

Abstract
Background
The prognostic implication of wild‐type APC (APC‐WT) in microsatellite stable (MSS) metastatic colorectal cancer (mCRC) is not well defined.
Materials and Methods
APC prognostic value was evaluated retrospectively in two independent cohorts of patient with MSS mCRC with a confirmatory analysis from a public data set from Memorial Sloan Kettering Cancer Center (MSKCC).
Results
In comparison with the APC‐mutant (APC‐MT) population (n = 255), APC‐WT patients (n = 86) tended to be younger (59% of age < 40 vs. 26% of age > 50), right‐sided (41.7% vs. 27%), BRAF V600E mutated (23.3% vs. 0.8%), and KRAS wild type (65.1% vs. 49.8%). Alternative WNT pathway alterations, RNF43 and CTNNB1, were over‐represented in the APC‐WT versus APC‐MT population (7% vs. 0.4% and 4.7% vs. 0.4%, respectively). APC‐WT patients had a worse overall survival (OS) than APC‐MT patients (22.6 vs. 45.6 months, p < .0001). Using a multivariate model correcting for primary tumor location, RAS and BRAF status, APC‐WT was predictive of poor survival (APC‐MT vs. APC‐WT, hazard ratio [HR], 0.62; 95% confidence interval [CI], 0.44–0.86, p = .0037). The prognostic implication of APC‐WT on OS was confirmed further in a similar multivariate model of 934 stage IV patients from MSKCC public database (APC‐MT vs. APC‐WT, HR, 0.63, 95% CI, 0.49–0.81, p < .0001).
Conclusion
APC‐WT is associated with poor OS in MSS mCRC regardless of RAS and BRAF status. Compared with APC‐MT mCRC tumors, APC‐WT tumors were associated with other Wnt activating alterations, including RNF43 and CTNBB1. Our data suggest alternative therapy needs to be investigated in APC‐WT patients.
Implications for Practice
Patients with microsatellite stable metastatic colorectal cancer with wild‐type APC had a worse overall survival than patients with mutated APC regardless of RAS/RAF status. APC status should be considered as a stratification factor in prospective trials, and novel therapeutic strategies need to be developed for this subgroup of patients.
Keywords: APC, Metastatic colorectal cancer, Microsatellite stable, prognostic, Wnt signaling
Short abstract
Despite advancements in therapy, colorectal cancer is still a leading cause of cancer‐related death in the U.S. This article evaluates the prognostic value of wild‐type APC in patients with microsatellite stable colorectal cancer.
Introduction
With an estimated 51,020 deaths in 2019, colorectal cancer is still a leading cause of cancer‐related death in the U.S. [1]. Despite the advancements in cytotoxic, biological, and targeted therapy over the last 2 decades, the overall survival of metastatic colorectal cancer at 5 years is 14% [2, 3]. It is increasingly acknowledged that colorectal cancer is a highly heterogeneous disease with diverse molecular and clinical features that affect therapeutic outcomes [4]. A comprehensive genomic disease classification‐beyond RAS, BRAF, and microsatellite stable (MSS) and microsatellite instable (MSI) is needed to better guide the management of this disease [5, 6].
Adenomatous polyposis coli (APC) is a tumor‐suppressor gene that acts as a gatekeeper of the Wnt/β‐catenin pathway by regulating β‐catenin phosphorylation [4]. Somatic APC mutations occur in approximately 75% of sporadic colorectal cancer and play a crucial role in the initiation of the adenoma‐carcinoma pathway [7, 8, 9]. A comparison of genomic alterations between early and late‐onset colorectal cancer showed a decreased incidence of APC mutations in younger patients [10]. Colorectal cancers with mutated APC conferred better overall survival (OS) than wild‐type tumors when treated with an epidermal growth factor receptor (EGFR) inhibitor [11]. These data suggest that APC mutation may account for EGFR inhibitor sensitivity and provide rationale for refining treatment guidelines in addition to extended RAS/RAF testing [11, 12].
Although several studies suggest that patients with colorectal cancer lacking APC mutation carry a worse outcome than APC‐mutated patients [13, 14, 15], no prior studies have focused on the impact of APC‐WT status on the survival of a large cohort of MSS metastatic CRC, especially when factoring in other established prognostic variables such RAS, BRAF, and sidedness. In this study, we analyzed the genomic profiles of 341 clinically characterized, sporadic, stage IV MSS colorectal tumors. A multivariate model including clinical covariates and RAS/BRAF status was deployed to further define the prognostic potential of APC. Our observations were further validated in a cohort of 934 patients with stage IV MSS colorectal cancer from Memorial Sloan Kettering Cancer Center (MSKCC) public data base.
Materials and Methods
Patients
We analyzed 287 patients with MSS stage IV colorectal cancer who had undergone treatment at the City of Hope National Medical Center (COH; Duarte, CA) between 2013 and 2019 and 54 patients with MSS stage IV colorectal cancer who had undergone treatment at University of California, Davis (UCD; Davis, CA) between 2012–2019. Patients’ demographics including gender, age, primary tumor location, and survival status were obtained from chart abstraction of each patient's electronic medical record. This study was approved by the Institutional Review Board IRB 14361 (COH) and IRB 1484151‐1 (UCD). Memorial Sloan Kettering Cancer Center public data set of 934 patients with metastatic colorectal cancer was used for the validation of our results. [15]
Genomic Analysis
Comprehensive genomic profiling of COH and UCD cohorts were conducted through next‐generation sequencing via FoundationOne (Foundation Medicine Inc., Cambridge, MA). The genomic analysis was conducted on DNA extracted form formalin‐fixed paraffin‐embedded tumor samples retrieved from surgical resection, or biopsies [16]. Samples from MSKCC were sequenced using the MSK‐IMPACT assay in the clinical laboratories of the Molecular Diagnostics Service at MSKCC [15].
Mutation Annotation
Somatic mutations were annotated based on OncoKB, a precision oncology knowledge base, for their mutation type and clinical implication (supplementary online Table 1) [17]. Variants implicated as oncogenic, likely oncogenic, and predicted oncogenic were recruited to this analysis. Variants with unknown significance, including likely neutral, inconclusive, and unknown, were filtered out.
Table 1.
Characteristics of patients with microsatellite stable metastatic colorectal cancer
| Characteristics | COH/UCD | MSKCC | ||||||
|---|---|---|---|---|---|---|---|---|
| Total (%)(n = 341) | APC‐WT (n = 86) | APC‐MT (n = 255) | p value | Total (%)(n = 934) | APC‐WT (n = 216) | APC‐MT (n = 718) | p value | |
| Age at diagnosis, median (range), yr | 56 (16–88) | 55 (16–85) | 56 (20–88) | .16 | 53 (13–93) | 53 (13–83) | 53 (22–93) | .25 |
| Gender, n (%) | ||||||||
| Male | 192 (56.3) | 44 (51.2) | 148 (58.0) | 494 (52.9) | 113 (52.3) | 381 (53.1) | ||
| Female | 149 (43.7) | 42 (48.8) | 107 (42.0) | .32 | 440 (47.1) | 103 (47.7) | 337 (46.9) | .88 |
| Site, a n (%) | ||||||||
| Right | 103 (30.7) | 35 (41.7) | 68 (27.0) | 241 (26.1) | 75 (35.5) | 166 (23.3) | ||
| Left | 233 (69.3) | 49 (58.3) | 184 (73.0) | .014 | 681 (73.9) | 136 (64.5) | 545 (76.7) | 6.60 × 10−04 |
| BRAF V600E, n (%) | ||||||||
| Mutated | 22 (6.5) | 20 (23.3) | 2 (0.8) | 49 (5.2) | 31 (14.4) | 18 (2.5) | ||
| Nonmutated | 319 (93.5) | 66 (76.7) | 253 (99.2) | 2.60 × 10−11 | 885 (94.8) | 185 (85.6) | 700 (97.5) | 6.98 × 10−10 |
| KRAS, n (%) | ||||||||
| Mutated | 158 (46.3) | 30 (34.9) | 128 (50.2) | 424 (45.4) | 92 (42.6) | 332 (46.2) | ||
| Nonmutated | 183 (53.7) | 56 (65.1) | 127 (49.8) | .017 | 510 (54.6) | 124 (57.4) | 386 (53.8) | .35 |
| NRAS, n (%) | ||||||||
| Mutated | 15 (4.4) | 3 (3.5) | 12 (4.7) | 38 (4.1) | 1 (.5) | 37 (5.2) | ||
| Nonmutated | 326 (95.6) | 83 (96.5) | 243 (95.3) | .77 | 896 (95.9) | 215 (99.5) | 681 (94.8) | 6.70 × 10−04 |
| TP53, n (%) | ||||||||
| Mutated | 257 (75.4) | 69 (80.2) | 188 (73.7) | 727 (77.8) | 169 (78.2) | 558 (77.7) | ||
| Nonmutated | 84 (24.6) | 17 (19.8) | 67 (26.3) | .25 | 207 (22.2) | 47 (21.8) | 160 (22.3) | .93 |
Data not available: 5 cases in the COH/UCD and 12 cases in theMSKCC cohort.
Abbreviations: APC‐MT, APC mutation; APC‐WT, wild‐type APC; COH, City of Hope National Medical Center; UCD, University of California, Davis.
Statistical Analysis
For baseline clinical and genomic characteristics, Fisher's exact test was used to compare proportions between patient groups based on APC mutation status. OS was examined from the date of documentation of metastatic disease to the date of death. Differences in OS between groups with or without clinically relevant mutations at a given gene of interest were compared using Kaplan‐Meier curves, with p values calculated via log‐rank test. Both univariate and multivariate Cox regression models were applied to estimate the hazard ratios and confidence intervals of survival based on mutation status and other clinical factors.
Results
Study Population
We analyzed a total of 341 patients with stage IV MSS colorectal adenocarcinomas (287 from COH, 54 from UCD). Median age was 56 years, 56.3% were male, and 43.7% were female. A total of 69.3% (233 of 336) of patients had a left‐sided primary tumor. The genomic features of our cohort were, in general, similar to those reported in the MSKCC database (Table 1). APC mutations were found in 74.8% (255 of 341) of tumors in the pooled COH/UCD cohort. The frequencies of mutations in KRAS, NRAS, BRAF, and TP53 were 46.3% (158 of 341), 4.4% (25 of 341), 6.5% (22 of 341), and 75.4% (257 of 341), respectively.
Wild‐type‐APC Was Associated with Right‐Sided Primary, BRAF V600E Mutation, and Younger Age
Wild‐type APC (APC‐WT) was present in 25.2% (86 of 341) of the COH/UCD cohort (Table 1). Right‐sided primary tumor was found in 41% of patients with APC‐WT versus 27% of patients with APC mutation (APC‐MT; p = .014). Right‐sided tumors were also more frequent in APC‐WT versus APC‐MT tumors within the MSKCC cohort (35.5% vs. 23.3%, p = .00066). BRAF V600E and APC mutations rarely co‐occurred. In the COH/UCD cohort, 23.3% of the APC‐WT tumors harbored BRAF V600E mutation, whereas only 0.8% of the APC‐MT tumors coexisted with BRAF V600E mutations (p < .0001). Similar findings were observed in the MSKCC cohort (14.4% BRAF mutations in APC‐WT vs. 2.5% in APC‐MT, p < .0001). Wild‐type KRAS was found in 65.1% of APC‐WT tumors and 49.8% of APC‐MT tumors (p = .017), whereas no significant difference was observed in the MSKCC cohort. No significant difference in NRAS mutations was observed between APC‐WT and APC‐MT tumors in the COH/UCD cohort, whereas a significant increase in NRAS mutations was noted in the APC‐MT population in MSKCC cohort (5.2% in APC‐MT vs. 0.5% in APC‐WT, p = .00067).The frequency of mutations in TP53 were similar between APC‐WT and APC‐MT population in both cohorts (Table 1). Interestingly, 59.3% of early onset patients (age < 40) from COH/UCD cohort were APC‐WT, whereas 25.4% of later onset patients (age > 50) were APC‐WT (p = .000563). MSKCC cohort analysis confirmed a similar trend, 30.3% of early onset patients were APC‐WT, whereas 22.2% of late onset patients were APC‐WT (p = .064; supplemental online Table 2).
Table 2.
Alterations in genes associated with Wnt signaling pathway
| COH/UCD | MSKCC | |||||||
|---|---|---|---|---|---|---|---|---|
| Characteristics | Total (%) (n = 341) | APC‐WT (n = 86) | APC‐MT (n = 255) | p value | Total (%) (n = 934) | APC‐WT (n = 216) | APC‐MT (n = 718) | p value |
| ARID1A | ||||||||
| Mutated | 13 (3.8) | 3 (3.5) | 10 (3.9) | 0.999 | 34 (3.6) | 3 (1.4) | 31 (4.3) | .059 |
| Nonmutated | 328 (96.2) | 83 (96.5) | 245 (96.1) | 900 (96.4) | 213 (98.6) | 687 (95.7) | ||
| CTNNB1 | ||||||||
| Mutated | 5 (1.5) | 4 (4.7) | 1 (0.4) | .015 | 11 (1.2) | 8 (3.7) | 3 (0.4) | .00063 |
| Nonmutated | 336 (98.5) | 82 (95.3) | 254 (99.6) | 923 (98.8) | 208 (96.3) | 715 (99.6) | ||
| FBXW7 | ||||||||
| Mutated | 24 (7.0) | 7 (8.1) | 17 (6.7) | .82 | 71 (7.6) | 7 (3.2) | 64 (8.9) | .005 |
| Nonmutated | 317 (93.0) | 79 (91.9) | 238 (93.3) | 863 (92.4) | 209 (96.8) | 654 (91.1) | ||
| RNF43 | ||||||||
| Mutated | 7 (2.1) | 6 (7.0) | 1 (0.4) | .0013 | 23 (2.5) | 21 (9.7) | 2 (0.3) | 3.26× 10−12 |
| Nonmutated | 334 (97.9) | 80 (93.0) | 254 (99.6) | 911 (97.5) | 195 (90.3) | 716 (99.7) | ||
| SOX9 | ||||||||
| Mutated | 27 (7.9) | 2 (2.3) | 25 (9.8) | .035 | 78 (8.4) | 10 (4.6) | 68 (9.5) | .024 |
| Nonmutated | 314 (92.1) | 84 (97.7) | 230 (90.2) | 856 (91.6) | 206 (95.4) | 650 (90.5) | ||
Abbreviations: COH, City of Hope National Medical Center; MSKCC, Memorial Sloan Kettering Cancer Center; MT, mutant; UCD, University of California, Davis; WT, wild type.
Wild‐type‐APC Was Associated with Alternative WNT Pathway Activating Mutations
Given that WNT pathway is activated in the majority of colorectal cancers, we analyzed mutation frequencies of WNT signaling‐related molecules other than APC in the COH/UCD cohort and MSKCC cohort. We identified five recurrently mutated genes (mutations present more than once in the COH/UCD and MSKCC cohort) involved in the WNT signaling pathway, which include ARID1A, CTNNB1, FBXW7, RNF43, and SOX9 (Table 2). No significant difference was observed in the frequencies of mutations in ARID1A and FBXW7 in the APC‐WT versus the APC‐MT tumors in the COH/UCD cohort, whereas the frequencies of mutations in FBXW7 was higher in the APC‐mutated tumors in the MSKCC cohort. The active mutation of CTNBB1, a key molecule of WNT pathway downstream of APC, was significantly more prevalent in the APC‐WT tumors compared with the APC‐MT tumors (COH/UCD cohort, 4.7% vs. 0.4%, p = .015; MSKCC cohort, 3.7% vs. 0.4%, p = .00063). Mutations in RNF43, a WNT signaling inhibitor, were significantly more frequent in the APC‐WT tumors compared with the APC‐MT tumors (COH/UCD cohort, 7% vs. 0.4%, p = .0013; MSKCC cohort, 9.7% vs. 0.3%, p < .0001). In contrast, SOX9, a positive regulator of WNT signaling, was significantly more mutated in APC‐MT tumors compared with APC‐WT tumors (COH/UCD cohort, 9.8% vs. 2.3%, p = .035; MSKCC cohort, 9.5% vs. 4.6%, p = .024). Within the APC‐WT population, BRAF V600E mutation was associated with higher RNF43 mutations, whereas non‐BRAF V600E was associated with higher RAS mutation frequency and a trend of higher alternative WNT pathway mutations, such as ARID1A, CTNNB1, and FBXW7 (supplemental online Table 3). In summary, alternative WNT pathway activating mutations were significantly more prevalent in the APC‐WT tumors compared with the APC‐MT tumors.
Table 3.
Univariate survival model
| Clinicopathologic variable | COH/UCD (n = 341) | MSKCC (n = 934) | ||
|---|---|---|---|---|
| HR (95%CI) | p value | HR (95%CI) | p value | |
| Age at diagnosis, continuous, yr | 1.00 (0.98–1.01) | .553 | 1.01 (1.00–1.02) | .294 |
| Gender, female vs. male | 1.00 (0.76–1.32) | .986 | 0.96 (0.78–1.19) | .713 |
| Primary tumor location, right vs. left | 1.61 (1.20–2.16) | .0017 | 1.64 (1.30–2.07) | 3.66 × 10−05 |
| APC, mutated vs. nonmutated | 0.54 (0.40–0.73) | 5.95 × 10−05 | 0.55 (0.44–0.70) | 8.36 × 10−07 |
| KRAS, mutated vs. nonmutated | 1.11 (0.85–1.46) | .447 | 1.37 (1.10–1.70) | .0044 |
| NRAS, mutated vs. nonmutated | 1.20 (0.59–2.45) | .622 | 1.75 (0.11–2.76) | .0153 |
| BRAF, mutated vs. nonmutated | 2.00 (1.30–3.08) | .0016 | 3.10 (2.27–4.24) | 9.67 × 10−13 |
| BRAF V600E, mutated vs. nonmutated | 2.56 (1.50–4.35) | 5.47 × 10−04 | 4.61 (3.22–6.61) | 8.17 × 10−17 |
| TP53, mutated vs. nonmutated | 1.33 (0.93–1.88) | .114 | 1.00 (0.77–1.30) | .989 |
| ARID1A, mutated vs. nonmutated | 1.11 (0.56–2.17) | .769 | 1.03 (0.56–1.87) | .934 |
| CTNNB1, mutated vs. nonmutated | 0.95 (0.35–2.55) | .913 | 1.67 (0.86–3.26) | .133 |
| FBXW7, mutated vs. nonmutated | 0.48 (0.25–0.91) | .025 | 1.20 (0.79–1.84) | .394 |
| RNF43, mutated vs. nonmutated | 4.18 (1.84–9.48) | 6.38 × 10−04 | 3.75 (2.23–6.32) | 6.8 × 10−07 |
| SOX9, mutated vs. nonmutated | 0.51 (0.28–0.93) | .029 | 0.75 (0.49–1.15) | .187 |
Abbreviations: CI, confidence interval; COH, City of Hope National Medical Center; HR, hazard ratio; MSKCC, Memorial Sloan Kettering Cancer Center, UCD, University of California, Davis.
Wild‐type‐APC Was Associated with Poor Prognosis in Metastatic MSS Colorectal Cancer
A systematic univariate survival analysis including patient age, gender, primary tumor location, and genetic alterations in our study showed significant differences in survival associated with tumor location, APC, BRAF and RNF43 mutation (Table 3). Similar to numerous prior studies, BRAF V600E mutation was associated with poor prognosis in the COH/UCD cohort (hazard ratio [HR], 2.56; 95% confidence interval [CI], 1.50–4.35; p < .0001) and MSKCC cohort (HR, 4.61; 95% CI, 3.22–6.61; p < .0001). Right‐sided tumors were associated with poor prognosis in both the COH/UCD and MSKCC cohort (COH/UCD cohort: HR, 1.61; 95% CI, 1.20–2.16; p = .0017; MSKCC cohort: HR, 1.64; 95% CI, 1.30–2.07; p < .0001). Although rare, RNF43 mutations were associated with poor prognosis in both cohorts (COH/UCD cohort: HR, 4.18; 95% CI, 1.84–9.18; p < .0001; MSKCC cohort: HR, 3.75; 95% CI, 2.23–6.32; p < .0001). APC‐WT was associated with inferior outcome in both cohorts (APC‐MT vs. APC‐WT, COH/UCD cohort, HR, 0.54; 95% CI, 0.40–0.73; p < .0001; MSKCC cohort, HR, 0.55; 95% CI, 0.44–0.70, p < .0001). The median overall survival of APC‐WT patients from the COH/UCD cohort was 22.6 months compared with 45.6 months in APC‐MT patients (p < .0001; Fig. 1A). In the MSKCC cohort, the median overall survival of APC‐WT patients was 42.6 months, whereas the median overall survival of APC‐MT patients is 70.0 months (p < .0001; Fig. 1B). In addition, we observed similar results when the COH cohort and UCD cohort were analyzed separately (supplemental online Fig. 1). The longer OS values of the MSKCC cohort are likely due to the high metastasectomy rate in the MSKCC cohort (>50%), whereas most of the patients seeking care at our clinic were not eligible for metastasectomy. On multivariate analysis that includes primary tumor location, RAS, and BRAF status and APC status, APC‐WT continued to be predictive of poor survival in the COH/UCD cohort (APC‐MT vs. APC‐WT: HR, 0.62; 95% CI, 0.44–0.86; p = .0037) and the MSKCC cohort (APC‐MT vs. APC‐WT: HR, 0.63; 95% CI, 0.49–0.81; p < .0001). Our multivariate model also confirmed the inferior outcome of right‐sided primary and BRAF mutant tumors (Table 4). Taken together, our analysis indicates that wild‐type APC is an independent poor prognostic marker for MSS metastatic colorectal cancer.
Figure 1.
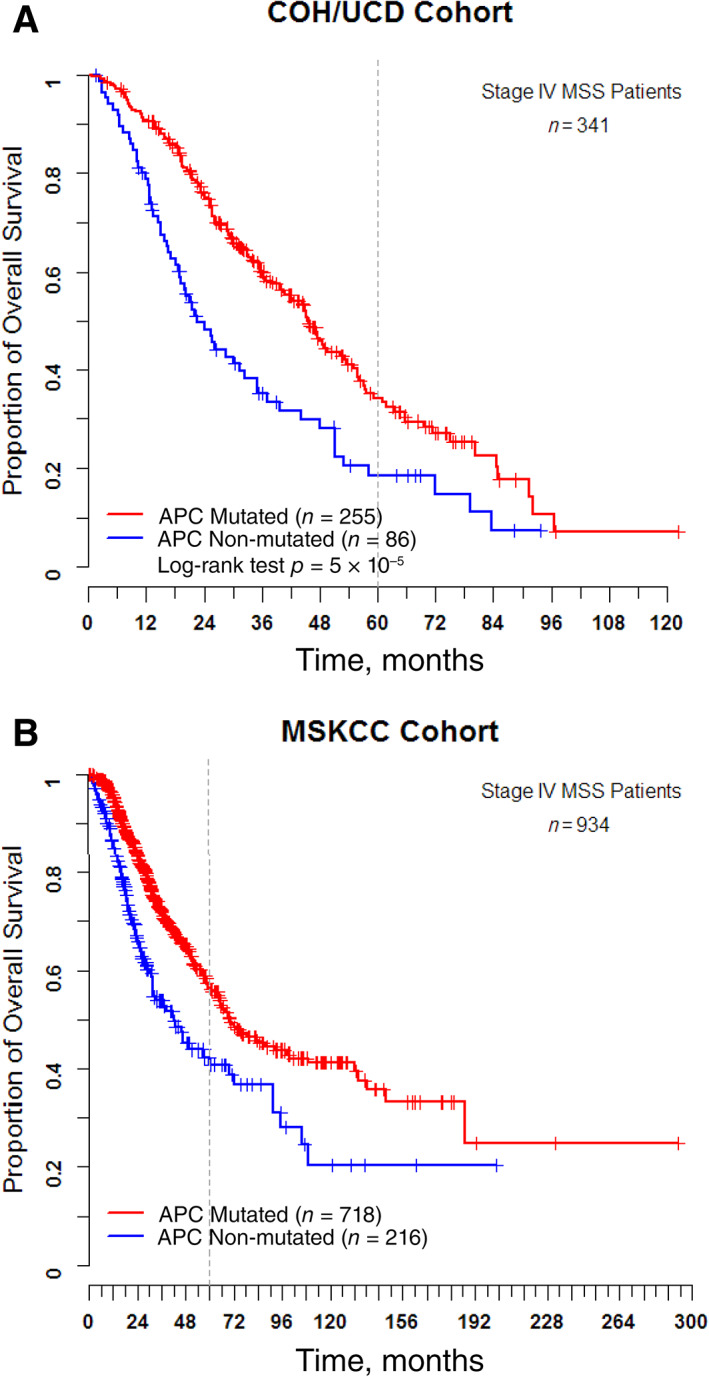
Kaplan‐Meier curves for overall survival of patients with MSS mCRC by APC status. (A): Kaplan‐Meier curves for overall survival of the COH/UCD cohort. (B): Kaplan‐Meier curves for overall survival of the MSCKK cohort.Abbreviations: COH, City of Hope National Medical Center; MSKCC, Memorial Sloan Kettering Cancer Center; MSS, microsatellite stable; UCD, University of California, Davis.
Table 4.
Multivariate survival model
| Clinicopathologic variable | COH/UCD (n = 341) | MSKCC (n = 934) | ||
|---|---|---|---|---|
| HR (95% CI) | p value | HR (95% CI) | p value | |
| Age at diagnosis, continuous, yr | 0.99 (0.98–1.01) | .245 | 1.00 (0.99–1.01) | .633 |
| Gender, female vs. male | 0.95 (0.71–1.27) | .735 | 0.96 (0.77–1.19) | .721 |
| Primary tumor location, right vs. left | 1.45 (1.05–2.02) | .026 | 1.16 (0.90–1.50) | .258 |
| APC, mutated vs. nonmutated | 0.62 (0.44–0.86) | .0037 | 0.63 (0.49–0.81) | 3.87× 10−04 |
| KRAS, mutated vs. nonmutated | 1.16 (0.86–1.58) | .330 | 1.72 (1.35–2.20) | 1.42× 10−05 |
| NRAS, mutated vs. nonmutated | 1.62 (0.77–3.40) | .203 | 2.70 (1.68–4.34) | 4.32× 10−05 |
| BRAF V600E, mutated vs. nonmutated | 1.73 (0.89–3.36) | .105 | 4.79 (3.10–7.39) | 1.55× 10−12 |
Abbreviations: CI, confidence interval; COH, City of Hope National Medical Center; HR, hazard ratio; MSKCC, Memorial Sloan Kettering Cancer Center, UCD, University of California, Davis.
Discussion
Somatic mutations in APC are present in approximately 70%–80% of colorectal cancers [9, 18]. Compared with MSS colorectal cancers, tumors with MSI have a lower frequency of APC mutations [19]. Although several studies have suggested worse outcomes for unselected patients with colorectal cancer with APC‐WT tumors, the prognostic implication of this genomic alteration in MSS metastatic colorectal cancer is not well defined [14, 15]. In this retrospective study, we focused our analysis on patients with MSS metastatic colorectal cancer from two independent institutions. We found APC‐WT is associated with right‐sided primary, BRAF mutation, and younger age. Using a multivariate model correcting for tumor location and RAS/RAF status, APC‐WT was an independent poor prognostic marker in MSS metastatic colorectal cancer. The poor prognostic value of APC‐WT was also significant when the COH cohorts and UCD cohorts were analyzed separately. In addition, our observations were validated using an independent cohort of 934 patients with metastatic colorectal cancer from the MSKCC data set.
There is no clear explanation for the worse outcome of APC‐WT colorectal cancers. The copresence of BRAF V600E mutation and the increased incidence of right colonic tumors in APC‐WT tumors may explain some of the poor outcomes associated with this group. However, APC‐WT maintains a poor prognostic value after correcting for these variables [8, 20, 21, 22, 23, 24]. This indicates that additional biological variables associated with APC‐WT status may contribute to the poor prognosis of this group. Consistent with prior reports, we showed that APC‐WT colorectal cancer occurs more often in younger patients than older patients [10]. A study of 24 first‐line clinical trials with metastatic colorectal cancer demonstrated that younger age was associated with worse progression‐free survival (PFS) and OS irrespective of type of therapy received and molecular status [25]. Therefore, additional biological variables that are yet to be defined and that contribute to a more aggressive histology in younger individuals may partly explain the poor prognosis of this population. In addition, APC‐WT tumors have been associated with an epithelial–mesenchymal transition (EMT) signature [13]. EMT has been suggested as one of the mechanisms leading to anti‐EGFR resistance and a worse overall colorectal cancer outcome [26, 27, 28]. Finally, APC‐WT status, as a standalone finding, has been recently associated with a diminished benefit from anti‐EGFR therapy in a retrospective analysis from the CALGB 80405 study. Because our data reflect a modern era of treatment that includes anti‐EGFR therapy, a differential outcome that is in favor of the APC‐MT group may reflect a benefit from anti‐EGFR driven within its RAS‐WT subpopulation [11]. Taken together, these results suggest that APC‐WT colorectal cancer may drive a distinct mechanism of tumorigenesis which then confers relative resistance to chemotherapy or targeted therapy, therefore leading to a worse overall outcome.
CTNNB1 and RNF43 were enriched within the APC‐WT colorectal cancer population, suggesting an alternative mechanism of WNT pathway activation in this subgroup. Prior studies and our own findings indicate that CTNNB1 and APC mutations are mutually exclusive, suggesting that CTNNB1 mutation could substitute for APC mutation as the initiator genomic alteration in colorectal tumor development [29]. As a tumor‐suppressor gene, RNF43 encodes a transmembrane ubiquitin ligase that downregulates the expression of membrane WNT receptor. Its mutation augments the activity of WNT ligands upon binding to WNT receptors [30, 31]. RNF43 mutations were mutually exclusive with APC mutation, and have correlated with BRAF V600E mutation and MSI [32, 33]. RNF43‐mutated tumors have conferred a worse OS in patients with RAS‐WT colorectal cancer when treated with cetuximab [11]. Indeed, in our data analysis, patients with RNF43‐mutated tumors exhibited earlier death within the APC‐WT population (supplemental online Fig. 2). It is possible that RNF43 led to more robust WNT activation than APC mutations, leading to a more sustained progrowth signaling and worse overall outcome.
We acknowledge the limitations of the retrospective analysis of our study. However, the large sample size and the reproducibility of our data within each institute (COH and UCD) as well as in an independent public data set lends strong support to our findings. Despite the potential patient heterogeneity across three institutes and the likely variable rate of metastasectomies that would explain the different OS among institutes, APC status was maintained as a negative prognostic marker across all centers. Few discrepancies in the occurrence rate of low frequency gene mutations were noted between the COH/UCD cohort and MSKCC cohort. These may be explained by sample power and, less likely, by variations in sequencing platform. Given the nature of retrospective study, and the heterogeneity of treatment in different lines of treatment, we did not analyze the impact of APC status on PFS or response rate. This study suggests APC status should be considered as a stratification biomarker in future prospective trials.
Conclusion
Our data demonstrated that APC‐WT is an independent poor prognostic marker for metastatic colorectal cancer. Our findings suggest that incorporating APC mutation assessment along with other known classifiers such as RAS and BRAF may improve colorectal cancer risk stratification. Our data suggest that alternative therapeutic interventions may be needed for patients with APC‐WT metastatic colorectal cancer.
Author Contributions
Conception/design: Chongkai Wang, Ching Ouyang, Ajay Goel, Michael Kahn, Marwan Fakih
Provision of study material or patients: Chongkai Wang, Ching Ouyang, May Cho, Jingran Ji, Marwan Fakih
Collection and/or assembly of data: Chongkai Wang, Ching Ouyang, May Cho, Jingran Ji, Jaideep Sandhu
Data analysis and interpretation: Chongkai Wang, Ching Ouyang, May Cho, Ajay Goel, Michael Kahn, Marwan Fakih
Manuscript writing: Chongkai Wang, Ching Ouyang, Ajay Goel, Michael Kahn, Marwan Fakih
Final approval of manuscript: Chongkai Wang, Ching Ouyang, May Cho, Jingran Ji, Jaideep Sandhu, Ajay Goel, Michael Kahn, and Marwan Fakih
Disclosures
May Cho: AstraZeneca, QED Biopharma, Bristol‐Myers Squibb, Taiho, HelioDx, I‐Mab Biopharma, Eisai, Incyte, Ipsen, Tempus (SAB), Immunocore Limited, Bristol‐Myers Squibb, Seattle Genetics, Exelexis, AstraZeneca, Incyte (RF); Marwan Fakih: Amgen (H), AstraZeneca, Amgen, Novartis (RF), Amgen, Array, Bayer, Pfizer (C/A), Amgen, Guardant 360 (Other‐Speaker’ bureau). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Supporting information
See http://www.TheOncologist.com for supplemental material available online.
Appendix S1: Supporting infomraiton
Acknowledgments
The data that support the findings of this study are available from the corresponding author upon reasonable request.
No part of this article may be reproduced, stored, or transmitted in any form or for any means without the prior permission in writing from the copyright holder. For information on purchasing reprints contact commercialreprints@wiley.com. For permission information contact permissions@wiley.com.
Disclosures of potential conflicts of interest may be found at the end of this article.
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019;69:7–34. [DOI] [PubMed] [Google Scholar]
- 2. Milano AF, Singer RB. The Cancer Mortality Risk Project ‐ Cancer mortality risks by anatomic site: Part 1 ‐ Introductory overview; part II ‐ Carcinoma of the colon: 20‐year mortality follow‐up derived from 1973‐2013 (NCI) SEER*STAT survival database. J Insur Med 2017;47:65–94. [DOI] [PubMed] [Google Scholar]
- 3. Siegel RL, Miller KD, Goding Sauer A et al. Colorectal cancer statistics, 2020. CA Cancer J Clin 2020;70:145–164. [DOI] [PubMed] [Google Scholar]
- 4. Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol 2011;6:479–507. [DOI] [PubMed] [Google Scholar]
- 5. Sadanandam A, Lyssiotis CA, Homicsko K et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med 2013;19:619–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. De Sousa E Melo F, Wang X, Jansen M et al. Poor‐prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat Med 2013;19:614–618. [DOI] [PubMed] [Google Scholar]
- 7. Christie M, Jorissen RN, Mouradov D et al. Different apc genotypes in proximal and distal sporadic colorectal cancers suggest distinct WNT/beta‐catenin signalling thresholds for tumourigenesis. Oncogene 2013;32:4675–4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990;61:759–767. [DOI] [PubMed] [Google Scholar]
- 9. Powell SM, Zilz N, Beazer‐Barclay Y et al. APC mutations occur early during colorectal tumorigenesis. Nature 1992;359:235–237. [DOI] [PubMed] [Google Scholar]
- 10. Lieu CH, Golemis EA, Serebriiskii IG et al. Comprehensive genomic landscapes in early and later onset colorectal cancer. Clin Cancer Res 2019;25:5852–5858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Innocenti F, Rashid N, Wancen M et al. Next‐generation sequencing (NGS) in metastatic colorectal cancer (MCRC): Novel mutated genes and their effect on response to therapy (Alliance). Ann Oncol 2019;30:4878a. [Google Scholar]
- 12. Yang M, Schell MJ, Loboda A et al. Repurposing EGFR inhibitor utility in colorectal cancer in mutant APC and TP53 subpopulations. Cancer Epidemiol Biomarkers Prev 2019;28:1141–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schell MJ, Yang M, Teer JK et al. A multigene mutation classification of 468 colorectal cancers reveals a prognostic role for APC. Nature Commun 2016;7:11743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jorissen RN, Christie M, Mouradov D et al. Wild‐type apc predicts poor prognosis in microsatellite‐stable proximal colon cancer. Br J Cancer 2015;113:979–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yaeger R, Chatila WK, Lipsyc MD et al. Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell 2018;33:125–136.e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Frampton GM, Fichtenholtz A, Otto GA et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 2013;31:1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chakravarty D, Gao J, Phillips S et al. OncoKB: A precision oncology knowledge base. JCO Precis Oncol 2017:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Segditsas S, Tomlinson I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene 2006;25:7531–7537. [DOI] [PubMed] [Google Scholar]
- 19. Salahshor S, Kressner U, Påhlman L et al. Colorectal cancer with and without microsatellite instability involves different genes. Genes Chromosomes Cancer 1999;26:247–252. [PubMed] [Google Scholar]
- 20. Jass JR. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology 2007;50:113–130. [DOI] [PubMed] [Google Scholar]
- 21. Chan TL, Zhao W, Leung SY et al. BRAF and KRAS mutations in colorectal hyperplastic polyps and serrated adenomas. Cancer Res 2003;63:4878–4881. [PubMed] [Google Scholar]
- 22. Kambara T, Simms LA, Whitehall VLJ et al. BRAF mutation is associated with DNA methylation in serrated polyps and cancers of the colorectum. Gut 2004;53:1137–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Missiaglia E, Jacobs B, D'Ario G et al. Distal and proximal colon cancers differ in terms of molecular, pathological, and clinical features. Ann Oncol 2014;25:1995–2001. [DOI] [PubMed] [Google Scholar]
- 24. Holch JW, Ricard I, Stintzing S et al. The relevance of primary tumour location in patients with metastatic colorectal cancer: A meta‐analysis of first‐line clinical trials. Eur J Cancer 2017;70:87–98. [DOI] [PubMed] [Google Scholar]
- 25. Lieu CH, Renfro LA, de Gramont A et al. Association of age with survival in patients with metastatic colorectal cancer: Analysis from the arcad clinical trials program. J Clin Oncol 2014;32:2975–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brand TM, Iida M, Wheeler DL. Molecular mechanisms of resistance to the EGFR monoclonal antibody cetuximab. Cancer Biol Ther 2011;11:777–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science 2011;331:1559–1564. [DOI] [PubMed] [Google Scholar]
- 28. Budinska E, Popovici V, Tejpar S et al. Gene expression patterns unveil a new level of molecular heterogeneity in colorectal cancer. J Pathol 2013;231:63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sparks AB, Morin PJ, Vogelstein B et al. Mutational analysis of the APC/β‐catenin/Tcf pathway in colorectal cancer. Cancer Res 1998;58:1130–1134. [PubMed] [Google Scholar]
- 30. Min BH, Hwang J, Kim NKD et al. Dysregulated wnt signalling and recurrent mutations of the tumour suppressor RNF43 in early gastric carcinogenesis. J Pathol 2016;240:304–314. [DOI] [PubMed] [Google Scholar]
- 31. Jiang XM, Hao HX, Growney JD et al. Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc Natl Acad Sci USA 2013;110:12649–12654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yan HHN, Lai JCW, Ho SL et al. RNF43 germline and somatic mutation in serrated neoplasia pathway and its association with braf mutation. Gut 2017;66:1645–1656. [DOI] [PubMed] [Google Scholar]
- 33. Giannakis M, Hodis E, Mu XJ et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat Genet 2014;46:1264–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
See http://www.TheOncologist.com for supplemental material available online.
Appendix S1: Supporting infomraiton