

Abstract
Disseminated tumor cells (DTCs) are known to enter a state of dormancy that is achieved via growth arrest of DTCs and/or a form of population equilibrium state, strongly influenced by the organ microenvironment. During this time, expansion of residual disseminated cancer is paused and DTCs survive to fuel relapse, sometimes decades later. This notion has opened a new window of opportunity for intervening and preventing relapse. Here we review recent data that have further augmented the understanding of cancer dormancy and discuss how this is leading to new strategies for monitoring and targeting dormant cancer.
In recent years, the understanding of metastasis biology and cancer cell dormancy has advanced considerably. The current knowledge of how cancer dormancy proceeds stemmed from pioneering work on angiogenesis1-3, on the immunoregulation of equilibrium states and antibody signaling4-13 and on how microenvironmental and signaling mechanisms control cellular dormancy through growth-arrest programs14,15. Key findings based on this body of work proposed that dormant cells might evade anti-proliferative therapies in a passive manner16. Since then, the field has expanded considerably to reveal the importance of the tissue microenvironment in driving dormancy and the reactivation of dormant cells. Developmental cues that contribute to tissue homeostasis by controlling the quiescence of normal adult stem cells, as well as various niche cells of different organs, have also been shown to drive dormancy17-19. Dormant cancer cells were also found to regulate active signaling mechanisms of adaptation and survival after therapies20-24, which demonstrated that dormant tumor cells do not merely survive chemotherapeutic drug insults in a passive manner. These findings have strengthened the notion that dormant DTCs persist over long periods of time by co-opting conserved growth-arrest and survival mechanisms that are active during development and in adult tissues.
In this Perspective, we focus on recent, mainly in vivo and human data and discuss how they inform evolving concepts about tumor dormancy, including the active role of the microenvironment.
Discussing definitions
The cancer dormancy field has worked on the basis of three definitions that are not necessarily mutually exclusive and may be complementary. These have led to the working hypothesis that asymptomatic minimal residual disease (MRD) can be defined and explained by the following three potential scenarios: (1) angiogenic dormancy, an impaired angiogenic response that maintains tumor mass constant in size by balancing proliferation and cell death;1-3 (2) immune-mediated dormancy, in which proliferative tumor-cell populations are constantly trimmed by cytotoxic immune-cell responses that also maintain an equilibrium between cell death and proliferation;4-13 and (3) cellular dormancy, in which solitary DTCs or small cell clusters enter a prolonged growth arrest with no increase in cell death19,24-27.
Equilibrium states, as described in the angiogenic dormancy program3, have long been proposed as a defining feature of dormant tumor-cell-mass dynamics, with evidence accumulating since the 1990s. This concept and the relevant literature have been reviewed extensively elsewhere18,26,28. The immune equilibrium hypothesis suggests that immunosurveillance keeps proliferating cell populations in check during MRD through T cell–mediated killing or anti-idiotypic antibody networks that cause growth suppression in mouse lymphoma; this hypothesis has also been reviewed previously26,29. Recent studies focused more on cellular dormancy and its relationship with immune cells indicate that quiescent DTCs are in fact evading CD8+ T cell– and natural killer cell (NK cell)–mediated detection and clearance27,30. Thus, as proposed in an integrated scenario26, quiescent DTCs may evade detection by the immune system, but as they switch into proliferation, they may be maintained in equilibrium by immune cytotoxic responses, which shows how these two processes can be complementary. To our knowledge there are no available studies showing that dormant solitary DTCs that become reactivated are then kept under tumor-mass dormancy by an angiogenic switch failure, but this possibility has also been proposed26. The large body of literature discussed in this Perspective has independently discovered that dormant cancer-cell populations consist of single solitary DTCs or small DTC clusters of 10–20 cells18,23,27,31-36. These DTCs are able to enter a reversible growth-arrest or quiescence state, in support of the proposal that this may be a common feature of solitary dormant cancer cells.
Whether all the scenarios discussed above can co-exist in patients remains an open question. The tissues that are sampled for further study may also bias the identification of the mechanisms involved. For example, several dormancy studies that complement the study of human DTCs from bone marrow (BM) with mouse models seem to point to quiescence as a feature of solitary DTCs17,19,26,37-39. This process seems to be governed by microenvironmental cues, resembling the lifelong quiescence program active in dormant adult stem cells, including hematopoietic, muscle, neural and hair-follicle stem cells17,26,37,38,40,41 (Fig. 1). However, mechanisms of senescence or differentiation have also been proposed to underlie DTC dormancy18,42,43. A confounding factor is that growth-arrest programs during quiescence, senescence and differentiation exhibit substantial overlap in their regulation of the cell-cycle machinery (Fig. 1). Nevertheless, senescence is by definition irreversible, and regulators of this program, such as p16 and p53, are commonly mutated or silenced early in tumorigenesis44. Additionally, senescence leads to the clearing of senescent cells by innate immune cells45, which has so far not been documented for potentially ‘senescent’ DTCs. Notably, several studies have documented that DTCs activate variations of pluripotency programs and a certain degree of chromatin remodeling linked to enhanced epigenetic plasticity46-51 (discussed below). On the contrary, senescent cells commonly show highly repressive heterochromatin, which limits plasticity52 (Fig. 1). However, the possibility that a hybrid state of senescence programs is activated by microenvironmental cues43 or therapeutic stress53 cannot be ruled out.
Fig. 1 ∣. Differences and commonalities among normal quiescent, senescent, differentiated cells and dormant cancer cells.
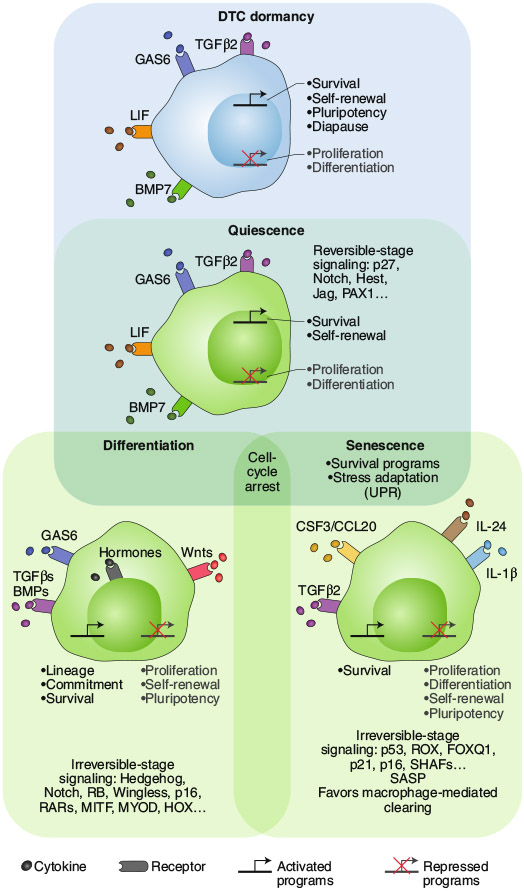
Cell-cycle arrest and survival programs are shared across all programs. Although differentiation and senescence are typically irreversible or highly stable stages, quiescence and dormancy are reversible. It would appear that normal quiescent cells and dormant cancer cells interpret the same microenvironmental factors in a similar manner, and this leads to the activation of survival and self-renewal programs. However, dormant DTCs also turn on diapause and pluripotency programs and an unfolded protein response (UPR) also observed in senescent cells. Normal quiescent, senescent and differentiated cells are green;44 the dormant cancer cell is blue26. SASP, senescence-associated secretory phenotype.
Indeed, a key attribute of dormant DTCs is their ability to retain a high degree of epigenetic and transcriptional plasticity and to reactivate different developmental programs to arrest growth and survive17,54 (discussed below). This high level of plasticity that enables cancer cells to employ various regulatory programs to induce a reversible growth arrest, which ensures persistence and confers an adaptive advantage, may be the true defining feature of dormancy. Thus, two key elements emerge as being necessary for better understanding of DTC dormancy. One is better definition of the identified programs functionally and molecularly, to understand how they resemble quiescence, senescence or differentiation and how they intersect with angiogenic dormancy and immune-mediated equilibrium. The second is definition of which of the programs identified in models are functionally relevant in patients.
Target-organ microenvironments
The tumor microenvironment consists of cellular and extracellular components that surround cancer cells. In the primary tumor site it can represent half or more of the tumor mass. However, in target organs in which solitary DTCs or small clusters of DTCs reside following dissemination, DTCs are initially a minor constituent of the tissue. Thus, how they interpret this new microenvironment and how the signals encoded’ in these initially normal tissue niches affect the induction of, maintenance of and escape from dormancy in cancer are important questions.
BM niches.
Animal and human biopsy studies support the notion that DTCs probably reach multiple organs but survive and eventually grow only in specific tissues and in a cancer-specific manner. Many studies have focused on the BM, as cancer cells can persist in this site for years without growing, and the detection of BM DTCs is commonly a marker of poor prognosis55-58. In the BM, osteoblasts have been suggested to induce solid-tumor or DTC dormancy59, with osteoclasts being involved in the escape from dormancy during the process of osteolytic bone metastases60 (Fig. 2). The role of osteoblasts in dormancy and bone metastasis, however, is more complex. Whereas the endosteal niche and osteoblasts can promote cancer-cell dormancy59, remodeling of this niche could cause the reactivation of or promote the survival of cancer cells61. For example, cancer cells depend on physical interactions with osteogenic cells through gap junctions to increase their intracellular calcium levels, and therefore the osteogenic niche, which includes osteoblasts, serves as a calcium reservoir for cancer cells to form micro-metastases62. Additionally, changes in osteoblast phenotype, such as the acquisition of an aged or senescent phenotype, could fuel metastasis63. Recently, considerable work has focused on vascular niches that induce sustained DTC dormancy in the BM23,31, brain35 and lungs34 (Fig. 2). BM vascular niches were shown to protect DTCs from chemotherapy through an integrin-mediated interaction between DTCs and molecules, including von Willebrand factor and the integrin ligand VCAM1, within the perivascular niches23. Disrupting these interactions with integrin-blocking antibodies results in a reduction in the DTC burden and the prevention of bone metastasis. Notably, although chemoprotection by the vascular niche seems to be cell cycle independent, inhibiting integrin β1 and/or integrin αvβ3 enhanced the chemotherapeutic response. How chemotherapy kills non-dividing DTCs with ablated integrin signaling is still unknown. Perhaps cancer cells become sensitized during the first divisions after their exit from the single-cell state. In the mouse brain, DTCs breach the blood–brain barrier and adhere to brain capillaries35. The position and morphology of DTCs is reminiscent of that of pericytes, as these cells spread on brain capillaries between pericytes and capillary surfaces, migrated along the vessels and remained dormant for long periods34. Although the mechanism of dormancy was not explored in that study34, the adhesion molecule L1CAM on DTCs was found to mediate access to the perivascular niche and drive transcriptional programs that awakened dormant DTCs, which led to the initiation of metastasis. Whether the reactivation of DTCs was caused by their pericyte-like behavior or changes in the niche itself is unclear. However, changes in the niche, as elicited by tip endothelial cells, are known to cause reactivation, whereas stalk endothelial cells promote dormancy64. Similarly, endothelial-cell expression of L1CAM ligands allows L1CAM+ DTCs to engage in proliferative programs, but it is possible that if the endothelium or other perivascular cells do not express such ligands, L1CAM+ DTCs may not be able to overcome dormancy signals. Therefore, the perivascular niche could induce or maintain DTC dormancy in a context-dependent manner.
Fig. 2 ∣. The various BM niches, cell types and cues that regulate the dormancy of DTCs and HSCs.

Both the osteoblastic niche and the perivascular niche in the BM are involved in DTC dormancy by producing a wide range of cues (cytokines, microRNA, extracellular vesicles, cell-cell contact signaling, etc.) that drive dormancy. Additionally, dormant cancer cells downregulate antigen presentation and upregulate immunosuppressive ligands in order to evade recognition by the immune system. Over time and reciprocally, cancer cells also remodel their surrounding microenvironment and hypothetically feed a proliferative positive loop. EVs, extracellular vesicles; ECs, endothelial cells; PD-1, receptor for PD-L1; MHCI, MHC class I; vWF, von Willebrand factor; MSC, mesenchymal stem cell.
Elegant conditional, tissue-specific knockout mouse models and bone-transplantation approaches revealed that perivascular Prx1+ mesenchymal stem cells expressing the chemokine CXCL12 maintained leukemic stem cells (LSCs) in a quiescent and treatment-resistant state in the BM65. Targeted deletion of CXCL12 in mesenchymal stem cells reduced the number of normal hematopoietic stem cell (HSCs) but promoted LSC expansion by increasing self-renewing cell divisions, with LSCs being eliminated by treatment with a tyrosine kinase inhibitor. Moreover, chronic myeloid leukemia (CML) cells induced a reduction in CXCL12-expressing mesenchymal stem cells and an increase in CXCL12-expressing endothelial cells, alterations that may provide a competitive advantage to LSCs over HSCs (Fig. 3). This work highlights how the perivascular niche may allow targeting of DTC–niche interactions in solid and hematopoietic cancer-cell dormancy to interfere with dormant DTCs that cause recurrence.
Fig. 3 ∣. Cues, receptors and cell types involved in DTC reactivation and pro-dormancy niches.

In the BM, a pro-dormancy niche involves the action of several factors (such as LIF, TGFβ2, GAS6, BMPs; additional details, Table 1) that leads to the induction of quiescence in both DTCs and HSCs. In contrast, reactivation niches in the lung and liver awaken dormant DTCs in a manner that involves formation of NETs, extracellular matrix stiffness, TGFβ1 and inhibitors of BMP molecules. NE, neutrophil elastase.
Open questions are whether DTCs move stochastically or are actively recruited into a perivascular niche, where they enter dormancy, or whether these are simply the niches that promote survival and quiescence. A recent study66 showed that similar to benign and malignant hematopoietic cells67, breast cancer DTCs enter the BM through sinusoidal vasculature that expresses the inflammatory molecules E-selectin and SDF-1, both in mice and in human samples66 (Fig. 3). Mice treated with an E-selectin inhibitor had fewer DTCs in the BM, indicative of decreased homing. In another model, E-selectin was identified as a pro-metastatic receptor of the bone vascular niche, with its pharmacological inhibition increasing survival and decreasing bone metastasis–associated bone degradation68. Thus, rather than suggesting homing, this study suggested a growth-promoting effect for E-selectin that was dependent on the Wnt signaling pathway and was bone specific. The differences between the specific findings of these two studies66,68 are unclear but may depend on differences in the models of BM colonization used and in the timing and type of endpoints assessed.
BM colonization and metastasis are common late relapse occurrences in ER+ breast cancer regulated by hormones (and thus the microenvironment). A recent study identified the kinase MSK1 as a potential regulator of dormancy in ER+ breast cancer69. Depletion of MSK1 was shown to lead to the epigenetic downregulation of genes encoding molecules that control luminal cell fate, such as FOXA1 and GATA3, and contribute to an increased metastasis-initiating potential. This effect was also observed when signaling via the kinase p38 was chemically inhibited, as MSK1 is a target of p38;69 this reproduced the role of this pathway in dormancy14. Anti-estrogen therapy is an effective therapy in human breast cancer70,71 but does not eradicate the disease in many patients71. Whether tamoxifen activates an MSK1–p38α (or other) signaling mechanism for dormancy is not clear, but understanding whether anti-estrogen therapies work through the induction of dormancy may be illuminating. Interestingly, dormant cell populations were recently shown to be prevalent during targeting of the estrogen receptor ERα71. A similar question was explored in humans72 through the sequential analysis of samples from patients undergoing extended neoadjuvant hormonal treatment. ‘Dormant’ tumors (i.e., tumors from patients with tumor-size reduction and no progression after long post-treatment periods of time) displayed enhanced DNA methylation and a more repressive chromatin state, whereas resistant tumors displayed the opposite profile. Although these data were not directly derived from DTCs, they may provide insights into the biomarkers to look for in DTCs or stroma in the adjuvant setting and to determine if anti-estrogens induce dormancy of residual DTCs. The BM is also a common site for dormancy for prostate cancer cells73. Wnt5a delivered systemically in in a prostate cancer model of metastasis induced a dormancy-like phenotype in cancer cells and a reduction in bone metastasis, potentially via ROR2 signaling72. This treatment also revealed that Wnt5a favored chemoresistance to docetaxel. Additional analyses may be needed to reveal whether these mechanisms are active in prostate cancer DTCs in the BM. Interestingly, Wnt5a has also been linked to a senescence- or dormancy-like phenotype in melanoma74.
Inflammation and extracellular matrix remodeling.
Given that dormancy is a reversible state regulated by the microenvironment, it is necessary to determine the microenvironmental changes that could trigger awakening from dormancy (Fig. 2). Bacterial lipopolysaccharide– or tobacco smoke–induced lung inflammation was recently shown to activate neutrophils to form neutrophil extracellular DNA traps (NETs)32. NETs were found to trigger the awakening of dormant DTCs through the release of neutrophil elastase and MMP9, enzymes that promote the laminin 111– and α3β1 integrin–dependent activation of a signaling axis involving the kinases FAK, ERK and MLCK and, ultimately, activation of the transcriptional regulator YAP Interestingly, for full awakening of DTCs, loss of perivascular thrombospondin TSP1 was also required. Metastatic breast cancer cells can themselves induce NETs32,75, which raises the possibility that they may create a positive pro-inflammatory proliferative loop. Surgical stress was also shown to trigger DTC awakening through an increase in NET formation, in a mouse model of liver ischemia–reperfusion76. Consistent with that, in a cohort of patients undergoing attempted curative liver resection for metastatic colorectal cancer, a correlation was observed between increased post-operative NET formation and a reduction in disease-free survival76. In a model of pancreatic ductual adenocarcinoma (PDAC), the TRAIL-R2 pathway was also suggested to lead to liver inflammation and the awakening of dormant DTCs during tumor resection77. Thus, it is possible that tissues that remain homeostatic may maintain DTC dormancy, whereas changes (such as inflammation) that alter the homeostatic balance (for example, through NETs and remodeling of the extracellular matrix) may trigger the awakening of DTCs from dormancy.
An important question is the link between aging and metastatic relapse and the age- and microenvironment-related signals that may regulate DTC dormancy and reactivation. Recently, in a mouse model of aging, metastatic cell lines were shown to be highly proliferative in aged BM, with the frequency of dormant DTCs decreasing in aged mice but not in young mice78. Mechanistic analysis revealed that several inflammatory cytokines known to promote cell proliferation (including the interleukins IL-1β, IL-6, IL-27 and IL-1F9 and factors such as CCL4, CCL5 and Tnfsf14) were upregulated in bones of aged mice, whereas proposed quiescence-inducing factors (including BMP4, BMP6, BMP7, Kitl, TGFβ2, Thbs2, Dkk1 and Dkk3) were downregulated78 (Table 1). Remodeling of the extracellular matrix may also stimulate metastasis and perhaps awakening from dormancy, as revealed by changes in age-related remodeling of the collagen matrix that stimulated metastasis in melanoma79-81.
Table 1 ∣.
Summary of dormancy- and reactivation-inducing Factor
| Factor | In vitro data |
In vivo data |
Human data |
Refs. | |
|---|---|---|---|---|---|
| Dormancy inducers | CXCL12 | Y | Y | Y | 65 |
| vWF | Y | Y | N | 23 | |
| TGFβ2 | Y | Y | N | 36 | |
| BMP4, BMP7 | Y | Y | N | 43,48 | |
| Hypoxia | Y | Y | Y | 33 | |
| GAS6 | Y | Y | N | 115,116 | |
| RA | Y | Y | N | 46 | |
| LIF | Y | Y | N | 97 | |
| TSP1 | Y | Y | N | 31 | |
| WNTs | Y | Y | N | 27,117 | |
| DNA methylation repressive chromatin state | Y | Y | Y | 46,72 | |
| Wnt5a | Y | Y | N | 72 | |
| Axolotl embryo | Y | N | N | 86 | |
| miR-126 | Y | Y | Y | 104 | |
| Jagged 1 | Y | Y | N | 118 | |
| Reactivation inducers | COCO | Y | Y | Y | 48 |
| Inflammation and NETs | Y | Y | Y | 32,75,76 | |
| Stiff collagen | Y | Y | N | 79 | |
| Aging | Y | Y | Y | 78,79,119 | |
| VCAM1 | Y | Y | N | 60 | |
| Periostin | Y | Y | N | 31 | |
| TGFβ1 | Y | Y | N | 36 |
Known cancer cell- and microenvironment-derived regulators that promote the dormancy or reactivation of dormant cancer cells in solid and liquid cancers. The type of data available for each factor is also indicated. These findings are compiled from different cancer types and thus these signaling pathways might not all be associated with the dormancy of every type of cancer or microenvironment. Y, yes; N, no.
The concept of reverting a malignant phenotype by normalizing the microenvironment is not new82-85 and supports the proposal that — in the appropriate context — cancer cells could remodel their epigenetic programs to silence mutated genomes. Axolotl embryos have been shown to fully reprogram malignant breast cancer cells into quiescence by inhibiting induction of the cell-cycle inhibitor p27 mediated by the kinases ERK1 and ERK2 and reducing signaling dependent on the transcription factor JUN and the tumor suppressor Rb86. These reprogrammed breast cancer cells also displayed a repressive chromatin state that was previously reported in retinoic acid–induced dormancy46. Although the mechanisms activated by the axolotl extract were not identified, this reveals how microenvironmental signals can epigenetically silence malignancy traits.
Such studies have expanded understanding of how the homeostasis of target-organ microenvironments may maintain dormancy, probably governed by adult stem-cell quiescence niches. They also support the proposal that alterations over time (in this case the organism’s age) or perturbations that alter target-organ microenvironments, such as inflammation and remodeling of the extracellular matrix, can control dormancy. A key question is how robust the dormancy-inducing microenvironments are. Given that, for example, in breast cancer, relapse that occurs more than 5 years after surgery is mostly stochastic and can be delayed for long periods, it is possible that multiple homeostatic barriers need to be eliminated for DTCs to awaken. Although patients will encounter situations that may activate inflammatory processes many times during their lives, many do not show immediate relapse.
Autophagy and metabolism.
Another important but poorly explored mechanism in dormancy biology is metabolic plasticity. For example, unfolded-protein-response pathways and signaling via the transcription factor HIF1α, which are intimately linked to metabolic changes, can enable residual cancer cells to persist and evade chemotherapy87. Recently, the glycerol biosynthesis enzyme GPD1 (glycerol-3-phosphate dehydrogenase 1) was shown to mark slow-cycling brain tumor stem cells with the ability to repopulate the tumor after temozolomide therapy in glioblastoma mouse models88. These GPD1+ cells were present in tumor margins, which suggested that they might escape resection and therapy. However, whereas GPD1+ cells were mostly negative for proliferation markers, they were the cells able to repopulate the lesions after therapy. This suggests that GPD1 is involved in the metabolic plasticity of slowly cycling cells that mediates reactivation but does not regulate long-term dormancy.
Autophagy is intimately linked to stress signaling and metabolic changes, as it is not only a source of metabolites for energy production but also a way to protect from proteotoxicity. Dormant cancer cells were shown to be more autophagic than their proliferating counterparts, which allowed them to survive in a quiescent state89. Notably, inhibiting autophagy specifically blocked the survival of dormant cells in the lungs in a manner dependent on the autophagy-related molecule ATG789. A separate study recently showed that the enzyme Pfkfb3 (6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3) is linked to metastatic relapse of breast cancer90. Interestingly, Pfkfb3 was downregulated by the onset of autophagy, which coincided with dormancy induction. Unlike an earlier study89, but consistent with other work reporting that autophagy may induce dormancy18,91, this paper90 showed that inhibition of autophagy restored PFKFB3 expression, which led to reactivation from dormancy. When autophagy functions only as a survival pathway and when it coordinates induction and maintenance of dormancy remains unclear. Elucidating this point is important because the second possibility supports the proposal that targeting autophagy could have deleterious effects for patients by causing reactivation from dormancy.
The immune system and DTC dormancy.
Major advances in immuno-oncology have fundamentally altered the understanding of the relationship between the adaptive immune system and cancer cells, which has led to the current breakthroughs in immunotherapies. A key question is whether dormant DTCs are restrained from expanding by an immune equilibrium mechanism and/or whether dormant DTCs evade recognition by the immune system (Fig. 2). Pioneering studies of carcinogenesis and disseminated cancer have provided support for the possibility of a role for immune equilibrium as a mechanism of dormancy13,92. Similarly, studies of a B cell lymphoma that establishes dormancy through a vaccination protocol revealed important components of how dormancy could be induced and maintained by CD8+ T cells and interferon-γ signaling6. Additionally, immunoevasion was shown to allow the persistence of dormant leukemic cells in mouse acute myeloid leukemia (AML) through upregulation of the checkpoint inhibitor B7-H1 (PD-L1) and the immunomodulatory receptor CTLA-4 ligand B7.1, but without involving a decrease in major histocompatibility complex (MHC) class I (ref. 93). More recently, it was shown that mice vaccinated against PDAC cells were protected against rechallenge with the same PDAC cells injected to colonize the mouse livers, as expected30. However, although the immune system eradicated most cancer cells, persistent single DTCs were detected. The immunoeva-sive DTCs were quiescent, did not express E-cadherin and upregulated the unfolded protein response, a pathway previously linked also to the drug resistance of dormant cancer cells22. The unfolded protein response was found to downregulate MHC class I, which reduced antigen presentation and rendered dormant DTCs invisible to CD8+ T cells. Although the molecular mechanisms differ, both leukemic cells and epithelial cancer cells that enter dormancy seem to find ways to escape detection by the immune system. As proposed earlier, immunoevasion by dormant DTCs may simply be reflective of the hijacking of developmental or tissue homeostasis mechanisms. For example, quiescent hair-follicle stem cells were shown to evade immune recognition by CD8+ T cells94. It would be interesting to test whether the signals that induce dormancy discussed in this Perspective (Table 1) modulate the expression of key immunoregulatory molecules such as MHC class I, PD-L1 and/or CTLA-4 and thereby influence immunodetection. Dormant lung cancer DTCs and HER2+ breast cancer DTCs were also shown to evade detection by NK cells and to be susceptible to eradication by NK cells only after escape from dormancy27. Although these studies support the proposal that DTCs are NK cell evasive during quiescence, they provide only partial insight into their immunoregulation, as they were performed in nude mice. Unlike the results reported in those papers, the immune system of mice carrying the EMT6 syngeneic triple-negative breast cancer model was shown to be able to fully eradicate dormant and proliferative DTCs, suggestive of their vulnerability to immunorecognition95. In contrast, a separate study in immunocompetent mice showed that D2A1 breast cancer cells were kept from forming metastases by immune-mediated killing, which led to an equilibrium that produced population dormancy96. However, when mice underwent sham surgery, the injury-associated inflammation and myeloid-cell mobilization led to immunosuppression, with the cancer-cell population progressing to metastasis. Studies in which no exogenous antigens were used95 may be relevant to understanding how patients with triple-negative breast cancer who survive beyond 5–8 years after surgery never relapse with metastasis. It would also be interesting to determine whether primary tumor removal, as is done routinely in patients, would yield the same results as sham surgery in mice96. Studies such as these, which formally investigated DTC biology and recognition by the immune system, raise new questions related to DTC dormancy and immunotherapies.
Commonalities of leukemia and DTC dormancy
The HSC and leukemia fields have made considerable advances in delineating how specific niches control the dormancy of HSCs and LSCs. At steady state, the BM niche is a tightly controlled microenvironment that regulates the proliferation, self-renewal, differentiation and migration of HSCs37. HSCs and DTCs in the BM seem to be cell-cycle arrested. Moreover, these two types of cells have been proposed to occupy the same BM niches and to respond to the microenvironmental cues similarly17,73 (Fig. 3). Both dormant HSCs and DTCs seem to adopt quiescence and survival mechanisms that enable them to avoid apoptosis and/or resist therapies. However, dormant solid-cancer cells share characteristics with embryonic and adult stem cells, such as upregulation of the pluripotency-related genes NR2F1, SOX9, SOX2, POU5F1 (which encodes OCT4) and NANOG17,27,46,48,97. This may be a distinction between the quiescence programs in HSCs and those in DTCs, whereby the latter cells may adopt developmental programs that provide additional cell plasticity and less ability to commit to differentiation states. However, other similarities are clear. For example, adult stem-cell niches have also been shown to be immune-protected sites94, which may be advantageous for DTCs. Adult stem cells and quiescent DTCs may also show downregulation of antigen presentation30,94, in support of the proposal that escaping immunosurveillance may also be a common attribute. Another example involves the ubiquitin ligase FBXW7, inhibition of which was shown to prevent the quiescence of LSCs98 and to be involved in the quiescence of lung adenocarcinoma cells99 and the awakening and subsequent chemotherapy-mediated killing of breast cancer DTCs100. These data suggest that some quiescence and/or dormancy mechanisms may be shared by LSCs and DTCs and thus provide novel potential anti-cancer targets for experimental exploration. Notably, the notion of awakening LSCs might work in some cases, given the unique oncogene addiction pathways of these cells65, but in solid cancers, awakening dormant cells may be too risky, given that chemotherapy is not uniformly effective and responses may vary depending on the organ in which metastases grow.
Hematopoietic malignancies may also inform cancer-cell heterogeneity during dormancy. Longitudinal BM sampling from patients to delineate clonal evolution from pre-leukemia to established AML has revealed that some rare, probably dormant, leukemic cell sub-clones were already present at diagnosis and were resistant to therapy, which led to relapse; this highlights the importance of understanding the dynamics of LSC heterogeneity101. Similar studies of solid-cancer DTCs, such as DTCs of melanoma, breast cancer and prostate cancer in patients at the M0 stage (no evidence of metastasis at diagnosis or surgery), were able to identify dormant DTCs that could be studied and targeted early on. A single-cell transcriptomic analysis of BM leukemic cells revealed that a specific sub-fraction among the heterogeneous population of LSCs, characterized by stem-cell and quiescence signatures, survived in the BM environment of patients with asymptomatic CML under treatment with a tyrosine kinase inhibitor and served as a reservoir for the emergence of resistant cells102. Treatment-induced quiescence of residual LSCs in this setting was demonstrated to rely on the activation of Jak2–Stat3 signaling dependent on non-canonical BMP4 signaling, with the ligand delivered by surrounding mesenchymal cells103. Dual targeting of BMP4 and Jak2 was efficient in reversing this tyrosine kinase inhibitor–dependent induced quiescence of the BMPR1B+ LSC sub-fraction adherent to stroma and allowed these cells to re-enter a differentiation process103. Such a strategy might contribute to elimination of the reservoir of dormant LSCs.
Another study of treatment-induced quiescence in CML showed that the microRNA miR-126 from endothelial or leukemic cells induced the quiescence of CML LSCs104. Interestingly, the tyrosine kinase BCR–ABL is known to inhibit the biogenesis of miR-126, and inhibition of BCR-ABL restored miR-126 expression in the cancer cells, which led to a quiescent LSC pool with enhanced survival. Moreover, combining inhibition of BCR-ABL and downregulation of miR-126 resulted in the elimination of LSCs, in support of the proposal that this microRNA may also coordinate survival during quiescence. Assessing if dormant DTCs from solid cancers are regulated by similar mechanisms when targeted therapies are used (for example, anti-estrogens, anti-androgen receptor, anti-HER2 or other kinases) could reveal novel cancer cell–intrinsic and/or microenvironmentally induced mechanisms of DTC quiescence and survival triggered by therapeutic interventions.
There is growing evidence not only that the niche influences cancer cells but also that leukemic cells can modulate their host BM microenvironment to survive and expand. For example, AML cells can remodel the vascular organization of the endosteal niche and thereby alter the number of stromal cells105 and render the niche less supportive of normal HSCs and their quiescence. Additionally, vascular permeability can be altered through the enhanced production of nitric oxide by endothelial cells106. Additional studies have highlighted the role of AML-derived extracellular vesicles in suppressing normal hematopoiesis by inhibiting protein synthesis107 through the internalization of miR-1246 in normal HSCs or by inducing the expression of DKK1, a suppressor of normal hematopoiesis108. In both studies, inhibiting this niche remodeling led to enhanced efficiency of chemotherapy107,108. These models may provide insight into how solid-cancer DTCs may change the niches in which they reside over time until they switch from a homeostatic pro-dormancy function to a reactivation state. Given that the molecules derived from these niches may be more abundant than the DTCs that lodged there, these soluble factors could serve as biomarkers that provide information on niches supportive of reactivation.
Future prospects
The treatment of metastasis has been limited to very few adjuvant therapies that rely on primary tumor data and aggressive targeting of metastatic lesions in patients at stage IV. The latter is a necessity, as these patients have few treatment options. However, the knowledge of tumor dormancy has revealed that MRD in solid cancers could be targeted earlier, even if DTCs are not actively proliferating109. Additionally, the notion that dormant DTCs co-exist with growing lesions in stage IV cancer has raised the possibility that these dormant DTCs may evade anti-proliferative treatments. Thus, strategies that combine anti-proliferative therapies with drugs that kill dormant cells may provide long-term benefit even in patients at stage IV.
However, the concepts noted above need to be tested in practice. Acceleration of the understanding of the processes that underlie cancer-cell dormancy and, consequently, ways to target it will require pairing of experimental models with human data, such as by having research scientists team up with clinicians to incorporate the measurement of endpoints related to dormancy in existing clinical trials. Such efforts are emerging; for example, NR2F1, a dormancy marker identified in experimental systems, has been used to stratify DTCs and to determine that patients with NR2F1hi DTCs had longer bone metastasis–free periods than those with NR2F1lo DTCs57. Notably, a proliferation marker such as Ki67 could not provide the same information, which supports the proposal that dormancy markers may provide additional insights beyond those provided by determining the snapshot proliferative status of DTCs. Such efforts, even if initiated in small populations of patients, would be key for the validation of experimental biology in patients.
The notion that dormant cancer cells activate autophagy18,89,90,110 has also led to a phase 2 clinical trial using autophagy inhibitors such as hydroxychloroquine and/or mTOR inhibitors in combination to treat patients with breast cancer and to prevent recurrence (clinical trial identifier NCT03032406). An additional clinical trial is focused on screening for the presence of BM DTCs in patients within 5 years of having completed breast cancer therapy (clinical trial identifier NCT02732171). Patients with BM DTCs will be offered the possibility of participating in other trials targeting DTCs, such as NCT03032406.
The concept of reprogramming malignant DTCs into dormancy46 has also led to a clinical trial testing this concept in disseminated prostate cancer that has not yet become detectable by imaging (clinical trial identifier NCT03572387). This clinical trial repurposed 5-azacytidine and all-trans retinoic acid (both drugs approved by the US Food and Drug Administration) to treat patients with prostate cancer after hormonal ablation. Although these studies are ongoing, they support the notion that foundational science can be used to develop innovative trials that can be executed despite potential cost issues24. Understanding how dormancy develops in patients may also help guide trials that were supposed to target micro-metastatic disease but failed to show therapeutic value. Preclinical data have identified RANKL (TNFSF11) as a tempting target with strong potential to prevent breast cancer bone metastasis111,112. A phase 3 study (D-CARE) was developed that combined a human blocking monoclonal antibody against the receptor for RANKL (denosumab) with standard-of-care adjuvant or neoadjuvant systemic therapy and locoregional treatments. However, this treatment did not improve disease-related outcomes for women with high-risk early breast cancer113. The reasons for this outcome may include the possibility that dosing schedules left a window for dormant DTC populations to become reactivated. There is also evidence from other studies that chemotherapy causes severe alterations of the BM microenvironment114 with clear signs of premature aging that could actually promote the reactivation of dormant DTCs. Unfortunately, the trial did not include intermediate endpoints for assessing the numbers or phenotypes of DTCs or circulating tumor cells and thus for anticipating what might have been happening to the residual disease. The development of markers and methods for detecting the DTCs that lead to MRD in such trials will enhance the understanding of how to target these residual cells on the basis of their unique biology.
Progress in on cancer dormancy research has expanded considerably and will probably yield innovative biomedical results in the future. The findings discussed in this Perspective highlight the fact that the time is ripe to start translating the knowledge gained from experimental models to the clinic.
Acknowledgements
We thank the Aguirre-Ghiso and Maguer-Satta labs for useful discussions during the preparation of this article. E.R. is supported by a doctoral fellowship from the University of Lyon, France, and by CLARA and IDEX Lyon mobility fellowships. E.R., A.R.N. and J.A.A.-G. are supported by grants CA109182, CA218024, CA216248 and CA196521 from the US National Institutes of Health; the Jimmy V Foundation; the Falk Medical Research Trust; HiberCell; and Metavivor. J.A.A.-G. is a Samuel Waxman Cancer Research Foundation Investigator. V.M.S. is supported by grants from ‘Fondation de France’ 2014-0047501 and 2017-00076282/Fondation Ramona Ehrman Amador, ‘Association Laurette Fugain’ ALF2014-03, Ligue contre le Cancer (Haute Savoie, Loire, Puy de Dôme and Rhone), ‘Association ALTE-SMP’ and the Institut Convergence PLASCAN.
Footnotes
Competing interests
J.A.A.-G. is a scientific co-founder of, scientific advisory board member and equity owner in HiberCell and receives financial compensation as a consultant for HiberCell, a Mount Sinai spin-off company focused on the research and development of therapeutics that prevent or delay the recurrence of cancer.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Almog N et al. Prolonged dormancy of human liposarcoma is associated with impaired tumor angiogenesis. FASEB J. 20, 947–949 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Folkman J Role of angiogenesis in tumor growth and metastasis. Semin. Oncol 29, 15–18 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Holmgren L, O’Reilly MS & Folkman J Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat. Med 1, 149–153 (1995). [DOI] [PubMed] [Google Scholar]
- 4.Uhr JW & Marches R Dormancy in a model of murine B cell lymphoma. Semin. Cancer Biol 11, 277–283 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Marches R, Hsueh R & Uhr JW Cancer dormancy and cell signaling: induction of p21waf1 initiated by membrane IgM engagement increases survival of B lymphoma cells. Proc. Natl Acad. Sci. USA 96, 8711–8715 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farrar JD et al. Cancer dormancy. VII. A regulatory role for CD8+ T cells and IFN-γ in establishing and maintaining the tumor-dormant state. J. Immunol 162, 2842–2849 (1999). [PubMed] [Google Scholar]
- 7.Marches R, Scheuermann RH & Uhr JW Cancer dormancy: role of cyclin-dependent kinase inhibitors in induction of cell cycle arrest mediated via membrane IgM. Cancer Res. 58, 691–697 (1998). [PubMed] [Google Scholar]
- 8.Vitetta ES et al. Tumor dormancy and cell signaling. V. Regrowth of the BCL1 tumor after dormancy is established. Blood 89, 4425–4436 (1997). [PubMed] [Google Scholar]
- 9.Uhr JW et al. Role of antibody signaling in inducing tumor dormancy. Adv. Exp. Med. Biol 406, 69–74 (1996). [DOI] [PubMed] [Google Scholar]
- 10.Racila E et al. Tumor dormancy and cell signaling: anti-mu-induced apoptosis in human B-lymphoma cells is not caused by an APO-1-APO-1 ligand interaction. Proc. Natl Acad. Sci. USA 93, 2165–2168 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Racila E et al. Tumor dormancy and cell signaling. II. Antibody as an agonist in inducing dormancy of a B cell lymphoma in SCID mice. J. Exp. Med 181, 1539–1550 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marches R et al. Tumour dormancy and cell signalling-III: Role of hypercrosslinking of IgM and CD40 on the induction of cell cycle arrest and apoptosis in B lymphoma cells. Ther. Immunol 2, 125–136 (1995). [PubMed] [Google Scholar]
- 13.Koebel CM et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature 450, 903–907 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Aguirre-Ghiso JA, Liu D, Mignatti A, Kovalski K & Ossowski L Urokinase receptor and fibronectin regulate the ERKMAPK to p38MAPK activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol. Biol. Cell 12, 863–879 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aguirre Ghiso JA, Kovalski K & Ossowski L Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. J. Cell Biol 147, 89–104 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Naumov GN et al. Ineffectiveness of doxorubicin treatment on solitary dormant mammary carcinoma cells or late-developing metastases. Breast Cancer Res. Treat 82, 199–206 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Aguirre-Ghiso JA & Sosa MS Emerging topics on disseminated cancer cell dormancy and the paradigm of metastasis. Ann. Rev. Cancer Biol 2, 377–393 (2018). [Google Scholar]
- 18.Sosa MS, Bragado P & Aguirre-Ghiso JA Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat. Rev. Cancer 14, 611–622 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giancotti FG Mechanisms governing metastatic dormancy and reactivation. Cell 155, 750–764 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schewe DM & Aguirre-Ghiso JA ATF6α-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc. Natl Acad. Sci. USA 105, 10519–10524 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ranganathan AC, Ojha S, Kourtidis A, Conklin DS & Aguirre-Ghiso JA Dual function of pancreatic endoplasmic reticulum kinase in tumor cell growth arrest and survival. Cancer Res. 68, 3260–3268 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ranganathan AC, Zhang L, Adam AP & Aguirre-Ghiso JA Functional coupling of p38-induced up-regulation of BiP and activation of RNA-dependent protein kinase-like endoplasmic reticulum kinase to drug resistance of dormant carcinoma cells. Cancer Res. 66, 1702–1711 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carlson P et al. Targeting the perivascular niche sensitizes disseminated tumour cells to chemotherapy. Nat. Cell Biol 21, 238–250 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghajar CM Metastasis prevention by targeting the dormant niche. Nat. Rev. Cancer 15, 238–247 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aguirre-Ghiso JA How dormant cancer persists and reawakens. Science 361, 1314–1315 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aguirre-Ghiso JA Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer 7, 834–846 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malladi S et al. Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell 165, 45–60 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aguirre-Ghiso JA The problem of cancer dormancy: understanding the basic mechanisms and identifying therapeutic opportunities. Cell Cycle 5, 1740–1743 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Linde N, Fluegen G & Aguirre-Ghiso JA The relationship between dormant cancer cells and their microenvironment. Adv. Cancer Res 132, 45–71 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pommier A et al. Unresolved endoplasmic reticulum stress engenders immune-resistant, latent pancreatic cancer metastases. Science 360, eaao4908 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ghajar CM et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol 15, 807–817 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Albrengues J et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 361, eaao4227 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fluegen G et al. Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat. Cell Biol 19, 120–132 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Er EE et al. Pericyte-like spreading by disseminated cancer cells activates YAP and MRTF for metastatic colonization. Nat. Cell Biol 20, 966–978 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Valiente M et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 156, 1002–1016 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bragado P et al. TGF-β2 dictates disseminated tumour cell fate in target organs through TGF-β-RIII and p38α/β signalling. Nat. Cell Biol 15, 1351–1361 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pinho S & Frenette PS Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol 20, 303–320 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goel AJ, Rieder MK, Arnold HH, Radice GL & Krauss RS Niche cadherins control the quiescence-to-activation transition in muscle stem cells. Cell Rep. 21, 2236–2250 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grzelak CA & Ghajar CM Metastasis ‘systems’ biology: how are macro-environmental signals transmitted into microenvironmental cues for disseminated tumor cells? Curr. Opin. Cell Biol 48, 79–86 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Horsley V, Aliprantis AO, Polak L, Glimcher LH & Fuchs E NFATc1 balances quiescence and proliferation of skin stem cells. Cell 132, 299–310 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kobielak K, Stokes N, de la Cruz J, Polak L & Fuchs E Loss of a quiescent niche but not follicle stem cells in the absence of bone morphogenetic protein signaling. Proc. Natl Acad. Sci. USA 104, 10063–10068 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sharma S et al. Secreted Protein Acidic and Rich in Cysteine (SPARC) mediates metastatic dormancy of prostate cancer in bone. J. Biol. Chem 291, 19351–19363 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kobayashi A et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J. Exp. Med 208, 2641–2655 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gorgoulis V et al. Cellular senescence: defining a path forward. Cell 179, 813–827 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Lujambio A et al. Non-cell-autonomous tumor suppression by p53. Cell 153, 449–460 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sosa MS et al. NR2F1 controls tumour cell dormancy via SOX9- and RARβ-driven quiescence programmes. Nat. Commun 6, 6170 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cackowski FC et al. Mer tyrosine kinase regulates disseminated prostate cancer cellular dormancy. J. Cell. Biochem 118, 891–902 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gao H et al. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell 150, 764–779 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Laughney AM et al. Regenerative lineages and immune-mediated pruning in lung cancer metastasis. Nat. Med 26, 259–269 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Metz EP & Rizzino A Sox2 dosage: A critical determinant in the functions of Sox2 in both normal and tumor cells. J. Cell. Physiol 234, 19298–19306 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jia Q et al. Low levels of Sox2 are required for melanoma tumorrepopulating cell dormancy. Theranostics 9, 424–435 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Duarte LF et al. Histone H3.3 and its proteolytically processed form drive a cellular senescence programme. Nat. Commun 5, 5210 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kishino E et al. Anti-cell growth and anti-cancer stem cell activity of the CDK4/6 inhibitor palbociclib in breast cancer cells. Breast Cancer 27, 415–425 (2019). [DOI] [PubMed] [Google Scholar]
- 54.Sosa MS, Bernstein E & Aguirre-Ghiso JA in Tumor Dormancy and Recurrence (eds. Wang Y & Crea F) 1–16 (Springer International Publishing, 2017). [Google Scholar]
- 55.Braun S et al. Cytokeratin-positive cells in the bone marrow and survival of patients with stage I, II, or III breast cancer. N. Engl. J. Med 342, 525–533 (2000). [DOI] [PubMed] [Google Scholar]
- 56.Chéry L et al. Characterization of single disseminated prostate cancer cells reveals tumor cell heterogeneity and identifies dormancy associated pathways. Oncotarget 5, 9939–9951 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Borgen E et al. NR2F1 stratifies dormant disseminated tumor cells in breast cancer patients. Breast Cancer Res. 20, 120 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Naume B et al. Clinical outcome with correlation to disseminated tumor cell (DTC) status after DTC-guided secondary adjuvant treatment with docetaxel in early breast cancer. J. Clin. Oncol 32, 3848–3857 (2014). [DOI] [PubMed] [Google Scholar]
- 59.Lawson MA et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat. Commun 6, 8983 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lu X et al. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging α4β1-positive osteoclast progenitors. Cancer Cell 20, 701–714 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zheng H et al. Therapeutic antibody targeting tumor- and osteoblastic niche-derived Jagged1 sensitizes bone metastasis to chemotherapy. Cancer Cell 32, 731–747.e736 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang H et al. The osteogenic niche is a calcium reservoir of bone micrometastases and confers unexpected therapeutic vulnerability. Cancer Cell 34, 823–839.e827 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Luo X et al. Stromal-initiated changes in the bone promote metastatic niche development. Cell Rep. 14, 82–92 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ghajar CM et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol 15, 807–817 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Agarwal P et al. Mesenchymal niche-specific expression of Cxcl12 controls quiescence of treatment-resistant leukemia stem cells. Cell Stem Cell 24, 769–784.e766 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Price TT et al. Dormant breast cancer micrometastases reside in specific bone marrow niches that regulate their transit to and from bone. Sci. Transl. Med 8, 340ra73 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sipkins DA et al. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature 435, 969–973 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Esposito M et al. Bone vascular niche E-selectin induces mesenchymal-epithelial transition and Wnt activation in cancer cells to promote bone metastasis. Nat. Cell Biol 21, 627–639 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gawrzak S et al. MSK1 regulates luminal cell differentiation and metastatic dormancy in ER+ breast cancer. Nat. Cell Biol 20, 211–221 (2018). [DOI] [PubMed] [Google Scholar]
- 70.Goss PE & Chambers AF Does tumour dormancy offer a therapeutic target? Nat. Rev. Cancer 10, 871–877 (2010). [DOI] [PubMed] [Google Scholar]
- 71.Hong SP et al. Single-cell transcriptomics reveals multi-step adaptations to endocrine therapy. Nat. Commun 10, 3840 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Selli C et al. Molecular changes during extended neoadjuvant letrozole treatment of breast cancer: distinguishing acquired resistance from dormant tumours. Breast Cancer Res. 21, 2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cackowski FC & Taichman RS Parallels between hematopoietic stem cell and prostate cancer disseminated tumor cell regulation. Bone 119, 82–86 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Webster MR et al. Wnt5A promotes an adaptive, senescent-like stress response, while continuing to drive invasion in melanoma cells. Pigment Cell Melanoma Res. 28, 184–195 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Park J et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci. Transl. Med 8, 361ra138 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tohme S et al. Neutrophil extracellular traps promote the development and progression of liver metastases after surgical stress. Cancer Res. 76, 1367–1380 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Miarka L et al. The hepatic microenvironment and TRAIL-R2 impact outgrowth of liver metastases in pancreatic cancer after surgical resection. Cancers 11, E745 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Singh A et al. Angiocrine signals regulate quiescence and therapy resistance in bone metastasis. JCI Insight 4, 125679 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kaur A et al. Remodeling of the collagen matrix in aging skin promotes melanoma metastasis and affects immune cell motility. Cancer Discov. 9, 64–81 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fane M & Weeraratna AT How the ageing microenvironment influences tumour progression. Nat. Rev. Cancer 20, 89–106 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ecker BL et al. Age-related changes in HAPLN1 increase lymphatic permeability and affect routes of melanoma metastasis. Cancer Discov. 9, 82–95 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Boudreau N & Bissell MJ Extracellular matrix signaling: integration of form and function in normal and malignant cells. Curr. Opin. Cell Biol 10, 640–646 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Weaver VM et al. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J. Cell Biol 137, 231–245 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Postovit LM et al. Human embryonic stem cell microenvironment suppresses the tumorigenic phenotype of aggressive cancer cells. Proc. Natl Acad. Sci. USA 105, 4329–4334 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mintz B & Illmensee K Normal genetically mosaic mice produced from malignant teratocarcinoma cells. Proc. Natl Acad. Sci. USA 72, 3585–3589 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Saad N et al. Cancer reversion with oocyte extracts is mediated by cell cycle arrest and induction of tumour dormancy. Oncotarget 9, 16008–16027 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nobre AR, Entenberg D, Wang Y, Condeelis J & Aguirre-Ghiso JA The different routes to metastasis via hypoxia-regulated programs. Trends Cell Biol. 28, 941–956 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rusu P et al. GPD1 specifically marks dormant glioma stem cells with a distinct metabolic profile. Cell Stem Cell 25, 241–257.e248 (2019). [DOI] [PubMed] [Google Scholar]
- 89.Vera-Ramirez L, Vodnala SK, Nini R, Hunter KW & Green JE Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat. Commun 9, 1944 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.La Belle Flynn A et al. Autophagy inhibition elicits emergence from metastatic dormancy by inducing and stabilizing Pfkfb3 expression. Nat. Commun 10, 3668 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sosa MS, Bragado P, Debnath J & Aguirre-Ghiso JA Regulation of tumor cell dormancy by tissue microenvironments and autophagy. Adv. Exp. Med. Biol 734, 73–89 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Eyles J et al. Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J. Clin. Invest 120, 2030–2039 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Saudemont A & Quesnel B In a model of tumor dormancy, long-term persistent leukemic cells have increased B7-H1 and B7.1 expression and resist CTL-mediated lysis. Blood 104, 2124–2133 (2004). [DOI] [PubMed] [Google Scholar]
- 94.Agudo J et al. Quiescent tissue stem cells evade immune surveillance. Immunity 48, 271–285.e275 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Piranlioglu R et al. Primary tumor-induced immunity eradicates disseminated tumor cells in syngeneic mouse model. Nat. Commun 10, 1430 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Krall JA et al. The systemic response to surgery triggers the outgrowth of distant immune-controlled tumors in mouse models of dormancy. Sci. Transl. Med 10, eaan3464 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Johnson RW et al. Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nat. Cell Biol 18, 1078–1089 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Takeishi S et al. Ablation of Fbxw7 eliminates leukemia-initiating cells by preventing quiescence. Cancer Cell 23, 347–361 (2013). [DOI] [PubMed] [Google Scholar]
- 99.Zhang W et al. Fbxw7 and Skp2 regulate stem cell switch between quiescence and mitotic division in lung adenocarcinoma. BioMed Res. Int 2019, 9648269 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 100.Shimizu H, Takeishi S, Nakatsumi H & Nakayama KI Prevention of cancer dormancy by Fbxw7 ablation eradicates disseminated tumor cells. JCI Insight 4, 125138 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shlush LI et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature 547, 104–108 (2017). [DOI] [PubMed] [Google Scholar]
- 102.Giustacchini A et al. Single-cell transcriptomics uncovers distinct molecular signatures of stem cells in chronic myeloid leukemia. Nat. Med 23, 692–702 (2017). [DOI] [PubMed] [Google Scholar]
- 103.Jeanpierre S et al. The quiescent fraction of chronic myeloid leukemic stem cells depends on BMPR1B, Stat3 and BMP4-niche signals to persist in patients in remission. Haematologica 10.3324/haematol.2019.232793 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang B et al. Bone marrow niche trafficking of miR-126 controls the self-renewal of leukemia stem cells in chronic myelogenous leukemia. Nat. Med 24, 450–462 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Duarte D et al. Inhibition of endosteal vascular niche remodeling rescues hematopoietic stem cell loss in AML. Cell Stem Cell 22, 64–77.e66 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Passaro D et al. Increased vascular permeability in the bone marrow microenvironment contributes to disease progression and drug response in acute myeloid leukemia. Cancer Cell 32, 324–341.e326 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Abdelhamed S et al. Extracellular vesicles impose quiescence on residual hematopoietic stem cells in the leukemic niche. EMBO Rep. 20, e47546 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kumar B et al. Acute myeloid leukemia transforms the bone marrow niche into a leukemia-permissive microenvironment through exosome secretion. Leukemia 32, 575–587 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Aguirre-Ghiso JA, Bragado P & Sosa MS Metastasis awakening: targeting dormant cancer. Nat. Med 19, 276–277 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Avivar-Valderas A, Wen HC & Aguirre-Ghiso JA Stress signaling and the shaping of the mammary tissue in development and cancer. Oncogene 33, 5483–5490 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sisay M, Mengistu G & Edessa D The RANK/RANKL/OPG system in tumorigenesis and metastasis of cancer stem cell: potential targets for anticancer therapy. Onco Targets Ther. 10, 3801–3810 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Esposito M & Kang Y Targeting tumor-stromal interactions in bone metastasis. Pharmacol. Ther 141, 222–233 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Coleman R et al. Adjuvant denosumab in early breast cancer (D-CARE): an international, multicentre, randomised, controlled, phase 3 trial. Lancet Oncol. 21, 60–72 (2020). [DOI] [PubMed] [Google Scholar]
- 114.Demaria M et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 7, 165–176 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yumoto K et al. Axl is required for TGF-β2-induced dormancy of prostate cancer cells in the bone marrow. Sci. Rep 6, 36520 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jung Y et al. Endogenous GAS6 and Mer receptor signaling regulate prostate cancer stem cells in bone marrow. Oncotarget 7, 25698–25711 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Harper KL et al. Mechanism of early dissemination and metastasis in Her2+ mammary cancer. Nature 540, 588–592 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bowers M et al. Osteoblast ablation reduces normal long-term hematopoietic stem cell self-renewal but accelerates leukemia development. Blood 125, 2678–2688 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kaur A et al. sFRP2 in the aged microenvironment drives melanoma metastasis and therapy resistance. Nature 532, 250–254 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]