

Molecular chirality of amino acids is linked to bacterial recognition by mammals to modulate IgA for bacterial symbiosis.
Abstract
Organisms use l-amino acids (l-aa) for most physiological processes. Unlike other organisms, bacteria chiral-convert l-aa to d-configurations as essential components of their cell walls and as signaling molecules in their ecosystems. Mammals recognize microbe-associated molecules to initiate immune responses, but roles of bacterial d-amino acids (d-aa) in mammalian immune systems remain largely unknown. Here, we report that amino acid chirality balanced by bacteria-mammal cross-talk modulates intestinal B cell fate and immunoglobulin A (IgA) production. Bacterial d-aa stimulate M1 macrophages and promote survival of intestinal naïve B cells. Mammalian intestinal d-aa catabolism limits the number of B cells and restricts growth of symbiotic bacteria that activate T cell–dependent IgA class switching of the B cells. Loss of d-aa catabolism results in excessive IgA production and dysbiosis with altered IgA coating on bacteria. Thus, chiral conversion of amino acids is linked to bacterial recognition by mammals to control symbiosis with bacteria.
INTRODUCTION
Natural chiral, asymmetric compounds, such as amino acids and sugars, have steric preferences for specific enantiomers. Proteinogenic amino acids other than glycine have chiral centers at the α carbon, with d- or l-configurations. Although d- and l-amino acids (d-/l-aa) have equivalent chemical properties, organisms exclusively use l-aa in ribosomal protein synthesis in all domains of life. To maintain l-aa predominance, metabolic pathways for amino acids are mostly chiral selective for l-enantiomers.
Unlike eukaryotes and archaea, bacteria have evolved a variety of amino acid racemases to stereo-convert l-aa into d-aa (1). Bacteria release d-aa to modulate diverse cellular processes, such as cell wall homeostasis, growth, and biofilm formation in the bacterial ecological niche (2). Also, d-aa are essential components of the peptides that cross-link sugar backbones in peptidoglycans in bacterial cell walls (1). Eukaryotes exploit these unique bacterial compounds to compete with bacteria for survival. Fungi produce penicillin, which targets d-Ala-d-Ala in peptidoglycans, to inhibit bacterial cell wall synthesis. On the other hand, bacteria tolerate bactericidal agents such as vancomycin, which also targets d-Ala-d-Ala, by replacing d-Ala with d-Ser or d-Asp (3). Furthermore, metazoans recognize partial structures of peptidoglycans for innate immunity against bacteria (4). Peptidoglycans are degraded by metazoan lysozyme, and degraded fragments, categorized as pathogen- or microbe-associated molecular pattern (PAMP or MAMP), activate innate immune responses via pattern recognition receptors (PRRs). These signals, mediated by PRRs, also modulate acquired immunity in mammals (5). Thus, bacterial structures containing d-aa are closely associated with antibacterial responses.
Free d-aa derived from bacteria are also involved in antibacterial responses in mammals. Symbiotic bacteria convert l-aa to d-aa in the intestines of mammalian hosts (6). In contrast, mammals maintain l-aa dominance by expressing two flavin adenine dinucleotide–dependent oxidoreductases that degrade d-aa, d-aa oxidase (DAO), and d-aspartate oxidase (DDO). DAO catalyzes oxidative deamination of neutral and basic d-aa, while DDO acts upon acidic d-aa, producing 2-oxo acids, hydrogen peroxide, and ammonia. At host-microbe interfaces, neutrophils (7) and the intestinal epithelium (6) produce DAO that oxidizes bacterial d-aa and generates hydrogen peroxide, which is toxic to pathogens such as Staphylococcus aureus and Vibrio cholerae. Furthermore, symbiotic bacteria regulate induction of intestinal DAO expression, and conversely, DAO shapes symbiont composition (6). Given that most d-aa are exclusively bacterial metabolites and are therefore types of PAMPs or MAMPs, this host-microbe interaction involving DAO not only maintains l-aa dominance in the host but also is likely related to development of host immunity.
In this study, we set out to test the in vivo effect of l-aa–to–d-aa conversion by symbiotic bacteria on development of mammalian immunity. Our results reveal unexpected roles of d-aa catabolism by host DAO in the adaptive immune system of the small intestine (SI) and provide a new perspective on host-microbe symbiosis via amino acid chirality.
RESULTS
Mammalian DAO degrades bacterial d-aa to maintain l-aa homochirality
Bacterial cell walls have unique structures, such as peptidoglycans and lipopolysaccharides, that trigger innate immune responses through PRRs (8). Peptidoglycans, found in both Gram-positive and Gram-negative bacteria, consist of repeating disaccharide backbones cross-linked with short amino acid chains that include principally d-alanine and d-glutamate (Fig. 1A). Bacteria widely express amino acid racemases, such as alanine racemase (Alr), glutamate racemase (Murl), and proline racemase (PrdF), to convert l-aa to d-aa (Fig. 1B). Because of its phylogenetic uniqueness to bacteria, d-alanine in peptidoglycans is recognized as a target for antibiotics. Serine/alanine racemase (vanT) is found only in bacteria (Fig. 1B) and replaces d-alanine with d-serine, conferring resistance to vancomycin (3). Notably, Mycoplasma and Chlamydia, which lack peptidoglycan (9), do not have amino acid racemases (Fig. 1B). Therefore, this broad taxonomic conservation of amino acid racemases among bacteria demonstrates that d-aa are produced primarily for peptidoglycan synthesis and constitute a bacterial biochemical “signature.”
Fig. 1. Phylogenetic distribution of d-aa synthetic and degradative enzymes.
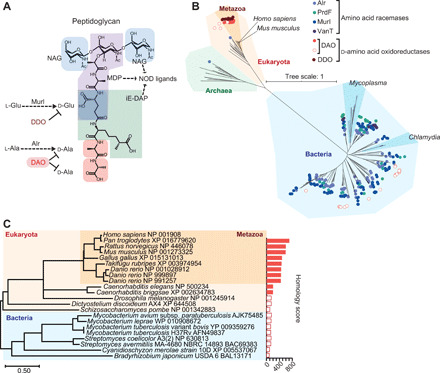
(A) The basic pattern of peptidoglycan (Gram-negative bacteria) shows incorporation of d-aa and innate ligands in the structure. NAG, N-acetylglucosamine; MDP, muramyl dipeptide; iE-DAP, γ-d-glutamyl-meso-diaminopimelic acid; NOD, nucleotide-binding oligomerization domain protein. (B) Amino acid racemases and d-aa oxidoreductases in the tree of life are shown. Circles indicate gene presence. Hollow circles indicate DAO genes with low homology (homology score < 200) to that of Homo sapiens. (C) A phylogenetic tree of DAO was analyzed using amino acid sequences of DAO. Bar graph represents BLAST homology scores. Hollow bars denote homology scores <200.
In contrast to bacterial amino acid racemases, eukaryotes, and especially metazoans, have well-conserved d-aa–degrading enzymes, DAO and DDO (Fig. 1B). Some species of bacteria, such as Mycobacterium and Streptomyces, also express DAO, but these bacterial enzymes exhibit little homology to metazoan DAO (Fig. 1C). This distinctive phylogenetic distribution of amino acid racemases and d-aa oxidoreductases led us to hypothesize that metazoans use DAO and DDO to degrade d-aa of symbiotic bacteria.
Because DAO has broader substrate specificity than DDO, we addressed DAO’s capacity to degrade bacterial d-aa. Mice with an inactivating point mutation in the DAO gene (DAOG181R/G181R), fed with a special diet that contains only l-aa (> 99% of total) as a protein source, showed significantly higher levels of blood d-serine, d-alanine, d-proline, and d-leucine compared to wild-type (WT) mice (Fig. 2A). In contrast, DAO mutant animals had blood l-aa levels comparable to those of WT mice (Fig. 2B). Together with our previous findings that symbiotic bacteria produce substantial amounts of d-aa (6), these results suggest that DAO plays a key role in maintenance of l-aa homochirality by degrading bacterial d-aa in mammals.
Fig. 2. DAO maintains l-aa homochirality and suppresses IgA production.
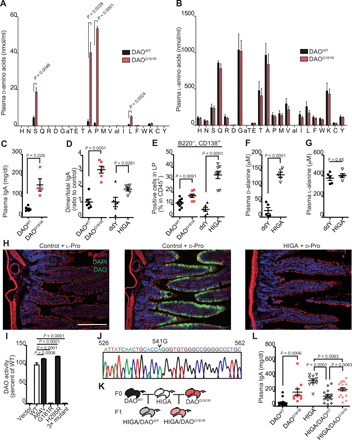
Blood d-aa (A) and l-aa (B) levels in DAOWT/WT and DAOG181R/G181R mice. Capital letters represent amino acids. “aT” and “aI” are allo-forms. (C) Plasma IgA levels in DAOWT/WT or DAOG181R/G181R mice. (D) The ratio of dimeric to total IgA analyzed by Western blot (n = 6 mice each). (E) IgA+ plasma cell percentage in the LP of SI (n = 8, 7, 6, and 8 mice); ddY, control. (F and G) Plasma free d-alanine (F) and l-alanine (G) in HIGA and ddY control mice (n = 5 mice each). (H) Activity-based labeling of DAO in SI of HIGA and control mice; active DAO (green), F-actin (red), nuclei (blue); scale bar, 100 μm. (I) Enzymatic activities of recombinant wild-type (WT), A64V, G181R, H295R, and triple-mutant DAO (n = 3). (J) Sequence electrograph showing a G541A point mutation (a G181R amino acid substitution) of DAO in HIGA mice. (K) HIGA mice were crossed with DAOWT/WT or DAOG181R/G181R mice. (L) Plasma IgA concentrations in parental and F1 mice (n = 16, 11, 10, 15, and 21 mice). Error bars, means ± SEM. Significance was analyzed with two-tailed unpaired t tests (A to G) and one-way analysis of variance (ANOVA) (I to L).
Metabolism of d-aa controls abundance of gut immunoglobulin A–producing plasma cells and blood immunoglobulin A levels
The epithelium of the SI produces DAO, which modulates the composition of symbiotic SI microbiota (6). The SI has the largest number of plasma cells, which are essential for secreting immunoglobulin A (IgA), the most abundant immunoglobulin isotype on mucosal surfaces in mammals (10). Intestinal microbiota are pivotal in stimulating production and diversification of IgA, which, in turn, regulates microbial population dynamics (11). Therefore, we hypothesized that DAO-mediated modulation of the SI microbiota affects IgA levels and/or diversity. We found that loss of DAO activity elevated levels of plasma IgA (Fig. 2C), which was the J-chain–containing dimeric form required for secretion onto mucosal surfaces (Fig. 2D and fig. S1, A to E). DAOG181R/G181R mice had higher numbers of IgA-producing plasma cells in the lamina propria (LP) of their SI than control animals (Fig. 2E). In contrast, numbers of IgA-positive B cells, which migrate to the LP and differentiate into IgA-producing plasma cells, in Peyer’s patches (PPs), spleens, and mesenteric lymph nodes (mLNs) did not differ in WT and DAO mutant mice (fig. S1, F to J). These results indicate a relationship between d-aa metabolism and dynamics of IgA immune responses in the SI mucosa.
To further clarify the relationship, we chose an animal model for IgA nephropathy, with elevated plasma and mucosal IgA [known as high-IGA (HIGA) mice] (fig. S1K) (12). To our surprise, these mice had 6.4-fold higher plasma d-alanine levels than control animals (Fig. 2F). In contrast, amounts of plasma l-alanine were comparable between the two groups of animals (Fig. 2G). DAO activity was markedly reduced in the SI epithelium of HIGA mice (Fig. 2H), suggesting that impaired DAO-mediated degradation of d-aa accounts for elevated plasma d-aa in these mice. Despite the marked reduction in DAO activity in HIGA mice, DAO expression was not impaired (fig. S1L). HIGA mice have three conserved missense mutations in the Dao gene, one of which (G181R) markedly reduces oxidative activity of DAO (Fig. 2, I and J, and fig. S1M). As in DAOG181R/G181R mice, in HIGA mice, increased plasma IgA was the J-chain–containing dimeric form (Fig. 2D and fig. S1, N to P), and their LP in SI contained higher numbers of IgA-producing plasma cells (B220−CD138+) than those of control mice (Fig. 2E). To test whether DAO deficiency in HIGA mice is linked to their elevated plasma IgA, we crossed them with DAOG181R/G181R or WT mice (Fig. 2K). Plasma IgA levels in F1 HIGA/DAOWT mice were 2.5× lower than those in F0 HIGA mice, whereas IgA levels in F1 HIGA/ DAOG181R were 1.7× higher than F1 HIGA/DAOWT mice (Fig. 2L). These results suggest that DAO deficiency contributes to IgA overproduction in HIGA mice, although the disparity between HIGA and DAOG181R/G181R mice does not exclude the possibility that additional factors are also involved. Thus, these observations strongly support our hypothesis that catabolism of microbial d-aa is associated with mucosal and systemic IgA homeostasis.
DAO-sensitive intestinal symbiotic bacteria trigger B cell differentiation
To address the mechanism by which d-aa metabolism influences IgA immune responses, we next tested the contribution of microbiota modulated by DAO to systemic IgA levels. Treatment of DAOG181R/G181R mice with oral antibiotics reduced plasma IgA to levels observed in treated control mice (Fig. 3A), consistent with the known role of intestinal bacteria in regulating mucosal IgA production (13). Reconstitution of the intestinal microbiome with microbiota from conventionally raised mice (CV-MB) restored plasma IgA (Fig. 3B). Mutant mice reconstituted with CV-MB developed 150-fold higher microbial densities in SI luminal contents than those of WT mice (Fig. 3C). This increased microbial population was not observed in the colon (fig. S2A), which lacks DAO expression (6), suggesting that the expansion of the SI microbiota consists largely of organisms that are DAO sensitive. In addition, microbial composition differed in reconstituted mutants and WT mice, with the DAOG181R/G181R mice exhibiting altered β-diversity and an enrichment of several bacterial genera such as Allobaculum (Fig. 3, D and E, and fig. S2, B to G).
Fig. 3. DAO-sensitive microbes promote IgA class switching.
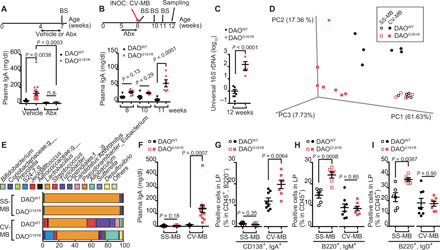
(A to I) DAOWT/WT and DAOG181R/G181R mice were treated with antibiotics or vehicle (A) (n = 9, 21, 12, and 9 mice), were treated with antibiotics followed by inoculation with CV-MB (B and C) (n = 7 and 6 mice), or were cross-fostered by dams colonized with SS-MB or CV-MB (D to I). BS, blood sampling; INOC, inoculation; Abx, antibiotics. (A, B, and F) Plasma IgA was measured with enzyme-linked immunosorbent assay (ELISA). (C) Amounts of SI microbiota were assessed with quantitative polymerase chain reaction (qPCR) of 16S ribosomal DNA (rDNA). (D) This plot indicates the principal coordinate analysis of unweighted UniFrac distances of SI microbiota that is detected by 16S rDNA metagenomic sequence. (E) Relative abundance of SI microbes is shown (average of n = 5 or 6 individuals per group). (G to I) Percentage of IgA+-plasma cells (G) or naïve B cells (H and I) in CD45+ cells in SI LP from DAOWT/WT and DAOG181R/G181R mice with indicated gut microbiota (n = 6, 6, 8, and 7 mice). Error bars, means ± SEM. Significance was analyzed with one-way ANOVA (A), two-way ANOVA (B), two-tailed unpaired t tests (C and G to I), and t tests with Welch’s correction (F).
To further investigate whether specific microbial taxa drive IgA production induced by DAO deficiency, pups of WT and DAOG181R/G181R mice born via cesarean section (C-section) were cross-fostered by dams populated with strain-selected microbiota (SS-MB), which is largely distinct from CV-MB (fig. S3, A to D). SS-MB comprises 3 strains of Lactobacillus, 20 strains of Bacteroidetes, and 47 strains of Clostridia and establishes a morphologically normal intestinal tract (14). In contrast to microbiome differences noted after reconstitution with CV-MB, colonization with SS-MB in WT and mutant mice led to development of very similar microbial contents and communities, predominantly composed of Lactobacilli (>90%) in both groups of mice (Fig. 3, D and E, and fig. S3, E and F). Moreover, after colonization with SS-MB, DAOG181R/G181R mice did not develop elevated plasma IgA or IgA-positive plasma cells in the SI epithelium, as after colonization with CV-MB (Fig. 3, F and G), supporting the view that DAO-sensitive microbes drive IgA production.
Upon activation by microbes, intestinal mature naïve B cells undergo class switching and differentiate into IgA-secreting plasma cells. Gut-associated lymphoid tissues (GALTs), including PPs, mLNs, and isolated lymphoid follicles (ILFs), are the primary sites for IgA class switching and then IgA-secreting plasma cells home to the gut LP (15). However, DAO deficiency did not increase the size or number of GALT or the number of IgA-positive cells in the GALT of SI (fig. S4, A to Q). On the other hand, IgA can also be induced in situ in the gut LP (16). Although there was no elevation of IgA-positive plasma cells in DAOG181R/G181R mice colonized with SS-MB, the LP of SI in these animals had increased numbers of mature naïve B cells (B220+IgM+ and B220+IgD+) compared to WT control mice (Fig. 3, H and I, and fig. S4R). These differences between mutant and control mice were not observed in animals colonized with CV-MB or in the bone marrow, spleens, or gut ILFs of mutant mice reconstituted with SS-MB (Fig. 3, H and I, and fig. S4, S to W). Apparently, the microbial community established in DAOG181R/G181R mice reconstituted with SS-MB does not stimulate signals that promote IgA class switching. Instead, in the absence of these signals and functional DAO, SS-MB appears to promote proliferation or survival of naïve B cell precursors in the LP of SI. Together, these observations suggest that DAO deficiency stimulates IgA production by increasing the number of naïve B cells and/or through class-switching signals induced by DAO-sensitive microbes in situ in the LP.
Intestinal metabolism of d-aa controls T cell–independent survival of naïve B cells
B cell proliferation and IgA class switching occur via both T cell–dependent (TD) and T cell–independent (TI) pathways (17). To investigate the requirement for T cells in promoting IgA production in DAO-deficient mice, DAOG181R/G181R mice were crossed with β and δ T cell receptor (TCRβ−/−,δ−/−) double knockout mice (fig. S5, A and B), which have no functional T cells and lack virtually all T cells (18). Triple-mutant pups, as well as controls, were cross-fostered by dams colonized with SS-MB (Fig. 4A). At 8 weeks of age, no animals had elevated plasma IgA (Fig. 4B), but there were increased numbers of naïve B cells in DAO mutant mice lacking T cells (Fig. 4, C and D, and fig. S5C). After the triple-mutant mice and controls were treated with antibiotics, CV-MB was introduced by coprophagy to test T cell dependency in IgA induction by DAO deficiency (Fig. 4A). In contrast to DAO mutant mice, in which increased plasma IgA levels were evident, there was minimal elevation of plasma IgA in DAO/TCRβδ triple-mutant animals (Fig. 4B). Furthermore, IgA-coating profiles on the SI microbiota also showed TD variation in several taxa, including segmented filamentous bacteria (SFB) (Candidatus Arthromitus), in the DAO mutant mice compared to WT controls (Fig. 4E and fig. S5D). Together, these observations suggest that DAO deficiency increases the abundance of naïve B cells via TI signals and stimulates IgA class switching as well as diversification in mature B cells, primarily through TD signals. Thus, DAO metabolism of microbial d-aa appears to control both the number and differentiation of B cells via distinct mechanisms.
Fig. 4. Both TD and TI pathways contribute to IgA regulation by intestinal DAO.
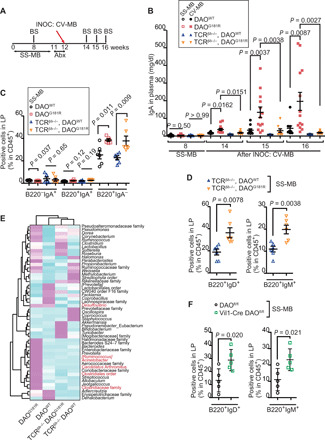
(A) DAOWT/WT, DAOG181R/G181R, TCRβ−/−,δ−/−/DAOWT/WT, and TCRβ−/−,δ−/−/DAOG181R/G181R mice colonized with SS-MB were treated with antibiotics, followed by inoculation with CV-MB. (B) Plasma IgA levels before and after inoculation with CV-MB were analyzed with ELISA (n = 10, 13, 7, and 9, respective groups). (C) Percentage of IgA+/− B220+/− cells among CD45+ cells in SI LP (n = 6, per group). (D) Percentages of naïve B cells among CD45+ cells in the SI LP at 8 weeks of age (n = 6, per group). (E) Abundance of IgA-coated bacteria on SI microbiota in mice colonized with CV-MB is depicted as averaged z scores. Taxa shown in red differ significantly among the four groups. (F) Percentages of naïve B cells in SI LP of DAOflox/flox and Vil1-Cre+/−/DAOflox/flox mice colonized with SS-MB (n = 5, per group). Error bars, means ± SEM. Significance was analyzed with two-tailed unpaired t tests (C, D, and F) and Kruskal-Wallis tests (B).
Intestinal B cells can arise in the bone marrow and migrate from a pool of naïve B cells in the peritoneal cavity (19), or they can develop in situ in the gut (20). Because DAO deficiency did not alter the number of naïve B cell precursors in the bone marrow or the peritoneal cavity (figs. S4V and S5, E to G), TI elevation in naïve B cell abundance in the SI LP of DAOG181R/G181R mice likely results from increased in situ replication. Mice with SI-specific knockout of DAO (fig. S5, H and K) colonized with SS-MB also showed increased numbers of naïve B cells in the SI LP (Fig. 4F), supporting the idea that intestinal DAO metabolism of microbial d-aa regulates number of naïve B cells in the SI.
DAO regulates expression of cytokines in intestinal epithelial cells
Transcriptomes of SI epithelium from TCRβ−/−,δ−/−/DAOWT/WT versus TCRβ−/−,δ−/−/DAOG181R/G181R mice colonized with SS-MB were compared so as to dissect pathways by which DAO modulates numbers of intestinal B cells (Fig. 5, A and B, and fig. S6, A and B). In DAO-deficient animals, in the distal SI, transcripts of immune-regulatory proteins were markedly elevated (Fig. 5B and fig. S6B). Up-regulated genes included transcripts for cytokines [interferon-γ (IFN-γ), interleukin-1β (IL-1β), and tumor necrosis factor–α (TNF-α)], alarmins (IL-33, S100a8, and S100a9), serum amyloid A1, A2, and A3 (SAA1, SAA2, and SAA3), and B cell–attracting chemokines (CCL19, CCL21, and CXCX13) (Fig. 5, C and D). Pathway analyses of up-regulated genes suggest that IFN-γ, IL-1β, and TNF-α, which can modulate B cell proliferation (21), are potential upstream drivers of aberrant immune cell development triggered by DAO deficiency (Fig. 5, E and F). On the other hand, absence of functional DAO did not affect B cell–activating factor (BAFF), a proliferation-inducing ligand (APRIL), or retinoic acid (RA), crucial factors that drive B cell survival/proliferation/differentiation (17, 22, 23), at transcription or protein/product levels (fig. S7). Because SS-MB colonization results in a similar microbial flora in DAOG181R/G181R and WT mice (Fig. 3, D and E, and fig. S3, E and F), these observations suggest the possibility that DAO substrates or metabolites themselves induce inflammatory cytokines, alarmins, and chemokines to recruit B cells to modulate their fate.
Fig. 5. DAO regulates cytokines in the SI epithelium.
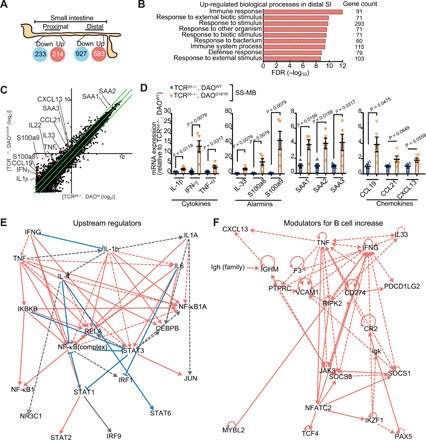
(A) Number of up-regulated or down-regulated genes in proximal and distal SI in TCRβ−/−,δ−/−/DAOG181R/G181R versus TCRβ−/−,δ−/−/DAOWT/WT mice colonized with SS-MB. (B) Functional classifications of significantly up-regulated genes in distal SI from TCRβ−/−,δ−/−/DAOG181R/G181R mice. FDR, false discovery rate. (C) Scatter plot showing the twofold or more up-regulated and down-regulated genes in log 2 scale. (D) mRNA expression of indicated cytokines, alarmins, serum amyloids, and chemokines in distal SI epithelium was analyzed by qPCR (n = 5 mice per group). (E) Top-scoring upstream regulators included several cytokines such as IFN-γ, IL-1β, IL-1α, IL-6, IL-4, and TNF, which were predicted to be extracellular upstream regulators. Solid line, leads to activation. Dashed line, leads to inhibition. Red line, findings consistent with state of downstream molecule. Blue, findings inconsistent with downstream. Gray, effect not predicted. (F) Genes associated with increased abundance of B cells suggest that TNF, IFN-γ, IL33, and CXCL13 are key regulators. Error bars, means ± SEM. Significance was analyzed with t test with Welch’s correction (D).
d-Alanine stimulates TNF-α release by M1 macrophages and prevents naïve B cell death
Amounts of d-alanine, the most active microbe-derived substrate of DAO in the SI (6), were elevated in the proximal and distal SI epithelium in DAOG181R/G181R mice colonized with SS-MB, relative to WT controls (Fig. 6A). In contrast, no microbe-specific molecules among more than 1200 metabolites examined showed significant differences (fig. S8 and data file S1). Addition of d-alanine to ex vivo SI LP cultures increased release of TNF-α (Fig. 6B), which was largely found in CD11b+ populations that include macrophages (Fig. 6C and fig. S9A). Furthermore, addition of d-alanine to cocultures of lymphocytes and macrophages enhanced survival of naïve B cells with induction of TNF-α and IL-1β (Fig. 6D and fig. S9, B to G), both of which, in turn, can serve as upstream drivers to control naïve B cell survival (fig. S9, H and I). As the coculture in the presence of either d-alanine or TNF-α increased expression of APRIL in macrophages (fig. S9J), enhanced survival of naïve B cells triggered by d-alanine may involve APRIL. d-Alanine stimulation of the proinflammatory M1 subtype of macrophages showed the most prominent increase in release of TNF-α (Fig. 6E) in both a dose- and time-dependent fashion (fig. S9, K and L). Such induction of TNF-α by d-alanine was fully blocked by selective potent inhibitors for nuclear factor κB (NF-κB) (QNZ and CAPE) and, in part, by those for mitogen-activated protein kinase kinase (MEK) (PD98059) or Janus kinase 2 (JAK2) (AG490) (Fig. 6F). Because signals that mediate MEK or JAK2 are also coupled to NF-κB in a number of stress-induced responses (24, 25), the partial inhibitory actions by MEK and JAK2 inhibitors may be associated with the NF-κB signaling pathway. Together, these observations suggest that DAO substrates, such as d-alanine, directly modulate survival of naïve B cells via a pathway that involves cytokine production by M1 macrophages.
Fig. 6. d-alanine stimulates M1 macrophages and promotes survival of naïve B cells.
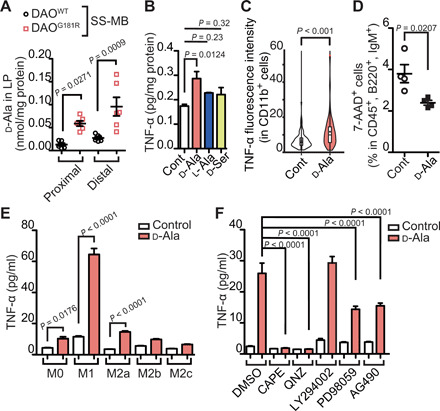
(A) Abundance of d-alanine in LP of proximal and distal SI epithelium in DAOWT/WT and DAOG181R/G181R mice colonized with SS-MB (n = 5 mice, per group). (B and C) TNF-α produced in ex vivo SI LP cultures treated with d-alanine or vehicle control were quantified absolutely in the LP cells (B, n = 3) or relatively in CD11b+ cells (C, n = 78 and 101). (D) Cell death of naïve B cells (%) among CD45+ cells in the presence of d-alanine or vehicle control in a mixed culture of lymphocytes and macrophages (n = 4, per group). (E) TNF-α amounts released in response to d-alanine from bone marrow–derived macrophages polarized with the indicated reagents. (F) TNF-α release from M1 macrophages stimulated by d-alanine with various signaling inhibitors (n = 4, per group). Error bars, means ± SEM. Significance was analyzed with two-tailed unpaired t tests (A and C to E) and one-way ANOVA (B and F).
DISCUSSION
The results presented here reveal the previously unrecognized significance of homeostatic regulation of amino acid chirality in mammalian physiology. Disturbance of mammalian amino acid homochirality by symbiotic bacterial stereoconversion is metabolically linked to two independent immune signals that regulate B cell survival and differentiation in the SI. Thus, in addition to beneficial effects on ribosomal protein synthesis, amino acid homochirality quantitatively and qualitatively regulates the adaptive immune system and contributes to symbiotic relationships with bacteria. It remains to be seen whether amino acid homochirality may be responsible for some other known modulatory effects of symbiotic bacteria on mammalian physiology.
Our results suggest that d-aa catabolism by DAO modulates IgA production in the LP of SI. The SI LP harbors the largest population of IgA-producing plasma cells, and high levels of free IgA, as well as IgA coating on microbes, occur in the SI (26). Therefore, the SI plays a critical role in IgA production. IgA is exceptionally abundant at mucosal surfaces and creates a homeostatic molecular barrier in concert with antimicrobial molecules to maintain symbiotic relationships with microbiota (26). DAO acts as an antimicrobial molecule by metabolism of bacterial d-aa on symbiotic and pathogenic bacteria (6) and inhibits IgA overproduction (Figs. 2 and 3). In a similar manner, loss of C-type lectins of the RegIII family, antibacterial molecules that attack bacterial peptidoglycans and limit invasion by bacteria, promotes IgA production (27). Therefore, IgA appears to compliment antimicrobial molecules, but the underlying mechanisms of the relationship remain poorly understood. Our findings reveal two distinct mechanisms by which DAO blocks B cells to regulate IgA production: (i) suppression of B cell number via regulation of d-aa and (ii) inhibition of B cell differentiation into plasma cells by shaping SI microbiota (fig. S10).
DAO controls the number of B cells in the LP of SI. Our results suggest that macrophage-derived TNF-α and IL-1β are upstream drivers of increased B cell number in DAO-null mice (Fig. 5, E and F). Macrophages are the most abundant mononuclear phagocytes in the LP and are thought to orchestrate mucosal inflammatory responses in addition to having phagocytic and bactericidal activities. Intestinal-resident macrophages release anti-inflammatory cytokines and are associated with immune tolerance (28), whereas intestinal macrophages recruited from blood monocytes release proinflammatory cytokines and contribute to pathogenic conditions, as in inflammatory bowel diseases (29). Therefore, proinflammatory cytokines detected in the absence of DAO may come from monocyte-derived macrophages and could be associated with deregulated, excessive inflammatory responses. Several lines of evidence support involvement of proinflammatory cytokines, such as IL-1 and TNF-α derived from macrophages, in B cell proliferation and differentiation (30, 31) and in intestinal IgA production (32). Effects on B cells triggered by loss of DAO do not mediate other B cell–proliferating signals, including BAFF/APRIL/RA (fig. S7), TD interaction of CD40-CD40L (Fig. 4), and TD release of IL-2 and IL-21 (Fig. 4), although we do not exclude potential involvement of APRIL (fig. S9H). Because some other cytokines or alarmins up-regulated by loss of DAO (Fig. 5, C and D) are also mitogenic to B cells (33, 34), there may be synergistic effects on B cells with TNF-α and IL-1β. Given involvement of monocyte-derived inflammatory macrophages in regulation of B cell number, our results suggest that DAO blocks activation of such macrophages to avoid excessive immune responses, by monitoring bacterial d-aa.
Our findings reveal a new proinflammatory function of d-alanine as a high-affinity substrate for DAO and as an exclusively bacterial metabolite in mammals. Consistent with our view that DAO influences proinflammatory macrophages, d-alanine stimulates proinflammatory M1 macrophages and induces release of TNF-α and IL-1β (fig. S9, F and G) in a similar manner to lipopolysaccharide, a component of Gram-negative bacterial cell walls. Although we did not identify the mechanism by which d-alanine stimulates macrophages, intracellular signaling involves NF-κB (Fig. 6F), a family of crucial transcription factors in development of innate and adaptive immune responses. Upon recognition of PAMPs or MAMPs, the NF-κB pathway is activated by PRRs, such as Toll-like receptors (35) and cytosolic receptors such as NOD (nucleotide-binding oligomerization domain containing) family proteins and RIG-I (retinoic-acid inducible gene I)–like helicases (36), to control transcription of cytokines and antimicrobial effectors, as well as to affect a diverse array of cellular processes. Therefore, d-alanine may be recognized by mammals as a bacterial signature, like PAMPs or MAMPs, to stimulate immune responses. Notably, d-alanine has high affinity for the GluN1 subunit of ionotropic N-methyl d-aspartate glutamate receptors (NMDARs) (37), which are expressed in the central nervous system and which may also be induced in certain macrophages. NMDARs are associated with innate immune response, and their inhibitors block NO production, Ca2+ entry, and release of proinflammatory cytokines, including TNF-α from macrophages (38). Beside NMDARs, G protein–coupled receptors may also serve as d-alanine receptors because of their affinity for many intestinal luminal molecules, such as short-chain fatty acids from bacteria, triglycerides, bile acids, and l-aa (39). Furthermore, several lines of evidence imply that sensing of amino acids by macrophages assists their development (40). The amino acid transporter CD98 plays an essential role in activation and development of macrophages (41, 42) and also facilitates B cell proliferation (43). Sensing systems of d-alanine or other bacterial d-aa and their immunomodulatory functions remain to be studied.
In addition to this antigen-independent regulation of B cell number, DAO also appears to influence IgA class switching of B cells, dependent on symbiotic SI microbiota (Fig. 3). These microbes induce production of polyreactive, low-affinity IgA (innate IgA) by promoting cross-talk between B cells and multiple components of the mucosal innate immune system (26). In innate IgA production, most symbiotic microbes trigger TI class switching that does not accompany somatic hypermutation (44), while only “aggressive” symbiotic bacteria, such as SFB, promote TD class switching (45). Our finding that modulation of SI microbiota by DAO inhibits TD innate IgA class switching (Figs. 3 and 4) may indicate that DAO controls IgA by targeting aggressive symbionts. Because DAO limits bacterial growth by degrading bacterial d-aa in an oxygen-dependent fashion (7), DAO-sensitive bacteria likely reside close to the intestinal epithelial surface, such that in the absence of DAO, they can contact antigen-presenting cells responsible for TD IgA induction. DAO inhibits growth and IgA coating of epithelium-attaching bacteria, such as SFB, and absence of DAO-sensitive bacteria does not promote TD IgA class switching (Fig. 4B). Given that TD class switching of B cells, which yields antigen-specific, high-affinity IgA, is suitable for responses against pathogens, repression of aggressive symbionts by DAO may protect the TD pathway required against pathogens from overload by symbiotic bacteria.
Bacteria evolved to convert l-aa to d-aa and to destroy l-aa homochirality despite its importance for ribosomal protein synthesis, whereas integrating d-aa into their cell walls enables them to protect themselves against degradation by most proteases of other organisms. d-aa are probably the simplest molecular features universally present in bacteria. We propose that d-aa function as bacterial signatures to promote mammalian immune responses to bacteria. Intestinal macrophages recognize bacterial d-aa to increase B cell numbers, and sampling bacteria close to the intestinal epithelium switches on TD B cell differentiation. DAO acts as a brake on both quantitative and qualitative processes by regulating d-aa abundance and bacterial growth, likely preventing excessive responses to symbiotic bacteria. Because d-aa in the gut largely reflect the presence of bacteria (6), amino acid chirality may function as a cue to modulate bacterial symbiosis. Given that mitochondria and plastids, bacterial endosymbionts in eukaryotes (46), have lost their ability to convert l-aa to d-aa, maintenance of l-aa homochirality may be crucial for eukaryotes to establish mutualistic relationships with bacteria.
MATERIALS AND METHODS
Study design
The aim of this study was to elucidate the roles of amino acid stereoconversion by symbiotic bacteria in the development of IgA-producing B cells. In vivo studies were carried out using age-matched adult mice colonized with either CV-MB or SS-MB in the same facility at Keio University. In vitro studies were performed in duplicate or triplicate within groups, and the experiments were repeated two or three times.
Animal studies
All experiments on animals were conducted in accordance with the institutional guidelines of Keio University and the Central Institute for Experimental Animals (CIEA), and all protocols were approved by the Animal Experiment Committee of Keio University and CIEA. All animals were maintained in a specific pathogen–free environment. TCRβ−/−,δ−/− mice (B6.129P2-Tcrbtm1Mom Tcrdtm1Mom/J) and Vil1-Cre transgenic mice [B6.Cg-Tg(Vil1-cre)1000Gum/J] were purchased from The Jackson Laboratory (ME, USA), and HIGA/NscSlc mice were from Japan SLC (Japan). Floxed DAO mice were generated by CRISPR-Cas9–mediated insertion of two loxP elements on introns 5 and 6 in the mouse DAO gene (Charles River Laboratories, Wilmington, MA) (fig. S5H). DAOG181R mice on the C57BL/6J background were described previously (47). DAOG181R/G181R mice and DAOWT/WT mice with CV-MB were the offspring of heterozygote breeders in the animal facility of Keio University. DAOG181R/G181R mice, DAOWT/WT mice, TCRβ−/−,δ−/−/ DAOG181R/G181R mice, TCRβ−/−,δ−/−/ DAOWT/WT mice, DAOflox/flox mice, and Vil1-Cre+/−/DAO flox/flox mice with SS-MB were generated by in vitro fertilization and transplantation to surrogate mothers, and pups C-sectioned on embryonic day 20 were fostered to CD-1 mothers with SS-MB, which originates from the microbiota resistant against Pseudomonas aeruginosa (14) and has been maintained in an isolator in the animal facility of CIEA.
For antibiotic treatments, a mixture of ampicillin (1.0 g/liter), neomycin (1.0 g/liter), and vancomycin (0.5 g/liter) was dissolved in drinking water and taken ad libitum for 1 to 3 weeks depending on the experiment. Inoculation of CV-MB was performed by placing mice in cages with bedding, which originates from a single cage of C57BL6 mice raised in the animal facility of Keio University.
Gene possession mapping
Enzymes were searched in the KEGG (Kyoto Encyclopedia of Genes and Genomes) Orthology (KO numbers: Alr, K01775; PrdF, K01777; MurI, K01776; VanT, K18348; DAO, K00273; and DDO, K00272) and obtained the taxa information that have genes for these enzymes. Gene possession was mapped on a phylogenetic tree, which was generated on the basis of 31 universal protein families and 192 organisms with complete genome sequences (48), using interactive Tree of Life version 4 (https://itol.embl.de/).
Molecular phylogenetic analysis by likelihood
The evolutionary history of DAO was inferred by using the maximum likelihood method and Whelan and Goldman model (49). The tree with the highest log likelihood (−5988.05) is shown. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using a Jones-Taylor-Thornton model and then selecting the topology with superior log likelihood value. A discrete gamma distribution was used to model evolutionary rate differences among sites [five categories (+G, parameter = 2.6204)]. The rate variation model allowed for some sites to be evolutionarily invariable [(+I), 2.53% sites]. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site (next to the branches). This analysis involved 22 amino acid sequences. All positions containing gaps and missing data were eliminated (complete deletion option). There were a total of 198 positions in the final dataset. Evolutionary analyses were conducted in Molecular Evolutionary Genetics Analysis X (www.megasoftware.net/).
Western blots
For DAO and APRIL detection, tissue homogenate from the kidney or distal SI was subjected to SDS–polyacrylamide gel electrophoresis (SDS-PAGE), transferred to polyvinylidene difluoride membrane, and blotted with a rabbit polyclonal antibody to DAO (1:1000) (6) or a biotinylated goat anti-APRIL antibody (1:1000) (BAF884, R&D Systems). For detection of IgA dimer, monomer, and J chain, 2 μl of plasma was mixed with a mouse monoclonal biotinylated antibody to IgA (clone RMM-1, BioLegend, CA, USA) conjugated with streptavidin-magnetic beads [New England Biolabs (NEB)] in 500 μl of phosphate-buffered saline (PBS) containing 1% bovine serum albumin (BSA) at 4°C with constant rotation for 2 hours. The beads were washed in 1 ml of PBS three times and applied to SDS-PAGE. Blots were blocked in PBS containing 0.1% Tween 20 and 1% BSA and immunolabeled with a goat polyclonal antibody to IgA (ab97233, Abcam; 1:1000) or a mouse monoclonal antibody to Ig J chain (clone B-6, sc-271967, Santa Cruz Biotechnology; 1:1000), followed by a donkey monoclonal horseradish peroxidase (HRP)–conjugated antibody to goat IgG or a goat monoclonal HRP-conjugated antibody to mouse IgG, respectively. Peroxidase activity was visualized with a Western blot enhanced chemiluminescence substrate (Cytiva, MA) and imaged with ImageQuant LAS 4000 mini (Cytiva, MA). For the quantification of protein levels, the density of each band was measured using ImageJ.
Preparation of SI LP mononuclear cells, SI epithelial cells, bone marrow cells, splenocytes, and peritoneal cavity cells
Mice were euthanized by cervical dislocation, and SI was collected without mesentery. The SI was opened longitudinally and washed with Ca2+, Mg2+-free Hanks’ balanced salt solution (HBSS) to remove intestinal content. After washing, the SI was further cut into small pieces and incubated with HBSS containing 1 mM dithiothreitol and 5 mM EDTA for 30 min at 37°C to remove the epithelial layer. After removal of the epithelial layer, the mucosal pieces were washed with HBSS and dissolved into solution by incubation with HBSS containing 1.5% fetal bovine serum (FBS) (Thermo Fisher Scientific), collagenase (1.0 to 3.0 mg/ml) (Wako, Japan), and deoxyribonuclease (DNase) (0.1 mg/ml; Sigma-Aldrich) for 1 hour at 37°C. The dissolved solution was centrifuged at 1700 rpm for 5 min, and the pellet was resuspended in 40% Percoll (GE Healthcare) and overlaid on 75% Percoll. Percoll gradient separation was performed by centrifugation at 2000 rpm for 20 min at 20°C. LP mononuclear cells (MNCs) were collected at the interphase between 40 and 75% Percoll layers. As for SI epithelial cells, the supernatant containing removed epithelial layer as described above was collected and centrifuged at 1700 rpm for 5 min at 4°C, and the pellet was resuspended in 40% Percoll. Percoll gradient separation was performed as well.
For isolation of BMC, femoral bone marrow was flushed out by FBS-HBSS using 23-gauge needle and syringe. The suspension was spun down at 50g for 30 s to remove debris and overlaid on 40 to 75% Percoll gradient. White blood cell layer was collected and used for analysis.
For splenocyte preparation, spleens were minced with scissors and mushed on 100-μm cell strainers that were filled with FBS-HBSS. Cells were collected by centrifugation and resuspended in red blood cell lysis buffer for 10 min. After centrifugation, splenocytes were purified on Percoll gradients.
For preparation of peritoneal cavity cells (PCCs), mice were euthanized under anesthesia and injected with 5 ml of 1% (w/v) BSA-PBS into the peritoneal cavity. Then, the peritoneal cavity was washed, and the suspension was collected.
Flow cytometry
The surface antigens of isolated single cell suspensions were preincubated with an Fcγ receptor (FcγR)–blocking monoclonal antibody (mAb) [anti-mouse CD16/32 (2.4G2), BD Biosciences, 553142] for 20 min before staining the cell surface antigens. After FcγR blocking, fluorescence-conjugated specific mAbs to surface molecules were incubated at 4°C protected from light for 30 min. After staining surface molecules, the cells were washed and resuspended with PBS containing 0.5% BSA (Nacalai Tesque, Japan). For flow cytometry analysis, FACSCanto II system (BD Biosciences) was used.
For cell death analysis of B cells, PCCs were cultured in RPMI 1640 containing 10% FBS and 1% penicillin and streptomycin (Thermo Fisher Scientific) with or without 2 mM d-alanine. At the seventh day of culture, floating cells were collected; stained with fluorescein isothiocyanate (FITC) anti-CD45, allophycocyanin (APC) anti-B220, and phycoerythrin (PE) anti-IgM for 30 min; washed; and resuspended with PBS containing 2% BSA and 2 mM EDTA. Dead cells were labeled with 7-aminoactinomycin D (7-AAD) (eBioscience) for 5 min before fluorescence-activated cell sorting analysis using BD Accuri C6 Plus (BD Biosciences).
Data were analyzed by FlowJo Vx software (Tree Star). The antibodies used for staining included the following. FITC anti-IgA (clone 10-3), APC-Cy7 anti-B220 (clone RA3-6B2), APC anti-CD138 (clone 281-2), and Brilliant Violet 510 (BV510) or FITC anti-CD45 clone (30-F11) were purchased from BD Biosciences; PE anti-IgM (clone RMM-1), PE-Cy7 or APC anti-B220 (clone RA3-6B2), anti-CD19 (clone 6C5), and BV421 anti-IgD (clone 11-26c.2a) were purchased from BioLegend.
Ex vivo survival assay of PCCs
Collection of PCCs was done as described above. As previously reported (50), 5 million to 10 million PCCs per mouse can be obtained from the peritoneal cavity. Among all live cells, 50 to 60% are B cells, 30% are macrophages, and 5 to 10% are T cells. The B cells consist of 65 to 70% B1 cells and 25 to 30% B2 cells. PCCs were resuspended in RPMI 1640 containing 10% FBS supplemented with penicillin and streptomycin (Gibco) and cultured on a 24-well plate with 2 mM d-alanine, TNF-α (10 ng/ml) (BioLegend), IL-1β (10 ng/ml) (BioLegend) for 5 days. These cells were harvested in PBS containing 1% BSA, blocked with FcγR-blocking mAb [anti-mouse CD16/32 (2.4G2), BD Biosciences, 553142] for 10 min, and stained with FITC anti-CD45 (30-F11), APC anti-B220 (RA3-6B2), and PE-IgM (RMM-1) at 4°C for 30 min. After being washed in 1% BSA-PBS, cells were stained with 7-AAD for 5 min before flow cytometry analysis.
Immunocytochemistry for intracellular TNF-α
LP MNCs were purified as written above. MNCs were cultured on μ-Dish ibiTreat (Nippon Genetics, Japan) in RPMI 1640 containing 10% FBS supplemented with penicillin–streptomycin–amphotericin B suspension (FUJIFILM Wako Pure Chemical Corporation, Japan) for 1 hour. Then, MNCs were treated with 10 mM d-alanine and brefeldin A (5 μg/ml) for 16 hours. The cells were blocked with an FcγR-blocking mAb for 10 min, stained with an APC anti-CD11b antibody (M1/70, BD Biosciences) at 4°C for 30 min, washed in PBS containing 1% BSA, fixed and permeabilized in BD Cytofix/Cytoperm solution (BD Biosciences) at 4°C for 20 min, and counterstained with PE anti–TNF-α (MP6-XT22, BioLegend). The cells were washed in PBS three times, and CD11b- and TNF-α–positive cells were visualized with an LSM 710 confocal microscope (Carl Zeiss, Germany). Fluorescence signal in CD11b-positive cells were analyzed using ImageJ software.
Quantification of cytokines by a bead-based immunoassay
CD19-negative cells were isolated from mouse PCCs using magnetic cell separation technology (Miltenyi Biotec). PCCs were preincubated with FcγR-blocking mAb for 5 min, labeled with biotin-conjugated anti-CD19 (clone REA749; Miltenyi Biotec) for 10 min, attached to streptavidin microbeads (Miltenyi Biotec), and suspended in PBS containing 2% FBS. CD19-positive cells were removed by magnetic column. CD19-negative PCCs were collected and cultured in RPMI 1640 containing 10% FBS with or without 2 mM d-alanine. Cytokines in the 48-hour cultured media were analyzed using a LEGENDplex mouse inflammation panel (BioLegend) according to the manufacturer’s protocol.
Polarization of bone marrow–derived macrophages
Mouse femoral and tibial bone marrow was flushed out by PBS containing 2% FBS and centrifuged at 1000g for 5 min. Red blood cells in the BMC were destroyed by resuspension with a red blood cell lysis buffer (155 mM NH4Cl, 12 mM NaHCO3, and 0.1 mM EDTA). To derive macrophages, after addition of RPMI 1640 medium containing 2% FBS and centrifugation at 1000g for 5 min, the BMC were cultured on noncoated plates in RPMI 1640 containing 10% FBS, 50 μM β-mercaptoethanol, macrophage colony-stimulating factor (10 ng/ml), and 1% penicillin–streptomycin–amphotericin B suspension (FUJIFILM Wako Pure Chemical Corporation) for 7 days. For polarization into M1 subtype, the macrophages were treated with lipopolysaccharide (10 ng/ml) and IFN-γ (50 ng/ml) for 24 hours. For polarization into M2a, M2b, or M2c subtypes, the macrophages were treated with IL-4 (20 ng/ml) and IL-13 (20 ng/ml), IL-1β (20 ng/ml), or transforming growth factor–β (50 ng/ml), respectively, for 24 hours. The polarized macrophages were treated with 2 mM d-alanine and cultured for 48 hours. TNF-α in the cultured media was quantified using a TNF-α mouse enzyme-linked immunosorbent assay (ELISA) kit (Thermo Fisher Scientific).
Bacterial 16S ribosomal RNA gene sequencing and analysis
Purification of bacterial genomic DNA (gDNA) was performed as described previously (6). The V1-V2 region of 16S ribosomal RNA (rRNA) gene was polymerase chain reaction (PCR)–amplified (primer pair: 27Fmod/338R with overhang adapters) (adapter-27Fmod: 5′-tcg tcg gca gcg tca gat gtg tat aag aga cag AGR GTT TGA TYM TGG CTC AG-3′; adapter-338R: 5′-gtc tcg tgg gct cgg aga tgt gta taa gag aca gTG CTG CCT CCC GTA GGA GT-3′) and indexed with the Nextera XT Index Kit (Illumina, San Diego, CA, USA) by PCR as described (6). The 16S rRNA amplicon with indices were column-purified, quantified with a Qubit 3.0 fluorometer (Thermo Fisher Scientific), pooled at a concentration of 4 nM, denatured, diluted to a final concentration of 8 pM, and sequenced using the MiSeq Reagent Kit v2 (500-cycle, paired-end reads; Illumina) on a MiSeq sequencer (Illumina). Paired-end reads were analyzed using QIIME (version 1.9) as described (6).
Isolation and characterization of IgA-bound bacteria
We followed IgA-seq protocol (51) with minor modifications. In short, streptavidin magnetic beads (SA beads) (50 μl per sample) (NEB) and goat biotinylated antibody against mouse IgA (10 μg per sample) (Abcam, ab97233) was mixed in 0.1% BSA-PBS at room temperature for 30 min. Contents of SI were suspended in 2 ml of Ca2+, Mg2+−free HBSS [HBSS(−)], mixed using vortex for 20 s, and spun down at 400g for 5 min. Supernatant was collected to other tube, and pellet was suspended in 1.5 ml of HBSS(−), vortexed, and spun down again. This step was done three times to collect bacteria. Then, collected supernatants were spun down at 5000g for 5 min. The pellet was suspended in 500 μl of 0.1% BSA-HBSS(−), mixed with anti–IgA-SA beads, and rotated at 4°C for 1 hour. The mixture was transferred to a 5-ml round tube and stood on a magnet. For collection of non-IgA bound bacteria, 150 μl of supernatant was collected and stocked at −80°C until use. For collection of IgA-bound bacteria, supernatant was removed, and beads were washed in 4 ml of 0.1% BSA-HBSS(−) four times. Bacteria-bound beads were suspended in 150 ml of HBSS(−) and stocked until use.
Bacterial gDNA was extracted according to the protocol written above. Data analysis was conducted as the same protocol above except for first PCR and operational taxonomic unit (OTU) picking. First PCR for IgA-seq, the number of cycles was 25 (WT versus DAO null) or 28 (DAOWT/WT mice versus DAOG181R/G181R versus TCRβ−/−,δ−/−/ DAOWT/WT mice versus TCRβ−/−,δ−/−/ DAOG181R/G181R). For IgA-seq OTU picking, samples were subsampled to 6849 reads (DAOWT/WT mice versus DAOG181R/G181R), 6241 reads (IgA bound: WT versus DAO null versus TCRβ−/−,δ−/−/ DAOWT/WT mice versus TCRβ−/−,δ−/−/ DAOG181R/G181R), and 11165 reads (IgA not bound: DAOWT/WT mice versus DAOG181R/G181R versus TCRβ−/−,δ−/−/ DAOWT/WT mice versus TCRβ−/−,δ−/−/ DAOG181R/G181R).
IgA+/IgA− bacteria were calculated as follows. The number of sequence reads about each genus was expressed as the ratio in each sample. Undetected samples were presumed 0.000001 (1 per 1,000,000 reads) for the purpose of calculation. The reads ratio of IgA+ sample was divided by the ratio of IgA− sample, and z score was estimated. For statistics, each sample of [IgA+/IgA− ratio] was tested using Kruskal-Wallis test.
Preparation of LP cells for ex vivo culture
Entire SIs, except for the proximal 5-cm portion, was dissected; lumens were flushed out with 20 ml of PBS, cut open longitudinally, and cut into 1-cm pieces. These pieces were washed in ice-cold PBS and incubated in a dissociation buffer [10 mM Hepes, 5 mM EDTA, and HBSS (−)] at 37°C with shaking for 20 min. After removal of supernatants containing the epithelial layer, tissues were washed in ice-cold PBS again and were incubated in a digestion buffer [collagenase D (500 μg/ml), DNase I (500 μg/ml), dispase (0.5 U/ml), and 2% FBS-HBSS (+)] at 37°C with shaking for 1 hour. The dissolved solution was passed through a 70-μm cell strainer (BD Biosciences), mixed with DMEM-10 medium [Dulbecco’s modified Eagle’s medium (Gibco), 10% FBS, 80 mM β-mercaptoethanol, l-glutamine (8 mg/ml), and 1% penicillin and streptomycin (Thermo Fisher Scientific)], and centrifuged at 800g for 20 min. The pellet was resuspended in 40% Percoll (GE Healthcare) and overlaid onto 80% Percoll. After centrifugation at 1300g for 20 min, the middle layer, which included lymphocytes, was collected and washed in DMEM-10 medium. Pelleted cells were cultured in a culture media (RPMI 1640, 10% FBS, and 1% penicillin–streptomycin–amphotericin B) for 24 hours before further experiments.
For assaying cytokine production, floating cells were removed by washing with PBS, and cells attached on culture plates were treated with 1 mM d- or l-alanine or d-serine for 24 hours. To inhibit release of cytokines into the media, brefeldin A (5 μg/ml) was added at 6 hours after the amino acid treatment and incubated for 18 hours. These cells were then washed in PBS, lysed in a lysis buffer [50 mM tris-HCl (pH 7.4), 15 mM NaCl, 20 mM EDTA, 1% Triton X-100, cOmplete EDTA-free (Roche)], and stored at −80°C until use. Cytokines in the lysate were analyzed by ELISA.
mRNA expression in SI epithelium
Proximal SI, defined as 5-cm portion from pylorus, and distal SI, defined as 5-cm portion from terminal ileum, were dissected, flushed with 10 ml of PBS, cut open longitudinally, and vortexed in 10 ml of PBS for 10 s. The inner layer of the SI was peeled off mechanically using glass plates and homogenized with stainless steel beads (3.2 mm in diameter, TOMY Seiko) in 1 ml of QIAzol (Qiagen) using Micro Smash (MS-100, TOMY Seiko) at 3500 rpm for 2 min. Total RNA was purified with an RNeasy microarray tissue mini kit (Qiagen) according to the manufacturer’s protocol, and gDNA was digested using ribonuclease-free DNase. Total RNA was converted to complementary DNA (cDNA) using a ReverTra Ace reverse transcription quantitative PCR (qPCR) kit (Toyobo). qPCR was conducted using a Thunderbird qPCR mix (Toyobo). PCR primers to amplify target genes used in this study are as follows: IL-1β, 5′-GCAACTGTTCCTGAACTCAACT-3′ (forward) and 5′-ATCTTTTGGGGTCCGTCAACT-3′ (reverse); TNFα, 5′-CCCTCACACTCAGATCATCTTCT-3′ (forward) and 5′- GCTACGACGTGGGCTACAG-3′ (reverse); IFN-γ, 5′-ATGAACGCTACACACTGCATC-3′ (forward) and 5’-CCATCCTTTTGCCAGTTCCTC-3′ (reverse); IL-33, ′-GGCTGCATGCCAACGACAAGG-3′ (forward) and 5′-AAGGCCTGTTCCGGAGGCGA-3′ (reverse); S100a8, 5′-AGTGTCCTCAGTTTGTGCAG-3′ (forward) and 5′-ACTCCTTGTGGCTGTCTTTG-3′ (reverse); S100a9, 5′-GTTGATCTTTGCCTGTCATGAG-3′ (forward) and 5′-AGCCATTCCCTTTAGACTTGG-3′ (reverse); SAA1, 5′-GAAGGAAGCTAACTGGAAAAACTC-3′ (forward) and 5′-CAGGCCCCCAGCACAACCTAC-3′ (reverse); SAA2, 5′-ATGAAGGAAGCTGGCTGGAAAGATG-3′ (forward) and 5′-CTCAGGACCCCAACACAGCCTTCT-3(reverse); SAA3, 5′-TGCCATCATTCTTTGCATCTTGA-3′ (forward) and 5′-CCGTGAACTTCTGAACAGCCT-3′ (reverse); BAFF, 5′-TGCCTTGGAGGAGAAAGAGA-3′ (forward) and 5′-GGAATTGTTGGGCAGTGTTT-3′ (reverse); APRIL, 5′-GCCTTCTGATCCTGACCGTGCC-3′ (forward) and 5′-AGTTTTGCGTTTGCCCGTGGA-3′ (reverse); 18S rRNA, 5′-GTAACCCGTTGAACCCCATT-3′ (forward) and 5′- CCATCCAATCGGTAGTAGCG-3′ (reverse); RALDH1, 5′-ATACTTGTCGGATTTAGGAGGCT-3′ (forward) and 5′-GGGCCTATCTTCCAAATGAACA-3′ (reverse); RALDH2, 5′-CAGAGAGTGGGAGAGTGTTCC-3′ (forward) and 5′-CACACAGAACCAAGAGAGAAGG-3′ (reverse); RALDH3, 5′-GGGTCACACTGGAGCTAGGA-3′ (forward) and 5′-CTGGCCTCTTCTTGGCGAA-3′ (reverse); CCL19, 5′-GGGGTGCTAATGATGCGGAA-3′ (forward) and 5′-CCTTAGTGTGGTGAACACAACA-3′ (reverse); CCL21, 5′-GTGATGGAGGGGGTCAGGA-3′ (forward) and 5′-GGGATGGGACAGCCTAAACT-3′ (reverse); CXCL12, 5′-TGCATCAGTGACGGTAAACCA-3′ (forward) and 5′-TTCTTCAGCCGTGCAACAATC-3′ (reverse); CXCL13, 5′-GGCCACGGTATTCTGGAAGC-3′ (forward) and 5′-GGGCGTAACTTGAATCCGATCTA-3′ (reverse).
Transcriptome analysis
Total RNA (10 μg) from each sample was mixed into one sample per group of animals. The quality of each RNA mixture was analyzed with NanoDrop (Thermo Fisher Scientific) and further evaluated using the Agilent 4200 TapeStation (Agilent Technologies, in which RNA integrity number was higher than 9.0. cDNA was constructed using the Low Input Quick Amp Labeling Kit (Agilent Technologies) and hybridized to SurePrint G3 Mouse GE Microarray 8x60K v2 (Agilent Technologies). Then, the signal intensity was calculated using a feature extraction software (Agilent Technologies), and differentially expressed genes were analyzed using a GeneSpring GX software (Agilent Technologies). Expression differences greater than or less than twofold were considered as “significant difference.” The up-regulated genes identified were further submitted to PANTHER v.14 (www.pantherdb.org/) for gene ontology analysis. Functional annotation chart was created on the basis of statistical significance with overall false discovery rate < 0.05.
The set of genes that was up-regulated more than twofold was analyzed and visualized using Ingenuity Pathway Analysis (IPA) software (Qiagen). The software calculated a significance of score (P value) for each network based on Ingenuity Knowledge Base in accordance with Fisher’s exact test. Enrichment of network-regulated genes in the datasets was ranked with activation z scores. The pathway shown in Fig. 5, E and F was drawn with up-regulated genes and revealed three upstream regulators: TNF-α (P = 9.35 × 10−12), IFN-γ (P = 4.7× 10−21), and IL-1β (P = 1.37× 10−19). The up-regulated genes, which were categorized in “increase of B cell,” were collected from the datasets, and networks between each gene were automatically drawn by IPA software.
Metabolome analysis
Contents of distal SI was collected and frozen by liquid nitrogen. For extracting ionic metabolites, approximately 20 mg of SI contents were dissolved in H2O containing internal standards [H3304-1002, Human Metabolome Technologies (HMT), Tsuruoka, Yamagata, Japan] with a ratio of 1:9 (w/v). After centrifugation, the supernatant was then centrifugally filtered through a Millipore 5000-Da cutoff filter (UltrafreeMC-PLHCC, HMT) to remove macromolecules (9100g, 4°C, 60 min) for subsequent analysis with capillary electrophoresis time-of-flight mass spectrometry (CE-TOFMS).
For extracting nonionic metabolites, approximately 20 mg of distal SI contents were dissolved in 1 ml of methanol containing internal standard (H3304-1002, HMT). After centrifugation, 10 μl of the supernatant was transferred into glass vials for evaporation under nitrogen gas and reconstituted with 200 μl of 50% isopropanol (v/v) for subsequent analysis with liquid chromatography TOFMS (LC-TOFMS).
Metabolome analysis was conducted at HMT with a Dual Scan package using CE-TOFMS and LC-TOFMS for ionic and nonionic metabolites, respectively. Hierarchical cluster analysis and principal components analysis were performed by HMT’s proprietary software, PeakStat and SampleStat, respectively (data file S1).
Determination of d-aa and l-aa levels by two-dimensional high-performance liquid chromatography
Plasma samples were mixed with ultrapure water and methanol at a 1:1:18 (v/v/v) ratio, vortexed for 10 s, and centrifuged at 4°C at 13,000g for 5 min. For the SI samples, the inner layers of proximal and distal SI were homogenized in PBS as described in the method for the “mRNA expression in SI epithelium” section. The PBS homogenate was centrifuged at 14,000g for 10 min. The supernatant was mixed with 18-fold (v/v) of methanol, vortexed for 2 min, and centrifuged at 4°C at 12,100g for 5 min. Supernatants were stored at −80°C until use. d-aa and l-aa concentrations in the methanol solution were determined with a two-dimensional high-performance liquid chromatography (HPLC) system (NANOSPACE SI-2 series, Osaka Soda, Japan) combining an octadecylsilyl column [KSAARP, 1.0-mm inner diameter (ID) × 400 mm] (designed by Kyushu University and KAGAMI Inc., Japan) and a Pirkle-type enantioselective column (KSAACSP-001S, 1.5 mm ID × 250 mm) (designed by Kyushu University and KAGAMI) as described (52).
Quantification of all-trans RA using an HPLC system
About 2 cm of the middle part of SI was cut out, and the luminal content was removed. Blood was collected from inferior vena cava and separated into plasma with EDTA. The tissues and plasma were stored at −80°C until use. Tissues were homogenized in acetonitrile (2 μl/mg tissue) using Micro Smash MS-100 (TOMY Seiko) at 3000 rpm for 2 min and centrifuged at 14,000g for 10 min. Plasma was mixed with the same volume of acetonitrile and centrifuged at 20,400g for 20 min. Twenty microliters of the supernatants was injected into the NANOSPACE system (Osaka Soda). Macromolecules were separated in a mobile phase (0.1% formic acid:methanol = 40:60) using a column (CAPCELL PAK MF Ph-1 SG80 S-5; 4.6 mm ID × 50 mm, Osaka Soda, Japan) and removed. The fraction including all-trans RA (ATRA) was collected with a column-switching technique. ATRA was concentrated with a column (CAPCELL PAK C18 MGII S-5; 2.0-mm ID × 35 mm, Osaka Soda), introduced into a mobile phase (0.1% formic acid:methanol = 10:90), and further separated using a column (CAPCELL PAK C18 MGII S-3; 2.0-mm ID × 150 mm). The absorbance at 350 nm was detected with an ultraviolet-visible detector (Osaka Soda, Japan). Peak area of the chromatogram was analyzed using EZChrom Elite software (Agilent, CA), and standard curve was drawn with results from 0.5 to 500 nM authentic ATRA (Sigma-Aldrich). The detection limit of this system was 100 fmol of ATRA.
Histology
Intestinal lumen was washed with PBS, cut open longitudinally, rolled up, embedded in optimal cutting temperature compound, and snap-frozen by liquid nitrogen. The tissue block was sliced into 10-μm sections using a cryostat (Leica, Germany) at −19°C. To observe ILFs, tissue section was stained with hematoxylin and 1% eosin, dehydrated with 70 to 100% ethanol, replaced with xylene, and embedded in DPX nonaqueous mounting medium (Merck Millipore).
For analysis of germinal center of PPs and mLNs, sections were fixed with methanol for 10 min and blocked in PBS containing 5% goat serum for 1 hour. These sections were stained with PE-labeled anti-IgD (11-26c, Thermo Fisher Scientific) and FITC-conjugated peanut agglutinin (Thermo Fisher Scientific) at room temperature for 1 hour, costained with 100 nM 4′,6-diamidino-2-phenylindole (DAPI) for 10 min, and coverslipped in a ProLong diamond antifade reagent (Thermo Fisher Scientific). Fluorescence was visualized using a BZ-9000 fluorescence microscope (Keyence).
To examine the cellularity of ILFs, sections of unfixed rolled SI were washed in PBS, blocked with PBS containing 5% goat serum at room temperature for 1 hour, and immunostained with FITC-conjugated anti-IgM (II/41, Thermo Fisher Scientific), PE anti-IgD (11-26c, Thermo Fisher Scientific), APC anti-B220 (RA3-6B2), and PE anti-CD4 (RM4-4, BioLegend) at 4°C overnight. After being washed, these sections were stained with 100 nM DAPI, washed in PBS, and coverslipped with the ProLong diamond antifade reagent. For detection of IgA and B220, frozen sections were fixed in methanol containing 0.3% H2O2 for 10 min, washed in PBS, blocked with 5% goat serum PBS at room temperature for 1 hour, and stained with biotinylated anti-B220 (RA3-6B2, BioLegend) and PE anti-IgA (mA-6E1, Thermo Fisher Scientific) at room temperature for 1 hour. These sections were washed in PBS, treated with streptavidin-HRP (Elite ABC kit, Vector Laboratories Inc.) and labeled with FITC-conjugated tyramid (PerkinElmer). Fluorescence signal was detected using the BZ-9000 fluorescence microscope (Keyence). To analyze frequency of T cells, B cells, and IgD+/IgM+/IgA+ cells, fluorescence intensities of PE anti-CD4, APC anti-B220, PE anti-IgD, FITC anti-IgM, and PE anti-IgA were measured using ImageJ software. Ratios of fluorescence intensities are described as arbitrary units.
For visualization of tissue DAO activity, HIGA, or ddY control mice were euthanized and their intestinal tissues were processed for cryo-sectioning as described (6). Tissue DAO activity was visualized by labeling with FITC-conjugated tyramide (PerkinElmer) activated by reaction of DAO with d-proline as described (47). Fluorescence signals were visualized using the BZ-9000 fluorescence microscope (Keyence). Each section being compared was imaged under identical conditions.
DAO activity assay of mouse tissue
DAO activity in tissue homogenates were detected using d-alanine as substrate as described (47).
Sequencing of DAO gene
Total RNA from kidney of HIGA mice was extracted with RNAiso (Takara) according to the manufacturer’s protocol. mRNA was reverse-transcribed into cDNA with oligo(dT) primers using a ReverTraAce reverse transcription PCR kit (Toyobo), and DAO gene was PCR-amplified with a set of primers [5′-ATGCGCGTGGCCGTGATCG-3′ (forward) and 5′-TCAGAGGTGGGAGGGAGGC-3′ (reverse)]. The PCR product was subcloned into pCR Blunt II TOPO cloning vector (Life Technologies). Sp6 and T7 primers were used for sequencing by the BigDye Terminator v3.1 Cycle Sequencing Kit. PCR product was cleaned up with a BigDye Xterminator purification kit. The fluorescence was analyzed with ABI PRISM 310 Genetic Analyzer (Applied Biosystems).
Activity assay of recombinant DAO mutants
Mouse WT Dao was cloned from a cDNA library of mouse kidney and subcloned into a pFLAG-CMV5a expression vector that includes C-terminal FLAG-tag (Sigma-Aldrich). Mouse Dao mutants containing individual A64V, G181R, or H295R substitutions or all three substitutions were generated from pFLAG-CMV5a-DAO using the QuickChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA). Primer sets used in mutagenesis were A64V-Dao [5′-agcaaccctcaggaggTggagtggagccagcaaacg-3′ (forward) and 5′-cgtttgctggctccactccAcctcctgagggttgct-3′ (reverse)], G181R-Dao [5′-attatcaactgcaccAgggtgtgggccggggccctg-3′ (forward) and 5′-cagggccccggcccacacccTggtgcagttgataat-3′ (reverse)], H295R-Dao [5′-gaaagagaatggcttcGttttggatcttcaagtgca-3′ (forward) and 5′-tgcacttgaagatccaaaaCgaagccattctctttc-3′ (reverse)]. COS7 cells were expressed with the FLAG-tagged WT or mutant DAO and cultured for 2 days. Then, the cells were harvested in a lysis buffer [50 mM tris-HCl (pH 7.4), 15 mM NaCl, 20 mM EDTA, and 1% Triton X-100), and the cell lysate was incubated with anti-FLAG M2 affinity gel (Sigma-Aldrich) at 4°C for 2 hours. The affinity gel was washed in an immunoprecipitation buffer [20 mM Hepes-NaOH (pH 7.4), 1 mM dithiothreitol, 1 mM EDTA, 150 mM NaCl, and 0.1% Triton X-100] three times and suspended in PBS. Then, the suspension was mixed with 10 mM d-proline, HRP (1.25 U/ml; Sigma-Aldrich), 50 mM Amplex Red (Thermo Fisher Scientific) and incubated at room temperature for 10 min. Fluorescence of excitation/emission at 540/590 nm was analyzed by SpectraMax Paradigm (Molecular Device), and relative intensity to WT DAO was calculated.
Statistical analysis
No statistical methods were used to predetermine sample size. Blinding was not possible for animal experiments. No randomization was used. Prism 8 (GraphPad Software) was used for data plotting and statistical analyses. Statistical significance was determined by two-sided unpaired t test to compare two groups or one-way analysis of variance (ANOVA) for multiple comparisons when data were normally distributed and had equal variance. If the variances of the data were not equal, then nonparametric tests were performed.
Acknowledgments
We thank S. D. Aird, B. M. Davis, and T. Zhang for comments on the manuscript; S. Aiso, M. Matsuoka, and A. Kurahashi for support; Y. Sugito for technical support on d-aa analysis; S. Fujie for RA analysis; and Y. Fukuchi and H. Nakajima for discussions. We appreciate the Collaborative Research Resources, School of Medicine, Keio University for technical support and reagents. Funding: This work was funded by Japan Agency for Medical Research and Development (AMED-PRIME) (18gm6010017h0001) (to J.S.), JSPS KAKENHI grant nos. 18 K07181 (to M.S.) and 16 K09327 (to J.S.), Research Grants for Life Sciences and Medicine from Keio University Medical Science Fund (to M.S.), Grant-in-Aid for JSPS Research Fellow (17 J10213) (to M.S.), Moritani Scholarship Foundation (to J.S.), Keio Gijuku Fukuzawa Memorial Fund for the Advancement of Education and Research (to J.S.), and NIH RO1 AI-043247 and HHMI (to M.K.W.). Author contributions: Conceptualization: M.S. and J.S. Data curation: M.S. and S.C. Formal analysis: M.S. and J.S. Funding acquisition: M.S., M.K.W., and J.S. Investigation: M.S., T.S., S.C., Y.H., and J.S. Methodology: K.H. and T.S. Project administration: J.S. Resources: M.G., R.T., M.M., M.I., and K.H. Supervision: T.K. and M.Y. Validation: J.S. Visualization: M.S. and J.S. Writing—original draft: M.S. and J.S. Writing—review and editing: M.K.W., M.Y., and J.S. Competing interests: M.M. is a founder and CEO of KAGAMI INC., a company working on analyses of chiral amino acids and research for medical applications. M.S. and J.S. are inventors on a patent, US16/340786 and JP2018-545077, related to this work filed by J.S., M.S., and S. Aiso on 13 October 2017 (priority date: 14 October 2016), under evaluation. The authors declare no other competing interests. Data and materials availability: 16S rRNA sequences, generated from intestinal samples in raw format before post-processing and data analysis, are deposited at the European Nucleotide Archive (accession nos. PRJEB35803 and PRJEB38154). Raw gene-expression data from transcriptome analyses are deposited to Gene Expression Omnibus (accession no. GSE142070).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/10/eabd6480/DC1
REFERENCES AND NOTES
- 1.Radkov A. D., Moe L. A., Bacterial synthesis of D-amino acids. Appl. Microbiol. Biotechnol. 98, 5363–5374 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Aliashkevich A., Alvarez L., Cava F., New insights into the mechanisms and biological roles of D-amino acids in complex eco-systems. Front. Microbiol. 9, 683 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zeng D., Debabov D., Hartsell T. L., Cano R. J., Adams S., Schuyler J. A., McMillan R., Pace J. L., Approved glycopeptide antibacterial drugs: Mechanism of action and resistance. Cold Spring Harb. Perspect. Med. 6, a026989 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Royet J., Dziarski R., Peptidoglycan recognition proteins: Pleiotropic sensors and effectors of antimicrobial defences. Nat. Rev. Microbiol. 5, 264–277 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Iwasaki A., Medzhitov R., Regulation of adaptive immunity by the innate immune system. Science 327, 291–295 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sasabe J., Miyoshi Y., Rakoff-Nahoum S., Zhang T., Mita M., Davis B. M., Hamase K., Waldor M. K., Interplay between microbial D-amino acids and host D-amino acid oxidase modifies murine mucosal defence and gut microbiota. Nat. Microbiol. 1, 16125 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cline M. J., Lehrer R. I., D-amino acid oxidase in leukocytes: A possible D-amino-acid-linked antimicrobial system. Proc. Natl. Acad. Sci. U.S.A. 62, 756–763 (1969). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolf A. J., Underhill D. M., Peptidoglycan recognition by the innate immune system. Nat. Rev. Immunol. 18, 243–254 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Otten C., Brilli M., Vollmer W., Viollier P. H., Salje J., Peptidoglycan in obligate intracellular bacteria. Mol. Microbiol. 107, 142–163 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cerutti A., Chen K., Chorny A., Immunoglobulin responses at the mucosal interface. Annu. Rev. Immunol. 29, 273–293 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sutherland D. B., Fagarasan S., IgA synthesis: A form of functional immune adaptation extending beyond gut. Curr. Opin. Immunol. 24, 261–268 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Imai H., Nakamoto Y., Asakura K., Miki K., Yasuda T., Miura A. B., Spontaneous glomerular IgA deposition in ddY mice: An animal model of IgA nephritis. Kidney Int. 27, 756–761 (1985). [DOI] [PubMed] [Google Scholar]
- 13.Macpherson A. J., Yilmaz B., Limenitakis J. P., Ganal-Vonarburg S. C., IgA function in relation to the intestinal microbiota. Annu. Rev. Immunol. 36, 359–381 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Itoh K., Urano T., Mitsuoka T., Colonization resistance against Pseudomonas aeruginosa in gnotobiotic mice. Lab. Anim 20, 197–201 (1986). [DOI] [PubMed] [Google Scholar]
- 15.Pabst O., New concepts in the generation and functions of IgA. Nat. Rev. Immunol. 12, 821–832 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Fagarasan S., Kinoshita K., Muramatsu M., Ikuta K., Honjo T., In situ class switching and differentiation to IgA-producing cells in the gut lamina propria. Nature 413, 639–643 (2001). [DOI] [PubMed] [Google Scholar]
- 17.Cerutti A., The regulation of IgA class switching. Nat. Rev. Immunol. 8, 421–434 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mombaerts P., Clarke A. R., Rudnicki M. A., Iacomini J., Itohara S., Lafaille J. J., Wang L., Ichikawa Y., Jaenisch R., Hooper M. L., Tonegawa S., Mutations in T-cell antigen receptor genes α and β block thymocyte development at different stages. Nature 360, 225–231 (1992). [DOI] [PubMed] [Google Scholar]
- 19.Fagarasan S., Shinkura R., Kamata T., Nogaki F., Ikuta K., Tashiro K., Honjo T., Alymphoplasia (aly)-type nuclear factor κB-inducing kinase (Nik) causes defects in secondary lymphoid tissue chemokine receptor signaling and homing of peritoneal cells to the gut-associated lymphatic tissue system. J. Exp. Med. 191, 1477–1486 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wesemann D. R., Portuguese A. J., Meyers R. M., Gallagher M. P., Cluff-Jones K., Magee J. M., Panchakshari R. A., Rodig S. J., Kepler T. B., Alt F. W., Microbial colonization influences early B-lineage development in the gut lamina propria. Nature 501, 112–115 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boussiotis V. A., Nadler L. M., Strominger J. L., Goldfeld A. E., Tumor necrosis factor alpha is an autocrine growth factor for normal human B cells. Proc. Natl. Acad. Sci. U.S.A. 91, 7007–7011 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Litinskiy M. B., Nardelli B., Hilbert D. M., He B., Schaffer A., Casali P., Cerutti A., DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat. Immunol. 3, 822–829 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mora J. R., Iwata M., Eksteen B., Song S.-Y., Junt T., Senman B., Otipoby K. L., Yokota A., Takeuchi H., Ricciardi-Castagnoli P., Rajewsky K., Adams D. H., von Andrian U. H., Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science 314, 1157–1160 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Ryan K. M., Ernst M. K., Rice N. R., Vousden K. H., Role of NF-κB in p53-mediated programmed cell death. Nature 404, 892–897 (2000). [DOI] [PubMed] [Google Scholar]
- 25.Digicaylioglu M., Lipton S. A., Erythropoietin-mediated neuroprotection involves cross-talk between Jak2 and NF-κB signalling cascades. Nature 412, 641–647 (2001). [DOI] [PubMed] [Google Scholar]
- 26.Bunker J. J., Bendelac A., IgA responses to microbiota. Immunity 49, 211–224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vaishnava S., Yamamoto M., Severson K. M., Ruhn K. A., Yu X., Koren O., Ley R., Wakeland E. K., Hooper L. V., The antibacterial lectin RegIIIγ promotes the spatial segregation of microbiota and host in the intestine. Science 334, 255–258 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Denning T. L., Wang Y.-c., Patel S. R., Williams I. R., Pulendran B., Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat. Immunol. 8, 1086–1094 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Rugtveit J., Nilsen E. M., Bakka A., Carlsen H., Brandtzaeg P., Scott H., Cytokine profiles differ in newly recruited and resident subsets of mucosal macrophages from inflammatory bowel disease. Gastroenterology 112, 1493–1505 (1997). [DOI] [PubMed] [Google Scholar]
- 30.Hoffmann M. K., Koenig S., Mittler R. S., Oettgen H. F., Ralph P., Galanos C., Hammerling U., Macrophage factor controlling differentiation of B cells. J. Immunol. 122, 497–502 (1979). [PubMed] [Google Scholar]
- 31.Falkoff R. J., Muraguchi A., Hong J. X., Butler J. L., Dinarello C. A., Fauci A. S., The effects of interleukin 1 on human B cell activation and proliferation. J. Immunol. 131, 801–805 (1983). [PubMed] [Google Scholar]
- 32.Kim Y.-I., Song J.-H., Ko H.-J., Kweon M.-N., Kang C.-Y., Reinecker H.-C., Chang S.-Y., CX3CR1+ macrophages and CD8+ T cells control intestinal IgA production. J. Immunol. 201, 1287–1294 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Romagnani S., Giudizi M. G., Biagiotti R., Almerigogna F., Mingari C., Maggi E., Liang C. M., Moretta L., B cell growth factor activity of interferon-gamma. Recombinant human interferon-gamma promotes proliferation of anti-mu-activated human B lymphocytes. J. Immunol. 136, 3513–3516 (1986). [PubMed] [Google Scholar]
- 34.Komai-Koma M., Gilchrist D. S., McKenzie A. N. J., Goodyear C. S., Xu D., Liew F. Y., IL-33 activates B1 cells and exacerbates contact sensitivity. J. Immunol. 186, 2584–2591 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Kawai T., Akira S., Signaling to NF-κB by Toll-like receptors. Trends Mol. Med. 13, 460–469 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Chamaillard M., Hashimoto M., Horie Y., Masumoto J., Qiu S., Saab L., Ogura Y., Kawasaki A., Fukase K., Kusumoto S., Valvano M. A., Foster S. J., Mak T. W., Nuñez G., Inohara N., An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat. Immunol. 4, 702–707 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Kleckner N. W., Dingledine R., Requirement for glycine in activation of NMDA-receptors expressed in Xenopus oocytes. Science 241, 835–837 (1988). [DOI] [PubMed] [Google Scholar]
- 38.Glezer I., Zekki H., Scavone C., Rivest S., Modulation of the innate immune response by NMDA receptors has neuropathological consequences. J. Neurosci. 23, 11094–11103 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Husted A. S., Trauelsen M., Rudenko O., Hjorth S. A., Schwartz T. W., GPCR-mediated signaling of metabolites. Cell Metab. 25, 777–796 (2017). [DOI] [PubMed] [Google Scholar]
- 40.van Beek A. A., Van den Bossche J., Mastroberardino P. G., de Winther M. P. J., Leenen P. J. M., Metabolic alterations in aging macrophages: Ingredients for inflammaging? Trends Immunol. 40, 113–127 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Tsumura H., Ito M., Li X.-K., Nakamura A., Ohnami N., Fujimoto J.-I., Komada H., Ito Y., The role of CD98hc in mouse macrophage functions. Cell. Immunol. 276, 128–134 (2012). [DOI] [PubMed] [Google Scholar]
- 42.Wuggenig P., Kaya B., Melhem H., Ayata C. K.; Swiss IBD Cohort Investigators, Hruz P., Sayan A. E., Tsumura H., Ito M., Roux J., Niess J. H., Loss of the branched-chain amino acid transporter CD98hc alters the development of colonic macrophages in mice. Commun. Biol. 3, 130 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cantor J., Browne C. D., Ruppert R., Féral C. C., Fässler R., Rickert R. C., Ginsberg M. H., CD98hc facilitates B cell proliferation and adaptive humoral immunity. Nat. Immunol. 10, 412–419 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Slack E., Balmer M. L., Fritz J. H., Hapfelmeier S., Functional flexibility of intestinal IgA -Broadening the fine line. Front. Immunol. 3, 100 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lécuyer E., Rakotobe S., Lengliné-Garnier H., Lebreton C., Picard M., Juste C., Fritzen R., Eberl G., McCoy K. D., Macpherson A. J., Reynaud C.-A., Cerf-Bensussan N., Gaboriau-Routhiau V., Segmented filamentous bacterium uses secondary and tertiary lymphoid tissues to induce gut IgA and specific T helper 17 cell responses. Immunity 40, 608–620 (2014). [DOI] [PubMed] [Google Scholar]
- 46.Archibald J. M., Endosymbiosis and eukaryotic cell evolution. Curr. Biol. 25, R911–R921 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Sasabe J., Miyoshi Y., Suzuki M., Mita M., Konno R., Matsuoka M., Hamase K., Aiso S., D-amino acid oxidase controls motoneuron degeneration through D-serine. Proc. Natl. Acad. Sci. U.S.A. 109, 627–632 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ciccarelli F. D., Doerks T., von Mering C., Creevey C. J., Snel B., Bork P., Toward automatic reconstruction of a highly resolved tree of life. Science 311, 1283–1287 (2006). [DOI] [PubMed] [Google Scholar]
- 49.Whelan S., Goldman N., A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 18, 691–699 (2001). [DOI] [PubMed] [Google Scholar]
- 50.Ray A., Dittel B. N., Isolation of mouse peritoneal cavity cells. J. Vis. Exp., e1488 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kubinak J. L., Petersen C., Stephens W. Z., Soto R., Bake E., O’Connell R. M., Round J. L., MyD88 signaling in T cells directs IgA-mediated control of the microbiota to promote health. Cell Host Microbe 17, 153–163 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Furusho A., Koga R., Akita T., Miyoshi Y., Mita M., Hamase K., Development of a highly-sensitive two-dimensional HPLC system with narrowbore reversed-phase and microbore enantioselective columns and application to the chiral amino acid analysis of the mammalian brain. Chromatography 39, 83–90 (2018). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/10/eabd6480/DC1