

Abstract
The coronavirus disease 2019 (COVID-19) pandemic, caused by the coronavirus severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has created an unprecedented threat to public health. The pandemic has been sweeping the globe, impacting more than 200 countries, with more outbreaks still lurking on the horizon. At the time of the writing, no approved drugs or vaccines are available to treat COVID-19 patients, prompting an urgent need to decipher mechanisms underlying the pathogenesis and develop curative treatments. To fight COVID-19, researchers around the world have provided specific tools and molecular information for SARS-CoV-2. These pieces of information can be integrated to aid computational investigations and facilitate clinical research. This paper reviews current knowledge, the current status of drug development and various resources for key steps toward effective treatment of COVID-19, including the phylogenetic characteristics, genomic conservation and interaction data. The final goal of this paper is to provide information that may be utilized in bioinformatics approaches and aid target prioritization and drug repurposing. Several SARS-CoV-2-related tools/databases were reviewed, and a web-portal named OverCOVID (http://bis.zju.edu.cn/overcovid/) is constructed to provide a detailed interpretation of SARS-CoV-2 basics and share a collection of resources that may contribute to therapeutic advances. These information could improve researchers’ understanding of SARS-CoV-2 and help to accelerate the development of new antiviral treatments.
Keywords: COVID-19, SARS-CoV-2, coronavirus, regulatory interactions, drug development, bioinformatics
Introduction
After the first cases found in Wuhan, China, in December 2019, the coronavirus disease 2019 (COVID-19) has now spread across the globe, posing a serious threat to global public health and the economy. The virus causing the pandemic is officially named as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) by the International Committee on Taxonomy of Viruses, considering its high sequence and pathological homology to SARS-CoV [1]. Currently, the diagnosis of SARS-CoV-2 infection has become easy and accurate. However, there are still no vaccines or approved drugs available at the time of the writing, and researchers are actively seeking a cure. Compared with its predecessors (SARS and MERS), SARS-CoV-2 is proven to be more transmissible. As a novel disease, the whole population is broadly susceptible, and infection of SARS-CoV-2 occurred in all age groups. According to the statistics provided by World Health Organization (WHO), as of 17 November 2020, globally, ~54.6 million people have been infected by COVID-19 (with over 250,000 new cases each day), including 1,320,148 deaths. Undeniably, the in-depth research on SARS-CoV-2 is and will long be vital to save human lives.
To accelerate the pace of research, this review aims to present a general and explicit landscape of SARS-CoV-2 at the molecular level, with emphasis on the key steps toward effective treatment of COVID-19, and provide useful resources and tools for researchers in relevant fields. For a better understanding, we also offer illustrative figures depicting the verified mechanisms of SARS-CoV-2, the phylogenetic information, a network presentation of current COVID-19 resources, and the latest status of current de novo drug development. All these sections provide information to promote drug development from different aspects. The online resources for each section are incorporated in http://bis.zju.edu.cn/overcovid, as long as the detailed information to help to gain a comprehensive understanding of SARS-CoV-2. We believe this paper may contribute to the easier accessibility of particular information and facilitate further research.
SARS-CoV-2 life cycle and the inhibitors in the process
The mechanism of cell entry is vital for the viral infectivity and pathogenesis of the virus. Compared with the other coronaviruses, a representative feature of SARS-CoV-2 is its higher infectivity, predominantly due to its higher receptor-binding affinity [2]. The cell entry of SARS-CoV-2 starts with the receptor recognition and binding, in which the receptor-binding domain (RBD) in the S1 subunit of the spike (S) protein regulates this process (Figure 1A and B). Similar to SARS-CoV, the canonical receptor for SARS-CoV-2 is angiotensin-converting enzyme 2 (ACE2). However, an additional main-chain hydrogen bond is in the receptor-binding motif of SARS-CoV-2, leading to a more compact attachment [3]. Surprisingly, the RBD of SARS-CoV-2 is mostly not in an efficient structural form, suggesting its unexploited potential to be more infectious. Currently, the functions and interaction partners of the majority of virus proteins have been identified for SARS-CoV-2. WikiPathways [4] provides a specific COVID-19 Pathway Collection, including a number of curated COVID-19-related pathways, pathways of other coronaviruses, and specific ACE2 pathways. The COVID-19 portal of WikiPathways also listed several external links relevant to COVID-19 pathways. COVID-19 disease map initiative [5] has integrated data from WikiPathways and other sources, providing more comprehensive knowledge of virus-host (V-H) interaction mechanisms. Users may check for data resources and best practices, or view and download pathway models (mostly in SMBL format). PubChem [6] provides a large collection of COVID-19 pathways from various sources, and other relevant information involving compounds, substances and bioassays.
Figure 1.
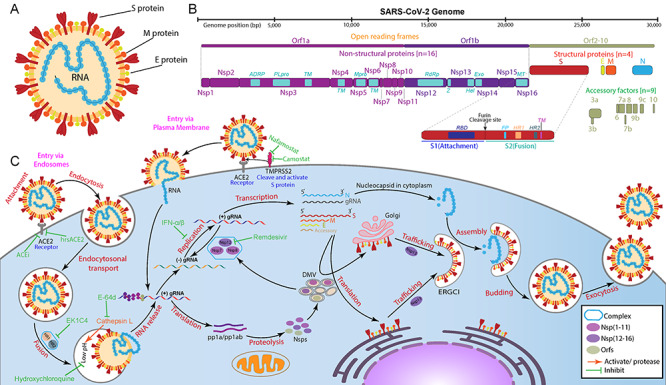
Structure, genome and replication cycle of SARS-CoV-2. (A) Structure of SARS-CoV-2, (B) It is a single-stranded positive-sense RNA virus with a length of 29.3 kb. SARS-CoV-2 genome with sixteen non-structural proteins, nine accessory factors and four structural proteins. (C) Depicting the replication cycle of SARS-CoV-2 with the potential inhibitors (green) against different stages (red).
During the replication cycle of SARS-CoV-2 (Figure 1C), multiple components at different stages can be targeted to inhibit or block the corresponding process [7]. A typical illustration of the SARS-CoV-2 life cycle is presented in the web-portal, together with the information of potential inhibitors and their targets. Several studies have targeted host cellular processing mechanisms, including endocytosis, autophagy and inflammatory response [7]. Besides, the non-canonical cell entry and complement activation mechanisms [8, 9] can also provide valuable targets for new therapy. With the increased understanding of SARS-CoV-2, more components are found to play roles in pathogenesis. For example, mitochondria are recognized to involve in host immune suppression [10] and the formation of double-membrane vesicles [11], suggesting the potential of mitochondrial proteins as drug targets.
SARS-CoV-2 phylogenies
The nsp14 exoribonuclease provides SARS-CoV-2 the ‘proofreading activity’, which will help in correcting the replication errors. Compared with influenza or HIV, the mutation rate of SARS-CoV-2 is much lower, making their genomes relatively stable. In addition, there is no existing immunity to SARS-CoV-2; thus, it is under very low evolutionary pressure. Therefore, SARS-CoV-2 is unlikely to develop many distinct subtypes.
By 17 November 2020, China National Center for Bioinformatics has collected 205,965 SARS-CoV-2 genomic sequences (Figure 2). Based on the public genomes, many researchers have performed analyses to study the phylogeny and epidemiology of SARS-CoV-2. Nextstrain [12] is an open-source project aiming to provide real-time tracking of pathogen evolution. A special SARS-CoV-2 portal is constructed (https://nextstrain.org/ncov/global) by Nextstrain, providing the phylogenic structure for different virus strains, epidemic spread features at the global and continental level, geography-based datasets and other analytics and visualizations. According to the phylogenetic information from Nextstrain, there are five main clades for SARS-CoV-2, each can be represented by specific marker mutations. The phylogenetic information provided by different groups or organizations vary, but they share a similar structure, and the major clades are mostly consistent [13]. The details of the main clades of SARS-CoV-2 are provided at http://bis.zju.edu.cn/overcovid/phylogenies.
Figure 2.
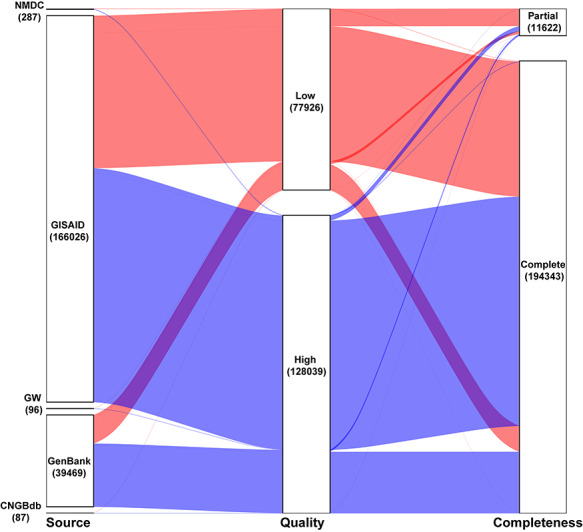
Properties of SARS-CoV-2 genome sequences curated by China National Center for Bioinformatics. The number in each box represents the amount of sequences from the corresponding source. The portal contains 205,965 sequences from five different sources (As of 17 November 2020). Of the sequences, 80% are from GISAID. Around 94% of the sequences are complete and the rest are partial and 33.63% of the sequences are of low quality.
A series of studies have focused on the mutation hotspots of SARS-CoV-2 (see http://bis.zju.edu.cn/overcovid/phylogenies), the number of identified recurrent mutations increases with the sample amount, but with decreasing speed, suggesting the existence of a limit. Also, Turakhia et al. [13] showed that recurrent mutations could be lab-associated systematic errors. A recent study [14] with 30,983 samples has identified 14 hotspot mutations, in which some are area-specific. It is known that some mutations may enhance expression (V367F and G502D) or ACE2 affinity (N501F, N501T, and Q498Y). However, these mutations are not exhibited as recurrent, and most mutations are either neutral or weakly deleterious. Among the recurrent mutations, only D614G has a significant functional consequence. D614G is validated to reduce S1 shedding, thus increase the stability of the S proteins [15]. As markers for the 20A clade, mutations D614G and P4715L are likely to occur simultaneously, but the P4715L mutation is not identified with significant functional consequence, and it is relatively rare for it to occur alone. The high overlap suggests a ‘founder effect’ leading to the broad spread of certain combinations of mutations.
Taken together, high stability is shown for SARS-CoV-2 phylogenies; it is possible that the first drug/vaccine will be effective for almost all patients.
RNA modification
RNA modification systems were suspected to impact the inference of divergence between SARS-CoV-2 and other RNA coronaviruses by evaluating the number of mismatches among the aligned RNA genomes [16]. Adenosine-to-inosine (A > I) transitions from ADAR (adenosine deaminases that act on RNA) deaminases and cytosine-to-uracil (C > T) changes from APOBEC (apolipoprotein B editing complex) deaminases may account for an observed A > G/C > T editing events. Single site RNA modification could be detected by nanopore direct RNA sequencing. At least 41 sites showed concrete evidence of RNA modification on SARS-CoV-2 transcripts, indicating RNA modifications mechanism may a hidden layer of regulation to escape from host immune response [17]. The most abundant motifs around these modifications were AAGAA and other A/G-rich sequences, which are highly present in the SARS-CoV-2 genome.
Conserved RNA motif and secondary structure
The available evidence suggests that conserved RNA motif present at the genomes of polyadenylated RNA virus families can be used for virus detection and characterization. Replication and transcription of coronaviruses RNA genomes involved with more proteins than virus-encoded proteins. Increasing evidence suggests the conservation of RNA structures was required for genome replication and packaging. Secretion-enhancing cis-regulatory targeting element (SECReTE) is ‘NYN’ or ‘NNY’ (where N is any nucleotide and Y is pyrimidine). The mutation of the SECReTE motif enhances or inhibits mRNA stability and association with the endoplasmic reticulum (ER). This motif was reported to promote mRNA stability, localization to ER, and translation in yeast [18]. It is interesting to found that the SARS-CoV-2 genome has 40 copies of SECReTE motifs at an abundance of ~1.3 SECReTEs/kilobase [19].
The RNA secondary structures play a significant role to enhance the stability and regulation of virus genome replication. The ScanFold approach can predict the minimum free energy (MFE) of the SARS-CoV-2 genome by using scanning window analysis [20]. The approach extracts a fixed window of RNA sequence and predicts native MFE through RNAfold; shuffling the RNA sequence with the same length by using either mononucleotide technique or Clote’s method and finally the average MFE of the randomized windows is used in the calculation of z-score. The z-score can be calculated as 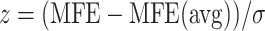 , where,
, where, 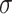 is the standard deviation of MFE values of random windows. The negative z-scores indicate more stability for the RNA structure. The results of z-score extracted from RNAStructuromeDB implies that SARS-CoV-2 is more ordered than ZIKV and HIV-1 and owns high structural regions with potential function (Figure 3A). Furthermore, we collected about 210 nt 5′-terminal genome sequences of five Betacoronaviruses (BCoV, HCoV-HKU1, SARS-CoV-Tor2, MHV-A59 and SARS-CoV-2) and predicted the alignment-based secondary structure using LocARNA. We found that several known stem-loop (SL) structures, including SL-1, SL-2 and SL-4, were highly conservated at the 5′-terminal of SARS-CoV-2 (as shown in Figure 3B). Instead of analyzing all Betacoronaviruses, other researchers focused on the conversation region of SARS-related Betacoronaviruses and reported that 79 highly conserved regions with at least 15 nt length over SARS-related bat coronaviruses genome. Moreover, 106 ‘SARS-CoV-2-conserved-structured’ regions were extracted as potential drug binding sites targets for antivirals in a recent study [21].
is the standard deviation of MFE values of random windows. The negative z-scores indicate more stability for the RNA structure. The results of z-score extracted from RNAStructuromeDB implies that SARS-CoV-2 is more ordered than ZIKV and HIV-1 and owns high structural regions with potential function (Figure 3A). Furthermore, we collected about 210 nt 5′-terminal genome sequences of five Betacoronaviruses (BCoV, HCoV-HKU1, SARS-CoV-Tor2, MHV-A59 and SARS-CoV-2) and predicted the alignment-based secondary structure using LocARNA. We found that several known stem-loop (SL) structures, including SL-1, SL-2 and SL-4, were highly conservated at the 5′-terminal of SARS-CoV-2 (as shown in Figure 3B). Instead of analyzing all Betacoronaviruses, other researchers focused on the conversation region of SARS-related Betacoronaviruses and reported that 79 highly conserved regions with at least 15 nt length over SARS-related bat coronaviruses genome. Moreover, 106 ‘SARS-CoV-2-conserved-structured’ regions were extracted as potential drug binding sites targets for antivirals in a recent study [21].
Figure 3.
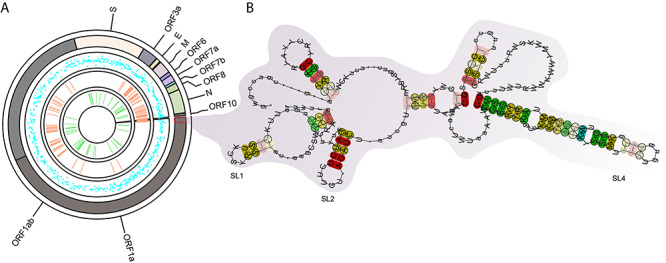
The conserved RNA structures in SARS-CoV-2. (A) The genome-wide RNA structure features were plotted on the SARS-CoV-2 genome. From outer to inner circle: 1, SARS-CoV-2 genome with annotation; 2, z-scores of RNA secondary structures predicted by Scanfold (blue dot). The less z-score, the RNA secondary structure more stable. 3, conversation regions between SARS, SARS-CoV-2 and SARS-related bat coronaviruses (light-salmon rectangle). 4, the predicted SECReTE RNA motifs (green rectangle). (B) the conservated SL-1, SL-2 and SL-4 predicted at the 5′-terminal of SARS-CoV-2.
Cell type-specific changes at single-cell resolution
The complex immune cell dysregulation could be associated with the susceptibility to infection by SARS-CoV-2. Single-cell RNA sequencing techniques were used to investigate the transcriptional changes in human infected by SARS-CoV-2. The single-cell co-expression spectrum of known receptors ACE2, TMPRSS2 and other genes were studied by mining the data from multiple data sources and different tissues [22, 23]. It is reported that lower ACE2 expression in young paediatric samples and upregulated ACE2 expression in airway epithelial and AT2 cells in the past and present smokers [23]. Also, the remarkable changes in the immune cell type proportions were found in patients with SARS-CoV-2 [24]. Bronchoalveolar lavage fluid immune cells from COVID-19 patients and healthy people were characterized by using single-cell RNA sequencing [25]. Pro-inflammatory monocyte-derived macrophages were highly observed in the bronchoalveolar lavage fluid from severe cases, whereas moderate patients were discriminated by the presence of highly clonally expanded CD8+ T cells. Liu X. et al. [22] investigated the potential risks of the SARS-CoV-2 virus on the male reproductive system from the embryonic stage to adulthood and demonstrated that it needs to evaluate the psychological effects of SARS-CoV-2 infection in the male reproductive system.
Using single-cell RNA sequencing, Cao et al. [26] identified 14 SARS-CoV-2-neutralizing monoclonal antibodies from 60 convalescent patients’ B cells. Peripheral blood mononuclear cell-type-specific transcriptional changes were detected during the recovery period of SARS-CoV-2 infection. These change of B cell-receptor (IGHV3-23 and IGHV3-7) were identified, and other B-cell-receptor isotypes (IGHV3-15, IGHV3-30 and IGKV3-11) previously used for other virus vaccine design and development were confirmed [27]. These results may offer new strategies for the development of vaccines and antibodies against SARS-CoV-2 in the future.
Various interactions help delineating SARS-CoV-2 pathogenesis
The RNA genome of SARS-CoV-2 can generate multiple viral sgRNAs and proteins, and their interactions with host cells have become a focus. Currently, researchers have confirmed the functions of the majority of these RNAs and proteins, but the mechanisms are not yet fully elucidated. In order to find and verify more interactions, laboratory experiments are being performed, as well as the computational approaches that predict interactions based on correlations and molecular characteristics.
Experimental identification of intra-viral and V-H protein–protein interactions
Protein–protein interactions (PPIs) between the host and virus plays an essential role during the course of viral infection and the development of the disease. Identification and systematic investigation of intra-viral and V-H PPIs are, therefore, crucial for understanding the molecular and cellular mechanisms of infection and subsequent development of antiviral therapies. A study conducted by Gordon et al. [28] reveals a blueprint of how SARS-CoV-2 hijacks host cellular pathways by mapping the V-H PPIs. In the systematic analysis, 26 tagged SARS-CoV-2 proteins were expressed in HEK293T kidney cells and used affinity-purified mass-spectrometry (AP-MS) method to account for PPIs. A map of 332 high confidence human protein interactions with viral proteins was discovered (Table 1), providing a first SARS-CoV-2 human protein interactome. Following that, some more research have been performed for quantification of the protein interaction between the virus and the human host as well as intra-viral interactions [29, 30]. Li et al. [30] identified 286 cellular proteins to interact with SARS-CoV-2 proteins, resulting in a total of 295 high-confidence viral-host PPIs. Besides, the study characterized the intra-viral PPI networks using genome-wide co-immunoprecipitation (co-IP) and yeast-two hybrid (Y2H) screen (Table 1). A significant difference was observed in the number of intra-viral PPIs detection between the two techniques. In detecting PPIs, the Y2H approach takes place in the nucleus; thus, the proteins that are not localized to the nucleus are likely not identified as interactors, which could result in the increase of false negatives, which is one of the demerits of the Y2H approach.
Table 1.
Experimental studies and computational prediction of V-H and intra-viral protein interaction of SARS-CoV-2
| Interaction types | Experimental technique | Cells | Number of PPIs | Number of proteins (host/virus) | Number of known druggable targets | References |
|---|---|---|---|---|---|---|
| Experimentally identified PPIs | ||||||
| V-H PPI | AP-MS | HEK293T kidney cells | 695 (V-H-H) 332 (V-H) | 332/26 | 66 druggable host factors targeted by 69 compounds | [28] |
| V-H PPI | AP-MS | A549 cells | 1484 (V-H) | 1086/ 24 (plus 27 SARS-1 proteins) | – | [29] |
| V-H PPI | AP-LC–MS | HEK293 cells | 295 (V-H) | 286/28 | NKRF-nsp9 as a possible drug target and IL8/IL6 antagonists as drugs | [30] |
| Intra-viral PPI | Y2H | Y2HGold yeast strain | 19 | NA/28 | [30] | |
| Intra-viral PPI | Co-IP | HEK293 cells | 52 | NA/28 | [30] | |
| Computationally predicted PPIs | ||||||
| Intra-viral PPI | Aggregated and evolution-based | – | 86 | NA/31 | – | [31] |
| V-H PPI | Structure-based and curated | – | 200 | 94/25 | – | [31] |
| V-H PPI | Sequence-based (PIPE4, SPRINT) | – | 279 | 225/14 | – | [32] |
LC, liquid chromatography; V-H-H, virus-host–host.
Computational prediction of SARS-CoV-2 intra-viral and human–virus PPIs
Besides using the experimental results of V-H PPIs, computationally predicted and curated PPIs are also being used for analyzing the pathogenesis of SARS-CoV-2 [33]. Several computational strategies, such as structure-, sequence- and evolution-based methods, have been conducted to analyze and quantify the intra-viral and V-H PPIs [31, 32, 34]. Structure-based methods for PPIs require the information on the 3D structure of both proteins. For example, the 3D structure of SARS-CoV-2 S protein has constructed by several research groups and conformed the interaction between S and human ACE2 protein [34, 35]. Similar structure-based predictions of interactions are also performed on other components such as nsp12-nsp7-nsp8 complex [36, 37]. However, the lacking of complete 3D structural information of all human proteins, as well as SARS-CoV-2 proteins, limit the structure-based protein interaction prediction. Sequence-based methods overcome the limitation and rely solely upon the sequence. Dick et al. [32] have used two sequence-based PPI prediction approaches such as PIPE4 (Protein-Protein Interaction Prediction Engine) and SPRINT (Scoring PRotein INTeractions) to predict host protein interactions with 14 SARS-CoV-2 proteins and identified 279 putative interactions connecting with 225 human proteins. To train the predictors and infer new putative V-H interactions, experimentally elucidated human-virus PPIs of several viral organisms were used those obtained from the VirusMentha database (https://virusmentha.uniroma2.it/). Furthermore, the study used the PIPE-Sites algorithm to predict the sub-sequence regions or physical binding site. Besides, the integrated bioinformatics approaches are also being used in structural genomics and interactomics to structurally characterize individual proteins and the information of evolutionary trajectories to identify V-H and intra-viral protein interactions [31].
SARS-CoV-2 regulatory network
Virus infection will interrupt the human regulatory network, leading to the dysfunction of specific cellular processes. To explore the complete V-H interactome, it is essential to incorporate data at the transcriptional/post-transcriptional levels. Many researchers have investigated the gene expression changes in SARS-CoV-2 infected cells to infer the regulatory pattern, find vital processes or key genes during host response. Through differential expression and functional enrichment analyses, dysregulation of particular pathways were uncovered [38, 39]. Prasad et al. [40] have performed a topological network analysis and discovered 15 candidate drug targets. At the post-transcription level, Srivastava et al. [39] has uncovered 22 human miRNAs and 51 proteins with the potential to bind with virus RNA and investigated the tissue distribution of these regulators. All these findings may guide the path toward effective therapeutics.
Aside from the SARS-CoV-2 specific information, many institutes/groups are utilizing general knowledge and data to help to infer the biological processes underlying viral pathogenesis. These data include known human PPI networks, interactions among diseases, drugs, genes, and specific knowledge like side effects, whose integration largely rely on approaches based on similarities, correlations and homologies. In addition to SARS-CoV-2 specific resources, several well-known general databases have also provided special pages/projects to aid SARS-CoV-2 study. Figure 4 illustrates current SARS-CoV-2-related databases/tools, and their research focuses on a network view to show the different scopes of the resources. The current effort of researchers is to infer the pathogenesis of SARS-CoV-2 through network-based approach by integrating V-H interactome of known HCoVs (SARS, MERS, HCoV-229E) and available human PPI data [33, 41]. These data involve different -omics and alternative pathways; thus, the total amount of components relating to one process could be huge. Current interactions from different resources are listed in http://bis.zju.edu.cn/overcovid/interactions. Appropriate integration of these data may deepen the understanding of V-H interaction, thus help further investigation on SARS-CoV-2.
Figure 4.
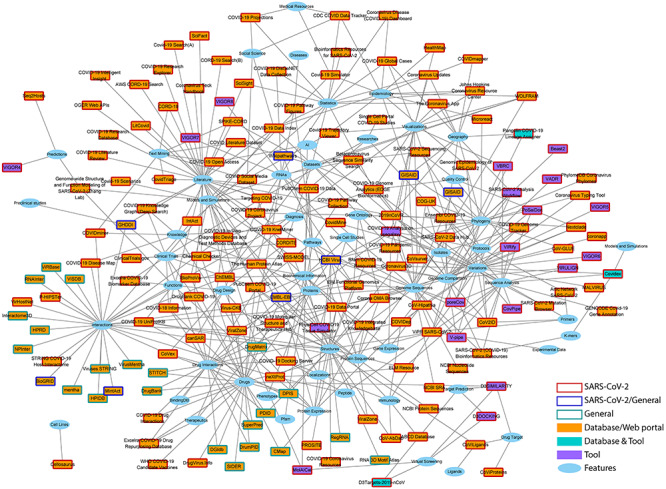
Various resources from different aspects to accelerate research of COVID-19. Squares represent the tools or databases/web platforms and the ellipses show the features (main focus areas) of the corresponding databases/tools. An interactable version of this network is available at http://bis.zju.edu.cn/overcovid/network.html. Users may zoom in/out, drag and click on the nodes to obtain more information and list of the features (for the selected resources) and resources (for the selected features). Users can also go directly to the corresponding page of the resources by simply clicking on thelist.
SARS-CoV-2 drug discovery
SARS-CoV-2 has caused nearly one and a half million deaths worldwide. Currently, no vaccine or antiviral treatment is approved or proven to be significantly effective for SARS-CoV-2. The rapid growth of infections and the lacking of treatments are presenting a severe challenge for public health, which urges researchers to develop new drugs or vaccines. However, it currently appears that acquired immunity is only for a very short period. In this way, the development of drugs is of critical importance. Like the typical approaches, the current drug discovery methods for COVID-19 can also be classified into three types, repurposable drug discovery, in silico chemical screening and de novo drug discovery (Figure 5A).
Figure 5.
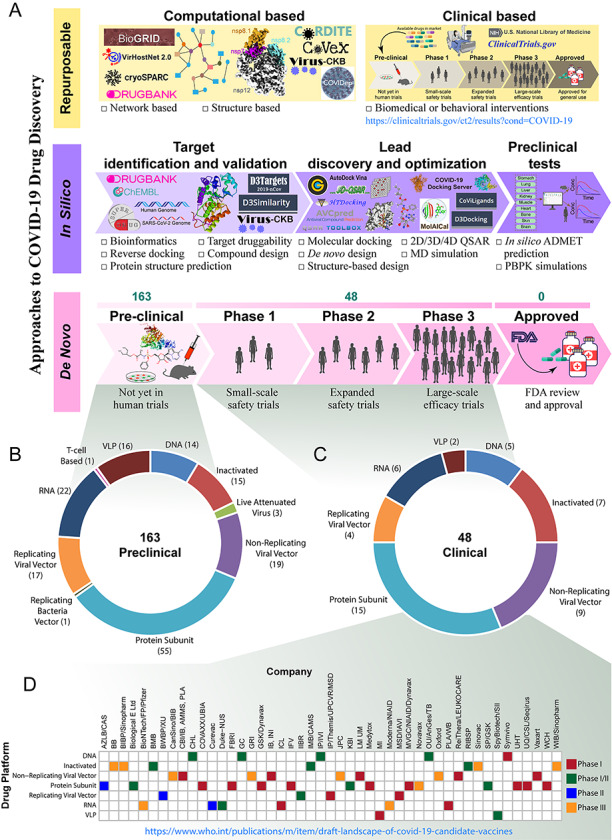
Current status of COVID-19 drug discovery. (A) Like the traditional approaches, the current drug discovery methods against COVID-19 also focuses on repurposable, in silico and de novo techniques. Repurposable drug discovery technique uses the existing drugs available in the market for the new indication. Both computational- and clinical trial-based methods are being used to find the repurposable drugs for COVID-19. Several webservers, databases and tools such as, CORDITE, CoVex, COVIDep and Virus-CKB have been developed to accelerate the drug repurposing in both the network- and structure-based approaches. Many clinical trials of the existing drugs are going on with different phases and ClinicalTrials.gov database includes the corresponding information. To facilitate in the field of in silico drug discovery research, several tools and databases like MolAICal, D3Similarity, D3Targets-2019-nCov, D3Docking, CoViLigands, COVID-19 docking server and others have developed. Several developers or manufacturers are developing novel drugs for COVID-19. According to the World health organization (WHO), (B) 163 drugs are in preclinical evaluation and (C) 48 are in clinical trials (as of 17 November 2020) and the drugs are based on different platforms. (D) Forty-Seven drug companies are involved in drug development, and the drugs are in clinical trials in different phases.
De novo drug discovery
The de novo drug discovery strategy is against the genetic and pathological characteristics, and design new chemicals from scratch. Despite the expenses and time dependency of this strategy, considering the urgent demand of the COVID-19 outbreak, many developers or manufacturers are developing vaccines against COVID-19, and the WHO are listing the drugs with various information, including vaccine platform, type of candidate vaccine, number of doses, the timing of doses and stage of clinical trials. According to the WHO, more than 211 candidate drugs against COVID-19 are being developed by the researchers and manufacturers worldwide (as of 17 November 2020). Of these drugs, 163 are in preclinical evaluation and 48 are in clinical evaluation (Figure 5B and C). The drugs are based on different platforms, including DNA, RNA, protein subunit, inactivated, non-replicating viral vector (Figure 5D).
In silico drug discovery
The in silico drug discovery strategy uses structural information of the compounds and prior knowledge to predict possible interactions with targets and their subsequent effects. In silico methods are facilitating in the primary stage of prediction and assessment of potential safety matters, permitting to increase the drug-discovery success rate and reduce time and costs associated with new drug development. Many tools, web servers, and databases have been developed for each step (such as identifying targets to lead optimization and preclinical test) of in silico drug discovery (Figure 5A). Bruno et al. [42] have provided a detailed review of the most common in silico tools and resources; those are widely used in drug discovery. Since the pandemic COVID-19 began, several research groups applying this strategy was published, and lists of candidates were provided [43]. Furthermore, some databases, web servers (Virus-CKB [44], D3Targets-2019-nCoV [45], COVID-19 Docking Server [46]) and tools (MolAICal [47]) have been developed to facilitate and accelerate the process of drug discovery against COVID-19.
Repurposable drug discovery
Drug repurposing (or drug repositioning) is a strategy to discover new indications of drugs from existing approved drugs [48]. Compared with the other two strategies, drug repurposing makes use of well-known compounds, promisingly reducing the risk, cost and, most important, the time. For a rare or novel disease like COVID-19, drug repurposing is a commonly recognized approach. High level of genomic sequence similarity of SARS-CoV-2 with other human coronaviruses such as SARS and MERS and others has also boosted the confidence, facilitating researchers to discover repurposable drug candidates. Current drug repositioning approaches can be roughly divided into computational-based and clinical-based approaches (Figure 5A).
Clinical based repositioning
For clinical based repositioning, the existing drugs have to go through several phases to obtain FDA approval. For example, Remdesivir—a viral RNA-dependent RNA polymerase inhibitor—is one such investigational broad-spectrum medication that has effective activity against Ebola virus disease in humans and has shown promise in preclinical studies against MERS and SARS [49]. A recent clinical study for the SARS-CoV-2 demonstrated that Remdesivir benefited hospitalized patients with lower respiratory tract infection [50]. For these drugs, although side effects and dosing regimens are known, they may not have particular anti-CoV effects and could associate with severe side effects. For example, chloroquine—antimalarial and autoimmune disease drug—was found highly effective in treating avian influenza A H5N1 virus [51]. Its antiviral activity against SARS-CoV-2 was suggested in vitro [52]. Nevertheless, a randomized, double-blind, placebo-controlled trial of hydroxychloroquine reported a high-risk exposure to a confirmed COVID-19 contact [53]. As of 17 November 2020; 3935 studies are included for clinical trials in the ClinicalTrials.gov database (Figure 5A), which provides various information on trials of the drugs.
Computational based repositioning
Computational prediction of a drug for repurposing is a suitable alternative to the laborious and costly experimental process. Computational methods and strategies for drug repurposing approaches of SARS-CoV-2 mainly focus on either chemical structures or network-based repurposing by using available diverse and heterogeneous genomics, biomedical and pharmacological data. Ongoing progress in the structural biology of SARS-CoV-2 is giving structure-based drug repurposing a robust basis. Recently, researchers have established the experimentally determined structures of different structural, non-structural, and accessory proteins of the virus [34–37]. The solved structures of the proteins are listed in the NCBI data respiratory (https://www.ncbi.nlm.nih.gov/Structure/SARS-CoV-2.html) with PDB ID. These structures are being used to perform molecular dynamics simulations and docking analyses to screen chemical binding of known compounds in the process of structure-based drug repurposing [54].
For rapid identification of candidate repurposable drugs, network-based computational repurposing approaches have been applied [28, 30, 40, 55], and several candidates are now under clinical trials [28, 56]. At the beginning of the COVID-19 pandemic and before the identification of experimentally validated SARS-CoV-2 V-H interactions, some efforts have been made to discover the drug targets and candidate drugs by using other HCoV-host interactomes through network-based drug repurposing [41, 55]. The identification and use of the experimentally validated SARS-CoV-2 V-H PPIs (Table 1) increase the confidence of accurately identifying drug targets and drug candidates against COVID-19 [28, 30]. Gordon et al. [28] have identified 66 druggable targets related to human proteins targeted by 69 drug candidates, 12 of which are in clinical trials, and the rest are in preclinical stages. Li et al. [30] identified NKRF-nsp9 as a possible drug target and IL8/IL6 antagonists as a drug that could be repurposed for COVID-19 treatment. Although these approaches can reduce the translational gap between preclinical testing and clinical outcomes, but the preclinical and clinical studies are necessary to validate these potential treatments, which is a significant barrier to the rapid development of drugs for the emerging COVID-19 outbreak.
Web platforms for target identification or/and drug repurposing against COVID-19
Currently, several web platforms and databases, particularly for SARS-CoV-2 drug development, has been constructed. CORDITE [57] is a curated corona drug interactions database for SARS-CoV-2 that collects and provides information on interactions of drugs and targets, computational prediction results, in vitro, in vivo study data, clinical trials and literature search. CORDITE enables researchers to access and download relevant data for conducting meta-analysis and designing new clinical studies via web server as well as an open API. CoVex [58] is another interactive web platform for network and system medicine data analysis as well as identification of putative drug repurposing candidates against SARS-CoV-2. It incorporated experimental V-H interaction data for SARS-CoV-2 and SARS-CoV-1, along with human PPIs as well as drug-target interactions and implemented several network analysis approaches to identify novel drug targets and drug repurposing candidates. The platform includes several tools that facilitate researchers and clinicians to find V-H interaction driven drugable targets as well as new repurposable drugs and visually explore druggable molecular mechanisms. For finding the candidate drugs, CoVex only accepts host proteins as seeds (does not integrate drugs that target viral proteins) and works at four steps (i) select proteins of interest, (ii) find drug targets, (iii) find drugs and (iv) inspect and download the results. COVIDep [59], a web-based platform, is designed to recommend real-time potential drug targets against COVID-19 based on B-cell and T-cell epitopes. It screens the experimentally-derived B-cell and T-cell epitopes from SARS-CoV through the ViPR database (www.viprbrc.org) and identifies those which are highly conserved within the latest SARS-CoV-2 sequence data available from GISAID (www.gisaid.org). The platform is developed based on Shiny app framework and comprising an intuitive graphical interface and interactive visualizations with flexible parameter setting. To automatically screen and discover drug candidates for COVID-19, an integrated web server called viral-associated disease-specific chemogenomics knowledgebase (Virus-CKB) has been developed [44]. The knowledgebase could be helpful for the prediction of the relevant protein targets as well as analysis data and visualized the outputs by using HTDocking, TargetHunter, BBB predictor, NGL Viewer, Spider Plot, etc. These web-platforms may facilitate the development of effective therapies.
The OverCOVID web-portal
To ensure that all investigators can have easy access to the resources, we have developed the OverCOVID web-portal (available at http://bis.zju.edu.cn/overcovid). OverCOVID contains more information with respect to this paper and separated the information into multiple pages representing different aspects. These pages can be accessed from any location of this website by clicking the corresponding buttons in the top navigation bar. First, in order to provide a comprehensive and basic understanding of SARS-CoV-2, we have presented the historical information of similar coronaviruses, their main hosts and the structural features in the ‘Home’ page. We have briefly described the life cycle of SARS-CoV-2 on this page, indicated the parts that can be targeted during the entire life cycle to assist in drug design, and listed several existing potential drugs from current studies. The ‘Phylogenies’ page uses the sequence data from GISAID and shows the development of each strain of the virus since the beginning of this year. We also provide a comparison of different nomenclature systems, including the marker gene variants representing for each virus strain. This page will be updated regularly so that investigators could stay up to date. For all the online databases and tools mentioned in this article, users can browse them by clicking on the ‘Resources’ button in the top navigation bar. A data table containing details for each online resource is presented on this page, including descriptions, links and relevant publications. The quick search function allows users to search for keywords, and the results can be filtered by resource type with a click on ‘database’, ‘web server’ or ‘tool’ placed in the left sidebar. Top features for these resources are also listed in the left sidebar. In the ‘Network’ page, an additional network view of the resources (Figure 4) is provided to visually indicate the scope of research for each database/tool and to intuitively present extra information (e.g. node degree may indicate the research focus). The ‘Interaction’ page provides information on the resources for each type of interaction as well as a corresponding download link or the link to the sourcesite.
Conclusion
To date, many countries and regions are still on the ‘upslope’ of the COVID-19 epidemic. With the infection rate showing an upward trend, the infection number is far from the levels of exposure required for ‘herd immunity’. To prevent COVID-19’s rampant spread, the rapid development of effective treatments is of utmost importance; thus, it is critical to enhancing the overall understanding of SARS-CoV-2. In this paper, we have reviewed several aspects that are closely related to drug development. First, we have provided detailed information on critical viral processes that can be targeted by drugs. Subsequently, the phylogenetic characteristics are analyzed to infer if one drug would work on a specific subtype or can serve as general therapy. The review of RNA conservation may help investigate accurate positions that can be targeted or are not suitable as drug targets. By reviewing existing tools and currently identified interactions, we have provided information that can help candidate target prioritization and drug repurposing using bioinformatic approaches. Finally, the basic knowledge, current trials and applicable methods for drug repositioning are listed.
The successful design of therapeutics largely relies on the complete dissection of relevant processes, the need for more comprehensive information and further data integration is constant. Using machine learning techniques, Gussow et al. [60] have compared a large amount of virus genomes and identified genomic features relating to a high fatality rate and the ability to infect human hosts. With more data incorporated and more algorithms applied, researchers will undoubtedly obtain results that may guide the clinical research. Recently, WHO has decided to discontinue hydroxychloroquine and lopinavir/ritonavir trial arms, as they have shown no observable effect toward hospitalized patients. The drugs failed to live up to their high expectations, suggesting that a list with enough number of candidates is essential to the success of efforts. We anticipate that the information provided in this paper may provide a useful starting point for addressing fundamental questions of SARS-CoV-2 and facilitating clinical research.
Key Points
COVID-19 is rampantly spreading worldwide. However, no specific effective treatment is developed. Rapid drug development is essential to meet urgent needs.
In this paper, we highlight data resources for the underlying molecular mechanisms, phylogenies, nucleotide-level modifications and conservations, potential drug targets, drug discovery and therapeutic strategies. All these data could provide assistance for future drug development.
This article provides the OverCOVID web-portal, which categorized and listed current SARS-CoV-2-related online resources, with easy-to-use searching and indexing functions. Other information, including virus mechanisms and phylogenic patterns, are also provided to help users with certain needs.
Md. Asif Ahsan is a postdoctoral research fellow at the Department of Bioinformatics, College of Life Sciences; The First Affiliated Hospital, School of Medicine, Zhejiang University, China. His research focuses on epistasis analysis or gene–gene interactions.
Yongjing Liu is a PhD candidate in Ming Chen's laboratory at Zhejiang University, China. His research interests include cancer genomics and multi-omics analysis.
Cong Feng is a PhD candidate in Ming Chen’s laboratory at Zhejiang University, China. His research focuses on DNA repeat detection and genome analysis.
Yincong Zhou is a postdoctoral researcher in Ming Chen’s laboratory at Zhejiang University, China. His research focuses on single-cell rna seq data analysis.
Guangyuan Ma is an undergraduate student at the College of Life Sciences, Zhejiang University, China. His research focuses on bioinformatics data analysis and deep learning.
Youhuang Bai is an associate professor at Fujian Agriculture and Forestry University. His research focuses on integrated approaches for RNA-mediated regulatory networks.
Ming Chen is the director of the Bioinformatics Lab of Zhejiang University and a professor at the Department of Bioinformatics, College of Life Sciences; The First Affiliated Hospital, School of Medicine, Zhejiang University, China.
Contributor Information
Md Asif Ahsan, Department of Bioinformatics, College of Life Sciences; The First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou 310058, China.
Yongjing Liu, Department of Bioinformatics, College of Life Sciences; The First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou 310058, China.
Cong Feng, Department of Bioinformatics, College of Life Sciences; The First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou 310058, China.
Yincong Zhou, Department of Bioinformatics, College of Life Sciences; The First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou 310058, China.
Guangyuan Ma, Department of Bioinformatics, College of Life Sciences; The First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou 310058, China.
Youhuang Bai, Department of Bioinformatics, Fujian Agriculture and Forestry University, Fuzhou, 350002, China.
Ming Chen, Department of Bioinformatics, College of Life Sciences; The First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou 310058, China.
Acknowledgments
We thank to Dr Andrew Harrison and Md. Selim Reza for helpful discussions.
Funding
National Key Research and Development Program of China (No. 2016YFA0501704, 2018YFC0310600), National Natural Sciences Foundation of China (No. 31771477, 31571366), and Jiangsu Collaborative Innovation Center for Modern Crop Production and Collaborative Innovation Center for Modern Crop Production co-sponsored by province and ministry, Foundation of Education Bureau of Fujian Province and Fujian Agriculture and Forestry University.
Author contributions
M.C. designed and supervised the project. M.A.A., Y.B., Y.L. and G.M. collected the data. M.A.A., Y.L. and Y.B. wrote the manuscript. M.A.A., Y.L. and Y.B. prepared the figures, tables and wrote the manuscript. C.F., Y.Z. and M.A.A. constructed the web-portal. All authors have read and approved the final manuscript.
Conflict of interest
None declared.
References
- 1. Coronaviridae Study Group of the International Committee on Taxonomy of V . The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol 2020;5:536–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang Q, Zhang Y, Wu L, et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020;181:894, e899–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shang J, Ye G, Shi K, et al. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020;581:221–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Slenter DN, Kutmon M, Hanspers K, et al. WikiPathways: a multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res 2018;46:D661–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ostaszewski M, Mazein A, Gillespie ME, et al. COVID-19 disease map, building a computational repository of SARS-CoV-2 virus-host interaction mechanisms. Scientific data 2020;7:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kim S, Chen J, Cheng T, et al. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res 2019;47:D1102–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sohag AAM, Hannan MA, Rahman S, et al. Revisiting potential druggable targets against SARS-CoV-2 and repurposing therapeutics under preclinical study and clinical trials: a comprehensive review. Drug Dev Res 2020;81:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Noris M, Benigni A, Remuzzi G. The case of complement activation in COVID-19 multiorgan impact. Kidney Int 2020;98:314–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang K, Chen W, Zhou Y-S, et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct Target Ther 2020;5:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Singh KK, Chaubey G, Chen JY, et al. Decoding SARS-CoV-2 hijacking of host mitochondria in COVID-19 pathogenesis. Am J Physiol Cell Physiol 2020;319:C258–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu KE, Fazal FM, Parker KR, et al. RNA-GPS predicts SARS-CoV-2 RNA residency to host mitochondria and nucleolus. Cell Syst 2020;11:102, e103–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hadfield J, Megill C, Bell SM, et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 2018;34:4121–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Turakhia Y, De Maio N, Thornlow B, et al. Stability of SARS-CoV-2 phylogenies. PLoS Genet 2020;16:1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alouane T, Laamarti M, Essabbar A, et al. Genomic diversity and hotspot mutations in 30,983 SARS-CoV-2 genomes: moving toward a universal vaccine for the "confined virus"? Pathogens 2020;9:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang L, Jackson CB, Mou H, et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat Commun 2020;11:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Di Giorgio S, Martignano F, Torcia MG, et al. Evidence for host-dependent RNA editing in the transcriptome of SARS-CoV-2. Sci Adv 2020;6:eabb5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim D, Lee JY, Yang JS, et al. The architecture of SARS-CoV-2 transcriptome. Cell 2020;181:914, e910–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cohen-Zontag O, Baez C, Lim LQJ, et al. A secretion-enhancing cis regulatory targeting element (SECReTE) involved in mRNA localization and protein synthesis. PLoS Genet 2019;15:e1008248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Haimovich G, Olender T, Baez C, et al. Identification and enrichment of SECReTE cis-acting RNA elements in the Coronaviridae and other (+) single-strand RNA viruses. bioRxiv 2020. doi: 10.1101/2020.04.20.050088. [DOI] [Google Scholar]
- 20. Andrews RJ, Peterson JM, Haniff HS, et al. An in silico map of the SARS-CoV-2 RNA Structurome. bioRxiv 2020. doi: 10.1101/2020.04.17.045161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rangan R, Zheludev IN, Hagey RJ, et al. RNA genome conservation and secondary structure in SARS-CoV-2 and SARS-related viruses: a first look. RNA 2020;26:937–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu X, Chen Y, Tang W, et al. Single-cell transcriptome analysis of the novel coronavirus (SARS-CoV-2) associated gene ACE2 expression in normal and non-obstructive azoospermia (NOA) human male testes. Sci China Life Sci 2020;63:1006–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Muus C, Luecken MD, Eraslan G, et al. Integrated analyses of single-cell atlases reveal age, gender, and smoking status associations with cell type-specific expression of mediators of SARS-CoV-2 viral entry and highlights inflammatory programs in putative target cells. bioRxiv 2020. [Google Scholar]
- 24. Wilk AJ, Rustagi A, Zhao NQ, et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat Med 2020;26:1070–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liao M, Liu Y, Yuan J, et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med 2020;26:842–4. [DOI] [PubMed] [Google Scholar]
- 26. Cao Y, Su B, Guo X, et al. Potent neutralizing antibodies against SARS-CoV-2 identified by high-throughput single-cell sequencing of convalescent patients' B cells. Cell 2020;182:73, e16–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wen W, Su W, Tang H, et al. Immune cell profiling of COVID-19 patients in the recovery stage by single-cell sequencing. Cell Discov 2020;6:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gordon DE, Jang GM, Bouhaddou M, et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020;583:459–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stukalov A, Girault V, Grass V, et al. Multi-level proteomics reveals host-perturbation strategies of SARS-CoV-2 and SARS-CoV. bioRxiv 2020. doi: 10.1101/2020.06.17.156455. [DOI] [Google Scholar]
- 30. Li J, Guo M, Tian X, et al. Virus-host interactome and proteomic survey reveal potential virulence factors influencing SARS-CoV-2 pathogenesis. Med (N Y) 2020;1:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Srinivasan S, Cui H, Gao Z, et al. Structural genomics of SARS-CoV-2 indicates evolutionary conserved functional regions of viral proteins. Viruses 2020;12:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dick K, Biggar KK, Green JR. Computational prediction of the comprehensive SARS-CoV-2 vs. human Interactome to guide the design of therapeutics. bioRxiv 2020. doi: 10.1101/2020.03.29.014381. [DOI] [PMC free article] [PubMed]
- 33. Messina F, Giombini E, Agrati C, et al. COVID-19: viral-host interactome analyzed by network based-approach model to study pathogenesis of SARS-CoV-2 infection. J Transl Med 2020;18:233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wrapp D, Wang N, Corbett KS, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020;367:1260–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lan J, Ge J, Yu J, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020;581:215–20. [DOI] [PubMed] [Google Scholar]
- 36. Gao Y, Yan L, Huang Y, et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020;368:779–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Peng Q, Peng R, Yuan B, et al. Structural and biochemical characterization of the nsp12-nsp7-nsp8 core polymerase complex from SARS-CoV-2. Cell Rep 2020;31:107774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Singh K, Chen Y-C, Judy JT, et al. Network analysis and transcriptome profiling identify autophagic and mitochondrial dysfunctions in SARS-CoV-2 infection. bioRxiv 2020. doi: 10.1101/2020.05.13.092536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Srivastava R, Daulatabad SV, Srivastava M, et al. Role of SARS-CoV-2 in Altering the RNA-Binding Protein and miRNA-Directed Post-Transcriptional Regulatory Networks in Humans. Int J Mol Sci 2020;21:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Prasad K, Khatoon F, Rashid S, et al. Targeting hub genes and pathways of innate immune response in COVID-19: a network biology perspective. Int J Biol Macromol 2020;163:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Guzzi PH, Mercatelli D, Ceraolo C, et al. Master regulator analysis of the SARS-CoV-2/human interactome. J Clin Med 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bruno A, Costantino G, Sartori L, et al. The in silico drug discovery toolbox: applications in lead discovery and optimization. Curr Med Chem 2019;26:3838–73. [DOI] [PubMed] [Google Scholar]
- 43. Wu C, Liu Y, Yang Y, et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B 2020;10:766–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Feng Z, Chen M, Liang T, et al. Virus-CKB: an integrated bioinformatics platform and analysis resource for COVID-19 research. Brief Bioinform 2020:1–14. doi: 10.1093/bib/bbaa155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shi Y, Zhang X, Mu K, et al. D3Targets-2019-nCoV: a webserver for predicting drug targets and for multi-target and multi-site based virtual screening against COVID-19. Acta Pharm Sin B 2020;10:1239–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kong R, Yang G, Xue R, et al. COVID-19 docking server: a meta server for docking small molecules, peptides and antibodies against potential targets of COVID-19. Bioinformatics 2020;36:5109–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bai Q, Tan S, Xu T, et al. MolAICal: a soft tool for 3D drug design of protein targets by artificial intelligence and classical algorithm. Brief Bioinform 2020:1–12. doi: 10.1093/bib/bbaa161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pushpakom S, Iorio F, Eyers PA, et al. Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov 2019;18:41–58. [DOI] [PubMed] [Google Scholar]
- 49. Sheahan TP, Sims AC, Graham RL, et al. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci Transl Med 2017;9:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Beigel JH, Tomashek KM, Dodd LE, et al. Remdesivir for the treatment of Covid-19 - final report. N Engl J Med 2020;383:1813–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yan Y, Zou Z, Sun Y, et al. Anti-malaria drug chloroquine is highly effective in treating avian influenza a H5N1 virus infection in an animal model. Cell Res 2013;23:300–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang M, Cao R, Zhang L, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res 2020;30:269–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Boulware DR, Pullen MF, Bangdiwala AS, et al. A randomized trial of hydroxychloroquine as postexposure prophylaxis for Covid-19. N Engl J Med 2020;383:517–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shah B, Modi P, Sagar SR. In silico studies on therapeutic agents for COVID-19: drug repurposing approach. Life Sci 2020;252:117652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhou Y, Hou Y, Shen J, et al. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov 2020;6:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tu YF, Chien CS, Yarmishyn AA, et al. A review of SARS-CoV-2 and the ongoing clinical trials. Int J Mol Sci 2020;21:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Martin R, Lochel HF, Welzel M, et al. CORDITE: the curated corona drug interactions database for SARS-CoV-2. iScience 2020;23:101297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sadegh S, Matschinske J, Blumenthal DB, et al. Exploring the SARS-CoV-2 virus-host-drug interactome for drug repurposing. Nat Commun 2020;11:3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ahmed SF, Quadeer AA, McKay MR. COVIDep: a web-based platform for real-time reporting of vaccine target recommendations for SARS-CoV-2. Nat Protoc 2020;15:2141–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gussow AB, Auslander N, Faure G, et al. Genomic determinants of pathogenicity in SARS-CoV-2 and other human coronaviruses. Proc Natl Acad Sci U S A 2020;117:15193–9. [DOI] [PMC free article] [PubMed] [Google Scholar]