

Abstract
Coronavirus disease 2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is a dreaded pandemic in lack of specific therapeutic agent. SARS-CoV-2 Mpro, an essential factor in viral pathogenesis, is recognized as a prospective therapeutic target in drug discovery against SARS-CoV-2. To tackle this pandemic, Food and Drug Administration-approved drugs are being screened against SARS-CoV-2 Mpro via in silico and in vitro methods to detect the best conceivable drug candidates. However, identification of natural compounds with anti-SARS-CoV-2 Mpro potential have been recommended as rapid and effective alternative for anti-SARS-CoV-2 therapeutic development. Thereof, a total of 653 natural compounds were identified against SARS-CoV-2 Mpro from NP-lib database at MTi-OpenScreen webserver using virtual screening approach. Subsequently, top four potential compounds, i.e. 2,3-Dihydroamentoflavone (ZINC000043552589), Podocarpusflavon-B (ZINC000003594862), Rutin (ZINC000003947429) and Quercimeritrin 6”-O-L-arabinopyranoside (ZINC000070691536), and co-crystallized N3 inhibitor as reference ligand were considered for stringent molecular docking after geometry optimization by DFT method. Each compound exhibited substantial docking energy >−12 kcal/mol and molecular contacts with essential residues, including catalytic dyad (His41 and Cys145) and substrate binding residues, in the active pocket of SARS-CoV-2 Mpro against N3 inhibitor. The screened compounds were further scrutinized via absorption, distribution, metabolism, and excretion - toxicity (ADMET), quantum chemical calculations, combinatorial molecular simulations and hybrid QM/MM approaches. Convincingly, collected results support the potent compounds for druglikeness and strong binding affinity with the catalytic pocket of SARS-CoV-2 Mpro. Hence, selected compounds are advocated as potential inhibitors of SARS-CoV-2 Mpro and can be utilized in drug development against SARS-CoV-2 infection.
Keywords: SARS-CoV-2; COVID-19; density functional theory; combinatorial molecular simulations; hybrid QM/MM calculations; 2,3-dihydroamentoflavone
Introduction
Human corona viruses (HCoVs), responsible for causing multiple respiratory diseases, have emerged periodically around the world and responsible for major pandemics [1]. In the present scenario, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent of Coronavirus disease 2019 (COVID-19), is the third major HCoVs explosion and a major health nuisance around the world [1, 2]. SARS-CoV-2 infection, initially originated in China’s Hubei Province, Wuhan, in December 2019, has diverged from the previously reported zoonotic pandemic viruses SARS-CoV and positioned under subgenera Sarbecovirus of the family Coronaviridae [1]. SARS-CoV-2 also contains a phospholipid bilayers envelope cored with tangled positive-sense single-stranded genomic RNA of ~30 kb length on phosphorylated nucleocapsid (N) protein [1, 3, 4]. Although, bats have been marked for major reservoir of HCoVs, but origin and transmission of SARS-CoV-2 to humans is not yet cleared while transmission between the people by respiratory droplets and direct contact with contaminated routes have been well established [1, 5]. SARS-CoV-2 infects people of all ages, but the older people and those with underlying medical conditions, such as heart complications, lung infection, and diabetes, are at a higher risk of getting severe COVID-19 [5]. On March 11, 2020, the WHO officially declared COVID-19 as pandemic based on more than 118 000 reported cases and 4291 deaths in 114 countries [6]. In absence of potential and specific therapeutics, COVID-19 cases are rapidly increasing and over 22 million cases with more than 0.7 million deaths, and about 15 million recoveries as of 19 August 2020 have been reported [7]. Therefore, the current circumstances demand an urgent need to identify and develop potential therapeutics for the treatment of infected people.
SARS-CoV-2 genome analysis (GenBank: MN908947.3) revealed translation of four structural proteins, including nucleocapsid (N) protein, membrane (M) protein, envelope (E) protein and Spike (S) protein, and four non-structural proteins, viz. papain-like protease (PLpro), 3-chymotrypsin-like protease (3CLpro or main protease Mpro), helicase, and RNA polymerase, along with accessory protein [8–11]. In SARS-CoV-2 as in other HCoVs, 3CLpro performs the autolytic cleavage from pp1a and pp1ab site translated by virus ORF1a/b, which shared 96% sequence similarity with Mpro of SARS-CoV strain [12, 13]. Remarkably, in vitro inhibition of Mpro in SARS-CoV by the selected inhibitors have shown anti-viral activity [14, 15]. Thus, the inhibition of viral Mpro, which is essentially required in viral infection cycle and lack of its homologues in the human, has been certified as standard antiviral therapeutic strategy [14]. Recent studies also recognized SARS-CoV-2 Mpro as one of the potential targets for the discovery and identification of potent drug candidates against SARS-CoV-2 [16–18]. Hence, crystal structures of SARS-CoV-2 Mpro have been solved at high resolution through X-ray crystallography technique, which is a significant achievement in COVID-19 research [17, 18].
In the field of drug discovery, natural compounds are used to treat a variety of diseases and infections, including HCoVs have been recently reviewed [19]. Approximately one-third of the top-selling medicines in the world are natural compounds or their derivatives [20, 21]. Moreover, natural compounds are widely recognized in the drug industry for their wide structural diversity, pharmacological activities, and less toxicity in contrast to synthetic drugs [20–22]. Additionally, larger fragments with reactive moieties were documented in natural compounds by comparison to synthetic compounds, hence, suggested as suitable hits in the drug development [23]. Under current COVID-19 pandemic, limited research has been conducted to screen the natural compounds against SARS-CoV-2 Mpro. Therefore, the identification of natural compounds with considerable potential to inhibit SARS-CoV-2 Mpro activity may results in the development of effective therapeutic against COVID-19 pandemic. Hence, in the present study, the natural compound collection at NP-lib database available via MTi-OpenScreen webserver was screened to identify potential inhibitors against SARS-CoV-2 Mpro using structure based virtual screening method. Following, the selected compounds were evaluated by combinatorial molecular simulations and quantum binding energy calculations to determine their potential against SARS-CoV-2 Mpro activity.
Materials and methods
Structure based virtual screening and ADMET analysis
Crystal structure of SARS-CoV-2 Mpro was retrieved from PDB database (PDB ID: 6 LU7) [17] for structure based virtual screening against natural compound database (NP-lib, which contained 1226 purchasable natural compounds) at MTi-OpenScreen webserver (https://bioserv.rpbs.univ-paris-diderot.fr/services/MTiOpenScreen/) [24]. It is noteworthy to mention that NP-lib database includes the compounds suitable for docking and purchasability according to the Zinc15 database after physico-chemical and toxicophore filtration [24]. For virtual screening, SARS-CoV-2 Mpro structure was prepared and minimized using default parameters of Dock Prep tool in UCSF Chimera-1.14 [25]. Following, prepared structure was uploaded at MTiOpenScreen webserver to perform virtual screening against natural compounds from NP-lib database under default parameters at the co-crystallized ligand N3 inhibitor binding pocket of coordinates 60 × 40 × 40 Å along three X, Y and Z axes, which covered all the essential residues center at −8.918, 17.918, 62.905 Å, as reported earlier [17, 26]. Following, top four ligands with highest docking score were considered for further analysis along with N3 inhibitor as reference ligand, as reported earlier [26].
Furthermore, the selected molecules were also studied for their physiochemical and medicinal chemistry properties using SwissADME server (http://www.swissadme.ch) [27].
Quantum chemical calculations
In Quantum chemical calculations, Becke’s three parameter exchange function (B3) with Lee-Yang-Parr hybrid density functional (LYP) approach has been evolved as the standard method for the structural geometry optimization and to precisely understand the chemical reactions [28]. While, a basis set is usually referred as a set of functions called basis functions (or atomic orbitals centered on atoms), linked in a linear combination to create molecular orbitals and selection of basis set depends on the types of elements involved in the system. Hence, the combination of B3LYP and 6-31G basis set (split-valence double-ζ basis set) in conjunction with the polarization functions on heavy and hydrogen atoms (d,p), is a well-established basis set to obtained the unprecedented accuracy for the organic molecules under consideration [29]. Although theoretical and practical limitations of B3LYP/6-31G(d,p) approach are obvious, and serious doubts arise when molecular thermochemistry was involved [30]. However, comparatively better results and less computational cost prompted to opt B3LYP/6-31G(d,p) in this study for the optimization of the screened natural compounds against SARS-CoV-2 Mpro. Briefly, the density functional theory (DFT) [31] treated by B3LYP [32, 33] in conjunction with 6-31G(d,p) basis sets was employed to optimized the geometry of screened potential natural compounds along with N3 inhibitor using Gaussian 03 software [34]. The respective ligand geometries were optimized at ground state (in gas) without including any imaginary frequencies, as reported earlier [35], and then utilized in molecular and quantum chemical calculation in Gaussian 03 software [34]. Additionally, these geometries were employed in re-docking analysis to achieve a significant stable ligand docked pose with receptor for the establishment of convincing intermolecular interactions in the respective complexes.
Re-docking and pose analysis
Herein, re-docking analysis was performed for DFT optimized geometries of selected natural compounds in the same active pocket of SARS-CoV-2 Mpro to get insight on the effect of geometry optimization and mode of protease inhibition by the selected ligands using MTi AutoDock (https://bioserv.rpbs.univ-paris-diderot.fr/services/MTiOpenScreen/) with default settings [24, 36]. Herein, at least 10 poses for each protein–ligand complex were produced and best pose with highest binding score at least root-mean-square deviation (RMSD) was selected for further interaction analysis in free academic Schrödinger-Maestro v12.4 suite [37]. Similar docking methodology was used on the crystal structure native ligand, i.e. Michael acceptor inhibitor-known as N3 inhibitor [17], to validate the docking procedure and for comparative analysis with the selected natural compounds. Later, both 3D and 2D interaction images were rendered using free academic Schrödinger-Maestro v12.4 suite [37].
Explicit-solvent MD simulation
The best poses obtained from re-docking studies were evaluated for the complex stability using 100 ns explicit-water molecular dynamics (MD) simulation using academic version of Desmond v5.6 [38] module in Schrödinger-Maestro v11.8 suite [39] under Linux environment on HP Z238 workstation, details are given in Supplementary Section S1.1.
Post-dynamics simulation analysis
Following MD simulation, the generated trajectories with coordinates at every 20 ps for viral protease docked with selected ligands were analyzed using Simulation Interaction Diagram of Desmond v5.6 module [38] in Schrödinger-Maestro v11.8 package [39]. Herein, RMSD, root-mean-square fluctuation (RMSF), and protein-ligand interaction profiling was extracted from the respective 100 ns MD simulation trajectory of each complex. Also, essential dynamics calculations were calculated using Bio3d package [40]; details are given in Supplementary Section S1.2.
MM-generalized born surface area calculations
For binding free energy calculations, molecular mechanics generalized Born surface area (MM/GBSA) method is comparatively popular and computationally less demanding with respect to alchemical free energy methods [41]. Hence, net binding free energy calculation was conducted using Prime MM/GBSA module [42] in Schrödinger-Maestro package by MM/GBSA protocol using default settings, as reported earlier [43].
QM/MM approach
The chemical reactivity and quantification of electronic interaction energies of biomolecular systems are the most challenging task. The hybrid QM/MM approaches may provide an alternate but demands high computational cost limits the applicability of these methods [44]. In hybrid QM/MM formalism, whole molecular system is fragmented into two parts, i.e. first smaller part, which is treated with quantum mechanical (QM) method, and the second larger part, i.e. environment, is preserved with a more efficient but less expensive MM method [45]. The total QM/MM energy of the system is considered as the sum of the energies of the model system (EQM), the environment (EMM), and the interactions between the model system and environment (EQM/MM) as obtained through high-level QM (model), low-level MM (environment) and medium level QM (interaction between the model system and environment) calculations, respectively (Equation 1).
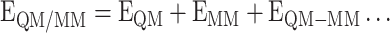 |
(1) |
As in many systems, the entropy of QM system is ignored [46], we have used an assumption in our present communication, to calculate the QM/MM binding energy (∆E) as the difference of QM/MM energy of the complex to the sum of the energies of the protein (environment) and the ligand (model), as represented by Equation (5).
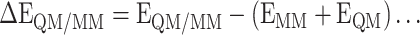 |
(2) |
Hence, QM/MM calculations on the final snapshot from 100 ns trajectory was performed using ONIOM (Own N-layered Integrated molecular Orbital and MM) method [47] with electronic embedding, as implemented in Gaussian 03 software [34]. The environment was treated as MM through AMBER force field while QM (model) part has been treated via exploiting B3LYP/6-31G(d,p), MP2/6-31G(d,p), and wb97xd/6-31g(d,p) density functional methods, respectively.
Results and discussion
Structure based virtual screening analysis and ADMET analysis
The virtual screening technique is used in the identification of small molecules from large compound databases, which hold considerable affinity with the active pocket of receptor [48–50]. Screening of natural compound libraries is of great interest because of high chemical diversity, biochemical specificity, and other molecular descriptor properties that make them favorable as lead ligands in drug discovery [51, 52]. Herein, virtual screening of SARS-CoV-2 Mpro against NP-lib database at MTiOpenScreen webserver results in collection of 654 compounds with docking score between −10 and −4 kcal/mol (Table S1).
Following, top four natural compounds, i.e. ZINC000043552589, ZINC000003594862, ZINC000003947429 and ZINC000070691536 with docking energy −10.5, −9.8, −9.5 and −9.4 kcal/mol, respectively, with favorable binding affinity in the active pocket of SARS-CoV-2 Mpro were considered for further analysis. Here, ZINC000043552589 was identified as 2,3-Dihydroamentoflavone, an amentoflavone-type bioflavonoid was first isolated from Cycas revoluta [53]. Likewise, ZINC000003594862 (Podocarpusflavon-B) was reported as an amentoflavone and extracted from Selaginella sinensis (Selaginellaceae) herb [54]. Interestingly, amentoflavone are well-known with antiviral activity against influenza A and B viruses, showed moderate anti-herpes simplex virus (HSV)-1 and anti-HSV-2 activities with EC50 values of 17.9 μg/ml (HSV-1) and 48.0 μg/ml (HSV-2), respectively [55]. Also, amentoflavone showed potent antiviral activity against respiratory syncytial virus (RSV) at IC50 of 5.5 mg/mL [55], and was isolated from Torreya nucifera; already reported in 2010 to possess micromolar range inhibitory activity IC50 of 8.3 μM against of SARS-CoV Mpro via FRET assay [56]. Moreover, amentoflavone has been reported with antiviral activities mediated by interaction with other viral targets in several viral infections, including dengue virus [57], coxsackievirus B3 [58], human immunodeficiency virus [59] and RSV [55]. Recently, high throughput virtual screening of chemical databases also screened amentoflavone as potent inhibitor of SARS-CoV Mpro [60]. This agreement with previously reported experimental observations specifically regarding Mpro target of SARS-CoV with a high similarity with SARS-CoV-2 provides an important validation for the screened compounds. Moreover, ZINC000003947429 and ZINC000070691536 identified as Vitamin P (Rutin) and Quercimeritrin 6″-O-L-arabinopyranoside were not documented for any in vitro antiviral activity. Thus, screened potential compounds were subjected to further stringent analysis for electronic properties and intermolecular interactions within active pocket of SARS-CoV-2 Mpro.
Moreover, the intrinsic properties of compounds along with drug-likeness and pharmacological characteristics have been suggested as an essential factor for medical applications [61, 62]. Herein, the potent molecules were evaluated for medical properties using SwissADME sever, as shown in Table S2. It is important to mention that druglikeness rules are not applicable to the natural products that are recognized by the active transport system when contemplating ‘druggable chemical entities’ [63, 64]. Additionally, Koehn in 2012 reviewed the 26 drugs approved between 1981 and 2011 stemmed from 18 natural products recognized to challenge the ‘rule of 5’ and its structures [63, 65]. Furthermore, based on number of electronegative species in the compound structure, each potent compound was studied for capacity for formation of hydrogen-bond acceptor and donor atoms. Interestingly, a maximum of 16 acceptor and 10 donor atoms were counted in ZINC000003947429 while a minimum of 10 acceptor and 4 donor atoms noted for ZINC000003594862 against N3 inhibitor (Table S2). In addition, other properties such as pharmacokinetics and medicinal chemistry friendliness were computed for the potent compounds (Table S2). Conclusively, the screened compounds were suggested with ideal medical properties by comparison to N3 inhibitor.
Theoretical calculations
Optimized geometry
The DFT calculations were used to establish the optimized structural parameters such as bond lengths, bond angles, and dihedral angles for potential compounds, i.e. ZINC000043552589, ZINC000003594862, ZINC000003947429 and ZINC000070691536, against N3 inhibitor using hybrid DFT B3LYP/6-31G(d,p) calculations (Tables S3–S7). Figure 1 shows the 2D structures and 3D optimized geometries of ZINC000043552589, ZINC000003594862, ZINC000003947429, ZINC000070691536 and N3 inhibitor plotted using atomic numbering scheme and without hydrogen atoms in GaussView 3.0.8.
Figure 1.
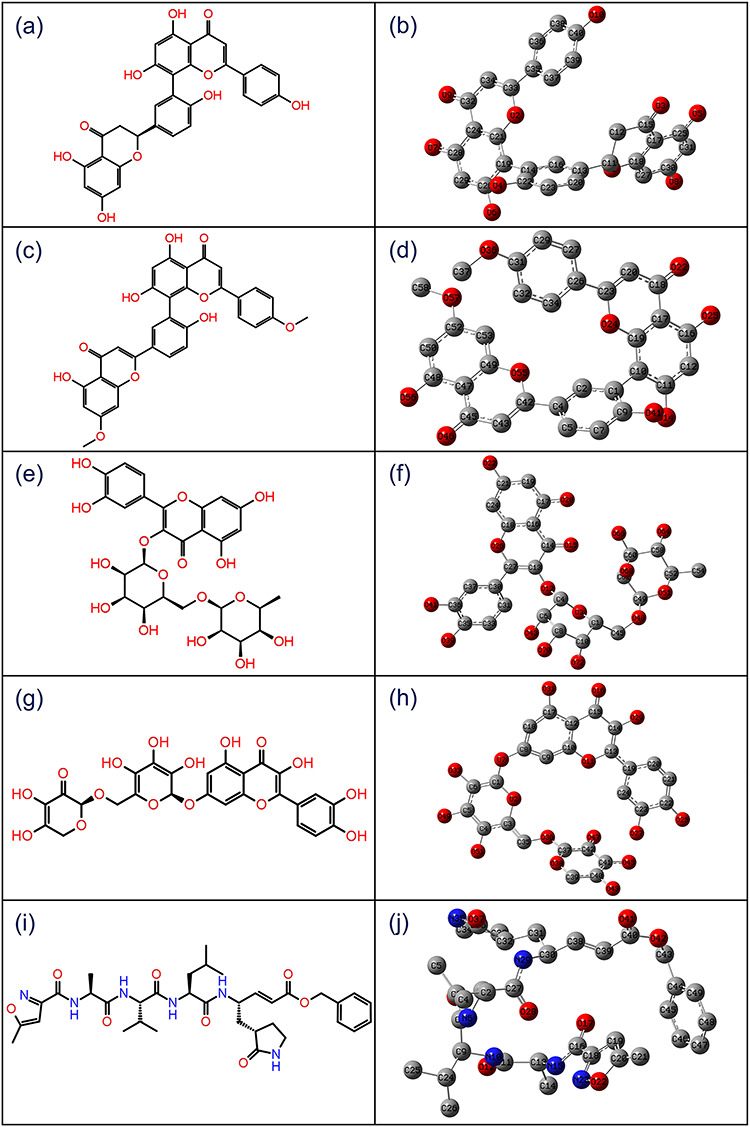
2D structures and 3D optimized molecular geometries of compounds, viz. (a–b) ZINC000043552589, (c–d) ZINC000003594862, (e–f) ZINC000003947429, (g–h) ZINC000070691536 and (i–j) reference compound N3 inhibitor, were calculated using B3LYP/6-31G(d,p) level of DFT calculations.
Natural charges and natural bond orbitals analysis
To better understand the electronic structure of potential natural compounds, natural bond orbital (NBO) calculation [66] was performed using NBO 3.1 program implemented in Gaussian 03 package at B3LYP/6-31G(d,p) method on the optimized structures. Herein, natural population analysis (NPA), was predicted for atomic summary of natural localized atomic charges on compounds [67]. Also, NPA analysis exhibited distribution of negative charges mostly on electronegative atoms, such as oxygen and on a few carbon atoms, located in both cyclic and non-cyclic chains; additionally, electrons accumulation in core, valence and Rydberg sub-shells for each compound were also considered (Tables S8–S12). According to an electrostatic point of view on the molecule, negative charged atoms have a tendency for electron donation to receptors while positive charged atoms (carbon) represent the capability to accept the electrons [68]. As a result, each compound was discovered with a tendency to donate and accept electrons. This reveals that NPA localized charges produced no substantial response for the respective systems; hence, further NBO analysis on selected compounds was considered (Tables S13–S17).
NBO calculations are useful to calculate the information about interactions in both filled and virtual orbital regions that can enhance the intra-and intermolecular interactions [69]. Thus, accepted convergence of NBO natural Lewis structures were considered to get better insights into structural description of each screened compound in terms of delocalization of electron density from the principal occupied Lewis-type (bond or lone pair) orbitals to unoccupied non-Lewis (anti-bonding or Rydberg) orbitals (Tables S13–S17). Moreover, highly occupied molecular orbitals (HUMO) and lower unoccupied molecular orbitals (LUMO) with high occupancy of NBO were predicted on each compound (Figure 2). The NBO analysis provides a convenient basis to study the conjugative interactions and efficient approach to predict an intra-and intermolecular bonding for a molecular system [66, 70]. Thus, respective high and lower occupied atoms stand for electron donor and acceptor species, respectively, were identified within each molecule (Figure 2), predicted as core sites for molecular contacts formation within active pocket of receptor.
Figure 2.
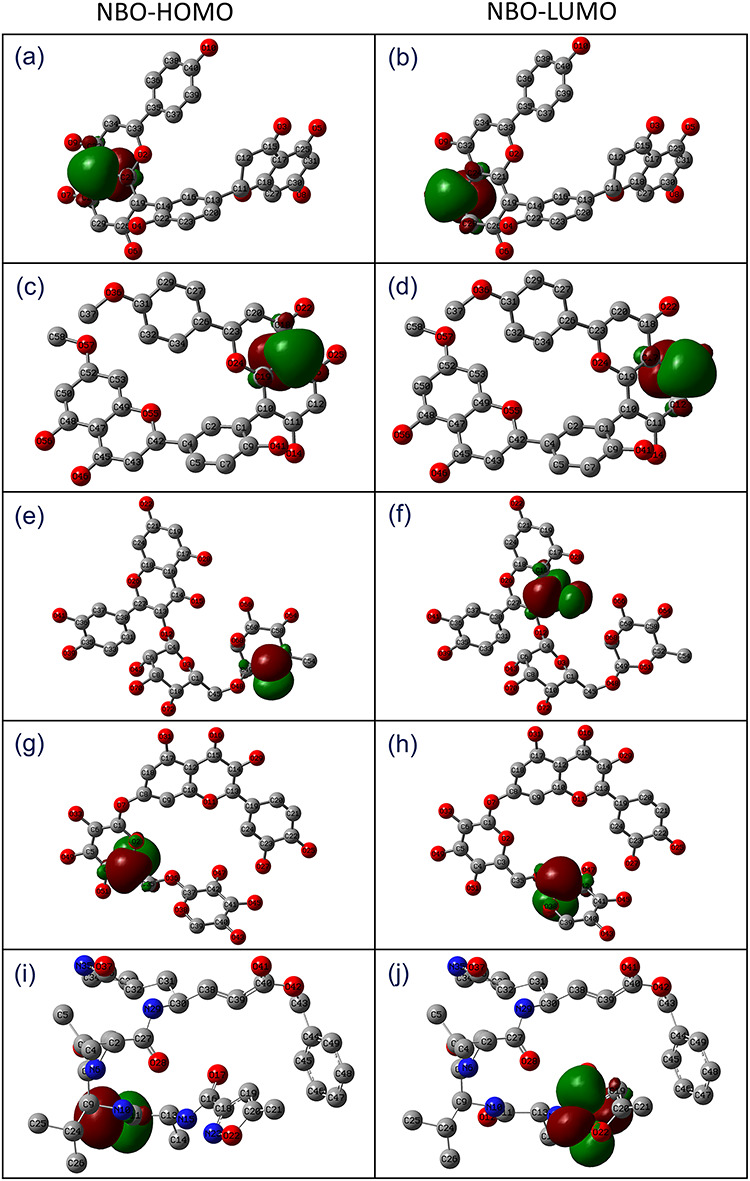
Natural bond orbitals for HOMO and LUMO estimated in the optimized geometries of selected natural compounds, i.e. (a–b) ZINC000043552589, (c–d) ZINC000003594862, (e–f) ZINC000003947429, (g–h) ZINC000070691536 and (i–j) reference compound N3 inhibitor.
Frontier molecular orbitals analysis
The frontier molecular orbitals play an important role in the electric and optical properties; hence, particularly frontier molecular orbitals energy, i.e. HOMO-LUMO band gap energy (EHOMO-ELUMO) is used to calculate the chemical reactivity, kinetic stability and chemical hardness of the molecule [71, 72]. A compound associated with a small band gap energy is generally classified with a high chemical reactivity and low kinetic stability, termed as soft molecule [73]. In this process, the reduction in energy gap indicates substantial intramolecular charge transfer within the molecule, which reflects the bioactivity of a molecule. Hence, EHOMO, ELUMO, and EHOMO-ELUMO values were calculated for potential compounds and reference ligand. Figure 3 shows asymmetric HOMO and LUMO for natural compounds and N3 inhibitor, where red and green color distribution signifies negative and positive phases, respectively, of the molecular frontier orbital wave function. Moreover, calculated energy band gap values showed considerable kinetic stability and a low chemical reactivity except for ZINC000070691536 against N3 inhibitor (Figure 3).
Figure 3.
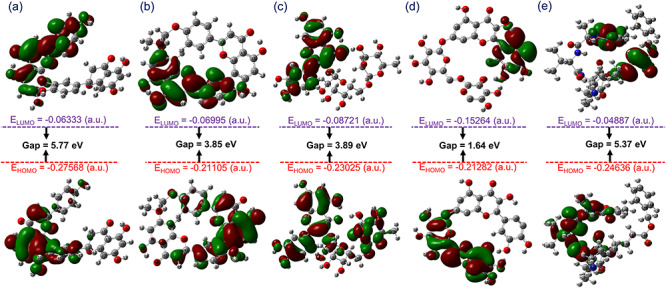
Frontier molecular orbitals predicted for (a) ZINC000043552589, (b) ZINC000003594862, (c) ZINC000003947429, (d) ZINC000070691536 and (e) N3 inhibitor calculated at B3LYP/6-31G(d,p) level.
Re-docking and intermolecular interaction analysis
The molecular docking algorithms are used in drug discovery to predict the potential ligand against drug-targetable receptors. Herein, DFT optimized geometries of potential compounds, ZINC000043552589, ZINC000003594862, ZINC000003947429 and ZINC000070691536, were re-docked in the active pocket of SARS-CoV-2 Mpro and analyzed for mechanistic interactions with active residues of viral protease. Remarkably, selected natural compounds showed substantial docking score >−12 kcal/mol and formation of considerable number of hydrogen bond with residues in the active pocket of SARS-CoV-2 Mpro (Figure 4).
Figure 4.
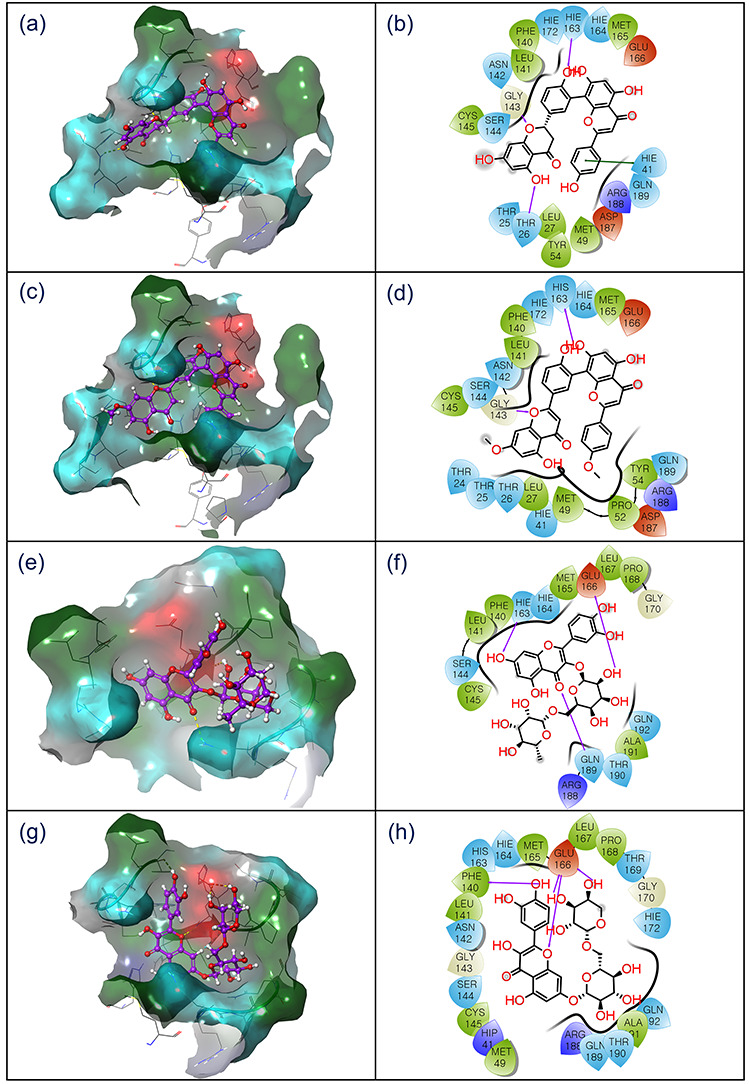
3D and 2D interaction diagram of SARS-CoV-2 Mpro-natural compounds; (a–b) ZINC000043552589, (c–d), ZINC000003594862, (e–f) ZINC000003947429 and (g–h) ZINC000070691536, docked complexes exhibiting interactions with respective ligands at 4 Å area in the active pocket of SARS-CoV-2 Mpro. In 3D interaction map, surface is generated based on the type of residue, and in 2D maps, hydrogen bond formation (pink arrows), hydrophobic (green), polar (blue), red (negative), violet (positive), glycine (grey) and π–π stacking (green line) interaction are logged for respective docked complexes.
The docked complex SARS-CoV-2 Mpro-ZINC000043552589 was recorded for significant binding affinity of −12.47 kcal/mol via three hydrogen bonds formation at residues Thr26 (N–H···O, 2.13 Å), Gly143 (N–H···O, 1.96 Å), and His163 (N–H···O, 2.2 Å). In addition, SARS-CoV-2 Mpro-ZINC000043552589 exhibited formation of π–π stacking by His41 residue. Likewise, SARS-CoV-2 Mpro-ZINC000003594862 complex exhibited considerable binding affinity of −13 kcal/mol and formed two hydrogen bonds with active residues Gly143 (N–H···O, 1.90 Å) and His163 (N···H–O, 2.31 Å). Whilst SARS-CoV-2 Mpro docked with ZINC000003947429 showed highest −15.63 kcal/mol docking score and formation of three hydrogen bonds at His163 (N–H···O, 2.14 Å), Glu166 (C=O···H, 2.47 Å), and Gln189 (N–H···O, 2.48 Å) residues. Moreover, SARS-CoV-2 Mpro-ZINC000070691536 complex also showed substantial docking score of −13.3 kcal/mol via formation of four hydrogen bonds at Phe140 (C=O···H, 1.96 Å) and Glu166 (C=O···H, 2.10 Å; N–H···O, 2.14 Å; and C=O···H, 2.17 Å) residues. Additionally, all the docked complexes were recorded for hydrophobic, polar, negative, positive, and glycine interactions (Table 1, Figure 4). The observed significant binding affinity and strong intermolecular interactions for natural compounds in the active pocket of SARS-CoV-2 Mpro suggested their substantial inhibitory activity against viral protease.
Table 1.
List of various interactions and interacting residues in the active pocket of SARS-CoV-2 Mpro with the potential natural compounds were logged from respective docked complexes
| S. no. | Drugs | Docking energy (kcal/mol) | H-bond | π–π stacking | Hydrophobic | Polar | Negative | Positive | Glycine |
|---|---|---|---|---|---|---|---|---|---|
| 1. | ZINC000043552589 | -12.47 | Thr26, Gly143, His163 | His41 | Leu27, Met49, Tyr54, Phe140, Leu141, Cys145, Met165 | Thr25, Thr26, His41, Asn142, Ser144, His163, His164, His172, Gln189 | Glu166, Asp187 | Arg188 | Gly143 |
| 2. | ZINC000003594862 | -13.0 | Gly143, His163 | — | Leu27, Met49, Pro52, Tyr54, Phe140, Leu141, Cys145, Met165 | Thr24, Thr25, Thr26, His41, Asn142, Ser144, His163, His164, His172, Gln189 | Glu166, Asp187 | Arg188 | Gly143 |
| 3. | ZINC000003947429 | -15.63 | His163, Glu166, Gln189 | — | Phe140, Leu141, Cys145, Met165, Leu167, Pro168, Ala191 | Ser144, His163, His164, Gln189, Thr190, Gln192 | Glu166 | Arg188 | Gly170 |
| 4. | ZINC000070691536 | -13.3 | Phe140, Glu166 (3) | — | Met49, Phe140, Leu141, Cys145, Met165, Leu167, Pro168, Ala191 | Gln92, Asn142, Ser144, His163, His164, Thr169, His172, Gln189, Thr190 | Glu166 | His41, Arg188 | Gly143, Gly170 |
| 5 | N3 inhibitor | -7.3 | Cys145, Glu166, Gln189(2) | — | Leu27, Met49, Tyr54 | Thr24, Thr25, Thr26, His41, Thr45, Ser46, Asn142, Ser144, His163, His164, His172, Gln189, Thr190, Gln192 | Glu166, Asp187 | Arg188 | Gly143 |
Note: Ala: Alanine; Arg: Arginine, Asn: Asparagine, Cys: Cysteine, Glu: Glutamic acid, His: Histidine, Leu: Leucine; Met: Methionine; Phe: Phenylalanine; Pro: Proline, Trp: Tryptophan; Tyr: Tyrosine, Val: Valine.
To further understand the inhibitory potential of selected natural compounds against SARS-CoV-2 Mpro, the molecular docking results were compared with the reference ligand N3 inhibitor under similar parameters. From docking analysis, N3 inhibitor (−7.3 kcal/mol) showed less binding affinity by comparison to potential natural compounds (maximum docking score −15.63 kcal/mol was recorded in SARS-CoV-2 Mpro-ZINC000003947429 complex) via four hydrogen bonds formation at Cys145 (N–H···O, 2.01 Å), Glu166 (N–H···O, 2.16 Å) and Gln189 (C=O···H, 2.30 Å and C=O···H, 3.06 Å) residues supplemented with other intermolecular interactions (Table 1, Figure S1). Remarkably, ZINC000003594862 exhibited a maximum of four hydrogen bonds followed by three hydrogen bonds for ZINC000003947429, and two hydrogen bonds for each ZINC000003594862 and ZINC000043552589 in the active pocket of SARS-CoV-2 Mpro (Table 1, Figure 4). Furthermore, respective SARS-CoV-2 Mpro-natural compounds complexes were noticed for one or more than one type of contacts with catalytic dyad (Cys145 and His41), substrate binding residues (Phe140, Leu141, Gly143, Ser144, His163, Met165 and Glu166), and other important residues in the viral protease active pocket (Thr24, Thr25, Met49, Phe140, Asn142, Asp187 and Gln189) (Table 1, Figures 4 and S1), as documented in the crystal structure with N3 inhibitor [17]. Interestingly, Gln189 residue, documented as essential site for the S1 subsite at the P1 position [17], was also logged for hydrogen formation (C=O···H, 2.30 Å and C=O···H, 3.06 Å) with ZINC000003947429 while exhibited polar interactions for other three docked selected natural compounds with SARS-CoV-2 Mpro (Table 1). The comparative intermolecular interaction analysis indicated that potent natural compounds have considerably occupied the catalytic dyad and substrate binding cleft of SARS-CoV-2 Mpro by strong hydrogen bond formation and substantial non-covalent interactions against N3 inhibitor. Hence, the computed interaction profiling supports the screened natural compounds as inhibitor against proteolytic function of SARS-CoV-2 Mpro, which is essentially required in viral pathogenesis [17]. Moreover, selected bioactive compounds show interactions with the highly conserved substrate-recognition pocket of SARS-CoV-2 Mpro, also directed the anti-proteolytic function of screened natural compounds against broad spectrum of HCoVs. To note, ZINC000003594862 (Podocarpusflavon-B) was recently identified to possess micromolar range inhibitory activity against SARS-CoV Mpro [56]. Hence, molecular docking and intermolecular interaction analysis deduced the potential inhibitory activity of screened natural compounds against SARS-CoV-2 Mpro by comparison to N3 inhibitor.
MD simulation analysis
In computational drug discovery, MD simulation is used to predict the docked complexes stability and formation of intermolecular interaction in reference to time [48, 49]. Herein, docked complexes of respective natural compounds with SARS-CoV-2 Mpro were analyzed via MD simulation to establish the respective complexes stability and intermolecular interactions between receptor and ligand against 100 ns interval. Initially, the last snapshots were extracted from the respective 100 ns MD trajectories and analyzed for intermolecular contacts. Remarkably, screened compounds exhibited hydrogen bonds formation supplemented with additional intermolecular interactions against N3 inhibitor (Table 2, Figures S2 and S3). Stimulatingly, each snapshot extracted from 100 ns simulation trajectories shared common molecular contacts with residues as logged in the respective docked poses (Table 1). These observations directed stability of natural compounds in the active pocket of viral protease during simulation by comparison to N3 inhibitor (Figures S2 and S3). Furthermore, three properties were extracted from MD trajectory for each complex with the aid of simulation interaction diagram in Maestro-Desmond interface, included (a) root mean square deviation (RMSD), (b) root mean square fluctuation (RMSF) and (c) protein-ligand contacts mapping.
Table 2.
List of various interactions and interacting residues in the active pocket of SARS-CoV-2 Mpro with the potential natural compounds were logged from the last pose of respective 100 ns MD trajectory
| S. no. | Drug | H-bond | π-π stacking | Hydrophobic | Polar | Negative | Positive | Glycine |
|---|---|---|---|---|---|---|---|---|
| 1. | ZINC000043552589 | Thr26, His41, Cys44, Glu166, Arg188 | — | Leu27, Val42, Cys44, Met49, Cys145, Met165, Leu167, Pro168 | Thr25, Thr26, His41, Asn142, Ser144, His163, His164, Gln189, Gln192 | Glu166, Asp187 | Arg188 | — |
| 2. | ZINC000003594862 | Thr24, Thr190 | His41 | Ile43, Cys44, Met49, Pro52, Cys145, Met165, Leu167, Pro168 | Thr24, Thr25, His41, Thr45, Ser46, His64, Gln189, Thr190, Gln192 | Glu166, Asp187 | Arg40, Arg188 | — |
| 3. | ZINC000003947429 | Glu166, Gln189 | — | Met49, Leu50, Met165, Leu167, Pro168, Ala191 | His41, Asn142, Gln189, Thr190 | Glu166 | Arg188 | — |
| 4. | ZINC000070691536 | Glu166(2), Thr190, Gln192 | His41 | Met49, Cys145, Met165, Leu167, Pro168, Val186, Ala191 | Asn142, Ser144, His163, His164, His172, Gln189, Thr190, Gln192 | Glu166, Asp187 | His41, Arg188 | Gly143 |
| 5. | N3 inhibitor | — | — | Leu50, Met165, Leu167, Pro168, Ala173, Phe181, Phe185, Val186 | His164, Gln189, Gln192 | Glu166, Asp187 | Arg188 | — |
Note: Ala: Alanine; Arg: Arginine, Asn: Asparagine, Cys: Cysteine, Glu: Glutamic acid, His: Histidine, Leu: Leucine; Met: Methionine; Phe: Phenylalanine; Pro: Proline, Trp: Tryptophan; Tyr: Tyrosine, Val: Valine.
RMSD and RMSF analysis
To measure the average change in displacement occurred in protein and ligand of the respective docked complexes with respect to initial frame, firstly RMSD values were computed for the protein structure (Cα, backbone, sidechain and heavy) and protein fit ligand from all the frames during 100 ns simulation trajectory, where RMSD for frame X can be expressed by following equation.
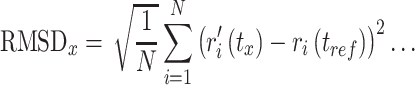 |
(3) |
where N is defined as the number of atoms in the atom selection; tref is marked as the reference time at 0 interval; r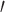 presented the position of the atoms under consideration in frame x after superimposition on the reference frame at interval
presented the position of the atoms under consideration in frame x after superimposition on the reference frame at interval 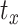 .
.
In all the docked complexes, Cα atoms of SARS-CoV-2 Mpro showed considerable fluctuations <3 Å, except in SARS-CoV-2 Mpro-ZINC000003594862 complex, which exhibited a higher variation of 3.8 Å still end of 100 ns simulation (Figure 5). These results indicated that viral protease experienced structural conformation changes induce by the binding of ZINC000003594862. These observations were further supported by acceptable deviations in the protein backbone, sidechain, and heavy atoms in docked SARS-CoV-2 Mpro crystal structure with an exception in SARS-CoV-2 Mpro-ZINC000003594862 complex (>3.5 Å). (Figure S4). Furthermore, RMSD analysis of protein fit ligands showed acceptable variations (<4 Å), except for ZINC000003594862 (>10 Å) at the end of 100 ns interval. Likewise, N3 inhibitor exhibited higher deviation >16 Å after 80 ns, which reflects non-stable behavior of the respective docked complex (Figure 5). However, it is important to mention that all the natural compounds docked with viral protease exhibited equilibrium within 20 ns, with an exception for SARS-CoV-2 Mpro-ZINC000003594862 complex, which exhibited state of equilibrium after 40 ns.
Figure 5.
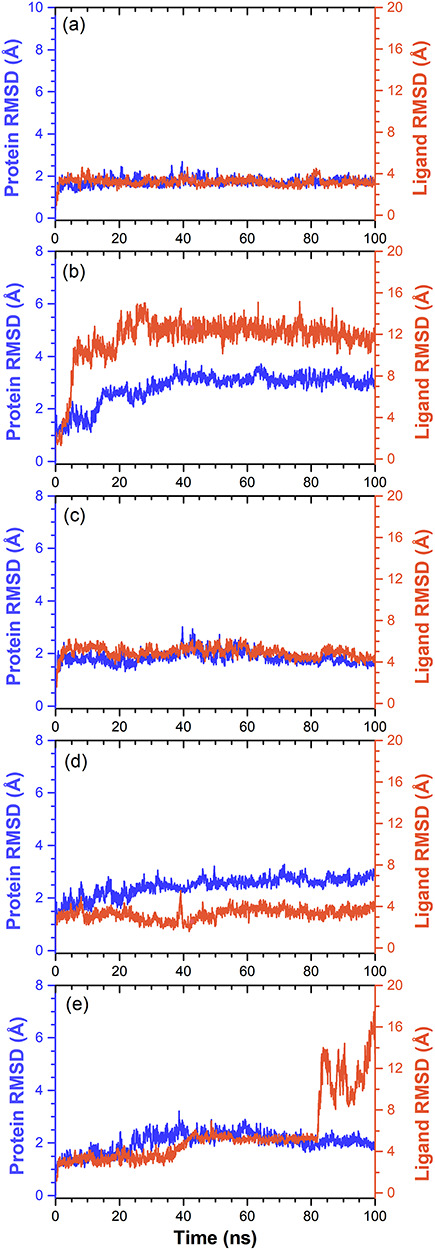
RMSD values extracted for alpha carbon atoms (blue curves) of SARS-CoV-2 Mpro and natural compounds (red curves) from the docked complexes, viz. (a) ZINC000043552589, (b), ZINC000003594862, (c) ZINC000003947429, (d) ZINC000070691536 and (e) N3 inhibitor, with respect to 100 ns simulation time.
Furthermore, RMSF calculations are useful in the characterization of local fluctuations along with protein chain and ligand molecule, which can be expressed from the simulation trajectory using the following equation.
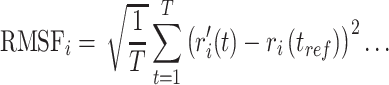 |
(4) |
where T presented the simulation interval over which the RMSF is calculated, tref defined as the reference time, 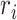 marked for the position of atom i in the reference time tref and
marked for the position of atom i in the reference time tref and 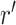 defined as the position of atom i at time t following superposition on the reference frame.
defined as the position of atom i at time t following superposition on the reference frame.
The local structural fluctuations in SARS-CoV-2 Mpro and docked ligands were calculated in terms of deviations contribute by residues of the protein and atoms of the ligands, respectively (Figure S5). Remarkably, residues of the protein in all the complexes showed acceptable RMSF values, except in C-terminal (>3.5 Å) and N-terminal (>4.5 Å) (Figure S5). Besides, atoms of the ligands were also recorded with considerable RMSF values (<4.5 Å), except for N3 inhibitor (<7.5 Å) (Figure S6). These RMSF values together with RMSD values for each complex supported the integration of screened potential compounds within the active pocket of viral protease against N3 inhibitor.
Protein-ligand interaction mapping
As hydrogen bonding properties in drug design are well known to exert strong influence on drug specificity, metabolism and adsorption; hence, intermolecular interactions, such as hydrogen bond formation, hydrophobic interactions, ionic interactions and water bridge formation, were extracted for the respective MD trajectories using default parameters of Desmond module.
All the selected compounds showed substantial interactions with catalytic residues, i.e. His41 and Cys145, and other substrate binding residues in the active pocket of SARS-CoV-2 Mpro against N3 inhibitor, which showed only significant interaction with substrate binding residue Glu166 (Figure 6). To note, these residues were also noticed in the respective docked complexes of selected compounds (Table 1). Moreover, screened compounds exhibited considerable intermolecular contacts such as hydrogen bond formation with SARS-CoV-2 Mpro active pocket residues during 30% of total MD simulation interval (Figure S7). Also, a total number of intermolecular contacts for the active residues and selected ligands, and their calculated density (dark area in orange color indicates the multiple contacts of ligand on that particular frame with the residues of protein) were extracted during 100 ns MD simulation interval (Figure S8). Again, these plots indicated the multiple interactions between the catalytic residues (His41 and Cys145) of SARS-CoV-2 Mpro and selected natural compounds during 100 ns simulation by comparison to N3 inhibitor. Conclusively, analysis of the respective MD trajectories established the considerable occupancy of screened natural compounds in the active pocket of SARS-CoV-2 Mpro against N3 inhibitor. Hence, the screened natural compounds can be arranged in the order of (a) ZINC000043552589, (b) ZINC000003947429, (c) ZINC000070691536 and (d) ZINC000003594862 as inhibitor of SARS-CoV-2 Mpro based on 100 ns MD simulation analysis.
Figure 6.
Protein-ligand interactions mapping for SARS-CoV-2 Mpro with potential natural compounds, i.e. (a) ZINC000043552589, (b) ZINC000003594862, (c) ZINC000003947429, (d) ZINC000070691536 and (e) N3 inhibitor, extracted from 100 ns MD simulations.
Essential dynamics analysis
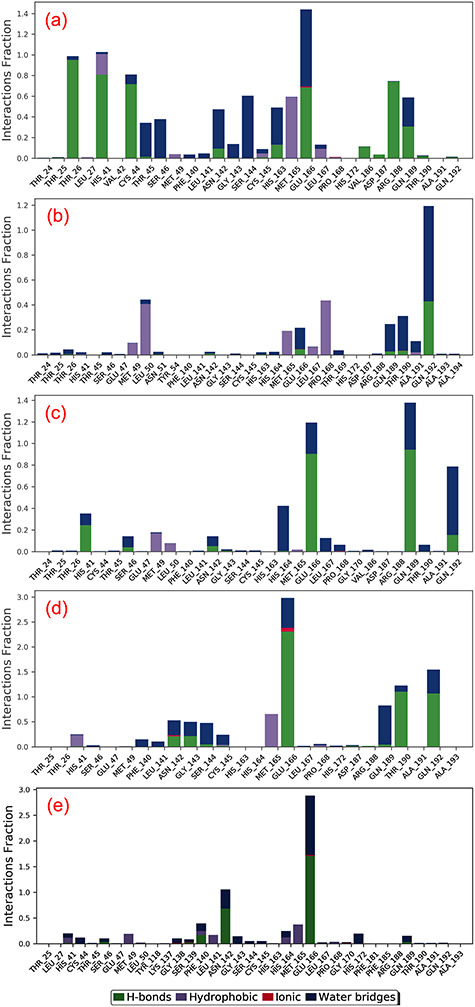
Principal component analysis (PCA), a covariance–matrix-based mathematical technique, is employed on the simulation trajectories to understand the domain dynamics and displacement of atoms in protein structure. Figure 7 shows percentage of variance (%) (eigen fraction) for the mean square positional variations in the covariance matrix as function of 20 eigen modes. All SARS-CoV-2 Mpro structures in each docked complex showed a sharp drop for percent in variance in reference to the initial three eigen modes, suggested a substantial conformational motion induced by docked ligand in the active pocket of protein. However, this variance was followed by instant elbow point and no further significant changes were observed in eigen fraction after 4 till 20 eigen values (Figure 7). These observations revealed the considerable flexibility of protein structure docked with respective ligands during initial stages of simulation which eventually reduced with simulation interval. Furthermore, percentage of contribution in eigen modes also sequentially decreases, suggested the localized fluctuations in SARS-CoV-2 structure in each docked complex tends to attain complex stability. Such motions in each protein-ligand complex during the simulations are suggested to contribute by docked ligands in the active pocket of viral protease direct to the formation of stable complex. Similar observations were observed previously in various complexes of Fructose transporter GLUT5 [74] and G-protein-coupled receptor 119 [75] docked with their respective ligands at the active pocket.
Figure 7.
Principal component analysis for the molecular dynamic simulation trajectories of SARS-CoV-2 Mpro docked with (a1–4) ZINC000043552589, (b1–4) ZINC000003594862, (c1–4) ZINC000003947429, (d1–4) ZINC000070691536 and (e1–4) N3 inhibitor. The percentage of total mean square displacement of residue positional variations recorded in each dimension is categorized by equivalent eigenvalue (PCs). The persistent color scale from blue to white to red directs the periodic jumps between the protein conformers duration the 100 ns simulation interval.
Figure 7 showed three PCA or eigen vectors for SARS-CoV-2 Mpro docked with selected potential compounds and N3 inhibitor extracted from respective MD trajectories and are represented in clusters. The analysis of eigen vectors supported the compact and the cluster motions in SARS-CoV-2 Mpro in respective complexes during MD simulation. In terms of essential dynamics, all the complexes of SARS-CoV-2 Mpro formed clusters between −35 and 30 along the direction of PC1, −35 and 40 for PC2 and −30 to 15 for PC3. In Figure 7, 2D plots demonstrate the variations in the ensemble distribution for each conformation of the docked complex during 100 ns simulation interval, where color gradient change from blue to red signifies the periodic jumps between the various conformational positions of SARS-CoV-2 Mpro structure in docked complexes with respective ligands. Altogether, correlated motion in SARS-CoV-2 structure in all the systems indicated the rigidity and considerable fluctuations induced at the active pocket by the binding of selected ligands during the simulation. Hence, these observations suggested the stability of selected natural compounds, i.e. ZINC000043552589, ZINC000003594862, ZINC000003947429 and ZINC000070691536, against N3 inhibitor in the active pocket of SARS-CoV-2 Mpro and eventually restricted the protein essential motion required for enzymatic activity, which leads to viral protease inhibition; supported by RMSD, RMSF, protein-ligand interaction profiling, and PCA analysis as well as substantial docking scores (Table 1, Figures 5 and 6).
MM generalized Born surface area analysis
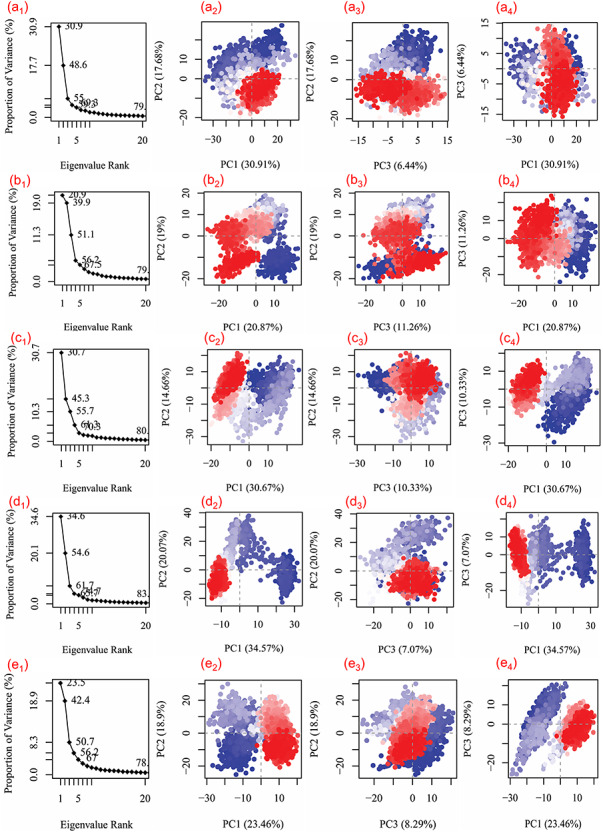
To calculate the net binding free energy and contribution of each energy component responsible for stability of potential compounds against N3 inhibitor docked in the active pocket of SARS-CoV-2 Mpro, MM generalized Born surface area (MM/GBSA) calculation was conducted on the collected snapshots from respective 100 ns simulation (Table S18). Figure 8 shows high net negative binding free energy values for SARS-CoV-2 Mpro docked with screened natural compounds against reference ligand N3 inhibitor (Figure S9). Additionally, analysis of recorded binding affinity values and physico-chemical components for each SARS-CoV-2 Mpro-natural compound docked complexes revealed significant contribution of ΔGBind Coulomb (Coulomb energy) and ΔGBind vdW (Van der Waals interaction energy) in the respective complex stability (Figure 8). These results advised the substantial affinity of potential compounds, i.e. ZINC000043552589, ZINC000003594862, ZINC000003947429 and ZINC000070691536, by comparison to N3 inhibitor with active residues in the catalytic pocket of SARS-CoV-2 Mpro. Hence, these binding free energy values advocated the potential of screened natural compounds as SARS-CoV-2 Mpro inhibitors against SARS-CoV-2 infection.
Figure 8.
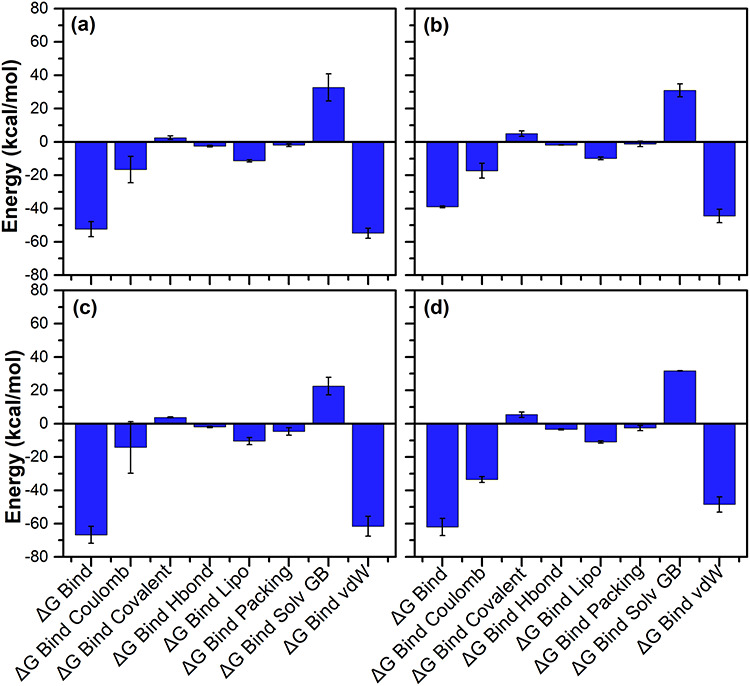
Calculated energy components and net MM/GBSA binding free energy (kcal/mol) with standard deviation values for extracted snapshots of SARS-CoV-2 Mpro complexes with selected natural compounds, i.e. (a) ZINC000043552589, (b) ZINC000003594862, (c) ZINC000003947429 and (d) ZINC000070691536, from respective 100 ns MD simulation trajectories.
Hybrid QM/MM results
The computation of docking affinity using molecular docking tools is based on scoring function with fixed dielectric charge for the protein and ligand atoms to be fast and accurate. As docking protocol lacks electronic interaction component, which is one of the essential components in the energy model, results in low accuracy of the docking results [44, 76]. Hence, the charge model can be improved for accurate binding energy prediction of a protein–ligand complex using the QM/MM methods, which can perform superior prediction of the electronic interactions [76]. In this study, the final snapshots extracted from 100 ns MD trajectories of respective docked SARS-CoV-2 Mpro-natural compounds complexes were used in QM/MM calculations. As each ligand exhibited exclusively non-covalent interactions with the protein; hence, ligand and protein were considered under the QM and MM region, respectively. The results of hybrid QM/MM calculations along with respective re-docking score and MM/GBSA binding free energy values for the potential natural compounds with SARS-CoV-2 Mpro are presented in Table 3. Among the considered three different hybrid QM/MM approaches for binding energy calculations, viz. B3LYP/6-31G(d,p), MP2/6-31G(d,p) and wb97xd/6-31g(d,p), the maximum binding energy values were obtained for each SARS-CoV-2 Mpro-natural compounds docked complex with wb97xd/6-31g(d,p) basis set treatment (Table 3). These results again suggested the significant contribution of dispersion energy (Van der Waals interactions) as observed from MM/GBSA calculations (Figure 8). Hence, QM/MM energy values advocate the stability of selected natural compounds with SARS-CoV-2 Mpro. However, reference ligand N3 inhibitor showed positive binding energy values through all the three considered quantum chemical methods, indicated the nonstable complex formation. Also, QM/MM binding energy calculations suggested that the density functional along with dispersion correction wb97xd/6-31g(d,p) has slightly better convergence properties compared to B3LYP and MP2 functionals (Table 3). Hence, it can be concluded that the dispersion corrected functional are suitable method to be used for the QM/MM binding energy estimations. Moreover, QM/MM binding energy revealed compounds with superior affinity with SARS-CoV-2 Mpro by comparison to calculated docking energy, which can be arranged in descending order, i.e. ZINC000043552589, ZINC000070691536, ZINC000003947429 and ZINC000003594862, as potent inhibitor of SARS-CoV-2 Mpro, supported by 100 ns MD simulation (Figure 5).
Table 3.
Comparison of re-docking scores, MM/GBSA binding free energy, and QM/MM binding energy for the selected natural compounds as SARS-CoV-2 Mpro inhibitors
| S. no. | SARS-CoV-2 Mpro-compound complex | Docking Energy (kcal/mol) | ∆G (kcal/mol) | ∆E (kcal/mol) | ||
|---|---|---|---|---|---|---|
| B3LYP/6-31G(d,p) | MP2/6-31G(d,p) | wb97xd/6-31g(d,p | ||||
| 1 | ZINC000043552589 | −12.47 | −52.35±4.54 | −215.79 | −218.18 | −319.04 |
| 2 | ZINC000003594862 | −13 | −38.92±0.59 | −81.55 | −53.81 | −82.93 |
| 3 | ZINC000003947429 | −15.63 | −70.41±4.81 | −156.41 | −128.51 | −156.60 |
| ZINC000070691536 | −13.3 | −62.01±5.15 | −216.96 | −144.39 | −217.34 | |
| 5 | Reference ligand N3 inhibitor | −7.3 | −54.84±8.92 | 696.65 | 699.84 | 50112.23 |
Note: ∆G: Binding free energy; ∆E: Binding energy.
Conclusion
With the advancement in SARS-CoV-2 infection and absence of any potential anti-viral therapeutic against its treatment, natural compounds are targeted as potential source for the anti-viral drug development. In this study, a total of four selected natural compounds, viz. ZINC000043552589, ZINC000003594862, ZINC000003947429 and ZINC000070691536, were selected based on highest docking score >-12 kcal/mol from the natural compound database NP-lib against SARS-CoV-2 Mpro. The respective complexes were further evaluated in terms of intermolecular interactions, complex stability and binding affinity against SARS-CoV-2 Mpro-N3 inhibitor as reference complex using computational methods. Based on collective analysis, selected four natural compounds were concluded with strong molecular contacts in the active pocket of SARS-CoV-2 Mpro. Hence, these compounds can be considered for further evaluation as SARS-CoV-2 Mpro inhibitors using in vitro studies for the drug development against SARS-CoV-2 infection.
Key Points
A total of 653 natural compounds were screened with SARS-CoV-2 Mpro, which showed energy between −10 and −4 kcal/mol.
Top four natural compounds were identified as 2,3-Dihydroamentoflavone, Podocarpusflavon-B, Rutin and Quercimeritrin 6″-O-L-arabinopyranoside with >−12 kcal/mol re-docking energy.
DFT calculations of selected natural compounds showed larger HOMO–LUMO gap refers to higher kinetic stability and lower chemical reactivity against N3 inhibitor.
Potential compounds established molecular contacts with catalytic dyad (Cys145 and His41) and substrate binding residues (Phe140, Leu141, Gly143, Ser144, His163, Met165 and Glu166).
MD simulation, MM/GBSA and QM/MM energy further supports the inhibitory potential of selected natural compounds against SARS-CoV-2 Mpro.
Supplementary Material
Acknowledgements
The authors would like to acknowledge Dr. Pranav Kumar, Director, Pathfinder Research and Training Foundation for his support during the study. Also, the authors are highly thankful to Dr. Amaresh Kumar Sahoo, Indian Institute of Information Technology, Prayagraj, India for providing his kind support in binding free energy calculation in Schrodinger 2019.2 suite.
Shiv Bharadwaj is an International Research Professor at Department of Biotechnology, Institute of Biotechnology, College of Life and Applied Sciences, Yeungnam University, Republic of Korea, who is working in the area of drug discovery and nanotechnology. He is also the member of World Society for Virology.
Amit Dubey is a researcher at Department of Biochemistry, University of Allahabad, Prayagraj, India, who is working in the area of structural bioinformatics and drug discovery.
Umesh Yadava is a Professor at Department of Physics, Deen Dayal Upadhyay Gorakhpur University, Gorakhpur, India, who is working in the area of drug discovery and X-ray crystallography.
Sarad Kumar Mishra is a Professor at Department of Biotechnology, D.D.U. Gorakhpur University, Gorakhpur, UP, India, who is working in the area of Enzyme Technology and Antiviral Research.
Sang Gu Kang is a Professor at Department of Biotechnology, Institute of Biotechnology, College of Life and Applied Sciences, Yeungnam University, Republic of Korea, who is working in the area of molecular genetics, bioinformatics and drug discovery.
Vivek Dhar Dwivedi is a Bioinformatics Scientist at Center for Bioinformatics, Computational and Systems Biology, Pathfinder Research and Training Foundation, Greater Noida, India, who is working in the area of viral informatics and drug discovery. He is also the member of World Society for Virology.
Contributor Information
Shiv Bharadwaj, Department of Biotechnology, Institute of Biotechnology, College of Life and Applied Sciences, Yeungnam University, Republic of Korea.
Amit Dubey, Department of Biochemistry, University of Allahabad, Prayagraj, India.
Umesh Yadava, Department of Physics, Deen Dayal Upadhyay Gorakhpur University, Gorakhpur, India.
Sarad Kumar Mishra, Department of Biotechnology, D.D.U. Gorakhpur University, Gorakhpur, UP, India.
Sang Gu Kang, Department of Biochemistry, University of Allahabad, Prayagraj, India.
Vivek Dhar Dwivedi, Bioinformatics Scientist at Center for Bioinformatics, Computational and Systems Biology, Pathfinder Research and Training Foundation, Greater Noida, India.
Data availability Statement
All the data cited in this manuscript is generated by the authors and available upon request from the corresponding authors.
Conflict of Interest
All the authors declared no conflict of interests.
Funding
The authors have received no financial support for the research, authorship, and/or publication of this article.
References
- 1. Kirtipal N, Bharadwaj S, Kang SG. From SARS to SARS-CoV-2, insights on structure, pathogenicity and immunity aspects of pandemic human coronaviruses. Infect Genet Evol 2020;85:104502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Coronaviridae Study Group of the International Committee on Taxonomy of V . The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol 2020;5:536–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 2020;395:565–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pal M, Berhanu G, Desalegn C, et al. Severe acute respiratory syndrome Coronavirus-2 (SARS-CoV-2): an update. Cureus 2020;12:e7423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jayaweera M, Perera H, Gunawardana B, et al. Transmission of COVID-19 virus by droplets and aerosols: a critical review on the unresolved dichotomy. Environ Res 2020;188:109819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cucinotta D, Vanelli M. WHO declares COVID-19 a pandemic. Acta Bio-Medica: Atenei Parmensis 2020;91:157–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Worldometer D. (2020). COVID-19 coronavirus pandemic. https://www.worldometers.info/coronavirus/ (18 May 2020, date last accessed).
- 8. Corman VM, Landt O, Kaiser M, et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill 2020;25:2000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cui J, Li F, Shi ZL. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol 2019;17:181–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Du Toit A. Outbreak of a novel coronavirus. Nat Rev Microbiol 2020;18:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li G, De Clercq E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat Rev Drug Discov 2020;19:149–50. [DOI] [PubMed] [Google Scholar]
- 12. Morse JS, Lalonde T, Xu S, et al. Learning from the past: possible urgent prevention and treatment options for severe acute respiratory infections caused by 2019-nCoV. Chembiochem 2020;21:730–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Perlman S, Netland J. Coronaviruses post-SARS: update on replication and pathogenesis. Nat Rev Microbiol 2009;7:439–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pillaiyar T, Manickam M, Namasivayam V, et al. An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J Med Chem 2016;59:6595–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zumla A, Chan JF, Azhar EI, et al. Coronaviruses - drug discovery and therapeutic options. Nat Rev Drug Discov 2016;15:327–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Torequl Islam M, Nasiruddin M, Khan IN, et al. A perspective on emerging therapeutic interventions for COVID-19. Front Public Health 2020;8:281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jin Z, Du X, Xu Y, et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020;582:289–93. [DOI] [PubMed] [Google Scholar]
- 18. Zhang L, Lin D, Sun X, et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 2020;368:409–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xian YF, Zhang J, Bian ZX, et al. Bioactive natural compounds against human coronaviruses: a review and perspective. Acta Pharmaceutica Sinica B 2020;10:1163–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Calixto JB. The role of natural products in modern drug discovery. Anais Da Academia Brasileira De Ciencias 2019;91(Suppl 3):e20190105. [DOI] [PubMed] [Google Scholar]
- 21. Beutler JAJCpip . Natural products as a foundation for drug discovery. Curr Protoc Pharmacol. 2019;86:e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Strohl WR. The role of natural products in a modern drug discovery program. Drug Discov Today 2000;5:39–41. [DOI] [PubMed] [Google Scholar]
- 23. Rodrigues T, Reker D, Schneider P, et al. Counting on natural products for drug design. Nat Chem 2016;8:531–41. [DOI] [PubMed] [Google Scholar]
- 24. Labbe CM, Rey J, Lagorce D, et al. MTiOpenScreen: a web server for structure-based virtual screening. Nucleic Acids Res 2015;43:W448–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pettersen EF, Goddard TD, Huang CC, et al. UCSF chimera–a visualization system for exploratory research and analysis. J Comput Chem 2004;25:1605–12. [DOI] [PubMed] [Google Scholar]
- 26. Bharadwaj S, Lee KE, Dwivedi VD, et al. Computational insights into tetracyclines as inhibitors against SARS-CoV-2 Mpro via combinatorial molecular simulation calculations. Life Sci 2020;257:118080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 2017;7:42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhao Y, Truhlar DG. Density functionals with broad applicability in chemistry. Acc Chem Res 2008;41:157–67. [DOI] [PubMed] [Google Scholar]
- 29. Forner W, Utz W. Correlated ab initio and density functional calculations on small model molecules for the unit cell of polyparaphenylene in its aromatic and quinoidal forms: equilibrium geometries and vibrational spectra. J Mol Structure-Theochem 2002;618:65–84. [Google Scholar]
- 30. Kruse H, Goerigk L, Grimme S. Why the standard B3LYP/6-31G* model chemistry should not be used in DFT calculations of molecular thermochemistry: understanding and correcting the problem. J Org Chem 2012;77:10824–34. [DOI] [PubMed] [Google Scholar]
- 31. Kohn W, Sham LJ. Self-consistent equations including exchange and correlation effects. Phys Ther Rev 1965;140:A1133. [Google Scholar]
- 32. Lee C, Yang W, RJJCP P. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density Phys. Rev. B 37 (1988) 785; AD Becke, density-functional thermochemistry. III. The Role of Exact Exchange 1993;98:5648. [DOI] [PubMed] [Google Scholar]
- 33. Miehlich B, Savin A, Stoll H, et al. Results obtained with the correlation energy density functionals of Becke and Lee. Yang and Parr 1989;157:200–6. [Google Scholar]
- 34. Fukuda R, Hasegawa J, Ishida M, et al. Gaussian 03, Revision B. 04; Gaussian, Pittsburgh, PA: Gaussian, Inc. 2003. [Google Scholar]
- 35. Yadava U, Gupta H, Roychoudhury M. A comparison of crystallographic and DFT optimized geometries on two taxane diterpenoids and docking studies with phospholipase A2. Med Chem Res 2012;21:2162–8. [Google Scholar]
- 36. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010;31:455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schrödinger Release 2020–2. New York, NY: Maestro, Schrödinger, LLC, 2020. [Google Scholar]
- 38. Bowers KJ, Chow DE, Xu H, et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In: SC'06: Proceedings of the 2006 ACM/IEEE Conference on Supercomputing. IEEE, 2006, 43–3. [Google Scholar]
- 39. Schrödinger Release 2018-4: Desmond Molecular Dynamics System, D.E . Shaw Research, New York, NY, 2018. Schrödinger, New York, NY: Maestro-Desmond Interoperability Tools, 2018. [Google Scholar]
- 40. Grant BJ, Rodrigues AP, ElSawy KM, et al. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics 2006;22:2695–6. [DOI] [PubMed] [Google Scholar]
- 41. Wang E, Sun H, Wang J, et al. End-point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design. Chem Rev 2019;119:9478–508. [DOI] [PubMed] [Google Scholar]
- 42. Schrödinger Release 2019–2. New York, NY: Prime, Schrödinger, LLC, 2019. [Google Scholar]
- 43. Adasme-Carreno F, Munoz-Gutierrez C, Caballero J, et al. Performance of the MM/GBSA scoring using a binding site hydrogen bond network-based frame selection: the protein kinase case. Phys Chem Chem Phys 2014;16:14047–58. [DOI] [PubMed] [Google Scholar]
- 44. Farrokhpour H, Pakatchian V, Hajipour A, et al. Protein-ligand interaction study of signal transducer smoothened protein with different drugs: molecular docking and QM/MM calculations. RSC Adv 2015;5:68829–38. [Google Scholar]
- 45. Senn HM, Thiel W. QM/MM methods for biomolecular systems. Angewandte Chem-Int Ed 2009;48:1198–229. [DOI] [PubMed] [Google Scholar]
- 46. Klahn M, Braun-Sand S, Rosta E, et al. On possible pitfalls in ab initio quantum mechanics/molecular mechanics minimization approaches for studies of enzymatic reactions. J Phys Chem B 2005;109:15645–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dapprich S, Komaromi I, Byun KS, et al. A new ONIOM implementation in Gaussian98. Part I. the calculation of energies, gradients, vibrational frequencies and electric field derivatives. J Mol Structure-Theochem 1999;461:1–21. [Google Scholar]
- 48. Bharadwaj S, Lee KE, Dwivedi VD, et al. Discovery of Ganoderma lucidum triterpenoids as potential inhibitors against dengue virus NS2B-NS3 protease. Scientific reports 2019;9:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bharadwaj S, Rao AK, Dwivedi VD, et al. Structure-based screening and validation of bioactive compounds as Zika virus methyltransferase (MTase) inhibitors through first-principle density functional theory, classical molecular simulation and QM/MM affinity estimation. 2020;1–14. [DOI] [PubMed]
- 50. Dwivedi VD, Arya A, Yadav P, et al. DenvInD: dengue virus inhibitors database for clinical and molecular research. Brief Bioinform 2020;bbaa098. doi: 10.1093/bib/bbaa098. [DOI] [PubMed] [Google Scholar]
- 51. Siddiqui AA, Iram F, Siddiqui S, et al. Role of natural products in drug discovery process. Int J Drug Dev Res 2014;6:172–204. [Google Scholar]
- 52. Dwivedi VD, Bharadwaj S, Afroz S, et al. Anti-dengue infectivity evaluation of bioflavonoid from Azadirachta indica by dengue virus serine protease inhibition. J Biomol Struct Dyn 2020;1–14. [DOI] [PubMed] [Google Scholar]
- 53. Anhut S, Seeger T, Zinsmeister H-D, et al. New dihydrobiflavones from the moss Plagiomnium cuspidatum. 1989;44:189–92. [Google Scholar]
- 54. Silva GL, Chai H, Gupta MP, et al. Cytotoxic biflavonoids from Selaginella willdenowii. Phytochemistry 1995;40:129–34. [DOI] [PubMed] [Google Scholar]
- 55. Ma SC, But PPH, Ooi VEC, et al. Antiviral amentoflavone from Selaginella sinensis. Biol Pharm Bull 2001;24:311–2. [DOI] [PubMed] [Google Scholar]
- 56. Ryu YB, Jeong HJ, Kim JH, et al. Biflavonoids from Torreya nucifera displaying SARS-CoV 3CL(pro) inhibition. Bioorg Med Chem 2010;18:7940–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Coulerie P, Nour M, Maciuk A, et al. Structure-activity relationship study of biflavonoids on the dengue virus polymerase DENV-NS5 RdRp. Planta Med 2013;79:1313–8. [DOI] [PubMed] [Google Scholar]
- 58. Wilsky S, Sobotta K, Wiesener N, et al. Inhibition of fatty acid synthase by amentoflavone reduces coxsackievirus B3 replication. Arch Virol 2012;157:259–69. [DOI] [PubMed] [Google Scholar]
- 59. Lin YM, Anderson H, Flavin MT, et al. In vitro anti-HIV activity of biflavonoids isolated from Rhus succedanea and Garcinia multiflora. J Nat Prod 1997;60:884–8. [DOI] [PubMed] [Google Scholar]
- 60. Olubiyi OO, Olagunju M, Keutmann M, et al. High throughput virtual screening to discover inhibitors of the main protease of the coronavirus SARS-CoV-2. Molecules 2020;25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Di L, Kerns EH. Drug-like properties: concepts, structure design and methods from ADME to toxicity optimization. Academic press, 2015. [Google Scholar]
- 62. Kramer C, Ting A, Zheng H, et al. Learning medicinal chemistry absorption, distribution, metabolism, excretion, and toxicity (ADMET) rules from cross-company matched molecular pairs analysis (MMPA) miniperspective. J Med Chem 2017;61:3277–92. [DOI] [PubMed] [Google Scholar]
- 63. Macarron R. Critical review of the role of HTS in drug discovery. Drug Discov Today 2006;11:277–9. [DOI] [PubMed] [Google Scholar]
- 64. Lipinski CAJDDTT . Lead-and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol. 2004;1:337–41. [DOI] [PubMed] [Google Scholar]
- 65. Darcel L, Djibo M, Gaillard M, et al. Trichormamide C structural confirmation through Total synthesis and extension to Analogs. Org Lett 2020;22:145–9. [DOI] [PubMed] [Google Scholar]
- 66. Glendening E, Reed A, Carpenter J, et al. Nbo Version 3.1, Tci, Vol. 65, Madison: University of Wisconsin. 1998. [Google Scholar]
- 67. Reed AE, Weinstock RB. Weinhold FJTJoCP. Natural Population Anal 1985;83:735–46. [Google Scholar]
- 68. Demircioglu Z, Kastas CA, Buyukgungor O. Theoretical analysis (NBO, NPA, Mulliken population method) and molecular orbital studies (hardness, chemical potential, electrophilicity and Fukui function analysis) of (E)-2-((4-hydroxy-2-methylphenylimino)methyl)-3-methoxyphenol. J Mol Struct 2015;1091:183–95. [Google Scholar]
- 69. Sheela N, Muthu S, SJSAPAM S, et al. Molecular orbital studies (hardness, chemical potential and electrophilicity), vibrational investigation and theoretical NBO analysis of 4-4′-(1H-1, 2, 4-triazol-1-yl methylene) dibenzonitrile based on abinitio and DFT methods. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy. 2014;120:237–51. [DOI] [PubMed] [Google Scholar]
- 70. Karabacak M, Sinha L, Prasad O, et al. The spectroscopic (FT-Raman, FT-IR, UV and NMR), molecular electrostatic potential, polarizability and hyperpolarizability, NBO and HOMO-LUMO analysis of monomeric and dimeric structures of 4-chloro-3,5-dinitrobenzoic acid. Spectrochim Acta A Mol Biomol Spectrosc 2012;93:33–46. [DOI] [PubMed] [Google Scholar]
- 71. Pearson RG. Electronic spectra and chemical reactivity. J Am Chem Soc 1988;110:2092–7. [Google Scholar]
- 72. Thanthiriwatte KS, de Silva KMN. Non-linear optical properties of novel fluorenyl derivatives - ab initio quantum chemical calculations. J Mol Structure-Theochem 2002;617:169–75. [Google Scholar]
- 73. Fleming I. Frontier orbitals and organic chemical reactions. Wiley, Hoboken, NJ, USA: 1977. [Google Scholar]
- 74. Ainsley J, Chaturvedi SS, Karabencheva-Christova TG, et al. Integrating molecular probes and molecular dynamics to reveal binding modes of GLUT5 activatory and inhibitory ligands. Chem Commun 2018;54:9917–20. [DOI] [PubMed] [Google Scholar]
- 75. Kaushik AC, Kumar A, Rehman AU, et al. Deciphering G-protein-coupled receptor 119 agonists as promising strategy against type 2 diabetes using systems biology approach. ACS Omega 2018;3:18214–26. [Google Scholar]
- 76. Khodarahmi G, Asadi P, Farrokhpour H, et al. Design of novel potential aromatase inhibitors via hybrid pharmacophore approach: docking improvement using the QM/MM method. RSC Adv 2015;5:58055–64. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data cited in this manuscript is generated by the authors and available upon request from the corresponding authors.