

This study presents a mechanism-based receptor-targeted cellular therapy approach to treat breast to brain metastasis.
Abstract
Basal-like breast cancer (BLBC) shows brain metastatic (BM) capability and overexpresses EGFR and death-receptors 4/5 (DR4/5); however, the anatomical location of BM prohibits efficient drug-delivery to these targetable markers. In this study, we developed BLBC-BM mouse models featuring different patterns of BMs and explored the versatility of estem cell (SC)–mediated bi-functional EGFR and DR4/5-targeted treatment in these models. Most BLBC lines demonstrated a high sensitivity to EGFR and DR4/5 bi-targeting therapeutic protein, EVDRL [anti-EGFR VHH (EV) fused to DR ligand (DRL)]. Functional analyses using inhibitors and CRISPR-Cas9 knockouts revealed that the EV domain facilitated in augmenting DR4/5-DRL binding and enhancing DRL-induced apoptosis. EVDRL secreting stem cells alleviated tumor-burden and significantly increased survival in mouse models of residual-tumor after macrometastasis resection, perivascular niche micrometastasis, and leptomeningeal metastasis. This study reports mechanism based simultaneous targeting of EGFR and DR4/5 in BLBC and defines a new treatment paradigm for treatment of BM.
INTRODUCTION
Breast cancer (BC) is the second most common cancer that can metastasize to the brain and, in fact, brain metastasis (BM) is a major cause of cancer-related deaths in patients with BC. Approximately 15 to 30% of patients with metastatic BC develop BM (1, 2). Along with the increase in the incidence of BC (3), the occurrence of BC-BM has also increased in recent years owing to improved extracranial disease control and poor central nervous system (CNS) penetration of drugs (4). Among the four main intrinsic subtypes of BC, basal-like breast cancer (BLBC) has a 70 to 80% overlap with triple-negative breast cancer (TNBC) and constitutes 12 to 15% of BC (5). BLBC has the poorest prognosis and the shortest survival among the BC subtypes (6), owing to the unavailability of specific therapeutic options including hormonal or molecular-targeted therapy. BLBC metastasizes to the brain more frequently than the other subtypes (7, 8), shortening patient survival (9).
BM continues to represent a formidable challenge in the clinical management of patients with cancer. Currently, stereotactic radiosurgery (SRS), surgical resection, and whole-brain irradiation are the most common treatment options for BM; however, these tumors are generally resistant to systemic chemotherapy because of the blood-brain barrier (BBB). For detectable tumors up to 3 cm, there is a favorable indication for SRS (10). However, residual invasive cancer cells following surgical resection of large tumors that were not eligible for SRS, undetectable dormant tumor cells in the perivascular niche (PVN), and leptomeningeal metastasis, also known as meningeal carcinomatosis, are challenging conditions and lack effective treatment options. Tumors in the eloquent areas of the brain are unresectable, and residual tumor cells after resection eventually cause recurrence. Because, compared to other cancers, BC is known to feature later recurrences (11), patients who have undergone treatment remain at a persistent risk even for decades (11, 12). Repeated chemotherapy is often unable to kill the PVN-dwelling cancer cells as they are known to establish a strict localization outside the vasculature (13, 14). Leptomeningeal metastasis is another devastating condition observed in 11 to 20% of patients with CNS metastasis of BC (15, 16). The lack of effective treatments leads to extremely short survival (median survival: 4 to 6 weeks) (17). Although intrathecal (IT) administration of anticancer agents is often attempted, there is no evidence for improvement in survival (18). Given the multistep and complex biological nature of BM, tumor models that recapitulate metastatic brain tumor features are limited. In this study, we first developed imageable mouse models for clinically challenging BLBC-BM conditions, including single intracranial metastasis resection, PVN micrometastasis, and leptomeningeal metastases, and used these models to investigate in detail the efficacy and mechanism of stem cell–based bifunctional BM treatments targeting overexpressed epidermal growth factor receptor (EGFR) and death receptors 4/5 (DR4/5).
EGFR is up-regulated in BLBC (19), and anti-EGFR therapies have been adopted (20). Given that EGFR is one of the most important mediators of BM in BC (21), and EGFR positive tumors are more frequent in BC-BM than primary tumors (22), targeting EGFR has the potential to be beneficial in BC-BM. However, in contrast to other EGFR–up-regulated cancers, EGFR-targeting therapies have not been successful in BLBC (20). On the other hand, BLBC cell lines are sensitive to DR4/5-induced death signaling triggered by tumor necrosis factor–related apoptosis-inducing ligand (TRAIL, herein as DRL) binding, which induces apoptosis selectively in cancer cells (23). However, DRL alone is not sufficient to treat BLBC (24), and even enhancing modifications of DRL have not translated into remarkable treatment benefits in patients (25). In a preliminary screen of BLBC-BM patient samples, EGFR and DR4/5 displayed a concurrent up-regulation in the tumor area. Therefore, it is of great interest to explore the strategy to simultaneously evaluate targeting EGFR and DR4/5 in BLBC-BM. A variable domain of camelid heavy-chain-only antibody (VHH), also known as nanobody is a small molecule, consisting solely of the antigen binding domain (26). We have previously engineered bivalent anti-EGFR VHH (EV) and shown their efficacy in mouse tumor models (27). We have recently developed a bi-functional molecule EV fused to DRL (EVDRL) that simultaneously targets EGFR and DR4/5, but its mechanism-based efficacy has not been fully understood.
Given the challenges related to systemic delivery of a majority of therapeutic agents across the BBB and short half-life and high hepatotoxicity of DRL (28), engineered stem cells offer an excellent platform to target CNS tumors. We and others have previously established use of neural stem cells (NSCs) and mesenchymal stem cells (MSCs) engineered to express tumor-specific biomolecules for treating primary brain neoplasms. Especially for tumors in the CNS, stem cell administration in the resection cavity has been shown to improve drug delivery (29). For micrometastasis at PVN, arterial delivery of therapeutic stem cells offers an advantage owing to the BBB penetration capability of stem cells (30–32). For leptomeningeal metastasis, the primary reason for the failure of IT drug administration is the difficulty of infusing drugs continuously and the incessant turnover of cerebrospinal fluid (CSF) that clears the drug (17). IT delivery of therapeutic stem cells offers an advantage as stem cells can survive in the CSF space and continuously secrete therapeutic molecules; however, there are no reports on stem cell therapy for leptomeningeal metastasis as a secondary CNS tumor. In this study, we characterized in detail the anti-BLBC efficacy of EVDRL and assessed the therapeutic efficacy of stem cell–delivered EVDRL in different mouse tumor models of breast to BM.
RESULTS
EGFR and DR4/5 are up-regulated in BLBC-BM
We analyzed the dataset from The Cancer Genome Atlas (TCGA) (33) and showed that BLBC [typically triple negative for estrogen receptor, progesterone receptor, and HER2 (TNBC); fig. S1A] has significantly higher expression of EGFR and DR4/5 mRNA compared to the other BC subtypes (Fig. 1A). In addition, a cohort of cell lines from TCGA (34) provided further evidence that BLBC cell lines have significantly higher expression of EGFR and DR5 mRNA compared to the non-BLBC subtypes (Fig. 1B). To confirm these results, we tested cellular and cell surface expression of EGFR and DR4/5 by Western blot and flow cytometry, respectively, in 15 human BC cell lines (HER2-enriched: SKBR3 and MDA-MB-453; luminal A: MCF7, HCC1500, ZR75-1, and HCC1428; luminal B: BT474, T47D, and MDA-MB-175VII; basal-like: BT549, Hs578T, SUM159, MDA-MB-231, MDA-MB-436, and MDA-MB-468) and three patient-derived BLBC-BM cell lines (BMET02, BMET05, and BMET15), which were confirmed by mRNA microarray (fig. S1B) (35). BLBC cell lines showed significantly higher expression of EGFR compared to the other BC subtypes (Fig. 1, C and D). Although higher expression of DR5 was observed in BLBC as compared to non-BLBC, the expression levels were not significant, most likely due to the insufficient number of cell lines tested (Fig. 1, C and D). In addition, immunohistochemistry of TNBC patient samples showed that BM tissue displayed a significantly higher expression of EGFR compared to the primary tumor (Fig. 1E and fig. S1C). Together, these data revealed that EGFR and DR4/5 are promising targets in BLBC-BM.
Fig. 1. EGFR and DR4/5 are up-regulated in BLBC-BM.
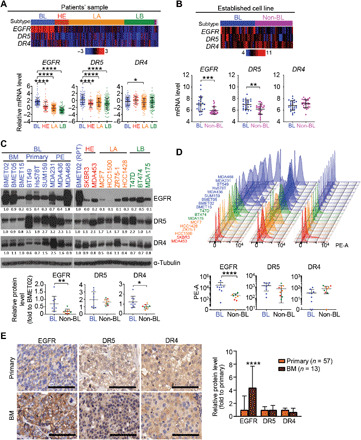
(A) Top: Heatmap of mRNA levels of EGFR, DR5, and DR4 in patient samples of four subtypes (BL, basal-like; HE, HER2-enriched; LA, luminal A; LB, luminal B) of BC from TCGA database (n = 526). Bottom: Comparison of EGFR, DR5, and DR4 mRNA levels between subtypes. (B) Top: Heatmap of mRNA levels of EGFR, DR5, and DR4 in cell lines of BLBC or non-BLBC from TCGA database (n = 52). Bottom: Comparison of EGFR, DR5, and DR4 mRNA levels between subtypes. (C) Top: Western blot (WB) of EGFR, DR5, and DR4 in 18 BC cell lines (PE, pleural effusion; RPT, repeat; loading control–adjusted ratios are provided under blots). Bottom: Relative expression of EGFR, DR5, and DR4 in BLBC and non-BLBC. (D) Top: Cell surface protein levels of EGFR, DR5, and DR4 analyzed by flow cytometry in 18 BC cell lines. Bottom: Comparison of cell surface expression of EGFR, DR5, and DR4 in BLBC and non-BLBC. PE-A, phycoerythrin-area. (E) Left: Representative micrograph of immunohistochemistry of EGFR, DR5, and DR4 in primary tumors and BM of TNBC. Scale bars, 100 μm. Right: Quantifications of immunohistochemical staining densities by ImageJ (primary, n = 57; BM, n = 13).
Development and characterization of mouse tumor models of BLBC-BM
We developed mouse models representing three major clinically relevant forms of BM: macrometastasis, micrometastasis, and leptomeningeal metastasis (Fig. 2A). First, we generated a patient-derived, BMET02 line expressing a bimodal firefly luciferase (Fluc)–mCherry (FmC) fusion protein (fig. S2A). We confirmed a direct correlation between Fluc signals and implanted BMET02-FmC cell numbers in vivo and show that BMET02-FmC has similar growth rate as parental BMET02 (fig. S2, B and C). Intracardiac injection, the most common method for development of experimental BM models, can lead to widespread tumor formation (36). Even “standard” intracarotid arterial (ICA) injection may reduce the rate of aberrant (nonbrain) metastasis, this route routinely requires the ligation of the external carotid artery,, thereby prohibiting repeated injections. Moreover, when using the standard ICA injection technique with ligation of the external carotid artery, we still observed extracranial metastasis, likely as a result of tumor cell distribution into small arterial feeder branches, such as the occipital artery (OA), pterygopalatine artery (PPA), and superior thyroid artery (STA) (fig. S3, A to C) (37–39). Therefore, we established a modified ICA injection technique, in which we ligated these feeder arteries, thereby greatly reducing the formation of extracranial tumors (fig. S3A and movie S1). In addition, partial preservation of the external carotid artery enabled multiple ICA injections from the same side; this allows us to efficiently test cell-based therapies in this model (movie S1). We injected BMET02-FmC into mice using this modified ICA injection technique and monitored tumor development by bioluminescence imaging (BLI). BLI signals of day 0 demonstrated the successful ICA injection of viable BMET02-FmC cells in mouse; however, the BLI signals then quickly dropped to undetectable levels, most likely due to only a very small portion of tumor cells completing extravasation and surviving in the brain (Fig. 2B). Chronological brain samples from ICA-injected BMET02-bearing mice showed multiple tumors in the brain parenchyma (Fig. 2C). Immunohistochemistry of brain blood vessels revealed that BC cells extravasated in the early phase (day 7), stayed alongside the blood vessel for a while (day 13), and then started growing along the vessels (day 20) (Fig. 2D). Immunohistochemistry showed late phase BLBC-BM tumors were highly proliferative and hypervascularized and surrounded by astrocytes (fig. S3D).
Fig. 2. Developing and characterizing clinically relevant mouse models for BLBC-BM.
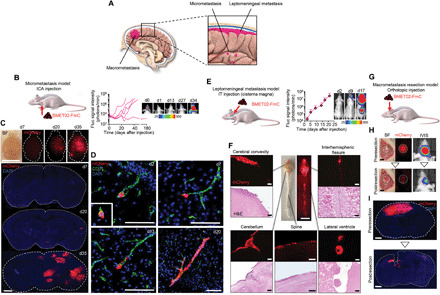
(A) Schematic representation of three clinical scenarios of BM. (B) Left: Schematic of micrometastasis model. Right: BLI signal curves of each mouse after ICA injection of BMET02-FmC and representative pictures. (C) Top: Chronological representative bright-field (BF) and fluorescence photographs of brain samples from ICA-injected BMET02-FmC–bearing mice. Scale bar, 10 mm. Bottom: Photomicrograph of coronal sections of the samples. Scale bar, 1 mm. (D) Chronological photomicrographs of immunohistochemistry of CD31 in brain sections that had ICA injection of BMET02-FmC. Scale bars, 100 μm. Critical moment of extravasation of cancer cells was observed on day 2 (inset of d2). (E) Left: Schematic of leptomeningeal metastasis model. Right: BLI signal curve of IT-injected BMET02-FmC–bearing mice (n = 2) and representative photographs. (F) Center: Representative photograph of brain and spine samples from mice 23 days after IT injection of BMET02-FmC. Scale bar, 10 mm. Surrounding: Representative microphotograph of fluorescence and hematoxylin and eosin (H&E) staining of the brain and spine samples. Scale bars, 100 μm. (G) Schematic of macrometastasis resection model. (H) Left: Representative intraoperative BF and fluorescence photographs of brain of pre- and postresection of BMET02-FmC tumor. Scale bars, 1 mm. Right: Representative pictures of BLI. (I) Representative photomicrograph of brain sections of pre- and postresection of tumor. Scale bars, 1 mm. Photo credit: Yohei Kitamura, Brigham and Women’s Hospital. DAPI, 4′,6-diamidino-2-phenylindole.
To develop a leptomeningeal metastasis model, we IT injected BMET02-FmC into the cisterna magna (fig. S3E). BLI showed tumor growth around the CNS (Fig. 2E). Brain and spine samples showed widely disseminated tumors in various areas of CSF space across the CNS (Fig. 2F). The tumor resection model was developed as we previously reported (Fig. 2G) (40). In short, following establishment of a cranial window, tumor cells were directly injected into the superficial brain parenchyma followed by bioluminescent imaging of tumor growth and microscopically assisted tumor resection. To confirm that orthotopically injected BMET02-FmC leads to a similar pattern of metastatic tumor formation as we had previously observed with ICA-injected tumor cells, we performed ex vivo brain sectioning followed by immunohistochemical staining for vascularization and surrounding astrocytes (fig. S3F). Fluorescence images and BLI showed that resection substantially reduced tumor size (Fig. 2, H and I). Together, these data demonstrate the establishment of three clinically relevant mouse models of BLBC-BM, which have the potential to facilitate development and preclinical testing of the next generation of BM therapies.
EVDRL induces apoptosis in BLBC cells
Clinical tissue obtained from patients with TNBC-BM and tumor tissues from ICA-injected patient-derived BMET02-FmC–bearing mice expressed both of EGFR and DR4/5 as revealed by immunofluorescence (Fig. 3A and fig. S4A). In addition, our flow cytometry analysis showed that each single tumor cell has both of EGFR and DR5 using a BLBC cell line (fig. S4B). To simultaneously target cell surface EGFR and DR4/5, we used two bi-functional proteins encoded by cDNAs for anti-EGFR VHH (EV) or anti-EGFR ScFv (ES) fused to DRL (EVDRL and ESDRL, respectively) (Fig. 3B and fig. S4C) and tested their efficacy in patient-derived BLBC-BM lines. Cell viability and protein assays showed that EVDRL induced significant cytotoxicity and caspase-mediated apoptosis compared to ESDRL (Fig. 3, C and D). We further assessed the antitumor effects of EVDRL in 18 BC cell lines. Most BLBC cell lines responded to DRL and EVDRL but not to EV. EVDRL consistently mediated more potent antitumor effects than DRL (Fig. 3E and fig. S4D). A correlation between the expression levels of DR5 (see Fig. 1D) and the efficacy of DRL was observed (Fig. 3F), and therapeutic effects of EVDRL relative to those of DRL were correlated with EGFR expression levels in BLBC cells (Figs. 1, C and D, and 3G and fig. S4E). These results suggest that the therapeutic sensitivity to EVDRL of BC cell is mainly determined by the expression levels of DR5 and EGFR (Fig. 3H).
Fig. 3. BLBC is sensitive to EVDRL, and EV domain of EVDRL enhances apoptosis-inducing effect of DRL depending on tumor cell surface EGFR expression.
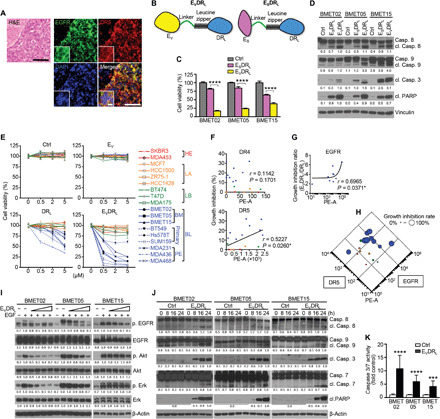
(A) Representative microphotograph of low and high (the insets) magnifications of H&E and immunohistochemistry of EGFR and DR4/5 in patient samples of TNBC- BM. Scale bars, 100 μm (main images) and 10 μm (insets). (B) Schematic showing the construction of anti-EGFR VHH-DRL (EVDRL) and anti-EGFR scFv-DRL (ESDRL) proteins. (C) Cell viability of BLBC-BM lines after 72-hour treatment with control media (Ctrl), ESDRL, or EVDRL. (n = 3, technical replicates). (D) WB showing cleavage of caspases and poly(ADP-ribose) polymerase (PARP) in BLBC-BM lines after 8-hour treatment with Ctrl, ESDRL, or EVDRL (n = 3, technical replicates). (Loading control–adjusted ratios are provided under blots; only cleaved part was quantified). (E) Cell viability of 18 BC cell lines after 72-hour treatment with different concentrations of Ctrl, EV, DRL, or EVDRL (n = 3, technical replicates). (F) Correlation between cell surface DR4/5 expression and growth inhibition effect of DRL at the time point of 24 hours. (G) Correlation between cell surface EGFR expression and growth inhibition ratio between DRL and EVDRL at the time point of 24 hours. (H) Correlation between cell surface DR5 and EGFR expression and the growth inhibition efficacy of EVDRL. (I) WB showing phosphorylation of EGFR and its downstream elements in BLBC-BM lines with EGF treatment after pretreatment with various concentrations of EVDRL (n = 3, technical replicates). (J) WB showing cleavage of caspases and PARP in BLBC-BM lines after 8-, 16-, and 24-hour treatment with Ctrl or EVDRL (n = 3, technical replicates). (K) Caspase-Glo 3/7 assay of BLBC-BM lines after 8-hour treatment with Ctrl or EVDRL (n = 3, technical replicates).
To explore candidate factors besides EGFR and DRs that might influence BLBC’s sensitivity to EVDRL, we first analyzed the difference in apoptosis-related genes among the subtypes based on the data from TCGA (fig. S5A). Among them, down-regulation of BCL2 and Bcl-xL (anti-apoptotic Bcl genes) and up-regulation of BID and BAX (pro-apoptotic Bcl gene) were found in BLBC (fig. S5A). Western blot analysis on different BLBC cell lines revealed differential expression of BCL2, Bcl-xL, and BID. However, no significant correlation between the protein expression of BCL-2, Bcl-xL, BID, and efficacy of DRL was observed (fig. S5B). Decoy death receptors, DcR1 and DcR2, are known to influence the sensitivity of cells to DRL (41). TCGA revealed down-regulation of DcR1 and up-regulation of DcR2 in BLBC (fig. S5A), and Western blot analysis of BLBC lines showed low expression of DcR1 and varying expression levels of DcR2 (fig. S5C). Furthermore, MYC, which is known to be related to both of apoptosis pathway and EGFR signaling pathway (42, 43), was down-regulated in BLBC cell lines (fig. S5A). Western blot analysis showed a negative correlation between protein expression of Myc and efficacy of EVDRL in the cell lines tested (fig. S5D). These results suggest that sensitivity of BLBC cells to EVDRL is mainly determined by EGFR and DR expression; however, other factors, such as apoptosis-related factors, might also influence their sensitivity to EVDRL.
As expected, we observed that EVDRL inhibited EGFR signaling (Fig. 3I) and induced caspase-mediated apoptosis in BLBC-BM tumor cells (Fig. 3, J and K). We also confirmed that EV alone and ESDRL also inhibited EGFR signaling (fig. S5, E and F). These data showed that EVDRL simultaneously targets EGFR and DR4/5 and consistently induces apoptosis in a cohort of BLBC lines.
EV domain of EVDRL enhances DRL-induced apoptosis
We initially hypothesized that the main function of EV domain of EVDRL would be to block EGFR signaling. However, EV alone showed marginal effects on the cell viability of BLBC tumor cells expressing high levels of EGFR (Fig. 3E), and the differences in efficacy between DRL and EVDRL were apparent very early ~24 hours after treatment (fig. S4D). This did not support our initial hypothesis and suggested that the EV domain has another mechanism beyond blocking EGFR signaling to enhance therapeutic efficacy of EVDRL. To identify the mechanism, we tested whether EV could sensitize tumor cells to DRL by modulating interactions between apoptosis pathways and EGFR downstream elements. However, we did not observe any changes when the cells were treated with EV (fig. S6A). Next, we combined EV and DRL to test whether this recapitulated the effects of EVDRL in BLBC-BM lines. Treatment with EV + DRL had a lower efficacy compared to EVDRL (Fig. 4, A and B), suggesting that fusing EV with DRL is necessary to enhance efficacy. Next, we tested the efficacy of EVDRL on another BLBC cell line, MDA-MB231-FmC, which has relatively low EGFR and high DR4/5. We observed similar findings in this line as well (fig. S6, B and C). Next, we assessed the proximity between EGFR and DR5 before and after treatment with EVDRL using real-time Förster resonance energy transfer (FRET) imaging on BMET02 and NIH-3T3 cells expressing recombinant EGFR–yellow fluorescent protein (YFP) and DR5–cyan fluorescent protein (CFP) (Fig. 4C). Both receptors were localized to the cell surface, and typical trace showed that treatment with EVDRL correlated with an increase in detectable FRET in the cells (Fig. 4D). These data suggest that EGFR and DR5 are in close proximity on the cell surface, and EVDRL binding further increases their association.
Fig. 4. EV domain of EVDRL enhances the apoptosis-inducing efficacy of DRL.
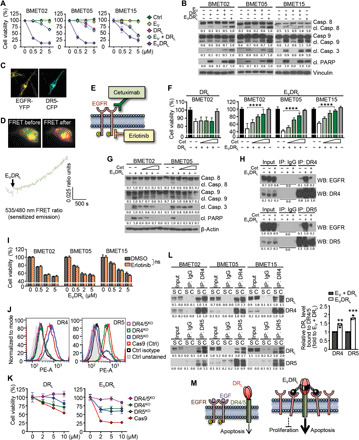
(A and B) Cell viability of BLBC-BM lines after 72-hour treatment (A) and WB of cleavage of caspases and PARP in BLBC-BM lines after 8-hour treatment (B) with control media (Ctrl), EV, DRL, EV + DRL, and EVDRL (n = 3, technical replicates). (C) Confocal images of unstimulated BMET02 cells stably expressing EGFR-YFP and transiently transfected with DR5-CFP. (D) Real-time FRET (sensitized emission) imaging in NIH-3T3 cells coexpressing DR5-CFP and EGFR-YFP. Ratio images depicting the bottom focal plane of the cell show FRET before (left image) and 30 min after treatment EVDRL (right image). (E) Schematic of EGFR inhibitors used for blocking experiments of EVDRL. (F and G) Cell viability (F) and WB showing cleavage of caspases and PARP (G) of BLBC-BM lines after 24-hour treatments with DRL and EVDRL after pretreatment with various concentrations of cetuximab (n = 3, technical replicates). (H) Coimmunoprecipitation (Co-IP) and WB analyses showing EGFR-EVDRL-DR4/5 complex formation in the presence of EVDRL and the attenuation of the complex by cetuximab in BMET02 (n = 2, technical replicates). (I) Cell viability of BLBC-BM lines treated by EVDRL for 24 hours with or without pretreatment by 1 μM erlotinib (n = 3, technical replicates). ns, not significant. (J) Flow cytometry showing reduction of cell surface expression of DR4/5 in BMET02 lines with CRISPR-Cas9 knockout (KO) of DR4, DR5, and DR4/5. (K) Cell viability of BMET02-DR4/5 KO lines after 72-hour treatment with DRL and EVDRL (n = 3, technical replicates). (L) Left: Co-IP and WB analyses showing levels of DRL bound to DR4/5 in BLBC-BM lines after 8-hour treatment with separated EV plus DRL (S) or combined EVDRL (C). Right: Quantification of levels of DRL bound to DR4/5 (n = 3). (M) Schematic showing functional difference between DRL (left) and EVDRL (right).
To confirm that EVDRL binding to EGFR and DR4/5 at the cell surface results in internalization of these receptors, we assessed the colocalization of EGFR with early, Rab5, and late, Rab7, regulators of endocytosis, posttreatment of cells with EVDRL. EGFR colocalized with early endosomal protein, Rab5, within 5 min and with late endosomal protein, Rab7, at 15-min time after EVDRL treatment as compared to control treated or in EGFR-negative cells, implying that EGFR was endocytosed into the cell cytoplasm (fig. S6D). Because this phenomenon did not take place when cells were treated with control media or when the treated cells had very low levels of EGFR, these findings imply that EGFR-bound EVDRL was endocytosed into the cell.
Next, we used cetuximab and erlotinib to block EGFR. Cetuximab, an anti-EGFR monoclonal antibody, blocks EVDRL-EGFR binding, extracellularly; and erlotinib, a receptor tyrosine kinase inhibitor, inhibits phosphorylation of EGFR, intracellularly (Fig. 4E). Consistent with previous reports (44), cetuximab and erlotinib alone had no effects on BLBC-BM cell proliferation (fig. S6E). As TNBC cell resistance to EGFR inhibitors could simply arise from relatively lower EGFR expression in TNBC than the other EGFR inhibitor–sensitive cancers, we compared EGFR expression of patients’ TNBC-BM samples with BM of non–small cell lung carcinoma (NSCLC), which is an EGFR inhibitor–sensitive cancer type. Similar levels of EGFR expression were seen in both TNBC-BM and NSCLC-BM (fig. S6F). In addition, a cell line cohort from TGCA database (34) showed that TNBC cell lines have the same or even higher EGFR mRNA level compared to NSCLC, pancreatic cancer, and colorectal cancer, which are all considered EGFR inhibitor–sensitive cancers (fig. S6G). Cetuximab significantly blocked EVDRL-mediated reduction of cell viability and induction of apoptosis in BLBC-BM lines but did not affect the effect of DRL (Fig. 4, F and G, and fig. S6H). Coimmunoprecipitation (Co-IP) studies showed that cetuximab interfered with formation of an EGFR-EVDRL-DR4/5 complex (Fig. 4H). After confirming that erlotinib sufficiently inhibits EGFR phosphorylation (fig. S6I), we treated erlotinib-pretreated BLBC-BM cells with EVDRL. Erlotinib pretreatment did not affect the efficacy of EVDRL (Fig. 4I). These results suggest that EGFR binding is critical for EVDRL therapeutic effects, but its efficacy is not mainly via blocking EGFR signaling.
To validate DR4/5 as the targets of EVDRL treatment, we generated DR4, DR5, and DR4/5 knockout (KO) BMET02 lines using CRISPR-Cas9 gene editing and treated them with DRL or EVDRL (Fig. 4J). We confirmed that the growth rate of BMET02-DR4/5KO is similar to its parental BMET02 cell line (fig. S2B). Both BMET02-DR4KO and BMET02-DR5KO lines were significantly less sensitive to DRL and EVDRL than the control line. KO of both DR4 and DR5 induced almost complete resistance to DRL and EVDRL (Fig. 4K and fig. S6J). These results suggested that DR4/5 expression is essential for EVDRL activity, which is enhanced by the interaction between EV domain and EGFR.
We tested whether DRL binding to DR4/5 differed when cells were exposed to EVDRL and EV + DRL. Co-IP assays showed 1.3- to 1.5- and 1.7- to 2.0-fold higher binding of EVDRL to DR4 and DR5, respectively, compared to DRL (Fig. 4L). These results indicated that the EV domain of EVDRL enhances the apoptosis-inducing function by increasing its binding to DR4/5 (Fig. 4M). ESDRL has lesser binding to DR5 compared to EVDRL resulting in its reduced apoptosis-inducing effect (fig. S6K).
EVDRL-secreting stem cells kill BLBC cells in vitro and in vivo
We generated human MSC (hMSC) expressing EVDRL and confirmed continuous secretion of EVDRL for 120 hours (Fig. 5A). hMSC expressed substantial levels of DR4/5 and high levels of DcR2 expression (fig. S5C). Coculture with BMET02-FmC showed that hMSC-EVDRL had more potent tumor-killing ability than hMSC-DRL (Fig. 5B and movie S2). In parallel, we also generated DRL- and EVDRL-secreting mouse NSC (mNSC), mouse MSC (mMSC), and induced pluripotent stem cell (iPSC)–derived NSCs (iPSC-NSC). All the EVDRL-secreting stem cells tested had significantly higher tumor cell killing activity compared with those secreting only DRL (Fig. 5C and fig. S7). Next, we encapsulated EVDRL-secreting stem cells in synthetic extracellular matrix (sECM) that enabled us to prevent “wash out” of therapeutic stem cells and retain them within the tumor resection cavity based on our previous work with glioblastoma resection model (40) and showed that they have efficient tumor killing abilities of BMET02-FmC cells in vitro (Fig. 5D and fig. S8A). We also confirmed that hMSC are viable in the mouse brain for at least 2 weeks after implantation (fig. S8B). In addition, we confirmed that hMSC-EVDRL treatment did not induce any significant toxicity, as indicated by stable body weight of treated mice as well as unremarkable histology of CNS and major organs (fig. S8, C to E).
Fig. 5. EVDRL-secreting stem cells have antitumor effects against BLBC in vitro and in vivo.
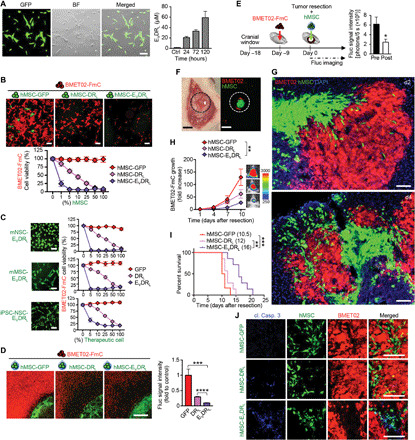
(A) Left: Photomicrograph of EVDRL-secreting hMSC. Scale bar, 100 μm. Right: Concentration of EVDRL in culture media of hMSC-EVDRL quantified by enzyme-linked immunosorbent assay (ELISA) (n = 2, technical replicates). (B) Top: Photomicrographs of BMET02-FmC cocultured with hMSC-GFP/DRL/EVDRL for 72 hours. Scale bars, 100 μm. Bottom: Cell viability of BMET02-FmC after 72-hour coculture with increasing percentages of hMSC-GFP, hMSC-DRL, or hMSC-EVDRL (n = 3, technical replicates). (C) Photomicrographs of different engineered stem cells (left) (scale bars, 100 μm) and cell viability of BMET02FmC cocultured with increasing percentages (0 to 100) of the stem cells (right) (n = 3, technical replicates). (D) Left: Photomicrograph of BMET02-FmC cocultured with sECM-encapsulated hMSC-GFP, hMSC-DRL, or hMSC-EVDRL. Scale bar, 1 mm. Right: Relative number of BMET02-FmC cells 72-hour following coculture with sECM-encapsulated hMSC-GFP, hMSC-DRL, or hMSC-EVDRL (n = 3, technical replicates). (E) Left: Experimental outline for testing efficacy of sECM-encapsulated hMSC-EVDRL in BMET02-FmC–bearing mice. Right: BLI signals before and after resection (n = 20). (F) Intraoperative photographs of light and fluorescence of mice implanted sECM-hMSC into the resection cavity of BMET02-FmC tumor. Scale bars, 1 mm. (G) Representative photomicrographs of brain section from mice 2 and 4 days after resection of BMET02-FmC tumor and implantation of sECM-hMSC. Scale bars, 100 μm. (H) Estimate of relative tumor volume after resection in treatment groups based on Fluc signal intensity of BMET02-FmC (hMSC-GFP, n = 6; hMSC-DRL, n = 7; hMSC-EVDRL, n = 7). (I) Kaplan-Meier survival curves of the mice with median survival (days) indicated in the legend. (J) Immunohistochemistry of cleaved caspase-3 of brain sections from treated and control mice. Scale bars, 100 μm. Photo credit: Yohei Kitamura, Brigham and Women’s Hospital
To explore the effect of EVDRL-secreting hMSC in vivo, we first used a macrometastasis mouse model. Nine days after stereotactic implantation of BMET02-FmC in the brain, the tumor was resected partially, and hMSCs expressing green fluorescent protein (GFP), DRL, or EVDRL were encapsulated in sECM and implanted in the resection cavity (Fig. 5, E and F). Brain histology showed that implanted sECM-encapsulated hMSCs migrated toward the tumor (Fig. 5G). BLI and histology of tissues revealed a significant decrease in tumor volumes in hMSC-EVDRL–treated mice as compared to the control group (Fig. 5H and fig. S8F). Also, the hMSC-EVDRL–treated group showed significantly longer survival time than hMSC-GFP– and hMSC-DRL–treated groups (Fig. 5I). Immunofluorescence showed that hMSC-EVDRL induced increased caspase-3 cleavage in tumor cells compared to hMSC-GFR or hMSC-DRL (Fig. 5J). These data reveal that stem cells engineered to simultaneously target EGFR and DR4/5 have potent efficacy in mouse tumor models of BM resection.
ICA-injected EVDRL-secreting stem cells effectively control PVN micrometastasis
To explore the therapeutic potential of stem cell delivery of EVDRL across BBB to micrometastatic cells within PVN, we used ICA injection of EVDRL-secreting stem cells. Since there is limited literature on the fate of ICA-injected stem cells, we first investigated the fate of five different types of stem cells (hMSC, hNSC, mNSC, mMSC, and iPSC-NSC) after ICA injection either in the presence or absence of tumors. Among the different stem cells tested, mNSCs survived in the brain for the longest period (fig. S9A). Since an extended survival of stem cells may increase their potential of tumor formation, we engineered mNSCs to coexpress EVDRL and a kill switch, prodrug-converting enzyme HSV-TK (TK) and confirmed that mNSC-TK and mNSC-EVDRL-TK cells were eradicated by ganciclovir (GCV) treatment (fig. S9B). Coculture with BMET02-FmC did not show additional therapeutic benefit of TK in mNSC-EVDRL-TK presumably because the highly efficient tumor killing by EVDRL masked other effects, but when cocultured with BT549-FmC, a relatively less sensitive BLBC line, an enhanced tumor killing effect was observed with mNSC-EVDRL-TK (fig. S9C). Next, we used BMET02-FmC micrometastatic models and ICA-injected mNSC-GFP or mNSC-EVDRL-TK and the BLI signal and monitored animal survival (Fig. 6A). Histology showed the presence of widely distributed stem cells along the brain vasculature that colocalized with BMET-02-FmC tumor cells (Fig. 6B). mNSC-EVDRL-TK treatment significantly extended both macrometastasis-free survival (Fig. 6, C and D) and overall survival (Fig. 6E) compared to mNSC-GFP. Further, GCV administration in mNSC-EVDRL-TK–treated mice resulted in similar therapeutic benefit compared with mNSC-EVDRL-TK alone, indicating enough safety of such therapeutic stem cells in a preclinical BM model, as well as little benefit of TK to mNSC-EVDRL in this cohort. Together, these studies reveal that ICA injection of EVDRL-secreting stem cells has therapeutic efficacy in mouse models of BC brain micrometastases.
Fig. 6. EVDRL-secreting stem cells show antitumor efficacy for micrometastasis of BLBC.
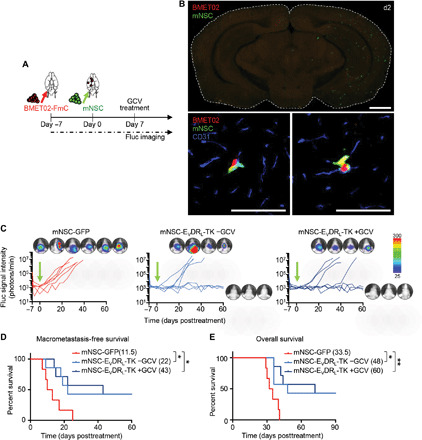
(A) Experimental outline for testing efficacy of ICA injection of mNSC-EVDRL-TK in mice that had ICA injection of BMET02-FmC 7 days before. (B) Top: Representative photomicrograph of whole brain section from ICA-injected BMET02-FmC–bearing mice 2 days after ICA injection of mNSC. Scale bar, 1 mm. Bottom: Representative photomicrograph of immunohistochemistry of CD31 of the brain section. Scale bars, 100 μm. (C) BLI signal curves and photographs of mice treated with mNSC-GFP/EVDRL-TK +/−GCV (mNSC-GFP, n = 6; mNSC-EVDRL-TK −GCV, n = 7; mNSC-EVDRL-TK +GCV, n = 7). (D) Kaplan-Meier curves of macrometastasis-free survival. The presence of macrometastasis was judged from the substantial BLI signal around 1 × 104 photons/min. Median macrometastasis-free survivals (days) are indicated in the legend. (E) Kaplan-Meier curves of overall survival of mice with median overall survival (days) indicated in the legend.
IT-injected EVDRL-secreting stem cells effectively treat leptomeningeal metastasis
To explore the efficacy of stem cell delivery of EVDRL for leptomeningeal metastasis, we tested IT injection of hMSC secreting EVDRL. First, we confirmed that hMSCs survived in the CSF space for at least 2 weeks after IT injection in naïve mice and were gradually cleared out (Fig. 7A). In addition, we confirmed that IT injection of stem cells did not affect mice body weight (fig. S10A). Next, we showed that IT-injected hMSC homed to the tumors of IT-injected BMET02-FmC that were growing in the CSF space of different CNS regions (Fig. 7B). hMSC-EVDRL secreted a substantial amount of EVDRL into the CSF (Fig. 7C). hMSCs engineered to express GFP and renilla luciferase (Rluc) (hMSC-GRl) or hMSC-EVDRL were IT-injected twice to BMET02-FmC–bearing mice and evaluated by BLI and survival monitoring (Fig. 7D). Rluc imaging detected accumulation of hMSC-GRl after two implantations, and Fluc imaging of tumor cells and harvested brain and spinal cord samples showed that hMSC-EVDRL significantly suppressed tumor growth (Fig. 7, E to G, and fig. S10B). Compared to the control, treatment with hMSC-EVDRL resulted in significant improvement in survival of tumor-bearing mice (Fig. 7H). We also tested the efficacy of hMSC-EVDRL on the tumors generated from another BLBC cell line, MDA231-BrM2-FmC (fig. S10C) (21). Mice bearing MDA231-BrM2-FmC tumors treated with IT-injected hMSC-EVDRL demonstrated a significant survival benefit (Fig. 7, I and J). Immunofluorescence of cleaved caspase-3 showed apoptosis induction in hMSC-EVDRL–treated tumors (Fig. 7K). These results clearly demonstrated the therapeutic efficacy of IT-injected hMSC-EVDRL in mouse models of BC leptomeningeal metastases.
Fig. 7. EVDRL-secreting stem cells have antitumor effects in leptomeningeal metastasis of BLBC.
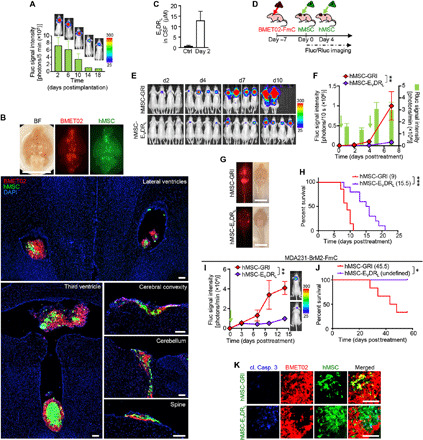
(A) BLI signal and photographs of IT-injected hMSC-GFP-Fluc (GFl)–bearing mice (n = 3). (B) Top: Representative photograph of whole-brain sample of an IT-established, BMET02-FmC leptomeningeal metastasis–bearing mice, 2 days after IT injection of hMSC. Scale bar, 10 mm. Bottom: Representative photomicrographs of brain and spine sections from the mice. Scale bars, 100 μm. (C) Concentration of EVDRL in CSF from mice before and 2 days after IT injection of hMSC-EVDRL quantified by ELISA (n = 3). (D) Experimental outline for testing efficacy of IT injections of hMSC-EVDRL in mice that had IT injection of BMET02-FmC 7 days before. (E) Representative BLI pictures of IT-injected BMET02-FmC–bearing mice treated with hMSC-GRl or hMSC-EVDRL. (F) Fluc signal curves of BMET02-FmC treated with hMSC-GRl or hMSC-EVDRL and Rluc signals of injected hMSC-GRl (hMSC-GRl, n = 7; hMSC-EVDRL, n = 10). (G) Representative photographs of whole-brain sample of IT-injected BMET02-FmC–bearing mice treated with hMSC-GRl or hMSC-EVDRL for 7 days. Scale bars, 10 mm. (H) Kaplan-Meier curves of overall survival of mice. Median survivals (days) are indicated in the legend. (I) Fluc signal curves and representative BLI images of mice bearing MDA231-BrM2-FmC tumors treated with hMSC-GRl or hMSC-EVDRL (hMSC-GRl, n = 6; hMSC-EVDRL, n = 5). (J) Kaplan-Meier curves of overall survival of mice with median survival (days) indicated in the legend. (K) Immunohistochemistry of cleaved caspase-3 in brain sections from IT-injected BMET02-FmC–bearing mice treated with hMSC-GRl or hMSC-EVDRL. Scale bars, 100 μm.
DISCUSSION
In this study, we developed different imageable mouse models of BLBC-BM and explored the versatility of stem cell–mediated bi-functional EGFR and DR4/5 therapeutics in these models. Our results show that the EV domain of EVDRL enhances DRL-induced apoptosis in a broad spectrum BLBC lines, and the stem cell–secreted EVDRL targets PVN micrometastasis and leptomeningeal metastasis, thus offering a promising therapeutic strategy for BLBC-BM.
Clinically relevant metastatic BM models are usually developed by intracardiac administration of cancer cells; however, this leads to widespread extracranial tumor formation (36, 45, 46). Standard ICA injections also cause extracranial tumors in the face (37–39). When using sagittal imaging as we show in fig. S3 (A to C), we observed extracranial tumor formation in about 80% of cases following standard ICA injections. Nevertheless, a number of previous studies have disregarded these findings. In these models, signal emitted from extracranial (often facial) tumors substantially confounds BLI evaluation of brain tumor progression and treatment benefits. In this study, we refined the technique of ICA injection, and our modified approach reduced the rate of extracranial tumor formation to less than 20% of cases, enabling accurate BLI monitoring of intracranial tumors and evaluation of treatment efficacy. Although creating our model requires advanced technical skills and is time consuming, we believe that creating and optimizing mouse tumor models of metastasis confer multiple advantages in advancing BM preclinical research.
Although promising, there may be some potential limitations in the models we developed and used in this study. Since BM is clinically developed by the cells via arterial extravasation, arterial injection is regarded as an optimal way to develop BM models (47). As ICA injection does not reproducibly induce resectable intracranial tumor formation (single large tumor at surgically accessible location), we had to use orthotopic BM cell line injections for developing BM resection model. Although not optimal, this model has some advantages, e.g., easier to develop models uniform in size and timing, especially for testing some therapeutic efficacy in limited living materials. Although we observed similar microenvironmental findings to arterially injected models, there is no doubt that arterial injection is the preferred way to create BM models for study when tumor resection is not considered.
DRL is a well-investigated molecule, which induces cancer-specific apoptosis. It is also known that BLBC is sensitive to DRL (23); however, the underlying mechanism has been unclear. We showed that the increased DRL sensitivity of BLBC is closely associated with up-regulated DR4/5 expression. However, DR-targeted therapies have shown poor efficacy (24, 25). Enhancement of the efficacy of DRL has been attempted by many researchers via approaches such as molecular modifications (to stabilize it or prolong its longevity), combination with other molecules, and sensitizing the target site to DRL (25, 48). Here, since our initial aim of fusing the EV domain to DRL was to block EGFR signaling, it was an unexpected discovery that the EV domain augmented DRL-DR4/5 binding and thereby enhanced the efficacy of EVDRL.
Because EGFR is up-regulated in BLBC (19) and is a marker of poor prognosis (49), it is a promising target for BLBC. However, EGFR-targeting therapy has failed to show any survival benefits (20). Consistently, BLBC cell lines were resistant to two EGFR inhibitors herein. However, we showed that the EV domain of EVDRL significantly improved the efficacy of DRL, suggesting that EGFR can be used as an anchor to increase therapeutic targeting of other cell surface molecules in BLBC and other EGFR-expressing cancers. We also showed that EGFR and DR5 expression levels are significantly related to the efficacy of EVDRL. These results imply that analysis of resected tumor tissue from patients could allow a prediction of efficacy of this treatment and thus offer personalized treatments. We show that VHH of anti-EGFR was more effective than scFv when fused to the N terminus of DRL. VHH composes of a single domain and is more stable and robust than scFv, which has a linker connecting two domains requiring a supramolecular assembly. Also, VHH has a unique epitope that is longer and contains a more flexible complementary determining region, which increases the affinity to corresponding receptors (50). These differences between VHH and scFv might contribute to the higher efficacy of EVDRL.
Our findings indicate that BLBC’s sensitivity to EVDRL is mainly determined by the expressions of EGFR and DRs. Although there are likely other apoptosis-related factors involved in BLBC’s sensitivity to EVDRL, e.g., Myc, our studies did not demonstrate how these factors could influence the sensitivity to EVDRL. Further investigations are needed to completely understand the influence of these factors on BLBC’s sensitivity to EVDRL. In this study, we have not compared the difference in efficacy between stem cell–delivered EVDRL therapy and systemic venous injection of EVDRL; however, given the short half-life of DRL and the inability of a majority of drugs to cross BBB, we anticipate that stem cell–delivered EVDRL will be a more effective treatment paradigm for BM.
BC often presents metastasis years or even decades after treatment and apparent good disease control (11, 12). This suggests that disseminated tumor cells of BC stays dormant within the PVN in the distant organs for a long period; these cancer cells outside the vasculature are difficult to target with systemically administered drugs (13, 14). We successfully developed cancer models with BC cells residing in the brain PVN and used stem cells to deliver therapeutic molecules. However, the treatment for dormant cancer cells in PVN is difficult due to the inability to detect such scattered cells in patients and drug delivery beyond BBB. Given the ability of engineered stem cells to migrate to cancer cells and penetrate BBB (30–32), our stem cell–based treatment has the potential to overcome that. Our results indicate that clinical trials of this therapy using selective arterial administration by neuroendovascular devices for patients with BC who had initial radical treatment are a feasible option to erase dormant cancer cells before growth, prevent future macrometastasis in the brain, and should be considered. Recent studies reported the existence of micrometastatic cancer cells in other organs, such as the lung, bone, and liver, and recurrence in these organs long periods after initial therapy is a considerable challenge (12). Our approach of stem cell delivery of potent therapeutics offers an immense potential of killing cancer cells dormant at the PVN in these organs.
We also showed that stem cell delivery is a promising approach for treating leptomeningeal metastasis, which is considered a terminal condition without any effective therapeutic strategies. We showed that IT-injected stem cells could stay alive in the niche for weeks and secrete therapeutic molecules into CSF without affecting the general health of the mouse. Clinical trials of this therapy should be considered for patients with BLBC leptomeningeal metastasis in the future. IT stem cell therapy has already been established as a safe treatment and tested in patients of trauma (51), stroke (52), epilepsy (53), and neurodegenerative diseases (54, 55). Two animal studies have reported IT stem cell therapies for disseminated primary brain tumor—glioma and medulloblastoma (56, 57). However, to our knowledge, there is no previous report showing efficacy of stem cell therapy for leptomeningeal metastasis, a secondary CNS tumor. The same strategy might be effective for leptomeningeal disease originating from other types of primary and metastatic cancers.
In conclusion, we demonstrate the efficacy of a stem cell–delivered therapeutics against EGFR and DR4/5 in mouse models representing three clinically challenging BM conditions. Our findings provide a scientific rationale that supports clinical trials of this strategy in patients with BLBC-BM.
MATERIALS AND METHODS
Antibodies and reagents
The following antibodies and reagents were used in this study. Antibodies against β-actin (#4970), phospho-AKT (Ser473, #4060), AKT (#9272), caspase-7 (#9492), caspase-8 (#9746), caspase-9 (#9508), cleaved caspase-3 (#9661), EGFR (#2646 and #4267), phospho-EGFR (Tyr1068, #3777), cleaved poly(ADP-ribose) polymerase (PARP; #9541), phospho-p44/42 mitogen-activated protein kinase (MAPK) (ERK1/2) (Thr202/Tyr204, #9101), p-44/42 MAPK (ERK1/2) (#9102), Fas-associated death domain protein (#2782), Bcl-2 (#2872), Bcl-xL (#2764), XIAP (#2042), cIAP2 (#3130), phospho–signal transducers and activators of transcription 3 (STAT3) (Tyr705, #9145), STAT3 (#4904), HER2 (#2242), horseradish peroxidase (HRP) anti-rabbit (#7074), Rab5 (#46449), Rab7 (#95746) (Cell Signaling Technology), anti–nuclear factor κB (#ab16502), anti-TRAIL (#ab9959), anti-CD31 (#ab28364), HRP anti-mouse (#ab205719) (Abcam), anti–α-tubulin (#T5168), anti-Vinculin (#V4505), NeuN (#MAB377), glial fibrillary acidic protein (GFAP) (#MAB3402) (Sigma-Aldrich), Alexa Fluor 488 anti-EGFR antibody (#352908), anti-DR4 (#1139), anti-DR5 (#2019) (ProSci), anti-DR4 (#sc-32255), anti-DR5 (#sc-166624), anti-cIAP1 (#sc-271419), normal mouse IgG (#sc-2025) (Santa Cruz), anti-Ki-67 (#180191Z), anti-GFAP (#180063), Alexa Fluor anti-rabbit 405 (#A-31556), Alexa Fluor anti-rabbit 488 (#A-11008), Alexa Fluor anti-mouse 555 (#A-21422), Alexa Flour anti-rabbit 647 (#A-21244), Phycoerythrin (PE) anti-DR4 (#12-6644-42), PE anti-DR5 (#12-9908-42), PE mouse IgG isotype (#12-4714-42) (Invitrogen), Cetuximab (ImClone Systems), Erlotinib (#SYN-1039, Selleck Chemicals), human recombinant EGF (R&D Systems), PE anti-EGFR (#352903, BioLegend), and IBA1 (#019-19741, FUJIFILM).
Immunohistochemistry for patient tissue samples
TNBC patient tissue samples were obtained from Massachusetts General Hospital as approved by institutional review board (IRB) at Harvard Medical School, Keio University Hospital as approved by IRB of Keio University School of Medicine, and US Biomax Inc. (#BR1901). Immunohistochemical analyses of patient samples were performed by Servicebio Inc. and iHisto Inc., using antibodies for EGFR (#GB13804), estrogen receptor (#GB11205), progesterone receptor (#GB11262) from Servicebio Inc. and the antibodies listed above.
Cell lines
Patient-derived BLBC-BM cell lines (BMET02, BMET05, BMET15) were obtained by dissociation of brain tumor samples from patients with metastatic breast carcinoma diagnosis and cultivated as described below. Brain tumor samples were obtained as approved by IRB at Harvard Medical School. Established BC cell lines MDA-MB-453, MCF7, HCC1500, HCC1428, ZR75-1, BT474, T47D, MDA-MB-175VII, and SUM159 were provided by A. Tilston-Lunel, Bob Varelas laboratory, Boston University. SKBR3 was provided by N. Wang, Massachusetts General Hospital. MDA-MB-231, MDA-MB-231-BrM2, MDA-MB-436, and MDA-MB-468 were provided by J. Massagué, Memorial Sloan Kettering Cancer Center. BT549, Hs578T, and NIH-3T3 were purchased from American Type Culture Collection. The immortalized hMSC line, hASC-TS, was a gift from L. Balducci (58). Immortalized mNSC line, C17.2, was provided by E. Y. Snyder (59). Bone marrow–derived mMSC line was obtained from D. Prockop, University of Texas. Immortalized human fetal NSC (hNSC) line, hNSC100, was provided by A. Martínez-Serrano, Autonomous University of Madrid. Mouse iPSC-NSC was generated from mouse embryonic fibroblasts as previously described (60).
BMET02, BMET05 BMET15, MDA-MB-453, MCF7, MDA-MB-175VII, MDA-MB-231, MDA-MB-231-BrM2, MDA-MB-436, and MDA-MB-468 were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (vol/vol) fetal bovine serum (FBS) and 1% (vol/vol) penicillin/streptomycin. Hs578T was grown in DMEM supplemented with 10% (vol/vol) FBS, insulin (0.01 mg/ml), and 1% (vol/vol) penicillin/streptomycin. SKBR3 was grown in McCoy’s 5a medium with 10% (vol/vol) FBS and 1% (vol/vol) penicillin/streptomycin. HCC1500, HCC1428, ZR75-1, BT474, and BT549 were grown in RPMI 1640 with 10% (vol/vol) FBS and 1% (vol/vol) penicillin/streptomycin. T47D was grown in RPMI 1640 with 10% (vol/vol) FBS, 1% (vol/vol) penicillin/streptomycin, and insulin (0.2 U/ml). SUM159 was grown in Ham’s F-12 with 5% (vol/vol) FBS, insulin (0.005 mg/ml), hydrocortisone (1 μg/ml), and 1% (vol/vol) penicillin/streptomycin. NIH-3T3 were cultured in DMEM supplemented with 10% NCS, penicillin (100 U/ml), and streptomycin (100 μg/ml). hMSC was grown in DMEM/F-12 supplemented with 10% (vol/vol) FBS, 1% (vol/vol) l-glutamine, 1% (vol/vol) penicillin/streptomycin, and recombinant human fibroblast growth factor (FGF) (40 ng/ml; R&D Systems, Minneapolis, MN). mNSC was grown in DMEM supplemented with 10% (vol/vol) FBS, 1% (vol/vol) l-glutamine, and 1% (vol/vol) penicillin/streptomycin. mMSC was grown in low-glucose DMEM supplemented with 15% (vol/vol) FBS, 1% (vol/vol) l-glutamine, 1% (vol/vol) nonessential amino acid solution, and 1% (vol/vol) penicillin/streptomycin. hNSC was cultured in 4:1 culturing medium [DMEM/F-12 (Invitrogen), 0.6% d-glucose (Sigma-Aldrich), 0.5% albumax (Invitrogen), 0.5% glutamine (Invitrogen), recombinant human FGF (40 ng/ml; R&D Systems), recombinant human EGF (40 ng/ml; R&D Systems), N2 supplements (Invitrogen), and 1% nonessential amino acid solution (Cellgro; Mediatech)] and growth medium [DMEM with 5% FBS, 1 mM sodium pyruvate (Cellgro; Mediatech), and 26 mM sodium bicarbonate]. iPSC-NSC was grown in NeuroCult basal medium (Stem Cell Technologies) supplemented with EGF (20 ng/ml; R&D Systems), FGF2 (20 ng/ml; R&D Systems), N2 supplement, Heparin, and 1% (vol/vol) penicillin/streptomycin on Geltrex (Fisher Scientific)–precoated flask.
Cell viability and caspase assays
Tumor cells were plated in 96-well plates and treated with different doses of anti-EGFR VHH (EV), DR ligand (DRL), EVDRL, or anti-EGFR scFv-TRAIL (ESDRL) and control media for 24, 48, and 72 hours. To obtain conditioned media containing these proteins, lentiviral plasmid vectors coding for EV, DRL, EVDRL, and ESDRL were transfected into 293T cells. Medium was changed the next day, collected 40 hours after transfection, concentrated using centrifugal filter (#UFC901024, MilliporeSigma), and stored at −80°C until future use. Their concentrations were quantified by enzyme-linked immunosorbent assay (ELISA). Control media were made from GFP control–transduced cells transduced in parallel with EV, DRL, EVDRL, and ESDRL. Cell viability was measured using an adenosine triphosphate–dependent luminescent reagent (CellTiter-Glo, #G755A, Promega; Glomax, Promega) according to the manufacturer’s instructions for non-Fluc–expressing cells or with d-luciferin (#122799, PerkinElmer) and coelenterazine h (#760506, PerkinElmer) for Fluc- and Rluc-expressing cells, respectively. Caspase-3/7 activity was determined using a DEVD-aminoluciferin assay (Caspase-Glo 3/7, #G8091, Promega) according to the manufacturer’s instructions. All experiments were performed in triplicates.
Western blot analysis
After treatment, cells were washed with cold phosphate-buffered saline (PBS) and then lysed with cold NP-40 lysis buffer (#BP-119, Boston BioProducts) supplemented with protease inhibitor (#A32965, Thermo Fisher Scientific) and phosphatase inhibitors (#P5726 and #P0044, Phosphatase Inhibitor Cocktail 2,3 from Sigma-Aldrich). Cells were scraped into tubes and centrifuged at 4°C at 13,000g for 10 min. Supernatant protein concentrations were determined using the Bio-Rad DC Protein Assay Kit (#500-0113, #500-0114, and #500-0115). The 6× SDS sample buffer (#BP-111R, Boston BioProducts) was added to the samples, which were then boiled for 5 min. Ten to forty micrograms of protein was loaded on SDS–polyacrylamide gel electrophoresis gel (#456-1086 and #456-1093, Bio-Rad), transferred to polyvinylidene difluoride membrane (#IPVH00010, Merck Millipore), and probed with primary antibodies overnight. After wash, the membrane was probed with secondary antibodies and developed with enhanced chemiluminescence (#1863096, #1863097, and #34095, Thermo Fisher Scientific).
Flow cytometry analysis of cell surface receptors
Cells were trypsinized, washed, and resuspended in stain buffer (#554657, BD Biosciences). Cells were stained with PE-conjugated anti-human EGFR, DR4, or DR5 antibodies in solution at 4°C for 30 min. For the double staining, PE-conjugated anti-DR5 antibody and Alexa Fluor 488–conjugated anti-EGFR antibody were used. Rinses were performed with stain buffer at 4°C. PE-conjugated isotype-specific IgG was used as a control. Flow cytometry was performed using FACSAria II (BD) cell sorter, and data were analyzed using FlowJo (BD).
Coimmunoprecipitation
After treatment, cells were washed with PBS twice and then lysed with radioimmunoprecipitation assay (RIPA) buffer (#BP-115, Boston BioProducts) supplemented with protease inhibitor (#A32965, Thermo Fisher Scientific). Cells were scraped into tubes and centrifuged at 4°C at 13,000g for 20 min. Supernatant protein concentrations were determined using the Bio-Rad DC Protein Assay Kit. A mixture of 1 mg of protein, 20 μl of anti-human DR4/5 antibodies, and 30 μl of Protein A/G agarose (#sc-2003, Santa Cruz) was incubated overnight at 4°C. After washing with RIPA buffer, samples were boiled with 6× SDS sample buffer (#BP-111R, Boston BioProducts) for 8 min. Samples were then centrifuged at 4°C at 13,000g for 2 min, and the supernatant was used for Western blot.
Lentiviral transductions and engineering of stable cell lines
Lentiviral vector of EVDRL was constructed by inserting cDNA encoding extracellular domain of DRL into LV-anti EGFR VHH (EV). Lentiviral vector of ESDRL was constructed by replacing EV domain of EVDRL with cDNA encoding anti EGFR scFv (Es). We used previously described lentiviral vectors of GFP, EV, and DRL (27, 61). Lentiviral packaging was performed by transfection of 293T cells as previously described (62), and cells were transduced with lentiviral vectors at multiplicity of infection of 2 in medium containing protamine sulfate (2 μg/ml). For BLI, cells were transduced with LV-Pico2-Fluc-mCherry, LV-Pico2-Rluc-mCherry, LV-Pico2-Fluc-GFP, or LV-Pico2-Rluc-GFP. They are selected by fluorescence-activated cell sorting (FACS) using a BD FACSAria Fusion cell sorter or by puromycin selection (1 μg/ml) in culture. GFP or mCherry expression was visualized by fluorescence microscopy.
Coculture and GCV treatment experiments
BMET02-FmC cells (2 × 103 cells per well) were cocultured with different numbers of therapeutic stem cells in 96-well plates. After 72 hours, the relative number of BMET02-FmC cells was determined by Fluc luminescence (Glomax, Promega). For coculture experiments with encapsulated stem cells, 5 × 104 of hMSC-GFP/DRL/EVDRL cells were encapsulated with 10-μl sECM (HyStem-C Hydrogels, #GS313, BioTime) and added at the center of the well of a 24-well plate. After 30 min, BMET02-FmC was seeded around the gel. After 72 hours, the relative number of BMET02-FmC cells was counted by Fluc luminescence (IVIS Lumina, PerkinElmer). For in vitro GCV treatment, cells were treated with GCV (5 μg/ml) for 96 hours, and the relative cell number of them was quantified by CellTiter-Glo (Promega).
CRISPR KO of DR4 and DR5
CRISPR KO of DR4 and DR5 was conducted as previously described (61, 63). To establish KO lines, cells were transduced with lentiviral Cas9 expression vectors coding for either tetracycline-inducible or constitutively expressed Cas9 protein as previously described (64, 65). Confirmed Cas9 lines were engineered with lentiviral single guide RNA (sgRNA) expression vector pLKO.DEST.hygro containing the sgRNA target sequences described above for DR4 or DR5, followed by selection with hygromycin (200 to 500 μg/ml). For generating the double KO lines, confirmed Cas9 lines were coengineered with pLKO.DEST.hygro and pLKO.DEST.egfp lentiviral expression vectors to express both DR4 and DR5 targeting sgRNAs followed by selection with hygromycin and FACS sorting for GFP.
ELISA of DRL
Concentrations of DRL released from therapeutic stem cell lines in culture medium or in CSF of mice that had IT injection of stem cells were quantified using a human-specific TRAIL antigen capture ELISA kit (#ab46074, Abcam).
Subtyping of cell lines
Genomic RNA from cell lines was extracted using the RNeasy Mini Kit (#74104, Qiagen). mRNA array was performed using a PAM50 plate (NanoString), and the results were analyzed by the company (35). Subtypes were decided on the basis of the algorithm from the company.
Real-time FRET imaging
Confocal images were acquired with a Leica TCS SP8 Falcon system equipped with a 440-nm pulsed SMD diode laser and a tunable white light laser (WLL). YFP constructs were imaged with 512-nm WLL excitation, and emissions were collected over the range of 533 to 565 nm. CFP constructs were imaged with a 440-nm excitation laser and an emission of 465 to 495. For sensitized emission FRET experiments, BMET02 or NIH-3T3 cells were plated on 22 × 50 mm glass coverslips and cotransfected with LV-EGFR-YFP and LV-DR5-CFP using Lipofectamine LTX transfection reagent. Coverslips were mounted onto a JG-23W HP flow-through perfusion chamber (Warner Instruments), and cells were maintained in Ringer’s solution containing 125 mM NaCl, 25 mM Hepes, 10 mM glucose, 5 mM K2HPO4, 1 mM MgSO4, and 1 mM CaCl2 (pH 7.40). Real-time FRET imaging experiments were performed using fluorescence ratio imaging systems built around a Nikon Eclipse TE2000-U inverted epifluorescence microscope equipped with an Andor Ultra 888 EM-CCD camera and a 60× Plan Apo TIRF (total internal reflection fluorescence) (numerical aperture, 1.45) oil immersion objective. Filter wheels (Sutter Instruments) were placed in the excitation and emission path, and image acquisition parameters were controlled by Metafluor software (Molecular Devices). FRET emission ratios (480 nm/535 nm; 440-nm excitation) were acquired every 10 s. After establishing baseline FRET levels for 3 to 10 min, EVDRL (2 μM) in Hepes-buffered Ringer’s solution was added manually to the chamber with a pipette, and FRET changes were followed for 30 to 40 min.
Immunofluorescence analyses of endocytosis proteins
BMET02 cells were treated with control media or EVDRL (2 μM) after 4-hour starvation and fixed on the plates 5 and 15 min after treatment. Cells were stained with primary antibodies (EGFR and Rab5/Rab7) and counter-stained with secondary antibodies (Alexa Fluor anti-rabbit 488 and Alexa Fluor anti-mouse 555, respectively).
Animal models
All in vivo procedures were approved by the Subcommittee on Research Animal Care at Brigham and Women’s Hospital. Mice that died or were euthanized for ethical reasons before defined experimental end points were excluded. Animals were randomly allocated to cages and experimental groups.
Macrometastasis resection model
Female nude mice (6 to 8 weeks of age, 20 to 25 g, Envigo) were immobilized on a stereotactic frame 9 days before tumor implantation. Using a stereomicroscope (SZX10, Olympus), a small circular portion of the skull covering the right cerebral hemisphere (3 mm by 5 mm) was removed to create a cranial window for subsequent tumor cell implantation and tumor debulking. Nine days later, the mice were again immobilized on a stereotactic frame, the previously established cranial window was exposed, and BMET02-FmC (5 × 104 cells per mouse) in 4 μl of PBS was superficially implanted into the right frontal cerebral cortex (2-mm lateral from bregma, 0.5-mm deep) using a microsyringe (Hamilton). Nine days after the implantation of tumor cells, the mice underwent fluorescence-guided tumor resection followed by implantation of hMSC into the resection cavity as previously described. hMSC (5 × 105 cells per mouse) was encapsulated in 10 μl of HyStem-C Hydrogels (#GS313, BioTime) 20 min before implantation to allow gel formation. Mice were then followed up for survival and sacrificed when neurological symptoms became apparent. Mice whose BLI signal disappeared completely after resection were excluded from this study.
Micrometastasis model
The detailed technique of ICA injection of tumor/stem cells is demonstrated in movie S1. Female nude mice (6 to 8 weeks of age) were anesthetized with ketamine-xylazine and fixed on the stage of a stereomicroscope (SZX10, Olympus). Midline skin incision was made to expose the right carotid arteries. Using 8-0 sutures, right OA, PPA, STA, and external carotid artery were ligated to prevent cells from going to extracranial parts. Internal and common carotid arteries were then ligated, and a catheter (#18000-10, Fine Science Tools) connected to a 1-ml syringe (Henke-Sass Wolf) was inserted into the external carotid artery. After releasing blood flow of common and internal carotid arteries, BMET02-FmC (5 × 104 cells per mouse) suspended in 100 μl of PBS was slowly injected through the catheter. After injection, external carotid artery was permanently ligated. Mice with apparent extracranial tumors that could disturb the evaluation were excluded from the study. Seven days after tumor injection, stem cells were injected into the same artery using the same technique as above without ligations of OA, PPA, and STA. mNSC (4 × 105 cells per mouse) in 100 μl of PBS was slowly injected.
Leptomeningeal metastasis model
IT injection of tumor was performed on the basis of previous literature (66) with slight modifications. Female nude mice (6 to 8 weeks of age) were immobilized on a surgical platform after anesthesia with ketamine-xylazine. Midline skin incision was made behind the neck, and occipital muscles were dissected. The dura mater between skull and atlas vertebra was exposed. Under observation of cerebellum and brainstem through the dura mater, a catheter connected to microsyringe (Hamilton) was inserted into cisterna magna. BMET02-FmC (2.5 × 104 cells per mouse) or MDA231-BrM2-FmC (5 × 104 cells per mouse) in 4 μl was injected slowly through the catheter. The hole in the dura mater was closed with a small muscle piece immediately after removing catheter. hMSC (5 × 105 cells per mouse) was injected in a similar manner via the same hole from the previous injection.
Orthotopic injection
For testing the fate of stem cells, and for creating tumor-bearing mice for screening of ICA-injected stem cells, orthotopic injection into brain was performed. Mice were immobilized on a stereotactic frame, BMET02-RmC cells (1 × 105 cells per mouse) or hMSC (5 × 105 cells per mouse) in 4 μl of PBS were implanted into the right frontal cerebrum (2-mm lateral and 1-mm anterior from bregma, 2.5-mm deep) using stereotactic frame.
In vivo imaging
BLI was used to follow in vivo growth of Fluc- or Rluc-engineered implanted tumor cells over time using a PerkinElmer IVIS Lumina system. For Fluc imaging, mice were imaged 7 min after intraperitoneal injection of d-luciferin (#122799, PerkinElmer). For Rluc imaging, mice were imaged 1 min after intravenous injection of coelenterazine h (#760506, PerkinElmer).
In vivo GCV treatment
For in vivo experiments involving mNSC elimination via the inducible suicide system HSV-TK, mice were treated daily with intraperitoneal injection of GCV (10 mg/kg) for 2 weeks starting 7 days after mNSC injection.
Tissue processing and immunohistochemistry of mouse samples
Tumor-bearing mice were perfused with PBS and subsequently with 4% paraformaldehyde. Brains and spines were harvested, followed by sectioning for histological analyses. Brain and spine sections on slides were washed in PBS and mounted with aqueous mounting medium (#H1000 and #H1200, Vector Laboratories) to be visualized with confocal microscopy (Axio Observer.Z1, Zeiss). For fluorescence immunohistochemistry, sections were incubated with primary antibodies overnight at 4°C. After wash, secondary antibodies were probed and detected by confocal microscope. For hematoxylin and eosin (H&E) staining, sections were incubated with H&E Y dye (1% alcohol), dehydrated with 70, 95, and 100% ethanol, and mounted in xylene-based mounting medium.
Statistical analyses
Data were analyzed by Student’s t test for comparison between two groups and by Pearson’s test for correlation. Data were plotted as means with SD for all in vitro data except Fig. 3E and fig. S4D, and with SEM for all in vivo data, Fig. 3E and fig. S4D. Survival curves were compared using the log-rank test. Analyses were done using Prism 7.0a and 8.3.1 (GraphPad). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Acknowledgments
We thank A. Tilston-Lunel and B. Varelas (Boston University), N. Wang (MGH), L. Balducci (Consorzio CARSO, Italy), E. Snyder (UCSD), J. Massagué (Memorial Sloan Kettering Cancer Center), D. Prockop (University of Texas), and A. Martínez-Serrano (Autonomous University of Madrid) for providing us cell lines. We thank D. Bhere (BWH) for helping with generation of iPSC-NSC, J. K. Khalsa (BWH) for helping with RNA extraction, and H. Wakimoto (MGH) for critical reading of the manuscript. Funding: This work was supported by NIH grants R01-CA201148 (to K.S.) and R01-NS107857 (to K.S.), Overseas Research Fellowships from Uehara Memorial Foundation, and Kanzawa Medical Research Foundation (Y.K.). Author contributions: Y.K.: Conception and design, provision of study material, collection and assembly of data, data analysis and interpretation, manuscript writing, and final approval of manuscript. N.K.: Collection and assembly of data, data analysis and interpretation, and final approval of manuscript. S.M.: Provision of study material, collection and assembly of data, data analysis and interpretation, and final approval of manuscript. W.D.: Conception and design, provision of study material, collection and assembly of data, and final approval of manuscript. C.R.: Provision of study material, collection and assembly of data, data analysis and interpretation, manuscript writing, and final approval of manuscript. N.A., E.R.L., and A.F.: Collection and assembly of data and final approval of manuscript. A.B., H.S., J.L.M., and P.K.B.: Provision of study material and final approval of manuscript. J.L.F., and A.M.H.: Provision of study material, collection and assembly of data, data analysis and interpretation, and final approval of manuscript. K.S.: Conception and design, provision of study material, data analysis and interpretation, manuscript writing, and final approval of manuscript. Competing interests: K.S. owns equity in and is a member of the Board of Directors of AMASA Therapeutics, a company developing stem cell–based therapies for cancer. K.S.’s interests were reviewed and are managed by Brigham and Women’s Hospital and Partners HealthCare in accordance with conflict of interest policies. P.K.B. has received grant/research support (to Massachusetts General Hospital) from Merck, BMS, and Lilly and honoraria from Merck, Genentech-Roche, and Lilly. The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/10/eabe8671/DC1
REFERENCES AND NOTES
- 1.Tabouret E., Chinot O., Metellus P., Tallet A., Viens P., Gonçalves A., Recent trends in epidemiology of brain metastases: An overview. Anticancer Res 32, 4655–4662 (2012). [PubMed] [Google Scholar]
- 2.Lin N. U., Bellon J. R., Winer E. P., CNS metastases in breast cancer. J. Clin. Oncol. 22, 3608–3617 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Siegel R. L., Miller K. D., Jemal A., Cancer statistics, 2018. CA Cancer J. Clin. 68, 7–30 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Mack F., Baumert B. G., Schäfer N., Hattingen E., Scheffler B., Herrlinger U., Glas M., Therapy of leptomeningeal metastasis in solid tumors. Cancer Treat. Rev. 43, 83–91 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Prat A., Adamo B., Cheang M. C. U., Anders C. K., Carey L. A., Perou C. M., Molecular characterization of basal-like and non-basal-like triple-negative breast cancer. Oncologist 18, 123–133 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Penault-Llorca F., Viale G., Pathological and molecular diagnosis of triple-negative breast cancer: A clinical perspective. Ann. Oncol. 23 ( Suppl 6), vi19–vi22 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Smid M., Wang Y., Zhang Y., Sieuwerts A. M., Yu J., Klijn J. G. M., Foekens J. A., Martens J. W. M., Subtypes of breast cancer show preferential site of relapse. Cancer Res. 68, 3108–3114 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Kennecke H., Yerushalmi R., Woods R., Cheang M. C. U., Voduc D., Speers C. H., Nielsen T. O., Gelmon K., Metastatic behavior of breast cancer subtypes. J. Clin. Oncol. 28, 3271–3277 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Oehrlich N. E., Spineli L. M., Papendorf F., Park-Simon T. W., Clinical outcome of brain metastases differs significantly among breast cancer subtypes. Oncol. Lett. 14, 194–200 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamamoto M., Serizawa T., Shuto T., Akabane A., Higuchi Y., Kawagishi J., Yamanaka K., Sato Y., Jokura H., Yomo S., Nagano O., Kenai H., Moriki A., Suzuki S., Kida Y., Iwai Y., Hayashi M., Onishi H., Gondo M., Sato M., Akimitsu T., Kubo K., Kikuchi Y., Shibasaki T., Goto T., Takanashi M., Mori Y., Takakura K., Saeki N., Kunieda E., Aoyama H., Momoshima S., Tsuchiya K., Stereotactic radiosurgery for patients with multiple brain metastases (JLGK0901): A multi-institutional prospective observational study. Lancet Oncol. 15, 387–395 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Goss P. E., Chambers A. F., Does tumour dormancy offer a therapeutic target? Nat. Rev. Cancer 10, 871–877 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Pan H., Gray R., Braybrooke J., Davies C., Taylor C., McGale P., Peto R., Pritchard K. I., Bergh J., Dowsett M., Hayes D. F.; EBCTCG , 20-year risks of breast-cancer recurrence after stopping endocrine therapy at 5 years. N. Engl. J. Med. 377, 1836–1846 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kienast Y., von Baumgarten L., Fuhrmann M., Klinkert W. E. F., Goldbrunner R., Herms J., Winkler F., Real-time imaging reveals the single steps of brain metastasis formation. Nat. Med. 16, 116–122 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Steeg P. S., Camphausen K. A., Smith Q. R., Brain metastases as preventive and therapeutic targets. Nat. Rev. Cancer 11, 352–363 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim H.-J., Im S.-A., Keam B., Kim Y.-J., Han S.-W., Kim T. M., Oh D.-Y., Kim J. H., Lee S.-H., Chie E. K., Han W., Kim D.-W., Kim T.-Y., Noh D.-Y., Heo D. S., Park I. A., Bang Y.-J., Ha S. W., Clinical outcome of central nervous system metastases from breast cancer: Differences in survival depending on systemic treatment. J. Neurooncol 106, 303–313 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Altundag K., Bondy M. L., Mirza N. Q., Kau S.-W., Broglio K., Hortobagyi G. N., Rivera E., Clinicopathologic characteristics and prognostic factors in 420 metastatic breast cancer patients with central nervous system metastasis. Cancer 110, 2640–2647 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Shapiro W. R., Johanson C. E., Boogerd W., Treatment modalities for leptomeningeal metastases. Semin. Oncol. 36, S46–S54 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Scott B. J., Kesari S., Leptomeningeal metastases in breast cancer. Am. J. Cancer Res. 3, 117–126 (2013). [PMC free article] [PubMed] [Google Scholar]
- 19.Reis-Filho J. S., Tutt A. N., Triple negative tumours: A critical review. Histopathology 52, 108–118 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Nakai K., Hung M. C., Yamaguchi H., A perspective on anti-EGFR therapies targeting triple-negative breast cancer. Am. J. Cancer Res. 6, 1609–1623 (2016). [PMC free article] [PubMed] [Google Scholar]
- 21.Bos P. D., Zhang X. H.-F., Nadal C., Shu W., Gomis R. R., Nguyen D. X., Minn A. J., van de Vijver M. J., Gerald W. L., Foekens J. A., Massagué J., Genes that mediate breast cancer metastasis to the brain. Nature 459, 1005–1009 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gaedcke J., Traub F., Milde S., Wilkens L., Stan A., Ostertag H., Christgen M., von Wasielewski R., Kreipe H. H., Predominance of the basal type and HER-2/neu type in brain metastasis from breast cancer. Mod. Pathol. 20, 864–870 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Rahman M., Davis S. R., Pumphrey J. G., Bao J., Nau M. M., Meltzer P. S., Lipkowitz S., TRAIL induces apoptosis in triple-negative breast cancer cells with a mesenchymal phenotype. Breast Cancer Res. Treat. 113, 217–230 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dilshara M. G., Jeong J.-W., Prasad Tharanga Jayasooriya R. G., Neelaka Molagoda I. M., Lee S., Park S. R., Choi Y. H., Kim G.-Y., Glutamine deprivation sensitizes human breast cancer MDA-MB-231 cells to TRIAL-mediated apoptosis. Biochem. Biophys. Res. Commun. 485, 440–445 (2017). [DOI] [PubMed] [Google Scholar]
- 25.de Miguel D., Lemke J., Anel A., Walczak H., Martinez-Lostao L., Onto better TRAILs for cancer treatment. Cell Death Differ. 23, 733–747 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams S. C., Small nanobody drugs win big backing from pharma. Nat. Med. 19, 1355–1356 (2013). [DOI] [PubMed] [Google Scholar]
- 27.van de Water J. A., Bagci-Onder T., Agarwal A. S., Wakimoto H., Roovers R. C., Zhu Y., Kasmieh R., Bhere D., Van Bergen en Henegouwen P. M. P., Shah K., Therapeutic stem cells expressing variants of EGFR-specific nanobodies have antitumor effects. Proc. Natl. Acad. Sci. U.S.A. 109, 16642–16647 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gores G. J., Kaufmann S. H., Is TRAIL hepatotoxic? Hepatology 34, 3–6 (2001). [DOI] [PubMed] [Google Scholar]
- 29.Kauer T. M., Figueiredo J. L., Hingtgen S., Shah K., Encapsulated therapeutic stem cells implanted in the tumor resection cavity induce cell death in gliomas. Nat. Neurosci. 15, 197–204 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Akiyama Y., Radtke C., Honmou O., Kocsis J. D., Remyelination of the spinal cord following intravenous delivery of bone marrow cells. Glia 39, 229–236 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Osaka M., Honmou O., Murakami T., Nonaka T., Houkin K., Hamada H., Kocsis J. D., Intravenous administration of mesenchymal stem cells derived from bone marrow after contusive spinal cord injury improves functional outcome. Brain Res. 1343, 226–235 (2010). [DOI] [PubMed] [Google Scholar]
- 32.Aleynik A., Gernavage K. M., Mourad Y. S., Sherman L. S., Liu K., Gubenko Y. A., Rameshwar P., Stem cell delivery of therapies for brain disorders. Clin. Transl. Med. 3, 24 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.The Cancer Genome Atlas Network , Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A. A., Kim S., Wilson C. J., Lehár J., Kryukov G. V., Sonkin D., Reddy A., Liu M., Murray L., Berger M. F., Monahan J. E., Morais P., Meltzer J., Korejwa A., Jané-Valbuena J., Mapa F. A., Thibault J., Bric-Furlong E., Raman P., Shipway A., Engels I. H., Cheng J., Yu G. K., Yu J., Aspesi P., de Silva M., Jagtap K., Jones M. D., Wang L., Hatton C., Palescandolo E., Gupta S., Mahan S., Sougnez C., Onofrio R. C., Liefeld T., MacConaill L., Winckler W., Reich M., Li N., Mesirov J. P., Gabriel S. B., Getz G., Ardlie K., Chan V., Myer V. E., Weber B. L., Porter J., Warmuth M., Finan P., Harris J. L., Meyerson M., Golub T. R., Morrissey M. P., Sellers W. R., Schlegel R., Garraway L. A., The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harris L. N., Ismaila N., McShane L., Andre F., Collyar D. E., Gonzalez-Angulo A. M., Hammond E. H., Kuderer N. M., Liu M. C., Mennel R. G., van Poznak C., Bast R. C., Hayes D. F.; American Society of Clinical Oncology , Use of biomarkers to guide decisions on adjuvant systemic therapy for women with early-stage invasive breast cancer: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 34, 1134–1150 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Daphu I., Sundstrøm T., Horn S., Huszthy P. C., Niclou S. P., Sakariassen P. Ø., Immervoll H., Miletic H., Bjerkvig R., Thorsen F., In vivo animal models for studying brain metastasis: Value and limitations. Clin. Exp. Metastasis 30, 695–710 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Liu Z., Wang Y., Kabraji S., Xie S., Pan P., Liu Z., Ni J., Zhao J. J., Improving orthotopic mouse models of patient-derived breast cancer brain metastases by a modified intracarotid injection method. Sci. Rep. 9, 622 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schackert G., Fidler I. J., Site-specific metastasis of mouse melanomas and a fibrosarcoma in the brain or meninges of syngeneic animals. Cancer Res. 48, 3478–3484 (1988). [PubMed] [Google Scholar]
- 39.Guo L., Ge J., Wang S., Zhou Y., Wang X., Wu Y., A novel method for efficient delivery of stem cells to the ischemic brain. Stem Cell Res. Ther. 4, 116 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi S. H., Stuckey D. W., Pignatta S., Reinshagen C., Khalsa J. K., Roozendaal N., Martinez-Quintanilla J., Tamura K., Keles E., Shah K., Tumor resection recruits effector t cells and boosts therapeutic efficacy of encapsulated stem cells expressing IFNβ in glioblastomas. Clin. Cancer Res. 23, 7047–7058 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O’Leary L., van der Sloot A. M., Reis C. R., Deegan S., Ryan A. E., Dhami S. P. S., Murillo L. S., Cool R. H., de Sampaio P. C., Thompson K., Murphy G., Quax W. J., Serrano L., Samali A., Szegezdi E., Decoy receptors block TRAIL sensitivity at a supracellular level: The role of stromal cells in controlling tumour TRAIL sensitivity. Oncogene 35, 1261–1270 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Pelengaris S., Khan M., Evan G., c-MYC: More than just a matter of life and death. Nat. Rev. Cancer 2, 764–776 (2002). [DOI] [PubMed] [Google Scholar]
- 43.Zhao K., Wang Q., Wang Y., Huang K., Yang C., Li Y., Yi K., Kang C., EGFR/c-myc axis regulates TGFβ/Hippo/Notch pathway via epigenetic silencing miR-524 in gliomas. Cancer Lett. 406, 12–21 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Dickler M. N., Cobleigh M. A., Miller K. D., Klein P. M., Winer E. P., Efficacy and safety of erlotinib in patients with locally advanced or metastatic breast cancer. Breast Cancer Res. Treat. 115, 115–121 (2009). [DOI] [PubMed] [Google Scholar]
- 45.Martinez-Aranda A., Hernandez V., Picon C., Modolell I., Sierra A., Development of a preclinical therapeutic model of human brain metastasis with chemoradiotherapy. Int. J. Mol. Sci. 14, 8306–8327 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Song H.-T., Jordan E. K., Lewis B. K., Liu W., Ganjei J., Klaunberg B., Despres D., Palmieri D., Frank J. A., Rat model of metastatic breast cancer monitored by MRI at 3 tesla and bioluminescence imaging with histological correlation. J. Transl. Med. 7, 88 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Steeg P. S., Targeting metastasis. Nat. Rev. Cancer 16, 201–218 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stuckey D. W., Shah K., TRAIL on trial: Preclinical advances in cancer therapy. Trends Mol. Med. 19, 685–694 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nielsen T. O., Hsu F. D., Jensen K., Cheang M., Karaca G., Hu Z., Hernandez-Boussard T., Livasy C., Cowan D., Dressler L., Akslen L. A., Ragaz J., Gown A. M., Gilks C. B., van de Rijn M., Perou C. M., Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin. Cancer Res. 10, 5367–5374 (2004). [DOI] [PubMed] [Google Scholar]
- 50.Gonzalez-Sapienza G., Rossotti M. A., Tabares-da Rosa S., Single-domain antibodies as versatile affinity reagents for analytical and diagnostic applications. Front. Immunol. 8, 977 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Santamaria A. J., Benavides F. D., Di Fede D. L., Khan A., Pujol M. V., Dietrich W. D., Marttos A., Green B. A., Hare J. M., Guest J. D., Clinical and neurophysiological changes after targeted intrathecal injections of bone marrow stem cells in a C3 tetraplegic subject. J. Neurotrauma 36, 500–516 (2018). [DOI] [PubMed] [Google Scholar]
- 52.Deng L., Peng Q., Wang H., Pan J., Zhou Y., Pan K., Li J., Wu Y., Wang Y., Intrathecal injection of allogenic bone marrow-derived mesenchymal stromal cells in treatment of patients with severe ischemic stroke: Study protocol for a randomized controlled observer-blinded trial. Transl. Stroke Res. 10, 170–177 (2018). [DOI] [PubMed] [Google Scholar]
- 53.Hlebokazov F., Dakukina T., Ihnatsenko S., Kosmacheva S., Potapnev M., Shakhbazau A., Goncharova N., Makhrov M., Korolevich P., Misyuk N., Dakukina V., Shamruk I., Slobina E., Marchuk S., Treatment of refractory epilepsy patients with autologous mesenchymal stem cells reduces seizure frequency: An open label study. Adv. Med. Sci. 62, 273–279 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Harris V. K., Vyshkina T., Sadiq S. A., Clinical safety of intrathecal administration of mesenchymal stromal cell-derived neural progenitors in multiple sclerosis. Cytotherapy 18, 1476–1482 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Sykova E., Rychmach P., Drahorádová I., Konrádová Š., Růžičková K., Voříšek I., Forostyak S., Homola A., Bojar M., Transplantation of mesenchymal stromal cells in patients with amyotrophic lateral sclerosis: Results of phase I/IIa clinical trial. Cell Transplant. 26, 647–658 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gu C., Li S., Tokuyama T., Yokota N., Namba H., Therapeutic effect of genetically engineered mesenchymal stem cells in rat experimental leptomeningeal glioma model. Cancer Lett. 291, 256–262 (2010). [DOI] [PubMed] [Google Scholar]
- 57.Shimato S., Natsume A., Takeuchi H., Wakabayashi T., Fujii M., Ito M., Ito S., Park I. H., Bang J. H., Kim S. U., Yoshida J., Human neural stem cells target and deliver therapeutic gene to experimental leptomeningeal medulloblastoma. Gene Ther. 14, 1132–1142 (2007). [DOI] [PubMed] [Google Scholar]
- 58.Balducci L., Blasi A., Saldarelli M., Soleti A., Pessina A., Bonomi A., Coccè V., Dossena M., Tosetti V., Ceserani V., Navone S., Falchetti M., Parati E., Alessandri G., Immortalization of human adipose-derived stromal cells: Production of cell lines with high growth rate, mesenchymal marker expression and capability to secrete high levels of angiogenic factors. Stem Cell Res. Ther. 5, 63 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ryder E. F., Snyder E. Y., Cepko C. L., Establishment and characterization of multipotent neural cell lines using retrovirus vector-mediated oncogene transfer. J. Neurobiol. 21, 356–375 (1990). [DOI] [PubMed] [Google Scholar]
- 60.Bhere D., Khajuria R. K., Hendriks W. T., Bandyopadhyay A., Bagci-onder T., Shah K., Stem cells engineered during different stages of reprogramming reveal varying therapeutic efficacies. Stem Cells 36, 932–942 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reinshagen C., Bhere D., Choi S. H., Hutten S., Nesterenko I., Wakimoto H., le Roux E., Rizvi A., du W., Minicucci C., Shah K., CRISPR-enhanced engineering of therapy-sensitive cancer cells for self-targeting of primary and metastatic tumors. Sci. Transl. Med. 10, eaao3240 (2018). [DOI] [PubMed] [Google Scholar]
- 62.Shah K., Hingtgen S., Kasmieh R., Figueiredo J. L., Garcia-Garcia E., Martinez-Serrano A., Breakefield X., Weissleder R., Bimodal viral vectors and in vivo imaging reveal the fate of human neural stem cells in experimental glioma model. J. Neurosci. 28, 4406–4413 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., Zhang F., Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sanjana N. E., Shalem O., Zhang F., Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang T., Wei J. J., Sabatini D. M., Lander E. S., Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80–84 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Choi S. A., Kwak P. A., Kim S.-K., Park S.-H., Lee J. Y., Wang K.-C., Oh H. J., Kim K., Lee D. S., Hwang D. W., Phi J. H., In vivo bioluminescence imaging for leptomeningeal dissemination of medulloblastoma in mouse models. BMC Cancer 16, 723 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/10/eabe8671/DC1