

Abstract
Harnessing the chemistry of onium ylide intermediates generated from transition metal catalysis is a powerful strategy to convert simple precursors into complex scaffolds. While the chemistry of onium ylides has been studied for over three decades, transformations of aziridinium ylides have just recently emerged as a versatile way to exploit the strain of these reactive intermediates to furnish densely functionalized N-heterocycles in a highly stereocontrolled manner. Herein, we provide a short overview of the key concepts and recent developments in this area, with a focus on how mechanistic studies to delineate the factors controlling the reactivity of aziridinium ylides can stimulate fruitful future investigations.
Keywords: ylide, ring expansion, cheletropic extrusion, carbene transfer, aziridine
Transition metal-catalyzed generation of ammonium and related ylides
The generation of ammonium ylides via inter- and intramolecular reactions of tertiary amines with thermally or photochemically generated carbenes (see Glossary) is well-established in the literature [1–6]. However, the high reactivity of “free” carbenes typically results in low yields and competing side reactions. To address these issues, methods for the catalytic generation of ammonium ylides have been developed that involve the attack of an amine lone pair on an electrophilic metal carbene complex [7]. These strategies are attractive alternatives to traditional base-promoted procedures, as ylides are formed under mild conditions and display attenuated reactivity. The resultant carbene-generated ylides have been employed in diverse synthetic transformations, including [2,3]-sigmatropic rearrangements, Stevens rearrangements, and 1,3-dipolar cycloadditions [8–14]. Creative ways to manipulate the reactivity of these unusual intermediates can lead to powerful methodologies to convert simple starting materials into stereochemically complex, densely functionalized heterocycles with high levels of diastereo- and enantiocontrol.
The structural features of ylides, particularly the nature of the onium group, play key roles in influencing reactivity [15–17]. Nitrogen ylides are the third most common type of onium ylide, behind phosphorus and sulfur [18–42]; however, the strongly Lewis basic nitrogen of tertiary amines presents a challenge, as it may inactivate the transition metals required to decompose diazoesters and other carbene precursors [43].
In contrast to the attention afforded to ammonium ylides, the aziridinium ylide subclass has been underexplored. The few published reports describing their reactivity highlight the potential for diverse pathways, including ring expansion via rearrangement or fragmentation by cheletropic extrusion [44–50]. If aziridinium ylides are to be viewed as truly versatile intermediates, the factors dictating their ultimate fate must be better understood and controlled.
Intramolecular Cu-catalyzed [2,3]-Stevens rearrangements of aziridinium ylides
In 2001, Clark explored the stereoselective synthesis of bicyclic amines through the ring expansion of ammonium ylides derived from various cyclic amines tethered to copper-supported carbenoids (Figure 1A) [44]. Cyclization precursor 1 was subjected to Cu(acac)2 in benzene at reflux. The bicyclic amine 2 (obtained in 24% isolated yield) was hypothesized to arise from a stereoselective, intramolecular [2,3]-rearrangement of the aziridinium ylide 3, where the ring strain imparted by the vinyl aziridine moiety facilitated productive ring expansion. Decomposition of indolizidine 2 was noted within one day of storage at −30°C, suggesting that product instability may have contributed to the modest yield.
Figure 1.
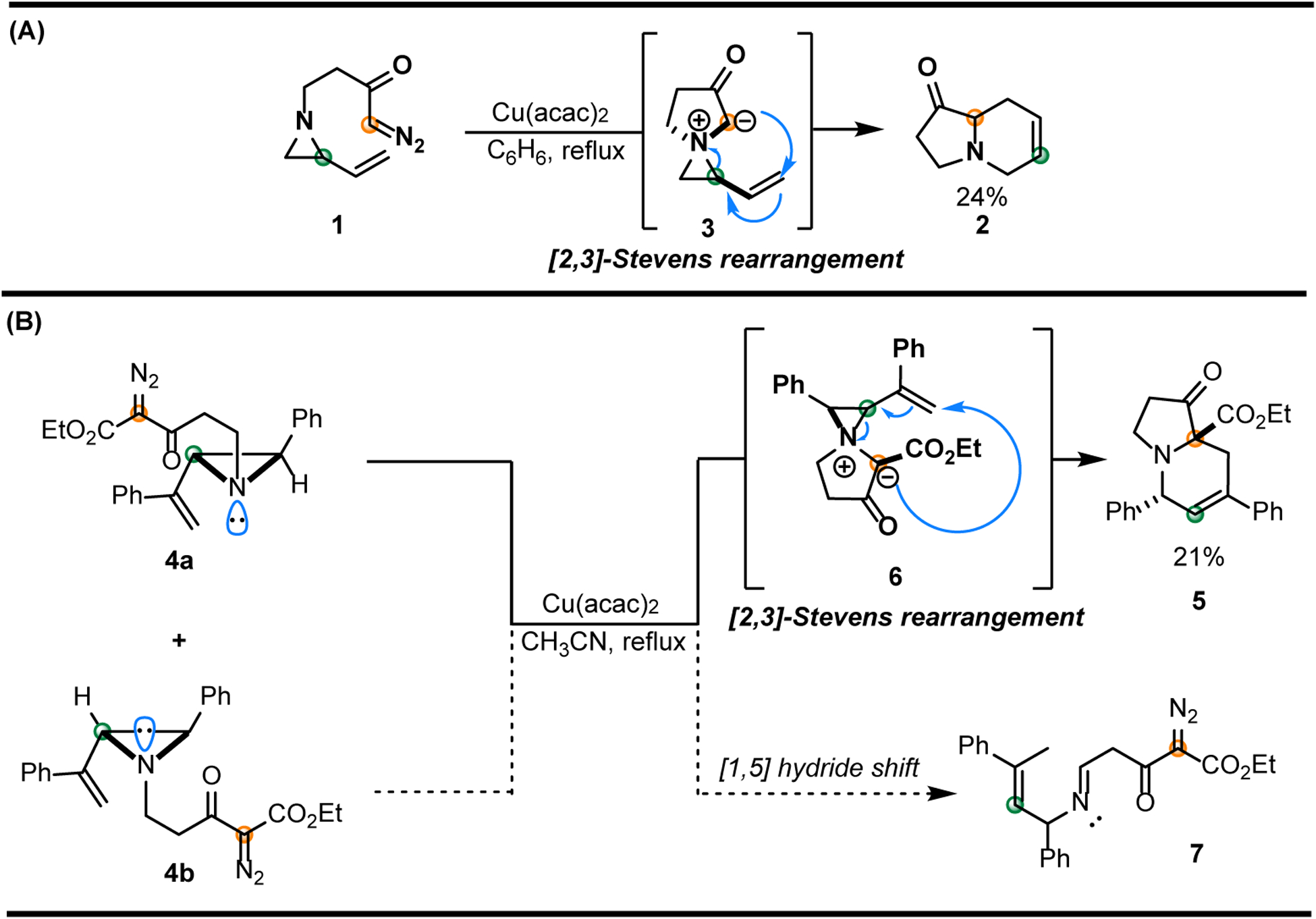
Intramolecular [2,3]-Stevens rearrangements of aziridinium ylides. (A) Clark’s attempted indolizidine synthesis from the [2,3]-rearrangement of spirocyclic ylide intermediate 3. (B) Rowlands’ Cu-catalyzed ring expansion attempt of aziridine invertomers 4a and 4b.
In 2004, Rowlands disclosed a single example of an aziridine ring expansion proposed to proceed through an intramolecular [2,3]-Stevens rearrangement of an aziridinium ylide intermediate (Figure 1B) [45]. In this study, a vinyl aziridine containing an internal diazoacetate tether was prepared as a 3:4 mixture of nitrogen invertomers, favoring 4b. The mixture was heated in the presence of catalytic Cu(acac)2 to furnish the bicyclic amine 5 in 21% isolated yield. The low yield was attributed to the orientations of the substituents in nitrogen invertomers 4a and 4b. Productive [2,3]-Stevens rearrangement requires a cis orientation between the alkene and the lone pair of electrons on the nitrogen prior to formation of the aziridinium ylide 6; thus, effective reaction occurs only from conformer 4a. In conformer 4b, the ineffective overlap between the anionic carbon and alkene pose steric and spatial constraints on the desired rearrangement. This leads to a [1,5]-hydrogen shift outcompeting nitrogen inversion in 4b to furnish imine 7, which degrades under the reaction conditions. This result highlights how productive ylide formation and subsequent rearrangement depend heavily on the presence of an accessible nitrogen lone pair and control over the stereochemistry at the newly pyramidalized ring nitrogen. The challenges encountered in this original study inspired recent investigations to expand the scope of chemistry involving aziridinium ylides by limiting nitrogen inversion through the use of tethers and electron-withdrawing groups within the aziridine scaffold.
Intermolecular Rh-catalyzed formal [3+1] ring expansion of bicyclic methyleneaziridines
The Schomaker group has extensively explored the chemistry of bicyclic methyleneaziridines (MAs), initially focusing on nucleophilic ring-opening and functionalization of the exocyclic alkene [51–58]. Methyleneaziridines have been readily transformed to other nitrogen-containing heterocycles, including aminated stereotriads, azetidin-3-ones, strained cyclooctynes, and aminated cycloheptenes. Interestingly, the constrained geometry and ring strain in MAs inhibits undesired nitrogen inversion and renders the nitrogen lone pair both sterically accessible and unusually nucleophilic, due to lack of conjugation with the carbamate tether. In 2017, the group exploited these features in a formal [3+1] ring expansion of MAs to methyleneazetidines with good scope, yields, and diastereoselectivities (Figure 2) [46]. The key aziridinium ylide intermediate was generated by nucleophilic addition of the ring nitrogen to a rhodium-bound carbene, where the bicyclic nature of the precursor and high E:Z ratios were key to successful ylide formation. A stereocontrolled [2,3]-Stevens rearrangement of the ylide ultimately delivered the azetidine products.
Figure 2.
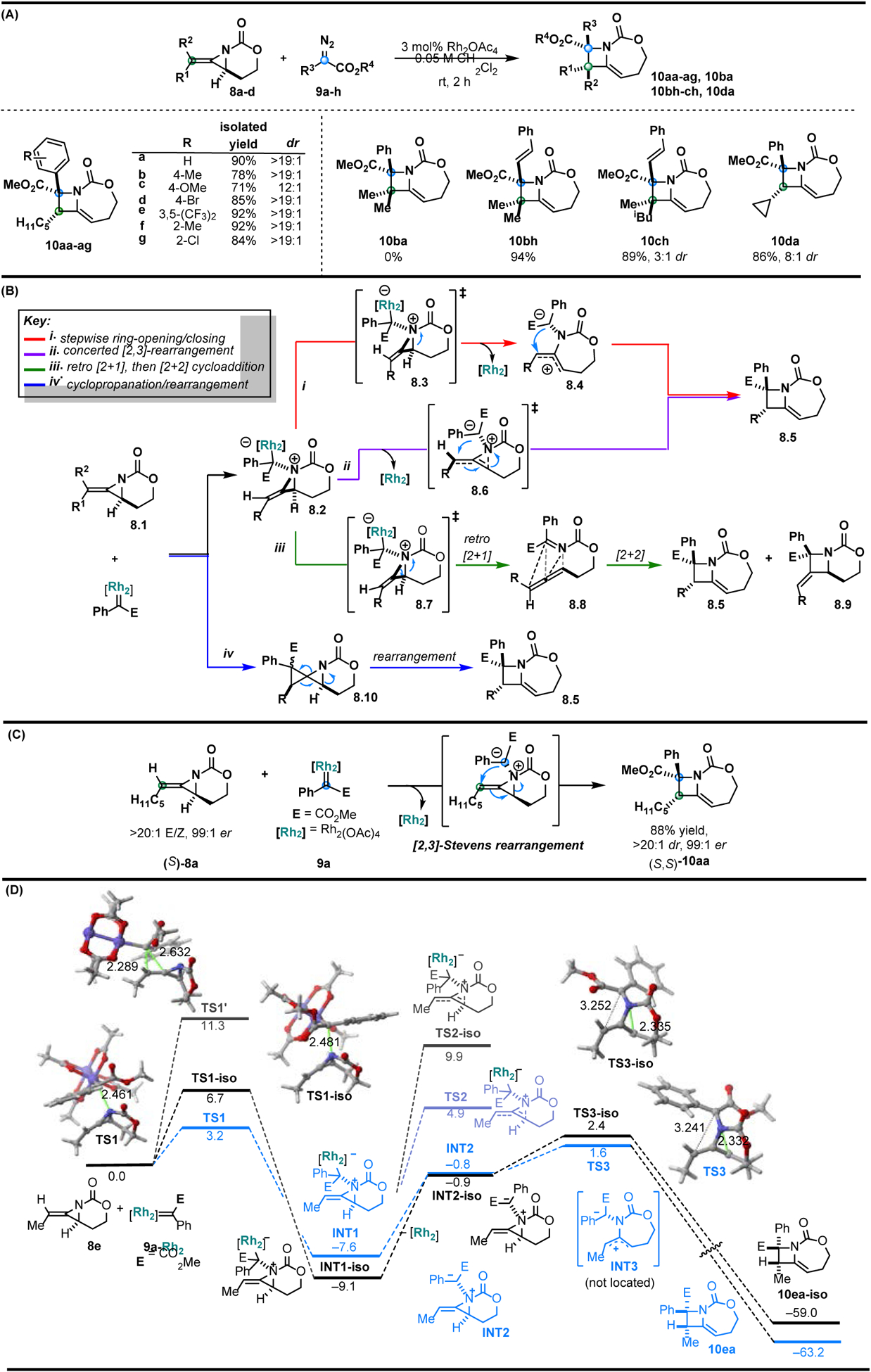
Intermolecular Rh-catalyzed synthesis of methyleneazetidines. (A) Select substrates from the diazoester and aziridine scopes of the [3+1] ring expansion of bicyclic methyleneazetidines. (B) Chirality transfer experiment using enantiopure methyleneaziridine (S)-8a. (C) Potential mechanisms for the [3+1] ring expansion. (D) Computed reaction profile for the process involving methyl-substituted methyleneazeridine 8e and dirhodium-bound carbene 9a-Rh2.
Experimental results showed that the electronics of the aryl substituents on the diazoester carbene precursors do not significantly affect the reaction outcome (Figure 2A). No larger N-heterocyclic ring expansion products were noted from ylides formed from vinyl-substituted diazoacetate 9h, despite the potential for competing vinylogous reactivity [59–62]. MAs containing a substituent cis to the aziridine nitrogen did not react with sterically hindered carbene 9a, as no formation of 10ba was observed; however, switching to the less-hindered styrenyl diazoester 9h delivered fully substituted methyleneazetidine 10bh. Adjacent quaternary stereocenters were successfully set in methyleneazetidine 10ch, which was obtained as a mixture of diastereomers with 89% yield and 3:1 dr; separation of the diastereomers gave the syn-Me/CO2Me isomer of 10ch in 54% yield and 15:1 dr.
Several possible pathways for the [3+1] ring expansion were studied both experimentally and computationally (Figure 2B). The functionalized methyleneazetidine product was initially proposed to result from a stepwise ring-opening, ring-closing sequence through 8.3 and 8.4 (Figure 2B i); this pathway would ablate any stereochemical information present in 8.1. In contrast, a concerted [2,3]-Stevens rearrangement through 8.6 was identified as an alternative mechanism that would result in enantioretention in 8.5 (Figure 2B ii). Cheletropic extrusion of the ylide 8.2 to give allenic intermediate 8.8, followed by a [2+2] cycloaddition, was also considered as a potential pathway (Figure 2B iii). However, the absence of 8.9, which would result from a [2+2] cycloaddition involving the proximal allene bond, renders this pathway unlikely. Rh-catalyzed alkene cyclopropanation is well-known [63]; in this system, cyclopropanation of 8.1, followed by rearrangement of the azaspiropentane intermediate 8.10, would yield methyleneazetidine 8.5 (Figure 2B iv). However, amines react readily with electrophiles and Lewis acids, supporting ylide formation over competing alkene cyclopropanation [51]. The possibility of a radical pathway was investigated using TEMPO as an intermolecular radical trap; no change was noted. In addition, methyleneazetidine 10da was successfully accessed without any ring opening of the cyclopropane substituent (Figure 2A), suggesting ring expansion is unlikely to proceed through a radical pathway. Excluding the Rh catalyst gave no azetidine product, highlighting the importance of metal-mediated decomposition of the diazoester prior to metallocarbene and subsequent ylide formation. Experimentally, the efficient transfer of chirality from enantioenriched (S)-8a to methyleneazetidine (S,S)-10aa indicates the ring expansion does not proceed through any intermediates that ablate the stereochemical information present in the aziridine precursor (Figure 2C). Thus, the most likely mechanism involves the concerted [2,3]-Stevens rearrangement shown in Figure 2B ii.
Follow-up computational studies by density functional theory (DFT) provided further support for this pathway (Figure 2D). Formation of aziridinium ylide INT1 from the nucleophilic addition of methyleneaziridine 8e to Rh-bound carbene 9a (via TS1, ΔG‡ = 3.2 kcal/mol) outcompeted an alternative concerted cyclopropanation pathway proceeding through TS1′ (ΔG‡ = 11.3 kcal/mol). Rh dissociation from INT1 to generate metal-free ylide INT2, followed by ring opening via TS3 (9.2 kcal/mol from INT1) was favored over a ring opening of the allylic C–N bond through TS2 (12.5 kcal/mol). Reports on the fates of rhodium and copper ylides employed in O–H insertion chemistry support this metal-free route, in spite of the energetically costly metal dissociation [64]. In the final step of the mechanism, a highly asynchronous, concerted [2,3]-Stevens rearrangement, which proceeds through TS3, yields the product methyleneazetidine 10aa. Overall, this efficient stereospecific reaction takes advantage of the strained bicyclic MA framework and the unique reactivity of aziridinium ylides to forge a new C-C bond, a new C-N bond, and two adjacent stereocenters.
N-Heterocycles from bicyclic aziridines via aziridinium ylides
Encouraged by the success of the [3+1] ring expansion, the Schomaker group sought to further develop the reactivity of aziridinium ylides by investigating aziridines lacking the exocyclic alkene present in MAs [47]. While no conversion was observed with the trans-11a aziridine isomer, the cis-11a aziridine isomer yielded imine 12 through a proposed cheletropic extrusion of an aziridinium ylide (Figure 3A,B). Similar aziridinium ylide reactivity was reported by Watanabe in 1972 [48], where Cu(acac)2-catalyzed addition of aziridine 13 to diazoester 14 furnished ethylene 15 and an α-imino ester 16 in quantitative yield, instead of the expected azetidine 18 (Figure 3C).
Figure 3.
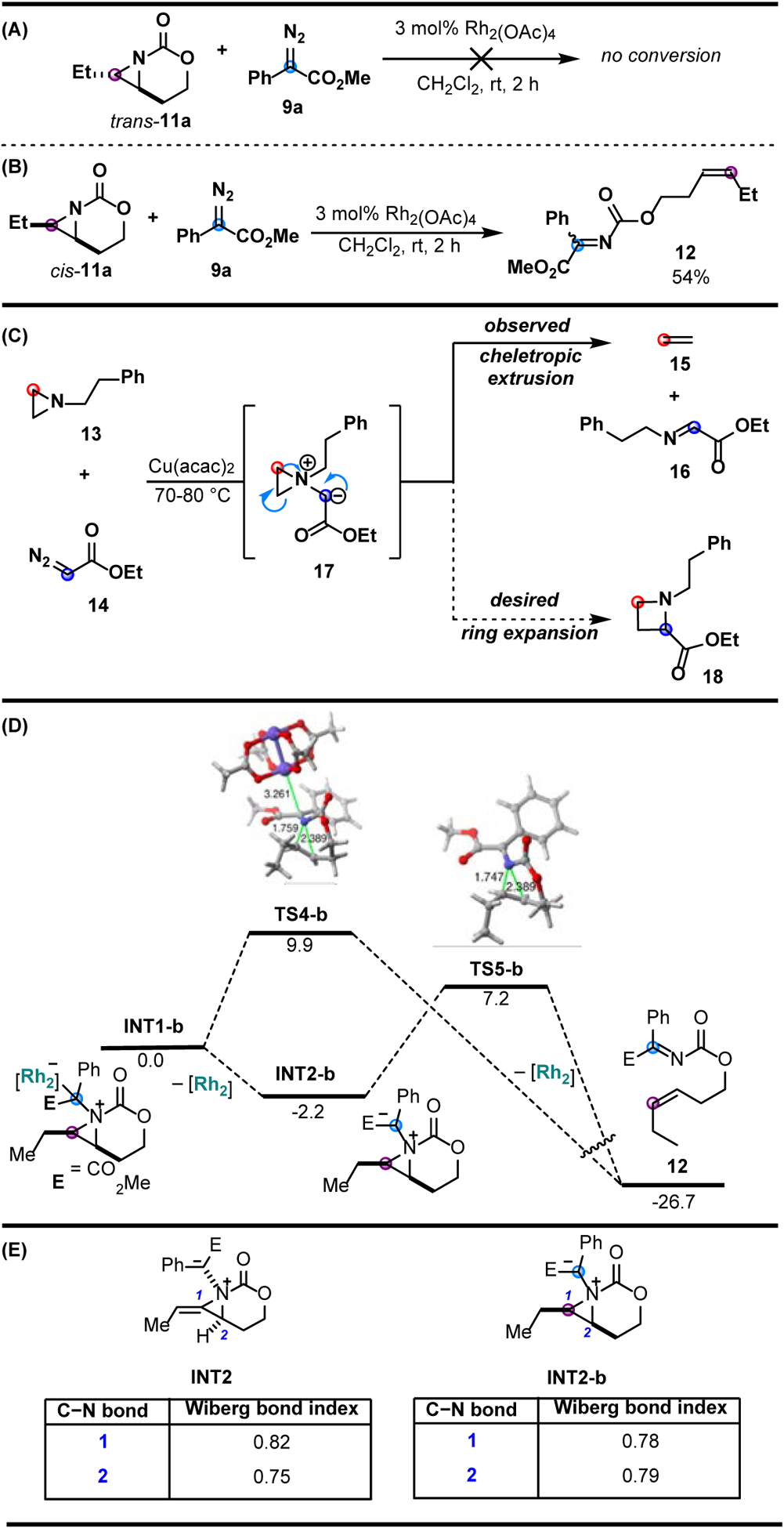
Exploration of unbiased bicyclic aziridines. (A) Unsucessful Rh-mediated carbene transfer between trans-11a and diazoacetate 9a. (B) Cheletropic extrusion from the Rh-mediated carbene transfer between cis-11a and diazoacetate 9a. (C) Cheletropic extrusion of an aziridinium ylide generated from a copper-mediated carbene transfer. (D) Computed reaction profile for the chelotropic extrusion of the aziridinium ylide formed in the reaction of 11a and dirhodium-bound carbene 9a-Rh2. (E) Computed Wiberg bond indices for the C-N bonds of INT2 and INT2-b.
The fragmentation of aziridinium ylide 17 indicates that the reaction proceeds via cheletropic extrusion of the aziridinium ylide intermediate, in lieu of the desired [1,2]-Stevens rearrangement.
Calculations on the bicyclic aziridine system show imine 12 results from cheletropic extrusion of either the rhodium-supported ylide INT1-b (via TS4-b) or the free ylide INT2-b (via TS5-b), with both pathways having favorable activation barriers of 9.9 and 7.2 kcal/mol, respectively (Figure 3D). However, both activation energy barriers for the cheletropic extrusion of INT1-b are significantly higher than the barrier of the analogous ring expansion of ylide INT2 (1.6 kcal/mol, via TS3) in the previously studied MA system (Figure 2B).
Features of aziridinium ylides arising from a methyleneaziridine (INT2, Figure 3E) and a bicyclic aziridine (INT2-b, Figure 3E) were compared. The differing bond strengths (0.82 and 0.75, respectively) of the vinylic and allylic C-N bonds in INT2 favor ring expansion through the initial rupture of the allylic C-N bond. In contrast, the nearly equivalent bond strengths (0.78 and 0.79, respectively) for the C-N bonds of the unbiased aziridine-derived ylide INT2-b favor a concerted extrusion over ring expansion. Based on this computational analysis, the absence of the exocyclic alkene in INT2-b contributes to cheletropic extrusion in the unbiased aziridine system. The increased ring strain (~4.5 kcal/mol) imparted by the exocyclic alkene of MA substrate 8e helps to differentiate the bond strengths of the two aziridine C-N bonds, thus biasing the allylic C-N bond in INT2 to break first.
Dehydropiperidines from bicyclic aziridines via aziridinium ylides
Building on the ability to control the fate of aziridinium ylide intermediates, the Schomaker group reported a formal [3+3] ring expansion of bicyclic aziridines to highly substituted dehydropiperidines with good yields and diastereoselectivities [49]. The aziridinium ylide was proposed to arise from the reaction of the bicyclic aziridine with a vinyl diazoacetate-derived rhodium carbene. Delocalization of the negative charge through the vinyl group of the diazo precursor was exploited to preclude competitive cheletropic extrusion and promote the desired ring expansion pathway to furnish the dehydropiperidine.
Bicyclic aziridine precursors were prepared from the corresponding homoallylic carbamates via Ag-catalyzed nitrene transfer [65], then subjected to the optimized Rh-catalyzed carbene transfer conditions. Carbene transfer was successful employing cis-substituted bicyclic aziridines, but no reaction was observed with the trans-aziridine isomers, likely due to steric congestion at the nitrogen lone pair that hindered productive ylide formation. A variety of substituents were tolerated in the cis-aziridine precursors, including alkyl groups, halides, and ethers (Figure 4A). A series of aryl-substituted diazoesters with varying steric and electronic features were surveyed to probe the impact on reaction outcome. Diazoesters with electron-donating and neutral substituents gave dehydropiperidines 20aa–20ac in similar yields, demonstrating that the electronics of the styrene in the carbene precursor do not heavily affect the reaction outcome. This was further confirmed with dehydropiperidine 20ad, which was obtained in good yield, despite the presence of an electron-withdrawing trifluoromethyl substituent. Dehydropiperidines 20ae and 20af were furnished in good yield and excellent dr from alkyl-substituted diazoacetates, which highlighted the extension of the chemistry beyond aryl diazoacetates.
Figure 4.
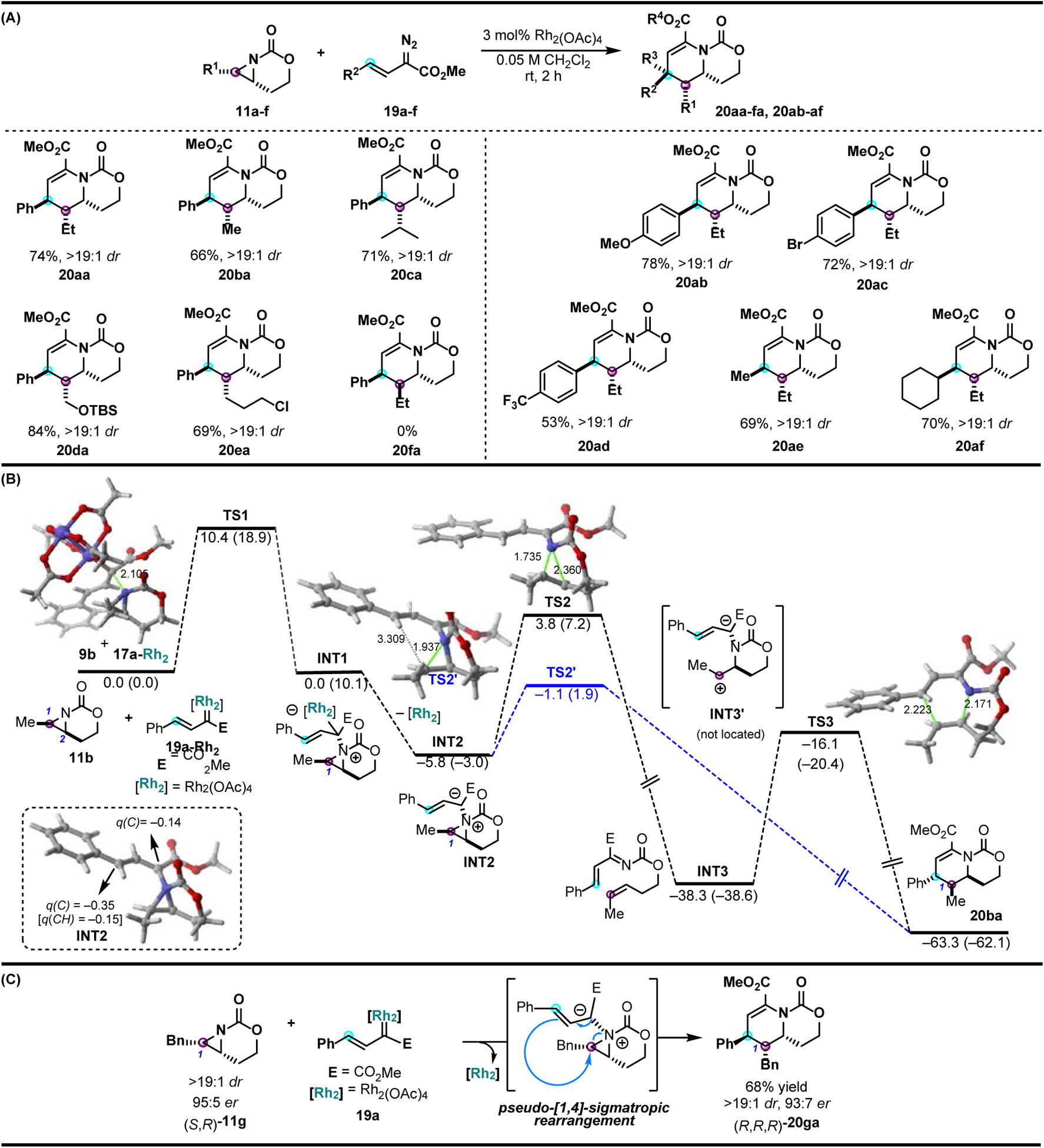
Intermolecular Rh-catalyzed synthesis of dehydropiperidines. (A) Select substrates from the aziridine and diazoester scopes of the [3+3] ring expansion of bicyclic aziridines. (B) Computed reaction profile for the process involving aziridine 11b and dirhodium-bound carbene 19a-Rh2. (C) Chirality transfer experiment using enantiopure aziridine (S,R)-11g.
DFT calculations supported formation of the aziridinium ylide INT1 from nucleophilic attack of the aziridine nitrogen on the Rh-supported carbene (Figure 4B); exergonic dissociation of the rhodium catalyst from INT1 gives zwitterion INT2. According to the computations, INT2 can undergo either a cheletropic extrusion pathway or ring expansion through a ring-opening/ring-closing cascade. In the former case, cheletropic extrusion from INT2 proceeding via TS2 would form azadiene INT3. A subsequent aza-Diels Alder cycloaddition through TS3 would then furnish the dehydropiperidine 20ba. However, this pathway was ruled out as the chirality of enantioenriched aziridine (S,R)-11g was transferred to (R,R,R)-20ga with excellent retention at C1 (Figure 4C). For the ring expansion pathway, direct formation of dehydropiperidine 20ba from INT2 was predicted. In this scenario, stereochemical information at C1 in 11b would be transferred with retention to C1 of the dehydropiperidine 20ba, a prediction that was confirmed experimentally. This latter pathway, considered to be a pseudo-[1,4]-sigmatropic rearrangement, represents the most energetically favorable pathway of the mechanisms that were studied computationally.
The retention of stereochemical information in the ring expansion further provided insight into the details of the rearrangement step. A proposed intramolecular SN2 attack of the benzylic carbon was invalidated, as an inversion of stereochemistry at C1 was not supported by the retention of chirality experiment or the X-ray crystal structure of 20ac. Computational insight into the observed experimental results suggests the rearrangement proceeds instead through a stereoretentive SN1-like mechanism. TS2 and TS2’ both present as low-barrier, early transition states, but C-N bond breakage in the two configurations is biased. In the lower energy TS2’, C-N bond breakage occurs at the external C1-N bond, which elongates to 1.937 Å. In the energetically disfavored TS2, the internal C2-N bond elongates to 2.360 Å, while the C1-N bond elongates to 1.735 Å. As predicted by TS2’ and confirmed by experimental results, the ring-opening of the C1-N aziridine bond and the subsequent C-C bond formation proceed with retention of stereochemistry at C1. This work represents the first examples of aziridinium ylides derived from unbiased aziridines that are able to bypass competitive cheletropic extrusion in favor of ring expansion.
Dehydropiperazines from bicyclic aziridines via aziridinium ylides
The Schomaker group explored other types of carbene precursors to further develop aziridinium ylide chemistry for the synthesis of complex N-heterocycles that are not easily prepared with current methods. Pyridotriazoles 21a have been reported to form α-imino metal carbenes 23a in the presence of a transition metal catalyst [66]. These metal-supported carbenes participate in transformations that include cyclopropanation, X-H insertion, and transannulations to form N-heterocycle derivatives [67–70]. However, in this case, reaction of bicyclic aziridine cis-11a with pyridotriazole 21a furnished a ketimine product 24aa through cheletropic extrusion of aziridinium ylide 26 (Figure 5A) [50]. DFT calculations were helpful in rationalizing the fate of the aziridinium ylide INT2 formed from nucleophilic addition of the aziridine to the electrophilic center of the Rh carbene and subsequent metal dissociation (Figure 5B). Two proposed fates of the ylide INT2 were investigated: cheletropic extrusion (via TS2) or ring expansion (via TS2ʹ). The cheletropic pathway ultimately terminates at imine INT3, as a subsequent aza-Diels-Alder reaction to afford the desired tricyclic ring expansion product 25aa is kinetically unfeasible, with a high barrier of >50 kcal/mol. This barrier is ascribed to the required loss of aromaticity of the pyridyl substituent prior to ring closure.
Figure 5.
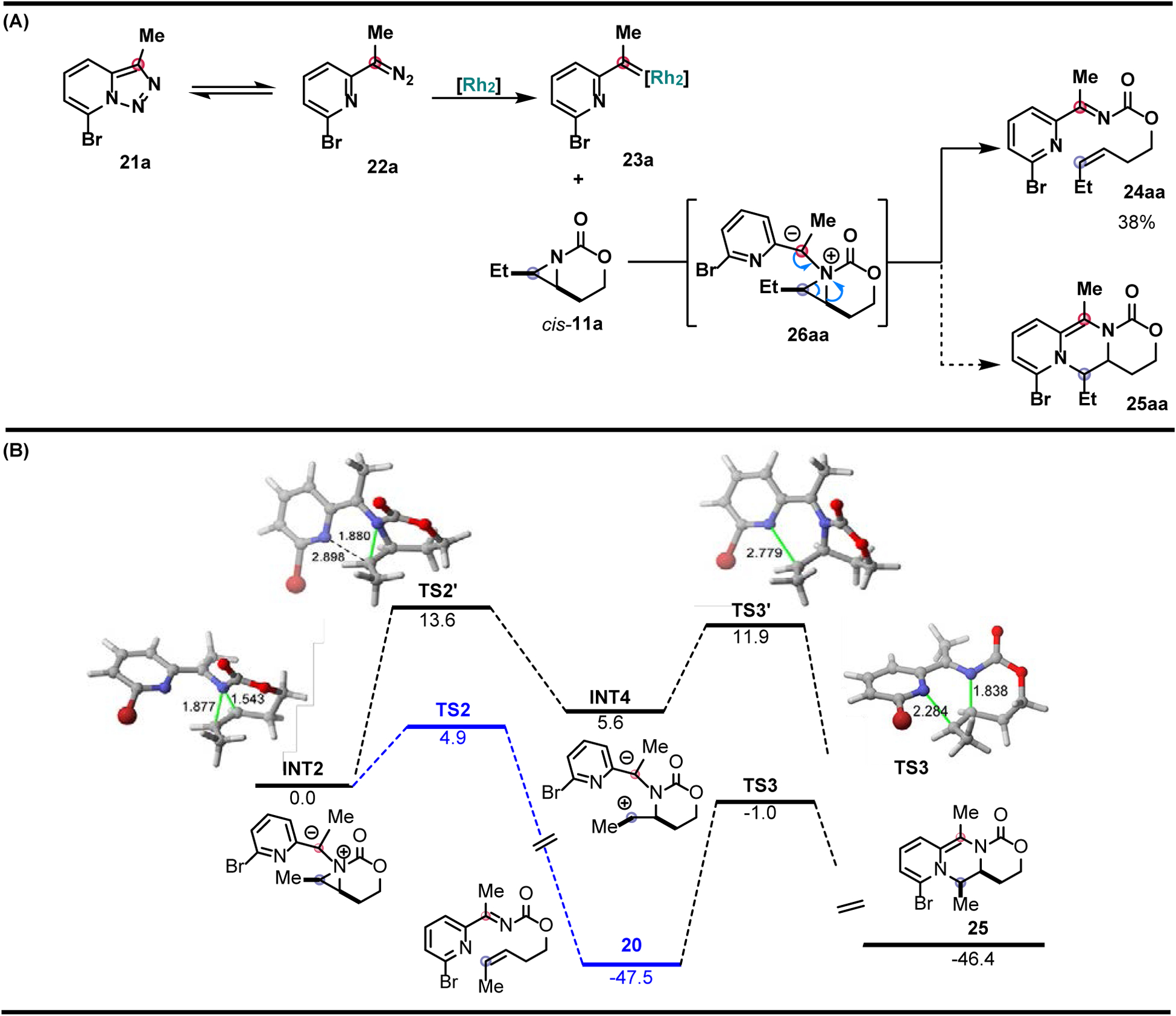
Exploring pyridotriazoles as carbene precursors toward the synthesis of N-heterocycles. (A) Attempted carbene transfer to access fused piperazines from the reaction between aziridine 9a and Rh-bound 23a-Rh2. (B) Computed reaction profile for the process involving cis-aziridine 11a and dirhodium-bound carbene 23a-Rh2.
Altering the identity of the carbene precursor was proposed as a solution to access the desired dehydropiperazine scaffold through a ring expansion pathway (Figure 6A). N-Sulfonyl-1,2,3-triazoles were chosen, as their utility as precursors for accessing metal-supported imino carbenes have been demonstrated in reactions that include transannulations, ring expansions, ylide formation, and C–H functionalization reactions [71–76]. N-Sulfonyl-1,2,3-triazoles 27a are reported to tautomerize in solution upon heating to afford α-diazo imines 28a in low concentrations, eliminating the need for slow addition of the carbene precursor to avoid dimerization [77]. Linear alkyl-substituted aziridines 11a–b and branched aziridine 11c gave the desired dehydropiperazines 31aa–ba and 31ca in good yield and excellent dr of >19:1. Heteroatom-containing substituents, including the alkyl chloride in 11d, were well-tolerated to deliver 31da in good yield as a single diastereomer. Dehydropiperazine 31ea was obtained in good yield, showing that substitution on the aziridine was not necessary for productive reaction. Tosyl-protected aryl N-sulfonyl-1,2,3-triazoles 27c–f were examined to evaluate the effect of altering the electronic and steric environment of the carbene precursor on reaction outcome (Figure 6B). N-sulfonyl-1,2,3-triazoles 27e–f, bearing electron-donating substituents, furnished the respective dehydropiperazines 31ae–af in good yields; however, strongly electron-withdrawing groups lowered the yield. As demonstrated with dehydropiperazine 31ag, the use of alkyl-substituted triazoles failed to furnish any desired product. Calculations showed aryl carbenes have a smaller HOMO-LUMO gap (3.40 eV versus 3.71 eV), suggesting stabilized carbenes of this type are more reactive towards nucleophilic bicyclic aziridines as compared to alkyl carbenes (Figure 6C). DFT calculations support the nucleophilic addition of the bicyclic aziridine 11f to the electrophilic center of the rhodium-supported carbene 29a, followed by Rh dissociation to furnish the aziridinium ylide INT2 (Figure 6D). A highly exergonic cheletropic extrusion of intermediate INT2 is predicted to produce alkene intermediate INT3 via TS2. INT3 may then undergo an aza-Diels Alder cycloaddition through TS3 to yield the corresponding dehydropiperazine 31fa. In contrast, an alternative reaction pathway directly produces the dehydropiperazine 6aa′ from ylide INT2 through a sigmatropic rearrangement in which breaking of the aziridine C-N bond and formation of a new C-N bond is concomitant, yet highly asynchronous. Calculations suggest the direct formation of dehydropiperazine 31fa through TS2′ is kinetically favored, though both reaction pathways are possible given the experimental reaction conditions.
Figure 6.
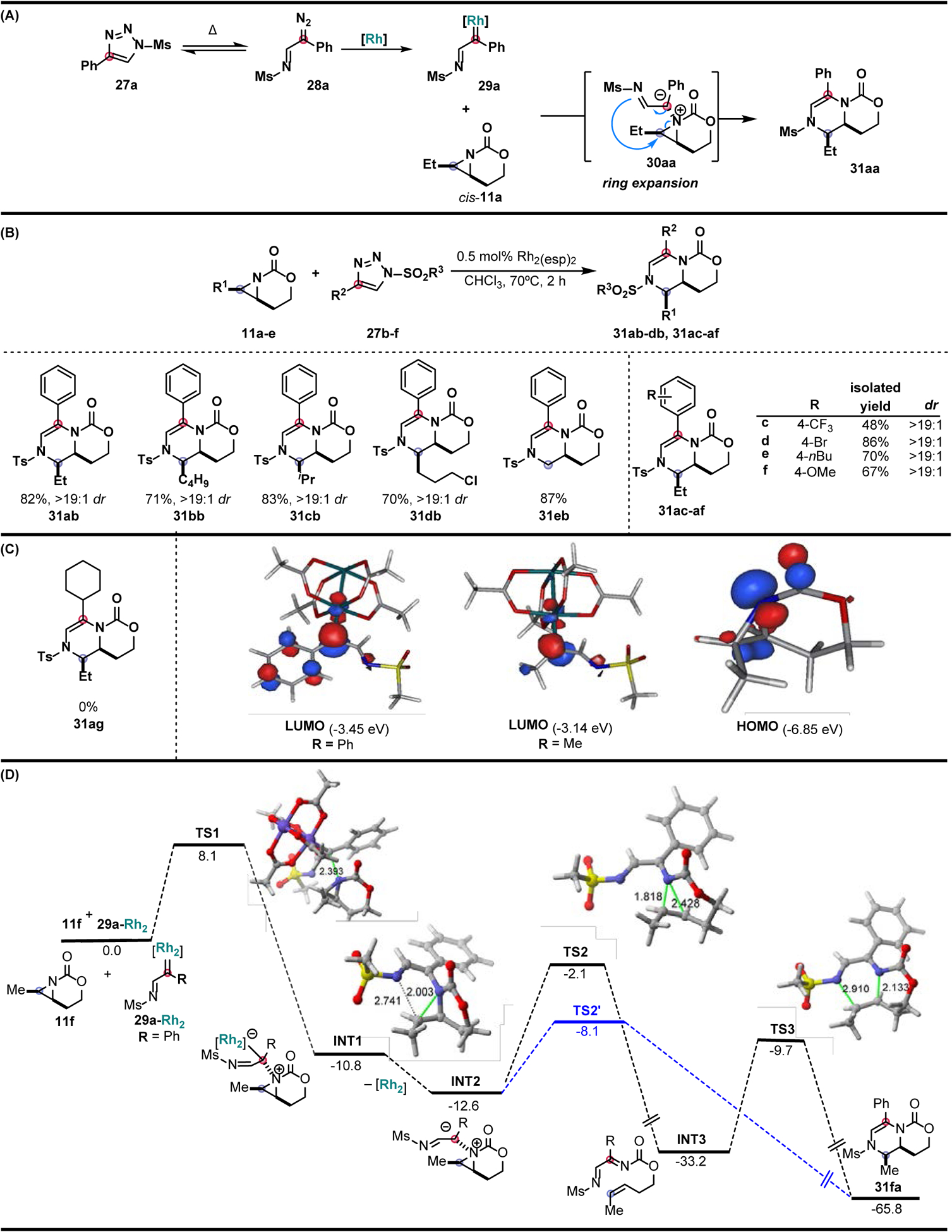
Intermolecular Rh-catalyzed synthesis of dehydropiperazines. (A) Dehydropiperazine synthesis via the ring expansion of an aziridinium ylide generated from a Rh-mediated carbene transfer. (B) Select dehydropiperazine products from the aziridine and diazoester scopes. (C) Left: Example of an inaccessible alkyl substrate. Right: Computed HOMO and LUMO energies for the bicyclic aziridine, aryl carbene, and alkyl carbene. (D) Computed reaction profile for the ring expansion process involving aziridine 11f and dirhodium-bound carbene 29a-Rh2.
Concluding Remarks
This review highlights recent strategies to exploit the unique features of aziridinium ylides as synthetic intermediates for the construction of densely substituted, stereochemically complex N-heterocycles. The key to expanding the utility of this chemistry hinges on obtaining a better mechanistic understanding of how to effectively form and control the subsequent reactivities of aziridinium ylide intermediates (see Outstanding Questions). A combination of experimental and computational studies has provided insight into some of the factors that contribute to the fate of diverse types of aziridinium ylides; continuing these investigations will enable the rational design of improved methods that furnish a broader range of complex nitrogen-containing heterocycles. Future work will aim to engage analogous onium ylides derived from smaller heterocycles as intermediates toward the synthesis of larger, highly functionalized heterocycles.
Supplementary Material
Glossary
- Aziridinium ylide
a subclass of N(sp3)-based ammonium ylides that contain a positively charged aziridinium nitrogen adjacent to an exocyclic nucleophilic carbanionic site; in this review, an ylide generated from the reaction between an aziridine and a metal-supported carbene
- Carbene
a compound containing a divalent carbon atom with a pair of nonbonding electrons
- Cheletropic extrusion
a pericyclic reaction in which two σ bonds terminating at the same atom are made or broken in a concerted fashion
- 1,3-Dipolar cycloaddition
a pericyclic chemical reaction between a 1,3-dipole and a dipolarophile leading to the formation of a five-membered ring
- Stereospecificity
a condition of a reaction in which the production of a single stereoisomer is directly determined by the stereochemistry of the starting material
- Stevens rearrangement
traditionally, a base-promoted transformation of a sulfonium or quaternary ammonium salt to a sulfide or tertiary amine, which is accompanied by the 1,2-mitrgration of an alkyl group from the central nitrogen or sulfur atom
References
- 1.Franzen V and Kuntze H (1959) Untersuchungen über Carbene, IV Reaktionen von Carbenen mit Aliphatischen Aminen. Liebigs Ann. Chem 627, 15–22. [Google Scholar]
- 2.Saunders N and Murray RW (1960) The reaction of dichlorocarbene with amines. Tetrahedron 11, 1–10. [Google Scholar]
- 3.Parham WE and Potoski JR (1967) Reactions of allylamines with dichlorocarbene. J. Org. Chem 32, 275–278. [Google Scholar]
- 4.Bamford WR and Stevens TS (1952) 911. A new reaction of aliphatic diazo-compounds. J. Chem. Soc 0, 4675–4678. [Google Scholar]
- 5.Tomioka H and Suzuki K (1989) Formal Activation of C–H Bonds Toward Carbene By Capto-dative Substituents. Tetrahedron Lett. 30, 6353–6356. [Google Scholar]
- 6.Tomioka H et al. (1995) Generation, Detection, and Reaction of Ammonium Ylides in Reactions of [2-[(Dimethylamino)alkyl]phenyl]phenylcarbenes. J. Org. Chem 60, 1298–1302. [Google Scholar]
- 7.Padwa A and Hornbuckle SF (1991) Ylide Formation from the Reaction of Carbenes and Carbenoids with Heteroatom Lone Pairs. Chem. Rev 91, 263–309. [Google Scholar]
- 8.Murphy GK and West FG (2006) [1,2]-or [2,3]-Rearrangement of Onium Ylides of Allyl and Benzyl Ethers and Sulfides via in Situ-Generated Iodonium Ylides. Org. Lett 8, 4359–4362. [DOI] [PubMed] [Google Scholar]
- 9.Xu B and Tambar UK (2017) Copper-Catalyzed Enantioselective, Diastereoselective, and Regioselective [2,3]-Rearrangements of Iodonium Ylides. Angew. Chem., Int. Ed 56, 9868–9871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boyer A (2014) Rhodium (II)-Catalyzed Stereocontrolled Synthesis of 2-Tetrasubstituted Saturated Heterocycles from 1- Sulfonyl-1, 2, 3-triazoles. Org. Lett 16, 5878–5881. [DOI] [PubMed] [Google Scholar]
- 11.West TH et al. (2015) Catalytic Stereoselective [2,3]-Rearrangement Reactions. ACS Catal. 5, 7446–7479. [Google Scholar]
- 12.Jaber DM et al. (2012) Competitive [2,3]-and [1,2]-Oxonium Ylide Rearrangements. Concerted or stepwise? Org. Lett 14, 1676–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hosseini SN et al. (2017) Evidence for Heterolytic Cleavage of a Cyclic Oxonium Ylide: Implications for the Mechanism of the Stevens [1,2]-Shift. Chem. Commun 53, 12654–12656. [DOI] [PubMed] [Google Scholar]
- 14.Li Z and Davies HML (2010) Enantioselective C-C Bond Formation by Rhodium-Catalyzed Tandem Ylide Formation/ [2,3]-Sigmatropic Rearrangement between Donor/Acceptor Carbenoids and Allylic Alcohols. J. Am. Chem. Soc 132, 396–401. [DOI] [PubMed] [Google Scholar]
- 15.Aggarwal VK et al. (2005) On the Importance of Leaving Group Ability in Reactions of Ammonium, Oxonium, Phosphonium, and Sulfonium Ylides. Angew. Chem., Int. Ed 44, 5468–5471. [DOI] [PubMed] [Google Scholar]
- 16.Fu Y et al. (2009) An Extensive Ylide Thermodynamic Stability Scale Predicted by First-Principle Calculations. J. Org. Chem 74, 810–819. [DOI] [PubMed] [Google Scholar]
- 17.Roiser L et al. (2018) Ammonium Ylide Mediated Cyclization Reactions. Asian J. Org. Chem 7, 852–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang K and Chen Y-C (2014) Organocatalytic reactions involving nitrogen-ylides. Tetrahedron Lett. 55, 2049–2055. [Google Scholar]
- 19.Qiu G et al. (2014) N-Imide Ylide-Based Reactions: C–H Functionalization, Nucleophilic Addition and Cycloaddition. Adv. Synth. Catal 356, 3483–3504. [Google Scholar]
- 20.Adrio J and Carretero JC (2014) Recent advances in the catalytic asymmetric 1,3-dipolar cycloaddition of azomethine ylides. Chem. Commun 50, 12434–12446. [DOI] [PubMed] [Google Scholar]
- 21.Tan WW and Yoshikai N (2015) Copper-catalyzed condensation of imines and α-diazo-β-dicarbonyl compounds: modular and regiocontrolled synthesis of multisubstituted pyrroles. Chem. Sci 6, 6448–6455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jing C et al. (2015) Divergent Synthesis of Multisubstituted Tetrahydrofurans and Pyrrolidines via Intramolecular Aldol-type Trapping of Onium Ylide Intermediates. Chem. Eur. J 21, 19202–19207. [DOI] [PubMed] [Google Scholar]
- 23.Medvedev JJ et al. (2015) Domino [4 + 1]-annulation of α,β-unsaturated δ-amino esters with Rh(II)–carbenoids – a new approach towards multi-functionalized N-aryl pyrrolidines. Org. Biomol. Chem 13, 2640–2651. [DOI] [PubMed] [Google Scholar]
- 24.Harada S et al. (2015) General Approach to Nitrogen-Bridged Bicyclic Frameworks by Rh-Catalyzed Formal Carbenoid Insertion into an Amide C-N Bond. J. Org. Chem 80, 10317–10333. [DOI] [PubMed] [Google Scholar]
- 25.Xu H-D et al. (2015) One-Pot Protocol to Functionalized Benzopyrrolizidine Catalyzed Successively by Rh2(OAc)4 and Cu(OTf)2: A Transition Metal-Lewis Acid Catalysis Relay. Org. Lett 17, 66–69. [DOI] [PubMed] [Google Scholar]
- 26.Zhu C et al. (2016) Gold-Catalyzed Formal [4+1]/[4+3] Cycloadditions of Diazo Esters with Triazines. Angew. Chem. Int. Ed 55, 11867 –11871. [DOI] [PubMed] [Google Scholar]
- 27.Nicolle SM et al. (2016) Stereoselective Synthesis of Functionalized Pyrrolidines by the Diverted N-H Insertion Reaction of Metallocarbenes with β-Aminoketone Derivatives. Angew. Chem. Int. Ed 55, 3749–3753. [DOI] [PubMed] [Google Scholar]
- 28.Day J et al. (2016) Stereoselective Synthesis of Tetrahydroindolizines through the Catalytic Formation of Pyridinium Ylides from Diazo Compounds. Angew. Chem. Int. Ed 55, 5809 –5813. [DOI] [PubMed] [Google Scholar]
- 29.Chen R et al. (2017) In Situ Generation of Quinolinium Ylides from Diazo Compounds: Copper-Catalyzed Synthesis of Indolizine. J. Org. Chem 82, 9291–9304. [DOI] [PubMed] [Google Scholar]
- 30.Douglas T et al. (2017) Iron-Catalyzed Indolizine Synthesis from Pyridines, Diazo Compounds, and Alkynes. Org. Lett 19, 6396–6399. [DOI] [PubMed] [Google Scholar]
- 31.Li H et al. (2018) [3 + 2] Cycloaddition of Nitrile Ylides with Diazonium Salts: Copper-Catalyzed One-Pot Synthesis of Fully Substituted 1,2,4-Triazoles. Org. Lett 20, 5224–5227. [DOI] [PubMed] [Google Scholar]
- 32.Zhang D et al. (2018) Asymmetric Synthesis of Tetrahydroindolizines by Bimetallic Relay Catalyzed Cycloaddition of Pyridinium Ylides. Angew. Chem. Int. Ed 57, 12323– 12327. [DOI] [PubMed] [Google Scholar]
- 33.Bosmani A et al. (2018) Polycyclic Indoline-Benzodiazepines through Electrophilic Additions of α-Imino Carbenes to Trçger Bases. Angew. Chem. Int. Ed 57, 7151 –7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baek Y et al. (2018) Regioselective Synthesis of Indolopyrazines through a Sequential Rhodium-Catalyzed Formal [3+3] Cycloaddition and Aromatization Reaction of Diazoindolinimines with Azirines. J. Org. Chem 83, 2349–2360. [DOI] [PubMed] [Google Scholar]
- 35.Koronatov AN et al. (2018) Rh(II)-Catalyzed Ring Expansion of Pyrazoles with Diazocarbonyl Compounds as a Method for the Preparation of 1,2-Dihydropyrimidines. J.Org. Chem 83, 9210–9219. [DOI] [PubMed] [Google Scholar]
- 36.Golubev AA et al. (2019) [2+1+1] Assembly of spiro β-lactams by Rh(II)-catalyzed reaction of diazocarbonyl compounds with azirines/isoxazoles. Org. Biomol. Chem 17, 6821–6830. [DOI] [PubMed] [Google Scholar]
- 37.Zheng H and Doyle MP (2019) Catalytic Desymmetric Cycloaddition of Diaziridines with Metalloenolcarbenes: The Role of Donor–Acceptor Cyclopropenes. Angew. Chem. Int. Ed 58, 12502 –12506. [DOI] [PubMed] [Google Scholar]
- 38.Cai W et al. (2019) Rh-Catalyzed Chemoselective [4+1] Cycloaddition Reaction toward Diverse 4-Methyleneprolines. J. Org. Chem 84, 10877–10891. [DOI] [PubMed] [Google Scholar]
- 39.He M et al. (2019) Rhodium-Catalyzed Regiodivergent [3+2] and [5+2] Cycloadditions of Quinolinium Ylides with Alkynes. Org. Lett 21, 5167–5171. [DOI] [PubMed] [Google Scholar]
- 40.Khaidarov AR et al. (2019) Synthesis of 1-(2-Aminovinyl)indoles and 1,3′-Biindoles by Reaction of 2,2-Diaryl-Substituted 2H-Azirines with α-Imino Rh(II) Carbenoids. J. Org. Chem 84, 3743–3753. [DOI] [PubMed] [Google Scholar]
- 41.Nickerson LA et al. (2020) Enantioselective synthesis of isochromans and tetrahydroisoquinolines by C–H insertion of donor/donor carbenes. Chem. Sci 11, 494–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou K et al. (2020) Iron-catalyzed [3 + 2]-cycloaddition of in situ generated N-ylides with alkynes or olefins: access to multi-substituted/polycyclic pyrrole derivatives. Org. Biomol. Chem 18, 409–414. [DOI] [PubMed] [Google Scholar]
- 43.Felthouse TR (1982) The Chemistry, Structure, and Metal-Metal Bonding in Compounds of Rhodium(II). Prog. Inorg. Chem 29, 73–166. [Google Scholar]
- 44.Clark JS et al. (2001) Rearrangement of ammonium ylides produced by intramolecular reaction of catalytically generated metal carbenoids. Part 2. Stereoselective synthesis of bicyclic amines. J. Chem. Soc., Perkin Trans. 1, 24, 3325–3337. [Google Scholar]
- 45.Barnes WK and Rowlands GJ (2004) Studies on the [2,3]-Stevens rearrangement of aziridinium ions. Tetrahedron Lett. 45, 5347–5350. [Google Scholar]
- 46.Schmid SC et al. (2017) A Stereoselective [3+1] Ring Expansion of the Synthesis of Highly Substituted Methylene Azetidines. Angew. Chem., Int. Ed 56, 12229–12233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schmid SC et al. (2018) Ring Expansion of Bicyclic Methyleneaziridines via Concerted, Near-Barrierless [2,3]-Stevens Rearrangement. ACS Catal. 8, 7907–7914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hata Y and Watanabe M (2012) Fragmentation Reaction of Aziridinium Ylides. Tetrahedron Lett. 46, 4659–4660. [Google Scholar]
- 49.Eshon J et al. (2020) Intermolecular [3 + 3] Ring-Expansion of Aziridines to Dehydropiperidines through the Intermediacy of Aziridinium Ylides. Nat. Commun 11, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dequina HJ et al. (2020) Rh-Catalyzed Aziridine Ring Expansions to Dehydropiperazines. Org. Lett 22, 3637–3641. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 51.Boralsky LA et al. (2011) Allene functionalization via bicyclic methyleneaziridines. Org. Lett 13, 1924–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rigoli JW et al. (2012) 1,4-Diazaspiro[2.2]pentanes as a flexible platform for the synthesis of diamine-bearing stereotriads. J. Org. Chem 77, 2446–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adams CS et al. (2012) Modular functionalization of allenes to aminated stereotriads. J. Am. Chem. Soc 134, 10807–10810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adams CS et al. (2014) Complete stereodivergence in the synthesis of 2-amino-1,3-diols from allenes. Chem. Sci 5, 3046–3056. [Google Scholar]
- 55.Burke EG and Schomaker JM (2015) Oxidative allene amination for the synthesis of azetidin-3-ones. Angew. Chem. Int. Ed 54, 12097–12101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gerstner NC et al. (2016) Stereocontrolled syntheses of seven-membered carbocycles by tandem allene aziridination/[4+3] reaction. Angew. Chem. Int. Ed 128, 13434–13437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Burke EG et al. (2017) Fine-Tuning Strain and Electronic Activation of Strain-Promoted 1,3-Dipolar Cycloadditions with Endocyclic Sulfamates in SNO-OCTs. J. Am. Chem. Soc 139, 8029–8037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Corbin JR et al. (2020) Biomimetic Imino-Nazarov Cyclizations via Eneallene Aziridination. J. Am. Chem. Soc 142, 5568–5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Davies HML et al. (1990) Anomalous reactivity of mono substituted rhodium stabilized vinylcarbenoids Tetrahedron Lett. 31, 6299–6302. [Google Scholar]
- 60.Davies HML et al. (1994) Carbenoid versus Vinylogous Reactivity in Rhodium (II)-Stabilized Vinylcarbenoids. J. Org. Chem 59, 4535–4541. [Google Scholar]
- 61.Lian Y and Davies HML (2012) Rh2(S-biTISP)2-Catalyzed Asymmetric Functionalization of Indoles and Pyrroles with Vinylcarbenoids. Org. Lett 14, 1934–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qin C and Davies HML (2013) Rh2(R-TPCP)4-Catalyzed Enantioselective [3+2]-Cycloaddition between Nitrones and Vinyldiazoacetates. J. Am. Chem. Soc 135, 14516–14519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Singh VK et al. (1997) Catalytic Enantioselective Cyclopropanation of Olefins Using Carbenoid Chemistry. Synthesis 2, 137–149. [Google Scholar]
- 64.Liang Y et al. (2009) Why Is Copper(I) Complex More Competent Than Dirhodium(II) Complex in Catalytic Asymmetric O-H Insertion Reactions? A Computational Study of the Metal Carbenoid O-H Insertion into Water. J. Am. Chem. Soc 131, 17783–17785. [DOI] [PubMed] [Google Scholar]
- 65.Ju M et al. (2017) Chemo- and Enantioselective Intramolecular Silver-Catalyzed Aziridinations. Angew. Chem. Int. Ed 56, 9944–9948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Abarca-González B (2002) The Chemistry of [1,2,3]Triazolo[1,5-a]pyridines. J. Enzym. Inhib. Med. Chem 17, 359–367. [DOI] [PubMed] [Google Scholar]
- 67.Chuprakov S et al. (2007) Rh-Catalyzed Transannulation of Pyridotriazoles with Alkynes and Nitriles. Angew. Chem., Int. Ed 46, 4757–4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Helan V et al. (2015) Cu-catalyzed transannulation reaction of pyridotriazoles with terminal alkynes under aerobic conditions: efficient synthesis of indolizines. Chem. Sci 6, 1928 –1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shi Y (2014) Rhodium-Catalyzed N–H Insertion of Pyridyl Carbenes Derived from Pyridotriazoles: A General and Efficient Approach to 2-Picolylamines and Imidazo[1,2-a]pyridines. Angew. Chem. Int. Ed 53, 14191–14195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chuprakov S and Gevorgyan V (2007) Regiodivergent Metal-Catalyzed Rearrangement of 3-Iminocyclopropenes into N-Fused Heterocycles. Org. Lett 9, 4463 –4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Parr BT et al. (2013) Rhodium-Catalyzed Conversion of Furans to Highly Functionalized Pyrroles. J. Am. Chem. Soc 135, 4716–4718. [DOI] [PubMed] [Google Scholar]
- 72.Spangler JE and Davies HML (2013) Catalytic Asymmetric Synthesis of Pyrroloindolines via a Rhodium(II)-Catalyzed Annulation of Indoles. J. Am. Chem. Soc 135, 6802–6805. [DOI] [PubMed] [Google Scholar]
- 73.Zibinsky M and Fokin VV (2013) Sulfonyl-1,2,3-Triazoles: Convenient Synthones for Heterocyclic Compounds. Angew. Chem., Int. Ed 52, 1507–1510. [DOI] [PubMed] [Google Scholar]
- 74.Chuprakov S et al. (2013) Transannulation of 1-Sulfonyl-1,2,3-triazoles with Heterocumulenes. J. Am. Chem. Soc 135, 4652–4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Muira T et al. (2013) Regiocontrolled Synthesis of Polysubstituted Pyrroles Starting from Terminal Alkynes, Sulfonyl Azides, and Allenes. Org. Lett 15, 3298–3301. [DOI] [PubMed] [Google Scholar]
- 76.Alford JS et al. (2013) Conversion of Cyclic Ketones to 2,3-Fused Pyrroles and Substituted Indoles. J. Am. Chem. Soc 135, 11712–11715. [DOI] [PubMed] [Google Scholar]
- 77.Horneff T et al. (2008) Rhodium-Catalyzed Transannulation of 1,2,3-Triazoles with Nitriles. J. Am. Chem. Soc 130, 14972–14974. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.