogenous miRNAs in mammal hosts may also function as a defense against viral infection, including against coronavirus strains [26, 27]. However, how the host miRNA milieu affects the evolution and host tropism of coronaviruses, especially in the host-jumping process of SARS-CoV-2, under natural conditions remains unclarified.
Here, we report a previously undiscovered catalog of adaptive mutations in human CoVs, which leads to the loss of RNAi-sensitive sites targeted by human miRNAs expressed in lung tissue and may help them escape the attacks from host miRNAs. We also introduce the essential parameters in predicting such RNAi-sensitive target sites that can induce effective suppression. It is expected that the knowledge about these adaptive mutations would be useful in understanding the epidemiology and pathogenesis of coronavirus infections, as well as in developing prophylactics and therapeutic drugs for better control of this life-threatening virus.
Results and discussion
Coronavirus strains have lost RNAi-sensitive target sites upon adopting into humans
To investigate the RNAi-sensitive sites on genomes of coronaviruses targeted by human miRNAs, we first downloaded the up-to-date human mature miRNA sequences from miRBase [28] with which we can infer candidate RNAi-sensitive targets for any given genome of coronavirus. As the strength of overall RNAi suppression against a virus should depend on the total amount of miRNA transcripts including all miRNA species that match their targets in the coronavirus genome, we therefore obtained the profile of miRNAs expressed in normal human lung, the common entryway of coronaviruses, from SEAweb [29], an up-to-date miRNA expression database. Using these data, we can estimate the total abundance of the coronavirus-targeting miRNA (coronavirus-targeting human miRNA) present in human lung targeting a given coronavirus genome; we hereafter refer to this information as CoVT-miRNA for convenience.
To identify candidate RNAi-sensitive targets in the genomes of coronavirus strains, we first scrutinize the genome sequences of all seven beta-coronavirus species known to infect humans. For each species, we examine specific strains for human hosts and their close relatives hosted by bats or other nonhuman animals (Table S1 and ‘Strain selection’ in the Experimental section for detail). Our initial analysis sought to obtain perfect matches to the entire collection of the human miRNAs resulted in no identifications. However, it is now firmly established that perfect matching is only essential for the so-called seed region of miRNA (the 5′-end; positions 2–8), hybridization and subsequent RNAi-based suppression can still occur despite the presence of mismatches or bulges in other miRNA positions [11].
A variety of algorithms have been developed for predicting targets of miRNA in mRNAs [30], but none of them resulted in precise prediction. As there is presently no algorithm for specifically predicting candidate RNAi-sensitive target sites in viral genomes, we here employed miRanda [31], one of the most popular tools in miRNA target prediction from mRNA sequences, instead. Initial analysis using default miRanda searching parameters identified many hundreds of potential target sites in each of the examined coronavirus genomes; moreover, when we used the human lung CoVT-miRNAs that putatively target these many sites, the tallied abundance encompassed more than 95% of all miRNA molecules present in normal lung cells. This obviously exceeds the required abundance of miRNAs for effective suppression. In a previous study, it was established that influenza proliferation and transmission were effectively inhibited via endogenous miRNA-based mechanisms when the abundance of matched miRNA species was as low as 1–2% [13].
The default parameters in our initial use of miRanda has allowed many false positives. Indeed, it included sites for which the predicted minimum free energy of the miRNA::target duplex was as high as −1 kcal/mol (calculated as previously described [32] where lower values indicate stronger hybridization). Given that thermodynamics fundamentally underlies hybridization—and is widely employed as a physical factor for various miRNA target prediction algorithms and siRNA design—we next tried to include the predicted miRNA::target duplex minimum free energy value as an additional criterion in an attempt to remove false positives from the list of target sites predicted by miRanda. To ensure strong hybridization of the miRNA::target duplex, we required that any candidate RNAi-sensitive targets must be both predicted by miRanda and exceeding a particular cutoff of minimum free energy value. To determine a suitable cutoff for the minimum free energy, we gradually relaxed the minimum free energy from the minimal value to −1 kcal/mol, and then assessed the accumulation of potential RNAi-sensitive target sites within a coronavirus genome and monitored the cumulative abundance for CoVT-miRNAs present in healthy human lungs, which finally resulted in a sigmoid accumulation curve.
When the curves for the human-adapted SARS coronavirus strains (including both SARS-CoV and SARS-CoV-2 strains) and their closely related nonhuman host strains (bat and civet) were plotted together, they entangled in most phases, and the curves of civet CoVs almost completely overlapped with the human CoVs (Figure 1a). However, the examination of the first inflection point of these sigmoid curves revealed a subtle distinction: the accumulated abundance for CoVT-miRNAs increased slightly faster for the bat-host coronavirus stains than for the human-host strains, forming a clear gap between the two sets of curves (Figure 1a, inset). This gap represents a free energy range of −26 to −25 kcal/mol in terms of hybridization energy and corresponds to a region where the accumulated CoVT-miRNA abundance reaches the 1–2% level, which falls into the sufficient abundance interval for effective RNAi against influenza viruses. We propose the following interpretation of this detected gap between the human versus bat CoVs: the human lung CoVT-miRNAs, when forming strong duplexes with free energy at least at the −26 to −25 kcal/mol level, are possible to achieve effective RNAi-based defense against the viruses. Thus the gap indicates that human CoVs have somehow deleted target sites that were otherwise hybridized by human CoVT-miRNAs strongly (with free energy lower than −26 to −25 kcal/mol).
Figure 1.
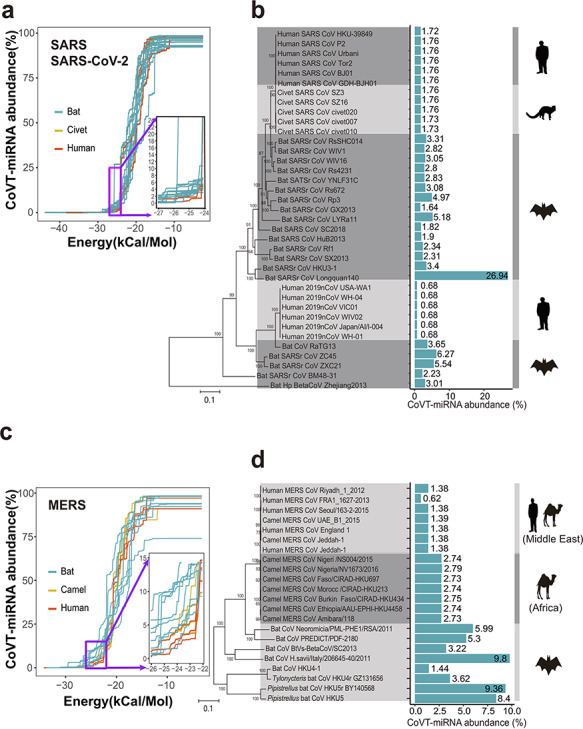
Comparison of the total abundance of coronavirus-targeting miRNAs (CoVT-miRNAs) between coronavirus strains of human and nonhuman hosts. (a and b) CoVT-miRNA abundance for strains in group 2b of beta-coronavirus, which includes SARS-CoV, SARS-CoV-2, and SARS-related coronaviruses. (a) The accumulation curves of CoVT-miRNA abundance for each strain when gradually relaxed based on minimal free energy predictions. Each curve indicates a coronavirus strain with red, yellow and blue curves, representing human, civet, and bat strains, respectively. Inset, an enlarged region showing the first inflection point of the predicted CoVT-miRNA abundance curves. (b) CoVT-miRNA abundance of each strain in a phylogenetic context. Blue bars indicate the CoVT-miRNA abundance of each strain in the dendrogram at the free energy cutoff of −25 kcal/mol with the values labeled alongside. The cartoons on the right show the animal species of the host of the strains in each clade. (c and d) CoVT-miRNA abundance for strains in group 2c of beta-coronavirus, which includes MERS-CoV and MERS-related CoVs. (c) The accumulation curves of CoVT-miRNA abundance for each strain. Red and blue curves represent strains of human and nonhuman host, respectively. The inset highlights details of the first inflection points of the accumulation curves. (d) CoVT-miRNA abundance for each strain of this group in a phylogenetic context.
We subsequently examined the total abundance of CoVT-miRNA (calculated with the cutoff of free energy at −25 kcal/mol) for the SARS-related coronaviruses (i.e. group 2b of beta-coronavirus) that includes the human-adapted SARS, SARS-CoV-2, and related coronaviruses adapted to nonhuman hosts in a phylogenetic context. In the dendrogram (Figure 1b), SARS-CoV and SARS-CoV-2, respectively separated from two distinct groups of bat CoVs [33]. The CoVT-miRNA abundance is very stable for both human SARS-CoV and SARS-CoV-2 but decreases by several folds compared to the closely related bat CoVs, indicating loss of RNAi-sensitive targets in human SARS-CoV. We noticed that the value of CoVT-miRNA abundance is even lower in SARS-CoV-2 than in SARS-CoV (0.68% versus 1.75%), which suggests that a comparatively lower abundance of miRNAs in the human lung can interfere with the SARS-CoV-2 genome, a finding which may contribute to the more rapid transmission of SARS-CoV-2 in the human population as compared to SARS-CoV. For strains hosted by civet (the candidate intermediate host animal for SARS-CoV), the CoVT-miRNA abundance levels are almost equal to those of the human-adapted SARS-CoV strains, suggesting that there is no obvious RNAi-related obstacles for viral transmission between civets and humans.
We performed a similar analysis for MERS-related coronaviruses, including human, camel and bat CoVs. In the accumulation curves, a similar gap between strains of human and nonhuman host was also observed at the first inflection point, again occurring at a similar free energy range of −26 to −25 kcal/mol and CoVT-miRNA abundance levels of 1–2%. However, we did detect exceptions with a few curves of the strains from Middle Eastern camels, which completely overlapped with human MERS-CoVs (Figure 1c).
In the dendrogram for MERS-related CoVs, i.e. group 2c of beta-coronavirus (Figure 1d), it seems that the human MERS-CoVs originated from strains of bat host, with camels serving as an intermediate host, following previous conclusions about MERS-CoV transmission [34]. The CoVT-miRNA abundance (also calculated at the free energy cutoff of −25 kcal/mol) is obviously lower for strains of human host than for strains of bat and African camel host, again clearly suggesting the loss of RNAi-sensitive targets in the human CoVs. In contrast to the strains of African camel host, which are clearly phylogenetically separated from human CoVs, the strains of Middle Eastern camel host are completely entangled with the human CoVs and have almost identical CoVT-miRNA abundance with human-host strains, suggesting free transfer between the two host species. These observations are in accordance with the reported fact that MERS-CoV strains isolated from Middle Eastern camels are genetically indistinguishable from human MERS-CoV strains, whereas strains from African camels are genetically distinct and have not been found to infect humans [35].
Expanding beyond SARS- and MERS-related coronaviruses, we conducted similar analyses for the four remaining human coronavirus species known to infect humans (the alpha-CoVs HCoV-229E and HCoV-NL63, and the group 2a beta-CoVs HCoV-OC43 and HCoV-HKU-1). We again detected gaps between strains of human and nonhuman host in the accumulation curves, again at a similar free energy level of −26 to −23 kcal/mol and with CoVT-miRNA abundance levels at 1–10% (Figure S1a–d). In addition, a comparison of CoVT-miRNA abundance in a phylogenetic context further supported that a decrease in CoVT-miRNA abundance levels (i.e. loss of RNAi-sensitive targets) occurred in coronaviruses when they transmitted within the human population (Figure S2a and b). The uniform decrease in the CoVT-miRNA abundance observed in all the seven coronaviruses has little possibility of being a coincidence. The shared cutoff in free energy and level of CoVT-miRNA abundance imply shared mechanisms for human miRNAs to confer an effective defense against coronaviruses. These findings strongly suggest that the loss of sensitive sites targeted by CoVT-miRNAs in the human lung might be a necessary step for the coronaviruses to adapt to humans as a new host.
Besides the positive strand, we also performed a similar analysis to identify RNAi-sensitive target sites on the minus-strand of the coronavirus following the same analysis steps. In the strains of group 2b and 2c beta-coronavirus, which include SARS-CoV, SARS-CoV-2, MERS-CoV and their zoonic relatives, similar analysis as described above observed no obvious distinctions between strains of human and nonhuman host (Table S2). Given that effective suppression executed by miRNAs interference relies on sufficient amount of miRNA::target duplexes, where the defensive mechanism depends on the concentration of both miRNAs and the matched targets on viral RNA molecules. The viral minus-strand RNA, which only functions as the template for replicating the virus genome, has much lower number of copies, that is, less cellular concentration than the positive strand. Thus, the sequence of minus-strand may have been experiencing less stress from host cellular miRNAs and shaped by the miRNAs to a much less extent.
Considering the known etiology of CoVs—which are transmitted via the respiratory tract [36]—lung cells are where CoVs initially infect and proliferate and should thus be the first tissue wherein they may face any RNAi-based defense. Following this assumption, the RNAi milieu (i.e. the total, tissue-specific miRNA population) in other tissues may have little impact on shaping coronavirus genomes. To explore this, we undertook a similar analysis as with our initial calculation of the CoVT-miRNAs abundance in the lung tissue, but this time used the expression data for miRNAs present in normal human plasma samples, where coronaviruses do not proliferate. As anticipated, we did not detect obvious gaps in the accumulation curves between strains of human and nonhuman host (Figure S3) and did not observe a decreasing trend for plasma CoVT-miRNA abundance in human CoVs as compared against related strains of nonhuman hosts (Figure S4).
Mutations as simple as point mutation can lead to the loss of RNAi-sensitive sites in coronavirus
We evaluated potential mutations that may have caused the loss of RNAi-sensitive targets in the human coronavirus strains. When comparing close strains of human and nonhuman host (when overall genome identity ≥95%), we found that there is often a substantial overlap of RNAi-sensitive target sites in-between. However, a few RNAi-sensitive target sites can be deleted by mutations as simple as a single-nucleotide transition (i.e. interchange of purines (A ↔ G) or pyrimidines (C ↔ U)). This kind of point mutation, especially when occurred in the region hybridized to the seed sequence of miRNA, had the potential to abolish the inhibitory effect of the corresponding miRNA, as experimentally demonstrated in previous studies [37, 38].
Examining SARS-CoV-2, by comparing the predicted target sites in the Wuhan-Hu-1 strain (the reference genome for SARS-CoV-2) to RaTG13, the up-till-now closest strain isolated from bat in 2013 with an overall genome identity of 96.1% (Table S3), we identified a total of 21 and 23 predicted target sites from the two genomes, with 12 shared by both, most of which are targets of low-abundance human lung CoVT-miRNAs. Notably, the top two targets of the most abundant miRNAs in RaTG13 are both absent from the Wuhan-Hu-1 genome: Site 1 is a target of miR-146b-5p (whose abundance is 2.95%), whereas Site 2 is a target of miR-200c-3p (0.27% in abundance). Thus, the human lung CoVT-miRNA abundance is 3.65% against the RaTG13 strain but only 0.68% against the Wuhan-Hu-1 strain. The loss of these two sites in SARS-CoV-2 has the potential to lead to the evasion of Wuhan-Hu-1 strain from RNAi attack triggered by the two abundant human lung CoVT-miRNAs. We also closely examined the two sites in the RaTG13 strain and the same genome positions in Wuhan-Hu-1. For Site 1 in Wuhan-Hu-1 genome, a single U → C transition in the binding site of the core 7-mer of the miRNA results in predicted loss of the target (Figure 2a). For Site 2, two complementary Cs in the RaTG13 genome are substituted by Us in Wuhan-Hu-1, forming G–U pairs which substantially weaken the original G–C pairs. An additional U → A transversion abolished a previous G–U pair, together impairing the strength of the miRNA::target duplex (Figure 2b).
Figure 2.
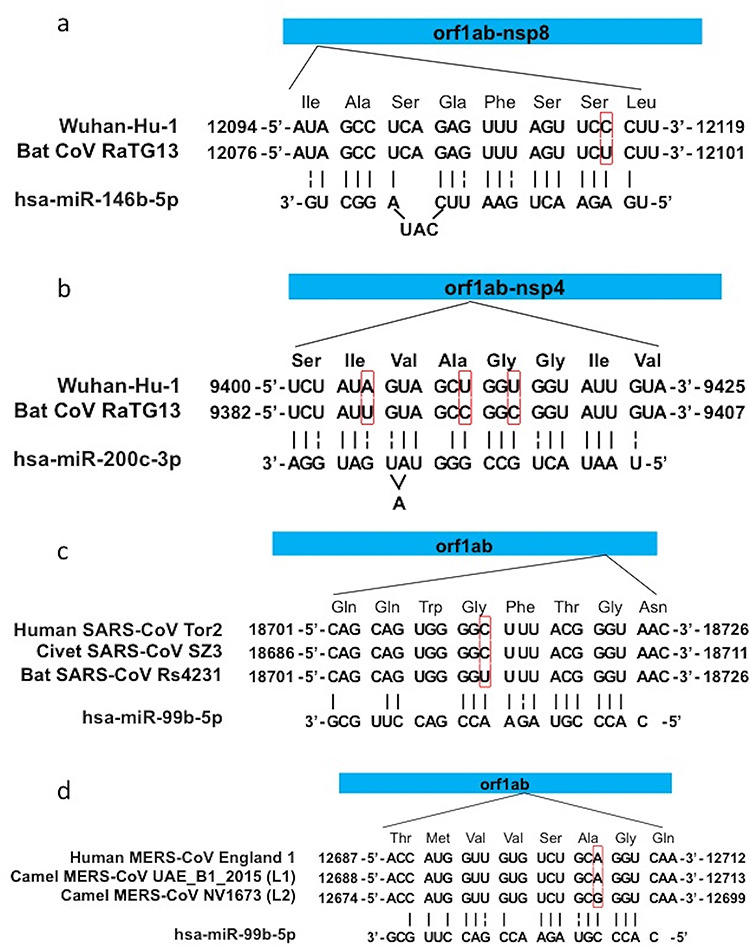
Examples of mutations that lead to the loss of RNAi-sensitive target sites. Target site for miR-146b-5p (Site 1) (a) and miR-200c-3p (Site 2) (b) in SARS-CoV-2, and sites targeted by miR-99b-5p in SARS-CoV (c) and MERS-CoV (d). The top bar indicates the position of target sites in the orf1ab gene, the three middle rows are the sequences of the target sites in the genome of corresponding strain which are aligned to their open-reading-frame with amino acid sequence labeled above, and the bottom row is the sequence of the miRNA matching the target. Strain names are labeled at the left, and red boxes highlight the mutated nucleotides. Vertical lines above miRNA sequences indicate the matching nucleotide pairs in the miRNAs::target duplexes, and dash lines indicate G–U pairs.
Broadly supporting the idea that adaptive mutations may result in the loss of RNAi-sensitive targets in coronaviruses and reduce the abundance of CoVT-miRNAs that may facilitate the process of host jump, we performed comparisons among SARS-related strains, including Tor2 (from human), SZ3 (from civet) and Rs4231 (from bat) (Table S4). The genome of Rs423 has a predicted target site for the human lung CoVT-miRNA miR-99b-5p (1.25% abundance), but the hybridization is not very strong as many mismatches in the 3′-end of the miRNA. At the position of the target site in the genomes of Tor2 and SZ3, there is a single U → C transition that potentially further weakens the hybridization and increases the free energy of the duplex over the cutoff of −25 kcal/mol (Figure 2c).
For the MERS-CoV strains, we did not observe any loss of RNAi-sensitive targets caused by single-nucleotide mutations in strains of human host when compared to those of bat host due to the low overall genome identities between these strains [39]. However, we did detect an informative point mutation in comparison among MERS-CoV strains from humans (England_1), Middle Eastern camel (UAE_B1_2015) and African camel (NV1673) [35] (Table S5). A site in the African camel strain is predicted to be targeted by the human lung CoVT-miRNA miR-99b-5p (1.25% abundance), but a single G → A transition in the region hybridized to the seed sequence of miR-99b-5p in the genomes of the human and Middle Eastern camel strains might lead to insensitivity of the site to the RNAi-based defense (Figure 2d).
Interestingly, all the aforementioned RNAi-target sites occur in the coding sequence of the orf1ab gene, which is highly conserved among coronaviruses. Moreover, all single-nucleotide mutations above mentioned in the human strains are synonymous (i.e. they do not change the amino acid they code for), indicating the narrow path that coronaviruses had to take to bypass the RNAi-based defense from the host. In contrast to the loss of RNA-sensitive targets, the gain of targets in human CoVs is also predictable due to the active recombination and error-prone replication in coronaviruses. However, among all the newly acquired RNAi-sensitive targets we detected in the human SARS-CoV-2, SARS-CoV, and MERS-CoV strains (Tables S3–S5), none of them correspond to miRNAs with abundance >0.2%, confirming that only those CoVT-miRNAs of high-abundance are capable of shaping the viral genomes as effective RNAi defense is depended on sufficient amount of miRNA::target duplexes.
Sustained selective pressure from microRNAs on the evolution of human coronaviruses
As host miRNAs are potential in shaping the genome of coronaviruses and select for strains of low CoVT-miRNA abundance, we examined whether the RNAi-sensitive targets were further abolished and whether any coronaviruses strains are being continuously selected for low CoVT-miRNA abundance during the transmission or outbreak of human coronaviruses. MERS-CoVs can serve as a representative case as they have been continuously sampled from worldwide patients for 8 years since its first recognition in 2012 [40], and there are now 254 strains with high-quality and publicly available genome sequences and information of collection date (Table S6).
We used seqTrack [41] to infer MERS-CoV strain haplotypes and generated a network that allows displaying all haplotypes according to their CoVT-miRNA abundance and sampling dates (Figure 3a). In general, the level of CoVT-miRNA abundance for the majority of these haplotypes is 1.384%, a level equals to that of the representative strains used in our earlier analysis (Figure 1d) and is largely sustained throughout the MERS epidemic. Due to the error-prone replication of coronaviruses and the high frequency of mutations, gain-and-loss of RNAi-sensitive targets continuously occurs as the virus propagated. Thus haplotypes with higher or lower CoVT-miRNA abundance have continuously emerged throughout the epidemic, and those of higher CoVT-miRNA abundance still have the chance to survive if they happen to be transferred to a patient in whom the abundance of corresponding CoVT-miRNAs are not sufficient to confer an effective suppression. However, haplotypes with reduced susceptibility to human RNAi defense, i.e. lower CoVT-miRNA abundance, would have better chance for transmission and undergo positive selection, thus yielding more progeny strains than those with increased CoVT-miRNA abundance (Figure 3b). Further supporting this, a linear regression of the haplotype data throughout the ongoing MERS epidemic revealed a conspicuous trend of gradual decrease in the CoVT-miRNA abundance level over time (Figure 3a). These results imply that selection pressure from host miRNAs has been continuously shaping the genomes of MERS-CoV, even after successful host jumping into humans.
Figure 3.
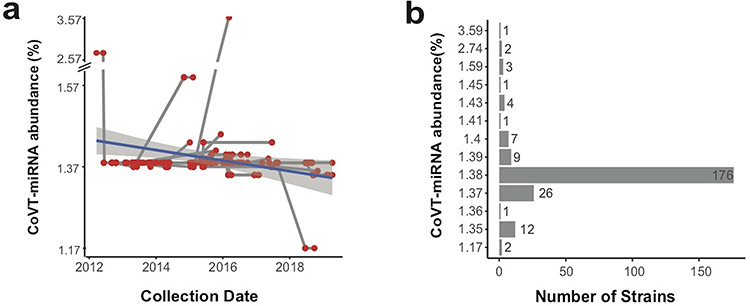
CoVT-miRNA abundance of MERS-Cov. (a) Transmission network of MERS-CoV haplotypes plotted according to their CoVT-miRNA abundance and sampling data throughout the course of the MERS epidemic. Each red dot indicates a haplotype, and the gray lines connecting haplotypes represent relationship of transmission. The navy line depicts the linear regression of the CoVT-miRNA abundance. (b) The number of MERS-CoV strains at different CoVT-miRNA abundance levels.
For the evolution of SARS-CoV-2 during transmission in humans, we downloaded genomic variation information from the all the up-to-date SARS-CoV-2 sequence database (19 June 2020 from the CNCB coronavirus database; https://bigd.big.ac.cn/ncov/) [42]. For all the 26 305 high-quality genomes, we inferred their RNAi-sensitive target sites on genome and calculated CoVT-miRNA abundance (Table S7). The RNAi-sensitive target sites are rather stable among these strains, and the list of RNAi-sensitive targets in the representative strain of Wuhan-Hu-1is shared by 24 209 genomes (92.03%) which sustain the low CoVT-miRNA abundance of 0.68% as well (Figure 4a). Within the strains with an altered value of CoVT-miRNA abundance, 1890 (7.18%) strains reduced the value to 0.441–0.679% via further loss of their RNAi-sensitive targets, whereas only 206 (0.78%) strains increased their CoVT-miRNA abundance to 0.681–9.752% by acquiring RNAi-sensitive targets.
Figure 4.
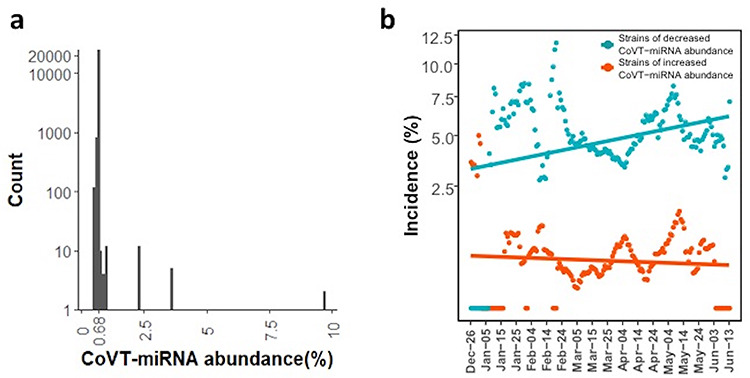
CoVT-miRNA abundance of SARS-CoV-2. (a) The number of SARS-CoV-2 strains as a function of CoVT-miRNA abundance. The highest peak is at 0.68%, accounted for 92.03% of the total. The number of strains with decreased and increased CoVT-miRNA abundance is 1890 (7.18%) and 206 (0.78%), respectively. (b) The incidence of strains with decreased (turquoise dots) and increased (red dots) CoVT-miRNA abundance. The incidence of the two strain types is calculated as the respective proportion of the total collection in a 10-day window and plotted against the SARS-CoV-2 epidemic since 26 December 2019 with a 1-day step. Turquois and red lines indicate the linear regression of the incidences of strains with decreased and increased CoVT-miRNA abundance, respectively. Pearson correlation is used to evaluate their correlations to the time course, and the incidence of decreased CoVT-miRNA abundance is significantly correlated (rho = 0.30, P = 8.7 × 10−5) but not that of the incidence of increased CoVT-miRNA abundance (rho = −0.083, P = 0.28).
The acquisition of RNAi-sensitive targets can also be the result of point mutations. For example, SNU01, a strain isolated from Korea, in which a reverse point mutation re-established the target site for miR-146b-5p (abundance 2.95%) that is carried by RaTG13 (from bat) and deleted in the predominant SARS-CoV-2 strains (Figure S5a). Another example is USA-CruiseA-11, isolated from a patient on the US cruise ship the Diamond Princess, for which a novel target site for human hsa-let-7b-5p (1.62% in abundance) was gained by a point mutation (Figure S5b). The existence of these strains seemingly inadaptive to human host with increased CoVT-miRNA abundance is more likely the result of the high mutation rate of coronavirus. Thriving with an average number of mutations around 6–7 per sequenced strains and an enormous viral population worldwide, SARS-CoV-2 is expected to be unprecedentedly diversified and fast-evolving with distinct mutation spectra [43, 44]. Recent evidence from the existence of quasispecies—a collection of closely related mutants [45]—in the viral population of SARS-CoV-2 sampled from a single patient [46] also confirmed the high mutation rate. Therefore, there would be more progenies with reduced CoVT-miRNA abundance and less with increased CoVT-miRNA abundance due to the positive natural selection, which is just we have observed in the population of SARS-CoV-2.
Accordingly, the incidence of strains with increased CoVT-miRNA abundance, if depends on the mutation rate of SARS-CoV-2, which is a constant, should keep stable over time and be much less than the incidence of strains with reduced CoVT-miRNA abundance due to less adaptation. The latter one, being more adaptive to the human host, should gradually increase along the epidemic course. So we plotted the incidence of strains with decreased and increased CoVT-miRNA abundance, i.e. the proportion of these strains in the total strains collected in a 10-day interval (Figure 4b), and it is clear that the incidence of strains with decreased CoVT-miRNA abundance was much higher than that of strains with increased CoVT-miRNA abundance. Moreover, the incidence of strains with increased CoVT-miRNA abundance showed no correlation to time (rho = 0.087, Pearson correlation, P = 0.26), whereas that of strains with decreased CoVT-miRNA abundance exhibited a conspicuous increasing trend over time (rho = 0.21, Pearson correlation, P = 0.0067). As the proportion of strains with decreased CoVT-miRNA abundance gradually increases, they might finally replace the current predominant strain due to increased adaptivity to humans. That is, the stress from the miRNA milieus in human lung cells continues to shape the evolution of SARS-CoV-2 during its transmission in humans.
Conclusion
Our study suggests that human coronaviruses, including the pandemic pathogen SARS-CoV-2, have lost RNAi-sensitive targets in their genomes, resulting in a reduced level of RNAi-based defense triggered by human miRNAs. Thus, selective pressure from host miRNAs could be a critical factor that constrains the host range of coronaviruses and profoundly affects the evolution of coronavirus even after transmission in humans. Our in-depth analysis also validated a previously unknown cutoff in minimum free energy (about −25 kcal/mol that corresponds to about 1–2% CoVT-miRNA abundance) for the miRNA::target duplex to induce effective RNAi-mediated suppression against coronavirus. The determination of this useful parameter can be very laborious and difficult if it had to be determined only from experimental research. All our findings rely on the availability of the abundant datasets of coronavirus genome sequences, in which seven species caused epidemics in humans due to possible zoonoses in recent decades.
It is important to note that the population of expressed miRNAs in lung tissue is variable among individuals and can be influenced by a variety of factors including age, gender, smoking, immunity, inflammation and other pathogenic conditions [47, 48]. Such variability in the human lung CoVT-miRNA milieu may help explain the apparently diverse susceptibility and clinical manifestations of coronaviruses among individuals [49]. Further investigations, including reverse genetics experiments, high-throughput sequencing of miRNAs in patients, and intensive sequencing of the genomes of diverse coronavirus strains can help to conclusively establish the specific functional contributions of host miRNAs in controlling host tropism and the infection process.
Adaptive mutations that cause the loss of RNAi-sensitive target sites, although can be as simple as a single-nucleotide transition, also constitute the complex virus–host interactions as important as the spike protein of coronavirus and should not be neglected. A deeper understanding of the complex process of host–virus interactions, such as the impact of RNAi-based host defense would almost certainly support more efficacious epidemiological modeling, public health policy measures, and even the development of novel prophylactic and therapeutic medicines to better control and treat the highly threatening and hugely disruptive infections from coronaviruses.
Material and methods
Coronavirus genome dataset
All genome sequences of examined coronavirus strains were downloaded from the NCBI viral genome database [https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/] and CNCB coronavirus database [https://bigd.big.ac.cn/ncov/]; all genome IDs in the phylogenetic analysis are listed in Table S1. In selecting strains for phylogenetic analysis, for species of HCoV-229E, NL63, OC43, HKU-1, MERS, and SARS, redundant strains (i.e. strains of the same genome length and collected at the same date, site and host species) were first removed, and then a dendrogram was inferred for each species from which at least one representative genome for each phylogenetic clade were selected; for SARS-CoV-2, we used the strains collected at the beginning of the epidemic to investigate the initial changes of this virus just after the host jumping onto humans. For the close relatives of human coronaviruses hosted by animals, both strains named as human coronaviruses related, such as ‘SARS-related strain’ from the NCBI viral genome database and strains reported in previous studies were included. In analysis of the effects of RNAi-based defense on viral evolution after transfer to humans, all available genomes of MERS-CoV and SARS-CoV-2 strains, i.e. 254 and 26 305 strains, respectively were used.
Predicting miRNA targets in viral genome sequences and calculating minimum free energy for miRNA::target duplex
The up-to-date sequences of mature human miRNAs were downloaded from the miRBase database [28]. We used miRanda [31] to predict the potential target sites matching human miRNAs in the genomes of all examined coronavirus strains with default parameters. The minimum free energy for each of the miRNA::target duplexes was calculated as previously described [32].
Calculation of CoVT-miRNA abundance
For a given viral strain, miRNA targets predicted by miRanda were further filtered by a cutoff of minimum free energy (at −25 kcal/mol) and regarded as RNAi-sensitive target sites. As each target can be interfered by an miRNA species at various expression levels in normal human lung, we defined the CoVT-miRNA abundance of the given strain as the sum of abundance of all the interfering miRNAs matching its targets. The expression level of all the miRNAs in different tissues were downloaded from SEAweb [29] and indicated as RPM (Reads of exon model per Million mapped reads). The abundance of each miRNA in a sample was calculated as the percentage of the miRNA’s RPM value in the total RPM of the sample, and final abundance of each miRNA species in each tissue was averaged among all samples of the same tissue type.
Phylogenetic tree construction
The complete genome sequence of each examined coronavirus strain was aligned using MAFFT (version 7) [50] with default parameters, and Maximum Likelihood phylogenetic trees were constructed in RAxML (version 0.9.0) [51] with the GTR + G substitution model and 1000 bootstrap replicates. The trees were visualized with iTOL v5 web [https://itol.embl.de] [52].
Transmission network of human MERS-CoV
The complete genome sequences of all human MERS-CoV strains were downloaded from NCBI with collection date and site, and then aligned using MAFFT (version 7) with default parameters. MERS-CoV haplotypes and transmission network were inferred and conducted with seqTrack [41] in the adegenet package [https://cran.r-project.org/web/packages/adegenet/index.html].
Statistical analysis
All statistical analyses were performed with R software [https://www.r-project.org]. Correlations were evaluated with linear regression and Pearson correlation with significance defined as P ≤ 0.05. The figures were plotted with the R package ggplot2 [https://cran.r-project.org/web/packages/ggplot2/] and ggpubr [https://cran.r-project.org/web/packages/ggpubr/].
Key Points
Suggesting the effects of miRNAs on the host tropism of coronaviruses in natural conditions, which provides convincing supportive evidence for the role of mammalian miRNA in the defense against viruses.
Illustrating the sustained effects of human RNAi on the evolution of coronaviruses, based on an analysis of 26 000+ high-quality SARS-CoV-2 and MERS-CoV genomes.
Providing algorithms for predicting miRNA targets on viral genome.
Identifying the miRNA target sites in coronaviruses, which might have been lost in their host-jumping process to humans.
Supplementary Material
Qingren Meng, PhD at Southern University of Science and Technology School of Medicine who performed the major analysis work in this paper.
Yanan Chu, assistant professor at Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing, China who participated in data processing for this work.
Changjun Shao is an engineer at Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing, China.
Jing Chen is an engineer at Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing, China.
Jian Wang is an engineer at Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing, China.
Zhancheng Gao, full professor at Peking University People’s Hospital, Beijing, China. As an experienced clinician in viral infection management, Dr Gao served as a counselor for WHO since 2006 and was a member of the writing committee for the WHO clinical management guidelines of influenza A (H5N1 and H1N1) pandemics.
Jun Yu, full professor at Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing, China. As an expert in the field of genomics, transcriptome and bioinformatics. Prof. Yu proposed the idea of escaping RNA interference in the process of viral host jumping.
Yu Kang, associate professor at Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing, China, and experienced in analysis of microbial genomics and development of bioinformatic tools.
Contributor Information
Qingren Meng, Southern University of Science and Technology School of Medicine, China.
Yanan Chu, Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing, China.
Changjun Shao, Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing, China.
Jing Chen, Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing, China.
Jian Wang, Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing, China.
Zhancheng Gao, Peking University People’s Hospital, Beijing, China.
Jun Yu, Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing, China.
Yu Kang, Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing, China.
Funding
National Natural Science Foundation of China (NSFC, 31671350, 31970568), National Science and Technology Major Project (2018ZX10712001-018-002), Programs of the Chinese Academy of Sciences (QYZDY-SSW-SMC017; Y8YZ02E001), Programs of Beijing Municipal Science and Technology Project (Z171100001317011), National Key R&D Program of China (2020YFC0848900).
Authors’ contribution
KY conceived and designed the project, performed data analysis, wrote and edited the manuscript; MQ performed the data analysis; YJ and ZG conceived and supervised the project; CY, SC, CJ and WJ did data collection and curation. All the authors have revised and approved the manuscript submission.
Data availability
The data underlying this article are available in [NCBI viral genome database], at https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/, and [CNCB coronavirus database, at https://bigd.big.ac.cn/ncov/. The datasets were derived from sources in the public domain: [NCBI, https://www.ncbi.nlm.nih.gov, and CNCB coronavirus database, https://bigd.big.ac.cn].
References
- 1. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature 2020;579:265–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Coronavirus Resource center of Johns Hopkins University and Medicine .
- 3. Cui J, Li F, Shi ZL. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol 2019;17:181–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Forni D, Cagliani R, Clerici M, et al. Molecular evolution of human coronavirus genomes. Trends Microbiol 2017;25:35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Corman VM, Muth D, Niemeyer D, et al. Hosts and sources of endemic human coronaviruses. Adv Virus Res 2018;100:163–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Andersen KG, Rambaut A, Lipkin WI, et al. The proximal origin of SARS-CoV-2. Nat Med 2020;26:450–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hartmann G. Nucleic acid immunity. Adv Immunol 2017;133:121–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ding SW, Voinnet O. Antiviral immunity directed by small RNAs. Cell 2007;130:413–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ghildiyal M, Zamore PD. Small silencing RNAs: an expanding universe. Nat Rev Genet 2009;10:94–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Berkhout B. RNAi-mediated antiviral immunity in mammals. Curr Opin Virol 2018;32:9–14. [DOI] [PubMed] [Google Scholar]
- 11. Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell 2009;136:642–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jacque JM, Triques K, Stevenson M. Modulation of HIV-1 replication by RNA interference. Nature 2002;418:435–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Langlois RA, Albrecht RA, Kimble B, et al. MicroRNA-based strategy to mitigate the risk of gain-of-function influenza studies. Nat Biotechnol 2013;31:844–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brettmann EA, Shaik JS, Zangger H, et al. Tilting the balance between RNA interference and replication eradicates Leishmania RNA virus 1 and mitigates the inflammatory response. Proc Natl Acad Sci USA 2016;113:11998–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maillard PV, Veen AG, Poirier EZ, et al. Slicing and dicing viruses: antiviral RNA interference in mammals. EMBO J 2019;38:e100941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Perez JT, Pham AM, Lorini MH, et al. MicroRNA-mediated species-specific attenuation of influenza a virus. Nat Biotechnol 2009;27:572–6. [DOI] [PubMed] [Google Scholar]
- 17. Weng KF, Hung CT, Hsieh PT, et al. A cytoplasmic RNA virus generates functional viral small RNAs and regulates viral IRES activity in mammalian cells. Nucleic Acids Res 2014;42:12789–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hussain M, Asgari S. MicroRNA-like viral small RNA from Dengue virus 2 autoregulates its replication in mosquito cells. Proc Natl Acad Sci USA 2014;111:2746–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guo Z, Li Y, Ding SW. Small RNA-based antimicrobial immunity. Nat Rev Immunol 2019;19:31–44. [DOI] [PubMed] [Google Scholar]
- 20. Li Y, Lu J, Han Y, et al. RNA interference functions as an antiviral immunity mechanism in mammals. Science 2013;342:231–4.24115437 [Google Scholar]
- 21. Qureshi A, Tantray VG, Kirmani AR, et al. A review on current status of antiviral siRNA. Rev Med Virol 2018;28:e1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu YP, Qiu Y, Zhang B, et al. Zika virus infection induces RNAi-mediated antiviral immunity in human neural progenitors and brain organoids. Cell Res 2019;29:265–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Qiu Y, Xu YP, Wang M, et al. Flavivirus induces and antagonizes antiviral RNA interference in both mammals and mosquitoes. Sci Adv 2020;6:eaax7989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bennasser Y, Le SY, Benkirane M, et al. Evidence that HIV-1 encodes an siRNA and a suppressor of RNA silencing. Immunity 2005;22:607–19. [DOI] [PubMed] [Google Scholar]
- 25. Adiliaghdam F, Basavappa M, Saunders TL, et al. A requirement for Argonaute 4 in mammalian antiviral defense. Cell Rep 2020;30:1690–1701.e1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ding S-W, Han Q, Wang J, et al. Antiviral RNA interference in mammals. Curr Opin Immunol 2018;54:109–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Qiu Y, Xu Y, Zhang Y, et al. Human virus-derived small RNAs can confer antiviral immunity in mammals. Immunity 2017;46:992–1004 e1005. [DOI] [PubMed] [Google Scholar]
- 28. Kozomara A, Birgaoanu M, Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic Acids Res 2019;47:D155–d162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rahman R-U, Liebhoff A-M, Bansal V, et al. SEAweb: the small RNA expression atlas web application. Nucleic Acids Res 2020;48:D204–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Riffo-Campos AL, Riquelme I, Brebi-Mieville P. Tools for sequence-based miRNA target prediction: what to choose? Int J Mol Sci 2016;17:1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Betel D, Wilson M, Gabow A, et al. The microRNA.Org resource: targets and expression. Nucleic Acids Res 2008;36:D149–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wuchty S, Fontana W, Hofacker IL, et al. Complete suboptimal folding of RNA and the stability of secondary structures. Biopolymers 1999;49:145–65. [DOI] [PubMed] [Google Scholar]
- 33. Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020;588:7836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Su S, Wong G, Shi W, et al. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol 2016;24:490–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chu DKW, Hui KPY, Perera R, et al. MERS coronaviruses from camels in Africa exhibit region-dependent genetic diversity. Proc Natl Acad Sci USA 2018;115:3144–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhou F, Yu T, Du R, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 2020;395:1054–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Boden D, Pusch O, Lee F, et al. Human immunodeficiency virus type 1 escape from RNA interference. J Virol 2003;77:11531–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sun Y, Zhang Y, Zhang X. Synonymous SNPs of viral genes facilitate virus to escape host antiviral RNAi immunity. RNA Biol 2019;16:1697–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Anthony SJ, Gilardi K, Menachery VD, et al. Further evidence for bats as the evolutionary source of Middle East respiratory syndrome coronavirus. MBio 2017;8:e00373–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Boheemen S, Graaf M, Lauber C, et al. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. MBio 2012;3:e00473–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jombart T, Eggo RM, Dodd PJ, et al. Reconstructing disease outbreaks from genetic data: a graph approach. Heredity (Edinb) 2011;106:383–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Song S, Ma L, Zou D, et al. The global landscape of SARS-CoV-2 genomes, variants, and haplotypes in 2019nCoVR. Genom Proteom Bioinform 2020. doi: 10.1016/j.gpb.2020.09.001. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Teng X, Li Q, Li Z, et al. Compositional variability and mutation spectra of monophyletic SARS-CoV-2 clades. Genom Proteom Bioinform 2020. doi: 10.1101/2020.08.26.267781. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yu J. From mutation signature to molecular mechanism in the RNA world: a case of SARS-CoV-2. Genom Proteom Bioinform 2020;S1672–0229(20)30092–9. doi: 10.1016/j.gpb.2020.07.003. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Presloid JB, Novella IS. RNA viruses and RNAi: Quasispecies implications for viral escape. Viruses 2015;7:3226–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shen Z, Xiao Y, Kang L, et al. Genomic diversity of SARS-CoV-2 in coronavirus disease 2019 patients. Clin Infect Dis 2020;71(15):713–20. doi: 10.1093/cid/ciaa203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ludwig N, Leidinger P, Becker K, et al. Distribution of miRNA expression across human tissues. Nucleic Acids Res 2016;44:3865–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lee EJ, Baek M, Gusev Y, et al. Systematic evaluation of microRNA processing patterns in tissues, cell lines, and tumors. RNA 2008;14:35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen N, Zhou M, Dong X, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet 2020;395:507–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nakamura T, Yamada KD, Tomii K, et al. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 2018;34:2490–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014;30:1312–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Letunic I, Bork P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res 2019;47:W256–w259. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in [NCBI viral genome database], at https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/, and [CNCB coronavirus database, at https://bigd.big.ac.cn/ncov/. The datasets were derived from sources in the public domain: [NCBI, https://www.ncbi.nlm.nih.gov, and CNCB coronavirus database, https://bigd.big.ac.cn].