

Abstract
Heart failure threatens the lives of patients and reduces their quality of life. Heart failure, especially heart failure with preserved ejection fraction, is closely related to systemic and local cardiac persistent chronic low-grade aseptic inflammation, microvascular damage characterized by endothelial dysfunction, oxidative stress, myocardial remodeling, and fibrosis. However, the initiation and development of persistent chronic low-grade aseptic inflammation is unexplored. Oxidative stress-mediated neutrophil extracellular traps (NETs) are the main immune defense mechanism against external bacterial infections. Furthermore, NETs play important roles in noninfectious diseases. After the onset of myocardial infarction, atrial fibrillation, or myocarditis, neutrophils infiltrate the damaged tissue and aggravate inflammation. In tissue injury, damage-related molecular patterns (DAMPs) may induce pattern recognition receptors (PRRs) to cause NETs, but whether NETs are directly involved in the pathogenesis and development of heart failure and the mechanism is still unclear. In this review, we analyzed the markers of heart failure and heart failure-related diseases and comorbidities, such as mitochondrial DNA, high mobility box group box 1, fibronectin extra domain A, and galectin-3, to explore their role in inducing NETs and to investigate the mechanism of PRRs, such as Toll-like receptors, receptor for advanced glycation end products, cGAS-STING, and C-X-C motif chemokine receptor 2, in activating NETosis. Furthermore, we discussed oxidative stress, especially the possibility that imbalance of thiol redox and MPO-derived HOCl promotes the production of 2-chlorofatty acid and induces NETosis, and analyzed the possibility of NETs triggering coronary microvascular thrombosis. In some heart diseases, the deletion or blocking of neutrophil-specific myeloperoxidase and peptidylarginine deiminase 4 has shown effectiveness. According to the results of current pharmacological studies, MPO and PAD4 inhibitors are effective at least for myocardial infarction, atherosclerosis, and certain autoimmune diseases, whose deterioration can lead to heart failure. This is essential for understanding NETosis as a therapeutic factor of heart failure and the related new pathophysiology and therapeutics of heart failure.
1. Introduction
Heart failure (HF) is a complex syndrome. Its typical symptoms are breathlessness, paroxysmal nocturnal dyspnea, reduced exercise tolerance, fatigue, tiredness, increased time to recover after exercise, and ankle swelling, resulting in decreased cardiac output and/or increased intracardiac pressure [1]. Currently, patients with HF are usually referred to as heart failure with reduced ejection fraction (HFrEF; LVEF < 40%), heart failure with midrange ejection fraction (HFmrEF; LVEF 40-49%), or heart failure with preserved ejection fraction (HFpEF; LVEF ≥ 50%) [1]. More than 64 million people in the world suffer from HF, with an estimated prevalence of 1-2% among adults in developed countries [2], while, in China, the HF prevalence of the Chinese adult population aged ≥35 years from 2012 to 2015 in China was 1.3% (estimated 13.7 million), which is a 44% increase compared to 2000. Among them, 1.4% of participants had left ventricular systolic dysfunction, and the prevalence of moderate/severe diastolic dysfunction was 2.7% [3]. The prevalence of HFpEF, HFmrEF, and HFrEF in China was 0.3%, 0.3%, and 0.7% [4]. Furthermore, among the 13687 patients with HF in 132 hospitals selected in the China-HF study from January 2012 to September 2015, the case fatality rate was 4.1% [4]. Moreover, the total number of HF patients in the world continues to increase because of the population growth and aging. HF has increased in low-income countries and shifted to HFpEF. Age, traditional risk factors for HF, sedentary lifestyle, and social deprivation are related to the occurrence of HF [5]. Many factors contributed to the development of HFpEF, such as inflammation, endothelial dysfunction, abnormal cardiac metabolism, cardiomyocyte hypertrophy, cardiac fibrosis, ventricular-vascular uncoupling, pulmonary hypertension, and chronotropic incompetence [6–13]. Although many studies have confirmed the correlation between inflammation and oxidative stress and the severity and prognosis of HF, except vitamin C, coenzyme Q10, and IL-1 antagonist anakinra, most of the clinical trials of anti-inflammatory and antioxidant therapy have been proved unsuccessful, indicating that we still have many unknowns about the mechanism of inflammation and oxidative stress in HF.
Neutrophils are powerful inducers of oxidative stress and inflammation in the immune system, but we know very little about their role and mechanism in HF. Recently, accumulating evidence shows that neutrophil extracellular traps (NETs) are an important way to be involved in the immune response. NETs are the last resort to control microbial infections released by neutrophils, and this unique cell death program of neutrophils is called “NETosis”. In this cell death process, citrullinated chromatin and bactericidal proteins from granules and cytoplasm are released and produce a network structure, which promotes the immobilization and killing of invading microorganisms in the extracellular environment. NETosis plays a vital role in host defense, autoimmunity, and blood coagulation [14, 15]. NETs can be activated through various disease-related stimuli, such as pathogens, antibodies and immune complexes, cytokines, microcrystals, and aging [16–19], and they also mediate tissue damage [20–22]. The induction of NETosis depends on the form of reactive oxygen species (ROS) via oxidative burst, and its main source is NADPH oxidase [23]. The structure of these NETs comprises various neutrophil-derived proteins such as myeloperoxidase (MPO), peptidylarginine deiminase 4 (PAD4), neutrophil elastase (NE), histones, neutrophil gelatinase-associated lipocalin (NGAL), proteinase-3, and DNA chains. In NETs, the enzymatic activity of MPO and NE may contribute to antibacterial activity or tissue damage [24, 25], and the MPO complex regulates NE release and actin dynamics [26]. Moreover, superoxide-dependent MPO-derived chlorinated lipids may be a mediator required for the formation of NETs (discussed in detail in the following section). PAD4 is important for NET-mediated antibacterial innate immunity [27], and it is also the nuclear button that triggers NETs in inflammatory diseases [28]. PAD4-mediated histone hypercitrullination promotes heterochromatin decondensation and chromatin unfolding to form NETs [29]. In addition to being a tool to defend against harmful foreign microbes, NETosis may also be the initiator of certain autoimmune and other noninfectious diseases [30]. NETs can also directly lead to acute and chronic inflammation, endothelial cell dysfunction, and thrombosis [31]. Some research results revealed that the formation of NETs occurs in the early or acute stage of noninfectious diseases, such as myocardial infarction and abdominal aortic aneurysms in cardiovascular diseases [32, 33], rheumatoid arthritis (RA) in autoimmune diseases, and antineutrophil cytoplasmic antibody- (ANCA-) associated vasculitis and inflammatory bowel disease [34–36]. Various facts indicate that NETosis may be involved in the process of HF, at least in the pathogenesis of heart failure-related diseases. Understanding, preventing, and targeting NETosis may contribute to the prevention and treatment of heart failure and improve the survival rate of patients.
2. NETosis Phenomena in HF
To our knowledge, the existence and function of NETs in HF, especially in HFpEF, have not been studied in detail. According to available data, in HF, especially in HFpEF, an aseptic inflammatory response with NETs as the core should be the necessary condition to enhance myocardial tissue damage, fibrosis, and ventricular remodeling.
More than a decade ago, several studies have reported that an increase in circulating MPO, as an independent risk factor, may play a key role in the occurrence and maintenance of chronic HF [37–39]. Furthermore, a recent study revealed that MPO is closely related to microvascular endothelial inflammation and dysfunction in HFpEF [40]. These findings indicate that circulating MPO derived from neutrophils may lead to the chlorination or nitration of protein tyrosine, causing protein dysfunction and vascular endothelial damage [39] and that the MPO-DNA complex may form NETs in HF tissues [41, 42]. NGAL, also known as lipocalin-2, is another component of NETs. As a siderophore-binding protein, it limits bacterial growth by sequestrating the iron-laden siderophore [43, 44]. Because it is also expressed in macrophages, endothelial cells, and cardiomyocytes, NGAL increase in the myocardial tissue of cardiac hypertrophy and experimental and clinical HF, although whether this source is related to NETs remains unclear [45–47]. Moreover, another index supports that neutrophils may be involved in the process of chronic HF. The neutrophil-to-lymphocyte ratio (NLR) is a risk factor for poor prognosis in elderly patients with chronic HF [48, 49] or HFpEF [50]. Furthermore, in animal models of HF, inflammatory infiltration of neutrophils was found in the myocardial tissue [51, 52]. These phenomena imply that NETs may promote the pathological process of HF.
A recent study reported that seipin/Bslc2 knockout mice, a model of Asian lean diabetes, developed HFpEF. The authors found that increased cardiac titin phosphorylation and reactive interstitial fibrosis associated with NETs lead to left ventricular stiffness. At the age of 23 weeks, cardiac hypertrophy and diastolic dysfunction were observed. The hearts of seipin/Bslc2 knockout mice displayed an increased end-diastolic pressure/volume relationship, increased end-diastolic pressure, and left atrial enlargement as well as exercise intolerance. NET was characterized by intact neutrophils with condensed nuclei (15 weeks old) and persisted as large amorphous extracellular structures, released DNA fibers decorated with MPO, and citrullinated histone (32 weeks old). By the age of 24 weeks, the mRNA and protein levels of the markers of cardiac inflammation and fibrosis, such as interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α), intercellular adhesion molecule-1, collagen 1, alpha-smooth muscle actin, and transforming growth factor beta family, were significantly increased in the heart of seipin/Bslc2 knockout mice [53]. In heart tissue of seipin/Bslc2 knockout mice, hyperphosphorylation of JAK2 and downstream STAT3 was also found [53], suggesting that JAK2 signal can be connected to myocardial dysfunction and NETosis. In another JAK2V617F allele conditional knock-in mouse model, clonal hematopoiesis accelerated pathological HF remodeling. After exposing these mice to stressors of coronary artery ligation-induced myocardial infarction and aortic constriction-induced pressure overload, researchers demonstrated that myeloid JAK2V617F mice showed accelerated cardiac inflammation and remodeling, larger infarct size, and HF. In addition, on stimulation with LPS, THP-1 cells transduced with JAK2V617F exhibited enhanced inflammatory responses, including transcripts of IL-6, IL-1β, TNF-α, and monocyte chemoattractant protein-1 (MCP-1), as well as AIM2 inflammasome component compared with JAK2WT cells [54]. Another study using the same model showed that JAK2V617F mutant neutrophils were prone to form NETs in vivo and lead to thrombosis events associated with myeloproliferative diseases. This study revealed that neutrophils expressing JAK2V617F increased the protein expression of peptidylarginine deiminase 4 (PAD4) required for NETosis and that NETosis and thrombosis in vivo driven by JAK2V617F require PAD4 [55]. Moreover, the PAD4 gene deletion in mice or the specific inhibitor GSK484 reduced myocardial infarction-induced neutrophil infiltration, citrulline histone H3 (citH3) expression, NET structure, and inflammatory cytokine secretion [56, 57]. A recent report reported that cardiac pressure overload induced NETosis and reduced left ventricular ejection fraction (LVEF) in WT mice but not in PAD4−/− mice [58]. The deletion or activation of genes listed in Table 1 shows the phenotypes of mice with HF and NETosis. Therefore, NETosis may be involved in the development of HF and hence may be a new target for treatment.
Table 1.
Effect of gene deletion or activation on NETosis and HF.
| Gene | NETosis | Heart failure |
|---|---|---|
| Seipin/Bslc2 | Compared with 15-week-old SKO (seipin/Bscl2 deficiency in developing adipocytes) mice, 32-week-old SKO mice had hearts with large amorphous NET structures, which released MPO and citrullinated histone-modified DNA fiber [53]. | Lipodystrophic mice with seipin/Bscl2 deletions were lean and diabetic and showed the main clinical manifestations and characteristics of HFpEF. Increased myocardial titin phosphorylation and reactive interstitial fibrosis associated with NETs led to left ventricular stiffness and HF [53]. |
| JAK2 | JAK2V617F-driven myeloproliferative neoplasm mouse models had a NET-rich, prothrombotic phenotype, whereas the JAK inhibitor ruxolitinib and DNase reduced the rate of venous thrombosis induced by the mouse model [55]. | At the 14th day of the myocardial infarction experiment, mice expressing JAK2V617F had enlarged infarct size, increased fibrosis, and significantly increased IL-6 and IL-1β levels. Ly6Chi monocytes, neutrophils, and macrophages in the infarcted area also showed an increasing trend. In another HF experiment in mice, 8 weeks after the operation, the heart mass and lung weight of the JAK2V617F experimental group increased significantly compared with the JAK2WT experimental group. Echocardiography revealed that the JAK2V617F experimental group displayed significantly increased cardiac posterior wall thickness and a progressive reduction of fractional shortening. TAC treatment induced more severe myocardial hypertrophy and cardiac fibrosis in the JAK2V617F group than in the JAK2WT group [54]. |
| PAD4 | After receiving ascending aorta contraction, wild-type mice showed neutrophils in the early stage of NETosis, extracellular H3Cit derived from Ly6G+ neutrophils, and citrullination of histone H3 in the nucleus; however, PAD4−/− mice did not show these characteristics [58]. | The heart functions of PAD4−/− mice in the early stage (3 d and 7 d) and late stage (28 d) were protected from the LVEF decrease observed in WT mice. Compared with WT animals, the collagen content of PAD4−/− mice was low. Furthermore, PAD4−/− mice had lower perivascular fibrosis than did WT mice [58]. |
| MPO | PMA induced significant ecDNA release in WT neutrophils but not in MPO−/− neutrophils compared with controls. However, 2-chlorofatty acids, as lipid metabolites and mediators of MPO, can bypass its physiological formation, resulting in a significant release of ecDNA from WT and MPO−/− neutrophils [220]. | Compared with wild-type mice, the peri-infarct zone of the ventricle of MPO-deficient (MPO−/−) mice showed less obvious conductivity heterogeneity and deceleration. Furthermore, MPO−/− mice showed decreased ventricular postischemic fibrosis, reflecting reduced accumulation of myofibroblasts [276]. |
| CXCR2 | Neutrophils cocultured with tumor cells and stimulated with CXCR2 receptor agonists, such as IL-8 and induced NETs [277]. | CXCR2 deficiency blocked angiotensin II-induced cardiac hypertrophy, fibrosis, and inflammation [278]. |
| MDK | The targeted midkine (MDK) not only inhibited the NETosis and infiltration of neutrophils in vivo but also reduced fibrosis occurrence in EAM and preserved the contractile function of the heart [169]. | The cardiac-specific MDK-overexpressing mice (MDK-TG) showed more severe cardiac hypertrophy and dysfunction and a lower survival rate after TAC than did WT mice [171]. |
| Sirt3 | Ex vivo LPS-induced NETosis increased in Sirt3−/− bone marrow-derived neutrophils. In vivo time to thrombotic occlusion in Sirt3−/− mice was reduced by half compared with Sirt3+/+ wild-type controls [226]. | Compared with wild-type mice, Sirt3−/− mice had a short lifespan. With increasing age, Sirt3−/− mice showed cardiac hypertrophy, fibrosis, and cardiac insufficiency. Sirt3 deficiency changed myocardial mitochondrial bioenergy, leading to the acetylation of OPA1, the target of Sirt3. Transfection of the deacetylated Opa1 gene improved the heart reserve capacity and protected the heart from hypertrophy and fibrosis [279]. |
| RIPK3 | Ripk3−/− neutrophils lacked Sytox green nucleic acid stain uptake into the nucleus, chromatin decondensation, NET formation, PicoGreen+ DNA release, and plasma membrane rupture as compared with Ripk3+/+ neutrophils [280]. | The haploinsufficiency of RIPK3 significantly attenuated Cops8-specific knockout mice in cardiomyocyte- (Cops8CKO-) induced cardiomyocyte necrosis and delayed mouse premature death [281]. |
3. NETosis in Diseases Inducing HF
Many diseases cause HF, such as myocardial infarction [59], atrial fibrillation [60], myocarditis [61], hypertrophic cardiomyopathy [62], chronic obstructive pulmonary disease [63], chronic kidney disease [64], diabetes [65], and autoimmune diseases. These examples show that many NET-related heart diseases and systemic diseases can together lead to HF (Figure 1). Here, the phenomenon and role of NETs in these diseases inducing HF will be discussed.
Figure 1.
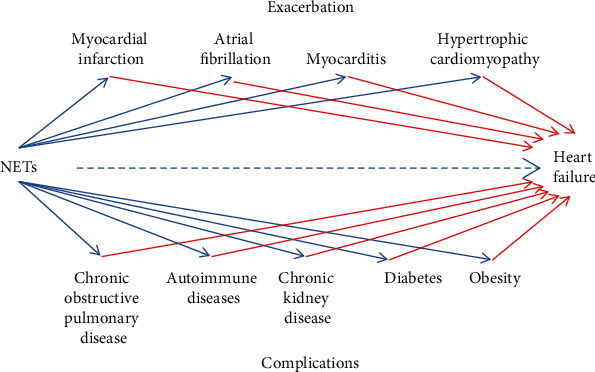
NETs' contribution to HF. NETs accelerate HF through promoting the deterioration of various heart diseases and systemic diseases. NETs: neutrophil extracellular traps.
NETs may be linked to aseptic inflammation and microthrombosis. Studies have demonstrated that NETs and NET-mediated microthrombosis can be induced during myocardial ischemia-reperfusion (I/R), which lead to myocardial “no-reflow” and myocardial infarction [66]. Multiple clinical investigations have revealed that in patients with ST-segment elevation myocardial infarction (STEMI), double-stranded DNA (dsDNA), citH3, or NE increased in the culprit lesion site obtained through percutaneous coronary intervention compared with the femoral site [67, 68]. Cardiac magnetic resonance imaging performed for evaluating microvascular obstruction (MVO) revealed that with an increase in serum dsDNA, STEMI patients were likely to develop MVO and their acute infarction area was larger, suggesting that NET components are associated with infarcted size, ventricular function, and clinical outcomes in STEMI [69]. Both humoral and cellular components are involved in plaque formation and the development of atherosclerosis, a cause of myocardial infarction and HF. Regardless of lipid changes, studies have shown that inflammatory cytokines and cellular components contribute to myocardial infarction and cardiac death. The formation of NETosis is considered to be a very important way in aseptic inflammation [70] and an important participant in promoting thrombosis in complex plaques with intraplaque haemorrhages and in adjacent vascular tissues of atherosclerosis [71]. Moreover, patients with a high serum NLR had a significantly high cumulative probability of developing atrial fibrillation [72–74]. Experimental and clinical evidence revealed that neutrophil MPO is involved in atrial fibrillation pathogenesis. Under right atrial electrophysiological stimulation, MPO-deficient mice were protected from atrial fibrillation, which was reversed when MPO was restored; this finding indicated that MPO is a key prerequisite for myocardial remodeling, leading to increased susceptibility to atrial fibrillation [75]. Furthermore, NETosis provides new prognostic information for adverse cardiovascular events in patients with atrial fibrillation [76]. In addition, in a model of hypertrophic and hypertensive cardiomyopathy caused by pressure overload, neutrophils could be seen infiltrating into the heart of mice treated with transverse aortic constriction (TAC), exhibiting inflammation and cardiac dysfunction [51]. The enrichment analysis of its Gene Ontology terminology showed that common upregulated differentially expressed genes were mainly enriched in neutrophil chemotaxis and the extracellular fibril tissue [77].
Recently, many case reports have described the clinical observation and pathology of “fulminant myocarditis” of coronavirus disease 2019 (COVID-19). The patients had increased levels of troponin I, creatine kinase-MB, and brain natriuretic peptide. In addition to the high concentrations of IL-1β, IFN-γ, IFN-inducible protein-10, and MCP-1, patients with COVID-19 had high IL-6 levels. Echocardiography revealed that the heart was enlarged and the LVEF was significantly decreased. Bedside chest radiographs showed typical ground-glass changes indicative of viral pneumonia [78–83]. Although no direct data are available showing the presence of NETs in the heart, increased levels of cell-free DNA, MPO-DNA complex, and citH3 in the blood and involvement of NETs in lung autopsy indicate that COVID-19 activates NETosis [84–86]. Furthermore, individual plasma or serum of isolated COVID-19 patients triggers NETosis [84, 85]. COVID-19 exacerbates excessive tissue inflammation and thrombotic microangiopathy, thereby increasing mortality [86–88]. Coxsackievirus B3 (CVB3) is another major factor inducing viral myocarditis [89, 90]. In the acute phase of viral myocarditis, CVB3 internalization leads to increased secretion of IL-6, IL-1β, TNF-α, and IL-8/C-X-C motif chemokine ligand 8, and infected neutrophils released MPO and triggered NETosis in the presence of TNF-α [91]. Neutrophils recognized CVB3 mainly through endosomal Toll-like receptor- (TLR-) 8, and the infection triggered NFκB activation [92]. Another report indicated that the mRNA expression of calcium-binding protein A8 and A9 (S100A8 and S100A9) derived from neutrophils in myocardial biopsy tissue of patients with CVB3-infected myocarditis increased by 13.0- and 5.1-fold, respectively. In CVB3-infected S100A8 and S100A9 knockout mice, left ventricular function improved and cardiac inflammatory and oxidative response decreased compared with those in wild-type CVB3-infected mice [93], suggesting that in addition to NETs, S100A8 and S100A9 derived from neutrophils play a key role in inducing myocarditis.
Patients with diabetes have an increased risk of HF, a high prevalence of HF and hospitalization, and poor prognosis [65]. Likewise, obesity and high-sensitivity cardiac troponin T (hs-cTnT) were both independently associated with incident HF, and individuals with severe obesity and a high hs-cTnT level had a significantly independent increased risk of incident HF [93]. This may be because increased cardiac output and hypertension lead to an increase in cardiac preload and afterload and left ventricular hypertrophy as well as the excessive deposition of myocardial collagen, abnormal protein glycosylation, and collagen cross-linking due to systemic inflammation. Therefore, obesity leads to HF through metabolic and inflammatory pathways [94]. Numerous studies have shown that the plasma or serum of patients with type 2 diabetes has increased levels of nucleosomes, human NE-DNA complex or MPO-DNA complex, IL-6, and TNF-α [95, 96]. Patients with previous myocardial infarction have increased citH3 and cell-free DNA levels [97]. Another study revealed that among 1572 patients with HF with decreased ejection fraction, 493 (31%) had diabetes. Compared with nondiabetic patients, diabetic patients have a high body mass index, severe symptoms and signs of HF, and hypertension history [98]. The authors performed the network analysis to determine whether the epidermal growth factor receptor and galectin-3 (GAL3) were the most prominent connecting proteins. The translation of these networks to biologic pathways revealed that diabetes was associated with inflammatory response and neutrophil degranulation [98]. By contrast, early studies have revealed that neutrophils infiltrate the capillaries of the subcutaneous and intra-abdominal fat tissue of obese patients [99, 100]. According to the plasma MPO-DNA complex, the NET level in the obesity group was higher than that in the healthy control group. In the medical history of patients with increased NETs, researchers observed increased thromboembolic events [101]. Prevention of NETosis with Cl-amidine, a cell-permeable pan PAD inhibitor, or dissolution of NETs with DNase restored endothelium-dependent vasodilation of the mesenteric arteries of diet-induced obese mice [102]. This indicates that obesity-induced NETs can impair the vasodilation function of mesenteric arterioles.
Accumulated evidence has confirmed that NETs are involved in the pathogenesis of autoimmune diseases such as RA, systemic lupus erythematosus (SLE), and vasculitis [103]. In clinical and biological observations, the overlap between NETs and RA, SLE, and vasculitis can usually be observed, which indicates that NETosis is the main triggering event of these autoimmune diseases. NETosis increases the possibility of association between citrullinated proteins or MPO autoantigens, and ANCA or anticitrullinated proteins/peptide antibodies (ACPA), triggering an autoimmune response [30, 104]. Numerous studies have revealed that RA and SLE can affect HF and other cardiovascular events (see review article References [105] and [106]). Furthermore, in RA patients, the plasma levels of ACPA and antimodified citrullinated vimentin antibody (AMCVA) are negatively correlated with LVEF [107]. Another study showed that higher ACPA and AMCVA levels are associated with higher adjusted mean left ventricular mass index (LVMI) compared with the lower antibody group [108]. An autopsy study of the heart found that the average and maximum intensity of anticitrulline staining in the myocardial interstitium of the RA group were 59% and 44% higher than those of the non-RA control group [109]. In a proteomics study targeting the citrullination of the heart protein of HF, 304 citrullination sites of 145 proteins were identified. Among them, citrullinated myosin decreased its intrinsic ATPase activity, and citrullinated tropomyosin leads to stronger F-actin binding; thus, the combined action of the two inhibits acto-heavy meromyosin ATPase activity, which indicates that citrullination of sarcomeric protein leads to an overwhelming reduction in Ca(2+) sensitivity in cardiomyocytes [110]. ANCA, as a specific antibody of proteinase-3 and MPO, is closely related to HF. In recent years, many cases have reported ANCA antibody-induced HFrEF, severe aortic regurgitation and mitral regurgitation, and a severe insufficiency of the aortic valve [111, 112]. One 20-year population-based cohort analysis recruited 58 patients diagnosed with ANCA-associated vasculitis (AAV); the risk of cardiovascular events (coronary artery disease, HF, and atrial fibrillation) was more than three times higher and the risk of cerebrovascular accident was eight times higher in AAV patients than in matched subjects [113]. The overlapping of antineutrophil citrullinated protein antibody or anti-MPO antibody with the heart damaged tissue suggests two possibilities: (1) with the infiltration of neutrophils in the cardiac tissue, NETosis is indeed a pathogenic factor of HF; (2) the existence of these two antibodies in the heart tissue is the embodiment of autoimmune disease eroding the heart function.
4. Promotion of NETosis and HF by Circulating Mitochondrial DNA
Mitochondrial DNA (mtDNA) leads to cardiac dysfunction in an aging heart. In elderly people aged >90 years, the plasma mtDNA level gradually increased with age, and the increase in the plasma mtDNA level was positively correlated with plasma TNF-α, IL-6, RANTES, and IL-1ra levels [114]. Moreover, the frequency of activated HLA-DR(+) neutrophils in elderly and frail elderly people was positively correlated with circulating mtDNA, which increased the expression of HLA-DR in neutrophils in a dose-dependent manner ex vivo [115]. Myocardial mtDNA content was positively correlated with the peripheral blood mtDNA content and left ventricular function in nonischemic HF patients [116]. Furthermore, during hypoxia reoxygenation, mtDNA induced inflammation and increased the expression of the proinflammatory cytokines IL-1β and TNF-α in mouse cardiomyocytes [117]. Circulating mtDNA may be used as a marker of increased mortality in patients with severe acute HF [118].
NETs represent extracellular structures that bind to and kill microorganisms. However, mtDNA released from damaged tissues can also induce NETosis, which may lead to tissue inflammation and thromboembolism. A decade-old study revealed that NETosis was induced by mtDNA released from cells [119]. mtDNA released from cancer, trauma, and tissue damage triggers NETosis through the TLR-9 signaling pathway [120, 121]. TLR-9, as one of the pattern recognition receptors (PRRs), senses unmethylated CpG dinucleotides that are relatively common in the genomes of most bacteria and DNA viruses but are suppressed and methylated in the genomes of vertebrate nuclei. The lysosomal localization of TLR-9 allows efficient detection of invading bacterial and viral nucleic acids while preventing accidental stimulation by CpG motifs within self-DNA [122]. Because mitochondria advance from prokaryotic bacteria, mtDNA retains molecular motifs similar to bacterial DNA. Therefore, TLR-9 can recognize mitochondrial unmethylated CpG sequences. The primary neutrophils of human and mouse express active and functional TLR-9 [123]. In the process of lung I/R, the release of acellular mtDNA triggers NETosis through the TLR-9 signal, and TLR-9 deficiency of neutrophils prevents mtDNA-induced NETosis [118, 119]. In HF, mtDNA escaping from autophagy cells can also induce myocarditis and dilated cardiomyopathy through TLR-9 signaling [124]. In a diastolic HF model established in cardiomyocyte-specific and inducible SERCA2a gene knockout C57Bl/6J mice, 4 weeks after conditional gene knockout, the mice were randomly given the TLR-9 agonist CpG-B. After 4 weeks, administering CpG-B shortened the life expectancy of SERCA2a gene knockout mice, reduced ventricular diastolic function, and increased heart and systemic inflammation, showing exacerbation of diastolic HF [125]. Stimulation of TLR-9 can induce inflammation and HF, whereas TLR-9 deficiency or long-term administration of the TLR-9 inhibitor E6446 or chloroquine can prevent left ventricular dilatation and cardiac insufficiency, fibrosis, and inflammation [126–128], providing a new perspective for the intervention and treatment of inflammatory-related diseases such as chronic HF. Another report revealed that cGAS-STING signaling is also included in NETosis induced by mtDNA. NETosis induced by mtDNA was attenuated in STING−/− and TLR9−/− mice in the area of tissue damage caused by I/R, suggesting that TLR-9 and STING pathways help mtDNA induce NET. Bone marrow neutrophils from STING−/− and TLR-9−/− mice showed a lower percentage of NET in mtDNA stimulation [123]. These findings provide a new perspective that circulating mtDNA may activate TLR-9- and cGAS-STING-mediated systemic and cardiac inflammation, chronic HF, and NETosis.
5. Damage-Associated Molecular Patterns and Cytokines Linking HF and NETosis
In addition to mtDNA, some other damage-associated molecular patterns (DAMPs) are used as important biomarkers and proinflammatory factors of HF, such as S100A8/A9 (calprotectin), high mobility box group box 1 (HMGB1), fibronectin extra domain A (FN-EDA), and GAL3 [129, 130]. Multiple studies have reported that serum S100A8/S100A9 levels derived from neutrophils were elevated in myocarditis [131], I/R injury [132], atrial fibrillation [133], or chronic HF [134], which can be used as a powerful potential biomarker. Thus, S100A8/A9 secreted by activated neutrophils is essential for proinflammatory function [135]. It is now understood that the induction of myocardial infarction leads to the rapid recruitment of neutrophils to the infarct, where they release the alarm protein S100A8/A9 and bind to TLR-4 to activate the naive neutrophil NLR family pyrin domain containing 3 inflammasome and promote IL-1β secretion. The released IL-1β stimulates the autonomous granulocyte production of hematopoietic stem cells in the bone marrow, which requires NETosis [136, 137].
In addition to the Kubota group, which used HMGB1 transgenic mice to show that cardiac nuclear HMGB1 exerts protective effects on myocardial infarction, cardiac hypertrophy, and HF [138–140], many studies have shown that HMGB1 is an independent predictor of death in HF [141–143]. By contrast, antagonistic HMGB1 box A, miR-129-5p, and the natural product HMGB1 inhibitor glycyrrhizic acid reduce infarct size and tissue damage of the heart; inhibit oxidative stress and inflammatory response; and prevent pressure overload-induced cardiac hypertrophy, heart fibrosis, and failure [144–147]. Importantly, HMGB1 promotes NETs and exacerbates tissue damage [148, 149]. Furthermore, soluble FN-EDA is a valuable biomarker in cardiac remodeling, and in patients with HF, the serum FN-EDA level was significantly elevated [150]. Studies have shown that in ApoE−/− mice, FN-EDA promoted myocardial I/R injury, increased infarct size, elevated plasma cardiac troponin I levels, induced neutrophil infiltration and extracellular traps, and caused cardiomyocyte apoptosis [151]. GAL3 is another important component of DAMPs and a biomarker for cardiac fibrosis and failure. A high GAL3 level was associated with HF severity, such as a high New York Heart Association HF class; high systolic blood pressure; high creatinine, N-terminal prohormone of brain natriuretic peptide, IL-6, and C-reactive protein levels; and lower maximal oxygen consumption [152, 153], as well as increased left ventricular weight and echocardiographic changes in the left ventricular end-diastolic volume [154, 155]. The GAL3 level is a strong and independent predictor of unfavorable outcomes, which can predict the long-term mortality of patients with severe chronic HF [156, 157]. GAL3 has a higher predictive value in patients with preserved LVEF than in patients with reduced LVEF [153, 156, 157]. In contrast, some early studies have revealed that GAL3 facilitate neutrophil recruitment and has the ability to activate neutrophils [158–160]. Other two PRRs, TLR-4 and receptor for advanced glycation end products (RAGE), appear to be DAMP receptors. Neutrophils express TLR-4 and RAGE [161–164]. HMGB1, FN-EDA, and GAL3 activate the inflammation of neutrophils and microglial cells through TLR-4 activity [153, 165, 166]. Moreover, HMGB1 mediates subsequent tissue damage amplification through neutrophil recruitment induced by RAGE [167]. In myocarditis, HMGB1 and its major receptor RAGE seem to be key factors in the pathogenesis of TnI-induced experimental autoimmune myocarditis (EAM) [168].
Dilated cardiomyopathy caused by myocarditis can develop into HF. A recent study reported that midkine induced cardiac inflammation by promoting neutrophil trafficking and NETosis in myocarditis. In EAM in mice, midkine as a target can not only inhibit NETosis and neutrophil infiltration in vivo but also reduce myocardial fibrosis and maintain cardiac function [169]. Midkine is a proproliferation and proinflammatory cytokine. A clinical investigation indicated that the serum midkine concentration of HF patients was significantly higher than that of controls. Patients with cardiac events had a higher midkine concentration than did patients without cardiac events [170]. After TAC, the midkine expression level was increased in the kidney and lungs but not in the heart. After TAC, compared with wild-type mice, transgenic mice with cardiac-specific overexpression of midkine showed more severe cardiac hypertrophy and dysfunction and a lower survival rate [171]. Moreover, renal ischemic injury could increase midkine expression in the proximal tubules of the kidney, leading to the recruitment of neutrophils to the tubule interstitium. The knockout of the midkine gene inhibited neutrophil infiltration [172] and reduced myocardial hypertrophy induced by subtotal nephrectomy compared with wild-type mice [173]. Many studies have demonstrated that LDL receptor-related protein 1 (LRP1) is an endocytic receptor of midkine [174, 175], and it facilitates neutrophil adhesion and trafficking through the interaction between LRP1 and β2 integrins during acute inflammation [176–178]. More importantly, midkine initiates NETosis through interaction with LRP1 [169]. The neutrophil activity mediated by DAMPs is shown in Table 2 and Figure 2. Furthermore, in a relatively early period, many studies have confirmed that high sensitivity C-reactive protein (hs-CRP) can be used as a potential indicator of risk stratification in HF patients [179]. The plasma hs-CRP level gradually increases with the deterioration of LV diastolic dysfunction [180]. Compared with HFrEF patients, CRP predicts mortality in HFpEF patients is significantly enhanced [181]. Recent data indicate that the higher the plasma hs-CRP is, the higher the mortality of HF patients is [182]. Thirty years ago, it was discovered that hs-CRP can bind to neutrophils [183]. Then, it was confirmed that CRP binds to IgG FcγRI (CD64) and IgG FcγRII (CD32) high-affinity receptors [184, 185]. Recent research results show that compared with patients with low CRP with STEMI, patients with high CRP are accompanied by elevated circulating IL-1β, NETosis, and NET-associated tissue factor plasma levels [186]. The serum CRP level of patients with HF or HF+type 2 diabetes (T2DM) is significantly higher than that of the healthy control group, and the NET release rate from HF or HF+T2DM patients is faster than that of T2DM and healthy control groups without stimuli [187]. Earlier studies pointed out that CRP stimulates the formation of oxidative bursts as a necessary condition for NET release [188–190]. Besides, studies from a decade ago have found a significantly higher level of platelet-derived soluble CD40L in patients with chronic HF [191, 192]. Recent researches also discovered a correlation between soluble CD40L and the release of NETs and the increase of oxidative burst [193]. The above summarized references indicate that DAMPs, soluble CD40L derived from platelets, and CRP in the adjacent tissues activate the formation of NETs.
Table 2.
Effect of inhibitors of enzymes activating NETosis on heart failure-related diseases.
| Classification | Therapeutic or experimental purposes | Drug | Animal | Model | Administration | Dose | Experimental period | Effects | References |
|---|---|---|---|---|---|---|---|---|---|
| PAD4 inhibitor | Myocardial infarction | GSK484 | 20–25 g male C57BL/6 mice | Permanent ligation of the left anterior descending coronary artery was performed to induce myocardial infarction in mice | Intraperitoneal injection (3 days before MI and 2 days after MI) | 4 mg/kg per day | 3 days before MI and 2 days after M I | Using GSK484 can moderately preserve the tissue structure and myocardial integrity of the ventricle after MI, reduce the infarct size, and reduce the level of myocardial CK-MB, LDH, and cTnT in the serum. PAD4 inhibition also effectively protects cardiomyocytes from MI-induced NET formation and secretion of inflammatory cytokines IL-1β, IL-6, and TNF-α and reduces cardiomyocyte apoptosis caused, thereby improving the overall heart function | [57] |
| Myocardial infarction and thrombus formation | Cl-amidine | C57BL6/J mice aged 8-14 weeks and weighing 21-25 g | (1) Use of wire injury or topical application of FeCl3 to induce thrombus in the mouse carotid artery; (2) the LAD was ligated and the PE-10 tube was withdrawn after 60 minutes to create a mouse cardiac ischemia-reperfusion model | In the myocardial ischemia-reperfusion model, Cl-amidine was administered intraperitoneally at the onset of ischemia and at time of reperfusion. A third dose of Cl-amidine was administered 12 hours after reperfusion | 10 mg/kg | FeCl3-induced mouse arterial thrombosis is highly consistent with the immune cell composition of coronary artery thrombosis in patients with myocardial infarction. Neutrophils are the most abundant cell type, and there are NETs and coagulation factors. Cl-amidine abrogated NET formation, reduced arterial thrombosis, and limited injury in a model of myocardial infarction | [252] | ||
| Atherosclerosis | Cl-amidine | Apolipoprotein-E (Apoe)−/− mice | Feeding high-fat food (42% from fat) | Daily subcutaneous injection | 10 mg/kg per day | Beginning at 7 weeks and through euthanasia at 18 weeks | Pharmacological inhibition can prevent the formation of NET, reduce the area of atherosclerotic lesions, and delay the time of carotid artery thrombosis, accompanied by decreased recruitment of networked neutrophils and macrophages to the artery, and decreased expression of interferon-α | [253] | |
| MPO inhibitor | Atherosclerosis | PF-06282999 | LDLR−/− male mice | Feeding a western diet (0.2% cholesterol), ad libitum | Daily gavage | 5 or 15 mg/kg | Starting from 8-10 weeks for 7-16 weeks | After 4 weeks of treatment with MPO inhibitors, it reduces the necrotic core area of the aortic root and decreases the activity of MPO and the uptake of [18F]-fluoro-deoxy-glucose in the aorta, as an indicator of plaque load and inflammation, but the inhibitory effect of MPO will not change the homing of white blood cells | [254] |
| Myocardial infarction | PF-1355 | 8-12 weeks old female C57Bl/6J mice | For ischemia-reperfusion injury (IRI) model, left coronary artery was transiently ligated for 30 minutes and then the ligation suture was removed | Twice daily by oral gavage | 50 mg/kg | 21 day | MPO inhibition for 7 days resulted in decreased MPO and CD11b expression, and inflammatory Ly6Chigh monocytes. Continuous MPO inhibition for more than 21 days can significantly improve the ejection fraction, the end-diastolic volume/end-systolic volume ratio, and the effect of cardiac magnetic resonance on the quality of the left ventricle | [255] | |
| NE inhibitor | Endotoxin-induced myocardial injury | Sivelestat | 9-12-week-old male mice | 20 mg/kg LPS intraperitoneal injection to induce endotoxin | Intraperitoneal injection | 0.2 mg/kg | 48 h after LPS treatment | The survival rate of mice injected with sivelestat was significantly higher, while the levels of serum troponin I and IL-6 were significantly lower than those of the control group. The vascular endothelium of mice treated with sivelestat clearly showed that the endothelial glycocalyx was well preserved at the ultrastructural level | [256] |
| Ischemia-reperfusion injury | SSR69071 | Male New Zealand white rabbit, weighing 2-3 kg | Coronary artery occlusion for 30 min followed by reperfusion for 120 min | Intravenous injection 15 min before coronary ligation or 25 min after coronary ligation (5 min before reperfusion) | 1, 3, and 10 mg/kg | Elastase activity in the heart was significantly increased in ischemia-reperfusion animals. The activity of elastase was significantly decreased after treatment with ssr69071 before reperfusion. Ssr69071 treatment significantly reduced myocardial infarction size | [257] |
LAD: left anterior descending artery; LPS: lipopolysaccharides; MI: myocardial infarction; MPO: myeloperoxidase; NE: neutrophil elastase; PAD4: peptidyl arginine deiminase-4; PCI: percutaneous coronary intervention.
Figure 2.

Initiation of NETs by PRRs. Various DMAPs and cytokines activate NET formation through PRRs. FN-EDA: fibronectin extra domain A; HMGB1: high mobility box group box 1; LRP1: LDL receptor-related protein 1; mtDNA: mitochondrial DNA; PRRs: pattern recognition receptors; RAGE: receptor for advanced glycation end products; ST2: a receptor for IL-33; TLR: Toll-like receptor.
6. Role of Oxidative Stress in HF and NETosis
As we all know, oxidative stress is closely related to inflammation. A large amount of evidence has shown that inflammation plays an important role in the onset and development of cardiovascular diseases [194]. Similarly, numerous evidences also fully reveal that oxidative stress and ROS-related low-grade chronic inflammation affect the occurrence and development of cardiac systolic and diastolic function, cardiac hypertrophy, and remodeling [195–197]. NETosis is a form of inflammation and cell death, while the formation of NET requires an oxidative burst of neutrophils [198, 199]. NETosis caused by various stimuli depends on the production of the oxidative burst, which is mainly produced by NADPH oxidase activation [200], but some studies have found that mitochondrial oxidative stress is indispensable for this process [201–203]. Previous studies have confirmed that the oxidative burst in the generation of NETosis involves various signals via NADPH oxidase, such as Rac and Pak signals [204], protein kinase C [205], Src family kinases [206], spleen tyrosine kinase [207], and Vav-PLCγ [208] (Figure 3). Oxidative burst is mediated by a variety of PRRs, such as TLR2, TLR3, TLR4, and TLR 9 [209–211]. Moreover, RAGE also activates oxidative burst in diabetes [212]. Interestingly, the priming of the neutrophil oxidative burst requires IL-8 to induce sequential recruitment of NADPH oxidase components into lipid rafts [213]. In this process, phospholipase D is also activated by IL-8 [214].
Figure 3.
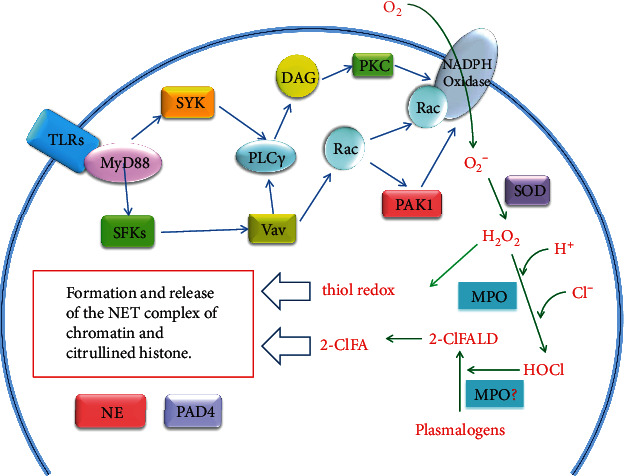
Possible pathways for the formation of NETs activated by oxidative stress. 2-ClFA: 2-chlorofatty acid; 2-ClFALD: 2-chlorofatty aldehyde; DAG: diacylglycerol; MPO: myeloperoxidase; NE: neutrophil elastase; PAD4: peptidylarginine deiminase 4; PAK1: P21 (Rac)-activated kinase 1; PLCγ2: a calcium-dependent phospholipase; PKC: protein kinase C; Rac: Rac GTPases, a small G-protein; SFKs: Src family protein tyrosine kinases, which include eight members: c-Src, c-Yes, Fyn, c-Fgr, Lyn, Hck, Lck, and Blk; SOD: superoxide dismutase; SYK: spleen tyrosine kinase; TLRs: Toll-like receptors; Vav: a guanine nucleotide exchange factor.
Recently, a study reported that the redox imbalance of neutrophils during the oxidative burst promotes the generation of NETosis. The authors believed that the generation of NETosis is partly regulated by the changes in the cytosolic thiol redox homeostasis in neutrophils and depends on the circumstances under which NETosis is generated [215]. Meanwhile, more research focuses on the role of MPO and MPO-derived HOCl in the uncontrolled NET formation (NETosis). Researchers found that completely neutralizing extracellular ROS is not enough to block PMA-triggered NETosis generation, but removing MPO-related ROS in azurophilic particles effectively prevents the suicidal NETosis [216]. Furthermore, in MPO-deficient neutrophils, the extracellular addition of MPO still cannot rescue the formation of NETosis, indicating that the intracellular granules are involved in [216]. MPO is the peroxidase in the lysosomal cyanophilic granules of neutrophils, and the target of HOCl derived from MPO has been a topic of concern in recent years. Up to now, it is known that plasmalogen phospholipids can be oxidized by HOCl to 2-chlorofatty aldehyde (2-ClFALD). Up to now, it is known that 2-ClFALD has four metabolic pathways, which can be oxidized to 2-chlorofatty acid (2-ClFA), reduced to 2-chlorofatty alcohol, formed Schiff base adducts with proteins and amines, or reacted with glutathione through nucleophilic attack of α-chlorinated carbon [217]. Because the plasma membrane of neutrophils, endothelial cells, vascular smooth muscle cells, myocardial cells and nerve cells enriched with plasmalogens as the source of 2-ClFALD, this provides the possibility of plasmalogen reacting with HOCl in biological systems [218]. In the heart of myocardial infarction rats, α-chloro fatty aldehyde 2-chlorohexadecanal (2-ClHDA), a 16-carbon chain chlorinated fatty aldehyde, accumulated in the hearts of rats with left anterior descending artery occlusion. Compared with sham-operated rats, levels of 2-ClHDA and neutrophil infiltration in myocardium of surgical infarction rats were increased [219]. In recent years, studies have revealed that human neutrophils are treated with physiological levels of 2-ClFAs to form NET structure, which is characterized by the binding of MPO to DNA and NE redistribution to the area around the nucleus. 2-ClFA can also induce NETosis of bone marrow-derived neutrophils in MPO-deficient mice [220] (Figure 3). Many studies have explored the effects of 2-ClFALD and 2-ClFA on the function of neutrophils, vascular endothelial cells, monocytes, and blood vessels, suggesting that MPO-derived chlorinated lipids can cause inflammation and vascular tension changes, and contribute to the occurrence and development of cardiovascular diseases [217].
Serum albumin is a powerful predictor of death or hospitalization in patients with HFpEF. Low serum albumin is the link between higher myocardial extracellular volume and higher levels of N-terminal pro-B-type natriuretic peptide (NT-proBNP) [221]. Low serum albumin is also associated with arterial stiffness, diastolic dysfunction, pulmonary hypertension, and inflammatory biomarkers such as white blood cell count, IL-6, and TNF-α in patients with HFpEF [222]. In patients with diabetic nephropathy, hypoalbuminemia, systemic inflammation, and oxidative stress are common. However, due to oxidized albumin, hypoalbuminemia in these patients may be underestimated, and increased oxidized albumin may lead to accelerated cycle between oxidative stress and neutrophil activation [223]. Recently, it has been reported that the oxidation of albumin, a major source of free thiol, is sufficient to trigger NETosis through accumulation of reactive oxygen species within neutrophils in pulmonary cancer metastasis, because the oxidation of albumin-derived free thiol leads to the redox imbalance in the blood [224]. The free thiol derived from albumin reflects the systemic redox state in the circulation. Studies have shown that patients with chronic HF with higher mean serum-free thiol concentration are younger, have better kidney function, have lower levels of NT-proBNP, and have reduced rehospitalization rates and increased patient survival rates [225]. These studies suggest that the redox imbalance of albumin oxidation through the serum-free thiol reaction is a way to trigger NETosis.
7. Role of SIRT3 in HF and NETosis
SIRT3 is a major mitochondrial deacetylase and an important factor in maintaining cardiac mitochondrial bioenergy. Multiple studies have revealed that SIRT3 deficiency or decreased expression can induce the excessive acetylation of pyruvate dehydrogenase, ATP synthase, cyclophilin D, and mitochondrial dynamic like GTPase OPA1; increase the opening of mitochondrial permeability transition pore; and change the process of oxidative phosphorylation, resulting in mitochondrial dysfunction [226–228]. Furthermore, SIRT3 deficiency or decreased expression caused changes in cardiomyocyte glycolysis, myocardial hypertrophy, and fibrosis and low cardiac reserve capacity, which impaired diastolic function and induced senile HF [226–228]. SirT3 expression was lower in STEMI and obese patients and hypertension model animals than in healthy donors [171, 227, 229]. In the LPS-excited laser-induced carotid artery thrombosis model, compared with the Sirt3+/+ wild-type control group, the thrombotic occlusion time in Sirt3−/− mice was reduced by half [171]. Moreover, ex vivo LPS-induced NETosis was increased in Sirt3−/− bone marrow-derived neutrophils, indicating that SIRT3 deletion in the inflammatory environment affected NETosis and arterial thrombosis in mice [171]. Another report indicated that long-term exposure to indoxyl sulfate, a uremic toxin that induces HF, promoted arterial thrombosis by reducing levels of SIRT1 and SIRT3 in the aorta [230].
8. Effect of NETosis on HF via Thromboembolism
In recent years, several studies have shown that NET promotes thrombus formation [55, 231, 232]. Neutrophil-derived PDP4 promotes vWF-platelet string formation [233]. The dense chromatin released by neutrophils constructs an extracellular DNA network. This network forms scaffolds in inflammatory vessels; promotes platelet adhesion, activation, and aggregation; absorbs red blood cells; leads to fibrin deposition; and induces thrombosis [234]. During NETosis, the released DNA also forms a network structure and interacts with vWF to promote the adhesion and activation of platelets [234]. Studies have shown that the new blood nucleic acid scavenger can inhibit thrombosis without increasing bleeding, which reversely demonstrated the role of the neutrophil DNA network structure in thrombosis [235]. Furthermore, neutrophils recruited to the tissue damage area release the specific alarm protein S100A8/A9 [236, 237] and stimulate their naive neutrophils to mature in a cell-autonomous manner [238]. Moreover, S100A8/A9 regulates thrombosis by binding to CD36 on platelets [239].
Recent studies have revealed that DMAPs that induce NETs also promote thrombosis, such as HMGB1, FN-EDA, GAL3, and C-X-C motif chemokine receptor 2 (CXCR2). In addition to activating neutrophils, HMGB1 also regulates platelet activation, granule secretion, adhesion, and diffusion through the TLR-4 pathway on platelets [240]. Similar to HMGB1, FN-EDA−/− mice showed prolonged time for thrombosis and complete occlusion, and the thrombus growth rate was significantly reduced compared with FN-EDA+/+ mice. TLR-4 gene deletion reversed accelerated thrombosis in FN-EDA+/+ mice [241]. Although no direct evidence exists regarding arterial thrombosis, in the deep venous thrombosis (DVT) model of GAL3+/+ mice, galectin-3-binding protein (GAL3BP) and GAL3 were localized in the vein wall, red blood cells, and platelets, whereas only GAL3 was expressed in neutrophils. GAL3 increased significantly during early DVT, and GAL3BP/GAL3 was co-located at the interface between neutrophils and endothelial cells. Some neutrophils attached to the vein wall, whereas the number of activated neutrophils in the vein wall of GAL3−/− mice significantly decreased. Thrombus size was associated with increased levels of GAL3 and IL-6 in the venous wall [242]. IL-8, neutrophil-activating peptide-2, and growth-related protein alpha trigger cell activation through binding to specific and different amino acid residues of CXCR2, which includes the induction of neutrophil migration [243]. Neutrophils migrate through a CXCR2-dependent mechanism to accumulate in the thrombus and stimulate the release of NETs to increase the frequency and size of thrombus [244]. The possible mechanism of DMAPs activating NET-dependent thrombosis is shown in Figure 4. As early as 1981, aortic thrombosis was reported to be present in congestive HF [245], and then, it was believed to be related to vWF and platelet receptor protein of glycoprotein [246]. In clinical practice, HF is considered to be associated with an increased incidence of thromboembolic events [247], and many causes of HF death are also related to thromboembolic events [248].
Figure 4.
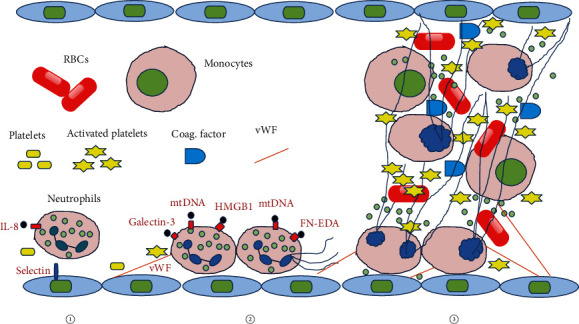
Possible mechanism of DMAPs activating NET-dependent thrombosis. (1) Migration of neutrophils driven by the chemokine receptor CXCR2 and ligand IL-8. (2) Various DMAPs induce thrombosis by activating NETs. (3) With the participation of activated platelets, endothelial cells, monocytes, red blood cells, and blood coagulation factors, neutrophils promote thrombosis. Coag. Factor: coagglutination factor; FN-EDA: fibronectin extra domain A; HMGB1: high mobility box group box 1; mtDNA: mitochondrial DNA; RBCs: red blood cells; vWF: von Willebrand factor.
9. Cardioprotective Effect of NETosis-Related Inhibitors
Neutrophil specific enzymes, MPO, PAD4, and NE, play a key role in the formation of NETs. Up to now, PAD4 inhibitors such as BMS-P5, GSK484, GSK199, and cl-amidine and MPO inhibitors such as PF-1355, PF-06282999, INV-315, and AZD5904 have been developed. MPO is a peroxidase containing heme, mainly expressed in neutrophils and a small amount in monocytes, where it catalyzes the production of reactive oxygen intermediates such as hypochlorous acid (HOCl) as antibacterial armory from hydrogen peroxide and Cl− ions. However, MPO is also associated with oxidative stress and tissue damage resulting from the potent oxidant HOCl, which plays a key pathogenic event in a variety of inflammatory states, including cardiovascular disease. MPO inhibitors, such as KYC, seem to prevent PMA-stimulated neutrophil HOCl without affecting superoxide production [249]. Another way to eliminate NET formation is PAD4 inhibitor. A report claimed that the PAD4 inhibitor Cl-amidine can inhibit leukocyte activation, oxidative burst from overactive leukocytes, and subsequent DNA damage of target epithelial cells in vitro and in vivo in an ulcerative colitis mouse model [250]. PAD4 and the cytoplasmic subunits of NADPH oxidase, p47phox and p67phox, can form a physical association, leading to citrullination of the cytoplasmic subunits of NADPH oxidase and oxidative explosion. The small molecule PAD4 inhibitors, such as BB-Cl-amidine and GSK484, can destroy the complex of PAD4 and the cytoplasmic subunits of NADPH oxidase and reduce the oxidative burst [251].
Recently, several studies have reported the effect of PAD4 inhibitors on cardiovascular disease (Figure 2). In a mouse myocardial ischemia model, GSK484, a PAD4 inhibitor, reduces infarct size and cardiomyocyte apoptosis, promotes the improvement of cardiac function, ameliorates cardiac neutrophil infiltration and NET formation, depresses PAD4 expression and cit-H3 level, and inhibits secretion of inflammatory cytokines [57]. Another study also supports that Cl-amidine abrogates NET formation, decreases arterial thrombosis, and limits injury in a mouse model of myocardial infarction [252]. Similar results have been observed in the ApoE−/− atherosclerosis mouse model. Treatment with Cl-amidine for 11 weeks decreases the atherosclerotic lesion area, reduces the recruitment of neutrophils and macrophages to the artery and prevents the formation of NET, and downregulates interferon-α expression in the arteries. In addition, Cl-amidine treatment delays carotid artery thrombosis in a photochemical injury model [253]. However, although the MPO inhibitor PF-06282999 cannot change the macrophage content and white blood cell homing in atherosclerotic plaques, it can reduce the necrotic core area, suggesting that PF-06282999 promotes the stability of atherosclerotic lesions in the LDLR−/− mouse model of atherosclerosis [254]. Likewise, another MPO inhibitor INV-315 results in reduced plaque burden and enhanced cholesterol efflux, decreased aortic iNOS gene expression, superoxide production, and nitrotyrosine content, and improved endothelial function of acetylcholine [255]. Further, gavage administration of the MPO inhibitor PF-1355 for 7 days also inhibits the increase in MPO activity in the myocardial infarction area of mice, reduces the number of inflammatory cells, and slows down the expansion of the left ventricle. After 21 days of administration, the mouse heart function and remodeling were improved (Figure 2) [256]. Moreover, specific NE from neutrophils is another key enzyme involved in the formation of NETs. Research reports have shown that its inhibitors Sivelestat and SSR69071 can improve myocardial injury caused by endotoxin or/and ischemia-reperfusion (Figure 2), respectively [257, 258]. Sivelestat treatment effectively maintained the structure of mouse vascular endothelium and endothelial glycocalyx [257], while SSR69071 can reduce infarct size [258]. Additionally, PAD4 inhibitor Cl-amidine or BB-Cl-amidine shows a good therapeutic effect on heart failure-associated autoimmune diseases such as rheumatoid arthritis, lupus erythematosus, MPO-ANCA-associated vasculitis, and Behçet's disease [259–263]. The MPO inhibitor PF-1355 also suppresses immune complex vasculitis and glomerular basement membrane nephritis [264].
Statins and metformin may effectively improve heart failure clinically (see review article References [265–268]). In recent years, a large number of studies have reported that both statins and metformin have shown effective anti-inflammatory effects. Evidence shows that simvastatin and metformin can inhibit the formation of NETs [269, 270]. Clinically, rosuvastatin significantly attenuates the plasma MPO level, platelets, and circulating neutrophils and monocytes [271, 272]. Similarly, metformin also decreased plasma MPO levels, reduces the number of peripheral blood neutrophils, and prevents infiltration to myocardium in clinical and animal experiments [273, 274]. These pharmacological and clinical studies have shown that resisting the formation of NETs is beneficial for improving heart failure.
10. Conclusion
In short, DAMPs, such as mtDNA, HMGB1, FN-EDA, and GAL3, released by damaged organs may activate neutrophils and form NETs through TLR-4/9 and RAGE, which requires the participation of oxidative stress, thereby causing or aggravating cardiac inflammation and fibrosis. NETs promote thrombosis of coronary microvessels and affect cardiac function. In some studies, neutrophil-specific MPO and PAD4 ablation or inhibition could improve the disease progression of HF, myocardial infarction, and atrial fibrillation, which all illustrate the fact that NETosis is a pathogenic factor of HF. Recently, Abrams et al. [275] have developed a new assay that may independently predict diffuse intravascular coagulation and mortality in critically ill patients caused by neutrophil extracellular traps. These facts are important for understanding the new pathophysiology of HF and will contribute toward the development of therapeutics and pharmacy.
Acknowledgments
This work was supported by grants from the Specialized Research Fund for the National Natural Science Foundation of China (81973511).
Abbreviations
- 2-ClFA:
2-Chlorofatty acid
- 2-ClFALD:
2-Chlorofatty aldehyde
- 2-ClHDA:
Alpha-chloro fatty aldehyde 2-chlorohexadecanal
- ACPA:
Anticitrullinated proteins/peptide antibody
- AMCVA:
Antimodified citrullinated vimentin antibody
- ANCA:
Antineutrophil cytoplasmic antibody
- citH3:
Citrulline histone H3
- COVID-19:
Coronavirus disease 2019
- hs-CRP:
High-sensitivity C-reactive protein
- hs-cTnT:
High-sensitivity cardiac troponin T
- CVB3:
Coxsackievirus B3
- CXCR2:
C-X-C motif chemokine receptor 2
- DAMPs:
Damage-associated molecular patterns
- dsDNA:
Double-stranded DNA
- DVT:
Deep venous thrombosis
- EAM:
Experimental autoimmune myocarditis
- FN-EDA:
Fibronectin extra domain A
- GAL3:
Galectin-3
- GAL3BP:
Galectin-3-binding protein
- HF:
Heart failure
- HFpEF:
Heart failure with preserved ejection fraction
- HMGB1:
High mobility box group box 1
- IL-1β:
Interleukin-1beta
- IL-6:
Interleukin-6
- IL-8:
Interleukin-8
- I/R:
Ischemia-reperfusion
- LRP1:
LDL receptor-related protein 1
- LVEF:
Left ventricular ejection fraction
- LVMI:
Left ventricular mass index
- MCP-1:
Monocyte chemoattractant protein-1
- MPO:
Myeloperoxidase
- mtDNA:
Mitochondrial DNA
- MVO:
Microvascular obstruction
- NE:
Neutrophil elastase
- NETs:
Neutrophil extracellular traps
- NETosis:
Neutrophil extracellular trap formation
- NGAL:
Neutrophil gelatinase-associated lipocalin
- NLR:
Neutrophil-to-lymphocyte ratio
- NT-proBNP:
N-terminal pro-B-type natriuretic peptide
- PAD4:
Peptidylarginine deiminase 4
- PRR:
Pattern recognition receptor
- RA:
Rheumatoid arthritis
- RAGE:
Receptor for advanced glycation end products
- ROS:
Reactive oxygen species
- S100A8/A9:
S100 calcium-binding protein A8/A9
- SLE:
Systemic lupus erythematosus
- STEMI:
ST-segment elevation myocardial infarction
- T2DM:
Type 2 diabetes mellitus
- TAC:
Transverse aortic constriction
- TLR4/9:
Toll-like receptor 4/9
- TNF-α:
Tumor necrosis factor-alpha.
Conflicts of Interest
The authors declare that they have no competing interests.
References
- 1.Ponikowski P., Voors A. A., Anker S. D., et al. 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: the task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) developed with the special contribution of the Heart Failure Association (HFA) of the ESC. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. European Heart Journal. 2016;37(27):2129–2200. doi: 10.1093/eurheartj/ehw128. [DOI] [PubMed] [Google Scholar]
- 2.Groenewegen A., Rutten F. H., Mosterd A., Hoes A. W. Epidemiology of heart failure. Epidemiology of heart failure. European Journal of Heart Failure. 2020;22(8):1342–1356. doi: 10.1002/ejhf.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hao G., Wang X., Chen Z., et al. Prevalence of heart failure and left ventricular dysfunction in China: the China Hypertension Survey, 2012–2015. European Journal of Heart Failure. 2019;21(11):1329–1337. doi: 10.1002/ejhf.1629. [DOI] [PubMed] [Google Scholar]
- 4.The Writing Committee of the Report on Cardiovascular Health and Disease in China. Report on cardiovascular health and disease in China 2019: an updated summary. Chinese Circulation Journal. 2020;35(9):833–854. [Google Scholar]
- 5.Uijl A., Koudstaal S., Direk K., et al. Risk factors for incident heart failure in age- and sex-specific strata: a population-based cohort using linked electronic health records. European Journal of Heart Failure. 2019;21(10):1197–1206. doi: 10.1002/ejhf.1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gomberg-Maitland M., Shah S. J., Guazzi M. Inflammation in Heart Failure With Preserved Ejection Fraction: JACC Heart Fail. 2016;4(4):325–328. doi: 10.1016/j.jchf.2015.11.013. [DOI] [PubMed] [Google Scholar]
- 7.Paulus W. J., Tschöpe C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. Journal of the American College of Cardiology. 2013;62(4):263–271. doi: 10.1016/j.jacc.2013.02.092. [DOI] [PubMed] [Google Scholar]
- 8.Schiattarella G. G., Rodolico D., Hill J. A. Metabolic inflammation in heart failure with preserved ejection fraction. Cardiovascular Research. 2021;117(2):423–434. doi: 10.1093/cvr/cvaa217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Curl C. L., Danes V. R., Bell J. R., et al. Cardiomyocyte functional etiology in heart failure with preserved ejection fraction is distinctive-a new preclinical model. Journal of the American Heart Association. 2018;7(11) doi: 10.1161/jaha.117.007451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Røe Å. T., Aronsen J. M., Skårdal K., et al. Increased passive stiffness promotes diastolic dysfunction despite improved Ca2+ handling during left ventricular concentric hypertrophy. Cardiovascular Research. 2017;113(10):1161–1172. doi: 10.1093/cvr/cvx087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gallet R., de Couto G., Simsolo E., et al. Cardiosphere-Derived Cells Reverse Heart Failure With Preserved Ejection Fraction in Rats by Decreasing Fibrosis and Inflammation. JACC: Basic to Translational Science. 2016;1(1-2):14–28. doi: 10.1016/j.jacbts.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakagawa A., Yasumura Y., Yoshida C., et al. Prognostic importance of right ventricular-vascular uncoupling in acute decompensated heart failure with preserved ejection fraction. Circulation: Cardiovascular Imaging. 2020;13(11) doi: 10.1161/CIRCIMAGING.120.011430. [DOI] [PubMed] [Google Scholar]
- 13.Sarma S., Stoller D., Hendrix J., et al. Mechanisms of chronotropic incompetence in heart failure with preserved ejection fraction. Circulation: Heart Failure. 2020;13(3, article e006331) doi: 10.1161/circheartfailure.119.006331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brinkmann V., Reichard U., Goosmann C., et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 15.Brinkmann V., Zychlinsky A. Neutrophil extracellular traps: is immunity the second function of chromatin? The Journal of Cell Biology. 2012;198(5):773–783. doi: 10.1083/jcb.201203170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang H., Tohme S., Al‐Khafaji A. B., et al. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology. 2015;62(2):600–614. doi: 10.1002/hep.27841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim S. W., Lee H., Lee H. K., Kim I. D., Lee J. K. Neutrophil extracellular trap induced by HMGB1 exacerbates damages in the ischemic brain. Acta Neuropathologica Communications. 2019;7(1) doi: 10.1186/s40478-019-0747-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin R., Xu J., Gao Q., et al. IL-33-induced neutrophil extracellular traps degrade fibronectin in a murine model of bronchopulmonary dysplasia. Cell Death Discovery. 2020;6(1):p. 33. doi: 10.1038/s41420-020-0267-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Branzk N., Lubojemska A., Hardison S. E., et al. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nature Immunology. 2014;15(11):1017–1025. doi: 10.1038/ni.2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sorvillo N., Cherpokova D., Martinod K., Wagner D. D. Extracellular DNA NET-works with dire consequences for health. Circulation Research. 2019;125(4):470–488. doi: 10.1161/CIRCRESAHA.119.314581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Knight J. S., Carmona-Rivera C., Kaplan M. J. Proteins derived from neutrophil extracellular traps may serve as self-antigens and mediate organ damage in autoimmune diseases. Frontiers in Immunology. 2012;3:p. 380. doi: 10.3389/fimmu.2012.00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carmona-Rivera C., Carlucci P. M., Goel R. R., et al. Neutrophil extracellular traps mediate articular cartilage damage and enhance cartilage component immunogenicity in rheumatoid arthritis. JCI Insight. 2020;5(13) doi: 10.1172/jci.insight.139388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Biermann M. H., Podolska M. J., Knopf J., et al. Oxidative burst-dependent netosis is implicated in the resolution of necrosis-associated sterile inflammation. Frontiers in Immunology. 2016;7:p. 557. doi: 10.3389/fimmu.2016.00557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Papayannopoulos V., Metzler K. D., Hakkim A., Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. The Journal of Cell Biology. 2010;191(3):677–691. doi: 10.1083/jcb.201006052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parker H., Albrett A. M., Kettle A. J., Winterbourn C. C. Myeloperoxidase associated with neutrophil extracellular traps is active and mediates bacterial killing in the presence of hydrogen peroxide. Journal of Leukocyte Biology. 2012;91(3):369–376. doi: 10.1189/jlb.0711387. [DOI] [PubMed] [Google Scholar]
- 26.Metzler K. D., Goosmann C., Lubojemska A., Zychlinsky A., Papayannopoulos V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Reports. 2014;8(3):883–896. doi: 10.1016/j.celrep.2014.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li P., Li M., Lindberg M. R., Kennett M. J., Xiong N., Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. The Journal of Experimental Medicine. 2010;207(9):1853–1862. doi: 10.1084/jem.20100239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wong S. L., Wagner D. D. Peptidylarginine deiminase 4: a nuclear button triggering neutrophil extracellular traps in inflammatory diseases and aging. FASEB Journal. 2018;32(12):6258–6370. doi: 10.1096/fj.201800691R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leshner M., Wang S., Lewis C., et al. PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structures. Frontiers in Immunology. 2012;3:p. 307. doi: 10.3389/fimmu.2012.00307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jorch S. K., Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nature Medicine. 2017;23(3):279–287. doi: 10.1038/nm.4294. [DOI] [PubMed] [Google Scholar]
- 31.Grayson P. C., Kaplan M. J. At the bench: neutrophil extracellular traps (NETs) highlight novel aspects of innate immune system involvement in autoimmune diseases. Journal of Leukocyte Biology. 2016;99(2):253–264. doi: 10.1189/jlb.5BT0615-247R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meher A. K., Spinosa M., Davis J. P., et al. Novel role of IL (interleukin)-1β in neutrophil extracellular trap formation and abdominal aortic aneurysms. Arteriosclerosis, Thrombosis, and Vascular Biology. 2018;38(4):843–853. doi: 10.1161/ATVBAHA.117.309897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou Z., Zhang S., Ding S., et al. Excessive neutrophil extracellular trap formation aggravates acute myocardial infarction injury in apolipoprotein e deficiency mice via the ROS-dependent pathway. Oxidative Medicine and Cellular Longevity. 2019;2019:15. doi: 10.1155/2019/1209307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen W., Wang Q., Ke Y., Lin J. Neutrophil function in an inflammatory milieu of rheumatoid arthritis. Journal of Immunology Research. 2018;2018:12. doi: 10.1155/2018/8549329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang H., Sha L. L., Ma T. T., Zhang L. X., Chen M., Zhao M. H. Circulating level of neutrophil extracellular traps is not a useful biomarker for assessing disease activity in antineutrophil cytoplasmic antibody-associated vasculitis. PLoS One. 2016;11(2, article e0148197) doi: 10.1371/journal.pone.0148197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.He Z., Si Y., Jiang T., et al. Phosphotidylserine exposure and neutrophil extracellular traps enhance procoagulant activity in patients with inflammatory bowel disease. Thrombosis and Haemostasis. 2017;115(4):738–751. doi: 10.1160/TH15-09-0710. [DOI] [PubMed] [Google Scholar]
- 37.Ng L. L., Pathik B., Loke I. W., Squire I. B., Davies J. E. Myeloperoxidase and C-reactive protein augment the specificity of B-type natriuretic peptide in community screening for systolic heart failure. American Heart Journal. 2006;152(1):94–101. doi: 10.1016/j.ahj.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 38.Tang W. H., Tong W., Troughton R. W., et al. Prognostic value and echocardiographic determinants of plasma myeloperoxidase levels in chronic heart failure. Journal of the American College of Cardiology. 2007;49(24):2364–2370. doi: 10.1016/j.jacc.2007.02.053. [DOI] [PubMed] [Google Scholar]
- 39.La Rocca G., Di Stefano A., Eleuteri E., et al. Oxidative stress induces myeloperoxidase expression in endocardial endothelial cells from patients with chronic heart failure. Basic Research in Cardiology. 2009;104(3):307–320. doi: 10.1007/s00395-008-0761-9. [DOI] [PubMed] [Google Scholar]
- 40.Hage C., Michaëlsson E., Kull B., et al. Myeloperoxidase and related biomarkers are suggestive footprints of endothelial microvascular inflammation in HFpEF patients. ESC Heart Failure. 2020;7(4):1534–1546. doi: 10.1002/ehf2.12700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang W., Peng W., Ning X. Increased levels of neutrophil extracellular trap remnants in the serum of patients with rheumatoid arthritis. International Journal of Rheumatic Diseases. 2018;21(2):415–421. doi: 10.1111/1756-185X.13226. [DOI] [PubMed] [Google Scholar]
- 42.Yazdani H. O., Chen H. W., Tohme S., et al. IL-33 exacerbates liver sterile inflammation by amplifying neutrophil extracellular trap formation. Journal of Hepatology. 2017;S0168-8278(17):32291–32292. doi: 10.1016/j.jhep.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Flo T. H., Smith K. D., Sato S., et al. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432(7019):917–921. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- 44.Saha P., Yeoh B. S., Olvera R. A., et al. Bacterial siderophores hijack neutrophil functions. Journal of Immunology. 2017;198(11):4293–4303. doi: 10.4049/jimmunol.1700261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yndestad A., Landro L., Ueland T., et al. Increased systemic and myocardial expression of neutrophil gelatinase-associated lipocalin in clinical and experimental heart failure. European Heart Journal. 2009;30(10):1229–1236. doi: 10.1093/eurheartj/ehp088. [DOI] [PubMed] [Google Scholar]
- 46.Nymo S. H., Ueland T., Askevold E. T., et al. The association between neutrophil gelatinase-associated lipocalin and clinical outcome in chronic heart failure: results from CORONA. Journal of Internal Medicine. 2012;271(5):436–443. doi: 10.1111/j.1365-2796.2011.02503.x. [DOI] [PubMed] [Google Scholar]
- 47.Marques F. Z., Prestes P. R., Byars S. G., et al. Experimental and human evidence for lipocalin-2 (neutrophil gelatinase-associated lipocalin [NGAL]) in the development of cardiac hypertrophy and heart failure. Journal of the American Heart Association. 2017;6(6) doi: 10.1161/JAHA.117.005971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Benites-Zapata V. A., Hernandez A. V., Nagarajan V., Cauthen C. A., Starling R. C., Wilson Tang W. H. Usefulness of neutrophil-to-lymphocyte ratio in risk stratification of patients with advanced heart failure. The American Journal of Cardiology. 2015;115(1):57–61. doi: 10.1016/j.amjcard.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yan W., Li R. J., Jia Q., Mu Y., Liu C. L., He K. L. Neutrophil-to-lymphocyte ratio compared to N-terminal pro-brain natriuretic peptide as a prognostic marker of adverse events in elderly patients with chronic heart failure. Journal of Geriatric Cardiology. 2017;14(2):127–134. doi: 10.11909/j.issn.1671-5411.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boralkar K. A., Kobayashi Y., Amsallem M., et al. Value of neutrophil to lymphocyte ratio and its trajectory in patients hospitalized with acute heart failure and preserved ejection fraction. The American Journal of Cardiology. 2020;125(2):229–235. doi: 10.1016/j.amjcard.2019.10.020. [DOI] [PubMed] [Google Scholar]
- 51.Wang Y., Sano S., Oshima K., et al. Wnt5a-mediated neutrophil recruitment has an obligatory role in pressure overload-induced cardiac dysfunction. Circulation. 2019;140(6):487–499. doi: 10.1161/CIRCULATIONAHA.118.038820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Valiente-Alandi I., Potter S. J., Salvador A. M., et al. Inhibiting fibronectin attenuates fibrosis and improves cardiac function in a model of heart failure. Circulation. 2018;138(12):1236–1252. doi: 10.1161/CIRCULATIONAHA.118.034609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bai B., Yang W., Fu Y., et al. Seipin knockout mice develop heart failure with preserved ejection fraction. JACC. Basic to Translational Science. 2019;4(8):924–937. doi: 10.1016/j.jacbts.2019.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sano S., Wang Y., Yura Y., et al. _JAK2_ _V617F_ -Mediated Clonal Hematopoiesis Accelerates Pathological Remodeling in Murine Heart Failure. JACC. Basic to Translational Science. 2019;4(6):684–697. doi: 10.1016/j.jacbts.2019.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wolach O., Sellar R. S., Martinod K., et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Science Translational Medicine. 2018;10(436):p. eaan8292. doi: 10.1126/scitranslmed.aan8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Savchenko A. S., Borissoff J. I., Martinod K., et al. VWF-mediated leukocyte recruitment with chromatin decondensation by PAD4 increases myocardial ischemia/reperfusion injury in mice. Blood. 2014;123(1):141–148. doi: 10.1182/blood-2013-07-514992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.du M., Yang W., Schmull S., Gu J., Xue S. Inhibition of peptidyl arginine deiminase-4 protects against myocardial infarction induced cardiac dysfunction. International Immunopharmacology. 2020;78:p. 106055. doi: 10.1016/j.intimp.2019.106055. [DOI] [PubMed] [Google Scholar]
- 58.Martinod K., Witsch T., Erpenbeck L., et al. Peptidylarginine deiminase 4 promotes age-related organ fibrosis. The Journal of Experimental Medicine. 2017;214(2):439–458. doi: 10.1084/jem.20160530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bahit M. C., Kochar A., Granger C. B. Post-myocardial infarction heart failure. JACC. Heart Failure. 2018;6(3):179–186. doi: 10.1016/j.jchf.2017.09.015. [DOI] [PubMed] [Google Scholar]
- 60.Packer M., Lam C. S. P., Lund L. H., Redfield M. M. Interdependence of atrial fibrillation and heart failure with a preserved ejection fraction reflects a common underlying atrial and ventricular myopathy. Circulation. 2020;141(1):4–6. doi: 10.1161/CIRCULATIONAHA.119.042996. [DOI] [PubMed] [Google Scholar]
- 61.Mavrogeni S., Spargias C., Bratis C., et al. Myocarditis as a precipitating factor for heart failure: evaluation and 1-year follow-up using cardiovascular magnetic resonance and endomyocardial biopsy. European Journal of Heart Failure. 2011;13(8):830–837. doi: 10.1093/eurjhf/hfr052. [DOI] [PubMed] [Google Scholar]
- 62.Maron B. J., Rowin E. J., Udelson J. E., Maron M. S. Clinical spectrum and management of heart failure in hypertrophic cardiomyopathy. JACC. Heart Failure. 2018;6(5):353–363. doi: 10.1016/j.jchf.2017.09.011. [DOI] [PubMed] [Google Scholar]
- 63.Canepa M., Franssen F. M. E., Olschewski H., et al. Diagnostic and therapeutic gaps in patients with heart failure and chronic obstructive pulmonary disease. JACC. Heart Failure. 2019;7(10):823–833. doi: 10.1016/j.jchf.2019.05.009. [DOI] [PubMed] [Google Scholar]
- 64.Dickhout J. G., Carlisle R. E., Austin R. C. Interrelationship between cardiac hypertrophy, heart failure, and chronic kidney disease: endoplasmic reticulum stress as a mediator of pathogenesis. Circulation Research. 2011;108(5):629–642. doi: 10.1161/CIRCRESAHA.110.226803. [DOI] [PubMed] [Google Scholar]
- 65.Kenny H. C., Abel E. D. Heart failure in type 2 diabetes mellitus. Circulation Research. 2019;124(1):121–141. doi: 10.1161/CIRCRESAHA.118.311371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ge L., Zhou X., Ji W. J., et al. Neutrophil extracellular traps in ischemia-reperfusion injury-induced myocardial no-reflow: therapeutic potential of DNase-based reperfusion strategy. American Journal of Physiology. Heart and Circulatory Physiology. 2015;308(5):H500–H509. doi: 10.1152/ajpheart.00381.2014. [DOI] [PubMed] [Google Scholar]
- 67.Mangold A., Alias S., Scherz T., et al. Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST-elevation acute coronary syndrome are predictors of ST-segment resolution and infarct size. Circulation Research. 2015;116(7):1182–1192. doi: 10.1161/CIRCRESAHA.116.304944. [DOI] [PubMed] [Google Scholar]
- 68.Hofbauer T. M., Mangold A., Scherz T., et al. Neutrophil extracellular traps and fibrocytes in ST-segment elevation myocardial infarction. Basic Research in Cardiology. 2019;114(5):1–15. doi: 10.1007/s00395-019-0740-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Helseth R., Shetelig C., Andersen G. Ø., et al. Neutrophil extracellular trap components associate with infarct size, ventricular function, and clinical outcome in STEMI. Mediators of Inflammation. 2019;2019:10. doi: 10.1155/2019/7816491.7816491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bonaventura A., Montecucco F., Dallegri F., et al. Novel findings in neutrophil biology and their impact on cardiovascular disease. Cardiovascular Research. 2019;115(8):1266–1285. doi: 10.1093/cvr/cvz084. [DOI] [PubMed] [Google Scholar]
- 71.Pertiwi K. R., van der Wal A. C., Pabittei D. R., et al. Neutrophil extracellular traps participate in all different types of thrombotic and haemorrhagic complications of coronary atherosclerosis. Thrombosis and Haemostasis. 2018;118(6):1078–1087. doi: 10.1055/s-0038-1641749. [DOI] [PubMed] [Google Scholar]
- 72.Gibson P. H., Cuthbertson B. H., Croal B. L., et al. Usefulness of neutrophil/lymphocyte ratio as predictor of new-onset atrial fibrillation after coronary artery bypass grafting. The American Journal of Cardiology. 2010;105(2):186–191. doi: 10.1016/j.amjcard.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 73.Friedrichs K., Adam M., Remane L., et al. Induction of atrial fibrillation by neutrophils critically depends on CD11b/CD18 integrins. PLoS One. 2014;9(2):p. e89307. doi: 10.1371/journal.pone.0089307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Berkovitch A., Younis A., Grossman Y., et al. Relation of neutrophil to lymphocyte ratio to risk of incident atrial fibrillation. The American Journal of Cardiology. 2019;123(3):396–401. doi: 10.1016/j.amjcard.2018.10.036. [DOI] [PubMed] [Google Scholar]
- 75.Rudolph V., Andrié R. P., Rudolph T. K., et al. Myeloperoxidase acts as a profibrotic mediator of atrial fibrillation. Nature Medicine. 2010;16(4):470–474. doi: 10.1038/nm.2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Arroyo A. B., de los Reyes-García A. M., Rivera-Caravaca J. M., et al. miR-146a regulates neutrophil extracellular trap formation that predicts adverse cardiovascular events in patients with atrial fibrillation. Arteriosclerosis, Thrombosis, and Vascular Biology. 2018;38(4):892–902. doi: 10.1161/ATVBAHA.117.310597. [DOI] [PubMed] [Google Scholar]
- 77.Gao J., Li Y., Wang T., et al. Analyzing gene expression profiles with preliminary validations in cardiac hypertrophy induced by pressure overload. Canadian Journal of Physiology and Pharmacology. 2018;96(8):701–709. doi: 10.1139/cjpp-2017-0585. [DOI] [PubMed] [Google Scholar]
- 78.Zeng J. H., Liu Y. X., Yuan J., et al. First case of COVID-19 complicated with fulminant myocarditis: a case report and insights. Infection. 2020;10:1–5. doi: 10.1007/s15010-020-01424-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hu H., Ma F., Wei X., Fang Y. Coronavirus fulminant myocarditis saved with glucocorticoid and human immunoglobulin. European Heart Journal. 2020;16 doi: 10.1093/eurheartj/ehaa190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim I. C., Kim J. Y., Kim H. A., Han S. COVID-19-related myocarditis in a 21-year-old female patient. European Heart Journal. 2020;41(19):p. 1859. doi: 10.1093/eurheartj/ehaa288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Irabien-Ortiz Á., Carreras-Mora J., Sionis A., Pàmies J., Montiel J., Tauron M. Miocarditis fulminante por COVID-19. Revista Espanola de Cardiologia (English ed.) 2020;73(6):503–504. doi: 10.1016/j.rec.2020.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Salamanca J., Díez-Villanueva P., Martínez P., et al. COVID-19 "Fulminant Myocarditis" Successfully Treated With Temporary Mechanical Circulatory Support. JACC. Cardiovascular Imaging. 2020;13(11):2457–2459. doi: 10.1016/j.jcmg.2020.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gill G. S., Vlacancich R., Mehta N., Chaturvedi M., Papolos A. Spectrum of cardiac involvement in COVID-19. Cureus. 2020;12(6) doi: 10.7759/cureus.8638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Middleton E. A., He X. Y., Denorme F., et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood. 2020;136(10):1169–1179. doi: 10.1182/blood.2020007008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zuo Y., Zuo M., Yalavarthi S., et al. Neutrophil extracellular traps in COVID-19. JCI Insight. 2020;5(11) doi: 10.1172/jci.insight.138999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Barnes B. J., Adrover J. M., Baxter-Stoltzfus A., et al. Targeting potential drivers of COVID-19: neutrophil extracellular traps. Journal of Experimental Medicine. 2020;217(6) doi: 10.1084/jem.20200652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Skendros P., Mitsios A., Chrysanthopoulou A., et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. The Journal of Clinical Investigation. 2020;130(11):6151–6157. doi: 10.1172/JCI141374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zuo Y., Zuo M., Yalavarthi S., et al. Neutrophil extracellular traps and thrombosis in COVID-19. Journal of Thrombosis and Thrombolysis. 2021;51(2):446–453. doi: 10.1007/s11239-020-02324-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tschöpe C., Müller I., Xia Y., et al. NOD2 (nucleotide-binding oligomerization domain 2) is a major pathogenic mediator of coxsackievirus B3-induced myocarditis. Circulation. Heart Failure. 2017;10(9) doi: 10.1161/CIRCHEARTFAILURE.117.003870. [DOI] [PubMed] [Google Scholar]
- 90.Gao X., Wei B., Deng Y., Huang Y. L., Wu W. Increased mobilization of CD45+CD34+VLA-4+ cells in acute viral myocarditis induced by coxsackievirus B3. Cardiology. 2017;138(4):238–248. doi: 10.1159/000477655. [DOI] [PubMed] [Google Scholar]
- 91.Rivadeneyra L., Charó N., Kviatcovsky D., de la Barrera S., Gómez R. M., Schattner M. Role of neutrophils in CVB3 infection and viral myocarditis. Journal of Molecular and Cellular Cardiology. 2018;125:149–161. doi: 10.1016/j.yjmcc.2018.08.029. [DOI] [PubMed] [Google Scholar]
- 92.Müller I., Vogl T., Pappritz K., et al. Pathogenic role of the damage-associated molecular patterns S100A8 and S100A9 in coxsackievirus B3-induced myocarditis. Circulation: Heart Failure. 2017;10(11, article e004125) doi: 10.1161/circheartfailure.117.004125. [DOI] [PubMed] [Google Scholar]
- 93.Ndumele C. E., Coresh J., Lazo M., et al. Obesity, subclinical myocardial injury, and incident heart failure. JACC. Heart Failure. 2014;2(6):600–607. doi: 10.1016/j.jchf.2014.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Karason K., Jamaly S. Heart failure development in obesity: mechanistic pathways. European Heart Journal. 2020;41(36, article 3485) doi: 10.1093/eurheartj/ehaa422. [DOI] [PubMed] [Google Scholar]
- 95.Carestia A., Frechtel G., Cerrone G., et al. NETosis before and after hyperglycemic control in type 2 diabetes mellitus patients. PLoS One. 2016;11(12, article e0168647) doi: 10.1371/journal.pone.0168647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Miyoshi A., Yamada M., Shida H., et al. Circulating neutrophil extracellular trap levels in well-controlled type 2 diabetes and pathway involved in their formation induced by high-dose glucose. Pathobiology. 2016;83(5):243–251. doi: 10.1159/000444881. [DOI] [PubMed] [Google Scholar]
- 97.Bryk A. H., Prior S. M., Plens K., et al. Predictors of neutrophil extracellular traps markers in type 2 diabetes mellitus: associations with a prothrombotic state and hypofibrinolysis. Cardiovascular Diabetology. 2019;18(1, article 49) doi: 10.1186/s12933-019-0850-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tromp J., Voors A. A., Sharma A., et al. Distinct pathological pathways in patients with heart failure and diabetes. JACC: Heart Failure. 2020;8(3):234–242. doi: 10.1016/j.jchf.2019.11.005. [DOI] [PubMed] [Google Scholar]
- 99.Shah T. J., Leik C. E., Walsh S. W. Neutrophil infiltration and systemic vascular inflammation in obese women. Reproductive Sciences. 2010;17(2):116–124. doi: 10.1177/1933719109348252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Elgazar-Carmon V., Rudich A., Hadad N., Levy R. Neutrophils transiently infiltrate intra-abdominal fat early in the course of high-fat feeding. Journal of Lipid Research. 2008;49(9):1894–1903. doi: 10.1194/jlr.M800132-JLR200. [DOI] [PubMed] [Google Scholar]
- 101.D’Abbondanza M., Martorelli E. E., Ricci M. A., et al. Increased plasmatic NETs by-products in patients in severe obesity. Scientific Reports. 2019;9(1, article 14678) doi: 10.1038/s41598-019-51220-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang H., Wang Q., Venugopal J., et al. Obesity-induced endothelial dysfunction is prevented by neutrophil extracellular trap inhibition. Scientific Reports. 2018;8(1, article 4881) doi: 10.1038/s41598-018-23256-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lee K. H., Kronbichler A., Park D. D., et al. Neutrophil extracellular traps (NETs) in autoimmune diseases: a comprehensive review. Autoimmunity Reviews. 2017;16(11):1160–1173. doi: 10.1016/j.autrev.2017.09.012. [DOI] [PubMed] [Google Scholar]
- 104.Valesini G., Gerardi M. C., Iannuccelli C., Pacucci V. A., Pendolino M., Shoenfeld Y. Citrullination and autoimmunity. Autoimmunity Reviews. 2015;14(6):490–497. doi: 10.1016/j.autrev.2015.01.013. [DOI] [PubMed] [Google Scholar]
- 105.Giles J. T., Post W., Blumenthal R. S., Bathon J. M. Therapy insight: managing cardiovascular risk in patients with rheumatoid arthritis. Nature Clinical Practice Rheumatology. 2006;2(6):320–329. doi: 10.1038/ncprheum0178. [DOI] [PubMed] [Google Scholar]
- 106.Dhakal B. P., Kim C. H., Al-Kindi S. G., Oliveira G. H. Heart failure in systemic lupus erythematosus. Trends in Cardiovascular Medicine. 2018;28(3):187–197. doi: 10.1016/j.tcm.2017.08.015. [DOI] [PubMed] [Google Scholar]
- 107.Norouzi S., Javinani A., Aminorroaya A., Masoumi M. Anti-modified citrullinated vimentin antibody: a novel biomarker associated with cardiac systolic dysfunction in patients with rheumatoid arthritis. BMC Cardiovascular Disorders. 2020;20(1, article 390) doi: 10.1186/s12872-020-01676-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Geraldino-Pardilla L., Russo C., Sokolove J., et al. Association of anti-citrullinated protein or peptide antibodies with left ventricular structure and function in rheumatoid arthritis. Rheumatology (Oxford) 2017;56(4):534–540. doi: 10.1093/rheumatology/kew436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Giles J. T., Fert-Bober J., Park J. K., et al. Myocardial citrullination in rheumatoid arthritis: a correlative histopathologic study. Arthritis Research & Therapy. 2012;14(1, article R39) doi: 10.1186/ar3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fert-Bober J., Giles J. T., Holewinski R. J., et al. Citrullination of myofilament proteins in heart failure. Cardiovascular Research. 2015;108(2):232–242. doi: 10.1093/cvr/cvv185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yatomi A., Mori S., Kawauchi H., et al. Predominant involvement of the aortic root in a patient with anti-neutrophil cytoplasmic antibody-associated vasculitis: congestive heart failure due to subacute severe aortic regurgitation and reversible conduction disturbance. Internal Medicine (Tokyo, Japan) 2020;59(5):663–671. doi: 10.2169/internalmedicine.3831-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Guterbaum T. J., Husic M., Voss A., Dahl J. S. Anti-neutrophil cytoplasmic antibody (ANCA) associated vasculitis causes aortic valve degeneration and severe aortic regurgitation. The American Journal of Case Reports. 2019;20:423–429. doi: 10.12659/AJCR.912693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Berti A., Matteson E. L., Crowson C. S., Specks U., Cornec D. Risk of cardiovascular disease and venous thromboembolism among patients with incident ANCA-associated vasculitis: a 20-year population-based cohort study. Mayo Clinic Proceedings. 2018;93(5):597–606. doi: 10.1016/j.mayocp.2018.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pinti M., Cevenini E., Nasi M., et al. Circulating mitochondrial DNA increases with age and is a familiar trait: implications for ‘inflamm-aging’. European Journal of Immunology. 2014;44(5):1552–1562. doi: 10.1002/eji.201343921. [DOI] [PubMed] [Google Scholar]
- 115.Verschoor C. P., Loukov D., Naidoo A., et al. Circulating TNF and mitochondrial DNA are major determinants of neutrophil phenotype in the advanced-age, frail elderly. Molecular Immunology. 2015;65(1):148–156. doi: 10.1016/j.molimm.2015.01.015. [DOI] [PubMed] [Google Scholar]
- 116.Knez J., Lakota K., Božič N., et al. Correlation between mitochondrial DNA content measured in myocardium and peripheral blood of patients with non-ischemic heart failure. Genetic Testing and Molecular Biomarkers. 2017;21(12):736–741. doi: 10.1089/gtmb.2017.0099. [DOI] [PubMed] [Google Scholar]
- 117.Mariero L. H., Torp M. K., Heiestad C. M., et al. Inhibiting nucleolin reduces inflammation induced by mitochondrial DNA in cardiomyocytes exposed to hypoxia and reoxygenation. British Journal of Pharmacology. 2019;176(22):4360–4372. doi: 10.1111/bph.14830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Krychtiuk K. A., Wurm R., Ruhittel S., et al. Release of mitochondrial DNA is associated with mortality in severe acute heart failure. European Heart Journal Acute Cardiovascular Care. 2020;9(5):419–428. doi: 10.1177/2048872618823405. [DOI] [PubMed] [Google Scholar]
- 119.Yousefi S., Mihalache C., Kozlowski E., Schmid I., Simon H. U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death and Differentiation. 2009;16(11):1438–1444. doi: 10.1038/cdd.2009.96. [DOI] [PubMed] [Google Scholar]
- 120.Liu L., Mao Y., Xu B., et al. Induction of neutrophil extracellular traps during tissue injury: involvement of STING and Toll-like receptor 9 pathways. Cell Proliferation. 2019;52(3, article e12579) doi: 10.1111/cpr.12579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mallavia B., Liu F., Lefrançais E., et al. Mitochondrial DNA stimulates TLR9-dependent neutrophil extracellular trap formation in primary graft dysfunction. American Journal of Respiratory Cell and Molecular Biology. 2020;62(3):364–372. doi: 10.1165/rcmb.2019-0140OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Barton G. M., Kagan J. C., Medzhitov R. Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nature Immunology. 2006;7(1):49–56. doi: 10.1038/ni1280. [DOI] [PubMed] [Google Scholar]
- 123.Lindau D., Mussard J., Wagner B. J., et al. Primary blood neutrophils express a functional cell surface Toll-like receptor 9. European Journal of Immunology. 2013;43(8):2101–2113. doi: 10.1002/eji.201142143. [DOI] [PubMed] [Google Scholar]
- 124.Oka T., Hikoso S., Yamaguchi O., et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485(7397):251–255. doi: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Dhondup Y., Sjaastad I., Scott H., et al. Sustained Toll-like receptor 9 activation promotes systemic and cardiac inflammation, and aggravates diastolic heart failure in SERCA2a KO mice. PLoS One. 2015;10(10, article e0139715) doi: 10.1371/journal.pone.0139715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lohner R., Schwederski M., Narath C., et al. Toll-like receptor 9 promotes cardiac inflammation and heart failure during polymicrobial sepsis. Mediators of Inflammation. 2013;2013:13. doi: 10.1155/2013/261049.261049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ye W., Tang X., Yang Z., et al. Plasma-derived exosomes contribute to inflammation via the TLR9-NF-κB pathway in chronic heart failure patients. Molecular Immunology. 2017;87:114–121. doi: 10.1016/j.molimm.2017.03.011. [DOI] [PubMed] [Google Scholar]
- 128.Ueda H., Yamaguchi O., Taneike M., et al. Administration of a TLR9 inhibitor attenuates the development and progression of heart failure in mice. JACC. Basic to Translational Science. 2019;4(3):348–363. doi: 10.1016/j.jacbts.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hartupee J., Patel R., Staloch L., et al. Diagnostic accuracy of damage-associated molecular patterns (DAMPs) in patients with heart failure with a reduced ejection fraction. Journal of Clinical and Translational Science. 2017;1(3):208–209. doi: 10.1017/cts.2017.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Turner N. A. Inflammatory and fibrotic responses of cardiac fibroblasts to myocardial damage associated molecular patterns (DAMPs) Journal of Molecular and Cellular Cardiology. 2016;94:189–200. doi: 10.1016/j.yjmcc.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 131.Müller I., Vogl T., Kühl U., et al. Serum alarmin S100A8/S100A9 levels and its potential role as biomarker in myocarditis. ESC Heart Failure. 2020;7(4):1442–1451. doi: 10.1002/ehf2.12760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Li Y., Chen B., Yang X., et al. S100a8/a9 signaling causes mitochondrial dysfunction and cardiomyocyte death in response to ischemic/reperfusion injury. Circulation. 2019;140(9):751–764. doi: 10.1161/CIRCULATIONAHA.118.039262. [DOI] [PubMed] [Google Scholar]
- 133.Bruhn L. V., Lauridsen K. G., Schmidt A. S., et al. Elevated calprotectin in patients with atrial fibrillation with and without heart failure. Scandinavian Journal of Clinical and Laboratory Investigation. 2017;77(3):210–215. doi: 10.1080/00365513.2017.1292364. [DOI] [PubMed] [Google Scholar]
- 134.Jensen L. J. N., Kistorp C., Bjerre M., Raymond I., Flyvbjerg A. Plasma calprotectin levels reflect disease severity in patients with chronic heart failure. European Journal of Preventive Cardiology. 2012;19(5):999–1004. doi: 10.1177/1741826711421078. [DOI] [PubMed] [Google Scholar]
- 135.Schenten V., Plançon S., Jung N., et al. Secretion of the phosphorylated form of S100A9 from neutrophils is essential for the proinflammatory functions of extracellular S100A8/A9. Frontiers in Immunology. 2018;9, article 447 doi: 10.3389/fimmu.2018.00447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sreejit G., Abdel-Latif A., Athmanathan B., et al. Neutrophil-derived S100A8/A9 amplify granulopoiesis after myocardial infarction. Circulation. 2020;141(13):1080–1094. doi: 10.1161/CIRCULATIONAHA.119.043833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nagareddy P. R., Sreejit G., Abo-Aly M., et al. NETosis is required for S100A8/A9-induced granulopoiesis after myocardial infarction. Arteriosclerosis, Thrombosis, and Vascular Biology. 2020;40(11):2805–2807. doi: 10.1161/ATVBAHA.120.314807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Takahashi T., Shishido T., Kinoshita D., et al. Cardiac nuclear high-mobility group box 1 ameliorates pathological cardiac hypertrophy by inhibiting DNA damage response. JACC. Basic to Translational Science. 2019;4(2):234–247. doi: 10.1016/j.jacbts.2018.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Funayama A., Shishido T., Netsu S., et al. Cardiac nuclear high mobility group box 1 prevents the development of cardiac hypertrophy and heart failure. Cardiovascular Research. 2013;99(4):657–664. doi: 10.1093/cvr/cvt128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kitahara T., Takeishi Y., Harada M., et al. High-mobility group box 1 restores cardiac function after myocardial infarction in transgenic mice. Cardiovascular Research. 2008;80(1):40–46. doi: 10.1093/cvr/cvn163. [DOI] [PubMed] [Google Scholar]
- 141.Volz H. C., Laohachewin D., Schellberg D., et al. HMGB1 is an independent predictor of death and heart transplantation in heart failure. Clinical Research in Cardiology. 2012;101(6):427–435. doi: 10.1007/s00392-011-0409-x. [DOI] [PubMed] [Google Scholar]
- 142.Volz H. C., Seidel C., Laohachewin D., et al. HMGB1: the missing link between diabetes mellitus and heart failure. Basic Research in Cardiology. 2010;105(6):805–820. doi: 10.1007/s00395-010-0114-3. [DOI] [PubMed] [Google Scholar]
- 143.Liu T., Zhang D. Y., Zhou Y. H., et al. Increased serum HMGB1 level may predict the fatal outcomes in patients with chronic heart failure. International Journal of Cardiology. 2015;184:318–320. doi: 10.1016/j.ijcard.2015.02.088. [DOI] [PubMed] [Google Scholar]
- 144.Andrassy M., Volz H. C., Igwe J. C., et al. High-mobility group box-1 in ischemia-reperfusion injury of the heart. Circulation. 2008;117(25):3216–3226. doi: 10.1161/CIRCULATIONAHA.108.769331. [DOI] [PubMed] [Google Scholar]
- 145.Xiao N., Zhang J., Chen C., Wan Y., Wang N., Yang J. miR-129-5p improves cardiac function in rats with chronic heart failure through targeting HMGB1. Mammalian Genome. 2019;30(9-10):276–288. doi: 10.1007/s00335-019-09817-0. [DOI] [PubMed] [Google Scholar]
- 146.Zhang L., Liu M., Jiang H., et al. Extracellular high-mobility group box 1 mediates pressure overload-induced cardiac hypertrophy and heart failure. Journal of Cellular and Molecular Medicine. 2016;20(3):459–470. doi: 10.1111/jcmm.12743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wu R. N., Yu T. Y., Zhou J. C., et al. Targeting HMGB1 ameliorates cardiac fibrosis through restoring TLR2-mediated autophagy suppression in myocardial fibroblasts. International Journal of Cardiology. 2018;267:156–162. doi: 10.1016/j.ijcard.2018.04.103. [DOI] [PubMed] [Google Scholar]
- 148.Maugeri N., Campana L., Gavina M., et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. Journal of Thrombosis and Haemostasis. 2014;12(12):2074–2088. doi: 10.1111/jth.12710. [DOI] [PubMed] [Google Scholar]
- 149.Wang Y., Du F., Hawez A., Hawez A., Mörgelin M., Thorlacius H. Neutrophil extracellular trap-microparticle complexes trigger neutrophil recruitment via high-mobility group protein 1 (HMGB1)-toll-like receptors(TLR2)/TLR4 signalling. British Journal of Pharmacology. 2019;176(17):3350–3363. doi: 10.1111/bph.14765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ziffels B., Ospel J., Grün K., et al. Detection of Soluble ED-A+ Fibronectin and Evaluation as Novel Serum Biomarker for Cardiac Tissue Remodeling. Disease Markers. 2016;2016:11. doi: 10.1155/2016/3695454.3695454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Chorawala M. R., Prakash P., Doddapattar P., Jain M., Dhanesha N., Chauhan A. K. Deletion of extra domain A of fibronectin reduces acute myocardial ischaemia/reperfusion injury in hyperlipidaemic mice by limiting thrombo-inflammation. Thrombosis and Haemostasis. 2018;118(8):1450–1460. doi: 10.1055/s-0038-1661353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Felker G. M., Fiuzat M., Shaw L. K., et al. Galectin-3 in ambulatory patients with heart failure: results from the HF-ACTION study. Circulation. Heart Failure. 2012;5(1):72–78. doi: 10.1161/CIRCHEARTFAILURE.111.963637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.de Boer R. A., Lok D. J. A., Jaarsma T., et al. Predictive value of plasma galectin-3 levels in heart failure with reduced and preserved ejection fraction. Annals of Medicine. 2010;43(1):60–68. doi: 10.3109/07853890.2010.538080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ho J. E., Liu C., Lyass A., et al. Galectin-3, a marker of cardiac fibrosis, predicts incident heart failure in the community. Journal of the American College of Cardiology. 2012;60(14):1249–1256. doi: 10.1016/j.jacc.2012.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lok D. J., Lok S. I., Bruggink-André de la Porte P. W., et al. Galectin-3 is an independent marker for ventricular remodeling and mortality in patients with chronic heart failure. Clinical Research in Cardiology. 2013;102(2):103–110. doi: 10.1007/s00392-012-0500-y. [DOI] [PubMed] [Google Scholar]
- 156.Carrasco-Sánchez F. J., Aramburu-Bodas O., Salamanca-Bautista P., et al. Predictive value of serum galectin-3 levels in patients with acute heart failure with preserved ejection fraction. International Journal of Cardiology. 2013;169(3):177–182. doi: 10.1016/j.ijcard.2013.08.081. [DOI] [PubMed] [Google Scholar]
- 157.Edelmann F., Holzendorf V., Wachter R., et al. Galectin-3 in patients with heart failure with preserved ejection fraction: results from the Aldo-DHF trial. European Journal of Heart Failure. 2015;17(2):214–223. doi: 10.1002/ejhf.203. [DOI] [PubMed] [Google Scholar]
- 158.Yamaoka A., Kuwabara I., Frigeri L. G., Liu F. T. A human lectin, galectin-3 (epsilon bp/Mac-2), stimulates superoxide production by neutrophils. Journal of Immunology. 1995;154(7):3479–3487. [PubMed] [Google Scholar]
- 159.Fernández G. C., Ilarregui J. M., Rubel C. J., et al. Galectin-3 and soluble fibrinogen act in concert to modulate neutrophil activation and survival: involvement of alternative MAPK pathways. Glycobiology. 2005;15(5):519–527. doi: 10.1093/glycob/cwi026. [DOI] [PubMed] [Google Scholar]
- 160.Bhaumik P., St-Pierre G., Milot V., St-Pierre C., Sato S. Galectin-3 facilitates neutrophil recruitment as an innate immune response to a parasitic protozoa cutaneous infection. Journal of Immunology. 2013;190(2):630–640. doi: 10.4049/jimmunol.1103197. [DOI] [PubMed] [Google Scholar]
- 161.Fan J., Malik A. B. Toll-like receptor-4 (TLR4) signaling augments chemokine-induced neutrophil migration by modulating cell surface expression of chemokine receptors. Nature Medicine. 2003;9(3):315–321. doi: 10.1038/nm832. [DOI] [PubMed] [Google Scholar]
- 162.Sabroe I., Prince L. R., Jones E. C., et al. Selective roles for Toll-like receptor (TLR)2 and TLR4 in the regulation of neutrophil activation and life span. Journal of Immunology. 2003;170(10):5268–5275. doi: 10.4049/jimmunol.170.10.5268. [DOI] [PubMed] [Google Scholar]
- 163.Collison K. S., Parhar R. S., Saleh S. S., et al. RAGE-mediated neutrophil dysfunction is evoked by advanced glycation end products (AGEs) Journal of Leukocyte Biology. 2002;71(3):433–444. [PubMed] [Google Scholar]
- 164.Touré F., Zahm J. M., Garnotel R., et al. Receptor for advanced glycation end-products (RAGE) modulates neutrophil adhesion and migration on glycoxidated extracellular matrix. The Biochemical Journal. 2008;416(2):255–261. doi: 10.1042/BJ20080054. [DOI] [PubMed] [Google Scholar]
- 165.Tadie J. M., Bae H. B., Jiang S., et al. HMGB1 promotes neutrophil extracellular trap formation through interactions with Toll-like receptor 4. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2013;304(5):L342–L349. doi: 10.1152/ajplung.00151.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Burguillos M. A., Svensson M., Schulte T., et al. Microglia-Secreted Galectin-3 Acts as a Toll-like Receptor 4 Ligand and Contributes to Microglial Activation. Cell Reports. 2015;10(9):1626–1638. doi: 10.1016/j.celrep.2015.02.012. [DOI] [PubMed] [Google Scholar]
- 167.Huebener P., Pradere J. P., Hernandez C., et al. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. The Journal of Clinical Investigation. 2015;125(2):539–550. doi: 10.1172/JCI76887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Bangert A., Andrassy M., Müller A. M., et al. Critical role of RAGE and HMGB1 in inflammatory heart disease. Proceedings of the National Academy of Sciences of the United States of America. 2016;113(2):E155–E164. doi: 10.1073/pnas.1522288113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Weckbach L. T., Grabmaier U., Uhl A., et al. Midkine drives cardiac inflammation by promoting neutrophil trafficking and NETosis in myocarditis. The Journal of Experimental Medicine. 2019;216(2):350–368. doi: 10.1084/jem.20181102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kitahara T., Shishido T., Suzuki S., et al. Serum midkine as a predictor of cardiac events in patients with chronic heart failure. Journal of Cardiac Failure. 2010;16(4):308–313. doi: 10.1016/j.cardfail.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 171.Netsu S., Shishido T., Kitahara T., et al. Midkine exacerbates pressure overload-induced cardiac remodeling. Biochemical and Biophysical Research Communications. 2014;443(1):205–210. doi: 10.1016/j.bbrc.2013.11.083. [DOI] [PubMed] [Google Scholar]
- 172.Sato W., Kadomatsu K., Yuzawa Y., et al. Midkine is involved in neutrophil infiltration into the tubulointerstitium in ischemic renal injury. Journal of Immunology. 2001;167(6):3463–3469. doi: 10.4049/jimmunol.167.6.3463. [DOI] [PubMed] [Google Scholar]
- 173.Honda Y., Shishido T., Takahashi T., et al. Midkine deteriorates cardiac remodeling via epidermal growth factor receptor signaling in chronic kidney disease. Hypertension. 2016;67(5):857–865. doi: 10.1161/HYPERTENSIONAHA.115.06922. [DOI] [PubMed] [Google Scholar]
- 174.Chen S., Bu G., Takei Y., et al. Midkine and LDL-receptor-related protein 1 contribute to the anchorage-independent cell growth of cancer cells. Journal of Cell Science. 2007;120(22):4009–4015. doi: 10.1242/jcs.013946. [DOI] [PubMed] [Google Scholar]
- 175.Sakamoto K., Bu G., Chen S., et al. Premature Ligand-Receptor Interaction during Biosynthesis Limits the Production of Growth Factor Midkine and Its Receptor LDL Receptor-related Protein 1. The Journal of Biological Chemistry. 2011;286(10):8405–8413. doi: 10.1074/jbc.M110.176479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ranganathan S., Cao C., Catania J., Migliorini M., Zhang L., Strickland D. K. Molecular Basis for the Interaction of Low Density Lipoprotein Receptor- related Protein 1 (LRP1) with Integrin αMβ2: The Journal of Biological Chemistry. 2011;286(35):30535–30541. doi: 10.1074/jbc.M111.265413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Spijkers P. P. E. M., da Costa Martins P., Westein E., Gahmberg C. G., Zwaginga J. J., Lenting P. J. LDL-receptor-related protein regulates beta2-integrin-mediated leukocyte adhesion. Blood. 2005;105(1):170–177. doi: 10.1182/blood-2004-02-0498. [DOI] [PubMed] [Google Scholar]
- 178.Weckbach L. T., Gola A., Winkelmann M., et al. The cytokine midkine supports neutrophil trafficking during acute inflammation by promoting adhesion via β2 integrins (CD11/CD18) Blood. 2014;123(12):1887–1896. doi: 10.1182/blood-2013-06-510875. [DOI] [PubMed] [Google Scholar]
- 179.Lamblin N., Mouquet F., Hennache B., et al. High-sensitivity C-reactive protein: potential adjunct for risk stratification in patients with stable congestive heart failure. European Heart Journal. 2005;26(21):2245–2250. doi: 10.1093/eurheartj/ehi501. [DOI] [PubMed] [Google Scholar]
- 180.Tang W. H. W., Shrestha K., van Lente F., et al. Usefulness of C-reactive protein and left ventricular diastolic performance for prognosis in patients with left ventricular systolic heart failure. The American Journal of Cardiology. 2008;101(3):370–373. doi: 10.1016/j.amjcard.2007.08.038. [DOI] [PubMed] [Google Scholar]
- 181.Koller L., Kleber M., Goliasch G., et al. C-reactive protein predicts mortality in patients referred for coronary angiography and symptoms of heart failure with preserved ejection fraction. European Journal of Heart Failure. 2014;16(7):758–766. doi: 10.1002/ejhf.104. [DOI] [PubMed] [Google Scholar]
- 182.Pellicori P., Zhang J., Cuthbert J., et al. High-sensitivity C-reactive protein in chronic heart failure: patient characteristics, phenotypes, and mode of death. Cardiovascular Research. 2020;116(1):91–100. doi: 10.1093/cvr/cvz198. [DOI] [PubMed] [Google Scholar]
- 183.Buchta R., Pontet M., Fridkin M. Binding of C-reactive protein to human neutrophils. FEBS Letters. 1987;211(2):165–168. doi: 10.1016/0014-5793(87)81429-1. [DOI] [PubMed] [Google Scholar]
- 184.Bharadwaj D., Stein M. P., Volzer M., Mold C., Clos T. W. D. The major receptor for C-reactive protein on leukocytes is fcγ receptor II. Journal of Experimental Medicine. 1999;190(4):585–590. doi: 10.1084/jem.190.4.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Stein M. P., Edberg J. C., Kimberly R. P., et al. C-reactive protein binding to FcgammaRIIa on human monocytes and neutrophils is allele-specific. Journal of Clinical Investigation. 2000;105(3):369–376. doi: 10.1172/JCI7817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Liberale L., Holy E. W., Akhmedov A., et al. Interleukin-1β mediates arterial thrombus formation via NET-associated tissue factor. Journal of Clinical Medicine. 2019;8(12, article 2072) doi: 10.3390/jcm8122072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Vulesevic B., Lavoie S. S., Neagoe P. E., et al. CRP induces NETosis in heart failure patients with or without diabetes. Immunohorizons. 2019;3(8):378–388. doi: 10.4049/immunohorizons.1900026. [DOI] [PubMed] [Google Scholar]
- 188.Zeller J. M., Sullivan B. L. C-reactive protein selectively enhances the intracellular generation of reactive oxygen products by IgG-stimulated monocytes and neutrophils. Journal of Leukocyte Biology. 1992;52(4):449–455. doi: 10.1002/jlb.52.4.449. [DOI] [PubMed] [Google Scholar]
- 189.Zeller J. M., Sullivan B. L. Monoclonal antibody to the type II Fc receptor for human IgG blocks potentiation of monocyte and neutrophil IgG-induced respiratory burst activation by aggregated C-reactive protein. Cellular Immunology. 1993;149(1):144–154. doi: 10.1006/cimm.1993.1143. [DOI] [PubMed] [Google Scholar]
- 190.Prasad K. C-reactive protein increases oxygen radical generation by neutrophils. Journal of Cardiovascular Pharmacology and Therapeutics. 2016;9(3):203–209. doi: 10.1177/107424840400900308. [DOI] [PubMed] [Google Scholar]
- 191.Chung I., Choudhury A., Patel J., Lip G. Y. Soluble CD40L, platelet surface CD40L and total platelet CD40L in congestive heart failure: relationship to platelet volume, mass and granularity. Journal of Internal Medicine. 2008;263(3):313–321. doi: 10.1111/j.1365-2796.2007.01891.x. [DOI] [PubMed] [Google Scholar]
- 192.Yan J. C., Liu P. J., du R. Z., et al. Relationship between CD40 ligand expression and B type natriuretic peptide levels in patients with chronic heart failure. Clinica Chimica Acta. 2008;392(1-2):17–20. doi: 10.1016/j.cca.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 193.Perazzio S. F., Soeiro-Pereira P. V., dos Santos V. C., et al. Soluble CD40L is associated with increased oxidative burst and neutrophil extracellular trap release in Behçet’s disease. Arthritis Research and Therapy. 2017;19(1, article 235) doi: 10.1186/s13075-017-1443-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Wenzel P., Kossmann S., Münzel T., Daiber A. Redox regulation of cardiovascular inflammation - immunomodulatory function of mitochondrial and Nox-derived reactive oxygen and nitrogen species. Free Radical Biology and Medicine. 2017;109:48–60. doi: 10.1016/j.freeradbiomed.2017.01.027. [DOI] [PubMed] [Google Scholar]
- 195.Goldenthal M. J. Mitochondrial involvement in myocyte death and heart failure. Heart Failure Reviews. 2016;21(2):137–155. doi: 10.1007/s10741-016-9531-1. [DOI] [PubMed] [Google Scholar]
- 196.van der Pol A., van Gilst W., Voors A. A., van der Meer P. Treating oxidative stress in heart failure: past, present and future. European Journal of Heart Failure. 2019;21(4):425–435. doi: 10.1002/ejhf.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Ayoub K. F., Pothineni N. V. K., Rutland J., Ding Z., Mehta J. L. Immunity, inflammation, and oxidative stress in heart failure: emerging molecular targets. Cardiovascular Drugs and Therapy. 2017;31(5-6):593–608. doi: 10.1007/s10557-017-6752-z. [DOI] [PubMed] [Google Scholar]
- 198.Schorn C., Janko C., Krenn V., et al. Bonding the foe - NETting neutrophils immobilize the pro-inflammatory monosodium urate crystals. Frontiers in Immunology. 2012;3, article 376 doi: 10.3389/fimmu.2012.00376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.de Bont C. M., Koopman W. J. H., Boelens W. C., Pruijn G. J. M. Stimulus-dependent chromatin dynamics, citrullination, calcium signalling and ROS production during NET formation. Biochimica et Biophysica Acta - Molecular Cell Research. 2018;1865(11):1621–1629. doi: 10.1016/j.bbamcr.2018.08.014. [DOI] [PubMed] [Google Scholar]
- 200.al-Khafaji A. B., Tohme S., Yazdani H. O., Miller D., Huang H., Tsung A. Superoxide induces neutrophil extracellular trap formation in a TLR-4 and NOX-dependent mechanism. Molecular Medicine. 2016;22(1):621–631. doi: 10.2119/molmed.2016.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Vorobjeva N., Galkin I., Pletjushkina O., et al. Mitochondrial permeability transition pore is involved in oxidative burst and NETosis of human neutrophils. Biochimica et Biophysica Acta - Molecular Basis of Disease. 2020;1866(5, article 165664) doi: 10.1016/j.bbadis.2020.165664. [DOI] [PubMed] [Google Scholar]
- 202.Wang Y., Wang W., Wang N., Tall A. R., Tabas I. Mitochondrial oxidative stress promotes atherosclerosis and neutrophil extracellular traps in aged mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2017;37(8):e99–e107. doi: 10.1161/atvbaha.117.309580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Lood C., Blanco L. P., Purmalek M. M., et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nature Medicine. 2016;22(2):146–153. doi: 10.1038/nm.4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Gavillet M., Martinod K., Renella R., Wagner D. D., Williams D. A. A key role for Rac and Pak signaling in neutrophil extracellular traps (NETs) formation defines a new potential therapeutic target. American Journal of Hematology. 2018;93(2):269–276. doi: 10.1002/ajh.24970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Wilson E., Olcott M. C., Bell R. M., Merrill A. H., Jr., Lambeth J. D. Inhibition of the oxidative burst in human neutrophils by sphingoid long-chain bases. Role of protein kinase C in activation of the burst. The Journal of Biological Chemistry. 1986;261(27):12616–12623. doi: 10.1016/S0021-9258(18)67135-2. [DOI] [PubMed] [Google Scholar]
- 206.Paul R., Obermaier B., van Ziffle J., et al. Myeloid Src kinases regulate phagocytosis and oxidative burst in pneumococcal meningitis by activating NADPH oxidase. Journal of Leukocyte Biology. 2008;84(4):1141–1150. doi: 10.1189/jlb.0208118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Van Ziffle J. A., Lowell C. A. Neutrophil-specific deletion of Syk kinase results in reduced host defense to bacterial infection. Blood. 2009;114(23):4871–4882. doi: 10.1182/blood-2009-05-220806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Graham D. B., Robertson C. M., Bautista J., et al. Neutrophil-mediated oxidative burst and host defense are controlled by a Vav-PLCgamma2 signaling axis in mice. Journal of Clinical Investigation. 2007;117(11):3445–3452. doi: 10.1172/JCI32729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Remer K. A., Brcic M., Jungi T. W. Toll-like receptor-4 is involved in eliciting an LPS-induced oxidative burst in neutrophils. Toll-like receptor-4 is involved in eliciting an LPS-induced oxidative burst in neutrophils. Immunology Letters. 2003;85(1):75–80. doi: 10.1016/S0165-2478(02)00210-9. [DOI] [PubMed] [Google Scholar]
- 210.Kuriakose T., Rada B., Watford W. T. Tumor Progression Locus 2-dependent Oxidative Burst Drives Phosphorylation of Extracellular Signal-regulated Kinase during TLR3 and 9 Signaling. The Journal of Biological Chemistry. 2014;289(52):36089–36100. doi: 10.1074/jbc.M114.587121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Hoarau C., Gérard B., Lescanne E., et al. TLR9 activation induces normal neutrophil responses in a child with IRAK-4 deficiency: involvement of the direct PI3K pathway. The Journal of Immunology. 2007;179(7):4754–4765. doi: 10.4049/jimmunol.179.7.4754. [DOI] [PubMed] [Google Scholar]
- 212.Ding Y., Kantarci A., Hasturk H., Trackman P. C., Malabanan A., van Dyke T. E. Activation of RAGE induces elevated O2- generation by mononuclear phagocytes in diabetes. Journal of Leukocyte Biology. 2007;81(2):520–527. doi: 10.1189/jlb.0406262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Guichard C., Pedruzzi E., Dewas C., et al. Interleukin-8-induced Priming of Neutrophil Oxidative Burst Requires Sequential Recruitment of NADPH Oxidase Components into Lipid Rafts. The Journal of Biological Chemistry. 2005;280(44):37021–37032. doi: 10.1074/jbc.M506594200. [DOI] [PubMed] [Google Scholar]
- 214.Sozzani S., Agwu D. E., Ellenburg M. D., et al. Activation of phospholipase D by interleukin-8 in human neutrophils. Blood. 1994;84(11):3895–3901. doi: 10.1182/blood.V84.11.3895.bloodjournal84113895. [DOI] [PubMed] [Google Scholar]
- 215.Xie K., Varatnitskaya M., Maghnouj A., et al. Activation leads to a significant shift in the intracellular redox homeostasis of neutrophil-like cells. Redox Biology. 2020;28, article 101344 doi: 10.1016/j.redox.2019.101344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Björnsdottir H., Welin A., Michaëlsson E., et al. Neutrophil NET formation is regulated from the inside by myeloperoxidase- processed reactive oxygen species. Free Radical Biology and Medicine. 2015;89:1024–1035. doi: 10.1016/j.freeradbiomed.2015.10.398. [DOI] [PubMed] [Google Scholar]
- 217.Palladino E. N. D., Hartman C. L., Albert C. J., Ford D. A. The chlorinated lipidome originating from myeloperoxidase-derived HOCl targeting plasmalogens: metabolism, clearance, and biological properties. Archives of Biochemistry and Biophysics. 2018;641:31–38. doi: 10.1016/j.abb.2018.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Nagan N., Zoeller R. A. Plasmalogens: biosynthesis and functions. Progress in Lipid Research. 2001;40(3):199–229. doi: 10.1016/S0163-7827(01)00003-0. [DOI] [PubMed] [Google Scholar]
- 219.Thukkani A. K., Martinson B. D., Albert C. J., Vogler G. A., Ford D. A. Neutrophil-mediated accumulation of 2-ClHDA during myocardial infarction: 2-ClHDA-mediated myocardial injury. American Journal of Physiology-Heart and Circulatory Physiology. 2005;288(6):H2955–H2964. doi: 10.1152/ajpheart.00834.2004. [DOI] [PubMed] [Google Scholar]
- 220.Palladino E. N. D., Katunga L. A., Kolar G. R., Ford D. A. 2-Chlorofatty acids: lipid mediators of neutrophil extracellular trap formation. Journal of Lipid Research. 2018;59(8):1424–1432. doi: 10.1194/jlr.M084731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Prenner S. B., Pillutla R., Yenigalla S., et al. Serum albumin is a marker of myocardial fibrosis, adverse pulsatile aortic hemodynamics, and prognosis in heart failure with preserved ejection fraction. Journal of the American Heart Association. 2020;9(3, article e014716) doi: 10.1161/JAHA.119.014716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Prenner S. B., Kumar A., Zhao L., et al. Effect of serum albumin levels in patients with heart failure with preserved ejection fraction (from the TOPCAT Trial) The American Journal of Cardiology. 2020;125(4):575–582. doi: 10.1016/j.amjcard.2019.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Michelis R., Kristal B., Zeitun T., et al. Albumin oxidation leads to neutrophil activation in vitro and inaccurate measurement of serum albumin in patients with diabetic nephropathy. Free Radical Biology and Medicine. 2013;60:49–55. doi: 10.1016/j.freeradbiomed.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 224.Inoue M., Nakashima R., Enomoto M., et al. Plasma redox imbalance caused by albumin oxidation promotes lung-predominant NETosis and pulmonary cancer metastasis. Nature Communications. 2018;9(1, article 5116) doi: 10.1038/s41467-018-07550-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Koning A. M., Meijers W. C., Pasch A., et al. Serum free thiols in chronic heart failure. Pharmacological Research. 2016;111:452–458. doi: 10.1016/j.phrs.2016.06.027. [DOI] [PubMed] [Google Scholar]
- 226.Gaul D. S., Weber J., van Tits L. J., et al. Loss of Sirt3 accelerates arterial thrombosis by increasing formation of neutrophil extracellular traps and plasma tissue factor activity. Cardiovascular Research. 2018;114(8):1178–1188. doi: 10.1093/cvr/cvy036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Zhang X., Ji R., Liao X., et al. MicroRNA-195 regulates metabolism in failing myocardium via alterations in sirtuin 3 expression and mitochondrial protein acetylation. Circulation. 2018;137(19):2052–2067. doi: 10.1161/CIRCULATIONAHA.117.030486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Castillo E. C., Morales J. A., Chapoy-Villanueva H., et al. Mitochondrial hyperacetylation in the failing hearts of obese patients mediated partly by a reduction in SIRT3: the involvement of the mitochondrial permeability transition pore. Cellular Physiology and Biochemistry. 2019;53(3):465–479. doi: 10.33594/000000151. [DOI] [PubMed] [Google Scholar]
- 229.Zeng H., He X., Chen J. X. Endothelial sirtuin 3 dictates glucose transport to cardiomyocyte and sensitizes pressure overload-induced heart failure. Journal of the American Heart Association. 2020;9(11, article e015895) doi: 10.1161/JAHA.120.015895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Grillon J. M., Johnson K. R., Kotlo K., Danziger R. S. Non-histone lysine acetylated proteins in heart failure. Biochimica et Biophysica Acta. 2012;1822(4):607–614. doi: 10.1016/j.bbadis.2011.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Karbowska M., Kaminski T. W., Znorko B., et al. Indoxyl sulfate promotes arterial thrombosis in rat model via increased levels of complex TF/VII, PAI-1, platelet activation as well as decreased contents of SIRT1 and SIRT3. Frontiers in Physiology. 2018;9, article 1623 doi: 10.3389/fphys.2018.01623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Arroyo A. B., Fernández-Pérez M. P., del Monte A., et al. miR-146a is a pivotal regulator of neutrophil extracellular trap formation promoting thrombosis. Haematologica. 2020 doi: 10.3324/haematol.2019.240226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Sorvillo N., Mizurini D. M., Coxon C., et al. Plasma peptidylarginine deiminase IV promotes VWF-platelet string formation and accelerates thrombosis after vessel injury. Circulation Research. 2019;125(5):507–519. doi: 10.1161/CIRCRESAHA.118.314571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Fuchs T. A., Brill A., Duerschmied D., et al. Extracellular DNA traps promote thrombosis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(36):15880–15885. doi: 10.1073/pnas.1005743107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Grassle S., Huck V., Pappelbaum K. I., et al. von Willebrand factor directly interacts with DNA from neutrophil extracellular traps. Arteriosclerosis, Thrombosis, and Vascular Biology. 2014;34(7):1382–1389. doi: 10.1161/ATVBAHA.113.303016. [DOI] [PubMed] [Google Scholar]
- 236.Jain S., Pitoc G. A., Holl E. K., et al. Nucleic acid scavengers inhibit thrombosis without increasing bleeding. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(32):12938–12943. doi: 10.1073/pnas.1204928109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Marinković G., Grauen Larsen H., Yndigegn T., et al. Inhibition of pro-inflammatory myeloid cell responses by short-term S100A9 blockade improves cardiac function after myocardial infarction. European Heart Journal. 2019;40(32):2713–2723. doi: 10.1161/CIRCULATIONAHA.119.043833. [DOI] [PubMed] [Google Scholar]
- 238.Sakuma M., Tanaka A., Kotooka N., et al. Myeloid-related protein-8/14 in acute coronary syndrome. International Journal of Cardiology Heart & Vessels. 2017;249:25–31. doi: 10.1016/j.ijcard.2017.09.020. [DOI] [PubMed] [Google Scholar]
- 239.Wang Y., Fang C., Gao H., et al. Platelet-derived S100 family member myeloid-related protein-14 regulates thrombosis. Journal of Clinical Investigation. 2014;124(5):2160–2171. doi: 10.1172/JCI70966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Vogel S., Bodenstein R., Chen Q., et al. Platelet-derived HMGB1 is a critical mediator of thrombosis. Journal of Clinical Investigation. 2015;125(12):4638–4654. doi: 10.1172/JCI81660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Prakash P., Kulkarni P. P., Lentz S. R., Chauhan A. K. Cellular fibronectin containing extra domain A promotes arterial thrombosis in mice through platelet Toll-like receptor 4. Blood. 2015;125(20):3164–3172. doi: 10.1182/blood-2014-10-608653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.DeRoo E. P., Wrobleski S. K., Shea E. M., et al. The role of galectin-3 and galectin-3-binding protein in venous thrombosis. Blood. 2015;125(11):1813–1821. doi: 10.1182/blood-2014-04-569939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Katancik J. A., Sharma A., de Nardin E. Interleukin 8, Neutrophil-Activating Peptide-2 and GRO-α Bind to and Elicit Cell Activation via Specific and Different Amino Acid Residues of CXCR2. Cytokine. 2000;12(10):1480–1488. doi: 10.1006/cyto.2000.0742. [DOI] [PubMed] [Google Scholar]
- 244.Yago T., Liu Z., Ahamed J., McEver R. P. Cooperative PSGL-1 and CXCR2 signaling in neutrophils promotes deep vein thrombosis in mice. Blood. 2018;132(13):1426–1437. doi: 10.1182/blood-2018-05-850859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Henry C. G., Gutierrez F., Lee J. T., et al. Aortic thrombosis presenting as congestive heart failure: an umbilical artery catheter complication. The Journal of Pediatrics. 1981;98(5):820–822. doi: 10.1016/S0022-3476(81)80857-8. [DOI] [PubMed] [Google Scholar]
- 246.Goto S., Sakai H., Ikeda Y., Hande S. Arterial thrombosis in heart failure. Lancet. 1998;351(9115):1558–1559. doi: 10.1016/S0140-6736(05)61124-5. [DOI] [PubMed] [Google Scholar]
- 247.di Pasquale G., Passarelli P., Ribani M. A., Borgatti M. L., Urbinati S., Pinelli G. Prophylaxis of thromboembolic events in congestive heart failure. Archives of Gerontology and Geriatrics. 1996;23(3):329–336. doi: 10.1016/S0167-4943(96)00728-5. [DOI] [PubMed] [Google Scholar]
- 248.de Macedo I. S., Dinardi L. F. L., Pereira T. V., et al. Thromboembolic findings in patients with heart failure at autopsy. Cardiovascular Pathology. 2018;35:23–28. doi: 10.1016/j.carpath.2018.04.004. [DOI] [PubMed] [Google Scholar]
- 249.Zhang H., Jing X., Shi Y., et al. N-acetyl lysyltyrosylcysteine amide inhibits myeloperoxidase, a novel tripeptide inhibitor1[S] Journal of Lipid Research. 2013;54(11):3016–3029. doi: 10.1194/jlr.M038273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Witalison E. E., Cui X., Hofseth A. B., et al. Inhibiting protein arginine deiminases has antioxidant consequences. Journal of Pharmacology and Experimental Therapeutics. 2015;353(1):64–70. doi: 10.1124/jpet.115.222745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Zhou Y., An L. L., Chaerkady R., et al. Evidence for a direct link between PAD4-mediated citrullination and the oxidative burst in human neutrophils. Scientific Reports. 2018;8(1, article 15228) doi: 10.1038/s41598-018-33385-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Novotny J., Chandraratne S., Weinberger T., et al. Histological comparison of arterial thrombi in mice and men and the influence of Cl-amidine on thrombus formation. PLoS One. 2018;13(1, article e0190728) doi: 10.1371/journal.pone.0190728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Knight J. S., Luo W., O’Dell A. A., et al. Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circulation Research. 2014;114(6):947–956. doi: 10.1161/CIRCRESAHA.114.303312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Roth Flach R. J., Su C., Bollinger E., et al. Myeloperoxidase inhibition in mice alters atherosclerotic lesion composition. PLoS One. 2019;14(3, article e0214150) doi: 10.1371/journal.pone.0214150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Liu C., Desikan R., Ying Z., et al. Effects of a novel pharmacologic inhibitor of myeloperoxidase in a mouse atherosclerosis model. PLoS One. 2012;7(12, article e50767) doi: 10.1371/journal.pone.0050767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Ali M., Pulli B., Courties G., et al. Myeloperoxidase inhibition improves ventricular function and remodeling after experimental myocardial infarction. JACC: Basic to Translational Science. 2016;1(7):633–643. doi: 10.1016/j.jacbts.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Fukuta T., Okada H., Takemura G., et al. Neutrophil elastase inhibition ameliorates endotoxin-induced myocardial injury accompanying degradation of cardiac capillary glycocalyx. Shock. 2020;54(3):386–393. doi: 10.1097/SHK.0000000000001482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Bidouard J. P., Duval N., Kapui Z., Herbert J. M., O'Connor S. E., Janiak P. SSR69071, an elastase inhibitor, reduces myocardial infarct size following ischemia-reperfusion injury. European Journal of Pharmacology. 2003;461(1):49–52. doi: 10.1016/S0014-2999(03)01298-6. [DOI] [PubMed] [Google Scholar]
- 259.Willis V. C., Gizinski A. M., Banda N. K., et al. N-α-benzoyl-N5-(2-chloro-1-iminoethyl)-L-ornithine amide, a protein arginine deiminase inhibitor, reduces the severity of murine collagen-induced arthritis. Journal of Immunology. 2011;186(7):4396–4404. doi: 10.4049/jimmunol.1001620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Kawaguchi H., Matsumoto I., Osada A., et al. Peptidyl arginine deiminase inhibition suppresses arthritis via decreased protein citrullination in joints and serum with the downregulation of interleukin-6. Modern Rheumatology. 2019;29(6):964–969. doi: 10.1080/14397595.2018.1532545. [DOI] [PubMed] [Google Scholar]
- 261.Knight J. S., Subramanian V., O'Dell A. A., et al. Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Annals of the Rheumatic Diseases. 2015;74(12):2199–2206. doi: 10.1136/annrheumdis-2014-205365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Kusunoki Y., Nakazawa D., Shida H., et al. Peptidylarginine deiminase inhibitor suppresses neutrophil extracellular trap formation and MPO-ANCA production. Frontiers in Immunology. 2016;7, article 227 doi: 10.3389/fimmu.2016.00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Safi R., Kallas R., Bardawil T., et al. Neutrophils contribute to vasculitis by increased release of neutrophil extracellular traps in Behçet's disease. Journal of Dermatological Science. 2018;92(2):143–150. doi: 10.1016/j.jdermsci.2018.08.010. [DOI] [PubMed] [Google Scholar]
- 264.Zheng W., Warner R., Ruggeri R., et al. PF-1355, a mechanism-based myeloperoxidase inhibitor, prevents immune complex vasculitis and anti-glomerular basement membrane glomerulonephritis. Journal of Pharmacology and Experimental Therapeutics. 2015;353(2):288–298. doi: 10.1124/jpet.114.221788. [DOI] [PubMed] [Google Scholar]
- 265.Bielecka-Dabrowa A., Fabis J., Mikhailidis D. P., et al. Prosarcopenic effects of statins may limit their effectiveness in patients with heart failure. Trends in Pharmacological Sciences. 2018;39(4):331–353. doi: 10.1016/j.tips.2018.02.003. [DOI] [PubMed] [Google Scholar]
- 266.Lee M. M. Y., Sattar N., McMurray J. J. V., Packard C. J. Statins in the prevention and treatment of heart failure: a review of the evidence. Current Atherosclerosis Reports. 2019;21(10, article 41) doi: 10.1007/s11883-019-0800-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Dludla P. V., Nyambuya T. M., Johnson R., et al. Metformin and heart failure-related outcomes in patients with or without diabetes: a systematic review of randomized controlled trials. Heart Failure Reviews. 2020 doi: 10.1007/s10741-020-09942-y. [DOI] [PubMed] [Google Scholar]
- 268.Eurich D. T., Weir D. L., Majumdar S. R., et al. Comparative safety and effectiveness of metformin in patients with diabetes mellitus and heart failure: systematic review of observational studies involving 34,000 patients. Circulation: Heart Failure. 2013;6(3):395–402. doi: 10.1161/circheartfailure.112.000162. [DOI] [PubMed] [Google Scholar]
- 269.al-Ghoul W. M., Kim M. S., Fazal N., Azim A. C., Ali A. Evidence for simvastatin anti-inflammatory actions based on quantitative analyses of NETosis and other inflammation/oxidation markers. Results in Immunology. 2014;4:14–22. doi: 10.1016/j.rinim.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Menegazzo L., Scattolini V., Cappellari R., et al. The antidiabetic drug metformin blunts NETosis in vitro and reduces circulating NETosis biomarkers in vivo. Acta Diabetologica. 2018;55(6):593–601. doi: 10.1007/s00592-018-1129-8. [DOI] [PubMed] [Google Scholar]
- 271.Andreou I., Tousoulis D., Miliou A., et al. Effects of rosuvastatin on myeloperoxidase levels in patients with chronic heart failure: a randomized placebo-controlled study. Atherosclerosis. 2010;210(1):194–198. doi: 10.1016/j.atherosclerosis.2009.10.046. [DOI] [PubMed] [Google Scholar]
- 272.Sexton T. R., Wallace E. L., Macaulay T. E., et al. The effect of rosuvastatin on thromboinflammation in the setting of acute coronary syndrome. Journal of Thrombosis and Thrombolysis. 2015;39(2):186–195. doi: 10.1007/s11239-014-1142-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Liu G., Wu K., Zhang L., et al. Metformin attenuated endotoxin-induced acute myocarditis via activating AMPK. International Immunopharmacology. 2017;47:166–172. doi: 10.1016/j.intimp.2017.04.002. [DOI] [PubMed] [Google Scholar]
- 274.Soraya H., Rameshrad M., Mokarizadeh A., Garjani A. Metformin attenuates myocardial remodeling and neutrophil recruitment after myocardial infarction in rat. Bioimpacts. 2015;5(1):3–8. doi: 10.15171/bi.2015.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Abrams S. T., Morton B., Alhamdi Y., et al. A novel assay for neutrophil extracellular trap formation independently predicts disseminated intravascular coagulation and mortality in critically ill patients. American Journal of Respiratory and Critical Care Medicine. 2019;200(7):869–880. doi: 10.1164/rccm.201811-2111OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Mollenhauer M., Friedrichs K., Lange M., et al. Myeloperoxidase mediates postischemic arrhythmogenic ventricular remodeling. Circulation Research. 2017;121(1):56–70. doi: 10.1161/CIRCRESAHA.117.310870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Teijeira Á., Garasa S., Gato M., et al. CXCR1 and CXCR2 chemokine receptor agonists produced by tumors induce neutrophil extracellular traps that interfere with immune cytotoxicity. Immunity. 2020;52(5):856–871.e8. doi: 10.1016/j.immuni.2020.03.001. [DOI] [PubMed] [Google Scholar]
- 278.Wang L., Zhang Y. L., Lin Q. Y., et al. CXCL1-CXCR2 axis mediates angiotensin II-induced cardiac hypertrophy and remodelling through regulation of monocyte infiltration. European Heart Journal. 2018;39(20):1818–1831. doi: 10.1093/eurheartj/ehy085. [DOI] [PubMed] [Google Scholar]
- 279.Benigni A., Cassis P., Conti S., et al. Sirt3Deficiency shortens life span and impairs cardiac mitochondrial function rescued byOpa1Gene transfer. Antioxidants and Redox Signaling. 2019;31(17):1255–1271. doi: 10.1089/ars.2018.7703. [DOI] [PubMed] [Google Scholar]
- 280.Desai J., Kumar S. V., Mulay S. R., et al. PMA and crystal-induced neutrophil extracellular trap formation involves RIPK1-RIPK3-MLKL signaling. European Journal of Immunology. 2016;46(1):223–229. doi: 10.1002/eji.201545605. [DOI] [PubMed] [Google Scholar]
- 281.Xiao P., Wang C., Li J., et al. COP9 signalosome suppresses RIPK1-RIPK3-mediated cardiomyocyte necroptosis in mice. Circulation. Heart Failure. 2020;13(8):p. e006996. doi: 10.1161/CIRCHEARTFAILURE.120.006996. [DOI] [PMC free article] [PubMed] [Google Scholar]