

Abstract
On August 16, 2018, the U.S. Food and Drug Administration approved lenvatinib (Lenvima, Eisai Inc.) for first‐line treatment of patients with unresectable hepatocellular carcinoma (HCC). Approval was based on an international, multicenter, randomized, open‐label, noninferiority trial (REFLECT; NCT01761266) conducted in 954 patients with previously untreated metastatic or unresectable HCC. Patients were randomized (1:1) to receive lenvatinib (12 mg orally once daily for patients with a baseline body weight ≥60 kg and 8 mg orally once daily for patients with a baseline body weight <60 kg) or sorafenib (400 mg orally twice daily) until radiological disease progression or unacceptable toxicity. REFLECT demonstrated that lenvatinib was noninferior but not statistically superior to sorafenib for overall survival (OS; hazard ratio, [HR] 0.92; 95% confidence intervals [CI], 0.79–1.06), with median OS of 13.6 and 12.3 months in the lenvatinib and sorafenib arms, respectively. REFLECT also demonstrated statistically significant improvements in investigator‐assessed progression‐free survival (PFS; HR, 0.66; 95% CI, 0.57–0.77]; p < 0.001), corresponding to median PFS of 7.4 and 3.7 months and overall response rate of 24.1% vs 9.2% per modified RECIST for HCC (mRECIST) in the lenvatinib and sorafenib arms, respectively. Consistent results were observed by an independent review facility per RECISTv1.1 and per mRECIST. The most common adverse reactions observed in the lenvatinib‐treated patients (≥20%) in decreasing frequency were hypertension, fatigue, diarrhea, decreased appetite, arthralgia/myalgia, decreased weight, abdominal pain, palmar‐plantar erythrodysesthesia syndrome, proteinuria, dysphonia, hemorrhagic events, hypothyroidism, and nausea.
Implications for Practice
This article describes the U.S. Food and Drug Administration's review of data from a single trial, REFLECT, that supported the approval of lenvatinib, as a single agent, for the first‐line treatment of unresectable hepatocellular carcinoma (HCC). REFLECT was an open‐label, noninferiority trial that randomized 954 patients with HCC who were ineligible for liver‐directed therapy with no prior systemic therapy for HCC to lenvatinib or sorafenib. REFLECT demonstrated that lenvatinib‐treated patients had similar survival, more responses, and longer time to progression than those receiving sorafenib. Serious side effects were more common among lenvatinib‐treated patients. Lenvatinib is an effective treatment for patients with previously untreated HCC.
Keywords: Hepatocellular carcinoma, Noninferiority, Lenvatinib, Sorafenib, Survival
Short abstract
This article reviews the data and basis for FDA approval of lenvatinib for the first‐line treatment of patients with unresectable hepatocellular carcinoma.
Introduction
Hepatocellular carcinoma (HCC) is the fourth most common global cancer, with an estimated 42,030 new cases (including intrahepatic bile duct cancers) diagnosed in the U.S. in 2019 and approximately 31,780 liver cancer‐related deaths [1, 2]. The estimated HCC 5‐year survival rate is 18%; the 5‐year survival rates are 33% for resectable disease, 11% for unresectable localized disease, and 2% for metastatic disease [3]. The most common risk factor for the development of HCC is chronic liver disease, including cirrhosis, due to hepatitis B virus (HBV) infection, hepatitis C virus (HCV) infection, alcohol abuse, or nonalcoholic steatohepatitis. Endemic HBV infection is the leading cause of HCC in Eastern Asian countries and most African countries, whereas in North America, Europe, and Japan, HCV infection is the leading virus‐related cause of HCC [2]. The Barcelona Clinic Liver Cancer (BCLC) is the most widely used staging system for HCC [4].
During review of this application, U.S. Food and Drug Administration (FDA)–approved systemic therapy for patients with locally advanced, unresectable, or metastatic HCC and Child‐Pugh A cirrhosis was limited to one drug, sorafenib. Approval of sorafenib in 2007 was based on results of the SHARP trial. The results of SHARP and the Asia‐Pacific trial, which also demonstrated the effect of sorafenib on survival in patients with HCC, were used to establish the noninferiority margin for REFLECT [5, 6, 7]. Baseline demographics and tumor characteristics for both trials are summarized in Tables 2 and 3.
Table 2.
Demographic characteristics in REFLECT, SHARP, and Asia‐Pacific trials
| Demographics | Lenvatinib, n (%) | Sorafenib, n (%) | SHARP, n (%) | Asia‐Pacific, n (%) |
|---|---|---|---|---|
| Randomized | 478 | 476 | 602 | 226 |
| Gender | ||||
| Male | 405 (85) | 401 (84) | 524 (87) | 193 (85) |
| Female | 73 (15) | 75 (16) | 78 (13) | 33 (15) |
| Race | ||||
| White | 135 (28) | 141 (30) | 534 (89) | NA |
| Nonwhite | 343 (72) | 335 (70) | 68 (11) | NA |
| Age, years | ||||
| <65 | 270 (56) | 283 (60) | 232 (39) | Median 51 yr |
| ≥65 | 208 (44) | 193 (40) | 370 (61) | |
| Region a | ||||
| Western | 157 (33) | 157 (33) | 602 (100) b | 0 |
| Asia‐Pacific | 321 (67) | 319 (67) | 0 | 226 (100) c |
Based on variables captured at stratification for randomization for REFLECT and as described in the Food and Drug Administration–approved package insert for Nexavar, supplement by publication of the SHARP trial, and the published results for the ASIA‐Pacific trial.
Western region: North America, Europe, Russia, and Israel.
Asia‐Pacific region: China, Hong Kong, Japan, Korea, Malaysia, Philippines, Singapore, Taiwan, and Thailand.
Western region: North and South America, Mexico, and Europe, Australia, and New Zealand.
Asia‐Pacific region: China, Taiwan, and South Korea.
Abbreviation: NA, not available.
Table 3.
Baseline characteristics of patients enrolled in REFLECT
| Baseline variable | Lenvatinib, n (%) | Sorafenib, n (%) | SHARP, n (%) | Asia‐Pacific, n (%) |
|---|---|---|---|---|
| Randomized | 478 | 476 | 602 | 226 |
| ECOG performance status at baseline | ||||
| 0 | 301 (63) | 299 (63) | 325 (54) | 59 (26) |
| 1 | 177 (37) | 177 (37) | 231 (38) | 155 (69) |
| Macroscopic portal vein invasion, extrahepatic spread, or both | ||||
| Yes | 297 (62) | 297 (62) | 421 (70) | 179 (79) a |
| No | 181 (38) | 179 (38) | 181 (30) | 47 (21) |
| Body weight | ||||
| <60 kg | 151 (32) | 148 (31) | NA | NA |
| ≥60 kg | 327 (68) | 328 (69) | NA | NA |
| Baseline Child‐Pugh Score | ||||
| 5 (Child‐Pugh A) | 368 (77) | 357 (75) | 581 (97) | 220 (97) |
| 6 (Child‐Pugh A) | 107 (22) | 114 (24) | 581 (97) | 220 (97) |
| 7 (Child‐Pugh B) | 3 (0.6) | 4 (0.8) | 20 (3.2) | 6 (3) |
| 8 (Child‐Pugh B) | 0 | 1 (0.2) | 20 (3.2) | 6 (3) |
| 9 (Child‐Pugh B) | 20 (3.2) | 6 (3) | ||
| 10–15 (Child‐Pugh C) | 1 (0.2) | ‐ | ||
| BCLC Staging | ||||
| Stage B | 104 (22) | 92 (19) | 105 (17) | |
| Stage C | 374 (78) | 384 (81) | 496 (82) | |
| Baseline AFP | ||||
| <200 ng/mL | 255 (53) | 286 (60) | NA | NA |
| ≥200 ng/mL | 222 (46) | 187 (39) | NA | NA |
| Missing | 1 (0.2) | 3 (0.6) | ||
| Number of disease site | ||||
| 1 | 207 (43) | 207 (44) | 25 (10) | |
| 2 | 167 (35) | 183 (38) | 79 (35) | |
| ≥3 | 103 (22) | 86 (18) | 122 (54) | |
| Missing | 1 (0.2) | 0 | ||
| Sites of disease involvement | ||||
| Liver | 441 (92) | 430 (90) | ||
| Lung | 163 (34) | 144 (30) | 112 (50) | |
| Bone | 51 (11) | 43 (9) | ||
| Lymph node | 127 (27) | 141 (30) | 72 (32) | |
| Other sites | 82 (17) | 97 (20) | ||
| Etiology of underlying liver disease | ||||
| Hepatitis B infection | 259 (52) | 244 (51) | 111 (18) | 165 (73) |
| Hepatitis C infection | 103 (22) | 135 (28) | 169 (28) | 19 (8.4) |
| Alcohol abuse | 33 (7) | 23 (4.8) | 159 (26) |
Based on variables captured at stratification for randomization for REFLECT and as described in the Food and Drug Administration–approved package insert for Nexavar, supplement by publication of the SHARP trial and the published results for the ASIA‐Pacific trial.
ClinicalTrials.gov [10].
Abbreviations: AFP, Alpha Fetoprotein; BCLC, Barcelona Clinic Liver Cancer; ECOG, Eastern Cooperative Oncology Group.
Lenvatinib was approved in the U.S. on February 13, 2015. Prior approvals for lenvantinib are summarized in Table 1. On August 15, 2018, the FDA approved lenvatinib for the first‐line treatment of patients with unresectable HCC based on the results of a single adequate and well‐controlled trial, Study 304 (REFLECT). The data reviewed and basis for this approval are discussed below.
Table 1.
Lenvatinib background information
| Structure |
|
|---|---|
| Mechanism of action | Inhibition of kinase activities of VEGF receptors VEGFR1 (FLT1), VEGFR2 (KDR), and VEGFR3 (FLT4). Inhibits other kinases implicated in pathogenic angiogenesis, tumor growth, cancer progression, and normal cellular functions, including FGF receptors FGFR1, 2, 3, and 4; platelet‐derived growth factor receptor α, KIT, and RET. |
| Pharmacokinetics | The maximum lenvatinib plasma concentration and the area under the concentration‐time curve in patients increased proportionally over the dose range of 3.2 mg (0.1 times the recommended clinical dose of 24 mg) to 32 mg (1.33 times the recommended clinical dose of 24 mg) with a median accumulation index of 0.96 (20 mg) to 1.54 (6.4 mg). |
| Prior approvals |
February 13, 2015: for the treatment of patients with locally recurrent or metastatic, progressive, radioactive iodine‐refractory differentiated thyroid cancer. May 13, 2016: for the treatment, in combination with everolimus, of patients with advanced renal cell carcinoma following one prior antiangiogenic therapy. |
Abbreviations: FGF, fibroblast growth factor; VEGF, vascular endothelial growth factor.
Clinical Trial Design
REFLECT is a randomized, international, open‐label, active‐controlled trial designed to establish the efficacy of lenvatinib for the first‐line treatment of patients with unresectable HCC. Key eligibility criteria were patients with HCC who were ineligible for local liver‐directed therapy; Child‐Pugh A and BCLC Stage C or Stage B, in which patients were ineligible for locoregional liver‐directed therapy; an Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 0 or 1; received no prior systemic therapy for HCC; and at least one measurable target lesion according to modified RECIST for HCC. Patients were randomized (1:1) to receive lenvatinib (12 mg orally, once daily, if baseline body weight ≥60 kg, or 8 mg orally once daily if baseline body weight <60 kg) or sorafenib 400 mg orally twice daily until clinical or radiological disease progression or unacceptable toxicity. Randomization was stratified by region (Western vs. Asia Pacific), presence of macroscopic portal vein invasion or extrahepatic spread (yes vs. no), ECOG PS (0 vs. 1), and body weight (<60kg vs. ≥60 kg). Tumor status was assessed every 8 weeks.
The primary efficacy endpoint was overall survival (OS); key secondary efficacy endpoints were progression‐free survival (PFS) and overall response rate (ORR) according to modified RECIST for HCC as assessed by the investigator. The primary analysis of OS was for noninferiority (NI); if the criterion for NI was met, then superiority of OS was tested. The primary analyses of PFS and ORR were for superiority.
The estimated sorafenib effect size for NI used in REFLECT was calculated from the results of two randomized, placebo‐controlled trials, the SHARP and Asia‐Pacific trials. The estimated hazard ratio (HR; 95% confidence interval [CI]) for survival was 0.69 (0.55–0.87) and 0.68 (0.50–0.93) in the SHARP and Asia‐Pacific trials, respectively. The pooled HR (two‐sided 95% CI) was 0.6865 (0.5709–0.8255). Because of the uncertainty regarding the validity of the constancy assumption (because the treatment effect of the active control drug was based on studies conducted approximately 10 years earlier), the FDA stated that Eisai would need to address the validity of the constancy assumption including the regional differences between the SHARP and Asia‐Pacific trials as compared with the REFLECT trial and potential differences in the diagnosis, treatment, and characteristics of patients with HCC since the SHARP and Asia‐Pacific trials were conducted. To account for this uncertainty, the FDA requested, and Eisai agreed, to reduce the magnitude of the NI margin from that calculated using a 50% retention of the treatment effect. Subsequently, Eisai designed the trial to use an NI margin of 1.08, corresponding to 60% of the treatment effect. Therefore, the criterion for NI was that the upper bound of the 95% CI for the OS HR of the REFLECT trial should be less than 1.08. The NI margin was calculated using the method described in Rothmann et al. [8].
The planned sample size of 940 patients used the following assumptions: accrual rate of 39 patients per month; HR of 0.8 favoring the lenvatinib arm; estimated median OS of 10 months and 12.5 months in the sorafenib and lenvatinib arms, respectively, approximately 97% power to meet criteria for NI, and approximately 82% power to demonstrate superiority with 700 death events. Two interim analyses were to be performed for futility to demonstrate NI, the first interim analysis was to be performed at 210 (30% information) deaths and the second interim analysis was at 490 (70% information) deaths. Overall survival was to be tested under a closed procedure and a fixed sequence procedure was used to control the overall type I error rate for the secondary endpoints of PFS and ORR at 0.05 (two‐sided), tested in this order after NI for OS was demonstrated.
Results
A total of 954 patients were enrolled from 183 sites across Asia, Europe, North America, Russia, and Israel. The baseline demographics and tumor characteristics for the REFLECT study population are summarized in Tables 2 and 3.
Efficacy
The efficacy results are summarized in Figure 1 and Table 4. REFLECT met its prespecified endpoint, demonstrating noninferior survival for lenvatinib as compared with sorafenib (HR, 0.92; 95% CI, 0.79–1.06) based on the prespecified NI margin of 1.08 but did not demonstrate superior overall survival for lenvatinib. REFLECT also demonstrated statistically significant improvements in investigator‐assessed PFS (HR, 0.66; 95% CI, 0.57–0.77; p < .001), corresponding to median PFS of 7.4 and 3.7 months and ORR of 24.1% versus 9.2% per modified RECIST for HCC (mRECIST) in the lenvatinib and sorafenib arms, respectively. Prior to initiation of the trial, the FDA expressed concerns regarding the reliable and accurate measurement of intrahepatic lesions in HCC; thus, the FDA recommended that the primary assessment of PFS and ORR be performed by an independent radiology facility (IRF). Given that the protocol‐specified primary analyses for investigator‐assessed PFS and ORR were significant, the FDA conducted exploratory analyses of PFS and ORR as assessed by an IRF according to RECIST v1.1, as well as mRECIST for HCC. These exploratory analyses were conducted because REFLECT was an open‐label trial with the potential for bias and to assess for constancy assumption and similarity of treatment effects in prior drug approvals for treatment of HCC. Whether measured according to mRECIST for HCC or RECIST v1.1, investigator assessment or IRF assessment, all analyses favored the lenvatinib arm. It is noted that the point estimates for ORR and the magnitude of the difference between arms were larger when using mRECIST for HCC rather than RECIST v1.1.
Figure 1.
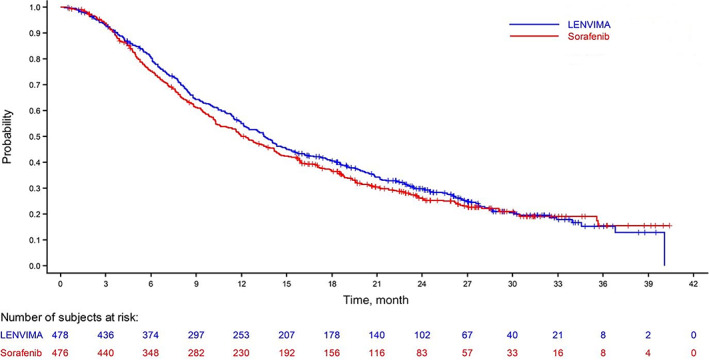
Kaplan‐Meier curves for overall survival in REFLECT trial.
Table 4.
Efficacy results in hepatocellular carcinoma in REFLECT
| Efficacy endpoint | Lenvatinib, n = 478 | Sorafenib, n = 476 |
|---|---|---|
| Overall survival | ||
| Number of deaths (%) | 351 (73) | 350 (74) |
| Median OS, (95% CI), mo | 13.6 (12.1–14.9) | 12.3 (10.4–13.9) |
| Hazard ratio (95% CI) a | 0.92 (0.79–1.06) | 0.92 (0.79–1.06) |
| Progression‐free survival by IRF per mRECIST for HCC | ||
| Number of events (%) | 311 (65) | 323 (68) |
| Median PFS (95% CI), mo | 7.3 (5.6–7.5) | 3.6 (3.6–3.7) |
| Hazard ratio (95% CI) a , b | 0.64 (0.55–0.75) | 0.64 (0.55–0.75) |
| Progression‐free survival by IRF per RECISTv1.1 | ||
| Number of events (%) | 307 (64) | 320 (67) |
| Median PFS in months (95% CI) | 7.3 (5.6–7.5) | 3.6 (3.6–3.9) |
| Hazard ratio (95% CI) a | 0.65 (0.56–0.77) | 0.65 (0.56–0.77) |
| Overall response rate by IRF per mRECIST for HCC, % (95% CI) | 41 (36–45) | 12 (10–16) |
| Overall response rate by IRF per RECIST v1.1, %, (95% CI) | 19 (15–22) | 7 (4–9) |
Stratified by region (Asia‐Pacific vs Western), macroscopic portal vein invasion or extrahepatic spread or both (yes, no), Eastern Cooperative Oncology Group performance status (0, 1), and body weight (<60 kg, ≥60 kg).
Nominal p values <.001
Abbreviations: CI, confidence interval; HCC, hepatocellular carcinoma; IRF, independent radiology facility; mRECIST, modified RECIST for HCC; OS, overall survival; PFS, progression‐free survival.
Safety
The analysis of safety use the “as‐treated” population comprising patients who received at least one dose of the study drugs, lenvatinib (n = 476) or sorafenib (n = 475). As predicted by the pharmacokinetic model, there was comparable exposure to lenvatinib among patients who received a starting dose of 8 mg and those who received a starting dose of 12 mg. Lenvatinib exposures were also similar between patients with mild and moderate hepatic impairment.
Forty‐three percent (43%) of lenvatinib‐treated patients and 30% of sorafenib‐treated patients experienced serious adverse events (SAEs), defined as events resulting in death, hospitalization, or serious morbidity (or requiring medical intervention to avoid these outcomes). Both fatal (12.8% vs. 7.6%) and nonfatal (39.7% vs. 26.9%) SAEs occurred more frequently in lenvatinib‐treated patients compared with sorafenib‐treated patients. The most frequently reported nonfatal SAEs were hepatic encephalopathy (5%), ascites (3%), hepatic failure (3%), and decreased appetite (2%). The most frequent adverse reactions or laboratory abnormalities leading to discontinuation of lenvatinib were fatigue (1%), hepatic encephalopathy (2%), hyperbilirubinemia (1%), and hepatic failure (1%). The majority of patients (62%) required dose modification; the most common (≥5%) adverse reactions leading to dose reduction or interruption of lenvatinib were fatigue (9%), decreased appetite (8%), diarrhea (8%), proteinuria (7%), hypertension (6%), and palmar‐plantar erythrodysesthesia syndrome (5%). The most common (incidence ≥10%) adverse reactions are listed in Table 5.
Table 5.
Adverse reactions occurring in ≥10% of patients in the lenvatinib‐treated arm in REFLECT
| Adverse reaction | LENVIMA, n = 476 | Sorafenib, n = 475 | ||
|---|---|---|---|---|
| Grade 1–4, % | Grade 3–4, % | Grade 1–4, % | Grade 3–4, % | |
| Endocrine | ||||
| Hypothyroidism | 21 | 0 | 3 | 0 |
| Gastrointestinal | ||||
| Diarrhea | 39 | 4 | 46 | 4 |
| Abdominal pain | 30 | 3 | 28 | 4 |
| Nausea | 20 | 1 | 14 | 1 |
| Vomiting | 16 | 1 | 8 | 1 |
| Constipation | 16 | 1 | 11 | 0 |
| Ascites | 15 | 4 | 11 | 3 |
| Stomatitis | 11 | 0.4 | 14 | 1 |
| General | ||||
| Fatigue | 44 | 7 | 36 | 6 |
| Pyrexia | 15 | 0 | 14 | 0.2 |
| Peripheral edema | 14 | 1 | 7 | 0.2 |
| Metabolism/nutrition | ||||
| Decreased appetite | 34 | 5 | 27 | 1 |
| Decreased weight | 31 | 8 | 22 | 3 |
| Musculoskeletal/connective Tissue | ||||
| Arthralgia/myalgia | 31 | 1 | 20 | 2 |
| Nervous system | ||||
| Headache | 10 | 1 | 8 | 0 |
| Renal/urinary | ||||
| Proteinuria | 26 | 6 | 12 | 2 |
| Respiratory, thoracic, and mediastinal | ||||
| Dysphonia | 24 | 0.2 | 12 | 0 |
| Skin/subcutaneous tissue | ||||
| Palmar‐plantar erythrodysesthesia syndrome | 27 | 3 | 52 | 11 |
| Rash | 14 | 0 | 24 | 2 |
| Vascular | ||||
| Hypertension | 45 | 24 | 31 | 15 |
| Hemorrhagic events | 23 | 4 | 15 | 4 |
Discussion
The FDA concluded that the overall risk‐benefit assessment of this supplemental application was favorable and that the evidence provided from the REFLECT trial demonstrated substantial evidence of the effectiveness of lenvatinib for the first‐line treatment of patients with unresectable hepatocellular carcinoma (Table 6). Approval of a new drug or new indication for treatment of cancer that is based on demonstration of noninferiority to another approved drug is an acceptable but rarely used strategy. The REFLECT trial demonstrated noninferior OS for lenvatinib compared with sorafenib in patients with unresectable or metastatic HCC who had not received prior systemic treatment for their disease. Thus, the FDA's review carefully considered the adequacy of the development program to support approval based on noninferiority.
Table 6.
Food and Drug Administration benefit‐risk summary
| Dimension | Evidence and uncertainties | Conclusions and reasons |
|---|---|---|
| Analysis of condition |
Approximately 42,220 new cases and approximately 30,200 deaths due to HCC estimated in the U.S. in 2018. a Estimated 5‐year survival rate is 17.7%. The 5‐year survival rates are 11% with regional (nodal) involvement and 2% with metastatic disease. |
Unresectable HCC is a serious and life‐threatening condition with unmet medical needs. |
| Current treatment options |
Available therapy for untreated unresectable or metastatic Child‐Pugh A HCC is sorafenib. Sorafenib approval based on a single (1:1) placebo‐controlled international trial demonstrating a significant improvement in OS (HR, 0.69; 95% CI, 0.55–0.87) and PFS (HR, 0.58; 95% CI, 0.45–0.74). |
One FDA‐approved drug for the first‐line treatment of unresectable HCC. |
| Benefit |
Lenvatinib's effectiveness is based on results of a single adequate and well‐controlled trial, Study 304 (REFLECT), randomizing 954 patients to lenvatinib (n = 478) or sorafenib (n = 476). REFLECT demonstrated that lenvatinib is NI to sorafenib for OS (HR, 0.92; 95% CI, 0.79–1.06) according to prespecified NI margin (≤1.08); median OS was 13.6 months (95% CI, 12.1–14.9) and 12.3 months (95% CI, 10.4–13.9) in lenvatinib and sorafenib arms, respectively. REFLECT also demonstrated a significant improvement in investigator‐assessed PFS according to mRECIST. Similar treatment effects on PFS (HR,0.65; 95% CI, 0.56–0.77) were observed by IRF‐assessed per RECISTv1.1 and mRECIST. REFLECT demonstrated a significant improvement in investigator‐assessed ORR per mRECIST (24.1% vs. 9.2%) for lenvatinib. Similar effects on ORR assessed by IRF were observed. The magnitude of ORR was greater when assessed using mRECIST than with RECISTv1.1 in both treatment arms. |
REFLECT demonstrated that lenvatinib has an effect on OS that is noninferior to sorafenib and demonstrated superior investigator‐assessed PFS and ORR. PFS and ORR effects supported by IRF assessment. |
| Risk and risk management |
The single‐agent lenvatinib safety profile was previously established in 1,421 patients with advanced cancers across multiple clinical trials. The safety profile in lenvatinib‐treated patients in REFLECT identified no new or unexpected adverse reactions. |
Product labeling adequately conveys risks of lenvatinib and mitigates risks of serious toxicities to facilitate a choice between lenvantinib or sorafenib treatment. |
ClinicalTrials.gov [10].
Abbreviations: CI, confidence interval; FDA, Food and Drug Administration; HCC, hepatocellular carcinoma; HR, hazard ratio; IRF, independent radiology facility; mRECIST, modified RECIST for HCC; NI, noninferiority; ORR, overall response rate; OS, overall survival; PFS, progression‐free survival.
In evaluating this application, the FDA considered whether REFLECT was adequate in design and conduct to support claims of noninferiority, whether this single trial provided substantial evidence of effectiveness, and whether the results can be extrapolated to the U.S. population.
Regarding specific considerations for noninferiority studies, the FDA evaluated whether the following inter‐linked elements in the FDA guidance for industry “Non‐Inferiority Clinical Trials to Establish Effectiveness” (NI guidance) had been met [9].
There is reliable information about the effect the active control drug had in past studies.
There is reason to believe the effect the active control drug has in the current NI study is similar to the effect observed in past studies.
The NI study provides reliable information about the effect of the test drug relative to the comparator.
The FDA concluded that the historical control effect size for the sorafenib arm was well‐estimated based on the meta‐analysis of two trials. The design and results of SHARP were assessed by the FDA in the application supporting approval of sorafenib for the first‐line treatment of unresectable HCC, concluding that SHARP was an adequate and well‐controlled trial. The FDA did not review the results of the Asia‐Pacific trial but relied on the published results in a peer‐reviewed journal, which discussed the prespecified plan for analysis of OS, supplemented by the information on this trial listed at www.clinicaltrials.gov [NCT00492752], and reached a conclusion that it was an adequate and well‐controlled trial capable of estimating the treatment effect [10]. Additionally, the FDA concluded that the observed relative treatment effect on OS, as reflected in the HR, was similar across the SHARP and Asia‐Pacific trials despite differences in the ethnic composition and other population factors and the meta‐analysis provided a reliable estimate of the historical treatment effect of sorafenib on OS.
In evaluating whether the treatment effect of sorafenib on survival in REFLECT was similar to that observed in the historical studies (i.e., meets the constancy assumption), the FDA focused on the similarity of the eligibility criteria, stratification variables for randomization, analysis plan for OS, and patient population enrolled in REFLECT and the studies used to generate the NI margin. There were no major advances in diagnostic criteria or ancillary/supportive care treatment that would be likely to affect survival in patients receiving initial systemic treatment for HCC during the conduct of these trials. All three trials used the same sorafenib dose and similar dose modification scheme for sorafenib toxicity; limitations on concomitant medications (concurrent antineoplastic drugs or those with potential drug interactions) were also similar. Differences in eligibility criteria across trials (e.g., enrollment of patients with ECOG PS 2 in SHARP and Asia‐Pacific trials but not REFLECT; limitation of enrollment to BCLC Stages A and B in REFLECT but absence of such restrictions in the SHARP and Asia‐Pacific trials) were also deemed unlikely to affect treatment outcomes given the small percentages of patients with ECOG PS of 2 (8% in SHARP and 5% in Asia‐Pacific), and no patients with HCC of BCLC stage A in the SHARP or Asia‐Pacific and unlikely to have adversely impacted the validity of the NI margin. The stratification variable for body weight in REFLECT was deemed unlikely to affect treatment outcomes. Although differences in baseline demographics (i.e., proportion of Asian patients, proportion of patients ≥65 years, and proportion of patients with ECOG PS 0) may have had potentially greater impact on the constancy assumption, the FDA concluded that prognostic effects on OS were smaller than treatment effects and noted the consistent relative treatment effects across trials.
The FDA further determined that REFLECT was an adequate and well‐controlled trial that provided reliable information about the relative treatment effects. In reaching this conclusion, The FDA noted that REFLECT was prospectively designed and provided independent evaluation of tumor‐based endpoints that confirmed investigator assessments, and potential sources of bias did not undermine confidence in the reliability of the results. The FDA reached this conclusion despite inadvertent unblinding of data sets during the conduct of the trial, because data integrity and reliability assessed by an audit conducted by a qualified independent third party verified key subject‐level data from the case report forms and IRF results were accurately captured in data sets.
The FDA concluded that the application provided substantial evidence of effectiveness based on REFLECT. According to the FDA guidance for industry, “Providing Clinical Evidence of Effectiveness for Human Drug and Biological Products,” a single adequate and well‐controlled study can support approval if the results provide statistically persuasive efficacy findings while demonstrating effects on secondary endpoints and consistency across subsets such that a second trial would be ethically or practically impossible to perform [11]. Furthermore, as stated in the NI guidance, a single trial acceptable for approval when there is availability of other relevant information. REFLECT demonstrated internal consistency for a treatment effect for lenvatinib across secondary efficacy endpoints, regardless of the assessor (investigator or IRF) or response criteria used.
Another key review issue for this application was the ability to extrapolate the results of REFLECT to the U.S. patient population, given that one‐third of the patients were enrolled in non‐Asian sites. Exploratory post hoc subset analyses of OS showed variability in treatment effects by country and region. Subgroup analyses of OS in the Western regions (HR, 1.08; 95% CI, 0.82–1.42) appeared inconsistent with OS results in the Asia‐Pacific region (HR, 0.86; 95% CI, 0.72–1.02), as did U.S. sites compared with other countries. The FDA noted that such subgroup analyses had several limitations. First, REFLECT was not designed to evaluate efficacy in any country or region, randomization was not stratified by country, and subgroup analyses by country resulted in small sample sizes with wide confidence intervals. The FDA concluded that observed differences were more likely due to chance or other unknown factors. Taking the limitations of post hoc analyses into consideration and that the results of the SHARP and Asia‐Pacific trials yielded similar relative treatment effects (HR) for OS for sorafenib, the FDA concluded that the results of REFLECT were applicable to the U.S. patient population.
Conclusion
The REFLECT trial demonstrated that treatment effect of lenvatinib on OS is noninferior to sorafenib. The upper and lower limits of the confidence interval around the hazard ratio indicate the potential for up to 6% higher immediate risk of death and up to 21% lower immediate risk of death for lenvatinib compared with sorafenib in patients with unresectable or metastatic HCC who have not received prior systemic treatment for their disease. The application was strengthened by demonstration of a statistically significant and large improvements in PFS and ORR according to both investigator and IRF assessment, irrespective of the response criteria used. The observed point estimates and magnitude of differences in ORR are dependent on the response criteria. These results demonstrate that lenvatinib provides clinically meaningful treatment effects to patients with unresectable HCC, a disease that confers a poor prognosis, which outweigh its toxicity and support a conclusion that lenvatinib is an acceptable alternative to sorafenib as the initial systemic treatment.
Author Contributions
Conception/Design: Abhilasha Nair, Kelie Reece, Martha B. Donoghue, Weishi (Vivian) Yuan, Lisa Rodriguez, Patricia Keegan
Collection and/or assembly of data: Abhilasha Nair, Martha B. Donoghue, Weishi (Vivian) Yuan, Lisa Rodriguez
Data analysis and interpretation: Abhilasha Nair, Martha B. Donoghue, Weishi (Vivian) Yuan, Lisa Rodriguez, Patricia Keegan
Manuscript writing: Abhilasha Nair, Kelie Reece, Martha B. Donoghue, Patricia Keegan
Final approval of manuscript: Abhilasha Nair, Kelie Reece, Martha B. Donoghue, Weishi (Vivian) Yuan, Lisa Rodriguez, Patricia Keegan, and Richard Pazdur
Disclosures
The authors indicated no financial relationships.
Acknowledgments
This is a U.S. Government work. There are no restrictions on its use.
Disclosures of potential conflicts of interest may be found at the end of this article.
No part of this article may be reproduced, stored, or transmitted in any form or for any means without the prior permission in writing from the copyright holder. For information on purchasing reprints contact Commercialreprints@wiley.com. For permission information contact permissions@wiley.com.
References
- 1. American Cancer Society . Cancer Facts and Figures: 2019. Atlanta, GA: American Cancer Society; 2019. Available at https://www.cancer.org/content/dam/cancer‐org/research/cancer‐facts‐and‐statistics/annual‐cancer‐facts‐and‐figures/2019/cancer‐facts‐and‐figures‐2019.pdf. Accessed January 18, 2020. [Google Scholar]
- 2. Yang JD, Hainaut P, Gores GJ et al. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol 2019;16:589–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cancer Stat Facts: Liver and intrahepatic bile duct cancer. National Cancer Institute Surveillance, Epidemiology, and End Results Program. Available at https://seer.cancer.gov/statfacts/html/livibd.html. Accessed January 18, 2020.
- 4. Puoti C. New insights on hepatocellular carcinoma: Epidemiology and clinical aspects. Hepatoma Res 2018;4:57. [Google Scholar]
- 5.Nexavar. Full prescribing information. Available at https://www.accessdata.fda.gov/drugsatfda_docs/label/2007/021923s004s005s006s007lbl.pdfAccessed December 10, 2019.
- 6. Llovet JM, Ricci S, Mazzaferro V et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008;359:378–390. [DOI] [PubMed] [Google Scholar]
- 7. Cheng AL, Kang YK, Chen Z et al. Efficacy and safety of sorafenib in patients in the Asia‐Pacific region with advanced hepatocellular carcinoma. Lancet Oncol 2009;10:25–34. [DOI] [PubMed] [Google Scholar]
- 8. Rothmann M, Li N, Chen G et al. Design and analysis of non‐inferiority mortality trials in oncology. Stat Med 2003;22:239–264. [DOI] [PubMed] [Google Scholar]
- 9. U.S. Department of Health and Human Services . Non‐Inferiority Clinical Trials to Establish Effectiveness. Guidance for Industry. Silver Spring, MD: Food and Drug Administration; 2016. Available at https://www.fda.gov/media/78504/download. Accessed December 11, 2019. [Google Scholar]
- 10. Clinicaltrials.gov. History of changes for study: NCT00492752: A randomized, double‐blinded, placebo‐controlled study of sorafenib in patients with advanced hepatocellular carcinoma. Available at https://clinicaltrials.gov/ct2/history/NCT00492752?V_15=View#StudyPageTop. Accessed December 11, 2019.
- 11. U.S. Department of Health and Human Services . Providing Clinical Evidence of Effectiveness for Human Drug and Biological Products. Guidance for Industry. Silver Spring, MD: Food and Drug Administration; 1998. Available at: https://www.fda.gov/media/71655/download. Accessed May 1, 2020. [Google Scholar]