

ABSTRACT
Meiotic recombination forms crossovers important for proper chromosome segregation and offspring viability. This complex process involves many proteins acting at each of the multiple steps of recombination. Recombination initiates by formation of DNA double-strand breaks (DSBs), which in the several species examined occur with high frequency at special sites (DSB hotspots). In Schizosaccharomyces pombe, DSB hotspots are bound with high specificity and strongly activated by linear element (LinE) proteins Rec25, Rec27 and Mug20, which form colocalized nuclear foci with Rec10, essential for all DSB formation and recombination. Here, we test the hypothesis that the nuclear localization signal (NLS) of Rec10 is crucial for coordinated nuclear entry after forming a complex with other LinE proteins. In NLS mutants, all LinE proteins were abundant in the cytoplasm, not the nucleus; DSB formation and recombination were much reduced but not eliminated. Nuclear entry of limited amounts of Rec10, apparently small enough for passive nuclear entry, can account for residual recombination. LinE proteins are related to synaptonemal complex proteins of other species, suggesting that they also share an NLS, not yet identified, and undergo protein complex formation before nuclear entry.
KEY WORDS: Meiotic recombination, Linear element proteins, Nuclear localization signal, S. pombe, DSB hotspot determinants, Synaptonemal complex proteins
Summary: We report the first identification of a nuclear localization signal for the meiotic recombination-promoting complex, the linear elements of fission yeast, related to the synaptonemal complex of other species.
INTRODUCTION
In sexually reproducing species, one generation gives rise to the next by formation of haploid gametes from diploid somatic cells; fusion of two gametes forms a diploid of the next generation. Gamete formation (meiosis) requires the accurate segregation of each pair of nearly identical chromosomes, one received from each of the parents. These chromosomes (homologs) replicate to form sister chromatids, align with each other, and exchange parts to form a physical connection (a crossover) holding the two homologs together. Along with sister chromatid cohesion, crossovers allow tension to form when the homologs begin to segregate from each other, as required for formation of viable gametes. In the absence of crossovers, and thus tension, homologs move nearly randomly in most species tested and often produce gametes with an improper (aneuploid) set of chromosomes that gives rise to inviable or disabled progeny. Meiotic crossovers are thus essential for the reproductive success of most species.
Formation of crossovers occurs by homologous recombination, which also generates novel combinations of genetic alleles to aid evolution of the species. During meiosis, recombination in the several species examined is initiated by the formation of DNA double-strand breaks (DSBs; reviewed in Nambiar et al., 2019); repair of a DSB with an intact homolog can generate a crossover, that is, reciprocal exchange of homologous chromosome sections flanking the DSB site. In the few species examined, DSBs do not occur at random across the genome; rather, they occur at higher than genome-average frequency at special sites, called DSB hotspots (Nambiar et al., 2019). Hotspots are determined by a complex interplay of DNA sequence, histone modification and binding of special proteins (Keeney et al., 2014). In the fission yeast Schizosaccharomyces pombe, the linear element (LinE) proteins Rec25, Rec27, and Mug20 bind DSB hotspots with remarkably high specificity and are required for formation of most DSBs at most hotspots (Fowler et al., 2013). These hotspot determinants act with another LinE protein Rec10, which is required for essentially all DSB formation and recombination, both at hotspots and in the intervening DSB cold regions (Lin and Smith, 1995; Ellermeier and Smith, 2005; Fowler et al., 2013). In the cases tested, these four proteins colocalize in the nucleus and form microscopically visible dots (foci); deletion of any one gene reduces or eliminates foci of the others, indicating that the four LinE proteins form a complex in the nucleus (Davis et al., 2008; Estreicher et al., 2012; Fowler et al., 2013). Other analyses, such as co-immunoprecipitation and yeast two-hybrid assays, also indicate that the LinE proteins form a complex (Estreicher et al., 2012).
Previous studies have shown special features of the LinE proteins. Three of them, Rec25, Rec27 and Mug20, contain only 125–151 amino acids, whereas Rec10 contains 791 amino acids (Lin and Smith, 1995; Martín-Castellanos et al., 2005; Estreicher et al., 2012). LinE proteins are related to the synaptonemal complex (SC) proteins of other species. Over limited regions, Rec10 shares amino acid sequence similarity with the SC protein Red1 of the budding yeast Saccharomyces cerevisiae (Lorenz et al., 2004). Rec27 and Mug20 share amino acid sequence similarity with the SC proteins SYP-2 and DDL-1, respectively, of the roundworm Caenorhabditis elegans (Fowler et al., 2013). SC proteins hold homologs in alignment from one end to the other, whereas LinE proteins appear by electron microscopy of nuclear spreads to align chromosomes over shorter distances, forming ‘linear elements’ (Bähler et al., 1993). In living cells, however, S. pombe meiotic chromosomes also appear aligned side-by-side from one end to the other, as seen by light microscopy of the sister chromatid cohesin Rec8 (Ding et al., 2016). The LinE complexes also form elongated structures mid-way through meiosis, as seen by light microscopy of living cells (Chuang, Y.-C. and G.R.S., unpublished observations). The S. pombe end-to-end alignment may be more fragile than the SC-mediated alignment of other species and partially fall apart upon opening of cells and spreading the nuclear contents for microscopic examination. The recombination deficiency of both LinE and SC mutants, and the sensitivity of both SC and LinE proteins to the chaotropic agent hexanediol, also suggest a similarity in their cellular functions and structures (Page and Hawley, 2004; Cromie and Smith, 2008; Chuang, Y.-C. and G.R.S., unpublished observations).
Rec10 has a set of amino acids potentially required for entry of the protein into the nucleus, where it is observed and acts (Lorenz et al., 2004; Spirek et al., 2010; Fowler et al., 2013). This nuclear localization signal (NLS) is predicted to bind to a protein complex involved in nuclear entry of many substrate proteins (reviewed in Soniat and Chook, 2015). Here, we experimentally identify a two-part NLS in Rec10. Our results show that its alteration or deletion blocks nuclear localization not only of Rec10 but also of other LinE proteins. Our data indicate that the LinE proteins form a complex even before nuclear entry, and that the other LinE proteins are cargos of Rec10. Without the Rec10 NLS, other LinE proteins form aberrant structures in the cytoplasm. The similarity between S. pombe LinE proteins and the SC of other species suggests that one of the SC proteins may also have an NLS, to our knowledge not shown in the SC of any species, and may also form a complex prior to nuclear entry.
RESULTS
Identifying the putative NLS of Rec10
Rec10 carries a predicted bipartite NLS at amino acids 497–500 and 516–519 (http://nls-mapper.iab.keio.ac.jp/cgi-bin/NLS_Mapper_form.cgi). Other programs, such as PROSITE, CAST, NLStradamus or NucPred, also predict an NLS in this region. This region is composed of clusters of positively charged amino acids, mostly lysine (K) and arginine (R). We designate amino acids 497–500 (KRKK) as site A, and amino acids 516–519 (KNKK) as site B (Fig. 1).
Fig. 1.

Schematic of Rec10 protein, showing NLS sites A and B defined and analyzed here. The solid blue line shows the full-length (791 amino acids) of Rec10. Positions of the NLS sites and their amino acid sequences are shown below; the position of the GFP fusion to the C-terminus of Rec10 is shown above.
To test the NLS hypothesis noted above, we mutated site A or B, or both, either by deletion or by change of each amino acid to alanine. Alanine is a small, non-charged amino acid, unlike lysine and arginine. If these sites are part of the NLS, importin α, part of the nuclear entry complex (Soniat and Chook, 2015), is predicted not to recognize the mutated Rec10 and not to carry it into the nucleus. Mutations were first made in rec10 on a plasmid, and strains carrying the plasmid-borne mutations were tested for localization of another LinE protein, Rec25, fused to green fluorescent protein (Rec25–GFP), and for recombination proficiency. The NLS mutations were then moved to the chromosome, where rec10 was fused to the GFP gene to visualize localization of Rec10 itself and to analyze DSB formation and recombination proficiency. Derivative strains with chromosomal rec10 without the GFP fusion were also tested for recombination proficiency and for localization of Rec25–GFP and, for the site A–site B double deletion mutant, localization of Rec27–GFP and Mug20–GFP. Similar results were obtained for each rec10 derivative, indicating that the plasmid versus chromosomal location of rec10 or fusion of Rec10 to GFP does not significantly interact with the NLS (Figs 2–4, Table 1).
Fig. 2.

Plasmid-borne rec10 NLS mutations block entry of other LinE proteins into the nucleus. Strains bearing rec25–GFP and a complete rec10 deletion on the chromosome and carrying a plasmid with rec10+ (WT), with the indicated rec10 NLS mutation, or without rec10 (rec10Δ) were harvested during asynchronous (h90) meiosis and examined by light microscopy. In the merged images, green indicates Rec25–GFP, and red indicates chromatin in the nucleus; yellow is their overlap. The dashed line outlines the cell. ΔA indicates deletion of Rec10 NLS site A, and AlaA indicates substitution of alanine for each of the four amino acids of site A; site B was similarly deleted or mutated, indicated as ΔB and AlaB, respectively. Combinations of these mutations are indicated by ΔA ΔB or AlaA AlaB. Images are representative of >50 cells from three separate cultures analyzed on different days. Cells are in the horsetail stage, during which the nucleus moves repeatedly from one end of the cell to the other; nuclei are longer when in the middle of the cell than when at either end, when the nucleus becomes nearly round (Robinow, 1977; Bähler et al., 1993; Chikashige et al., 1994). Scale bars: 5 μm.
Fig. 4.

Chromosomal rec10 NLS mutations block entry of Rec25–GFP, Rec27–GFP and Mug20–GFP into the nucleus. (A) Strains bearing rec25–GFP and without (WT) or with the indicated rec10 NLS mutations were harvested during asynchronous (h90) meiosis and examined by light microscopy. (B) Strains bearing rec27–GFP were analyzed as in panel A. (C) Strains bearing mug20–GFP were analyzed as in panel A. In the merged images, green indicates GFP, and red indicates chromatin in the nucleus; yellow is their overlap. See Fig. 2 for examples of separate images for Rec25–GFP and chromatin. The dashed line outlines the cell. NLS mutations are designated as in Fig. 2. Images are representative of >50 cells from three separate cultures analyzed on different days. Cells are in the horsetail stage, as in Fig. 2. Scale bars: 5 μm.
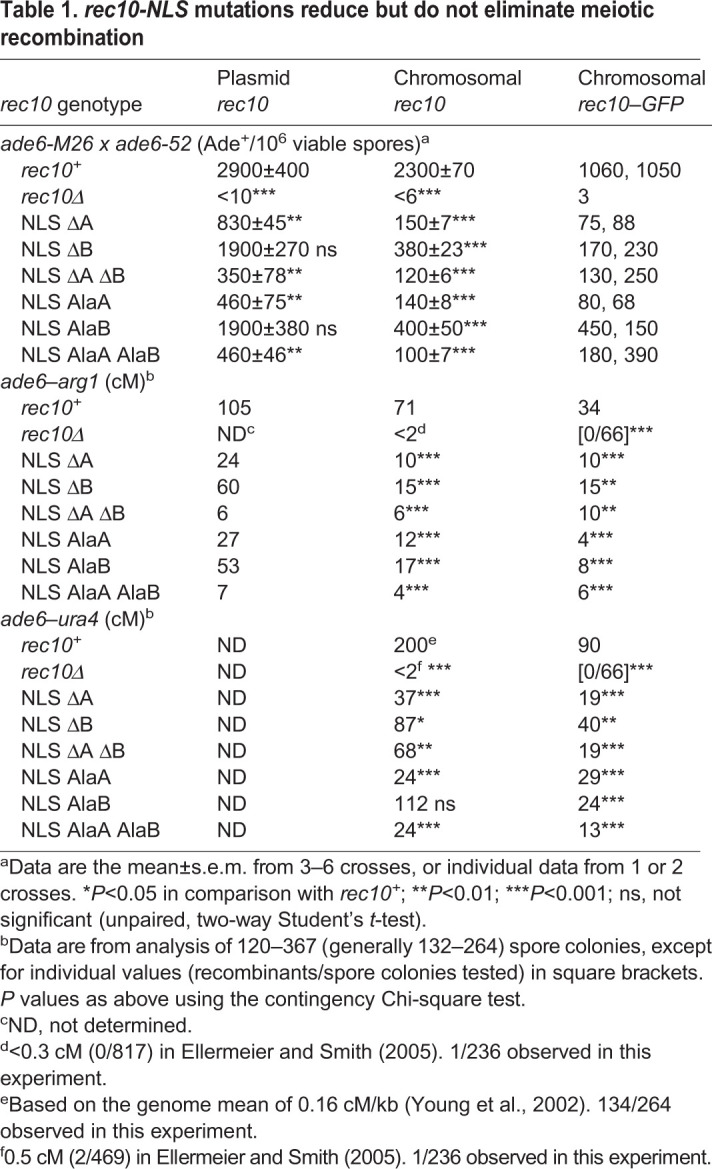
Table 1.
rec10-NLS mutations reduce but do not eliminate meiotic recombination
Single and double Rec10 NLS mutants have aberrant cytoplasmic localization of linear element proteins
To assess the importance of each putative NLS site in Rec10, we determined the cellular localization of Rec25–GFP, a partner protein of Rec10 (Davis et al., 2008). We tested strains carrying rec10 mutated (or not) on a plasmid with an estimated copy number of 1–2 (Wahls and Davidson, 2008). For the NLS site A and site B mutants, sharp foci (‘speckles’) of Rec25–GFP were prominent in the cytoplasm, and occasional faint dots were observed in the nucleus (Fig. 2). Wild-type Rec10 produced strong nuclear foci of Rec25–GFP without detectable cytoplasmic foci. rec10Δ produced less fluorescence, nearly uniformly distributed in the nucleus, but still significantly above the mitotic level (Fig. S1), consistent with previous reports of abundant Rec25, as assayed by western blot analysis, in rec10Δ cells (Davis et al., 2008). Rec25–GFP was readily visible in all other situations (wild-type cells and NLS rec10 mutants), implying that Rec25–GFP likely forms a complex with wild-type and NLS-mutant forms of Rec10 (see Discussion). Similar results were seen with both deletion and alanine substitution mutations at each NLS site and with the double mutations.
Rec10 NLS mutations reduce meiotic recombination
Rec10 is essential for recombination throughout the genome (Ellermeier and Smith, 2005). To test whether the NLS, and thus nuclear import, of Rec10 is required for recombination, we measured the frequency of intragenic recombination (gene conversion, or non-reciprocal recombination) at ade6 and the frequency of intergenic recombination (crossing-over, or reciprocal recombination) between ade6 and arg1. In assays with plasmid-borne rec10, conversion was reduced in all the NLS mutants by factors ranging from ∼2 to ∼8, but in no case did recombination approach the very low level of rec10Δ (∼300 times lower than that of wild-type cells) (Table 1). Crossovers were reduced by factors of up to 15, but again were not reduced as much as in rec10Δ (Ellermeier and Smith, 2005). NLS deletions and alanine substitutions behaved similarly. In all four cases, site A mutations reduced recombination more than site B mutations. These results show that the Rec10 NLS is important for recombination but is not as essential as Rec10 itself. The residual recombination in rec10 NLS mutants may reflect nuclear entry of small amounts of Rec10 (see Discussion).
Chromosomal Rec10 NLS mutations reflect plasmid-borne mutations in recombination and localization phenotypes
To ensure levels of Rec10 protein more similar to those in wild-type cells, and to assay Rec10 localization itself, we moved the rec10 NLS mutations to the chromosome and fused the rec10 gene to GFP (Rec10-NLS–GFP). The NLS mutant proteins formed cytoplasmic speckles, much like those of Rec25–GFP described above for plasmid-borne rec10 NLS mutations, or larger clumps (Fig. 3). The rec10 NLS deletion and alanine substitution mutations had similar phenotypes, as did the single and double site mutations. The similar appearance of Rec25–GFP (Fig. 2) and Rec10–GFP (Fig. 3) is consistent with these proteins forming a complex, either in the nucleus (wild type) or in the cytoplasm (NLS mutants) (see Discussion).
Fig. 3.

Chromosomal rec10 NLS mutations block entry of Rec10–GFP into the nucleus. Strains without (WT) or with a rec10 NLS mutation were harvested during asynchronous (h90) meiosis and examined by light microscopy. Green indicates Rec10, and red indicates chromatin in the nucleus; yellow is their overlap. The dashed line outlines the cell. NLS mutations are designated as in Fig. 2. Images are representative of >50 cells from three separate cultures analyzed on different days. Cells are in the horsetail stage, as in Fig. 2. Scale bars: 5 μm.
Recombination was also reduced similarly in chromosomal and plasmid-borne rec10 NLS mutants (Table 1). Recombinant frequencies for ade6 gene conversion and ade6–arg1 crossing over, as well as for ade6–ura4 crossing over, were reduced by factors of ∼3 to ∼15 by the chromosomal NLS mutations. The reductions were slightly greater than those caused by the plasmid-borne alleles, but again not as great as those caused by rec10Δ. In 15 of 16 cases tested (Table 1), site A mutations reduced recombination more than site B mutations; double site A–site B mutations reduced recombination at least as much as a single mutation.
To compare directly plasmid-borne and chromosomal rec10 NLS mutations (without GFP fusion), we assayed Rec25–GFP localization and recombination in both sets of strains. For each single and double NLS mutant, Rec25–GFP formed abundant cytoplasmic speckles and was depleted in the nucleus (Figs 2 and 4). As with the plasmid-borne mutations, deletion and alanine substitution mutations, as well as single and double mutations, behaved similarly. This outcome is as expected if the rec10 gene is regulated similarly on the low copy-number plasmid and on the chromosome. It also allows interchangeable consideration of the plasmid- and chromosomal-borne rec10 NLS mutations. Rec27–GFP and Mug20–GFP behaved much like Rec25–GFP in cells with wild-type Rec10 or Rec10 deleted for both NLSs (Fig. 4B,C). This result supports the view that the three small LinE proteins (Rec25, Rec27 and Mug20) obligatorily interact, consistent with the nearly identical phenotypes of deletion of each small LinE (Davis et al., 2008; Estreicher et al., 2012; Fowler et al., 2013).
Based on the cytoplasmic localizations of these three LinE–GFPs, Rec10 appears to aggregate in the cytoplasm when its NLS is mutated (as directly seen above with Rec10–GFP). Rec10 may self-interact (Ma et al., 2017). The cytoplasmic speckles appear to be formed by complexes of Rec10 and the other LinEs, just as they colocalize in the nuclear foci during wild-type meiosis (Davis et al., 2008; Estreicher et al., 2012; Fowler et al., 2013). The cytoplasmic speckles in the mutants were less numerous and larger than the nuclear foci in the wild-type strain, suggesting different structures for the cytoplasmic and nuclear LinE complexes (see Discussion).
The chromosomal rec10 alleles with NLS mutations were tested for recombination proficiency to allow direct comparison with wild-type rec10 (chromosomal with no GFP fusion) (Table 1). As with rec10 in the other situations (plasmid-borne or fused to GFP), recombination was substantially reduced by the NLS mutations, by factors of ∼6 to ∼23 for ade6 gene conversion and by factors of up to 18 for crossovers flanking ade6. In no case, however, was recombination reduced as much as with rec10Δ. In 13 of 21 cases with chromosomal rec10 alleles, the GFP fusion reduced recombination proficiency by a factor of ∼2, presumably a result of the GFP fusion interfering slightly with function of the LinE complex.
Rec10 NLS mutations reduce meiotic DNA break formation at DSB hotspots
The reduced levels of recombination in the rec10 NLS mutants noted above could reflect reduced DSB formation; all four LinE deletion mutants have reduced DSB levels at DSB hotspots (Ellermeier and Smith, 2005; Martín-Castellanos et al., 2005; Fowler et al., 2013). We assayed DSB formation in strains with the chromosomal rec10–GFP fusion, with and without NLS mutations (Fig. 5). As expected from the ∼2-fold reduction of recombinant frequencies by the GFP fusion (Table 1), DSB levels in the rec10–GFP (wild-type NLS) strain were about half of that in rec10+ (Fig. S2; Table S1). The rec10 NLS mutations ΔA and ΔA ΔB in the rec10–GFP strains further reduced DSB formation at most of the hotspots tested by a factor of four or more (Fig. 5; Fig. S2, Table S1). These data show directly that the Rec10 NLS is important for the initiation of meiotic recombination by the formation of DSBs.
Fig. 5.

Rec10–NLS mutations reduce DSB formation at DSB hotspots. Strains (pat1-as1 rad50S) with the indicated chromosomal rec10 genotype were induced for meiosis and harvested at the indicated times. DNA was analyzed by Southern blotting for DSBs at the ade6-3049 hotspot on the 74 kb PmeI fragment of chromosome 3 (A) and at multiple hotspots (hotspot A, mbs1, mbs2) on the 501 kb NotI fragment of chromosome 1 (B). NLS mutations are designated as in Fig. 2. See Fig. S2 for additional data.
Rec10 abundance is not altered by NLS mutations
Retention of Rec10 in the cytoplasm rather than entry into the nucleus could lead to its partial degradation and reduce, but not eliminate, meiotic recombination. To test this possibility, we induced into meiosis strains carrying the chromosomal rec10 alleles with the NLS mutations and the GFP fusion. Protein abundance was assayed by polyacrylamide gel electrophoresis and western blot analysis using antibody against GFP. The results (Fig. 6) show that Rec10 is meiotically induced to the same level in wild-type strains and in each of the six NLS mutants. Thus, the relocalization of Rec10–GFP and the other LinE–GFPs, and the reduction of DSBs and recombination by the NLS mutations, cannot be attributed to aberrant expression of rec10 or instability of the Rec10–GFP protein.
Fig. 6.

Rec10 protein is present at wild-type levels in rec10-NLS mutants. Strains with chromosomal rec10-NLS–GFP fusions were induced for meiosis (pat1-as1) and analyzed by western blotting for Rec10 abundance in cell extracts at the indicated times. NLS mutations are designated as in Fig. 2. M, protein markers with the indicated masses.
DISCUSSION
Our results show the importance of the NLS of Rec10 for entry of LinE proteins into the nucleus and their promotion of DSB formation and recombination. These results imply that Rec10 forms a complex with the other LinE proteins before nuclear entry. The conservation of LinE proteins among widely divergent species suggests that one or another related protein of the synaptonemal complex of other species has an NLS with similar function.
Both of the two predicted Rec10 NLS sites are important for Rec10 and the three other LinE proteins to enter the nucleus. Site A, amino acids KRKK (497–500), appears to be more important than site B, amino acids KNKK (516–519) (Fig. 1), at least for recombination, the most quantitative assay used here (Table 1). The double mutant, whether deletions or substitutions to alanine, behaved in most cases like single-site mutants, suggesting that the two sites act together. Two-site NLSs in other proteins behave similarly (Soniat and Chook, 2015), suggesting that the Rec10 NLS is canonical. The small LinE proteins (Rec25, Rec27 and Mug20) were visible at only low levels in the nucleus in the NLS mutants (Figs 2 and 4), showing that their nuclear entry mainly depends on the Rec10 NLS. Rec10 and the other LinEs colocalize in the nucleus in wild-type Rec10 strains (Davis et al., 2008; Estreicher et al., 2012; Fowler et al., 2013). One interpretation of these results is that Rec10 forms a complex with the other LinEs in the cytoplasm and only then does this complex enter the nucleus. An alternative is that the small LinE proteins enter the nucleus on their own and associate with Rec10 if Rec10 has entered the nucleus (with a wild-type NLS); otherwise, the small LinE proteins remain in the cytoplasm if Rec10 remains in the cytoplasm (with a mutant NLS). In NLS mutants this complex still forms but remains mainly in the cytoplasm, consistent with the similar appearance of Rec10–GFP and Rec25–GFP (or Rec27–GFP or Mug20–GFP) cytoplasmic speckles in the NLS mutants (compare Figs 2 and 4 with Fig. 3). These speckles appear qualitatively larger and less numerous than the nuclear foci in wild-type NLS strains. We propose that the cytoplasmic NLS complex forms aggregates when it cannot enter the nucleus, much as SC proteins of other species form aggregates (‘polycomplexes’) when they cannot form proper nuclear structures (Sym and Roeder, 1995). LinE proteins share amino acid sequence similarity with SC proteins (see Introduction), consistent with this view.
Residual recombination in all the NLS mutants implies that a small amount of Rec10 still enters the nucleus in these mutants. The LinE complex, with a molecular mass of 139.0 kDa assuming one of each subunit, may be unable to enter the nucleus without the aid of importin, whereas a Rec10 monomer, with a molecular mass of 89.9 kDa, may be able to enter, as shown for other proteins in this size range (Wang and Brattain, 2007). Complete deletion of rec10 reduces recombination as much as does deletion of the DSB-forming protein Rec12 – up to 500 times less recombination than in wild-type cells (DeVeaux et al., 1992; Ellermeier and Smith, 2005). Residual recombination in the Rec10 NLS mutants may occur without participation of the other LinE proteins (Rec25, Rec27 and Mug20), because these proteins are not as stringently required for recombination as Rec10 (Davis et al., 2008; Estreicher et al., 2012). In the NLS mutants, nuclear Rec10 levels may be below or near the level of detection by microscopy but still be sufficient to promote low, but detectable, levels of recombination.
The bipartite NLS determined here is conserved among Schizosaccharomyces and other species. Both parts of the NLS are present, with only small differences, at the same positions in an alignment of the Schizosaccharomyces species S. kambucha, S. octosporus and S. cryophilus (Ma et al., 2017). The more distantly related Schizosaccharomyces japonicus has a bipartite NLS, predicted by NLS Mapper, about 70 amino acids toward the N-terminus. This outcome suggests that these species, which contain orthologs of all four LinE proteins, also control DSB hotspot activity by formation and nuclear import of a LinE protein complex. Although S. cerevisiae Red1 has a bipartite NLS, predicted by NLS Mapper, at amino acids 551–574 (of 827 total), this has not, to our knowledge, been experimentally verified. The fruit fly Drosophila melanogaster SC protein C(2)M contains a predicted bipartite NLS (Manheim and McKim, 2003), but this NLS has not been confirmed experimentally (K. McKim, personal communication). Making and testing NLS-deficient mutants in other species would allow determination of the extent to which formation of a large protein complex is required for nuclear entry of proteins critical for meiotic recombination. We anticipate that the observations reported here will extend to many and perhaps most species.
MATERIALS AND METHODS
Strains, plasmids and oligonucleotides
S. pombe strains and their genotypes are in Table S2; plasmids are in Table S3. Oligonucleotides, purchased from Integrated DNA Technologies and used for mutagenesis and DNA sequencing, are in Table S4. Genetic manipulations and culture media were as described previously (Smith, 2009).
Mutagenesis and strain constructions
Q5 Site-Directed Mutagenesis kit (New England BioLabs) was used to make substitutions, and Quickchange Site-Directed Mutagenesis kit (Agilent) was used to make deletions, following the protocols provided by the companies. Mutagenesis was done on pYL176, a plasmid carrying rec10, an ampicillin-resistance gene for selection in E. coli, and the S. cerevisiae URA3 gene for selection in S. pombe ura4 mutants (Lin and Smith, 1995). Plasmids were extracted from four to eight separate E. coli transformants for each mutant and sequenced. Plasmids with the desired mutations were introduced into S. pombe strain GP9806 (h90 rec10-175::kanMX6 ura4-294 rec25-303::GFP-hphMX6) by transformation to Ura+. Table S5 lists the NLS mutations, the oligonucleotides for their construction, the initial plasmids, and the chromosomal derivatives.
The rec10 NLS mutations were transferred to the chromosome by digestion of these plasmids with MluI and NsiI to create a 2.67 kb fragment containing the rec10 gene and ∼0.1 kb to each side. The digested DNA was used to transform strain GP7301 (rec10-260::ura4+) to 5-fluoro-orotic acid (FOA)-resistance (Boeke et al., 1984).
To create rec10-NLS-GFP chromosomal derivatives, strain GP9845 (rec10-306::ura4+-GFP-natMX6; a gift from Yu-Chien Chuang and Randy Hyppa, Fred Hutchinson Cancer Research Center, Seattle, WA) was similarly transformed to FOA-resistance, using the 2.17 kb MluI–XhoI fragment containing most of rec10 and its NLS mutation. rec10-306 was made in two steps – substitution of natMX6 for kanMX6 (in rec10-203::GFP-kanMX6 to make rec10-301::GFP-natMX6) (Hentges et al., 2005), followed by substitution of ura4+ for the rec10 NLS region. For the second substitution, DNA flanking the left and right sides of the Rec10 NLS sites was generated with oligonucleotides OL4278 and OL4279 (left) and OL4280 and OL4281 (right) using S. pombe rec10+ DNA as template; OL4279 and OL4280 have 20 nucleotides of ura4+ sequence. ura4+ DNA was generated by a PCR with OL4282 and OL4283 using pFY20 as template; OL4282 and OL4283 have 20 nucleotides of rec10+ sequence. The three PCR reactions were diluted 1:10 and used as template in a PCR using OL4278 and OL4281, which generated the rec10 coding sequence substituted from nucleotides 373 to 1632 with ∼1.8 kb bearing ura4+. Strain GP9836 (rec10-301::GFP-natMX6) was transformed with this DNA, with selection for Ura+ to generate the chromosomal rec10-306::ura4+-GFP-natMX6 allele. rec10-306 has the GFP gene fused to the C-terminus of the rec10 gene and the nourseothricin-resistance determinant natMX6 inserted near the 3′ end of the GFP gene (Fowler et al., 2013). Integration at rec10 was verified by PCR specific for rec10+ and rec10-306 (Table S4).
Drug-resistance genes nourseothricin N-acyltransferase (nat) and hygromycin B phosphotransferase (hph) were PCR-amplified from plasmids pFA6a-natMX6 and pFA6a-hphMX6 (Table S3) using oligonucleotides and conditions similar to those in Sato et al. (2005). The resultant fragments were used to transform strains to the new drug resistance, thereby replacing the resident drug-resistance gene (Bähler et al., 1998). Strains GP8762 (h90 rec10-203::GFP-kanMX6) and GP8766 (h90 rec25-204::GFP-kanMX6) were transformed with natMX6 and hphMX6, respectively, to make strains GP9747 (h90 rec10-301::GFP-natMX6) and GP9745 (h90 rec25-303::GFP-hphMX6).
Recombination assays
Plasmid-borne rec10-NLS mutations
Strain GP6994 (h− ade6-52 ura4-D18 rec10-175::kanMX6) and GP9775 (h+ ade6-M26 ura4-D18 rec10-175::kanMX6 arg1-14) were transformed with the mutated pYL176 plasmids. For each mutant and pYL176 (rec10+) control, the two strains were crossed. After two days on sporulation medium (SPA) plus adenine and arginine, spores were harvested, treated sequentially with glusulase and ethanol, and washed. Appropriate dilutions of spore suspensions were spread on yeast extract medium (YEA) medium, to assay total viable spore titer, and on YEA medium supplemented with guanine (YEAG), which inhibits adenine uptake, to assay Ade+ recombinant titers; Ade+ recombinant frequency is expressed as the ratio of these titers. Colonies were picked from YEA, noting their color (light red for ade6-52 and dark red for ade6-M26), to YEA plus adenine medium, incubated at 32°C for one day, and replicated using velvet onto minimal nitrogen base agar (NBA) plus adenine medium supplemented or not with arginine to score arg1. Intergenic recombinant frequencies were converted to genetic distance using Haldane's equation. See Smith (2009) for details.
Chromosomal rec10-NLS mutations
To test rec10 NLS mutants, FOA-resistant transformants of strain GP7301 (h− ade6-52 rec10-260::ura4+ ura4-D18) (Table S5) were mated with strain GP4414 (h+ ade6-M26 arg1-14 rec10-175::kanMX6). Control strains were GP935 (rec10+) and GP6994 (rec10-175::kanMX6). Spores were analyzed as above, plus replication onto minimal NBA plus adenine and arginine medium supplemented or not with uracil to score ura4.
Chromosomal rec10-NLS–GFP mutations
To test rec10–GFP NLS mutants, FOA-resistant transformants of strain GP9845 (h90 rec10-306::ura4+-GFP-natMX6 ura4-D18) were mated with strain GP4914 (h+ ura4-D18 ade6-M26 arg1-14) to obtain h+ ura4-D18 ade6-M26 arg1-14 rec10-NLS-GFP-natMX6 derivatives. These isolates (Table S5) were mated with strain GP4625 (h− ade6-52 rec10-175::kanMX6). Control strains were GP4914 (rec10+) and GP9775 (rec10-175::kanMX6). Spores were analyzed as above.
Each cross was conducted with independent cultures on one to three days. Intragenic (ade6) recombinant frequencies from each cross were analyzed separately; intergenic (ade6–ura4 and ade6–arg1) data from the crosses were pooled and analyzed (Table 1).
Fluorescence microscopy
Homothallic (h90) cells were spotted on SPA without supplements; after 16 h of incubation at 25°C, cells were harvested, stained with Hoechst 33342, and spotted on poly-lysine-coated microscope slides. Cells with an elongated nucleus (in the horsetail stage) were scored for localization of green fluorescence (GFP). Images are of a single focal plane (Figs 2 and 4) or maximal projections of 9–12 sections, step size 0.2 µm, to cover the whole cell (Fig. 3). Images were obtained with an Evos FL Auto 2 microscope (Thermo Fisher Scientific) at 60× magnification (Figs 2 and 4) or with a DeltaVision microscope (GE Healthcare) at 100× magnification (Fig. 3). Images taken using the DeltaVision microscope were deconvolved and projected using softWorX software program (Applied Precision). All images were analyzed with ImageJ software. Images from three cultures were obtained on different days.
Protein abundance by western blot analysis
Synchronous meiosis was induced as described previously (Hyppa and Smith, 2009) using the pat1-as1 (L95G) allele at 25°C (Guerra-Moreno et al., 2012). In brief, haploid h− pat1-as1 (L95G) rec10-NLS–GFP strains were grown in liquid minimal medium EMM2, washed, and starved for nitrogen. After 18–22 h of starvation, when the cultures had reached OD600 of 0.6–0.8, NH4Cl and 3-MB-PP1 (Sigma Aldrich) were added to 0.50% (w/v) and 25 µM, respectively, to start meiosis. Culture samples (25 ml) were collected at the indicated times. Cells were collected by centrifugation and washed once with water and once with 20% trichloroacetic acid (TCA). Cell pellets were frozen on liquid nitrogen and kept at −20°C until protein extraction. Proteins were extracted using TCA and glass beads. Protein extracts from the entire 25 ml were suspended in 50 µl of NuPAGE LDS Sample buffer (Thermo Fisher Scientific). Samples (5 µl each) were loaded into wells of a NuPAGE Tris-acetate 3–8% polyacrylamide gel (Thermo Fisher Scientific) and electrophoresed for 60 min at 15 V/cm. Proteins were transferred to Immobilon-FL PVDF membranes (Millipore), which were probed sequentially with rabbit anti-GFP antibodies (Clontech Living Colors full-length; Takara Bio) and IRDye680 goat anti-rabbit IgG antibodies (Li-Cor). Membrane images were taken using a Li-Cor Odyssey NIR Scanner.
DSB assays
Cells (h− rad50S pat1-as1 haploids, with chromosomal rec10+, rec10-GFP, or rec10-NLS-GFP) were induced for meiosis at 25°C as in the preceding section, harvested at 0, 6 and 7 h, and embedded in agarose plugs (cells from 3 ml per plug). Cells were lysed and treated with RNase and proteinase K. DNA was digested with a restriction enzyme (PmeI for Fig. 5A, Fig. S2A,B; NotI for Fig. 5B and Fig. S2C); the fragments were separated by pulsed-field gel electrophoresis and analyzed by Southern blot hybridization, as described previously (Hyppa and Smith, 2009). Probes for hybridization are described in Young et al. (2002) and Fowler et al. (2018).
Supplementary Material
Acknowledgements
We are grateful to Emma Chen for excellent technical assistance; Yu-Chien Chuang and Randy Hyppa for construction of strain GP9845 (rec10-306::ura4+-GFP-natMX6) and much helpful advice; Yu-Chien Chuang for help with microscopy; Kim McKim and Scott Hawley for information about D. melanogaster C(2)M and other SC proteins; and Sue Amundsen for helpful comments on the manuscript.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: M.W., M.-C.N., G.R.S.; Methodology: M.W., M.-C.N.; Software: M.W., M.-C.N.; Validation: M.W., M.-C.N., G.R.S.; Formal analysis: M.W., M.-C.N.; Investigation: M.W., M.-C.N., G.R.S.; Resources: G.R.S.; Data curation: M.W., M.-C.N.; Writing - original draft: G.R.S.; Writing - review & editing: M.W., M.-C.N., G.R.S.; Supervision: G.R.S.; Project administration: G.R.S.; Funding acquisition: G.R.S.
Funding
This work was supported by research grant R35 GM118120 from the National Institutes of General Medical Sciences and philanthropic contributions to the Fred Hutchinson Cancer Research Center. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at https://jcs.biologists.org/lookup/doi/10.1242/jcs.253518.supplemental
Peer review history
The peer review history is available online at https://jcs.biologists.org/lookup/doi/10.1242/jcs.253518.reviewer-comments.pdf
References
- Bähler, J., Wyler, T., Loidl, J. and Kohli, J. (1993). Unusual nuclear structures in meiotic prophase of fission yeast: a cytological analysis. J. Cell Biol 121, 241-256. 10.1083/jcb.121.2.241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bähler, J., Wu, J.-Q., Longtine, M. S., Shah, N. G., McKenzie, A., III, Steever, A. B., Wach, A., Philippsen, P. and Pringle, J. R. (1998). Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast 14, 943-951. [DOI] [PubMed] [Google Scholar]
- Boeke, J. D., LaCroute, F. and Fink, G. R. (1984). A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol. Gen. Genet. 197, 345-346. 10.1007/BF00330984 [DOI] [PubMed] [Google Scholar]
- Chikashige, Y., Ding, D. Q., Funabiki, H., Haraguchi, T., Mashiko, S., Yanagida, M. and Hiraoka, Y. (1994). Telomere-led premeiotic chromosome movement in fission yeast. Science 264, 270-273. 10.1126/science.8146661 [DOI] [PubMed] [Google Scholar]
- Cromie, G. A. and Smith, G. R. (2008). Meiotic recombination in Schizosaccharomyces pombe: a paradigm for genetic and molecular analysis. In Recombination and Meiosis: Models, Means, and Evolution (ed. Egel R. and Lankenau D.-H.), pp. 195-230. Berlin: Springer-Verlag. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, L., Rozalén, A. E., Moreno, S., Smith, G. R. and Martín-Castellanos, C. (2008). Rec25 and Rec27, novel linear-element components, link cohesin to meiotic DNA breakage and recombination. Curr. Biol. 18, 849-854. 10.1016/j.cub.2008.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVeaux, L. C., Hoagland, N. A. and Smith, G. R. (1992). Seventeen complementation groups of mutations decreasing meiotic recombination in Schizosaccharomyces pombe. Genetics 130, 251-262. 10.1093/genetics/130.2.251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, D.-Q., Matsuda, A., Okamasa, K., Nagahama, Y., Haraguchi, T. and Hiraoka, Y. (2016). Meiotic cohesin-based chromosome structure is essential for homologous chromosome pairing in Schizosaccharomyces pombe. Chromosoma 125, 205-214. 10.1007/s00412-015-0551-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellermeier, C. and Smith, G. R. (2005). Cohesins are required for meiotic DNA breakage and recombination in Schizosaccharomyces pombe. Proc. Natl. Acad. Sci. USA 102, 10952-10957. 10.1073/pnas.0504805102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estreicher, A., Lorenz, A. and Loidl, J. (2012). Mug20, a novel protein associated with linear elements in fission yeast meiosis. Curr. Genet. 58, 119-127. 10.1007/s00294-012-0369-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler, K. R., Gutiérrez-Velasco, S., Martín-Castellanos, C. and Smith, G. R. (2013). Protein determinants of meiotic DNA break hot spots. Mol. Cell 49, 983-996. 10.1016/j.molcel.2013.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler, K. R., Hyppa, R. W., Cromie, G. A., and Smith, G. R. (2018). Physical basis for long-distance communication along meiotic chromosomes. Proc. Natl. Acad. Sci. USA115, E9333-E9342. 10.1073/pnas.1801920115 [DOI]
- Guerra-Moreno, A., Alves-Rodrigues, I., Hidalgo, E. and Ayté, J. (2012). Chemical genetic induction of meiosis in Schizosaccharomyces pombe. Cell Cycle 11, 1621-1625. 10.4161/cc.20051 [DOI] [PubMed] [Google Scholar]
- Hentges, P., Van Driessche, B., Tafforeau, L., Vandenhaute, J. and Carr, A. M. (2005). Three novel antibiotic marker cassettes for gene disruption and marker switching in Schizosaccharomyces pombe. Yeast 22, 1013-1019. 10.1002/yea.1291 [DOI] [PubMed] [Google Scholar]
- Hyppa, R. W. and Smith, G. R. (2009). Using Schizosaccharomyces pombe meiosis to analyze DNA recombination intermediates. In Meiosis (ed. Keeney S.), pp. 235-252. Totowa, NJ: Humana Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney, S., Lange, J. and Mohibullah, N. (2014). Self-organization of meiotic recombination initiation: general principles and molecular pathways. Annu. Rev. Genet. 48, 187-214. 10.1146/annurev-genet-120213-092304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y. and Smith, G. R. (1995). Molecular cloning of the meiosis-induced rec10 gene of Schizosaccharomyces pombe. Curr. Genet. 27, 440-446. 10.1007/BF00311213 [DOI] [PubMed] [Google Scholar]
- Lorenz, A., Wells, J. L., Pryce, D. W., Novatchkova, M., Eisenhaber, F., McFarlane, R. J. and Loidl, J. (2004). S. pombe meiotic linear elements contain proteins related to synaptonemal complex components. J. Cell Sci. 117, 3343-3351. 10.1242/jcs.01203 [DOI] [PubMed] [Google Scholar]
- Ma, L., Fowler, K. R., Martín-Castellanos, C. and Smith, G. R. (2017). Functional organization of protein determinants of meiotic DNA break hotspots. Sci. Rep. 7, 1393. 10.1038/s41598-017-00742-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manheim, E. A. and McKim, K. S. (2003). The synaptonemal complex component C(2)M regulates meiotic crossing over in Drosophila. Curr. Biol. 13, 276-285. 10.1016/S0960-9822(03)00050-2 [DOI] [PubMed] [Google Scholar]
- Martín-Castellanos, C., Blanco, M., Rozalén, A. E., Pérez-Hidalgo, L., García, A. I., Conde, F., Mata, J., Ellermeier, C., Davis, L., San-Segundo, P.et al. (2005). A large-scale screen in S. pombe identifies seven novel genes required for critical meiotic events. Curr. Biol. 15, 2056-2062. 10.1016/j.cub.2005.10.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nambiar, M., Chuang, Y.-C. and Smith, G. R. (2019). Distributing meiotic crossovers for optimal fertility and evolution. DNA Repair 81, 102648. 10.1016/j.dnarep.2019.102648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page, S. L. and Hawley, R. S. (2004). The genetics and molecular biology of the synaptonemal complex. Annu. Rev. Cell Dev. Biol. 20, 525-558. 10.1146/annurev.cellbio.19.111301.155141 [DOI] [PubMed] [Google Scholar]
- Robinow, C. F. (1977). The number of chromosomes in Schizosaccharomyces pombe: light microscopy of stained preparations. Genetics 87, 491-497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato, M., Dhut, S. and Toda, T. (2005). New drug-resistant cassettes for gene disruption and epitope tagging in Schizosaccharomyces pombe. Yeast 22, 583-591. 10.1002/yea.1233 [DOI] [PubMed] [Google Scholar]
- Smith, G. R. (2009). Genetic analysis of meiotic recombination in Schizosaccharomyces pombe. In Meiosis (Keeney S.), pp. 65-76. Totowa, NJ: Humana Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soniat, M. and Chook, Y. M. (2015). Nuclear localization signals for four distinct karyopherin-β nuclear import systems. Biochem. J. 468, 353-362. 10.1042/BJ20150368 [DOI] [PubMed] [Google Scholar]
- Spirek, M., Estreicher, A., Csaszar, E., Wells, J., McFarlane, R. J., Watts, F. Z. and Loidl, J. (2010). SUMOylation is required for normal development of linear elements and wild-type meiotic recombination in Schizosaccharomyces pombe. Chromosoma 119, 59-72. 10.1007/s00412-009-0241-5 [DOI] [PubMed] [Google Scholar]
- Sym, M. and Roeder, G. S. (1995). Zip1-induced changes in synaptonemal complex structure and polycomplex assembly. J. Cell Biol. 128, 455-466. 10.1083/jcb.128.4.455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahls, W. P. and Davidson, M. K. (2008). Low-copy episomal vector pFY20 and high-saturation coverage genomic libraries for the fission yeast Schizosaccharomyces pombe. Yeast 25, 643-650. 10.1002/yea.1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, R. and Brattain, M. G. (2007). The maximal size of protein to diffuse through the nuclear pore is larger than 60 kDa. FEBS Lett. 581, 3164-3170. 10.1016/j.febslet.2007.05.082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young, J. A., Schreckhise, R. W., Steiner, W. W. and Smith, G. R. (2002). Meiotic recombination remote from prominent DNA break sites in S. pombe. Mol. Cell 9, 253-263. 10.1016/S1097-2765(02)00452-5 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.