

Abstract
In early January 2020, the novel coronavirus (SARS-CoV-2) responsible for a pneumonia outbreak in Wuhan, China, was identified using next-generation sequencing (NGS) and readily available bioinformatics pipelines. In addition to virus discovery, these NGS technologies and bioinformatics resources are currently being employed for ongoing genomic surveillance of SARS-CoV-2 worldwide, tracking its spread, evolution and patterns of variation on a global scale. In this review, we summarize the bioinformatics resources used for the discovery and surveillance of SARS-CoV-2. We also discuss the advantages and disadvantages of these bioinformatics resources and highlight areas where additional technical developments are urgently needed. Solutions to these problems will be beneficial not only to the prevention and control of the current COVID-19 pandemic but also to infectious disease outbreaks of the future.
Keywords: SARS-CoV-2, COVID-19, pathogen discovery, bioinformatics, phylogenetic analysis, next-generation sequencing
Introduction
In late December 2019, pneumonia of unidentified cause was first reported in Wuhan, China [1]. Clinical diagnosis using various commercialized assays targeting multiple common respiratory pathogens failed to identify the causative agent [2]. However, the next-generation sequencing (NGS) of clinical samples, particularly bronchoalveolar lavage fluid from the first group of patients, soon identified a novel coronavirus [1–3], later named severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [4]. In this review article, we briefly summarize the NGS technologies and bioinformatics resources employed in the discovery and surveillance of SARS-CoV-2, as well as their advantages and disadvantages, and highlight areas where future work will be particularly profitable.
NGS approaches for SARS-CoV-2 discovery and surveillance
NGS, also termed high-throughput sequencing, has become one of the most widely used approaches in virus research, especially in the areas of the diagnosis of infectious diseases of unidentified cause [1], virus evolution [5] and virus discovery [6, 7]. Multiple sequencing strategies have been applied to discover and monitor the causative agent of the ongoing COVID-19 pandemic—SARS-CoV-2 (Figure 1). Among these, metagenomics has proven itself to be a simple, unbiased and highly efficient approach to virus discovery [7, 8, 9]. The metagenomic approach works best when the abundance of the target virus (SARS-CoV-2) is relatively high and other microorganisms in the samples also need to be analyzed (Figure 1A). Importantly, the proportion of virus-related reads can be greatly increased if the total RNA of clinical samples from COVID-19 patients is subject to ribosomal RNA (rRNA) depletion during the library preparation step [10]. Alternatively, a hybrid capture method can be used to enrich SARS-CoV-2 by using a mixture of RNA probes corresponding to SARS-CoV-2-specific fragments following library construction (Figure 1B).
Figure 1.
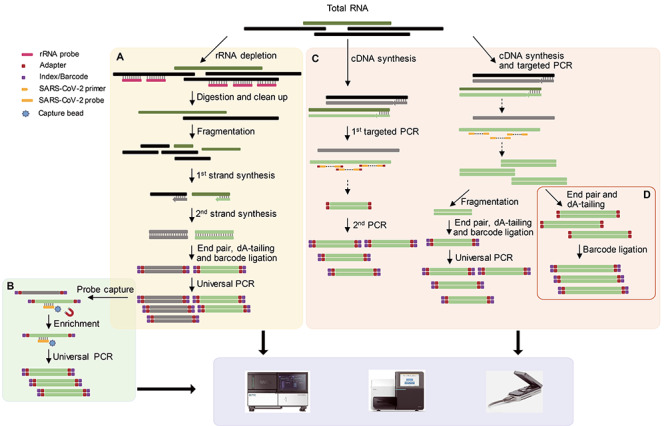
The workflow of different NGS sequencing approaches currently available for virus discovery and genomic surveillance. The library construction scheme employed in (A) metatranscriptomic sequencing, (B) a hybrid capture-based approach based on a metatranscriptomic library, (C) multiplex PCR amplification for NGS platforms and (D) the Oxford Nanopore sequencing platform.
After many SARS-CoV-2 genomes were obtained using metagenomics sequencing during the early stages of the outbreak, a multiplex polymerase chain reaction (PCR) amplification technology targeting SARS-CoV-2 was developed (Figure 1C and D): total RNA is reverse transcribed to synthesize cDNA, and a PCR is then run using multiple amplification primer pairs targeting SARS-CoV-2, followed by ligation reaction to add the indexes/barcodes. The libraries are subsequently sequenced on Illumina, MGI or Nanopore platforms (https://artic.network/ncov-2019) [11]. In particular, the multiplex PCR amplification technology is efficient in cases of samples with low viral load [12, 13], when the cycle threshold (Ct) value of SARS-CoV-2 quantitative real-time (qRT)-PCR ranges from 24.5 to 31.8 (1–100 viral genome copies per microliter) [12]. Facilitated with the multiplex PCR amplification technology, the MinION device is widely used to diagnose and identify SARS-CoV-2 within hours with high sensitivity [14, 15]. Importantly, however, multiplex PCR amplification sequencing cannot be used to sequence highly diverse or recombinant viruses because the primers are designed according to the reference genomes. PCR may also be limited by the primer dimer formation and the non-optimized reaction system. In addition, the error rate of this technology is higher than most other NGS platforms, with many of the deletions accumulating in the homopolymers [16–18]. It may therefore be preferable to use the Oxford Nanopore platform supplemented with Sanger or Illumina and MGI platforms to obtain viral genomes with higher accuracy and coverage [19].
Bioinformatics resources for SARS-CoV-2 discovery
As NGS will usually generate millions of sequencing reads with or without a priori knowledge of SARS-CoV-2, the efficiency of virus discovery is heavily dependent on the downstream bioinformatics tools employed. Unfortunately, there is still not a fully integrated bioinformatics pipeline available that is able to automatically analyze NGS data and identify those reads potentially related to viruses. In the context of virus discovery, a typical NGS data analysis workflow consists of several essential steps, including quality control of the NGS data, removal of host/rRNA data, reads assembly, taxonomic classification and virus genome verification (Figure 2). Fortunately, numerous applications are now available for every step (Table 1).
Figure 2.
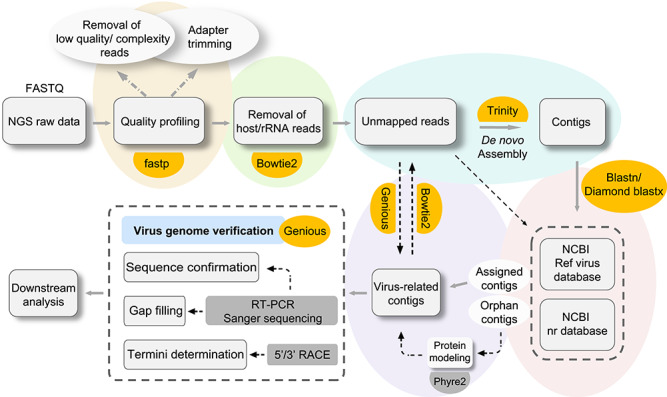
A schematic workflow and the bioinformatics resources used in novel virus discovery. Each key step in the workflow is shown with different backgrounds. Computational tools used in the SARS-CoV-2 discovery by our group are colored in orange.
Table 1.
Summary of the available bioinformatics resources for SARS-CoV-2 discovery and genomic surveillance
Quality profiling
The quality control and preprocessing of raw FASTQ files is critical for subsequent analyses, especially for degraded samples, and involves removing adapter sequences, filtering low quality/complexity reads, error correction, etc. Sequence matching-based adapter trimming tools like Trimmomatic [20], Cutadapt [21] and SOAPnuke [22] (Table 1) can be employed as adapter trimmers and can also perform sliding window or maximum information quality filtering. Recently, all-in-one FASTQ preprocessors, such as AfterQC [23] and fastp [24] (Table 1), provide a variety of functions, including quality profiling, adapter and polyG/polyX tail trimming, base correction, per-read quality pruning and unique molecular identifier preprocessing. These efficient computer applications can support both single-end and paired-end (PE) sequencing data, with the exception of PE data requiring some additional steps based on overlapping analysis. For instance, adapter sequences are detected using the overlapping detection algorithm of each pair and can be trimmed with even only one base in the tail, whereas most sequence matching-based tools require at least three bases. Fastp also performs sequence matching-based adapter trimming when setting specific adapter sequences. In addition, for multiplex PCR amplification technology that targets SARS-CoV-2, multiple amplification primer pairs will be removed after general quality control steps using a particular Python script (Cut_Multi_Primer.py) (Table 1) developed by MGI Tech (https://github.com/MGI-tech-bioinformatics/SARS-CoV-2_Multi-PCR_v1.0).
Removal of host/rRNA data
The next challenge is to efficiently process immense amount of data and identify potential virus-related sequences after the quality control of the raw data. As virus genetic material will normally only comprise a tiny proportion of the total nucleic acids present in any sequencing run, the (more) abundant host reads need to be removed by mapping all reads to a host reference genome (if available) using mapping and alignment tools (such as Hisat2 [25], BWA [26], Bowtie2 [27] or KMA [28]) (Table 1). rRNA also needs to be removed using Bowtie2 or SortmeRNA [29] (Table 1), although it is also possible to perform rRNA depletion at the library preparation stage. However, for samples with low concentration or low quality, the host/rRNA depletion step can be skipped to increase the chances of obtaining viral reads.
Reads assembly
Without a priori knowledge of a novel virus genome, a routine approach is to de novo assemble the reads into contigs. Generally, there are two different assembly algorithms [30]: (i) the de Bruijn graph approach is usually used to assemble short reads by converting them to k-mers, which is employed in programs like Trinity [31], Megahit [32], SPAdes [33] and Trans-ABySS [34] (Table 1); and (ii) the overlap–layout–consensus (OLC) approach, which is normally used for the assembly of long reads and is applicable to highly similar genomes such as different viral variants or haplotypes, and employed in programs like PEHaplo [35] and SAVAGE [36] (Table 1). De novo assembly is the best approach in the context of emerging infectious diseases where no reference genomes are available, such as COVID-19. Read assembly can be challenging under virus discovery settings because both sequence divergence and background noise may be extensive, and no tools are always guaranteed to give the best results [37]. Recently, a specialized assembler, coronaSPAdes [38] (Table 1), was developed to recover genome sequences of the Coronaviridae (including both novel and known species), employing algorithmic assembly from rnaviralSPAdes and the HMM-guided algorithms of biosyntheticSPAdes, based on the genome organization from fragmented assemblies.
Taxonomic classification
Once the reads are assembled into contigs, the next step is to assign contigs to a specific taxon (i.e. species, genus and family) as a means of taxonomic classification. The most common approach is to blast the individual contigs against a nucleotide/protein database, such as the non-redundant protein sequence database (nr) or the reference virus sequence database (RefSeq_viruses). Diamond [39] (Table 1) is one of the most popular tools used for aligning translated short reads against the nr database and is much faster than Blastx (a gold standard tool for protein alignment) with a similar degree of sensitivity. Blastn [40] (Table 1), as a traditional nucleotide-to-nucleotide search program, is still widely used for nucleotide sequence alignment. As there will be tens of thousands of contigs, it is usually advisable to create a local database that contains protein sequences of all known reference viruses to further reduce the computational burden and accelerate the blast process. However, the results from local blast searches should be interpreted with caution because of false hits to non-viral proteins that share homology with viral counterparts. It is therefore important to perform a confirmatory Blastx search against the nr database to avoid false positives. Another strategy is to directly align all remaining reads to reference databases using alignment tools for contigs: this will greatly reduce the computational resources required for data analysis, especially with libraries constructed using human-related samples. Reads from the virus-positive library are then de novo assembled as described above. The remaining unannotated contigs are tentatively assigned as ‘orphan’ contigs [41]. Although divergent in primary sequence, such orphaned contigs can be further analyzed using protein structure-informed approaches such as that implemented in Phyre2 [42] (Table 1).
Virus genome verification
After virus-associated contigs are extracted, the quality of the contigs can be examined by read mapping. The reliable contigs with unassembled overlaps, or those from the same scaffold, are then merged to form longer viral contigs using contig assembly tools (SEQMAN or Geneious), followed by iterative read mapping for further extension of the genome at both ends. Results in sam/bam format can be visualized using programs like IGV [43], Geneious (http://www.geneious.com) or QIAST [44] (Table 1). If necessary, gaps can be filled by RT-PCR and Sanger sequencing, and genome termini can be determined by RNA circularization or 5′/3′ RACE kits. The consensus sequence determined from the final assembly of the mapped reads can be used as the newly identified virus genome for downstream analyses.
For sequence data generated from third-generation sequencing platform (e.g. Oxford Nanopore sequencing), the data analysis workflow is basically the same as that described above, but using different programs at each step due to the production of longer reads. For instance, NanoPack [45] (Table 1) is a comprehensive preprocessing tool with several individual scripts for long-read sequencing data, providing multiple quality profiling features (NanoStat, NanoPlot and NanoComp), read filtering and trimming (NanoFilt) and the removal of contamination (NanoLyse). Porechop (https://github.com/rrwick/Porechop) (Table 1) functions as an adapter trimmer and is able to find and remove adapters from Oxford Nanopore reads through alignment-based strategies, even with low sequence identity. In addition, new alignment tools (i.e. Minimap2 [46], NGMLR [47], MarginAlign [48]) and multiple de novo assembly tools (i.e. Canu [49], Falcon (https://github.com/PacificBiosciences/falcon), Miniasm [50]) (Table 1) based on OLC approaches have now also been developed specifically for long-read data.
Bioinformatics resources for genomic and evolutionary analyses
SARS-CoV-2-related databases
There have been a number of SARS-CoV-2-related databases, such as Global Initiative on Sharing All Influenza Data (GISAID, https://www.gisaid.org/) [51], the National Center for Biotechnology Information (NCBI, https://www.ncbi.nlm.nih.gov/sars-cov-2/) [52], Genome Warehouse (https://bigd.big.ac.cn/gwh/), National Bioinformatics Center (CNCB)/National Genomics Data Center (NGDC) database (https://bigd.big.ac.cn/ncov/) [53] and Virus Pathogen Resource (https://www.viprbrc.org/) [54]. Among them, GISAID deposited the largest number of SARS-CoV-2 genome sequences. These databases play important roles in sequence archive, homology searching, variation discovery, disease phenotype association, etc.
Multiple sequence alignment
Accurate multiple sequence alignment (MSA) is the foundation of all comparative genome sequence analyses. The number of available MSA methods has increased in recent decades [55], although they can be classified into three major categories: (i) progressive-based methods (including CLUSTALW [56], MAFFT [57] and MUSCLE [58]), (ii) consistency-based methods (including T-Coffee [59], ProbCons [60] and some versions of MAFFT [61]) and (iii) evolution-based methods (including PRANK [62], Bali-Phy [63] and StatAlign [64]) (Table 1). JABAWS [65] integrates a variety of MSA tools (e.g. MUSCLE, MAFFT and ClustalW) (Table 1), which can be conveniently packaged to run locally. Web services, such as EMBL-EBI [66], also provide free access to online applications of popular sequence analysis tools (e.g. MUSCLE, MAFFT, ClustalW, T-Coffee and webPRANK [67]) (Table 1). Different MSA algorithms can produce different alignments, obviously impacting all downstream analyses [68]. Fortunately, because SARS-CoV-2 genomes are so similar in sequence, with few insertion–deletion events (indels), MSA is normally straightforward. The resulting alignment can be then visualized, analyzed, annotated and manually edited using Jalview [69], MSAViewer [70], AliView [71], Bioedit [72] or Geneious (https://www.geneious.com) (Table 1).
Phylogenetic and evolutionary analyses
Phylogenetic trees are central to understanding the emergence and evolution of SARS-CoV-2 and can be estimated using a variety of approaches, particularly distance-based methods such as neighbor joining (NJ) [73] and character-based methods including maximum parsimony (MP) [74], maximum likelihood (ML) [75] and Bayesian inference (BI) [76]. The NJ, ML and BI methods use explicit statistical models of nucleotide or amino acid substitution (that can be compared and evaluated using programs such as jMODELTEST [77] and ProtTest [78] (Table 1). Although they are both based on substitution models, BI methods differ from ML methods in that they use statistical distributions to quantify uncertainties (posterior distributions) both in the tree and model parameters [79]. Substitution models are not employed in MP which instead attempts to minimize the number of evolutionary changes across the tree in accord with the parsimony principle [80]. Bayesian methods have been extended to determine the patterns of virus spread in both space (i.e. phylogeography) and time [81]. Both applications require sequence evolution to proceed at an approximately constant rate, the so-called molecular clock of evolution. Before running analyses assuming a molecular clock, it is advisable to test its presence through a regression of root-to-tip genetic distances against date of sampling (e.g. using the TempEst program [82]). A number of computer programs and packages implementing these phylogenetic methods have been developed, such as BIONJ [83] for NJ; PhyML [84], RAxML [85] and IQ-TREE [86] for ML; and MrBayes [87], PhyloBayes [88], BEAST1 [89] and BEAST2 [90] for BI (Table 1). Multiple methods are included in packages such as PAUP [91] and MEGA [92] (Table 1). Recently, a novel integrated desktop platform, PhyloSuite [93] (Table 1), has been developed for streamlined molecular sequence data management and phylogenetics studies. A variety of approaches are also available for visualization of the resultant phylogeny, such as Dendroscope v3.5.10 [94], FigTree v1.4.3 (http://tree.bio.ed.ac.uk/software/figtree/) and ggtree [95] (Table 1). Online services such as iTOL [96] and Evolview [97] (Table 1) can also be used to annotate phylogenetic trees.
Both ML and BI approaches have been used extensively to study the evolution of SARS-CoV-2 [98, 85, 99–103]. When a large number of SARS-CoV-2 genome sequences are being analyzed, IQ-TREE v1.6.8 and RAxML v8.2.9 (Table 1) are recommended as they can utilize many computation nodes with high efficiency and hence are applicable to large datasets. For example, a BI-based analysis platform, Nextstrain [104], analyzes the latest SARS-CoV-2 data from GISAID [51] and visualizes the spread and evolution of all available SARS-CoV-2 strains in real time (https://nextstrain.org/ncov/global/zh).
Finally, although alignment-free approaches have recently been proposed to enable genome-scale phylogenetic inference, such as the average common subsequence [105], composition vector (CVTree) [106], k-mer [107] methods, to the best of our knowledge, they have not been used in the phylogenetic analysis of SARS-CoV-2.
Virus genome annotation and analysis
Genome annotation
For a new virus, genome annotation can be challenging. The open reading frames of SARS-CoV-2 were initially predicted using Geneious v11.1.5 and annotated using the Conserved Domain Database (https://www.ncbi.nlm.nih.gov/cdd/) [108]. Subsequently, online services, such as the popular genome browser UCSC (http://genome.ucsc.edu/covid19.html), Ensembl (https://covid-19.ensembl.org/index.html), or NCBI SARS-CoV-2 Resources (https://www.ncbi.nlm.nih.gov/sars-cov-2/) [52] were developed to facilitate SARS-CoV-2 genome annotation. For multiple SARS-CoV-2 sequences, cross-referencing on the reference virus genome, NC_045512.3 (strain Wuhan-Hu-1), from GenBank (https://www.ncbi.nlm.nih.gov/), simplifies gene annotation. In addition, there are several offline applications(e.g. GFF2PS [109], Vectro NTI [110] and IBS [111]) (Table 1) that are able to annotate virus genomes.
Detection of genetic variation
Levels of genetic identity provide a simple impression of the relationships between viruses. Several computer programs can be used to calculate pairwise sequence identities between sequences, such as Geneious v11.1.5 [2]. The DNADIST program of PHYLIP v3.697 [112] (Table 1) can also be used to estimate the genetic distance matrix of SARS-CoV-2.
When the number of sequences is small, visual inspection of the alignment is sufficient to identify mutational changes. For example, by inspecting the alignment of full-length SARS-CoV-2 genomes, mutational sites including nucleotide substitutions and indels could be readily identified in each virus genome using MEGA X [102, 103]. However, when the number of sequences to be analyzed is large, visual inspection becomes challenging. Online resources, including the CNCB/NGDC database (https://bigd.big.ac.cn/ncov/), Nextstrain website [104] and the UCSC Genome Browser for SARS-CoV-2 (http://genome.ucsc.edu/covid19.html), can display the single-nucleotide polymorphisms at more than 10 000 sites across the SARS-CoV-2 genome.
Detecting coronavirus recombination
Coronaviruses are well known to have undergone frequent recombination [113] so that the occurrence of this process should be considered carefully. For example, we used SimPlot v3.5.1 [114] (Table 1) to detect potential inter-lineage recombination in betacoronaviruses, in which a sliding window analysis is employed to determine the changing patterns of sequence similarity between sequences which can then be verified by phylogenetic analysis. RDP4 [115] (Table 1) is a popular package to detect recombination events in a specific dataset and contains a number of common and important algorithms used for recombination detection, such as RDP, GENECONV, 3Seq, Chimaera, SiScan, MaxChi and LARD. Generally, a recombination event is regarded as reliable when it is detected by multiple independent methods.
Homology modeling
Homology modeling is a useful tool to predict protein structures depending on the degree of similarity between the target sequence and the template sequences available in the databases (e.g. PDB), thereby helping to make inferences on protein function. The three-dimensional structures of SARS-CoV-2 have been modeled using the website Swiss-Model program [116] and displayed using PyMOL v2.1 [117] (Table 1): these studies revealed that SARS-CoV-2 may also use human ACE2 as binding receptor [2]. Similarly, by using homology modeling, we showed that the Spike protein of a bat coronavirus, RmYN02, might be not able to bind to human ACE2 [118].
Current challenges and future directions
A combination of NGS technology and available bioinformatics tools successfully identified SARS-CoV-2 within days of the report of a novel pneumonia. In addition, genomic epidemiology—based on a solid bed rock of phylogenetic analysis—has played an irreplaceable role in investigating the origins, tracing the spread, monitoring the evolution and variation of SARS-CoV-2, and will clearly play a key role in helping to contain the COVID-19 pandemic as well as future outbreaks.
Along with the development of sequencing technologies, the output of sequencing devices such as the Illumina Novaseq6000 reaches terabases per flow cell. Sample multiplexing should therefore be employed to maximize the efficiency and reduce costs, although this also leads to index hopping/swapping that can wrongly assign viruses to samples [119, 120]. The misassignment rates from Exclusion Amplification (ExAmp) chemistry (HiSeqX, HiSeq4000 and NovaSeq) instruments are estimated 0.2 to be 6%, approximately 10-fold higher than random cluster amplification instruments such as MiSeq [120, 121]. A high input of adapters and no dilution of PCR-free libraries often result in a high ratio of residual-free adapters. Therefore, index swapping rates using the PCR-free library construction method are higher than PCR-based methods, and one must be careful when interpreting those viruses found in pooled samples sequenced on a single lane [119]. It is also advisable that libraries with comparably high viral load are constructed in the same batch and loaded in the same lane to help eliminate amplicon contamination in other samples with low viral loads [11]. In contrast to Illumina technologies, MGI sequencers utilize the DNA nanoball technology that can reduce the misassignment rate to 0.0001–0.0004% under recommended procedures [120], although single indexed adapters somewhat limit these advantages. To mitigate cross-contamination, a non-redundant dual-indexing approach has been designed to be used on the Illumina sequencing platform, which would sharply reduce the index hopping rate.
Challenges remain when employing NGS data for novel virus discovery, in particular when the proportion of virus reads is very low. The situation is especially complex for highly divergent viruses that exhibit such little similarity in primary sequence and hence that they need to be identified by homology-based or protein structure-based methods. As a consequence, a better understanding of virosphere clearly requires more adequately validated methods and advances in computational analyses.
At the time of writing, there are >144 000 full-length genome sequences of SARS-CoV-2 available from GISAID. The generation of such an enormous amount of data represents both an achievement and a challenge. In particular, phylogenetic analysis and visualization of SARS-CoV-2 genomes is a cumbersome exercise with such a huge amount of data. In this case, both offline applications and online services cannot work efficiently due to the high demand of computational resources (memory/CPU). Splitting the whole dataset into several smaller sub-datasets, aligning the sub-datasets independently and combining them after MSA is a simple but feasible way to proceed in these circumstances. Even if phylogenetic analysis is feasible (e.g. [122]), such large trees are extremely difficult to visualize and interpret. Subsampling may therefore be the optimal way to proceed, and algorithms for this purpose are urgently needed.
In sum, technical advances in NGS and bioinformatics enabled us rapidly identify the causative agent of COVID-19 and track its global spread. However, the marked increase in the SARS-CoV-2 genome sequences has highlighted the technical obstacles in analyzing such ‘big’ datasets. Solutions to these problems will assist not only in the control of the current COVID-19 pandemic but also in those future outbreaks of infectious disease that will undoubtedly occur.
Key Points
A variety of next-generation sequencing technologies have been applied for the discovery and genomic surveillance of SARS-CoV-2, with metatranscriptomic sequencing suitable for virus discovery and amplicon/probe hybridization-based approaches more effective in genomic surveillance.
No fully integrated bioinformatics pipeline is currently available for virus discovery. However, there are many tools available for each component step, from quality control of the raw genomic sequence data to virus genome verification.
Current bioinformatics resources in multiple sequence alignment, phylogenetics, tree visualization and genomic analysis are robust and reliable. However, the sharp increase in the amount of SARS-CoV-2 genome sequence data available poses serious challenges for data storage and analysis that urgently need to be resolved.
Tao Hu is an associate professor at Shandong First Medical University, China. His major interest is applying computational biology approaches to analyzing ‘big’ biological data.
Juan Li is a lecturer at Shandong First Medical University, China. She received a PhD in veterinary microbiology and her research interest is the molecular evolution of RNA viruses.
Hong Zhou is a lecturer at Shandong First Medical University. She received a PhD in microbiology and works on the discovery of novel viruses from wildlife using next-generation sequencing.
Cixiu Li is an associate professor at Shandong First Medical University. She received a PhD in pathogen biology and her research interests include viromics and virus discovery.
Edward C. Holmes is an ARC Australian Laureate Fellow and professor at The University of Sydney, Australia. His research interests include virus evolution, metagenomics and the emergence and spread of novel infectious diseases.
Weifeng Shi is a professor at Shandong First Medical University. He received a PhD in bioinformatics and his research interests include the origins, evolution and spread of emerging viruses.
Contributor Information
Tao Hu, Shandong First Medical University, China.
Juan Li, Shandong First Medical University, China.
Hong Zhou, Shandong First Medical University, China.
Cixiu Li, Shandong First Medical University, China.
Edward C Holmes, University of Sydney, Australia.
Weifeng Shi, Shandong First Medical University, China.
Funding
Key research and development project of Shandong province (2020SFXGFY01 and 2020SFXGFY08); National Major Project for Control and Prevention of Infectious Disease in China (2018ZX10101004 and 2017ZX10104001); Academic Promotion Programme of Shandong First Medical University (2019QL006); National Key Research and Development Programme of China (grant no. 2020YFC0840800); Taishan Scholars Programme of Shandong Province (ts201511056 to W.S.).
References
- 1. Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China. 2019. N Engl J Med 2020;382:727–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 2020;395:565–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature 2020;579:265–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rambaut A, Holmes EC, O'Toole A, et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol 2020;5(11):1403–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ni M, Chen C, Qian J, et al. Intra-host dynamics of Ebola virus during 2014. Nat Microbiol 2016;1:16151. [DOI] [PubMed] [Google Scholar]
- 6. Shi M, Lin XD, Tian JH, et al. Redefining the invertebrate RNA virosphere. Nature 2016;540:539–43. [DOI] [PubMed] [Google Scholar]
- 7. Zhang YZ, Shi M, Holmes EC. Using metagenomics to characterize an expanding virosphere. Cell 2018;172:1168–72. [DOI] [PubMed] [Google Scholar]
- 8. Palacios G, Druce J, Du L, et al. A new arenavirus in a cluster of fatal transplant-associated diseases. N Engl J Med 2008;358:991–8. [DOI] [PubMed] [Google Scholar]
- 9. Wilson MR, Naccache SN, Samayoa E, et al. Actionable diagnosis of neuroleptospirosis by next-generation sequencing. N Engl J Med 2014;370:2408–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kraus AJ, Brink BG, Siegel TN. Efficient and specific oligo-based depletion of rRNA. Sci Rep 2019;9:12281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Quick J, Grubaugh ND, Pullan ST, et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat Protoc 2017;12:1261–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xiao M, Liu X, Ji J, et al. Multiple approaches for massively parallel sequencing of SARS-CoV-2 genomes directly from clinical samples. Genome Med 2020;12:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Paden CR, Tao Y, Queen K, et al. Rapid, sensitive, full-genome sequencing of severe acute respiratory syndrome coronavirus 2. Emerg Infect Dis 2020;26(10):2401–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fauver JR, Petrone ME, Hodcroft EB, et al. Coast-to-coast spread of SARS-CoV-2 during the early epidemic in the United States. Cell 2020;181:990–6e995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang M, Fu A, Hu B, et al. Nanopore targeted sequencing for the accurate and comprehensive detection of SARS-CoV-2 and other respiratory viruses. Small 2020;e2002169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sarkozy P, Jobbágy Á, Antal P. Calling homopolymer stretches from raw nanopore reads by analyzing k-mer dwell times. In: EMBEC & NBC 2017, Singapore, 2018. p. 241–4. Springer, Singapore.
- 17. Gu W, Miller S, Chiu CY. Clinical metagenomic next-generation sequencing for pathogen detection. Annu Rev Pathol 2019;14:319–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang Y, Yang Q, Wang Z. The evolution of nanopore sequencing. Front Genet 2014;5:449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim D, Lee JY, Yang JS, et al. The architecture of SARS-CoV-2 transcriptome. Cell 2020;181:914–21 e910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30:2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Martin M. CUTADAPT removes adapter sequences from high-throughput sequencing reads. EMBnet J 2011;17:10–2. [Google Scholar]
- 22. Chen Y, Chen Y, Shi C, et al. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 2018;7:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen S, Huang T, Zhou Y, et al. AfterQC: automatic filtering, trimming, error removing and quality control for fastq data. BMC Bioinformatics 2017;18:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen S, Zhou Y, Chen Y, et al. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018;34:i884–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim D, Paggi JM, Park C, et al. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 2019;37:907–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li H, Durbin R. Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics 2010;26:589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods 2012;9:357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Clausen P, Aarestrup FM, Lund O. Rapid and precise alignment of raw reads against redundant databases with KMA. BMC Bioinformatics 2018;19:307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kopylova E, Noe L, Touzet H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012;28:3211–7. [DOI] [PubMed] [Google Scholar]
- 30. Li Z, Chen Y, Mu D, et al. Comparison of the two major classes of assembly algorithms: overlap-layout-consensus and De Bruijn graph. Brief Funct Genomics 2012;11:25–37. [DOI] [PubMed] [Google Scholar]
- 31. Grabherr MG, Haas BJ, Yassour M, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 2011;29:644–U130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li D, Luo R, Liu CM, et al. MEGAHIT v1.0: a fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016;102:3–11. [DOI] [PubMed] [Google Scholar]
- 33. Bankevich A, Nurk S, Antipov D, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 2012;19:455–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Robertson G, Schein J, Chiu R, et al. De novo assembly and analysis of RNA-seq data. Nat Methods 2010;7:909–12. [DOI] [PubMed] [Google Scholar]
- 35. Chen J, Zhao YC, Sun YN. De novo haplotype reconstruction in viral quasispecies using paired-end read guided path finding. Bioinformatics 2018;34:2927–35. [DOI] [PubMed] [Google Scholar]
- 36. Baaijens JA, El Aabidine AZ, Rivals E, et al. De novo assembly of viral quasispecies using overlap graphs. Genome Res 2017;27:835–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Holzer M, Marz M. De novo transcriptome assembly: a comprehensive cross-species comparison of short-read RNA-Seq assemblers. Gigascience 2019;8:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Meleshko D, Korobeynikov A. coronaSPAdes: from biosynthetic gene clusters to coronaviral assemblies. bioRxiv 2020. 10.1101/2020.07.28.224584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods 2015;12:59–60. [DOI] [PubMed] [Google Scholar]
- 40. Camacho C, Coulouris G, Avagyan V, et al. BLAST+: architecture and applications. BMC Bioinformatics 2009;10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ortiz-Baez AS, Eden JS, Moritz C, et al. A divergent articulavirus in an Australian gecko identified using meta-transcriptomics and protein structure comparisons. Viruses 2020;12:613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kelley LA, Mezulis S, Yates CM, et al. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 2015;10:845–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Robinson JT, Thorvaldsdottir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol 2011;29:24–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gurevich A, Saveliev V, Vyahhi N, et al. QUAST: quality assessment tool for genome assemblies. Bioinformatics 2013;29:1072–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. De Coster W, D'Hert S, Schultz DT, et al. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 2018;34:2666–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 2018;34:3094–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sedlazeck FJ, Rescheneder P, Smolka M, et al. Accurate detection of complex structural variations using single-molecule sequencing. Nat Methods 2018;15:461–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jain M, Fiddes IT, Miga KH, et al. Improved data analysis for the MinION nanopore sequencer. Nat Methods 2015;12:351–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Koren S, Walenz BP, Berlin K, et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 2017;27:722–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li H. Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics 2016;32:2103–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shu Y, McCauley J. GISAID: global initiative on sharing all influenza data - from vision to reality. Euro Surveill 2017;22:30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hatcher EL, Zhdanov SA, Bao Y, et al. Virus variation resource - improved response to emergent viral outbreaks. Nucleic Acids Res 2017;45:D482–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhao WM, Song SH, Chen ML, et al. The 2019 novel coronavirus resource. Yi Chuan 2020;42:212–21. [DOI] [PubMed] [Google Scholar]
- 54. Zhang Y, Zmasek C, Sun GY, et al. Hepatitis C virus database and bioinformatics analysis tools in the virus pathogen resource (ViPR). Methods Mol Biol 1911;2019:47–69. [DOI] [PubMed] [Google Scholar]
- 55. Notredame C. Recent progress in multiple sequence alignment: a survey. Pharmacogenomics 2002;3:131–44. [DOI] [PubMed] [Google Scholar]
- 56. Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 1994;22:4673–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Katoh K, Misawa K, Kuma K, et al. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 2002;30:3059–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 2004;32:1792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Notredame C, Higgins DG, Heringa J. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol 2000;302:205–17. [DOI] [PubMed] [Google Scholar]
- 60. Do CB, Mahabhashyam MS, Brudno M, et al. ProbCons: probabilistic consistency-based multiple sequence alignment. Genome Res 2005;15:330–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Katoh K, Toh H. Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform 2008;9:286–98. [DOI] [PubMed] [Google Scholar]
- 62. Loytynoja A. Phylogeny-aware alignment with PRANK. Methods Mol Biol 2014;1079:155–70. [DOI] [PubMed] [Google Scholar]
- 63. Suchard MA, Redelings BD. BAli-Phy: simultaneous Bayesian inference of alignment and phylogeny. Bioinformatics 2006;22:2047–8. [DOI] [PubMed] [Google Scholar]
- 64. Novak A, Miklos I, Lyngso R, et al. StatAlign: an extendable software package for joint Bayesian estimation of alignments and evolutionary trees. Bioinformatics 2008;24:2403–4. [DOI] [PubMed] [Google Scholar]
- 65. Troshin PV, Procter JB, Barton GJ. Java bioinformatics analysis web services for multiple sequence alignment--JABAWS:MSA. Bioinformatics 2011;27:2001–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. McWilliam H, Li W, Uludag M, et al. Analysis tool web services from the EMBL-EBI. Nucleic Acids Res 2013;41:W597–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Loytynoja A, Goldman N. webPRANK: a phylogeny-aware multiple sequence aligner with interactive alignment browser. BMC Bioinformatics 2010;11:579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Blackburne BP, Whelan S. Class of multiple sequence alignment algorithm affects genomic analysis. Mol Biol Evol 2013;30:642–53. [DOI] [PubMed] [Google Scholar]
- 69. Waterhouse AM, Procter JB, Martin DM, et al. Jalview version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009;25:1189–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yachdav G, Wilzbach S, Rauscher B, et al. MSAViewer: interactive JavaScript visualization of multiple sequence alignments. Bioinformatics 2016;32:3501–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Larsson A. AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014;30:3276–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hall T. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 1999;41:95–8. [Google Scholar]
- 73. Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 1987;4:406–25. [DOI] [PubMed] [Google Scholar]
- 74. Fitch WM. Toward defining the course of evolution: minimum change for a specific tree topology. Syst Biol 1971;20:406–16. [Google Scholar]
- 75. Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol 1981;17:368–76. [DOI] [PubMed] [Google Scholar]
- 76. Larget B, Simon DL. Markov Chasin Monte Carlo algorithms for the Bayesian analysis of phylogenetic trees. Mol Biol Evol 1999;16:750–0. [Google Scholar]
- 77. Posada D. jModelTest: phylogenetic model averaging. Mol Biol Evol 2008;25:1253–6. [DOI] [PubMed] [Google Scholar]
- 78. Abascal F, Zardoya R, Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics 2005;21:2104–5. [DOI] [PubMed] [Google Scholar]
- 79. Kapli P, Yang Z, Telford MJ. Phylogenetic tree building in the genomic age. Nat Rev Genet 2020;21:428–44. [DOI] [PubMed] [Google Scholar]
- 80. Bos DH, Posada D. Using models of nucleotide evolution to build phylogenetic trees. Dev Comp Immunol 2005;29:211–27. [DOI] [PubMed] [Google Scholar]
- 81. Baele G, Suchard MA, Rambaut A, et al. Emerging concepts of data integration in pathogen phylodynamics. Syst Biol 2017;66:e47–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Rambaut A, Lam TT, Max Carvalho L, et al. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol 2016;2:vew007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Gascuel O. BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol Biol Evol 1997;14:685–95. [DOI] [PubMed] [Google Scholar]
- 84. Guindon S, Lethiec F, Duroux P, et al. PHYML online--a web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res 2005;33:W557–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014;30:1312–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Nguyen LT, Schmidt HA, Haeseler A, et al. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 2015;32:268–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003;19:1572–4. [DOI] [PubMed] [Google Scholar]
- 88. Lartillot N, Lepage T, Blanquart S. PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 2009;25:2286–8. [DOI] [PubMed] [Google Scholar]
- 89. Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 2007;7:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Bouckaert R, Heled J, Kuhnert D, et al. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol 2014;10:e1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Wilgenbusch JC, Swofford D. Inferring evolutionary trees with PAUP*. Curr Protoc Bioinformatics 2003;Chapter 6:Unit 6.4. [DOI] [PubMed] [Google Scholar]
- 92. Tamura K, Peterson D, Peterson N, et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 2011;28:2731–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zhang D, Gao F, Jakovlic I, et al. PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol Ecol Resour 2020;20:348–55. [DOI] [PubMed] [Google Scholar]
- 94. Huson DH, Scornavacca C. Dendroscope 3: an interactive tool for rooted phylogenetic trees and networks. Syst Biol 2012;61:1061–7. [DOI] [PubMed] [Google Scholar]
- 95. Yu G, Lam TT, Zhu H, et al. Two methods for mapping and visualizing associated data on phylogeny using Ggtree. Mol Biol Evol 2018;35:3041–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Letunic I, Bork P. Interactive tree of life (iTOL) v4: recent updates and new developments. Nucleic Acids Res 2019;47:W256–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Subramanian B, Gao S, Lercher MJ, et al. Evolview v3: a webserver for visualization, annotation, and management of phylogenetic trees. Nucleic Acids Res 2019;47:W270–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020;579:270–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lam TT-Y, Jia N, Zhang YW, et al. Identification of 2019-nCoV related coronaviruses in Malayan pangolins in southern China. bioRxiv 2020;583(7815):282–285. [DOI] [PubMed] [Google Scholar]
- 100. Boni MF, Lemey P, Jiang X, et al. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat Microbiol 2020;5(11):1408–1417. [DOI] [PubMed] [Google Scholar]
- 101. Covid-Investigation Team . Clinical and virologic characteristics of the first 12 patients with coronavirus disease 2019 (COVID-19) in the United States. Nat Med 2020;26:861–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kumar S, Stecher G, Li M, et al. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 2018;35:1547–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Stefanelli P, Faggioni G, Lo Presti A, et al. Whole genome and phylogenetic analysis of two SARS-CoV-2 strains isolated in Italy in January and February 2020: additional clues on multiple introductions and further circulation in Europe. Euro Surveill 2020;25(13):2000305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Hadfield J, Megill C, Bell SM, et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 2018;34:4121–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Ulitsky I, Burstein D, Tuller T, et al. The average common substring approach to phylogenomic reconstruction. J Comput Biol 2006;13:336–50. [DOI] [PubMed] [Google Scholar]
- 106. Sun J, Xu Z, Hao B. Whole-genome based Archaea phylogeny and taxonomy: a composition vector approach. Chin Sci Bull 2010;55:2323–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Kurtz S, Narechania A, Stein JC, et al. A new method to compute K-mer frequencies and its application to annotate large repetitive plant genomes. BMC Genomics 2008;9:517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Marchler-Bauer A, Bo Y, Han L, et al. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res 2017;45:D200–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Abril JF, Guigo R. gff2ps: visualizing genomic annotations. Bioinformatics 2000;16:743–4. [DOI] [PubMed] [Google Scholar]
- 110. Lu G, Moriyama EN. Vector NTI, a balanced all-in-one sequence analysis suite. Brief Bioinform 2004;5:378–88. [DOI] [PubMed] [Google Scholar]
- 111. Liu W, Xie Y, Ma J, et al. IBS: an illustrator for the presentation and visualization of biological sequences. Bioinformatics 2015;31:3359–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Felsenstein J. PHYLIP (phylogeny inference package), version 3.5c. March 1993 https://csbf.stanford.edu/phylip/index.html, 1993.
- 113. Su S, Wong G, Shi W, et al. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol 2016;24:490–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Lole KS, Bollinger RC, Paranjape RS, et al. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol 1999;73:152–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Martin DP, Murrell B, Golden M, et al. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol 2015;1:vev003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Waterhouse A, Bertoni M, Bienert S, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 2018;46:W296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Janson G, Zhang C, Prado MG, et al. PyMod 2.0: improvements in protein sequence-structure analysis and homology modeling within PyMOL. Bioinformatics 2017;33:444–6. [DOI] [PubMed] [Google Scholar]
- 118. Zhou H, Chen X, Hu T, et al. A novel bat coronavirus closely related to SARS-CoV-2 contains natural insertions at the S1/S2 cleavage site of the spike protein. Curr Biol 2020;30:2196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Costello M, Fleharty M, Abreu J, et al. Characterization and remediation of sample index swaps by non-redundant dual indexing on massively parallel sequencing platforms. BMC Genomics 2018;19:332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Li Q, Zhao X, Zhang W, et al. Reliable multiplex sequencing with rare index mis-assignment on DNB-based NGS platform. BMC Genomics 2019;20:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Valk T, Vezzi F, Ormestad M, et al. Index hopping on the Illumina HiseqX platform and its consequences for ancient DNA studies. Mol Ecol Resour 2020;20(5):1171–1181. [DOI] [PubMed] [Google Scholar]
- 122. Lanfear R. A global phylogeny of SARS-CoV-2 from GISAID data, including sequences deposited up to 31-July-2020. 2020. Zenodo . doi: 10.5281/zenodo.3958883. [DOI] [Google Scholar]