

Abstract
Motivation
The outbreak of novel severe acute respiratory syndrome coronavirus (SARS-CoV-2, also known as COVID-19) in Wuhan has attracted worldwide attention. SARS-CoV-2 causes severe inflammation, which can be fatal. Consequently, there has been a massive and rapid growth in research aimed at throwing light on the mechanisms of infection and the progression of the disease. With regard to this data science is playing a pivotal role in in silico analysis to gain insights into SARS-CoV-2 and the outbreak of COVID-19 in order to forecast, diagnose and come up with a drug to tackle the virus. The availability of large multiomics, radiological, bio-molecular and medical datasets requires the development of novel exploratory and predictive models, or the customisation of existing ones in order to fit the current problem. The high number of approaches generates the need for surveys to guide data scientists and medical practitioners in selecting the right tools to manage their clinical data.
Results
Focusing on data science methodologies, we conduct a detailed study on the state-of-the-art of works tackling the current pandemic scenario. We consider various current COVID-19 data analytic domains such as phylogenetic analysis, SARS-CoV-2 genome identification, protein structure prediction, host–viral protein interactomics, clinical imaging, epidemiological research and drug discovery. We highlight data types and instances, their generation pipelines and the data science models currently in use. The current study should give a detailed sketch of the road map towards handling COVID-19 like situations by leveraging data science experts in choosing the right tools. We also summarise our review focusing on prime challenges and possible future research directions.
Contact
hguzzi@unicz.it, sroy01@cus.ac.in
Keywords: data science, SARS-CoV-2, COVID-19, artificial intelligence, network science
1 Introduction
The massive outbreak of SARS-CoV-2 viral infections in the world has led to a life-threatening pathogenic disease, which has been named COVID-19 (COronaVIrus Disease 2019) by the World Health Organization (WHO) [1]. The surprisingly rapid human-to-human transmission has created an alert due to the exponential increase in the number of cases in relatively short time [2] (https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports). Since December 2019, the novel coronavirus has had a surprisingly high diffusion rate among humans, hence the WHO has declared it a pandemic in March 2020. It had already infected more than 17 million people and has spread to over 213 countries in less than six months and is continuing. Unlike other severe acute respiratory syndromes coronaviruses (order Nidovirales, family Coronaviridae, subfamily Coronavirinae) like SARS-CoV or Middle East Respiratory Syndrome (MERS) coronavirus, SARS-CoV-2 spread is global and so far uncontained. The unprecedented scenario of the COVID-19 pandemic triggered scientific communities to rapidly develop vaccines and study how to contain the outbreak. Moreover, the explosion of COVID-19-related scientific literature confirms that scientific communities are actively contributing to the understanding of SARS-CoV-2. Figure 1 depicts a clear growth trend. According to Nature Index updates on 27 June 2020 (https://www.natureindex.com/news-blog/the-top-coronavirus-research-articles-by-metrics), 67 753+ papers on COVID-19 have been published so far (see also [3]).
Figure 1.
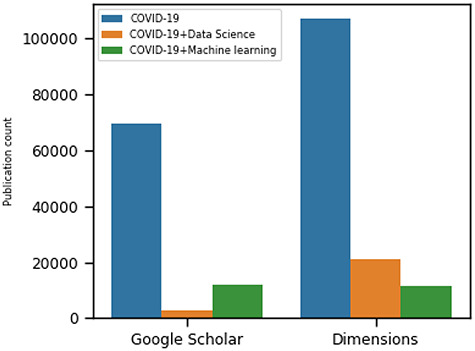
The trends of COVID-19 related research publications from two sources: Dimensions [4] and Google Scholar [5] as of 28 September 2020. We searched by using the following keywords: COVID-19, COVID-19 and Machine Learning and COVID-19 and Data Science. The search filter includes published articles, preprints, edited books, monographs, proceedings and chapters.
A large proportion of the scientific community, comprising almost all disciplines, is working on developing vaccines, therapies, as well as the management of patients and resources in order to combat the virus. As a consequence, we observe an increasing availability of freely available COVID-19 related omics and clinical data. For instance, the GISAID database (https://www.gisaid.org/) has collected more than 67 000 viral genomic sequences in a very short time. The Johns Hopkins dashboard (https://coronavirus.jhu.edu/map.html) has become one of the primary data sources for disease monitoring from an epidemiological perspective. The rapid accumulation of data and the need to support wet-lab investigations has implied an increasing effort in exploiting computational-based approaches (e.g. deep learning, artificial intelligence, network medicine) on COVID-19 related datasets [6]. These methods help to understand the pathogenesis of the disease and hopefully will lead to the development of a vaccine or new drugs. This, however, has resulted in an accumulation of data, algorithms, software and tools that need to be categorised and organised. A complete and exhaustive categorisation of all the approaches is undoubtedly a tough task due to the high publication rate.
We aim therefore to present the main characteristics of the current landscape, as depicted in Figure 2: (i) data sources, (ii) repositories, (iii) data science models, (iv) decision-making and (v) interpretation. Figure 2 also illustrates data analysis processes. Some processes return data and/or models that can be used as input (feedback) for the data analysis workflow. The integration and analysis step uses data science models to infer knowledge from the results of the previous steps (see Decision row in Figure 2). For instance, the drug–disease association needs a network biology approach to determine the relationship between drug molecules and their impact on patients. Many laboratories are producing a massive amount of heterogeneous data, considering both format and content. Viral sequences are represented as strings. Usually, raw clinical data (i.e. clinical records, biological analyses) are highly unstructured and heterogeneous, while medical images are more standardised data. Such data are accumulated into public databases or websites, which can be integrated with other existing databases (e.g. virus–host interaction databases, clinical and epidemiological databases) to enrich knowledge or correlate information. Such an approach can be useful in the drug–disease association, screening possible candidate vaccines or supporting healthcare decisions (e.g. management of resources such as intensive care units [ICUs]).
Figure 2.
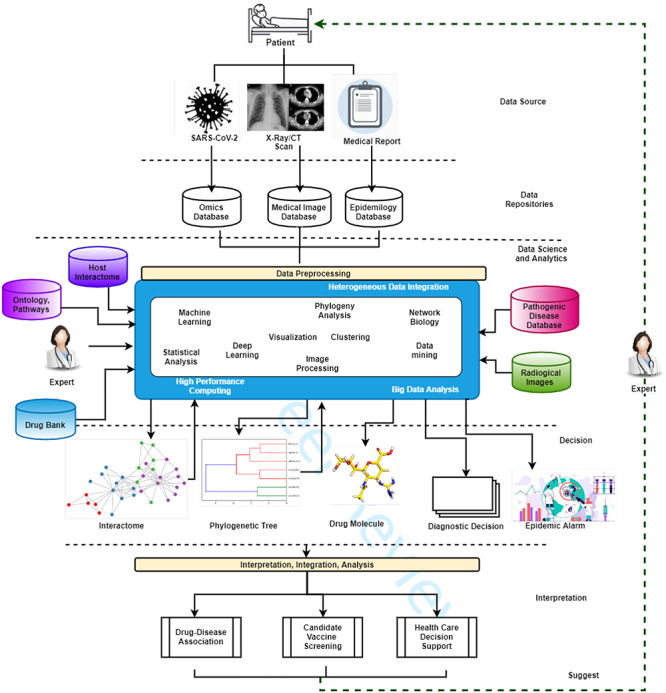
A data science landscape for SARS-CoV-2 and COVID-19 studies. Many different technologies produce a large quantity of data related to patients at different scales (e.g. molecular data, medical images and clinical data and epidemiological data). The accumulation of this data is the pre-requisite for a substantial rise of data science approaches (e.g. deep-learning and classical data mining) that often integrate existing data stored in databases or a priori knowledge (e.g. domain experts or ontologies). Such approaches produce new information about molecular interactions, phylogenetic analysis, in silico design of drugs or healthcare management decisions. The output may guide the execution of novel experiments closing the loop of the whole process.
Contributions of the current paper can be summarised as follows:
 we present a comprehensive review of the in silico approaches adopted so far to handle COVID-19 in genomics, proteomics, interactomics, epidemics, clinical imaging and drug design;
we present a comprehensive review of the in silico approaches adopted so far to handle COVID-19 in genomics, proteomics, interactomics, epidemics, clinical imaging and drug design; we present data analytics tasks related to COVID-19;
we present data analytics tasks related to COVID-19; we present an overall landscape suggesting strategies to integrate heterogeneous COVID-19 data sources;
we present an overall landscape suggesting strategies to integrate heterogeneous COVID-19 data sources; we report data sources, models and tools, which can be used to study SARS-CoV-2 and COVID-19;
we report data sources, models and tools, which can be used to study SARS-CoV-2 and COVID-19; we take a look at computational biology and bioinformatics approaches available in the literature.
we take a look at computational biology and bioinformatics approaches available in the literature.
In conclusion, we aim to offer to data scientists, medical doctors, healthcare advisers and drug/vaccine designers a landscape on data and tools that are useful for their activities on COVID-19.
2 COVID-19 virology and data science: background
The recent pandemic has promoted the collaboration across different research communities (e.g. virologists, computational biologists, medicine specialists, data scientists, etc.) to shed light on the pathogenesis and contrast strategies for the COVID-19 disease. A large number of recent research efforts has generated bulk experimental data. In order to unveil the basic molecular mechanism of the disease, there is the need to use data science-related technologies to improve knowledge on virus. Thus, it is crucial to have the right tools and data for understanding the biology behind SARS-CoV-2. Here, we try to briefly introduce the SARS-CoV-2 virus genetically related to COVID-19 and how omics, bioimages and epidemiological data related to COVID-19 are generated and stored in publicly available data repositories. We introduce the concept of data science as a useful set of tools and methodology to gain new insights from the available COVID-19 related datasets.
2.1 COVID-19: a novel coronavirus disease
COVID-19, a highly infectious viral disease caused by SARS-CoV-2, was discovered at the end of 2019. The symptomatic COVID-19 patients usually experience mild to moderate respiratory problems together with fever, dry cough and tiredness. A few non-severe patients also experience aches, pains, sore throat, diarrhoea, skin rashes, conjunctivitis, headaches, discolouration of fingers or toes and most significantly loss of taste or smell. Infection is transmitted through close proximity of an infected person, droplets generated by infected persons through coughs, sneezes or exhaling or touching contaminated surface. It enters through eyes, nose or mouth. Trend shows [7] that patients on and above 65 years of age with comorbidities are more vulnerable and may need ICU admission.
2.2 Virus biology of SARS-CoV-2
Viruses are small microorganisms that use living cells to replicate. Viruses cause many infectious diseases responsible for millions of deaths every year [8]. They exist in the form of small independent particles named virions. Each virion consists of two main components: (i) the genetic information, encoded as DNA or RNA, and (ii) a protein coat, named capsid, which wraps the genetic material. Sometimes the capsid is surrounded by an envelope of lipids. Virions have different shapes that are used in their classification [9]. As viruses are not able to replicate by themselves, they need to use the cell metabolism of a host organism. The virus replication cycle may be summarised in the following six steps [10]:
Attachment. Viruses bind the surface of host cells.
Penetration. Viruses enter the host cell through receptor-mediated endocytosis or membrane fusion.
Uncoating. The viral capsid is removed, and virus genomic materials are released.
Replication. Viruses use the host cells to replicate their genomic information. During this step, viral proteins are synthesized and possibly assembled. Viral proteins may interact with each other and with the host proteins to perform their function (e.g. regulate the protein expression).
Assembly. Virus particles may self-assembly with host proteins, causing the modification of some proteins.
Release. Viruses can be released from the host cell by lysis, a process that kills the cell.
Viruses are classified into major classes using phenotypic characteristics, such as morphology, nucleic acid type (e.g. RNA or DNA), etc. The International Committee on Taxonomy of Viruses (ICTV) is in charge of updating the viral taxonomy. The Baltimore classification system is also used where viruses are grouped into seven groups based on mRNA synthesis.
SARS-CoV-2 belongs to 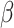 -coronaviruses that are a subgroup of the coronavirus family. These are giant enveloped positive-stranded RNA viruses that are usually able to infect a wide variety of mammals and avian species. All the members of the family cause respiratory or digestive and enteric diseases [11]. The infection mechanism is based on the action of surface spikes constituted by glycoproteins (named S or spike proteins), responsible for binding to host cell receptors. The literature reports seven
-coronaviruses that are a subgroup of the coronavirus family. These are giant enveloped positive-stranded RNA viruses that are usually able to infect a wide variety of mammals and avian species. All the members of the family cause respiratory or digestive and enteric diseases [11]. The infection mechanism is based on the action of surface spikes constituted by glycoproteins (named S or spike proteins), responsible for binding to host cell receptors. The literature reports seven 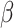 -coronaviruses that are responsible for causing diseases in humans. Four strains cause mild respiratory apparatus infection, which can be usually treated without lethal consequences (HCoV-229E, HKU1, NL63 and OC43). More recently, three strains of betacoronaviruses have severe and potentially fatal consequences: SARS-CoV, MERS-CoV and SARS-CoV-2 [12]. SARS-CoV caused an outbreak in China in 2002 characterised by a severe respiratory syndrome. MERS-CoV caused an outbreak in the Middle East in 2012. Both viruses caused similar disease symptoms, which led to pneumonia. MERS-CoV infected patients also presented gastrointestinal complications and kidney failure. The third member of the family, SARS-CoV-2, appeared in December 2019 in Wuhan, Hubei Province, China [13]. From the initial steps, it presented a surprisingly rapid diffusion rate. Until now, COVID-19 has killed more people than SARS and MERS combined, despite a lower fatality rate [14]. By the end of April 2020, the COVID-19 virus had already caused over 1500 000 confirmed cases around the world, of which around 350 000 hospitalised, and over 94 000 deaths. In China there has been more than 80 000 confirmed cases, with more than 3000 deaths.
-coronaviruses that are responsible for causing diseases in humans. Four strains cause mild respiratory apparatus infection, which can be usually treated without lethal consequences (HCoV-229E, HKU1, NL63 and OC43). More recently, three strains of betacoronaviruses have severe and potentially fatal consequences: SARS-CoV, MERS-CoV and SARS-CoV-2 [12]. SARS-CoV caused an outbreak in China in 2002 characterised by a severe respiratory syndrome. MERS-CoV caused an outbreak in the Middle East in 2012. Both viruses caused similar disease symptoms, which led to pneumonia. MERS-CoV infected patients also presented gastrointestinal complications and kidney failure. The third member of the family, SARS-CoV-2, appeared in December 2019 in Wuhan, Hubei Province, China [13]. From the initial steps, it presented a surprisingly rapid diffusion rate. Until now, COVID-19 has killed more people than SARS and MERS combined, despite a lower fatality rate [14]. By the end of April 2020, the COVID-19 virus had already caused over 1500 000 confirmed cases around the world, of which around 350 000 hospitalised, and over 94 000 deaths. In China there has been more than 80 000 confirmed cases, with more than 3000 deaths.
The sequence and structural analysis revealed a marked similarity between SARS-CoV and SARS-CoV-2, as confirmed by the evidence that the new coronavirus binds with the ACE2 receptor. Unfortunately, it presents a closer affinity than the previous virus [14]. Moreover, the expression pattern of ACE2 in human respiratory epithelia and oral mucosa may represent the cause of human-to-human transmission. Clinical manifestations of COVID-19 may be severe since they seem to impact all of the tissues and organs that express the ACE2 receptor. Some of the clinical conditions are severe pneumonia, kidney failure, anaemia, neurological problems, cardiovascular complications and also a severe inflammation known as cytokine storm, occurring in the most serious cases [15–17].
2.3 COVID-19 data generation and sources
COVID-19 pandemic has contributed to massive, unprecedented and rapid growth in data generation and research publications across the world. Figure 1 illustrates publication trend considering keywords related to COVID-19, whereas Figure 3 depicts a snapshot of the distribution of publications considering biological related issues. As reported in the academic search engine named Dimension (https://app.dimensions.ai/discover/publication), a total of 730 datasets related to COVID-19 are publicly available. Published data can be primary (e.g. sequences, clinical images, medical reports) or secondary data (e.g. protein structures, interactomes, epidemiological). We consider data to be primary when it is directly generated from the virus or the patient. From the primary data, more refined, summarised and inferred outputs are elaborated and then stored as secondary data. During COVID-19 data analysis and inference play a significant role as a reference set to extract or infer new knowledge. Next, we briefly discuss primary and secondary data types, the ways they are generated and published in available repositories for COVID-19-related data mining processes for further information.
Figure 3.
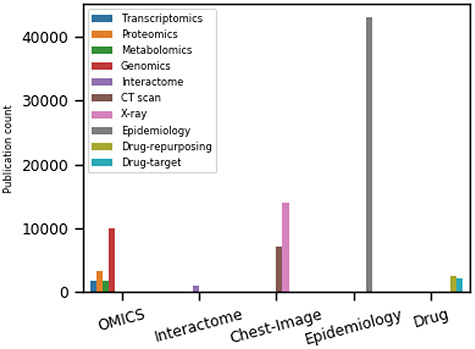
The trends of COVID-19-related research articles on five major topics (OMICS, interactome, chest imaging, epidemiology and drug repurposing) based on the search hits from the Google Scholar as of 28 September 2020. The search filter includes published articles, preprints, edited books, monographs, proceedings and chapters.
2.3.1 Omics data
High-throughput omics technologies (http://omics.org) use biochemical assays (i.e. analytical procedure to detect and quantify cellular processes) to discover molecules in the biological samples. Omics data fall (but are not limited to) into the following classes: genomics, transcriptomics, proteomics and metabolomics. Both omics technologies and data are used for insights into new unknown viruses. Thus, a preliminary activity to study COVID-19 disease has been the sequencing of the genome of the SARS-CoV-2 to elucidate how the virus grows, mutates and replicates [18]. Blood or throat swab specimens are collected both from population and patients showing compatible symptoms or suspected to have been infected. The extracted RNA material is then further sequenced, for instance, by using next generation sequencing (i.e. NGS). The output of NGS is the SARS-CoV-2 genome, which is usually stored in public repositories (see available SARS-CoV-2 nucleotide and protein sequence repositories in Table 1). NGS enables the retrieval of complete RNA information, including transcription and expression levels, functions, locations, trafficking and degradation. In a recent study, the architecture of SARS-CoV-2 transcriptome [19, 20] is reported. Transcriptomic data are highly effective in furthering understanding of the processes of cellular differentiation, carcinogenesis, transcription regulation and SARS-CoV-2, followed by the discovery of important biomarkers. The two main sources of SARS-CoV-2 RNA expression can be found in NCBI (http://www.ncbi.nlm.nih.gov/geo/) and OmicsDI (https://www.omicsdi.org/). Finally, metabolomic data analysis may be used to help in identifying potential chemical biomarkers for COVID-19. Metabolomics is used for the analysis of phenotypes and to gain insights into the metabolic state of biological systems. However, there are very few works in the literature providing metabolic data from COVID-19 samples. For instance, Shen et al. [21] reported proteomic metabolomic profiling of sera from 46 COVID-19 and 53 control patients data extracted from ProteomeXchange Consortium dataset (https://www.iprox.org/).
Table 1.
Popularly used omics, interactomics, chest image, epidemic and repurposed drug molecules data repositories for COVID-19 data science research
| Data type | Repositories | Description | Source |
|---|---|---|---|
| Omics | |||
| Nucleotide/protein | GISAID | More than 75 000 viral genomic sequences of SARS-CoV-2 (updating) | https://www.gisaid.org/ |
| Nucleotide/protein | NCBI | More than 25 000 nucleotide, 250 401 protein (updating) | https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/ |
| Structure | RCSB PDB | SARS-CoV-2 proteins (updating) | https://www.rcsb.org/covid19 |
| Structure | SWISS-MODEL | SARS-CoV-2 proteins (updating) | https://swissmodel.expasy.org/repository/species/2697049 |
| Heterogeneous | COVID-19 hg | https://www.covid19hg.org | |
| Metabolomics | iProX | Integrated proteome resources center | https://www.iprox.org/ |
| Transcriptomics | NCBI | RNA expression data | http://www.ncbi.nlm.nih.gov/geo/ |
| Transcriptomics | OmicsDI | RNA expression data | https://www.omicsdi.org/ |
| Interactomics | |||
| Interactions, network | BioGRID | More than 800 interacting proteins (updating) | https://thebiogrid.org/ |
| Interaction, graph | IntAct | 4479 binary interactions (updating) | https://www.ebi.ac.uk/intact/ |
| Interacting protein | HPA | More than 200 interacting human proteins with SARS-CoV-2 (updating) | https://www.proteinatlas.org/humanproteome/SARS-CoV-2∼ |
| Chest Imaging | |||
| X-Ray | github | More than 800 images (updating) | https://github.com/ieee8023/covid-chestxray-dataset |
| CT | github | 349 images from 216 patients | https://github.com/UCSD-AI4H/COVID-CT |
| github | 63 849 images from 377 patients | https://github.com/mr7495/COVID-CTset | |
| github | 104 009 CT images from 1489 patients | https://github.com/lindawangg/COVID-Net/ | |
| MosMED | Chest CT scans with COVID-19 related findings | https://mosmed.ai/en/ | |
| Both | BIMCV-COVID19+ | X-ray images CXR (CR, DX), 1380 CX, 885 DX and 163 CT | https://osf.io/nh7g8/ |
| Epidemiological | |||
| Information | CIDRAP | Cases of coronavirus disease, situation report, epidemiology, virology, clinical features | https://www.cidrap.umn.edu/COVID-19∼/epidemiology |
| WHO | Information regarding COVID-19 | https://covid19.who.int/ | |
| Italian Civil Protection | https://github.com/pcm-dpc/COVID-19∼ | ||
| SCIENTIFIC DATA [42] | Curated individual-level data from national, provincial and municipal health reports and online reports | https://doi.org/10.6084/m9.figshare.11974344 | |
| Drug repurposing | |||
| Molecule | Drugbank | Contains around 13 606 drug entries | https://www.drugbank.ca/COVID-19∼ |
| PubChem | World largest database: more than 350 million Compounds, Substances, BioAssay | https://pubchem.ncbi.nlm.nih.gov/ | |
| ChEMBL | SARS-CoV-2-related bioactive molecules with drug-like properties | https://www.ebi.ac.uk/chembl/ | |
| Excelra | COVID-19-related drugs that are ‘clinical, pre-clinical and experimental’ stage. | https://www.excelra.com/COVID-19∼-drug-repurposing-database/ | |
| CAS | Anti-viral drugs and related chemical compounds for COVID-19 disease | https://www.cas.org/ | |
| Pharmaceutical | Drugs in all stages of preclinical and clinical development for COVID-19 indication | https://www.pharmaceutical-technology.com/coronavirus-drug-trials-studies/ |
2.3.2 Interactome data
Interactomics is the study of biochemical interactions among biological molecules (e.g. proteins, transcription factors, small molecules). Since these interactions are the elementary building blocks of almost all cellular processes, their elucidation appears as an essential step in describing SARS-CoV-2 mechanisms of infection and replication [22]. There are many experimental platforms for deriving physical interactions among proteins [23], such as affinity purification mass-spectrometry (AP-MS) and yeast-two-hybrid (Y2H). Gordon et al. [24] have expressed 26 out of 29 SARS-CoV-2 proteins and used an AP-MS to identify 332 human proteins to which the viral proteins bind. There are several bioinformatics tools enabling the prediction of interactions using biological information coupled with network science (see for instance [25] for a more detailed comparison). Different proteomic technologies can be used to study the complete set of interactions for several viruses [26–28]. For instance, research projects have elucidated quite a large map of interactions for SARS-CoV-2. Such interactions are usually modelled by using graphs and stored in a growing number of databases, such as Virus Mint [29], String Viruses [30], HpiDB [31], Virus Mentha [32] and VirHostNet [33].
Despite the existence of such platforms, the rapid diffusion of the SARS-CoV-2 virus makes the extraction of reliable information (e.g. correlations, interactions) from these databases particularly difficult. Consequently, the first studies mainly used interaction predictions performed by using bioinformatic tools. For instance, in [34], the homology among SARS-CoV-2 and other coronaviruses has been used to infer putative interactions among viral proteins and host–viral proteins. Differently, a wet-lab approach [24] has been used where SARS-CoV-2 proteome is cloned and AP-MS is used to identify 332 protein interactions between SARS-CoV-2 and human cells. Finally, in [35], a preliminary tool consisting of a curated SARS-CoV-2 interactions dataset extracted by the IMEx consortium is presented.
2.3.3 Chest images data
X-ray and computer tomography (CT) technologies can be used to detect lungs and respiratory tracks infected by COVID-19. Ground-glass (GGO) pattern is the most common finding in a chest CT image of a COVID-19 infected patient. Patterns are usually multifocal, peripheral and bilateral. However, during COVID-19, GGO may appear as a unifocal lesion, most commonly located in the inferior lobe of the right lung [36]. Chest X-ray images have been observed to be insensitive in the early phase of the disease. However, they become useful in tracking the progression of the disease.
The urgent need for an automatic diagnostic tool for the rapid detection of COVID-19 patients encourages the data science community to develop novel machine learning-based diagnostic frameworks. TrainingData.io (https://www.trainingdata.io/) is a platform offering a free collaborative tool that allows data scientists and radiologists to share training data annotations that can be used for developing machine learning models. We report a non-exhaustive list of repositories containing annotated COVID-19 infected chest images in Table 1.
2.3.4 Epidemiological data
Epidemiological data are a collection of non-experimental observations obtained by gathering any health-related data source by domain experts, where such data include environmental, clinical and laboratory data, geographic spread and so on. Thus these data can be associated with the geographic spread as well as the risk associated with co-morbidities. Consequently, many independent groups have started to collect epidemiological data produced and made available by healthcare providers. Dong et al. [37] have designed and developed the first dashboard hosted at the Johns Hopkins University, providing free access to health data collected from almost all nations. Data are related to COVID-19 reported cases, i.e. infected, dead and recovered patients. For instance, the Italian government has provided a similar dashboard together with raw data related to COVID-19 [38]. Similarly, Xu et al. [39] have realised an open-access database for storing patient information produced in laboratories. Stored data are related to movements (for retrieving travel history), symptoms and demographics. All of these projects share some common characteristics: (i) the use of simple formats (e.g. tabular formats), (ii) the possibility of export in standard data sharing format (e.g. comma-separated values), (iii) simple query interfaces, (iv) the integration of geographic data and (v) demographic information [39], as reported in Table 1.
2.3.5 Drug-target databases
A drug is a small organic molecule [40] that can activate or inhibit the function of a therapeutically relevant protein during the onset of a disease. The discovery of a new novel drug and the subsequent approval by the Food and Drug Administration (FDA) is a complex, expensive and time-consuming process. Drug discovery involves two main steps: (i) drug–target identification and (ii) development of small molecules able to interact with the target [41]. The approved drug molecules and targets are often stored in publicly available databases, usually in the simplified molecular input line entry system (SMILE) format, for drug repurposing or commercial development. We report some drug databases that are useful for drug-discovery process. For example, DrugBank is a repository containing both drug and drug target information. The latest release contains 13 596 drug entries, including 2640 approved small molecule drugs, 1389 approved biological molecules (i.e. proteins, peptides, vaccines and allergenic), 131 nutraceuticals and over 6377 experimental drugs still in discoveryphase. Additionally, 5225 non-redundant protein sequences (i.e. drug target/enzyme/transporter/carrier) are linked to these drug entries. PubChem is a collection of freely accessible chemical information that stores chemical and physical properties, biological activities, safety and toxicity information, patents and literature citations. It contains  million compounds,
million compounds,  million substance descriptions and details,
million substance descriptions and details,  million of bioactivities conducted on compounds and over
million of bioactivities conducted on compounds and over 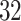 million of pieces information relating to drugs with published papers and
million of pieces information relating to drugs with published papers and 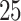 million patent descriptions. Excelra is an open-source COVID-19 Drug Repurposing Database that stores a list of approved small molecules being at an early stage of experimentation. Pharmaceutical Technology (https://www.pharmaceutical-technology.com/coronavirus-drug-trials-studies/) is a coronavirus drug tracker that lists drugs at all stages of pre-clinical and clinical development (from discovery through to preregistration) for COVID-19. This list is updated dynamically, based on the Global Data Pharma Intelligence Center Drugs database (https://www.globaldata.com/). CAS (https://www.cas.org/) contains a connection of nearly 50 000 chemical substances, stored in the SD file (.sdf) format, along with related metadata such as CAS Registry Number and physical properties for each element. Other relevant non-exhaustive drug database instances are listed in Table 1.
million patent descriptions. Excelra is an open-source COVID-19 Drug Repurposing Database that stores a list of approved small molecules being at an early stage of experimentation. Pharmaceutical Technology (https://www.pharmaceutical-technology.com/coronavirus-drug-trials-studies/) is a coronavirus drug tracker that lists drugs at all stages of pre-clinical and clinical development (from discovery through to preregistration) for COVID-19. This list is updated dynamically, based on the Global Data Pharma Intelligence Center Drugs database (https://www.globaldata.com/). CAS (https://www.cas.org/) contains a connection of nearly 50 000 chemical substances, stored in the SD file (.sdf) format, along with related metadata such as CAS Registry Number and physical properties for each element. Other relevant non-exhaustive drug database instances are listed in Table 1.
2.4 The data science pipeline
Data science is a novel interdisciplinary research field that leverages methods, processes and algorithms supporting the extraction of relevant knowledge from (big)-data. The term data science was introduced for the first time in 2008 by DJ Patil and Jeff Hammerbacher [43]. Data science pipelines are made of four major phases: (i) raw data collection, (ii) preprocessing, (iii) descriptive or predictive modelling and (iv) interpretation. An illustrative representation of a typical data science workflow for COVID-19 management is reported in Figure 4.
Figure 4.
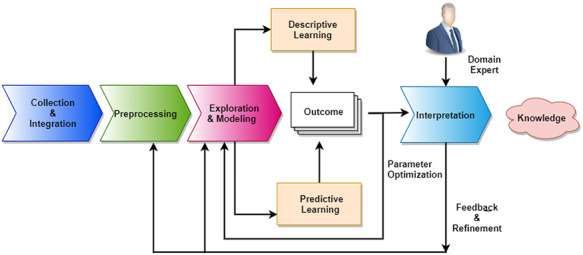
Major phases of data science pipeline towards decision making and analysis. Data initially collected and integrated from many sources. Then they need to be pre-processed to filter uninformative or possibly misleading values (e.g. outliers or noise). Then existing models are used to explain data or extract relevant patterns describing data or predicting associations. Finally, results need to be interpreted and explained by domain experts. Each step of analysis may generate corrections or refinements that are applied to precedent steps.
The integration of heterogeneous data is a crucial step in data science. The success of any data science model depends on the quality of data. Due to flaws, noise or errors in data generation, the outcome of a data science workflow can lead to incorrect results and/or interpretations. It is crucial therefore to apply different scrubbing and cleaning processeses on the data. Data standardisation and transformation are required if data have been generated by multiple and varying sources. Collectively, all of the above steps are called preprocessing [44]. Data exploration, which consists in feeding data to the computing model, can then be performed. Any data science process should include the statistical description (e.g. type, distribution, significant features, relationship among the data variables) of the input dataset, which leads to a better understanding of the data itself and of the preprocessing and analysis phases. Feature selection helps in identifying relevant attributes in the dataset. Visualisation aims to explore the possible relationship between features in the dataset or among different datasets. Dimensionality and data reduction help to make the data science workflow more efficient and resistant to noise.
Data can be analysed through the use of both descriptive (unsupervised) and predictive (supervised) models that often may be merged together by ensembling [45]. It is worth mentioning a few deep learning frameworks that could be helpful for COVID-19 predictive data analysis. Convolutional neural networks (CNNs) [46] are extensively used to analyse radiological chest images of COVID-19 patients. A different kind of CNN model, specifically designed for graph or network data, is a graph convolution network (GCN) [47]. Due to the lack of adequate training samples during COVID-19 to train deep models, the synthetic data play a significant role [48, 49]. A recent breakthrough in deep model architecture is given by the generative adversarial networks (GANs) [50] for generating synthetic data akin to real data. A GAN is made of two simultaneously trained neural networks: generator and discriminator. A discriminator recognises training samples, whereas a generator creates fake instances to challenge the discriminator, which enhances both modules in learning crucial discriminating features in the original dataset. Regression analysis is a supervised model that can also be used to predict pandemic trends [51]. Clustering [52] is a well-known unsupervised learning model that describes and summarises the hidden pattern inside data based on certain proximity metrics. Results of the previous methods have to be validated by domain experts. Expert feedbacks can also be iteratively suggested to refine the phases of the data science pipeline (see Figure 2).
3 SARS-CoV-2 omics data analysis
The main objective of the omics study is to discover the proximal origin of SARS-CoV-2, its mutational variants, and develop a predictive model for identifying SARS-CoV-2 from an isolated strain or sequence or effective chemical biomarker identification through transcriptomics and metabolomics studies. In addition to that, nucleotide sequences are used to determine the SARS-CoV-2 viral genome and 3D protein structure prediction as depicted in Figure 5.
Figure 5.
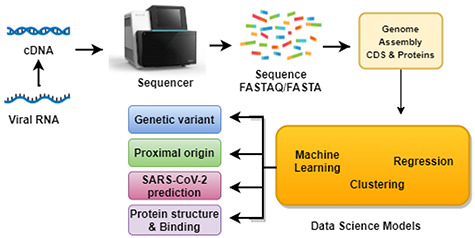
Omics data generation and data analysis workflow. Fragments of nucleic acid sequences of the virus, extracted form the host organism, are used as input for data processing algorithms. Many goals of these analyses are (i) analysis of genetic variants, (ii) analysis of genomes of viruses infecting different species, (iii) prediction of protein interactions and interactome and (iv) gather structure and dynamics of viral proteins.
3.1 Phylogeny and mutant variation analysis
Discovering the evolutionary origin of SARS-CoV-2 is currently the most urgent research topic. It aims to generate an evolutionary tree by using its nearest species [53]. Clustering is one of the most popular techniques to create a phylogeny among the coronavirus family-like SARS-COV and MERS-COV [54–56]. The evolution of viruses is mapped onto in silico algorithms using phylogenetic analysis. In general, the creation of a phylogenetic tree primarily relies on the analysis of sequence through alignment. Several algorithms have been developed to produce high-quality sequence alignments for both global alignments (focusing on the whole sequence) and local alignments (focusing on local regions of the sequence). Few common established methods and software have been used for SARS-CoV-2 genome alignment, such as DNAMAN (https://www.lynnon.com/dnaman.html), ClustalW [57], MUSCLE [58], Jalview [59] and MAFFT [60]. From alignment data, the evolutionary history is inferred by using the neighbour-joining (or maximum likelihood) method [57, 61]. Alignment approaches are also frequently used for the identification of single nucleotide polymorphisms (SNPs) of rapidly evolving viruses like SARS-CoV-2 [12, 12, 62]. On the other hand, alignment-free methods use feature-based approaches and compare the sequences by using the derived features. Due to the low mutation rate and a high degree of similarity among SARS-CoV-2 genomes, very few studies have been performed using alignment-free methods [63, 64]. Studies reveal that SARS-CoV-2 genome is similar to SARS-related coronaviruses (https://www.ecohealthalliance.org/2020/01/) found in the Pangolin [65] or Bat [66, 67]. Scientists are studying the new strains variants of coronavirus to understand its mutant variants. Despite the close similarity of the different SARS-CoV-2 genomes [68, 69], significant variations are also reported in the literature [70]. These studies, important for an understanding of the spread of the disease, can be useful for antiviral drug design [71].
3.2 Genome detection
For the elucidation of the infection mechanism it is important to individuate the complete sequence of virus genome and its circulating variants by means of machine learning models coupled with comparative genomics. Both the alignment and alignment-free methods are applied to generate features. In order to train machine learning models, some of the most used features are k-mers (i.e. subsequences of length k) and N-grams (i.e. a contiguous sequence of N items from a given sample), amino acid chemical properties and mutation information extracted by alignment methods.
In [63] an integrated approach is used to identify key genomic features that differentiate SARS-CoV-2 from SARS-COV and MERS-COV [72] coupled with decision trees that are also applied for sequence classification based on over 5000 unique viral genomic sequences, totaling 61.8 million bp (base pairs) that include 29 COVID-19 virus sequences. Recent works have sought to combine five well-known classification models in selecting features derived from a set of genomes belonging to a large set of coronavirus families and genomes of SARS-CoV-2 for detecting novel SARS-CoV-2. For instance, Fang et al. use a bi-path CNN (BiPathCNN) [73].
3.3 Protein structure prediction
Mutations of the genome may alter the encoded amino acid sequence, and the so-called non-synonymous mutations can influence the structure and function of the resulting protein [71]. Understanding the protein structures is required for identifying functional motifs and elucidating the possible binding mechanisms with the host proteins and for discovering antiviral drugs [74, 75]. The elucidation of protein structures by wet lab experiments requires a considerable amount of time. Structure prediction therefore is performed by using computational methods. Recently, SARS-CoV-2 proteins have been predicted (https://deepmind.com/) by the AlphaFold and the structures are deposited in the Protein Data Bank (https://www.rcsb.org/). AlphaFold [76] is a deep two-dimensional dilated convolutional residual network that predicts the inter-residue distances between pairs of amino acids and the angles between chemical bonds that connect those amino acids. trRosetta [77] is also used to predict SARS-CoV-2 protein structures. In addition to this, other existing protein structure and homology, modelling tools like COMPOSER [78], SWISS-MODEL [79], PyMOL c [80] and I-Tasser [81] are used for rapid prediction and comparison of Spike (S) protein [82, 83], Envelope (E) protein [2] and ab initio homology modelling [81].
3.4 Transcriptomics and metabolomics data analysis
Alongside sequence data and structural analysis, several researchers have focused on transcriptomics and metabolomics data analysis to design better therapeutic strategies for COVID-19. The aims of the transcriptomic data analysis are to investigate the activity of the set of genes in different organs and functional pathways and their possible role in causing infections and the regulation of various immunological factors inside the cell of SARS-CoV-2 patients during COVID-19 disease.
A study of transcriptomic data analysis of COVID-19 lungs and bronchoalveolar lavage fluid samples revealing predominant B-cell activation responses to infection is presented in [84]. The authors have used Metascape [85] for functional enrichment analysis of experimental data to determine the transcriptomic signature of lung tissues from COVID-19 patients. Further, xCell software [86] is used for computational deconvolution analysis to evaluate the relative proportions of immune cell subsets inCOVID-19 and healthy control samples.
For a better understanding of the pathophysiology of COVID-19, Gardinassi et al. [87] analysed public transcriptome datasets. They have considered the transcriptional signature of COVID-19 infected with SARS-CoV-1 and Influenza A (IAV) viruses. A core transcriptional signature induced by the respiratory viruses in peripheral leukocytes has been identified and the absence of significant type I interferon/antiviral responses has also been noted for SARS-CoV-2 infected. They also have identified the higher expression of genes involved in metabolic pathways including heme biosynthesis, oxidative phosphorylation and tryptophan metabolism.
Based on the publicly available high throughput gene expression data of several respiratory infection viruses, including SARS-CoV-2, a host transcriptome-based drug repurposing strategy has also been proposed [88]. The two main areas are interaction network construction using functional enrichment analysis and drug repurposing. STRING data repository is used for interaction network construction and functional enrichment analysis. For drug repurposing, the authors have used DrugBank and Connectivity Map (CMap) provided by the CLUE (https://clue.io/cmap) web tool. Finally, they suggested six approved PTGS2 inhibitor drugs for the treatment of COVID-19. Similar studies [89] were conducted, where analysis of major histocompatibility complexes and innate immune system gene expression from SARS-CoV-2 infected RNA-seq data of human cell line and virus transcriptome data is utilised to predict T-and B-cell epitopes.
To the best of our knowledge, few studies involve metabolomic data on COVID-19 patients. A metabolomic data analysis coupled with proteomics data is conducted for metabolomic characterization of SARS-CoV-2 patient sera in [21]. A random forest based learning model is applied on proteomic and metabolomic data derived from 18 non-severe and 13 severe patients. Finally, the authors find 29 important variables including 22 proteins and 7 metabolites. In another attempt, metabolomic and lipidomic data [90] are used revealing that metabolite and lipid alterations are correlated during COVID-19 disease.
3.5 Discussion
The majority of the COVID-19 related research relies on genomics and proteomics data of SARS-CoV-2 and other coronaviruses more than transcriptomic and metabolomic data. Existing tools have been extensively used to analyse SARS-CoV-2 omics data, and very few innovative approaches have been developed. Of course, the omics data generation and free distribution have made the major contribution to omics research. With the availability of high throughput and high resolution omics data it is now possible to perform a micro-level investigation of COVID-19 pathogenesis. Multi-omics data integration [91] together with effective data science and machine learning models [92, 93] is one possible way to improve understanding of the pathogenesis of COVID-19 or other viral diseases.
3.6 Interactomics
Interactomics research related to SARS-CoV-2 has two main goals: (i) development of possible therapies for helping affected people, and (ii) introduction of a novel vaccine for blocking the spread. Despite the existence of many different laboratories that have sequenced the whole genome and the availability of such data, the above-described issue may be successfully addressed only by looking at a molecular scale through the elucidation of interactions of viral and host proteins. As aforementioned, during the replication step, the virus proteins use the host environment, interact with each other and the host proteins, causing loss of function or even the death of the cells. The complete elucidation of the whole set of such interactions is therefore a crucial step for combatting the viruses. Understanding the interplay between host and virus proteins is crucial to identify virus-related diseases and potential targets for therapeutic strategies. Such information may clarify the viral molecular machinery during the viral infection, survival within the host and replication. This knowledge can also help to discern the protein interactions that are crucial for transmission and replication [34].
The literature contains many examples of the use of mass spectrometry for determining SARS-CoV-2 protein interactions [24, 94]. For instance, AP-MS based SARS-CoV-2 -host interactomes reported for 26 SARS-CoV-2 proteins with 332 host proteins [24]. The study aimed to identify possible drug targets. Therefore they isolate 66 possible drug targets in human proteins suggesting potential 69 compounds (of which, 29 drugs are approved by the US Food and Drug Administration, 12 are in clinical trials and 28 are pre-clinical compounds). As it is a time-consuming and expensive task to elucidate experimentally validated complete host protein interactions with viral protein. The in silico prediction is the only viable alternative. Some studies presented the investigation of virus–host interactomes using tools and methodologies from graph theory [27, 95], demonstrating the importance of studying virus–host interplay at network level [96–100]. Data related to the interactions (or functional associations) among biologically relevant macromolecules (e.g. proteins, genes, etc.) are usually modelled by means of graph theory and its related formalism [101, 102]. Consequently, biological entities are represented as nodes, while edges model their associations [103]. Such networks may contain a single kind of molecule, such as protein–protein Interactions (PPI), or gene–gene interactions [23, 104]. More recently, it has been shown that biological processes are formed by the synergistic interplay of different molecules (i.e. genes, non-coding RNA, proteins, mi-RNA, etc.) [105]. Consequently, novel models that integrate such diverse aspects and describe the interplay of the heterogeneous actor have been introduced. The use of more complex network models comprising different nodes and the various interactions is growing [106, 107]. The SARS-CoV-2 scenario, as we describe in the following, also contains such models (e.g. [10]). In Figure 6 we report a summarised view of in silico interactome graph inference workflow, involving different interactome and omics data sources.
Figure 6.
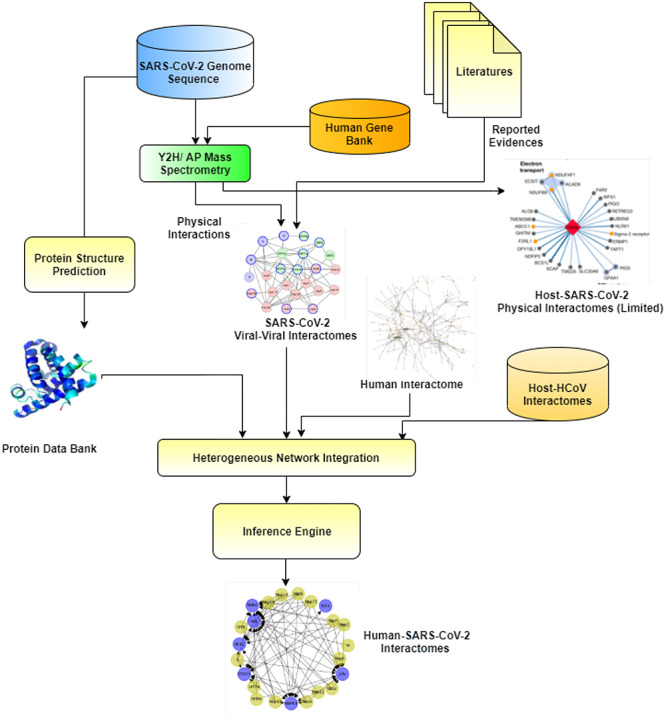
Data integration process to build a host–SARS-CoV-2 Interactome graph. The building of the integrated host–viral interactome starts with the analysis of the viral genome. Then viral proteins and the interactions with host protein are determined. The determination of such interactions is often performed by integrating experimental data with knowledge extracted from literature. Furthermore, protein structures are also predicted. All of this information (structures, virus–host interactions and viral interactions) is integrated by using heterogeneous networks. The final product of the process is an interactome.
From a bioinformatics perspective, a few key questions need to be addressed [9]:
 Are the infected proteins central or peripheral?
Are the infected proteins central or peripheral? Do all of the viruses attach to similar proteins from a network point of view?
Do all of the viruses attach to similar proteins from a network point of view? What happens in an infected host interactome?
What happens in an infected host interactome?
These considerations guided the first attempts to produce an interactome-wide map of SARS-CoV-2 proteins and their interactions with human proteins enabling scientists to answer the above questions. Thus, the interactions may be categorised in two main classes: (i) intra-viral interactions, i.e. interactions that occur among viral proteins that are in general limited and easy to determine; (ii) host–virus interactions, i.e. interactions that occur among viral and host proteins, which may potentially be numerous. One of the main challenges in this area is represented by the different speeds between the spread of SARS-CoV-2 and the time needed for wet-lab experiments. Therefore, all of the approaches we discuss below integrate both in silico and wet-lab experiments.
One of the first approaches of building a SARS-CoV-2 interactome is described in [34]. The authors have derived the first map of both intra-viral and host–viral proteins using a bioinformatics approach based on the homology between SARS-CoV-2 and the previous 2002 SARS-CoV virus. The hypothesis underlying the work is that the similarity between two viruses is also preserved at the interactome level. Thus many interactions of 2002 SARS-CoV may be preserved in SARS-CoV-2. Consequently, they derived a whole SARS-CoV-2 interactome containing both intra-viral and virus–host interactions. The authors derived a 2002 SARS-CoV interactome by analysing the available literature. Such data are integrated with a genome-wide analysis through Y2H on SARS-CoV ORFeome, obtaining a resulting intra-viral interaction network consisting of 31 proteins and 86 unique interactions. Then, the authors used both Y2H interaction data and literature mining to derive the viral–host interactions. The final virus–host interaction network consisting of 118 proteins, 93 host proteins and 114 unique virus–host interactions.
Multiple interactome analysis is another method used to integrate data obtained from heterogeneous protein or gene networks. In a similar attempt the authors in [108] proposed the integration of PPIs and gene expression data that are both obtained from available databases. Authors started with data related to three existing viruses (SARS-CoV, MERS-CoV, HCoV-229E) to infer the interactome of SARS-CoV-2. They also integrated an additional PPI database in order to reconstruct the action of SARS-CoV-2 at the proteome level, obtaining a network consisting in 13 020 nodes and 71 496 interactions. In parallel, the authors inferred a gene co-expression network using random walk with restart (RWR) algorithm and S-glycoproteins of SARS-CoV, MERS-CoV and HCoV-229E as seeds. Similarly, the HCoV–host interactome network was built by assembling known networks (e.g. SARS-CoV, MERS-CoV, HCoV-229E, HCoV-NL63, mouse MHV, avian IBV) obtaining a SARS-CoV-2 phylogenetically close interactome. As a novel attempt [109], the codon pattern is used to infer possible interactions between 26 SARS-CoV-2 proteins and selective host proteins involved in 17 major cell signalling pathways.
3.7 Discussion
For the described reasons above, and those connected with the previous studies into the SARS-CoV-2 virus, the number of known protein interactions is constantly growing. Discovering these interactions constitutes the first step in determining targeted therapies. Despite the large number of investigations in the literature, virus–host interactomes are far from exhaustive and the impact of mutations in both virus and humans has not yet been completed unravelled. Therefore, research in this field benefits from any increase in the discovery of novel protein interactions. Nonetheless the development of targeted therapies suffer from certain limitations. Finally, it should be noted that the integration of data collected from different omics sources [110] and medical images may contribute to understanding the evolution of the disease.
4 Chest image analysis for diagnosis and monitoring of COVID-19
Image analysis transforms digital images into measurements providing meaningful information of the images themselves. Automated chest image analysis may help in the early diagnosis of COVID-19 thereby assisting physicians in case of emergency. Two kinds of chest radiography images, obtained through X-ray and CT scanners, are recognised as useful for diagnosing pneumonia in COVID-19 patients. Data manipulation techniques can be used for the automatic (or semi-automatic) analysis of large amounts of images requiring substantial quantitative assessment and computation. Chest images can be used in many COVID-19-related scenarios, e.g. predicting the need for ICU resources, predicting survival rates, studying the patient’s trajectories during treatment [1]. Imaging is a fast, non-invasive, relatively cheap and already a widely adopted clinical practice that can be used to monitor the evolution of the disease. The ultimate goals are improving patient healthcare, biomarker design for the COVID-19 and, most importantly, early COVID-19 detection in patients.
Machine learning methods or CNN-based methods have also been used for this aim. For instance, COVID-Net [111] is the first open source CNN-based framework designed using deep learning techniques for COVID-19 detection. It has been used to analyse X-ray chest images; authors developed COVIDx, an open-access benchmark dataset composed of 13 975 CXR images from 13 870 patients. Model performance has been evaluated with other deep neural network architectures for comparative purposes. The model predicts three possible outcomes for each input image: (a) healthy, (b) non-COVID-19 (e.g. viral, bacterial, etc.) infection and (c) viral COVID-19 infection.
A transfer learning-based CNN has also been applied in [1] for detecting various anomalies in small medical image datasets. The authors collected 1427 X-ray images (224 COVID-19, 700 common pneumonia and 504 normal cases) from several sources such as Cohen (https://github.com/ieee8023/covid-chestxray-dataset), Radiological Society of North America (RSNA), Radiopaedia and the Italian Society of Medical and Interventional Radiology (SIRM) (https://www.kaggle.com/andrewmvd/convid19-xrays).
In [112], a deep-learning based method has been applied to pulmonary CT images to distinguish patients affected pneumonia related COVID-19 from healthy cases. At first, candidate infection regions have been isolated from the pulmonary CT image set by using Residual CNN (ResNet-23), a pre-trained neural network to identify image features. A combined CNN- long short term memory (LSTM) architecture is also used to detect infected patients X-ray images in [49]. CNN is used for extracting features from images, while LSTM is used for analysing these features. Similarly, in Inf-Net [113], a CNN has been used to perform the segmentation of lung CT images of COVID-19 patients. Moreover, in the absence of training images, synthetic Chest X-Ray (CXR) images can be generated by using the GAN model proposed in [48] or by using the statistical techniques described in [114, 115]. A binary classifier, using manta-ray foraging optimization (MRFO) for features extraction and KNN for classification, has been used to classify COVID-19 affected chest X-ray images in [116].
4.1 Discussion
Deep learning is one of the most commonly adopted choices by the data science community. Due to the availability of deep models and easy-to-use frameworks, researchers are able to use them to set up and develop methods for helping in COVID-19 diagnosis. The integration of both X-ray and CT scans may improve the quality of detection. Due to the similar lung damage and symptoms between COVID-19 and common pneumonia or influenza, the chances of false positives are quite high. One option for more accurate COVID-19 diagnosis, which is yet to be fully explored, is to integrate information extracted from chest images with transcriptomic and metabolomic data [110]. Despite the increasing demand of rapid COVID-19 diagnostic systems, most of the data science approaches described above suffer from low data availability. The larger the sample data volume, the more reliable the diagnostic system should be. In order to compensate for the data scarcity, both GAN and statistical models can be successfully used the hand the issue.
5 Epidemiological data analysis
From the beginning of the outbreak, a significant source of information has come from observation of novel COVID-19 cases and has been used to predict the evolution of the disease diffusion. Such data have been used with both existing and ad hoc mathematical models [39, 117]. The main aims of these approaches are (i) controlling the diffusion of COVID-19; (ii) supporting healthcare providers in allocating resources (e.g. planning ICUs); and (iii) evaluating the impact of containment measures.
From a data science perspective view, almost all of these efforts use real data to build and fit both predictive and observatory models. Most of them use deterministic models based on classical epidemiological studies. Consequently, real data are used to calculate model parameters based on ordinary differential equations (ODE) [118–120]. The diffusion of such works has been very rapid; for instance, simple queries on Google Scholar or on preprint servers returned more than two thousands papers on average.
In [121] authors integrated information of existing data sources provided by the Johns Hopkins University, WHO, Chinese Center for Disease Control and Prevention, National Health Commission and Dingxiangyuan (DXY, a Chinese epidemiological database). The proposed tool allows scientists to perform exploratory data analyses, using visualisation techniques to highlight differences in the reported cases (e.g. infected, dead and recovered people), in different countries.
Moreover, more sophisticated models have tried to integrate epidemiological data with other data to study the impact of other information (e.g. environmental, geographical, clinical) [122, 123]. A COVID-19 outbreak forecasting model has been developed using LSTM networks [124]. The John Hopkins University and Canadian health datasets have been used to extract significant features able to predict trends and possible duration of the current global COVID-19 pandemic.
5.1 Discussion
The COVID-19 outbreak has been characterised by a notable accumulation of epidemiological data publicly available on the web. The data are mainly related to infected patients as well as to the number of deaths. However, it should be noted that such datasets present some main drawbacks: (i) there is in general little attention paid to their reliability and (ii) there has been little effort in data format standardisation and semantics. For instance, there is no standardised way to determine the exact number of infected; moreover, variations in diagnosis the causes of death at country level may skew mortality figures at different countries. The vast majority of the described approaches integrating epidemiological and environmental data shows the above-mentioned limitations. From a computer science point of view, as far as principled data integration is concerned, the use of technologies and methodologies from the data-warehouse and on-line analytical processing (OLAP) communities may constitute a valid and stable choice, since most of the data are available in text format and could be stored on a relational or NoSQL database.
6 Drug repurposing and target prediction
Drug discovery aims to identify small molecules that potentially modulate the functions of target proteins. The development of a new drug molecule for COVID-19 is a time-consuming and costly task. In the COVID-19 era, the long process for the determination of a novel drug is not feasible, due to the rapid spread of the virus. It is of utmost importance to identify candidate anti-viral drugs more rapidly; these may control the adverse effects of COVID-19, thereby hopefully reducing the mortality rate. The best alternative is to look for already FDA-approved drugs that may bind with the therapeutic target (viral or host) proteins.
Data analysis for discovering possible candidates from the existing drugs is a well-known process referred to as ‘drug repurposing’. It involves the identification of new uses for approved (or experimental) drugs as a possible cure for novel pathologies. The process, as depicted in Figure 7, is based on the integration of molecular data (e.g. interactomes, co-expression networks), concerning the existing drug–disease association.
Figure 7.
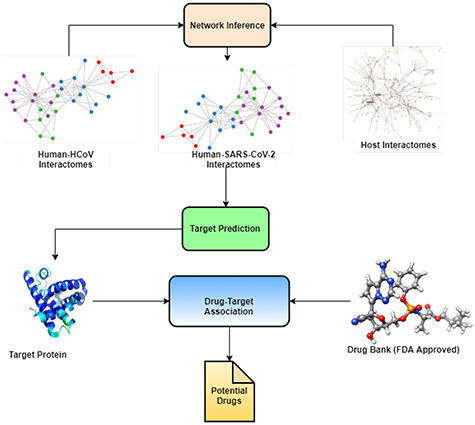
Drug repurposing process. The process is based on the integration of molecular data and drug-disease associations. The analysis is often performed by deep-learning or network embedding. The output list of candidate drugs is then confirmed via wet-lab experiments and clinical trials.
The availability of high-resolution proteomics, interactomics and drug–target association data makes it now feasible to quickly find a suitably small molecule in silico with the help of advanced (deep) neural network models. A good number of deep learning-based drug–target associations and repurposing tools are available for other viral diseases and thus can also be used for COVID-19 data analysis (see Table 2).
Table 2.
Data science tools and techniques for SARS-CoV-2 data analysis
| Task | Data type | Data science models | Available tools |
|---|---|---|---|
| Phylogeny/ alignment | Nucleotide/protein sequence | UPGMA, WPGMA, neighbour-joining, maximum likelihood, Fitch–Margoliash method, maximum parsimony, Bayesian inference | ClustalW, Clustal , MAFFT, MUSCLE, T-Coffee https://www.ebi.ac.uk/Tools/ https://www.genome.jp/tools-bin/clustalw DNAMAN https://www.lynnon.com/dnaman.html , MAFFT, MUSCLE, T-Coffee https://www.ebi.ac.uk/Tools/ https://www.genome.jp/tools-bin/clustalw DNAMAN https://www.lynnon.com/dnaman.html
|
| Structure prediction | Protein sequence | Deep neural network (NeBcon, ResPRE, ResTriplet and TripletRes), QSQE, supervised machine learning (SVM), multiple regression | SWISS-MODEL [79], PyMOL [80], I-Tasser [81], COMPOSER [78] |
| SARS-CoV-2 predictor | Nucleotide sequence | Conventional models (Naïve Bayes, K-nearest neighbors, artificial neural networks, decision tree and support vector machine), deep models CNN, Bi-path CNN (BiPathCNN) | COVID-Predictor [132] |
| Protein interactions | Protein sequence, PPI networks, protein structure | Graph analysis | Cytoscape https://apps.cytoscape.org/ |
| Chest imaging analysis | Chest x-ray or CT image | Deep learning models (VGG19), Mobile Net, Inception, Xception and Inception ResNet (v2,18,23,50), GAN, Dice similarity coefficient (DSC) | TrainingData.io https://www.trainingdata.io/ |
| Epidemic trend analysis | Experimental and observational | LSTM statistical models (SIR, Bayesian imputation, linear and polynomial regression) | Worldometers-coronavirus https://www.worldometers.info/coronavirus/ WHO-COVID19-report https://www.who.int/emergencies/diseases/novel-coronavirus-2019 COVID-19 Projections https://covid19-projections.com/ |
| Drug interaction and repurposing | Protein sequence, drug molecules, protein structure | Graph analysis, graphical convolution network | DeepDR [133], kGCN [134], DeepChem [135], D3Targets-2019-nCoV [136], CoVex [137] |
Recent trends have adopted network-embedding techniques [125] and DNN to produce lists of possible candidate drugs that will be confirmed through wet-lab experiments and clinical trials. It should be noted that, after the in silico identification, the drug repurposing process requires time and funds for the subsequent clinical trials, but the overall time required is shorter than developing a new molecule from scratch [126].
The authors in [24] have used an experimentally validated host–viral network to test 69 existing drug compounds constituting potential drug targets to treat COVID-19. Multiple network-based strategies coupled with GCNs have been explored to rank drug repurposing candidates. At first, COVID-19 interactome modules have been identified, considering 56 different human tissues. Existing drug molecules have then been ranked by means of a proximity measure based on their ability to interact with their protein targets. In [125] network proximity analyses have been performed on drug targets and HCoV–host interactions and 16 potential anti-HCoV repurposable drugs have been selected. They have used host proteins from four known HCoVs (SARS-CoV, MERS-CoV, HCoV-229E and HCoV-NL63) based on phylogeny analysis and performed functional enrichment followed by drug association analysis. Another network-based approach for deriving possible drug targets has been attempted in [10], where both protein interaction and gene co-expression networks have been used to identify master regulators [127] involved during SARS-CoV-2 infection. Physical interactions of proteins were extracted from [34]. The co-expression network has been generated by using SARS-CoV-2-human interactome proteins, derived from [128] and the largest human lung RNA-Seq dataset available from the GTEX consortium (www.gtexportal.org). The authors identified a number of key proteins involved during an infection such as ACE2, TMPRSS and MOCK. They hypothesised that these proteins may be potential therapeutics targets, evidencing that COVID-19 is characterised by a large inflammation process, not limited to the respiratory apparatus.
6.1 Discussion
As discussed in Section 6, drug repurposing is a crucial methodology for COVID-19-related therapies, since the adoption of the classical and very time-consuming drug-discovery approach [129] is unfeasible. Data science and computational intelligence are the two most useful building blocks of all the other drug repurposing approaches. Unfortunately, drug repurposing process needs, to the best of our knowledge, the introduction of up-to-date medical guidelines to become widely adopted. Despite the fact that data science will clearly help in accelerating drug repurposing, some challenges remain.
For instance, in silico drug repurposing, being based on simplified models for both humans and viruses, may not reproduce all of the possible side effects. Moreover, the vast majority of drug repurposing approaches do not consider the impact of dosage or the responses on different tissues (the original drugs could be optimised for other scenarios). Therefore, clinical tests, which are mandatory for candidate molecules, may well lead to a slowdown in the process, since short term trials may not have sufficient statistical power (e.g. due to the small number of patients). Moreover, existing drug repurposing approaches do not often consider, among other factors: (i) heterogeneous populations with different genetic backgrounds, (ii) the existence of different phenotypes (e.g. patients with a different level of illness), (iii) as well as genetic differences on the SARS-CoV-2 circulating variants. Nevertheless, some examples have produced positive results such as [130, 131].
7 Summary and challenges
As evidenced before, the potential applications for data science, deep-learning and artificial intelligence are countless in this field. However, due to the speed of the spread of the virus and the number of novel approaches proposed worldwide, it could seem that data science may fail to slow down the pandemic, hence the urgent need for a comprehensive vademecum for practitioners, industry experts, as well as researchers. In this work, we provided an in-depth overview of the data sources and methods that are currently used to elucidate the primary mechanism of pathogenesis and development of COVID-19. We included data types from the molecular scale to patient (medical imaging) and population-scale (epidemiological data) and discussed the main approaches for modelling COVID-19 infection, drug repurposing, population surveillance, disease and treatment. Table 2 provides an overall summary of data science models, types of tasks and data and various software tools. We also discussed some relevant challenges for data science applications in healthcare, including the need to introduce more standards and the need for more straightforward data integration. Finally, we firmly believe that data science can be valuable support in fighting COVID-19.
7.1 Current challenges
 Ontology-based federation of data: The current scenario is characterised by many data formats that differ both in schemas and content; there is, therefore, the need to introduce a novel data federation mechanism that is able to integrate data both horizontally and vertically.
Ontology-based federation of data: The current scenario is characterised by many data formats that differ both in schemas and content; there is, therefore, the need to introduce a novel data federation mechanism that is able to integrate data both horizontally and vertically. Development of graph-based models: The integration of data into a unified model (ideally including patients molecular and clinical information) could be a key feature in gaining more precise and effective modelling the diffusion of the virus [91, 110] and the definition of more ‘models’;
Development of graph-based models: The integration of data into a unified model (ideally including patients molecular and clinical information) could be a key feature in gaining more precise and effective modelling the diffusion of the virus [91, 110] and the definition of more ‘models’; Leveraging the use of efficient and high-throughput analysis workflows: The rapid spread of the virus and the unprecedented production of data require the introduction of novel efficient and high-throughput data analysis environments, possibly structured as virtual laboratories federated by means of cloud infrastructures [138].
Leveraging the use of efficient and high-throughput analysis workflows: The rapid spread of the virus and the unprecedented production of data require the introduction of novel efficient and high-throughput data analysis environments, possibly structured as virtual laboratories federated by means of cloud infrastructures [138]. Analysis of circulating variants and their impact: Due to the rapid mutation rate of the virus, a large number of mutations are constantly appearing for SARS-CoV-2 which may alter its protein structures. Structure-based drug development depends on the structural coherence between drug molecules and target proteins; hence the study of viral structure variations is essential for stable anti-viral drug development. By predicting strain variations with machine learning methods, domain experts will be able to design anti-viral drugs or reuse those known to be effective in similar contexts.
Analysis of circulating variants and their impact: Due to the rapid mutation rate of the virus, a large number of mutations are constantly appearing for SARS-CoV-2 which may alter its protein structures. Structure-based drug development depends on the structural coherence between drug molecules and target proteins; hence the study of viral structure variations is essential for stable anti-viral drug development. By predicting strain variations with machine learning methods, domain experts will be able to design anti-viral drugs or reuse those known to be effective in similar contexts. Low data deep models for drug discovery: The accuracy of deep-learning models depends on the availability of well-sized training datasets. Unfortunately, these large datasets are often unavailable, or unbalanced (e.g. far more positive examples than negative ones). Therefore, there is a need for generating reliable and statistically sound synthetic datasets. For instance, synthetic sample data (X-ray) is generated by using generative adversarial networks during the training phase of the model. In a recent attempt, a one-shot LSTM framework [139] has been proposed [140] for repurposed drug discovery in cases of low data availability [141]. A similar method has yet to be designed and implemented for COVID-19.
Low data deep models for drug discovery: The accuracy of deep-learning models depends on the availability of well-sized training datasets. Unfortunately, these large datasets are often unavailable, or unbalanced (e.g. far more positive examples than negative ones). Therefore, there is a need for generating reliable and statistically sound synthetic datasets. For instance, synthetic sample data (X-ray) is generated by using generative adversarial networks during the training phase of the model. In a recent attempt, a one-shot LSTM framework [139] has been proposed [140] for repurposed drug discovery in cases of low data availability [141]. A similar method has yet to be designed and implemented for COVID-19. Explainable artificial intelligence for a more reliable diagnostic systems: Diagnosis and drug discovery are two of the most sensitive tasks requiring very high predictive accuracy. Due to the phenotype similarity between COVID-19 infection and pneumonia, differentiating early symptoms of COVID-19 chest infection can be a challenging task. Explainable artificial intelligence [142] is an innovative concept enabling both researchers and domain experts to trace back the results obtained from the AI model from output features to the input features, thus allowing for a clearer interpretation of data and information. Explainable AI models may be implemented in COVID-19 image-based clinical diagnostic systems for earlier and more reliable prediction.
Explainable artificial intelligence for a more reliable diagnostic systems: Diagnosis and drug discovery are two of the most sensitive tasks requiring very high predictive accuracy. Due to the phenotype similarity between COVID-19 infection and pneumonia, differentiating early symptoms of COVID-19 chest infection can be a challenging task. Explainable artificial intelligence [142] is an innovative concept enabling both researchers and domain experts to trace back the results obtained from the AI model from output features to the input features, thus allowing for a clearer interpretation of data and information. Explainable AI models may be implemented in COVID-19 image-based clinical diagnostic systems for earlier and more reliable prediction.
Author contributions
Pietro H. Guzzi and Swarup Roy conceived and designed the study and equally shared work responsibility and guidance. Swarup Roy defined the manuscript structure. Jayanta Kumar Das and Giuseppe Tradigo equally contributed to the definition of the study and writing. Pierangelo Veltri supervised the study, and he is responsible in reviewing the final version.
Key Points
We present a comprehensive review of the in silico approaches adopted so far to handle COVID-19 in genomics, proteomics, interactomics, epidemics, clinical imaging and drug design;
We present data analytics tasks related to COVID-19;
We present an overall landscape suggesting strategies to integrate heterogeneous COVID-19 data sources;
We report data sources, models and tools, which can be used to study SARS-CoV-2 and COVID-19;
We take a look at computational biology and bioinformatics approaches available in the literature.
Acknowledgments
This work has been partially carried out at NetRA Lab, Sikkim University, with the support of Department of Science and Technology (DST), Govt. of India under DST-ICPS Data Science program [DST/ICPS/Cluster/Data Science/General]. This work has been partially supported by VQA PON from Italian Ministry of Economic Development (MISE). The authors thank Rina Mary Mazza and Konrad Arkadiusz Urbanek for suggestions on final copy editing of the manuscript.
Jayanta Kumar Das Department of Pediatrics, School of Medicine, Johns Hopkins University, Maryland, USA.
Giuseppe Tradigo eCampus University, Via Isimbardi 10, 22060 Novedrate, CO, Italy.
Pierangelo Veltri and Pietro H Guzzi Department of Surgical and Medical Sciences, Magna Graecia University, Catanzaro, 88100, Italy.
Swarup Roy Network Reconstruction & Analysis (NetRA) Lab, Department of Computer Applications, Sikkim University, Gangtok, India
Contributor Information
Jayanta Kumar Das, Department of Pediatrics, School of Medicine, Johns Hopkins University, Maryland, USA.
Giuseppe Tradigo, eCampus University, Via Isimbardi 10, 22060 Novedrate, CO, Italy.
Pierangelo Veltri, Department of Surgical and Medical Sciences, Magna Graecia University, Catanzaro, 88100, Italy.
Pietro H Guzzi, Department of Surgical and Medical Sciences, Magna Graecia University, Catanzaro, 88100, Italy.
Swarup Roy, Network Reconstruction & Analysis (NetRA) Lab, Department of Computer Applications, Sikkim University, Gangtok, India.
Funding
Pietro H. Guzzi and Pierangelo Veltri has been partially funded by PON-MISE VQA.
References
- 1. Apostolopoulos ID, Mpesiana TA. Covid-19: automatic detection from x-ray images utilizing transfer learning with convolutional neural networks. Phys Eng Sci Med 2020;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bianchi M, Benvenuto D, Giovanetti M, et al. Sars-cov-2 envelope and membrane proteins: structural differences linked to virus characteristics? Biomed Res Int 2020;2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Effenberger M, Kronbichler A, Shin JI, et al. Association of the covid-19 pandemic with internet search volumes: a Google trendstm analysis. Int J Infect Dis 2020;95:192–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hook DW, Porter SJ, Herzog C. Dimensions: building context for search and evaluation. Front Res Metr Anal 2018;3:23. [Google Scholar]
- 5. Noruzi A. Google scholar: the new generation of citation indexes. Libri 2005;55(4):170–80. [Google Scholar]
- 6. Barabási A-L, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet 2011;12(1):56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sanyaolu A, Okorie C, Marinkovic A, et al. Comorbidity and its impact on patients with covid-19. SN Compr Clin Med 2020;1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. World Health Organization, et al. Human viruses in water, wastewater and soil. In: Report of a WHO Scientific Group. Switzerland: Geneva, 1979. [PubMed] [Google Scholar]
- 9. Cannataro M, Guzzi PH, Sarica A. Data mining and life sciences applications on the grid. WIREs Data Min Knowl Discov 2013;3(3):216–38. [Google Scholar]
- 10. Guzzi PH, Mercatelli D, Ceraolo C, et al. Master regulator analysis of the sars-cov-2/human interactome. J Clin Med 2020;9(4):982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu Y, Ho W, Huang Y, et al. Sars-cov-2 is an appropriate name for the new coronavirus. Lancet 2020;395(10228):949–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Andersen KG, Rambaut A, Lipkin WI, et al. The proximal origin of sars-cov-2. Nat Med 2020;26(4):450–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Phelan AL, Katz R, Gostin LO. The novel coronavirus originating in Wuhan, China: challenges for global health governance. JAMA 2020;323(8):709–10. [DOI] [PubMed] [Google Scholar]
- 14. Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (covid-19) outbreak in China: summary of a report of 72 314 cases from the Chinese Center for Disease Control and Prevention. JAMA 2020;323(13):1239–42. [DOI] [PubMed] [Google Scholar]
- 15. Steardo L, Luca Steardo RZ, Jr, Verkhratsky A. Neuroinfection may contribute to pathophysiology and clinical manifestations of covid-19. Acta Physiol 2020;e13473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zheng Y-Y, Ma Y-T, Zhang J-Y, et al. Covid-19 and the cardiovascular system. Nat Rev Cardiol 2020;17(5):259–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Atri D, Siddiqi HK, Lang J, et al. Covid-19 for the cardiologist: a current review of the virology, clinical epidemiology, cardiac and other clinical manifestations and potential therapeutic strategies. JACC Basic Transl Sci 2020;5(5):518–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Veltri P, Cannataro M, Tradigo G. Sharing mass spectrometry data in a grid-based distributed proteomics laboratory. Inf Process Manag 2007;43(3):577–91. cited By 19. [Google Scholar]
- 19. Kim D, Lee J-Y, Yang J-S, et al. The architecture of sars-cov-2 transcriptome. Cell 2020;181(4):914–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guzzi PH, Agapito G, Cannataro M. Coresnp: parallel processing of microarray data. IEEE Trans Comput 2013;63(12):2961–74. [Google Scholar]
- 21. Shen B, Yi X, Sun Y, et al. Proteomic and metabolomic characterization of covid-19 patient sera. Cell 2020;182(1):59–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nassa G, Tarallo R, Guzzi PH, et al. Comparative analysis of nuclear estrogen receptor alpha and beta interactomes in breast cancer cells. Mol Biosyst 2011;7(3):667–76. cited By 21. [DOI] [PubMed] [Google Scholar]
- 23. Cannataro M, Guzzi PH, Veltri P. Protein-to-protein interactions: technologies, databases, and algorithms. ACM Comput Surv 2010;43(1):1–36. [Google Scholar]
- 24. Gordon DE, Jang GM, Bouhaddou M, et al. A sars-cov-2 protein interaction map reveals targets for drug repurposing. Nature 2020;583(7816):459–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guzzi PH, Roy S. Biological Network Analysis: Trends, Approaches, Graph Theory, and Algorithms. Elsevier, 2020. [Google Scholar]
- 26. De Chassey B, Navratil V, Tafforeau L, et al. Hepatitis c virus infection protein network. Mol Syst Biol 2008;4(1):230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Calderwood MA, Venkatesan K, Xing L, et al. Epstein–Barr virus and virus human protein interaction maps. Proc Natl Acad Sci 2007;104(18):7606–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Friedel CC, Haas J. Virus–host interactomes and global models of virus-infected cells. Trends Microbiol 2011;19(10):501–8. [DOI] [PubMed] [Google Scholar]
- 29. Chatr-Aryamontri A, Ceol A, Peluso D, et al. Virusmint: a viral protein interaction database. Nucleic Acids Res 2008;37(suppl_1):D669–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cook H, Doncheva N, Szklarczyk D, et al. Viruses. string: a virus–host protein–protein interaction database. Viruses 2018;10(10):519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ammari MG, Gresham CR, McCarthy FM, et al. Hpidb 2.0: a curated database for host–pathogen interactions. Database 07 2016;2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Calderone A, Licata L, Cesareni G. Virusmentha: a new resource for virus–host protein interactions. Nucleic Acids Res 2014;43(D1):D588–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Guirimand T, Delmotte S, Navratil V. Virhostnet 2.0: surfing on the web of virus/host molecular interactions data. Nucleic Acids Res 2014;43(D1):D583–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Srinivasan S, Cui H, Gao Z, et al. Structural genomics of sars-cov-2 indicates evolutionary conserved functional regions of viral proteins. Viruses 2020;12(4):360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Perfetto L, Pastrello C, Del-Toro N, et al. The imex coronavirus interactome: an evolving map of coronaviridae–host molecular interactions. BioRxiv 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhou S, Wang Y, Zhu T, et al. Ct features of coronavirus disease 2019 (covid-19) pneumonia in 62 patients in Wuhan, China. Am J Roentgenol 2020;214(6):1287–94. [DOI] [PubMed] [Google Scholar]
- 37. Dong E, Hongru D, Gardner L. An interactive web-based dashboard to track covid-19 in real time. Lancet Infect Dis 2020;20(5):533–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Italian Data . https://github.com/pcm-dpc/COVID-19.
- 39. Xu B, Moritz UG, Kraemer BG, et al. Open access epidemiological data from the covid-19 outbreak. Lancet Infect Dis 2020;20(5):534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Imming P, Sinning C, Meyer A. Drugs, their targets and the nature and number of drug targets. Nat Rev Drug Discov 2006;5(10):821–34. [DOI] [PubMed] [Google Scholar]
- 41. Réda C, Kaufmann E, Delahaye-Duriez A. Machine learning applications in drug development. Comput Struct Biotechnol J 2020;18:241–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xu B, Gutierrez B, Mekaru S, et al. Epidemiological data from the covid-19 outbreak, real-time case information. Sci Data 2020;7(1):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Voytek B. Social media, open science, and data science are inextricably linked. Neuron 2017;96(6):1219–22. [DOI] [PubMed] [Google Scholar]
- 44. Roy S, Sharma P, Nath K, et al. Pre-processing: a data preparation step. Encyclop Bioinform Comput Biol ABC Bioinform 2018;463. [Google Scholar]
- 45. Jha M, Guzzi PH, Veltri P, et al. Functional module extraction by ensembling the ensembles of selective module detectors. Int J Comput Biol Drug Design 2019;12(4):345–61. [Google Scholar]
- 46. Schmidhuber J. Deep learning in neural networks: an overview. Neural Netw 2015;61:85–117. [DOI] [PubMed] [Google Scholar]
- 47. Kipf, TN, Welling, M. Semi-supervised classification with graph convolutional networks. arXiv preprint arXiv:1609.02907, 2016.
- 48. Waheed A, Goyal M, Gupta D, et al. Covidgan: data augmentation using auxiliary classifier gan for improved covid-19 detection. IEEE Access 2020;8:91916–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Islam MZ, Islam MM, Asraf A. A combined deep CNN-LSTM network for the detection of novel coronavirus (covid-19) using x-ray images. Inform Med Unlock 2020;100412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Goodfellow I, Pouget-Abadie J, Mirza M, et al. Generative adversarial nets. Adv Neural Inf Process Syst 2014;2672–80. [Google Scholar]
- 51. Ghosal S, Sengupta S, Majumder M, et al. Linear regression analysis to predict the number of deaths in India due to sars-cov-2 at 6 weeks from day 0 (100 cases—March 14th 2020). Diab Metab Syndrome Clin Res Rev 2020;14(4):311–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Roy S, Bhattacharyya DK. An approach to find embedded clusters using density based techniques. In: International Conference on Distributed Computing and Internet Technology. Springer, 2005, 523–35. [Google Scholar]
- 53. Ceraolo C, Giorgi FM. Genomic variance of the 2019-ncov coronavirus. J Med Virol 2020;92(5):522–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gonzalez JM, Gomez-Puertas P, Cavanagh D, et al. A comparative sequence analysis to revise the current taxonomy of the family coronaviridae. Arch Virol 2003;148(11):2207–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yavarian J, Rezaei F, Shadab A, et al. Cluster of Middle East respiratory syndrome coronavirus infections in Iran, 2014. Emerg Infect Dis 2015;21(2):362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Penzes Z, González JM, Calvo E, et al. Complete genome sequence of transmissible gastroenteritis coronavirus pur46-mad clone and evolution of the purdue virus cluster. Virus Genes 2001;23(1):105–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wu C, Yang L, Yang Y, et al. Analysis of therapeutic targets for sars-cov-2 and discovery of potential drugs by computational methods. Acta Pharm Sinica B 2020;10(5):766–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhang T, Wu Q, Zhang Z. Probable pangolin origin of sars-cov-2 associated with the covid-19 outbreak. Curr Biol 2020;30(7):1346–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Waterhouse AM, Procter JB, Martin DMA, et al. Jalview version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009;25(9):1189–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Katoh K, Standley DM. Mafft multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 2013;30(4):772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Turista DDR, Islamy A, Kharisma VD, et al. Distribution of covid-19 and phylogenetic tree construction of sars-cov-2 in Indonesia. J Pure Appl Microbiol 2020;14(suppl 1):1035–42. [Google Scholar]
- 62. Yin C. Genotyping coronavirus sars-cov-2: methods and implications. Genomics 2020;112(5):3588–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gussow AB, Auslander N, Faure G, et al. Genomic determinants of pathogenicity in sars-cov-2 and other human coronaviruses. Proc Natl Acad Sci 2020;117(26):15193–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Das J, Roy S. Comparative analysis of human coronaviruses focusing on nucleotide variability and synonymous codon usage pattern. BioRxiv 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lopes LR, de Mattos Cardillo G, Paiva PB. Molecular evolution and phylogenetic analysis of sars-cov-2 and hosts ace2 protein suggest Malayan pangolin as intermediary host. Brazil J Microbiol 2020;1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Uddin M, Mustafa F, Rizvi TA, et al. Sars-cov-2/covid-19: viral genomics, epidemiology, vaccines, and therapeutic interventions. Viruses 2020;12(5):526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Li X, Song Y, Wong G, et al. Bat origin of a new human coronavirus: there and back again. Sci China Life Sci 2020;63(3):461–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tabibzadeh A, Zamani F, Laali A, et al. Sars-cov-2 molecular and phylogenetic analysis in covid-19 patients: a preliminary report from Iran. Infect Genet Evol 2020;104387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Forster P, Forster L, Renfrew C, et al. Phylogenetic network analysis of sars-cov-2 genomes. Proc Natl Acad Sci 2020;117(17):9241–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Islam MR, Hoque MN, Rahman MS, et al. Genome-wide analysis of sars-cov-2 virus strains circulating worldwide implicates heterogeneity. Sci Rep 2020;10(1): 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tiwari M, Mishra D. Investigating the genomic landscape of novel coronavirus (2019-ncov) to identify non-synonymous mutations for use in diagnosis and drug design. J Clin Virol 2020;104441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Randhawa GS, Soltysiak MPM, Roz HE, et al. Machine learning using intrinsic genomic signatures for rapid classification of novel pathogens: Covid-19 case study. PLoS One 2020;15(4):e0232391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Fang Z, Tan J, Wu S, et al. Ppr-meta: a tool for identifying phages and plasmids from metagenomic fragments using deep learning. GigaScience 2019;8(6) giz066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Huang Y, Yang C, Xu X-f, et al. Structural and functional properties of sars-cov-2 spike protein: potential antivirus drug development for covid-19. Acta Pharmacol Sin 2020;1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Liu Z, Xiao X, Wei X, et al. Composition and divergence of coronavirus spike proteins and host ace2 receptors predict potential intermediate hosts of sars-cov-2. J Med Virol 2020;92(6):595–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Senior AW, Evans R, Jumper J, et al. Improved protein structure prediction using potentials from deep learning. Nature 2020;577(7792):706–10. [DOI] [PubMed] [Google Scholar]
- 77. Yang J, Anishchenko I, Park H, et al. Improved protein structure prediction using predicted interresidue orientations. Proc Natl Acad Sci 2020;117(3):1496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sutcliffe MJ, Haneef I, Carney D, et al. Knowledge based modelling of homologous proteins, part i: three-dimensional frameworks derived from the simultaneous superposition of multiple structures. Protein Eng Design Select 1987;1(5):377–84. [DOI] [PubMed] [Google Scholar]
- 79. Waterhouse A, Bertoni M, Bienert S, et al. Swiss-model: homology modelling of protein structures and complexes. Nucleic Acids Res 2018;46(W1):W296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Janson G, Zhang C, Prado MG, et al. Pymod 2.0: improvements in protein sequence-structure analysis and homology modeling within pymol. Bioinformatics 2017;33(3):444–6. [DOI] [PubMed] [Google Scholar]
- 81. Yang J, Yan R, Roy A, et al. The i-tasser suite: protein structure and function prediction. Nat Methods 2015;12(1):7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Walls AC, Park Y-J, Tortorici MA, et al. Structure, function, and antigenicity of the sars-cov-2 spike glycoprotein. Cell 2020;181(2):281–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Romero-López JP, Carnalla-Cortés M, Pacheco-Olvera DL, et al. Arturo Reyes-Sandoval, et al. Prediction of sars-cov2 spike protein epitopes reveals HLA-associated susceptibility. Researchsquare 2020. [Google Scholar]
- 84. Cavalli E, Petralia MC, Basile MS, et al. Transcriptomic analysis of covid-19 lungs and bronchoalveolar lavage fluid samples reveals predominant b cell activation responses to infection. Int J Mol Med 2020;46(4):1266–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zhou Y, Zhou B, Pache L, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 2019;10(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Aran D, Hu Z, Butte AJ. xcell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 2017;18(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Gardinassi LG, Souza COS, Sales-Campos H, et al. Immune and metabolic signatures of covid-19 revealed by transcriptomics data reuse. Front Immunol 2020;11:1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Loganathan T, Ramachandran S, Shankaran P, et al. Host transcriptome-guided drug repurposing for covid-19 treatment: a meta-analysis based approach. PeerJ 2020;8:e9357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kushwaha SK, Kesarwani V, Choudhury S, et al. Sars-cov-2 transcriptome analysis and molecular cataloguing of immunodominant epitopes for multi-epitope based vaccine design. Genomics 2020;112(6):5044–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wu D, Shu T, Yang X, et al. Plasma metabolomic and lipidomic alterations associated with COVID-19. Nat Sci Rev 04 2020;7(7):1157–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Subramanian I, Verma S, Kumar S, et al. Multi-omics data integration, interpretation, and its application. Bioinform Biol Insights 2020;14:1177932219899051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ahmed Z, Mohamed K, Zeeshan S, et al. Artificial intelligence with multi-functional machine learning platform development for better healthcare and precision medicine. Database 2020;2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Acharjee A, Ament Z, West JA, et al. Integration of metabolomics, lipidomics and clinical data using a machine learning method. BMC Bioinform 2016;17(15):440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Chi X, Liu X, Wang C, et al. Humanized single domain antibodies neutralize sars-cov-2 by targeting the spike receptor binding domain. Nat Commun 2020;11(1):1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Uetz P, Dong Y, Zeretzke C, et al. Herpesviral protein networks and their interaction with the human proteome. Science 2006;311:239–42. [DOI] [PubMed] [Google Scholar]
- 96. Dyer MD, Murali TM, Sobral BW. The landscape of human proteins interacting with viruses and other pathogens. PLOS Pathog 2008;4(2) e32+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Uetz P, Dong Y-A, Zeretzke C, et al. Herpesviral protein networks and their interaction with the human proteome. Science 2006;311(5758):239–42. [DOI] [PubMed] [Google Scholar]
- 98. Vidal M, Cusick ME, Barabási A-L. Interactome networks and human disease. Cell 2011;144(6):986–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Cannataro M, Guzzi PH, Mazza T, et al. Preprocessing of mass spectrometry proteomics data on the grid. In: 18th IEEE Symposium on Computer-Based Medical Systems (CBMS’05). IEEE, 2005, 549–54. [Google Scholar]
-
100.
Guzzi PH, Cannataro M.
 -cs: an extension of the tm4 platform to manage affymetrix binary data. BMC Bioinform 2010;11(1):315. [DOI] [PMC free article] [PubMed] [Google Scholar]
-cs: an extension of the tm4 platform to manage affymetrix binary data. BMC Bioinform 2010;11(1):315. [DOI] [PMC free article] [PubMed] [Google Scholar] - 101. Cowen L, Ideker T, Raphael BJ, et al. Network propagation: a universal amplifier of genetic associations. Nat Rev Genet 2017;18:551–62. [DOI] [PubMed] [Google Scholar]
- 102. Vijayan V, Milenković T. Multiple network alignment via multimagna++. IEEE/ACM Trans Comput Biol Bioinform 2017;15(5):1669–82. [DOI] [PubMed] [Google Scholar]
- 103. Carrington, PJ, Scott J, Wasserman, S. Models and Methods in Social Network Analysis, vol. 28. Cambridge University Press, 2005. [Google Scholar]
- 104. Crawford J, Milenković T. Cluenet: clustering a temporal network based on topological similarity rather than denseness. PLoS One 2018;13(5):e0195993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Di Martino MT, Guzzi PH, Caracciolo D, et al. Integrated analysis of micrornas, transcription factors and target genes expression discloses a specific molecular architecture of hyperdiploid multiple myeloma. Oncotarget 2015;6(22):19132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Navarro C, Martínez V, Blanco A, et al. ProphTools: general prioritization tools for heterogeneous biological networks. GigaScience 2017;6(12):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Gligorijevic V, Malod-Dognin N, Przulj N. Integrative methods for analyzing big data in precision medicine. Proteomics 2016;16(5):741–58. [DOI] [PubMed] [Google Scholar]
- 108. Messina F, Giombini E, Agrati C, et al. Covid-19: viral–host interactome analyzed by network based-approach model to study pathogenesis of sars-cov-2 infection. J Transl Med 2020;18(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Das J, Chakrobarty S, Roy S. Impact analysis of sars-cov2 on signaling pathways during covid19 pathogenesis using codon usage assisted host–viral protein interactions. bioRxiv 2020. [Google Scholar]
- 110. Antonelli L, Guarracino MR, Maddalena L, et al. Integrating imaging and omics data: a review. Biomed Signal Process Control 2019;52:264–80. [Google Scholar]
- 111. Kim M, Kang J, Kim D, et al. Hi-covidnet: deep learning approach to predict inbound covid-19 patients and case study in South Korea. In: Proceedings of the 26th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, 2020, 3466–73.
- 112. Butt C, Gill J, Chun D, et al. Deep learning system to screen coronavirus disease 2019 pneumonia. Appl Intell 2020;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Fan D-P, Zhou T, Ji G-P, et al. Inf-net: automatic covid-19 lung infection segmentation from ct images. IEEE Trans Med Imaging 2020. [DOI] [PubMed] [Google Scholar]
- 114. Li Y, Dong W, Chen J, et al. Efficient and effective training of covid-19 classification networks with self-supervised dual-track learning to rank. IEEE J Biomed Health Inf 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Pan Y, Guan H, Zhou S, et al. Initial ct findings and temporal changes in patients with the novel coronavirus pneumonia (2019-ncov): a study of 63 patients in Wuhan, China. Eur Radiol 2020;1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Elaziz MA, Hosny KM, Salah A, et al. New machine learning method for image-based diagnosis of covid-19. PLoS One 2020;15(6):e0235187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Giordano G, Blanchini F, Bruno R, et al. Modelling the covid-19 epidemic and implementation of population-wide interventions in Italy. Nat Med 2020;1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Liu Y, Gayle AA, Wilder-Smith A, et al. The reproductive number of covid-19 is higher compared to sars coronavirus. J Travel Med 2020;37(2):taaa021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Mazza A, Fruci B, Guzzi P, et al. In PCOS patients the addition of low-dose spironolactone induces a more marked reduction of clinical and biochemical hyperandrogenism than metformin alone. Nutrition Metab Cardiovasc Dis 2014;24(2):132–9. cited By 21. [DOI] [PubMed] [Google Scholar]
- 120. Grasselli G, Pesenti A, Cecconi M. Critical care utilization for the covid-19 outbreak in Lombardy, Italy: early experience and forecast during an emergency response. JAMA 2020;323(16):1545–6. [DOI] [PubMed] [Google Scholar]
- 121. Dey SK, Rahman MM, Siddiqi UR, et al. Analyzing the epidemiological outbreak of covid-19: a visual exploratory data analysis approach. J Med Virol 2020;92(6):632–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Onder G, Rezza G, Brusaferro S. Case-fatality rate and characteristics of patients dying in relation to covid-19 in Italy. JAMA 2020;323(18):1775–6. [DOI] [PubMed] [Google Scholar]
- 123. Khalili M, Karamouzian M, Nasiri N, et al. Epidemiological characteristics of covid-19: a systemic review and meta-analysis. MedRxiv 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Chimmula VKR, Zhang L. Time series forecasting of covid-19 transmission in Canada using LSTM networks. Chaos Solitons Fractals 2020;109864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Zhou Y, Hou Y, Shen J, et al. Network-based drug repurposing for novel coronavirus 2019-ncov/sars-cov-2. Cell Discov 2020;6(1):1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Harrison C. Coronavirus puts drug repurposing on the fast track. Nat Biotechnol 2020;38(4):379–81. [DOI] [PubMed] [Google Scholar]
- 127. Lim WK, Lyashenko E, Califano A. Master regulators used as breast cancer metastasis classifier. In: Biocomputing 2009. World Scientific, 2009, 504–15. [PMC free article] [PubMed] [Google Scholar]
- 128. Li Y, Wan Y, Liu P, et al. A humanized neutralizing antibody against mers-cov targeting the receptor-binding domain of the spike protein. Cell Res 2015;25(11): 1237–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Zhou Y, Wang F, Tang J, et al. Artificial intelligence in covid-19 drug repurposing. Lancet Digit Health 2020;2(12):e667–e76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Zeng X, Song X, Ma T, et al. Repurpose open data to discover therapeutics for covid-19 using deep learning. J Proteome Res 2020;19(11):4624–36. [DOI] [PubMed] [Google Scholar]
- 131. Richardson P, Griffin I, Tucker C, et al. Baricitinib as potential treatment for 2019-ncov acute respiratory disease. Lancet 2020;395(10223) e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Sarkar JP, Saha I, Seal A, et al. Covid-predictor: RNA sequence based prediction of coronavirus. Researchsquare 2020. [Google Scholar]
- 133. Zeng X, Zhu S, Liu X, et al. deepdr: a network-based deep learning approach to in silico drug repositioning. Bioinformatics 2019;35(24):5191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Kojima R, Ishida S, Ohta M, et al. kgcn: a graph-based deep learning framework for chemical structures. J Cheminform 2020;12:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Ramsundar, B, Eastman, P, Walters, P, Pande, V. Deep Learning for the Life Sciences: Applying Deep Learning to Genomics, Microscopy, Drug Discovery, and More. O’Reilly Media, Inc., 2019. [Google Scholar]
- 136. Shi Y, Zhang X, Kaijie M, et al. D3targets-2019-ncov: a webserver for predicting drug targets and for multi-target and multi-site based virtual screening against covid-19. Acta Pharm Sinica B 2020;10(7):1239–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Sadegh S, Matschinske J, Blumenthal DB, et al. Exploring the sars-cov-2 virus-host-drug interactome for drug repurposing. Nat Commun 2020;11(1):3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Yang, CC, Veltri, P. Intelligent healthcare informatics in big data era. Artif Intell Med, 65(2):75 – 77, 2015. Intelligent healthcare informatics in big data era. [DOI] [PubMed] [Google Scholar]
- 139. Baskin II. Is one-shot learning a viable option in drug discovery? Exp Opin Drug Discov 2019;14(7):601–3. [DOI] [PubMed] [Google Scholar]
- 140. Altae-Tran H, Ramsundar B, Pappu AS, et al. Low data drug discovery with one-shot learning. ACS Central Sci 2017;3(4):283–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Abbasi K, Poso A, Ghasemi J, et al. Deep transferable compound representation across domains and tasks for low data drug discovery. J Chem Inf Model 2019;59(11):4528–39. [DOI] [PubMed] [Google Scholar]
- 142. Gunning D. Explainable artificial intelligence (xai). Defense Advanced Research Projects Agency (DARPA), and Web 2017;2:2. [Google Scholar]