

Abstract
Human activity is an important driver of ecological and evolutionary change on our planet. In particular, domestication and biological introductions have important and long-lasting effects on species’ genomic architecture and diversity. However, genome-wide analysis of independent domestication and introduction events within a single species has not previously been performed. The Pacific kelp Undaria pinnatifida provides such an opportunity because it has been cultivated in its native range in Northeast Asia but also introduced to four other continents in the past 50 years. Here we present the results of a genome-wide analysis of natural, cultivated and introduced populations of U. pinnatifida to elucidate human-driven evolutionary change. We demonstrate that these three categories of origin can be distinguished at the genome level, reflecting the combined influence of neutral (demography and migration) and non-neutral (selection) processes.
Subject terms: Ecology, Evolution, Genetics, Ecology
Human-driven evolutionary changes are captured by comparison of the genomes of natural, cultivated and introduced populations of a globally distributed Pacific kelp.
Main
Unprecedented evolutionary experiments have resulted from the spread of humans on our planet. Initially considered anecdotal and rare1, human-driven evolutionary change is now reported at increasing rates2,3. These processes leave footprints in the genomes of many species that are cultivated4,5, domesticated6,7 or transported across different biogeographic regions (that is, biological introductions)8,9. In-depth examination and understanding of these changes is an important research area, because of their considerable ecological, economic and health implications10.
Domestication is a form of co-evolution between a species (that is, human) and another species it controls (in terms of growth and reproduction) for its own benefit. This process has shaped the evolution of hundreds of plants and animals11,12. As far back as 12,000 years ago, and the transition in human behaviour from food gathering to cultivation13, agricultural societies depended on the domestication and diversification of wild species, notably through selective breeding, hybridization or inbreeding14,15. As a result of the application of these methods, there was selection for so-called ‘domestication phenotypes’. These traits can arise through conscious selection (intentional choice made by humans of preferred phenotypes in cultivated species for use and propagation) or unintended selection (natural selection in crop species as a result of human cultivation practices in agro-ecological environments), challenging the clear discrimination of traits directly selected during domestication and other traits.
In a globalized environment, where borders are crossed legally and illegally every day by hundreds of thousands of humans and goods, plant and animal species are dispersed knowingly or unwittingly around the world. Since the end of the twentieth century, nonindigenous species have become a major concern in our societies16,17. These human-driven migrations bring into contact populations or species that have evolved in isolation in their respective native range, exacerbating evolutionary changes, notably through admixture and hybridization18–20, as well as by selective pressure on the introduced species in its new range21. Biological introductions thus represent a fascinating opportunity to understand major evolutionary processes, such as genotype by environment interactions22 or speciation dynamics23.
Only a handful of case studies offer the opportunity to simultaneously address human-driven evolutionary change due to domestication and introduction. The Pacific kelp Undaria pinnatifida (Harvey) Suringar (Laminariales, Phaeophyceae) provides such an opportunity. In its native range of Northeast Asia, this brown edible seaweed was exploited for centuries before being cultivated, and its farming represents 6.9% of worldwide seaweed production24. The transition from the harvesting of natural populations to the cultivation on ‘long lines’ happened during the 1950s25 with the development of seaweed cultivation techniques and their application to U. pinnatifida in Japan, then Korea and finally China26. During this period, farmers selected desired phenotypes and only recently were breeding techniques used to develop cultivars26–28. Parallel to cultivation, U. pinnatifida has been intentionally and unintentionally transported by humans across the planet. Since its first report outside its native range (that is, along the Mediterranean coast of France in the 1970s29), U. pinnatifida has become established along the coastlines of 14 countries across 4 continents30–32. This kelp presents the rare characteristic of being independently cultivated in its native range and introduced in four continents outside its native range. This situation contrasts with other well-studied cases such as Oryza33 and Sorghum34 in which domestication preceded escape to the wild. Recent studies have demonstrated the importance of genome-wide analyses, based on whole-genome sequencing data, to understand human-driven evolutionary change, notably with respect to domestication4–7, with fewer studies examining invasive species8,9, and none addressing both aspects at the same time. Here we report the genome sequence of a Korean cultivar of U. pinnatifida, sequenced independently from the genome sequence of the Chinese gametophyte35. On the basis of whole‐genome sequencing of multiple individuals sampled in native, cultivated and introduced populations, we compared genome architecture across these different categories. We argue that the differences observed among them probably result from the combined influence of demography and selection.
Results and discussion
The nuclear genome of U. pinnatifida Kr2015
We extracted genomic DNA from a cultivated U. pinnatifida sporophyte harvested in November 2015 in Wando, Korea and generated a nuclear genome assembly using PacBio long reads with ~100× sequence coverage (Supplementary Note and Supplementary Table 1). The assembled contigs were polished with ~32× coverage of Illumina paired-end reads. The resulting assembly consisted of 3,876 contigs with a total size of 634 megabases (Mb) with N50 (minimum contig length to cover 50 percent of the assembly) of 406 kb (Supplementary Figs. 1 and 2 and Supplementary Table 2). A genetic map36 was used to anchor and order 72.7% of the assembly (461 Mb; 1,325 contigs) into 30 linkage groups corresponding to the number of chromosomes in U. pinnatifida37 (Fig. 1, Supplementary Note, Supplementary Fig. 1 and Supplementary Tables 3–5). The genomes of U. pinnatifida from China35 and Kr2015 were largely comparable in length, composition and organization (Supplementary Note). Synteny analysis revealed discrepancies between the two assemblies that could represent recombination events or artefacts resulting from how these data were assembled (Supplementary Note and Supplementary Fig. 3). We also compared the Kr2015 genome to other brown algal genomes. The Kr2015 genome was annotated using a combination of transcript- and homology-based methods (Supplementary Note, Supplementary Fig. 4 and Supplementary Table 1). The annotation pipeline predicted 20,716 complete protein-coding genes, of which 78.25% were supported by transcriptome data (Supplementary Note).
Fig. 1. Pseudochromosome-level assembly of the U. pinnatifida genome and E. siliculosus.

The outermost circle (blue) represents the density of genes in percentage of coverage in 500-kb windows. The middle circle (red) represents the density of repeated elements in percentage of coverage in 500-kb windows. The centre shows syntenic links between genes of the two species. The units (Mb) and tick marks on the axis for LG05 are common for all such axes.
The genome of U. pinnatifida Kr2015 is the largest reported thus far for brown algae. It is comprised of 52.1% (330.3 Mb) repeated elements, of which at least 19.14% are transposable elements, representing 121 Mb of the genome (Supplementary Note and Supplementary Table 6). Genomes of Laminariales are larger than those of Ectocarpales (for example, Ectocarpus siliculosus; see Supplementary Table 7). This genome expansion is driven by the differential rate of repeated element insertion (Supplementary Note and Supplementary Fig. 5). Insertion of repeated elements was homogeneous along the pseudochromosomes and resulted in a significantly (Wilcoxon rank sum test P value < 2.2 × 10−16) reduced gene density in Kr2015 when compared to E. siliculosus (Fig. 1). The traditional repeat-rich heterochromatin and gene-rich euchromatin could not be clearly differentiated in Kr2015 (refs. 38–40). Therefore, organization of brown algal chromosomes appears to be similar to that in fungi41, but different from that in plants42. This differential insertion of repeated elements does not appear to have disturbed the gene order: synteny is largely conserved between U. pinnatifida Kr2015 and E. siliculosus (Fig. 1). Despite the deep split of these two lineages 128.9–220.2 million years ago43,44, large chromosomal rearrangements are rare. Overall, 16 U. pinnatifida Kr2015 pseudochromosomes share synteny with one chromosome from E. siliculosus (Fig. 1 and Supplementary Note). The chromosome number discrepancy between E. siliculosus (28 chromosomes) and U. pinnatifida (30 chromosomes) may be explained by 4 splitting and fusion events involving 5 and 7 chromosomes, respectively (Fig. 1).
The gene inventory of U. pinnatifida is largely shared with other brown algae; however, the Laminariales common ancestor contains expanded gene families that encode nuclear-targeted proteins with transcription regulation functions (Supplementary Note). This suggests that following the split with Ectocarpales, the Laminariales may have evolved a more sophisticated control of gene expression (Supplementary Note).
Genome polymorphism across individuals
We resequenced the genomes of 41 individuals of U. pinnatifida from 9 populations in 3 categories: 2 natural kelp beds; 2 cultivated populations from the native range; and 5 introduced populations from France and New Zealand (Fig. 2a). We generated a total of 853.77 Gb of cleaned–trimmed paired-end sequence from the 41 individuals (average 20.69 Gb per individual). These reads were mapped to the reference genome assembly of U. pinnatifida. We obtained an average sequencing depth of 30.67× and average genome coverage of 94.77% (Supplementary Table 8). Using GATK45 to call variants, we identified 6,123,124 high-quality single-nucleotide polymorphisms (SNPs) and 1,130,417 high-quality insertions or deletions (indels) shared across the 9 populations (Supplementary Note and Supplementary Fig. 6). A large proportion of the variants were found in intergenic regions (53.81%) and only 3.07% were present in exons (Supplementary Fig. 7).
Fig. 2. Genetic structure according to geographic locations.

a, The sampling strategy for the study of the impact of human activities on the genome architecture and diversity of U. pinnatifida. Green box, natural populations from the native range: Goseong (Korea) and Tongyeong (Korea). Blue box, cultivated populations from the Wando (Korea) farming area harvested in 2015 and 2017. Red box, natural populations introduced in New Zealand and France, outside the native range: Lyall Bay (New Zealand), Thau (France), Roscoff (France) and Wellington Harbour (New Zealand) sampled in 1987 and 2017. b, PCA of 7,253,541 variants called in 41 individuals of U. pinnatifida. The left plot shows PC1 versus PC2; the right plot shows PC2 versus PC3. c, Maximum-likelihood phylogenetic tree reconstruction of the 9,777 high-quality SNPs shared by all 41 individuals. The support values shown near the nodes were estimated by 1,000 parametric bootstrap replications. A blue background highlights Korean populations, a red background highlights French populations and a green background highlights New Zealand populations. d, Admixture analysis showing the membership (ancestry proportion) to five identified clusters (K = 5) that best explained the overall genetic variance of the dataset.
The nuclear variant data were used to explore genetic diversity among U. pinnatifida individuals. Both principal component analysis (PCA) and phylogenetic reconstruction revealed a clear segregation of individuals according to their geographic locations (Fig. 2b,c). The admixture analysis performed with the R package LEA46 revealed consistent clusters for the number of groups (K) best explaining the genetic variance (K = 4 and 5; Extended Data Fig. 1, Supplementary Note and Supplementary Fig. 8). Only the CUL_Kr_Wando2015_4 individual was inconsistent with geographic clustering because it grouped with the Tongyeong individuals in the PCA analysis and the phylogenetic tree (Fig. 2b,c). This could be the outcome of either introgression between natural and cultivated populations, or cryptic genetic diversity within the cultivated accession (not detected here because of the limited number of individuals studied). As it had the highest level of admixture with a large number of SNPs/indels shared with Tongyeong individuals (Fig. 2d and Extended Data Fig. 1), admixture analysis supports the introgression hypothesis. This singular individual was excluded from subsequent analyses.
Extended Data Fig. 1. Admixture analysis.
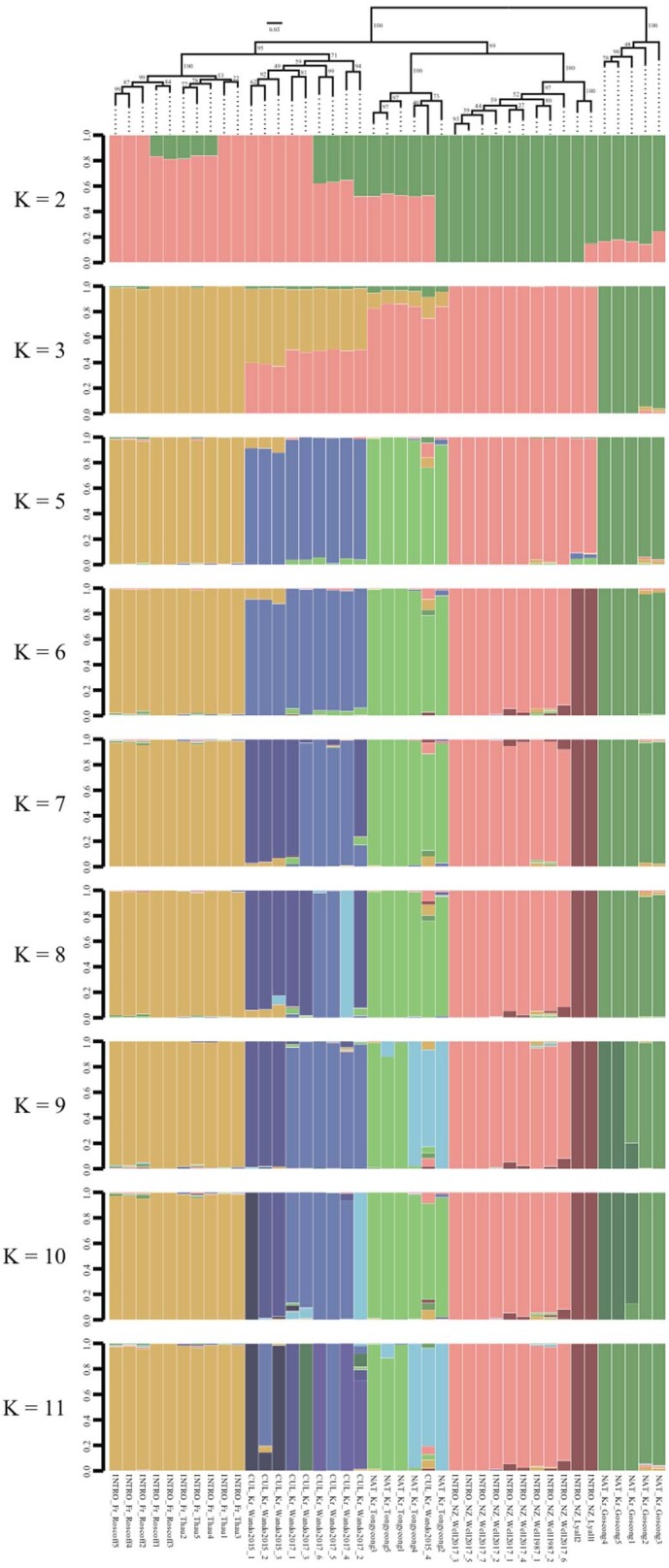
Ancestry proportion obtained with snmf for number of clusters K ranging from 2 to 11. The phylogenetic tree of Fig. 2c is reproduced on top.
Time (albeit of short duration) appears to have little to no influence, because populations remain stable over time. For instance, the individuals sampled in 1987 and 2017 in the introduced population of Wellington (New Zealand) were indistinguishable in the three analyses (Fig. 2b–d and Extended Data Fig. 1). Similar observations based on double-digest restriction-site-associated DNA sequencing have been made over about 20 generations in populations introduced in France47. We examined two introduction ranges, which had been previously reported to have distinct introduction histories48,49. Our data are in agreement with these reports: the 21 introduced individuals formed 2 distinct clusters, corresponding to France and New Zealand. The clustering of the Lyall Bay and Wellington Harbour populations was consistent with local spreading by human vectors (for example, leisure boating), as observed in France47.
Overall, population structure analysis shows that the study populations do not present a cryptic substructure, confirms the temporal genetic stability of U. pinnatifida48 and marks the French and New Zealand introductions as two independent examples of how human activity has impacted the U. pinnatifida genome.
Genomic landscape based on place of origin
To further explore the genome-wide impact of human activity, we characterized the genomic landscape in the different populations. Natural populations were characterized by high genetic diversity (mean π = 0.0044; Fig. 3a,b, Extended Data Fig. 1, Supplementary Fig. 9 and Supplementary Table 9) and high recombination rates (linkage disequilibrium (LD) half-maximum decay at 3.95 kb in natural; Fig. 3c) but relatively high homozygosity (natural mean total runs of homozygosity (ROH) length = 80.9 Mb and average ROH length = 1.13 Mb; Fig. 3d and Supplementary Table 10). Compared to natural populations, cultivated and introduced populations display contrasting features regarding their genomic landscape, for population diversity, LD and ROH. We might have expected that both cultivation and introduction processes would lead to a reduction in diversity through demographic bottleneck/founder events, associated with the introduction or the selection of few individuals; however, we did not observe such a pattern. Indeed, introduced populations of U. pinnatifida behaved as expected with low genetic diversity (France mean π = 0.0015; New Zealand mean π = 0.0022; Fig. 3a,b, Extended Data Fig. 1, Supplementary Fig. 9 and Supplementary Table 9), low recombination rates (LD half-maximum decay at 10.47 kb in New Zealand and at 27.33 kb in France; Fig. 3c) and high levels of homozygosity (France mean total ROH length = 338.5 Mb and average ROH length = 1.79 Mb; New Zealand mean total ROH length = 201.2 Mb and average ROH length = 1.08 Mb; Fig. 3d and Supplementary Table 10). In contrast, cultivated populations were characterized by high genetic diversity (cultivated mean π = 0.0040; Fig. 3a,b, Extended Data Fig. 1, Supplementary Fig. 9 and Supplementary Table 9), high recombination rates (LD half-maximum decay at 3.14 kb in cultivated; Fig. 3c) and low homozygosity (cultivated mean total ROH length = 87.9.5 Mb and average ROH length = 0.96 Mb; Fig. 3d and Supplementary Table 10).
Fig. 3. Genomic landscape in different population types.

a, Violin plot of genetic diversity estimated using π in non-overlapping 10-kb windows. b, Manhattan plot of genetic diversity (π) estimated in 250-kb windows for the natural (green), cultivated (blue), New Zealand (red) and French (yellow) populations. Local polynomial regression fittings are shown on the plots. c, LD decay in the four different types of population, with the thin line indicating the distance at which LD is one-half of its maximum. d, ROH in the 41 individuals. Natural, cultivated, New Zealand and French are shown in green, blue, red and yellow, respectively.
Influence of introduction history on the genomic landscape
When compared to those of populations from its native range, the characteristics of the French and New Zealand U. pinnatifida populations probably reflect founder events, whereby a small number of individuals were introduced to the new habitat. Such founder events are uncommon in marine introduced species, when compared to their terrestrial counterparts20. However, we have evidence of such a founder effect that is known to reduce genetic diversity and the rate of LD decay, and to increase inbreeding (here, ROH), particularly in selfing species such as U. pinnatifida47.
However, additional comparisons between the French and New Zealand introduced populations revealed different patterns between the two regions. The two French populations display a lower genetic diversity (Fig. 3a,b) and a higher LD (Fig. 3b) than the New Zealand populations. The populations introduced to New Zealand waters display properties closer to those observed in the natural population in Korea than in the populations introduced to France. These features are in agreement with the supposed introduction history and vectors in these two regions. On the basis of field and genetic studies47–51, it has been hypothesized that the introduction occurred as a result of aquaculture in France and shipping in New Zealand. In France, U. pinnatifida would have been first introduced in the Thau Lagoon (Thau population here) with Pacific oyster imports from Asia, and then transported to Brittany (that is, the Roscoff population). Two sequential founder events thus probably occurred, from the same source in the native range49. Conversely, repeated introductions probably occurred with shipping in New Zealand, leading to a moderate decrease in genetic diversity when compared to the native range and the French populations, as found using mitochondrial haplotype analysis49. This scenario was further supported by individuals sampled in Wellington in 1987 at the time of the first report of this alga in New Zealand52. These seaweeds had a genetic diversity that was slightly higher than in the French population (Supplementary Fig. 10a and Supplementary Table 9), and the length of the ROH was shorter than in the French population (Supplementary Fig. 10b). Finally, the decrease of ROH length in 30 years (about 60 generations) suggests that repetitive introductions provided the potential for admixture in New Zealand.
Effect of cultivation on the genomic landscape
The cultivation process, through selective breeding, is expected to create a genetic bottleneck, resulting in cultivars that have low diversity and a suppressed recombination rate. The cultivated populations of U. pinnatifida in Korea, however, deviated from these predictions, with genetic diversity (mean π = 0.0040; Supplementary Table 9) and LD disequilibrium decay (LD half-maximum decay at 3.14 kb; Fig. 3c) comparable to those of natural populations (Fig. 3a–c). Interestingly, in France, cultivated populations of U. pinnatifida have a genetic diversity that is lower than that of natural populations (for example, twofold to threefold lower47) in accordance with expectations of the cultivation process (with some exceptions53). This observed discrepancy might be explained by the difference in the scale of the cultivation in these two countries. In France, U. pinnatifida cultivation remains limited to a few farms, whereas in Korea, its culture averages 0.5 million wet weight tonnes annually24–26. Owing to this large scale, the fertilization of culture ropes is carried out in large indoor pools in which multiple sporangia of individuals with valued phenotypes are placed together (Extended Data Fig. 2). Frequently, individuals from different natural populations are mixed with cultivated individuals from previous years. In this artificial environment, the mixing of genetically distinct zoospores is favoured, resulting in the observed high diversity and low LD; both of which are comparable to those of native natural populations. Furthermore, this cultivation methodology prevents the naturally high selfing rate of natural U. pinnatifida populations47. This selfing rate probably results from the low motility and short life span of the zoospores of this species54,55, traits that are mitigated by the large fertilization pool present in the water circulation system. This results in cultivated individuals having the highest level of heterozygosity and the lowest coverage of ROH in our study, even lower than in natural populations. Furthermore, these low values are consistent across all cultivated individuals (Fig. 3d and Extended Data Fig. 3), supporting the idea that they are a consequence of farming practices, the breeding method in particular.
Extended Data Fig. 2. Large scale cultivation facility of Undaria pinnatifida in Korea.

Indoors pools where multiple sporangia of Undaria pinnatifida are placed to enable recruitment on culture ropes (left).
Extended Data Fig. 3. Heterozygosity and run of homozygosity.
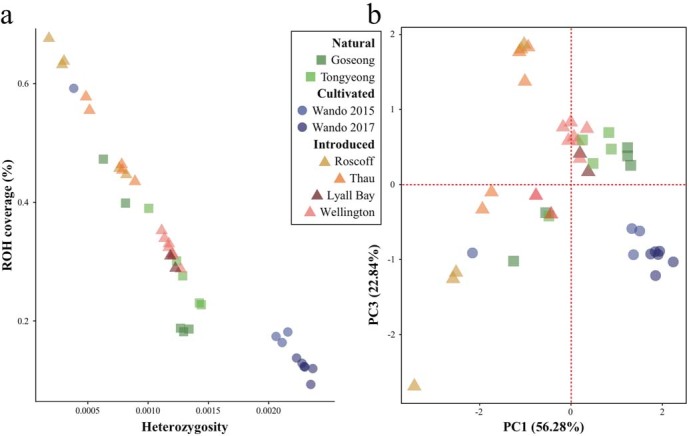
(a) Average genome-wide level of heterozygosity in the individuals as a function of the coverage of run of Homozygosity (ROH) in the genomes of the 41 individuals. (b) Principal Component Analysis (PCA) calculated on the number of ROH, total length of ROH and average length of ROH of each of the 41 individuals.
This unexpected genomic landscape resulting from large-scale cultivation of U. pinnatifida could be of great interest for conservation biologists. U. pinnatifida populations have been declining in its native range56, as have kelp forests globally57. It is therefore clear that conservation efforts are needed to protect these valuable marine species. In this regard, our analysis suggests that if kelp cultivation is designed to maintain high genetic variation, then farmed individuals could act as reservoirs of evolutionary potential. U. pinnatifida provides a model for such an approach. Such genetic rescue approaches have previously been used in allogamous mammals58,59.
Catalogue of regions under putative selection
The distinct genetic structures of U. pinnatifida populations (Fig. 2) could be explained by positive selection on many genomic regions. However, the history of these populations (that is, bottlenecks and reduced population effective sizes (that is, neutral evolutionary processes60)) could also produce patterns of genetic diversity that resemble selective sweeps. Furthermore, the putative functions of loci implicated in putative selective sweeps can be overinterpreted61, and in the absence of other evidence (for example, expression level and phenotype), they should be considered with caution. We identified putative selection signals associated with cultivated or introduced populations of U. pinnatifida by using a decorrelated composite of multiple signals (DCMS)62 method on three different statistics calculated in 50-kb windows along the genome (Fig. 4 and Supplementary Note).
Fig. 4. Signatures of selection in Undaria pinnatifida.

a, A close-up of population genetic statistics (FST, reduction of diversity (ROD) and Tajima’s D) in the linkage group LG16. The blue box highlights the genomic region under putative positive selection, black boxes represent annotated protein-coding genes encoded in this genomic region, and the red box represents a mannitol 1-phosphate dehydrogenase coding gene. b, Manhattan plot of the DCMS score calculated in non-overlapping 50-kb windows in the comparison between natural and cultivated populations. c, Manhattan plot of the DCMS score calculated in non-overlapping 50-kb windows in the comparison between individuals sampled in Wellington Harbour in 1987 and individuals sampled in Wellington Harbour in 2017.
A comparison of natural and cultivated individuals from the native range revealed that the 508 genes encoded in the 224 genomic windows identified to be under selection (DCMS score P value < 0.025) were enriched in several biological processes such as glycolipid biosynthesis and cytokinetic process (Supplementary Note and Supplementary Tables 11 and 12). Intentional selection for increased yield in U. pinnatifida culture could explain this enrichment in genes related to carbohydrate biosynthesis. These genes have functions in the alginate, mannitol and sulfate fucan pathways (Supplementary Table 11). However, in total, these pathways contain >150 genes, greatly exceeding the number of genes under putative selection (8 in total). Genomic analysis of Saccharina japonica also identified genes involved in carbohydrate metabolism (for example, fructose-1,6-bisphosphate aldolase) in genomic regions under putative selection63, but interestingly, different pathways were recovered in the two species. More generally, the biological process selected during cultivation in the two species differed greatly (Supplementary Note). This could indicate that the traits of interest selected by the farmers differ between the two species or that the cultivation processes are at different levels of completion.
The important morphological differences between natural and cultivated individuals suggest that developmental processes have diverged in these two groups (Extended Data Fig. 4). However, gene families that might play a role in development, such as the Cupin-like, C2H2 zinc-finger or imm upregulated genes, were absent from the regions under putative selection despite the large number of copies encoded in the genome of U. pinnatifida (19, 34 and 10, respectively). Similarly, with the exception of one copy, peptidase s8 and s53, which were proposed as blade length and width quantitative trait loci in S. japonica64, were not detected by the DCMS analysis (Supplementary Table 11). The high phenotypic plasticity observed in brown algae65,66, and in U. pinnatifida in particular67, could help explain this absence and support a polygenic basis for the phenotypic differences, perhaps underpinned by differential regulation and post-transcriptional modification. In this context, the enrichment of a variety of genes having regulatory and kinase activities could be linked to selection of regulatory networks underlying development (Supplementary Table 11). Exploratory transcriptome analysis of genes within regions under positive selection revealed that they could potentially have different expression levels when compared to genes of similar functions encoded elsewhere in the genome (Supplementary Note, Supplementary Fig. 11 and Supplementary Table 12). However, these are preliminary results (Supplementary Note) and a more comprehensive transcriptomic analysis is needed to better understand the effect of positive selection on gene expression in the cultivated U. pinnatifida.
Extended Data Fig. 4. Morphotypes of Undaria pinnatifida in Korea.

Observed phenotypes of Undaria pinnatifida individuals collected in (a) a farm in Wando (Korea) and in (b) a natural population in Tongyeong (Korea).
In contrast to the cultivated versus natural population comparisons, the analysis across 30–60 generations (about 1–2 generations per year55) in Wellington Harbour between 1987 and 2017 did not reveal the enrichment of a particular biological function (Supplementary Note and Supplementary Tables 13 and 14). The high variance in allelic frequency resulting from the founding effect during the initial introduction and the insufficient time of 30–60 generations for selection to operate on standing genetic variation could indicate that the signals detected by our analysis result mostly from neutral effects. It is also possible that the relatively wide ecological niche of U. pinnatifida and the comparable environments in Korea and New Zealand have suppressed divergence. For example, none of the genes involved in defence mechanisms against infection, such as the vanadium-dependent bromoperoxidases and iodoperoxidases68 or the LRR-GTPases of the ROCO family69, is encoded in the regions under putative selection. However, a number of the genes under selection appeared to have roles in stress, homeostasis and membrane functions, suggesting adaptation to the New Zealand environment (Supplementary Table 14).
Conclusion
The generation of a high-quality genome assembly combined with resequencing data from 41 individuals provides a detailed picture of the effect of human activity on genome evolution in U. pinnatifida. Our results strongly support the introduction scenario proposed for France and New Zealand and reveal how genome architecture is shaped by introduction history. For individuals in the native range, our analysis revealed unexpected effects of cultivation on the genomic landscape and provided insights into how natural selection may impact these individuals. Furthermore, our study offers a foundation on which future analyses of dispersal and adaptation in new environments can be designed.
In the future, targeted sampling and an explicit experimental design are needed to better connect genetic and phenotypic information. In particular, quantitative trait locus mapping in crosses between cultivars from breeding lines and natural individuals could help elucidate the domestication process in U. pinnatifida. In the introduced populations, phenotypic comparisons and environmental measurements in native and introduced sites could allow a genome-wide association study to identify the genetic variants underlying regional phenotypic differences. In particular, the connectivity between the cultivated and natural populations should be assessed to test the role of escaped cultivars in the generation of genetic novelty in nature.
Methods
Algal material, genome sequencing and annotation
The U. pinnatifida individual used for reference genome sequencing was collected from a longline rope in a culture farm in Wando, Korea on 23 January 2015. High-quality DNA was extracted using a modified cetyl trimethylammonium bromide method (Supplementary Note). According to the instructions of the manufacturers, Illumina paired-end sequencing (PE: 101 bp) and long PacBio reads were sequenced and processed for error correction and quality filtration (Supplementary Note). The sequencing reads were assembled and polished, and finally superscaffolding was performed using data from Shan et al.36. The final assembly of the genome (Kr2015) was assessed by alignment of the proteins encoded in the genomes of E. siliculosus and S. japonica and core eukaryotic genes (Supplementary Table 5 and Supplementary Note).
Transposable elements and repeats were masked in the Kr2015 assembly using a combination of RepeatModeler and RepeatMasker, and their insertion time was estimated (Supplementary Note). For the gene prediction, a collection of proteins from seven species was mapped on the masked Kr2015 assembly and eight complementary DNA libraries were generated and sequenced (Supplementary Note). Genes were predicted using a homology-based and transcriptome-based in-house pipeline and the predicted genes were functionally annotated (Supplementary Note).
Comparative analysis
Orthologous analysis was conducted on the genome data from a selection of 19 taxa representing the diversity of the stramenopiles. The sequences of orthologous single genes found in all species were aligned and used to reconstruct a maximum-likelihood phylogenetic tree that was used as the backbone of a Dollo parsimony analysis (Supplementary Note). Syntenic analyses were conducted with the Kr2015 gene model against the gene model of the Chinese assembly of U. pinnatifida and the gene model of E. siliculosus (Supplementary Note)
Additional U. pinnatifida genomes and variant calling
Genomic DNA for a total of 41 U. pinnatifida individuals sampled in Korea (natural and cultivated populations), France (introduced populations) and New Zealand (introduced populations) was extracted, and for each individual, ~30× coverage of short-read data were generated (Supplementary Table 8, Supplementary Note). For each individual, after trimming of sequencing adapters and low-quality bases, the reads were mapped in the Kr2015 genome. These mapping data were used to call variants for each individual before all variants were combined to form the primary variant dataset containing 25,414,685 variants (21,619,805 SNPs and 3,794,880 indels). The variants were filtered for quality thresholds, allele frequency and genotyping rate to produce a final dataset of 7,253,541 (6,123,124 SNPs and 1,130,417 indels) variants.
Population genomics
The population structure was investigated with PCA, phylogenetic tree reconstruction and admixture analyses (Supplementary Note). For each type of population, the expected heterozygosity (He), π, fixation index (FIS), LD (estimated from r2) and ROH (that is, chromosome fragments within a single individual that have shared parental ancestry) were estimated (Supplementary Note). Detection of regions affected by selection was conducted using a combination of statistics calculated in non-overlapping 50-kb windows: reduction of diversity, delta Tajima’s D and population differentiation (FST). For each window and each statistic, a P value was determined and these were used to calculate the DCMS value of each window (Supplementary Note).
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Supplementary Figs. 1–17 and Note.
Supplementary Tables 1–19.
Acknowledgements
This study was supported by the Collaborative Genome Program of the Korea Institute of Marine Science and Technology Promotion funded by the Ministry of Oceans and Fisheries (20180430), the National Research Foundation of Korea (NRF-2017R1A2B3001923, 2020R1C1C1008173) and the Next-generation BioGreen21 Program (PJ01389003) from the Rural Development Administration, Korea. Sampling in the Wando farms was supported by the National Institute of Fisheries Sciences (R2020004, to I.K.H.). Sampling in New Zealand was supported by NIWA SSIF funds to W.N. We thank the staff of the Herbarium of the Museum of New Zealand Te Papa Tongarewa for access to specimens. Sampling in France, and research by F.V. on U. pinnatifida, benefited from the support of the French National Research Agency (ANR) with regards to the IDEALG project (ANR-10-BTBR-04). We thank F. Riquet and A. Simon for the sampling in the Thau Lagoon. This is publication ISE-M 2020-295.
Extended data
Author contributions
L.G., F.V. and H.S.Y. designed the projects. L.G., J.W.C., I.K.H., W.N. and F.V. collected the samples. L.G. and J.H.Y. conducted the experiments (DNA and RNA extractions). L.G., Y.S., D.B., F.V. and H.S.Y. analysed and interpreted the data. L.G., F.V. and H.S.Y. wrote the draft and all authors read the manuscript.
Data availability
The raw sequencing reads were deposited in the National Center for Biotechnology Information database under the BioProject accession code PRJNA646283. The assemblies, gene model and functional annotation were deposited in the Marine Genome Information Center (http://www.magic.re.kr/) database under the accession code MA00358.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information Nature Ecology & Evolution thanks Thomas Mock and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
is available for this paper at 10.1038/s41559-020-01378-9.
Supplementary information
is available for this paper at 10.1038/s41559-020-01378-9.
References
- 1.Kettlewell, B. The Evolution of Melanism: the Study of a Recurring Necessity; with Special Reference to Industrial Melanism in the Lepidoptera (Clarendon Press, 1973).
- 2.Stockwell CA, Hendry AP, Kinnison MT. Contemporary evolution meets conservation biology. Trends Ecol. Evol. 2003;18:94–101. [Google Scholar]
- 3.Slabbekoorn H, Ripmeester EAP. Birdsong and anthropogenic noise: implications and applications for conservation. Mol. Ecol. 2008;17:72–83. doi: 10.1111/j.1365-294X.2007.03487.x. [DOI] [PubMed] [Google Scholar]
- 4.Fan W, et al. Sequencing of Chinese castor lines reveals genetic signatures of selection and yield-associated loci. Nat. Commun. 2019;10:3418. doi: 10.1038/s41467-019-11228-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshida GM, et al. Genome-wide patterns of population structure and linkage disequilibrium in farmed Nile tilapia (Oreochromis niloticus) Front. Genet. 2019;10:745. doi: 10.3389/fgene.2019.00745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang G-D, Xie H-B, Peng M-S, Irwin D, Zhang Y-P. Domestication genomics: evidence from animals. Annu. Rev. Anim. Biosci. 2014;2:65–84. doi: 10.1146/annurev-animal-022513-114129. [DOI] [PubMed] [Google Scholar]
- 7.Stein JC, et al. Genomes of 13 domesticated and wild rice relatives highlight genetic conservation, turnover and innovation across the genus Oryza. Nat. Genet. 2018;50:285–296. doi: 10.1038/s41588-018-0040-0. [DOI] [PubMed] [Google Scholar]
- 8.Lee Y, et al. Genome-wide divergence among invasive populations of Aedes aegypti in California. BMC Genom. 2019;20:204. doi: 10.1186/s12864-019-5586-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Puzey J, Vallejo-Marín M. Genomics of invasion: diversity and selection in introduced populations of monkeyflowers (Mimulus guttatus) Mol. Ecol. 2014;23:4472–4485. doi: 10.1111/mec.12875. [DOI] [PubMed] [Google Scholar]
- 10.Pyšek P, Richardson DM. Invasive species, environmental change and management, and health. Annu. Rev. Environ. Resour. 2010;35:25–55. [Google Scholar]
- 11.Larson G, Fuller DQ. The evolution of animal domestication. Annu. Rev. Ecol. Evol. Syst. 2014;45:115–136. [Google Scholar]
- 12.Meyer RS, Purugganan MD. Evolution of crop species: genetics of domestication and diversification. Nat. Rev. Genet. 2013;14:840–852. doi: 10.1038/nrg3605. [DOI] [PubMed] [Google Scholar]
- 13.Devore, I. & Lee, R. B. Man the Hunter (Aldine de Gruyter, 1999).
- 14.Hill WG, Kirkpatrick M. What animal breeding has taught us about evolution. Annu. Rev. Ecol. Evol. Syst. 2010;41:1–19. [Google Scholar]
- 15.Kingsbury, N. Hybrid: The History and Science of Plant Breeding (Univ. Chicago Press, 2009).
- 16.Van Driesche, J. & Van Driesche, R. Nature Out of Place: Biological Invasions in the Global Age (Island Press, 2000).
- 17.Pimentel D, et al. Economic and environmental threats of alien plant, animal, and microbe invasions. Agric. Ecosyst. Environ. 2001;84:1–20. [Google Scholar]
- 18.Rius M, Darling JA. How important is intraspecific genetic admixture to the success of colonising populations? Trends Ecol. Evol. 2014;29:233–242. doi: 10.1016/j.tree.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 19.McFarlane SE, Pemberton JM. Detecting the true extent of introgression during anthropogenic hybridization. Trends Ecol. Evol. 2019;34:315–326. doi: 10.1016/j.tree.2018.12.013. [DOI] [PubMed] [Google Scholar]
- 20.Viard F, David P, Darling JA. Marine invasions enter the genomic era: three lessons from the past, and the way forward. Curr. Zool. 2016;62:629–642. doi: 10.1093/cz/zow053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sax D, et al. Ecological and evolutionary insights from species invasions. Trends Ecol. Evol. 2007;22:465–471. doi: 10.1016/j.tree.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 22.Zenni RD, et al. Evolutionary dynamics of tree invasions: complementing the unified framework for biological invasions. AoB Plants. 2017;9:plw085. doi: 10.1093/aobpla/plw085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Viard, F., Riginos, C. & Bierne, N. Anthropogenic hybridization at sea: three evolutionary questions relevant to invasive species management. Phil. Trans. R. Soc. B10.1098/rstb.2019.0547 (2020). [DOI] [PMC free article] [PubMed]
- 24.The State of World Fisheries and Aquaculture (FAO, 2020).
- 25.Yamanaka R, Akiyama K. Cultivation and utilization of Undaria pinnatifida (wakame) as food. J. Appl. Phycol. 1993;5:249–253. [Google Scholar]
- 26.Hwang EK, Yotsukura N, Pang SJ, Su L, Shan TF. Seaweed breeding programs and progress in eastern Asian countries. Phycologia. 2019;58:484–495. [Google Scholar]
- 27.Pang S, Hu X, Wu C, Hirosawa A, Ohno M. Intraspecific crossings of Undaria pinnatifida (Harv.) Sur.—a possible time-saving way of strain selection. Chin. J. Oceanol. Limnol. 1997;15:227–235. [Google Scholar]
- 28.Shan TF, Pang SJ, Li J, Gao SQ. Breeding of an elite cultivar Haibao No. 1 of Undaria pinnatifida (Phaeophyceae) through gametophyte clone crossing and consecutive selection. J. Appl. Phycol. 2016;28:2419–2426. [Google Scholar]
- 29.Perez R, Lee JY, Juge C. Observations sur la biologie de l’algue japonaise Undaria pinnatifida (Harvey) Suringar introduite accidentellement dans l’étang de Thau. Sci. Pêche. 1981;315:1–12. [Google Scholar]
- 30.James K, Kibele J, Shears NT. Using satellite-derived sea surface temperature to predict the potential global range and phenology of the invasive kelp Undaria pinnatifida. Biol. Invasions. 2015;17:3393–3408. [Google Scholar]
- 31.Epstein G, Smale DA. Undaria pinnatifida: a case study to highlight challenges in marine invasion ecology and management. Ecol. Evol. 2017;7:8624–8642. doi: 10.1002/ece3.3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shan T, et al. Genetic analysis of a recently established Undaria pinnatifida (Laminariales: Alariaceae) population in the northern Wadden Sea reveals close proximity between drifting thalli and the attached population. Eur. J. Phycol. 2019;54:154–161. [Google Scholar]
- 33.Qiu J, et al. Genome re-sequencing suggested a weedy rice origin from domesticated indica-japonica hybridization: a case study from southern China. Planta. 2014;240:1353–1363. doi: 10.1007/s00425-014-2159-2. [DOI] [PubMed] [Google Scholar]
- 34.Paterson AH, et al. The evolution of an invasive plant, Sorghum halepense L. (‘Johnsongrass’) Front. Genet. 2020;11:317. doi: 10.3389/fgene.2020.00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shan T, et al. First genome of the brown alga Undaria pinnatifida: chromosome-level assembly using PacBio and Hi-C technologies. Front. Genet. 2020;11:140. doi: 10.3389/fgene.2020.00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shan T, Pang S, Li J, Li X, Su L. Construction of a high-density genetic map and mapping of a sex-linked locus for the brown alga Undaria pinnatifida (Phaeophyceae) based on large scale marker development by specific length amplified fragment (SLAF) sequencing. BMC Genom. 2015;16:902. doi: 10.1186/s12864-015-2184-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yabu H, Yasui H, Notoya M. Chromosome numbers of Undaria pinnatifida f. distans. Bull. Fac. Fish. Hokkaido Univ. 1988;39:6–13. [Google Scholar]
- 38.Verma, R. S. Heterochromatin: Molecular and Structural Aspects (Cambridge Univ. Press 1988).
- 39.Zhang X, et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell. 2006;126:1189–1201. doi: 10.1016/j.cell.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 40.Vergara Z, et al. Retrotransposons are specified as DNA replication origins in the gene-poor regions of Arabidopsis heterochromatin. Nucleic Acids Res. 2017;45:8358–8368. doi: 10.1093/nar/gkx524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Castanera R, Borgognone A, Pisabarro AG, Ramírez L. Biology, dynamics, and applications of transposable elements in basidiomycete fungi. Appl. Microbiol. Biotechnol. 2017;101:1337–1350. doi: 10.1007/s00253-017-8097-8. [DOI] [PubMed] [Google Scholar]
- 42.Kim S, et al. Genome sequence of the hot pepper provides insights into the evolution of pungency in Capsicum species. Nat. Genet. 2014;46:270–278. doi: 10.1038/ng.2877. [DOI] [PubMed] [Google Scholar]
- 43.Silberfeld T, et al. A multi-locus time-calibrated phylogeny of the brown algae (Heterokonta, Ochrophyta, Phaeophyceae): investigating the evolutionary nature of the ‘brown algal crown radiation’. Mol. Phylogenet. Evol. 2010;56:659–674. doi: 10.1016/j.ympev.2010.04.020. [DOI] [PubMed] [Google Scholar]
- 44.Kawai H, Hanyuda T, Draisma SGA, Wilce RT, Andersen RA. Molecular phylogeny of two unusual brown algae, Phaeostrophion irregulare and Platysiphon glacialis, proposal of the Stschapoviales ord. nov. and Platysiphonaceae fam. nov., and a re-examination of divergence times for brown algal orders. J. Phycol. 2015;51:918–928. doi: 10.1111/jpy.12332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DePristo MA, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frichot E, François O. LEA: an R package for landscape and ecological association studies. Methods Ecol. Evol. 2015;6:925–929. [Google Scholar]
- 47.Guzinski J, Ballenghien M, Daguin-Thiébaut C, Lévêque L, Viard F. Population genomics of the introduced and cultivated Pacific kelp Undaria pinnatifida: marinas—not farms—drive regional connectivity and establishment in natural rocky reefs. Evol. Appl. 2018;11:1582–1597. doi: 10.1111/eva.12647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Uwai S, et al. Genetic diversity in Undaria pinnatifida (Laminariales, Phaeophyceae) deduced from mitochondria genes - origins and succession of introduced populations. Phycologia. 2006;45:687–695. [Google Scholar]
- 49.Voisin M, Engel CR, Viard F. Differential shuffling of native genetic diversity across introduced regions in a brown alga: aquaculture vs. maritime traffic effects. Proc. Natl Acad. Sci. USA. 2005;102:5432–5437. doi: 10.1073/pnas.0501754102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Floc’h JY, Pajot R, Wallentinus I. The Japanese brown alga Undaria pinnatifida on the coast of France and its possible establishment in European waters. ICES J. Mar. Sci. 1991;47:379–390. [Google Scholar]
- 51.South PM, Floerl O, Forrest BM, Thomsen MS. A review of three decades of research on the invasive kelp Undaria pinnatifida in Australasia: an assessment of its success, impacts and status as one of the world’s worst invaders. Mar. Environ. Res. 2017;131:243–257. doi: 10.1016/j.marenvres.2017.09.015. [DOI] [PubMed] [Google Scholar]
- 52.Hay CH, Luckens PA. The Asian kelp Undaria pinnatifida (Phaeophyta: Laminariales) found in a New Zealand harbour. N. Z. J. Bot. 1987;25:329–332. [Google Scholar]
- 53.Salamon M, Lévêque L, Ballenghien M, Viard F. Spill-back events followed by self-sustainment explain the fast colonization of a newly built marina by a notorious invasive seaweed. Biol. Invasions. 2020;22:1411–1429. [Google Scholar]
- 54.Forrest BM, Brown SN, Taylor MD, Hurd CL, Hay CH. The role of natural dispersal mechanisms in the spread of Undaria pinnatifida (Laminariales, Phaeophyceae) Phycologia. 2000;39:547–553. [Google Scholar]
- 55.Schiel DR, Thompson GA. Demography and population biology of the invasive kelp Undaria pinnatifida on shallow reefs in southern New Zealand. J. Exp. Mar. Biol. Ecol. 2012;434–435:25–33. [Google Scholar]
- 56.Tamaki H, Kusaka K, Fukuda M, Arai S, Muraoka D. Undaria pinnatifida habitat loss in relation to sea urchin grazing and water flow conditions, and their restoration effort in Ogatsu Bay, Japan. J. Water Environ. Technol. 2009;7:201–213. [Google Scholar]
- 57.Wernberg, T., Krumhansl, K., Filbee-Dexter, K. & Pedersen, M. F. in World Seas: an Environmental Evaluation (ed. Sheppard, C.) 57–78 (Elsevier, 2019).
- 58.Johnson WE, et al. Genetic restoration of the Florida panther. Science. 2010;329:1641–1645. doi: 10.1126/science.1192891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Whiteley AR, Fitzpatrick SW, Funk WC, Tallmon DA. Genetic rescue to the rescue. Trends Ecol. Evol. 2015;30:42–49. doi: 10.1016/j.tree.2014.10.009. [DOI] [PubMed] [Google Scholar]
- 60.Jensen JD, Kim Y, DuMont VB, Aquadro CF, Bustamante CD. Distinguishing between selective sweeps and demography using DNA polymorphism data. Genetics. 2005;170:1401–1410. doi: 10.1534/genetics.104.038224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pavlidis P, Jensen JD, Stephan W, Stamatakis A. A critical assessment of storytelling: gene ontology categories and the importance of validating genomic scans. Mol. Biol. Evol. 2012;29:3237–3248. doi: 10.1093/molbev/mss136. [DOI] [PubMed] [Google Scholar]
- 62.Ma Y, et al. Properties of different selection signature statistics and a new strategy for combining them. Heredity. 2015;115:426–436. doi: 10.1038/hdy.2015.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ye N, et al. Saccharina genomes provide novel insight into kelp biology. Nat. Commun. 2015;6:6986. doi: 10.1038/ncomms7986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang X, et al. High-density SNP-based QTL mapping and candidate gene screening for yield-related blade length and width in Saccharina japonica (Laminariales, Phaeophyta) Sci. Rep. 2018;8:13591. doi: 10.1038/s41598-018-32015-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Demes KW, Graham MH, Suskiewicz TS. Phenotypic plasticity reconciles incongruous molecular and morphological taxonomies: the giant kelp, Macrocystis (Laminariales, Phaeophyceae), is a monospecific genus. J. Phycol. 2009;45:1266–1269. doi: 10.1111/j.1529-8817.2009.00752.x. [DOI] [PubMed] [Google Scholar]
- 66.Charrier B, Le Bail A, de Reviers B. Plant Proteus: brown algal morphological plasticity and underlying developmental mechanisms. Trends Plant Sci. 2012;17:468–477. doi: 10.1016/j.tplants.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 67.Shibneva SY, Skriptsova AV, Shan TF, Pang SJ. The different morphs of Undaria pinnatifida (Phaeophyceae, Laminariales) in Peter the Great Bay (Sea of Japan) are phenotypic variants: direct evidence. J. Appl. Phycol. 2013;25:1909–1916. [Google Scholar]
- 68.Küpper FC, Carrano CJ. Key aspects of the iodine metabolism in brown algae: a brief critical review. Metallomics. 2019;11:756–764. doi: 10.1039/c8mt00327k. [DOI] [PubMed] [Google Scholar]
- 69.Zambounis A, Elias M, Sterck L, Maumus F, Gachon CMM. Highly dynamic exon shuffling in candidate pathogen receptors. What if brown algae were capable of adaptive immunity? Mol. Biol. Evol. 2012;29:1263–1276. doi: 10.1093/molbev/msr296. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figs. 1–17 and Note.
Supplementary Tables 1–19.
Data Availability Statement
The raw sequencing reads were deposited in the National Center for Biotechnology Information database under the BioProject accession code PRJNA646283. The assemblies, gene model and functional annotation were deposited in the Marine Genome Information Center (http://www.magic.re.kr/) database under the accession code MA00358.