

Abstract
Background: Recently, 5-aminolevulinic acid (ALA) has been reported to modulate inflammatory development via an antioxidant effect. Hence, the aim of this study was to determine the anti-atherosclerotic effect of ALA.
Methods and Results: Low-density lipoprotein (LDL) receptor knockout mice were fed the following diets for 24 weeks: normal diet (n=6); 1.25% cholesterol diet (high-cholesterol diet, HCD; n=7); HCD+ALA (46 mg/kg/day; n=10); and HCD+ezetimibe (5 mg/kg/day; n=10). At 40 weeks, HCD+ALA had reduced LDL cholesterol (320±68 vs. 379±49 mg/dL), triglyceride (141±44 vs. 195±49 mg/dL) and oxidized LDL (380±40 vs. 422±64 pg/mL) compared with HCD only. En face lesion area for the entire aortic surface was significantly smaller in mice that received HCD+ALA than in mice that received only HCD (32±5% vs. 39±4%, P<0.05). ALA intake exogenously increased tissue heme oxygenase-1 (HO-1) level in plaque composite tissue of the carotid arterial wall compared with HCD only (18±8 vs. 12±3 pg/μL, P<0.05), and HO-1-positive plaque showed modest NADPH oxidase 4 expression.
Conclusions: ALA intake induces exogenous production of HO-1 at plaque sites, and improves lipid profiles and attenuation of atherosclerotic plaque progression in vivo.
Key Words: 5-Aminolevulinic acid, Atherosclerosis, Heme oxygenase-1, NADPH oxidase 4, Plaque
The pathogenesis of atherosclerosis involves several pathophysiological steps based on several inflammatory pathways. Oxidative stress has been implicated in the pathogenesis of various cardiovascular diseases including atherosclerosis.1–3 Indeed, all risk factors for cardiovascular disease, including hypertension, hypercholesterolemia, diabetes mellitus and cigarette smoking, as well as cardiovascular disease itself, are associated with increased production of reactive oxygen species (ROS) in the vascular wall, a situation that eventually culminates in oxidative stress.4,5
Excessive production of ROS by NADPH oxidases (NOX) mediates oxidative stress associated with increased inflammation and is crucial for atherosclerotic disease.6 Vendrov et al found that increased NOX4 expression is also correlated with the severity of atherosclerosis in human subjects.7 More recently Lozhkin et al reported that upregulation of NOX4 in vascular smooth muscle cells (VSMC) also contributes to inflammation during aging and in atherosclerosis.8 Additionally, several antioxidant agents have been shown to attenuate plaque formation in animal studies.9
Heme oxygenase-1 (HO-1), which is induced under a condition of hyperlipidemia, functions as an intrinsic protective factor against atherosclerotic lesion formation, possibly by inhibiting lipid peroxidation and influencing the nitric oxide pathway,10 and HO-1 induction has been reported to suppress oxidative stress and prevent vulnerable plaque formation.11 Datla et al showed that HO-1 overexpression inhibits NADPH oxidase activity in VSMC.12
Recently, 5-aminolevulinic acid (ALA) itself has been reported to have the potential to modulate metabolic disease.13 ALA is synthesized from glycine and succinyl-coenzyme A (succinyl-CoA) in mitochondria and is metabolized to heme, which induces the antioxidant HO-1.14
We hypothesized that exogenous ALA intake would augment HO-1 induction and attenuate atherosclerotic plaque progression via regulation of NOX. Hence, the aim of this study was to determine the anti-atherosclerotic effect of ALA via the antioxidant effect of exogenous HO-1 induction in vivo.
Methods
Animals
Inbred female low-density lipoprotein (LDL) receptor knockout mice were housed under specific pathogen-free conditions in an environmentally controlled clean room under a 12-h light/dark schedule with lights on at 8:30 p.m. in a temperature- and humidity-controlled room (22±1℃ and 55±5%, respectively). The experiments were performed from 9 a.m. to 4 p.m. The animal experimental protocol was in accord with the ARRIVE guidelines. This study was conducted according to the guidelines of the Institutional Review Board for the Care of Animal Subjects of the National Defense Medical College. The Institutional Review Board for the Care of Animal Subjects of the National Defense Medical College approved this study (ethics approval no. 15049).
Experiment Protocol
Four groups of LDL receptor knockout mice (n=33 in total) were fed the following diets until 40 weeks of age (Figure 1): a normal diet until 16 weeks of age, after which 3 groups were then switched to the following diets for 24 weeks, while 1 group continued on the normal diet (CLEA Rodent Diet-2 [CE-2], n=6): a 1.25% cholesterol diet (high-cholesterol diet [HCD]: 82.7% CE-2, 15% beef tallow, 1.25% cholesterol, 1.046% α-cornstarch, n=7); an HCD+ALA diet (82.7% CE-2, 15% beef tallow, 1.25% cholesterol, 1.0% α-cornstarch, 0.046% ALA [purity >98%, SBI pharma, Tokyo, Japan], n=10); and an HCD+ezetimibe diet (82.7% CE-2, 15% beef tallow, 1.25% cholesterol, 1.041% α-cornstarch, 0.005% ezetimibe; CLEA Japan, Tokyo, Japan, n=10). All of the diets were purchased from CLEA Japan (Tokyo, Japan). The mice were able to access each diet any time until 40 weeks of age. Body weight was measured every month and average diet intake volume was measured every week. The dose of ALA was set to 50 mg/kg/day, a dose that has been reported to have a sufficient metabolic benefit without any adverse effect,15–18 and that of ezetimibe was set to 5 mg/kg/day.19,20
Figure 1.

Experiment protocol. Sampling of whole blood was carried out via the orbital sinus. ALA, 5-aminolevulinic acid.
Whole blood samples obtained on orbital sinus blood sampling were analyzed for serum total cholesterol, LDL cholesterol (LDL-C), triglyceride (TG), total bilirubin, aspartate transaminase (AST) and alanine transaminase (ALT) at 16 weeks, 24 weeks, 32 weeks and 40 weeks of age. Serum total cholesterol, LDL-C, TG, total bilirubin, AST and ALT were quantified using routine laboratory methods (Nagahama Life Science Laboratory, Shiga, Japan). At the end of the breeding period, whole blood samples were taken from the inferior vena cava and again analyzed for serum total cholesterol, LDL-C, TG, total bilirubin, AST and ALT as well as oxidized LDL. Oxidized LDL was measured using a Mouse Oxidized Low Density Lipoprotein (OxLDL) enzyme-linked immunosorbent assay (ELISA) kit (KT-32067, Kamiya Biomedical Company, Seattle, WA, USA).
Mice were killed and the aortas and bilateral carotid arteries were carefully removed and stored at −80℃.
Atherosclerotic Plaque Area
Aortas were excised at 40 weeks (24 weeks after the start of each treatment), and plaque volume was determined on oil red O staining. In brief, the thoracic ascending and abdominal aorta was removed from the heart. After cutting off minor branching arteries, the aorta was fixed in formal sucrose (4% paraformaldehyde, 5% sucrose, 20 mM EDTA, pH 7.4). After the adventitial and adipose tissue had been removed, each of the aortas was cut open longitudinally. For neutral lipid visualization, sections were rinsed in 60% isopropanol and incubated in a saturated, filtered solution of oil red O in 60% isopropanol for 1 h. The stained aorta was pinned open, and the ratio of the intimal surface stained with oil red O in the arch and thoracic regions was measured morphometrically (Win ROOF version 5.5; Mitsuya-Shoji, Tokyo, Japan).
Tissue HO-1
At 40 weeks, left carotid arteries (along with the common carotid to bifurcation of internal and external carotid arteries; approximately 2 cm in length) were excised and immediately stored at −80℃. To measure HO-1, the tissue samples were homogenized in 0.3 mL radio-immunoprecipitation assay (RIPA) lysis buffer with addition of 3 μL Halt Protease inhibitor cocktail (100×; Thermo Fisher Scientific, Tokyo, Japan) for 30 min and then homogenized (NS-310E3, 30,000 rpm for 20 s, Microtec, Tokyo, Japan) followed by disruption in an ultrasonic cell disruptor (Bioruptor, 3 times for 30 s each time with intervals of 30 s, Cosmo Bio, Tokyo, Japan) at 4℃. Thirty min later, the supernatant was ultracentrifuged (100,000×g, 10 min, twice, Tomy MX-301, Tomy Seiko, Tokyo, Japan) at 4℃. The supernatant was used to determine HO-1.
Tissue HO-1 concentration was measured in the carotid artery atherosclerotic plaque composition and in the liver using an HO-1 Mouse SimpleStep ELISA kit (Abcam 204524, Cambridge, MA, USA).
Histopathology
For histopathology, right carotid artery sections were fixed in 10% formalin and embedded in paraffin. One of 2 series of 5-μm-thick slices of paraffin-embedded tissue was placed on a slide for immunohistochemistry. Antigen retrieval was performed by heating in 10 mmol/L citrate buffer (pH 6.0) followed by treatment with 0.6% H2O2 in methanol. The section was incubated with a primary antibody at 4℃ overnight. After washing with phosphate-buffered salts, the section was incubated with a secondary antibody (goat anti-rabbit IgG HRP; Abcam 6721) at room temperature for 1 h. Finally, the section was washed and then immunostained with DAB solution (Abcam 64238). The primary antibodies were a rabbit polyclonal antibody to HO-1 (Abcam 13243) and a rabbit polyclonal antibody to NOX4/NADPH oxidase 4 (Bioss bs-3684R, Woburn, MA, USA). The other series of slices was deparaffinized and stained with hematoxylin and eosin (HE). Using the plaque immunohistochemical samples in which an atherosclerotic plaque had formed, the ratio of the HO-1-positive area and the ratio of the NOX4-positive area in the vessel wall area were measured morphometrically for quantitative analysis (Win ROOF version 5.5, Mitsuya-Shoji, Tokyo, Japan).
Statistical Analysis
Statistical analysis was done using 1-way ANOVA–Bonferroni test. Data are presented as mean±SD, with P<0.05 considered to be statistically significant.
Results
Mouse Body Weight and Serum Lipid Profile
The HCD diet augmented mouse body weight (33±5 g at 40 weeks) compared with the normal diet (25±3 g at 40 weeks) with time (Figure 2A). From the measurement of volume of consumed diet, ALA intake was determined to be 46 mg/kg/day. ALA supplementation (46 mg/kg/day) as well as ezetimibe supplementation (5 mg/kg/day) had no influence on the body weight increase caused by HCD intake (31±4 g and 34±7 g, respectively). ALA supplementation also had no adverse effect, as indicated by the serum total bilirubin, AST and ALT being maintained in the normal range during the experiment (Figure 2B–D).
Figure 2.

Change in (A) body weight, (B) total serum bilirubin, (C) aspartate aminotransferase (AST) and (D) alanine aminotransferase (ALT) in mice according to diet. *P<0.05, normal diet vs. the other groups. ALA, 5-aminolevulinic acid.
The HCD-only diet increased serum total cholesterol and LDL-C after 8 weeks of feeding (at 24 weeks of age) as shown in Figure 3. Ezetimibe supplementation completely inhibited the elevation of serum total cholesterol and LDL-C (Figure 3A,C). ALA supplementation for 24 weeks resulted in a significant decrease in serum LDL-C (320±68 mg/dL) at 40 weeks of age (Figure 3C). Until 32 weeks of age, TG remained in the normal range in all 4 groups, but HCD and HCD+ALA increased serum TG at 40 weeks of age. The HCD+ALA group, however, had a small increase in TG (141±44 mg/dL) compared with that in the HCD-only group (195±49 mg/dL; Figure 3B).
Figure 3.

Change in (A) total cholesterol, (B) triglyceride, (C) low-density lipoprotein (LDL) cholesterol and (D) oxidized LDL in mice according to diet. *P<0.05; HCD vs. normal diet group and HCD+ezetimibe group, **P<0.05; HCD vs. the other groups, ***P<0.05. ALA, 5-aminolevulinic acid.
HCD and HCD+ezetimibe resulted in an increase in serum oxidized LDL at 40 weeks of age (422±64 pg/mL and 402±59 pg/mL, respectively). ALA supplementation inhibited the elevation of serum oxidized LDL (380±40 pg/mL), and the serum oxidized LDL level was not significantly different from that on the normal diet (345±60 pg/mL; Figure 3D).
Aortic Atherosclerotic Plaque Area
Intake of the HCD-only diet for 24 weeks resulted in the development of lipid-rich atherosclerotic plaque in the aorta (Figure 4A). Plaque was dominantly seen at the aortic arch and bifurcating sites of several major branches. The ratio of en face lesion area for the entire aortic surface reached 0.39±0.04 in the HCD-only group, but the HCD+ALA group had a decreased ratio of 0.32±0.05. Ezetimibe supplementation prevented atherosclerotic plaque accumulation and the ratio was 0.12±0.05, as low as that in mice fed a normal diet (Figure 4B).
Figure 4.

Aortic plaque area in (A) representative photographs of the aorta on oil red O stain and (B) ratio of en face plaque area to the entire aortic surface in mice, according to diet. *P<0.05. Scale bar, 2 mm. ALA, 5-aminolevulinic acid; HCD, high-cholesterol diet.
Antioxidant Effect
HCD intake for 24 weeks resulted in compensatory induction of endogenous HO-1 production in the carotid artery, and HO-1 was recruited to plaque, which was often seen at the bifurcation site of the common carotid artery. HO-1 level in carotid tissue of HCD mice was 2-fold higher than in the normal diet mice at 40 weeks (12±3 pg/μL vs. 6.5±2 pg/μL, P<0.05, Figure 5A). ALA supplementation exogenously increased HO-1 in the carotid plaque-rich bifurcation site (18±8 pg/μL, Figure 5A) compared with the HCD-only group (Figure 5A). The HCD+ezetimibe group had less HO-1 production, probably due to smaller plaque formation.
Figure 5.

Heme oxygenase-1 (HO-1) in (A) the bifurcation site of the common carotid artery; (B) a plaque-poor site of the common carotid artery; and (C) in the liver, in mice according to diet. *P<0.05. ALA, 5-aminolevulinic acid; HCD, high-cholesterol diet.
Figure 5C shows HO-1 level in the liver. Liver HO-1 in the normal diet mice was regarded as basal HO-1 production (32±4 pg/μL). Liver HO-1 level was not increased by HCD only (36±8 pg/μL) or by HCD+ALA (38±8 pg/μL). Taken together, these results indicate that exogenous HO-1 induction seemed to be specifically augmented in atherosclerotic plaque tissue.
Histopathology
On HE staining of the carotid artery in HCD-only mice, typical atherosclerotic plaque consisting of thickened intima with foam cell formation was seen. These pathology findings were in accordance with human atherosclerotic grade 3 histology showing pathologic intimal thickening with extracellular lipids underneath a layer of foam cell macrophages (Figure 6A).21
Figure 6.

(A) Hematoxylin and eosin (HE) staining and (B) heme oxygenase-1 (HO-1) immunostaining of the carotid arteries in mice according to diet. (A) A thickened intima with foam cells is seen in both the high-cholesterol diet (HCD) mice and the HCD+5-aminolevulinic acid (HCD+ALA) mice. (B) Expression of immunoreactive HO-1 was markedly increased in medial and intimal smooth muscle cells, especially at the carotid atherosclerotic lesions of HCD+ALA mice (arrow). Scale bar, 0.3 mm.
In line with the quantitative HO-1 results, expression of immunoreactive HO-1 was markedly enhanced at carotid atherosclerotic lesions in the HCD+ALA mice, particularly in lesions in medial and intimal smooth muscle cells (Figure 6B). HCD+ezetimibe prevented plaque deposition in vessels and resulted in almost no HO-1-positive areas (Figure 6B).
Given that the core of the plaque consisted of foam cells, cells with stronger HO-1 expression had weaker expression of immunoreactive NOX4 (Figure 7). Quantitatively, there was an inverse relationship between the ratio of the HO-1-positive area and the ratio of the NOX4-positive area (Figure 7, Bottom).
Figure 7.
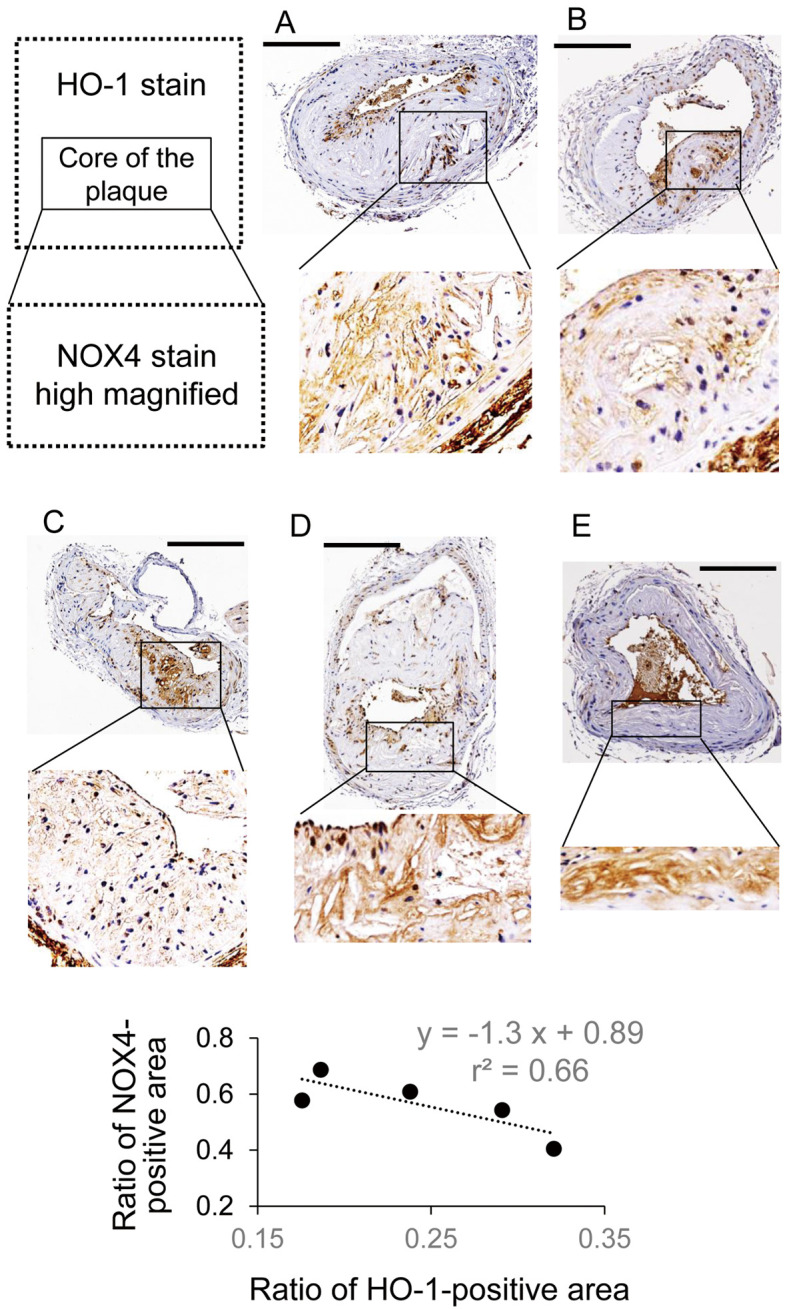
Heme oxygenase-1 (HO-1) and NADPH oxidase (NOX)-4 immunostaining of carotid arteries from mice treated with (A,B) a high-cholesterol diet (HCD) and (C–E) an HCD+5-aminolevulinic acid (HCD+ALA) diet. Scale bar, 0.3 mm. (Bottom) Analytic result from the immunohistochemistry samples.
Discussion
In this study, ALA supplementation improved serum lipid profiles with augmentation of HO-1 induction and reduction of the atherosclerotic plaque area in the whole aorta in mice fed an HCD.
ALA is naturally synthesized from glycine and succinyl-CoA and is finally metabolized to heme in mitochondria in the human body. Initially, ALA was developed industrially for plant fertilizer producing chlorophyll. An et al reported that ALA inhibits abscisic acid-induced stomatal closure by reducing H2O2 and calcium levels in guard cells, and that it simultaneously improves plant drought tolerance.22 This antioxidant mechanism as an inhibitor of H2O2-generating NADPH oxidase improved mitochondrial function in animals.
Cheng et al reported that HO-1 induction by cobalt protoporphyrin (CoPP) impeded lesion progression into vulnerable plaques, indicated by a reduction in necrotic core size and intraplaque lipid accumulation, whereas cap thickness and VSMC were increased.11 In their study, HO-1 protein level was 20-fold higher than in normal mice.11 Several researchers have reported, however, that high overdose expression of HO-1 exacerbates inflammation: when HO-1 expression is low, low cellular heme and iron levels may allow for decreased oxidative injury and upregulation of important enzymes, whereas excessive accumulation of reactive iron when HO-1 expression is high would result in increased oxidative stress, cytotoxicity, and abnormal cellular proliferation.23 Excessive HO-1 not only leads to enhanced cell injury but also prolongs the repair process of injured endothelial tissue.24 From these viewpoints, long-term ALA may be practical for treating a chronic disease in a clinical situation given that, in the present study, the level of HO-1 at plaque-rich sites in mice on the HCD+ALA diet was 3-fold higher than in mice fed a normal diet.
In the present study, HO-1 was overexpressed in the atherosclerotic plaque composite of the carotid arterial wall in mice given exogenous ALA: the HCD+ALA group had a 1.5-fold higher HO-1 level than the HCD-only group. In line with this, chronic CoPP treatment induced a 5-fold higher HO-1 expression compared with that in the normal diet rats and attenuated the coronary constrictor response to ischemia-reperfusion.25 Modest and chronic expression of HO-1 resulted in a significant increase in serum adiponectin.26 The HO-1-mediated increase in adiponectin provides the heart and vascular system with tolerance and resistance not only to the oxidative stress generated in diabetes, but also to other types of vascular stress.26,27
As a positive control group, ezetimibe supplementation completely inhibited atherosclerotic plaque formation and kept the serum lipid profile normal. It did not cause body weight gain or oxidative stress, indicated by elevation of serum oxidized LDL. These results are compatible with results obtained by Umemoto et al and Tie et al showing that ezetimibe inhibited cholesterol absorption in the intestine and kept the lipid profile normal without an anti-inflammatory effect.19,28
In contrast, in the present study, ALA intake decreased oxidized LDL to within the normal range via induction of HO-1 production. Oxidized LDL has been shown to play a major role in atherogenesis, and, as an antioxidant, reduces production of mitochondrial ROS and accumulation of oxidative damage to mitochondrial DNA, proteins, and lipids.29,30
Recently, NADPH oxidase has been shown to be a key component for modifying metabolic diseases. In the present study, HO-1 expression in the core of the plaque composed of foam cells attenuated NOX4 expression. Taken together, this indicates that ALA has an antioxidant action as an inhibitor of H2O2-generating NOX4.22
In the present study, the antioxidant agent ALA induced HO-1 production along with improvements in systemic inflammatory and lipid profiles such as plasma oxidized LDL and TG.
Study Limitations
In this study the anti-atherosclerotic effect of ALA was modest compared with the anti-atherosclerotic effect of ezetimibe, which completely inhibited plaque progression. Further study is necessary to elucidate the adjunctive efficacy of ALA added to statin therapy and also its effectiveness for regression of plaque and maintenance of vascular function.
In the present study, ezetimibe-treated mice were used as positive controls without the use of statins because the condition of LDL receptor knockout would crucially affect the pharmacological mechanism of a statin, which inhibits Hydroxymethylglutaryl-CoA and upregulates the LDL receptor. We also considered that statins have pleiotropic effects including several antioxidant effects on atherosclerosis, but the aim of this study was to elucidate the effect of HO-1 induced by ALA on atherosclerosis formation.
We evaluated local NOX4 expression in the plaque area as an essential antioxidant pathway of ALA, but NADPH oxidase subunits such as NOX2 and p22phox are also important for atherosclerotic plaque formation. Hence further studies are expected.
Conclusions
ALA exogenously induced the production of HO-1 at plaque sites, improving lipid profiles and attenuating atherosclerotic plaque progression in vivo.
Financial Support
This work was supported in part by JSPS KAKENHI Grant Number 16K12885 (to K.H. and Y.M.).
Disclosures
The authors declare no conflicts of interest.
Author Contributions
K.H. and Y.M. contributed to the conception and design of the study; K.H. and Y.M. carried out the experiments and acquired data; Y.M. performed data analysis; K.H. wrote the first draft of the manuscript; M.N., M.A. and K.I. contributed to manuscript revision and supervised the project. All authors read and approved the submitted version.
Acknowledgments
The excellent technical assistance of Ms. Nozomi Saiki, Ms. Masami Konno (blood sampling), Ms. Wakako Kayukawa, Ms. Miho Ushida and Mr. Taro Takee (immunostaining, ELISA measurement) is gratefully acknowledged.
References
- 1. Li H, Horke S, Förstermann U.. Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis 2014; 237: 208–219. [DOI] [PubMed] [Google Scholar]
- 2. Stocker R, Keaney JF Jr.. Role of oxidative modifications in atherosclerosis. Physiol Rev 2004; 84: 1381–1478. [DOI] [PubMed] [Google Scholar]
- 3. Harrison D, Griendling KK, Landmesser U, Hornig B, Drexler H.. Role of oxidative stress in atherosclerosis. Am J Cardiol 2003; 91: 7A–11A. [DOI] [PubMed] [Google Scholar]
- 4. Förstermann U.. Oxidative stress in vascular disease: Causes, defense mechanisms and potential therapies. Nat Clin Pract Cardiovasc Med 2008; 5: 338–349. [DOI] [PubMed] [Google Scholar]
- 5. Li H, Förstermann U.. Uncoupling of endothelial NO synthase in atherosclerosis and vascular disease. Curr Opin Pharmacol 2013; 13: 161–167. [DOI] [PubMed] [Google Scholar]
- 6. Salazar G.. NADPH oxidases and mitochondria in vascular senescence. Int J Mol Sci 2018; 19: e1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vendrov AE, Vendrov KC, Smith A, Yuan J, Sumida A, Robidoux J, et al.. NOX4 NADPH oxidase-dependent mitochondrial oxidative stress in aging-associated cardiovascular disease. Antioxid Redox Signal 2015; 23: 1389–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lozhkin A, Vendrov AE, Pan H, Wickline SA, Madamanchi NR, Runge MS.. NADPH oxidase 4 regulates vascular inflammation in aging and atherosclerosis. J Mol Cell Cardiol 2017; 102: 10–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Siti HN, Kamisah Y, Kamsiah J.. The role of oxidative stress, antioxidants and vascular inflammation in cardiovascular disease (a review). Vascul Pharmacol 2015; 71: 40–56. [DOI] [PubMed] [Google Scholar]
- 10. Ishikawa K, Sugawara D, Wang Xp, Suzuki K, Itabe H, Maruyama Y, et al.. Heme oxygenase-1 inhibits atherosclerotic lesion formation in ldl-receptor knockout mice. Circ Res 2001; 88: 506–512. [DOI] [PubMed] [Google Scholar]
- 11. Cheng C, Noordeloos AM, Jeney V, Soares MP, Moll F, Pasterkamp G, et al.. Heme oxygenase 1 determines atherosclerotic lesion progression into a vulnerable plaque. Circulation 2009; 119: 3017–3027. [DOI] [PubMed] [Google Scholar]
- 12. Datla SR, Dusting GJ, Mori TA, Taylor CJ, Croft KD, Jiang F.. Induction of heme oxygenase-1 in vivo suppresses NADPH oxidase derived oxidative stress. Hypertension 2007; 50: 636–642. [DOI] [PubMed] [Google Scholar]
- 13. Ota U, Hara T, Nakagawa H, Tsuru E, Tsuda M, Kamiya A, et al.. 5-Aminolevulinic acid combined with ferrous ion reduces adiposity and improves glucose tolerance in diet-induced obese mice via enhancing mitochondrial function. BMC Pharmacol Toxicol 2017; 18: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nishio Y, Fujino M, Zhao M, Ishii T, Ishizuka M, Ito H, et al.. 5-Aminolevulinic acid combined with ferrous iron enhances the expression of heme oxygenase-1. Int Immunopharmacol 2014; 19: 300–307. [DOI] [PubMed] [Google Scholar]
- 15. Koganei M, Saitou Y, Tsuchiya K, Abe F, Tanaka T, Horinouchi I, et al.. Effects of 5-aminolevulinic acid on a murine model of diet-induced obesity. J Clin Biochem Nutr 2015; 57: 145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sato T, Yasuzawa T, Uesaka A, Izumi Y, Kamiya A, Tsuchiya K, et al.. Type 2 diabetic conditions in Otsuka Long-Evans Tokushima fatty rats are ameliorated by 5-aminolevulinic acid. Nutr Res 2014; 34: 544–551. [DOI] [PubMed] [Google Scholar]
- 17. Takahashi J, Misawa M, Murakami M, Mori T, Nomura K, Iwahashi H.. 5-Aminolevulinic acid enhances cancer radiotherapy in a mouse tumor model. Springerplus 2013; 2: 602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Perez M, Rodriguez B, Shintani T, Watanabe K, Miyanari S, Harrigan R.. 5-Aminolevulinic acid (5-ALA): Analysis of preclinical and safety literature. Food Nutr Sci 2013; 4: 1009–1013. [Google Scholar]
- 19. Umemoto T, Subramanian S, Ding Y, Goodspeed L, Wang S, Han CY, et al.. Inhibition of intestinal cholesterol absorption decreases atherosclerosis but not adipose tissue inflammation. J Lipid Res 2012; 53: 2380–2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nakagami H, Osako MK, Takami Y, Hanayama R, Koriyama H, Mori M, et al.. Vascular protective effects of ezetimibe in ApoE-deficient mice. Atherosclerosis 2009; 203: 51–58. [DOI] [PubMed] [Google Scholar]
- 21. Nakashima Y, Fujii H, Sumiyoshi S, Wight TN, Sueishi K.. Early human atherosclerosis: Accumulation of lipid and proteoglycans in intimal thickenings followed by macrophage infiltration. Arterioscler Thromb Vasc Biol 2007; 27: 1159–1165. [DOI] [PubMed] [Google Scholar]
- 22. An Y, Liu L, Chen L, Wang L.. ALA inhibits ABA-induced stomatal closure via reducing H2O2 and Ca(2+) levels in guard cells. Front Plant Sci 2016; 7: 482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Suttner DM, Dennery PA.. Reversal of HO-1 related cytoprotection with increased expression is due to reactive iron. FASEB J 1999; 13: 1800–1809. [DOI] [PubMed] [Google Scholar]
- 24. Maruhashi K, Kasahara Y, Ohta K, Wada T, Ohta K, Nakamura N, et al.. Paradoxical enhancement of oxidative cell injury by overexpression of heme oxygenase-1 in an anchorage-dependent cell ECV304. J Cell Biochem 2004; 93: 552–562. [DOI] [PubMed] [Google Scholar]
- 25. L’Abbate A, Neglia D, Vecoli C, Novelli M, Ottaviano V, Baldi S, et al.. Beneficial effect of heme oxygenase-1 expression on myocardial ischemia-reperfusion involves an increase in adiponectin in mildly diabetic rats. Am J Physiol Heart Circ Physiol 2007; 293: H3532–H3541. [DOI] [PubMed] [Google Scholar]
- 26. Li M, Kim DH, Tsenovoy PL, Peterson SJ, Rezzani R, Rodella LF, et al.. Treatment of obese diabetic mice with a heme oxygenase inducer reduces visceral and subcutaneous adiposity, increases adiponectin levels, and improves insulin sensitivity and glucose tolerance. Diabetes 2008; 57: 1526–1535. [DOI] [PubMed] [Google Scholar]
- 27. Abraham NG, Kappas A.. Pharmacological and clinical aspects of heme oxygenase. Pharmacol Rev 2008; 60: 79–127. [DOI] [PubMed] [Google Scholar]
- 28. Tie C, Gao K, Zhang N, Zhang S, Shen J, Xie X, et al.. Ezetimibe attenuates atherosclerosis associated with lipid reduction and inflammation inhibition. PLoS One 2015; 10: e0142430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Witztum JL, Steinberg D.. Role of oxidized low density lipoprotein in atherogenesis. J Clin Invest 1991; 88: 1785–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mikhed Y, Daiber A, Steven S.. Mitochondrial oxidative stress, mitochondrial DNA damage and their role in age-related vascular dysfunction. Int J Mol Sci 2015; 16: 15918–15953. [DOI] [PMC free article] [PubMed] [Google Scholar]