

Abstract
Background
Chronic inflammation and residual HIV transcription persist in people living with HIV (PLWH) receiving antiretroviral therapy (ART), thus increasing the risk of developing non-AIDS co-morbidities. The mechanistic target of rapamycin (mTOR) is a key regulator of cellular metabolism and HIV transcription, and therefore represents an interesting novel therapeutic target.
Methods
The LILAC pilot clinical trial, performed on non-diabetic ART-treated PLWH with CD4+/CD8+ T-cell ratios <0.8, evaluated the effects of metformin (12 weeks oral administration; 500-850 mg twice daily), an indirect mTOR inhibitor, on the dynamics of immunological/virological markers and changes in mTOR activation/phosphorylation in blood collected at Baseline, Week 12, and 12 weeks after metformin discontinuation (Week 24) and sigmoid colon biopsies (SCB) collected at Baseline and Week 12.
Findings
CD4+ T-cell counts, CD4+/CD8+ T-cell ratios, plasma markers of inflammation/gut damage, as well as levels of cell-associated integrated HIV-DNA and HIV-RNA, and transcriptionally-inducible HIV reservoirs, underwent minor variations in the blood in response to metformin. The highest levels of mTOR activation/phosphorylation were observed in SCB at Baseline. Consistently, metformin significantly decreased CD4+ T-cell infiltration in the colon, as well as mTOR activation/phosphorylation, especially in CD4+ T-cells expressing the Th17 marker CCR6. Also, metformin decreased the HIV-RNA/HIV-DNA ratios, a surrogate marker of viral transcription, in colon-infiltrating CD4+ T-cells of 8/13 participants.
Interpretation
These results are consistent with the fact that metformin preferentially acts on the intestine and that mTOR activation/phosphorylation selectively occurs in colon-infiltrating CCR6+CD4+ T-cells. Future randomized clinical trials should evaluate the benefits of long-term metformin supplementation of ART.
Keywords: Metformin, ART, HIV reservoirs, Th17, mTOR
Research in context.
Evidence before the study
Although antiretroviral therapy (ART) controls HIV replication at undetectable plasma levels, ART does not block HIV transcription, a step in the viral replication cycle under the control of the cellular machinery. Residual HIV transcription occurs in ART-treated PLWH and fuels a state of chronic immune activation and inflammation that leads to the occurrence of non-AIDS co-morbidities. Thus, there is a need for additional therapeutic interventions to specifically target HIV transcription.
Added value of this study
In this context, the LILAC trial NCT02659306 was performed to monitor the effects of metformin, a widely used drug for the treatment of diabetes and metabolic disorders, on multiple immunological and virological parameters in a cohort of non-diabetic ART-treated PLWH, with CD4/CD8 ratios below 0.8. The originality of our study is represented by state-of-the-art immunological and virological investigations performed not only in the blood but also in sigmoid colon biopsies before and after metformin treatment. Metformin is an indirect negative modulator of the mechanistic target of rapamycin (mTOR), a metabolic checkpoint documented to facilitate HIV transcription. The idea to target mTOR with metformin is justified by our previous studies demonstrating that mTOR activation preferentially occurs in Th17-polarized CCR6+ T-cells, a subset of CD4+ T-cells highly permissive to HIV infection and enriched in HIV reservoirs in the blood and colon of ART-treated PLWH.
Implications of all the available evidence
The results included in this manuscript provide a proof-of-concept that metformin can be safely used in ART-treated PLWH to decrease mTOR-mediated immune activation and inflammation in the colon. This study emphasizes the importance of performing investigation in colon biopsies when assessing the effects of HIV cure/remission interventions. Our study also provides a strong rational for future randomized clinical trials to assess the effects of metformin on residual HIV transcription.
Alt-text: Unlabelled box
1. Introduction
Antiretroviral therapies (ART) efficiently control HIV-1 replication and improve the health of people living with HIV (PLWH) [1]. Nevertheless, viral reservoirs persist in cellular/anatomic sanctuaries during ART, with viral rebound occurring rapidly upon ART interruption, thus requiring a life-long treatment [2], [3], [4]. The persistence of HIV reservoirs in ART-treated PLWH is associated with chronic inflammation/immune activation, alterations of the intestinal mucosal homoeostasis, and metabolic disorders [5], [6], [7]. One mechanism underlying these alterations is the residual HIV transcription leading to the subsequent HIV RNA and protein expression in cells carrying viral reservoirs. Indeed, HIV transcription, a process controlled by Tat and the transcriptional machinery of the host cell, is not currently not targeted by ART [1,[7], [8], [9]]. Also, a fraction of PLWH exhibit nonsuppressible viremia despite adherence to ART [10], [11], [12]. Thus, the identification of additional therapeutic interventions targeting HIV transcription is required.
Early studies documented the intestinal tropism of HIV infection, with a massive depletion of CD4+ T-cells early upon infection [13], [14], [15], [16] and functional alterations of the intestinal barrier, leading to microbial translocation [17], [18], [19]. These alterations are mainly linked to the depletion of Th17-polarized cells [20], [21], [22], a subset of CD4+ T-cells specialized in the maintenance of mucosal barrier homeostasis [23]. By their location at mucosal sites [24], Th17 cells represent an important target for infection [25,26]. The depletion of Th17 cells occurs rapidly and their frequency/function is not restored even when ART is initiated during early acute stages [27]. Despite their depletion, a fraction of Th17 cells is long lived [28] and carry HIV reservoirs [29,30], mainly in the colon of ART-treated PLWH [31], [32], [33]. Th17 cells exhibit unique transcriptional and metabolic features [34,35] that render them highly permissive to infection [36], [37], [38], [39], [40]. One particularity of Th17 cells is their dependency on the mechanistic target of rapamycin (mTOR) for lineage commitment/polarization [22,41,42]. Noteworthy, our group previously reported preferential mTOR activation (reflected by its phosphorylation level) in Th17-polarized CCR6+CD4+ T-cells infiltrating the colon of ART-treated PLWH, and in CD4+ T-cells exposed in vitro to retinoic acid, a gut-homing tropism mediator [22,43].
The mTOR is a key regulator of energy balance and metabolism at the cellular level [44], [45], [46]. This conserved serine/threonine kinase forms two complexes with different protein components [mTOR complex 1 (mTORC1) and 2 (mTORC2)]; it acts as a nutrient sensor that stimulates nucleotide synthesis, glucose uptake and glycolysis, while it inhibits the autophagy process [44], [45], [46], [47]. Of note, mTOR activation via phosphorylation was reported to positively regulate HIV replication by acting directly at the level of viral entry [48] and transcription [49,50], as well as indirectly through the inhibition of autophagy-mediated viral particle degradation [51]. Most recent studies demonstrated that mTOR activation facilitates HIV reverse transcription and subsequent intracellular trafficking by modulating the metabolic status of TCR-activated T-cells [52]. This provides a molecular explanation for the preferential infection and persistence of HIV-DNA in gut-homing CCR6+CD4+ T-cells of ART-treated PLWH [31,33], and points to mTOR activation as a key regulator of residual HIV transcription. Indeed, studies by our group and others demonstrated that mTOR inhibitors reduce HIV transcription [49,50] and viral outgrowth in vitro [43].
Metformin (dimethylguanide), one of the most widely used drugs for type II diabetes, has well-established immuno-metabolic regulator properties [53,54]. Mechanistically, metformin impedes mTOR activation/phosphorylation by inhibiting the mitochondrial complex I enzyme, which results in a decreased ATP production and an increased AMP:ATP ratio that subsequently activates the energetic sensor AMP-activated protein kinase (AMPK), finally leading to the inhibition of mTORC1 signaling pathway. Alternatively, metformin inhibits mTOR activation independently of AMPK, via the inhibition of Rag GTPase (RAG) activity, an mTORC1 activator [55], [56], [57]. By using such mechanism, metformin limits intestinal glucose absorption and hepatic glucose production, while improving glucose uptake in peripheral tissues, like muscles [54]. In addition, metformin has been shown to reduce age-related diseases in humans, likely via the modulation of microbiota composition and microbial metabolism [58]. Finally, metformin was reported to inhibit Th17 polarization, and, as a result, to decrease tumor growth [59] and autoimmunity symptoms [60]. However, the effect of metformin on HIV replication, especially in gut-homing Th17 cells expressing the highest levels of phosphorylated mTOR and representing key HIV infection/persistence targets [22,31,43], remains to be explored.
In this manuscript, we evaluated the immunological and virological effects of metformin supplementation of ART for 12 weeks in non-diabetic PLWH presenting relatively low CD4/CD8 ratios (<0.8). This is a sub-study of the LILAC pilot clinical trial (NCT02659306) [61,62] performed on matched blood and sigmoid colon biopsies.
2. Methods
2.1. Ethics
This study was approved by the Research Ethics Boards of the Research Institute of the McGill University Health Centre (MUHC) number MP-37-2016-2456, and by the Health Canada Therapeutic Products Directorate. The study was also approved by the Internal Review Board (IRB) of the Ottawa Hospital Research Institute, ON, Canada (IRB No. 20160433-01H) and the IRB of the CHUM Research Centre, Montréal, QC, Canada (IRB No. 17.074). This study was conducted in accordance with the Declaration of Helsinki of 1975. Each participant provided written informed consent before any study procedure. The CIHR/CTN protocol CTNPT027 Trial registration is NCT02659306.
2.2. Study design and participants
The LILAC study was conducted between October 2016 and August 2018. Study participant enrollment and biological sample collection were performed at the McGill University Health Centre (MUHC), Glen site, Montréal, QC, Canada, for matched blood and SCB (n=13), and at The Ottawa Hospital, Ottawa, ON, Canada, for blood collection (n=9) (Fig. 1). Research investigations were performed at the CHUM Research Centre, Montréal, QC, Canada. For this study, n=22 HIV-infected individuals (20 males, 2 females; Supplemental Table 1) were recruited, based on the inclusion criteria recently published by our group [61]. Briefly, participants were virologically suppressed (<40 copies per mL, at least two measurements per year) under ART for a minimum of 2 years and exhibited CD4/CD8 ratio <0.8 (indicative of high risk of inflammation and non-AIDS events) [61]. Participants were non-diabetic, as defined by a HbA1c < 5.9% [61]. To assess tolerability, participants received orally 500 mg metformin twice daily (bis in die; b.i.d) (Glucophage®; Sanofi-Aventis Canada Inc.) during the first week, following which the dose was increased to 850 mg b.i.d (Weeks 2-12) [61], reaching a similar dose to the one taken by diabetic individuals, with metabolic improvement being observed [53]. The treatment duration was chosen based on a previous clinical study in non-diabetic patients treated for polycystic ovary syndrome [63]. Consistent with reported ability of dolutegravir to increase metformin plasma exposure [64], study participants using dolutegravir maintained the 500 mg metformin b.i.d. throughout the study. Blood was collected at Baseline, after 12 weeks of metformin administration (Week 12) and 12 weeks post-metformin discontinuation (Week 24) (n=22), while in a sub-study performed in Montreal, n=13 participants consented to optional endoscopic collection of SCB at Baseline and Week 12 (Fig. 1). Study participants benefited from colon cancer screening at the time of SCB collection [61].
Fig. 1.

Flow chart of the clinical trial. Depicted is the schematic representation of the metformin treatment and the sample collection. Briefly, blood and plasma from HIV-infected individuals with undetectable plasma viral load (<40 HIV-RNA copies/mL) under ART were collected at Baseline, after 12 weeks of metformin treatment (500 to 850 mg b.i.d.) (Week 12) and after 12 week of metformin discontinuation (Week 24) in Montreal (n=13) and in Ottawa (n=9). In the sub-study performed in Montreal, matched sigmoid colon biopsies (SCB) were collected at Baseline and Week 12 (n=13).
2.3. Blood and SCB collection
Blood was collected and PBMCs isolated and cryopreserved until use, as we previously described [43]. Matched SCB (≈32 biopsies/donor) were collected from HIV-infected individuals receiving ART (Supplemental Table 1) and processed using Liberase DL (Roche Diagnostics), as previously described [31,43]. Matched peripheral blood (20 mL/donor) was collected the same day from biopsy donors and immediately processed.
2.4. Plasma biomarker measurements
Plasma was collected from peripheral blood and subsequently cryopreserved. Thawed plasma was inactivated with disruption buffer (PBS, 0.05% Tween-20, 2.5% Triton X-100, 1% trypan blue, 0.02% thimerosal) at 37°C for 1 hour. The concentration of 45 biomarkers was quantified using Human XL Cytokine Discovery Luminex (R) High Performance Assay (R&D Systems) according to the manufacturer's instructions. In parallel, plasma levels of sCD14, LBP, and I-FABP were quantified by ELISA using commercially available kits (R&D Systems).
2.5. Flow cytometry staining and analysis
Surface and intracellular staining (using the Fixation/Permeabilization Solution Kit, BD) were performed with specific fluorochrome-conjugated Abs (Supplemental Table 2), as previously described [43]. Cells were analyzed using a LSRII cytometer (Diva, version 6, BD), and FlowJo (version 10.0.6, Tree Star Inc.). All Abs were titrated for an optimal noise/signal ratio and Ab cocktails were validated by comparing single with multiple staining. Positive gates were placed based on fluorescence minus one control [43].
2.5.1. Magnetic and flow cytometry cell sorting
Total CD4+ T cells were isolated from cryopreserved PBMCs by negative selection using magnetic beads (Stem Cell Technologies, Vancouver, Canada). Alternatively, memory CD4+ T-cells from matched SCB and PBMCs were sorted and simultaneously phenotypically analyzed by flow cytometry upon staining with specific Abs (Supplemental Table 2), as previously described [43]. Briefly, blood memory CD4+ T-cells (CD45RA−CD3+CD4+) and colon memory CD3+ (CD45RA−CD3+CD4+/low) lacking CD8+ T-cell (CD8), B cell (CD19), NK cell (CD56), and epithelial cell (CD326) lineage markers were sorted using the BD-FACSAriaIII (BD Biosciences) in the Biosafety level 3 facility. The viability dye LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (Invitrogen) was used to exclude dead cells. Flow cytometry gates were defined using the fluorescence minus one strategy, as previously described [43].
2.5.1. Quantification of integrated and total HIV-DNA
Levels of integrated and total (Gag) HIV-DNA were quantified in cell lysates by nested real-time PCR (105 cells/test in triplicate; detection limit, 3 HIV-DNA copies), as previously reported [43]. The ACH2 cells carrying one copy of integrated HIV-DNA per cell (NIH AIDS Reagent Program) were used as a standard curve, as we reported [43]. Primers and probes are listed in Supplemental Table 3. For Gag HIV-DNA amplification, the external/internal primers and the probe used were the same as those used for LTR-Gag HIV-RNA quantification (Supplemental Table 3). For CD3 amplification, the external primers were HCD3OUT 5’ and HCD/OUT 3’; the internal primers for the real-time PCR were HCD3IN 5’ and HCD3IN 3’; and the probe used was: CD3 FamZen (Supplemental Table 3). All measures were performed in triplicate.
2.6. Quantification of CA US HIV-RNA in total blood CD4+ T-cells
Total RNA was extracted from total CD4+ T-cells using RNeasy Mini Kit with on-column DNase treatment (Qiagen), according to the manufacturer's instructions. The quality (260nm/280nm ratio) and quantity of RNA collected were evaluated by spectrophotometry on a Nanodrop instrument. Quantification of LTR-Gag HIV-RNA was performed by nested real-time RT-PCR. The external primers for the first amplification were ULF1 and UR1; the internal primers for the second amplification PCR were Lambda (λ) T (Forward) and UR2 (Reverse); and the probe used was UHIV FamZen (Supplemental Table 3). The first amplification was performed on Proflex PCR System (Applied Biosystem), while the second amplification was performed on the RotorGene instrument (Qiagen). US LTR-gag HIV-RNA standards were generated from plasmids by in vitro transcription (MEGAscript™ T7 Transcription Kit, ThermoFisher). The number of copies of each transcript was normalized to the levels of the reference gene beta glucuronidase (GUSB). The oligonucleotides used for GUSB amplification were GusbF1 and GusbR1 for the first amplification and GusbF2 and GusbR2 for the second amplification; and the probe sequence was GUSB-HEX (Supplemental Table 3). All measurements were performed in triplicate.
2.6.1. Quantification of CA US HIV-RNA in colon memory CD4+ T-cells
Total DNA and RNA were dually extracted from colon memory CD4+ T-cells using the AllPrep DNA/RNA Mini Kit (Qiagen), according to the manufacturer's instructions, as we reported [65]. Cell-associated HIV-RNA and HIV-DNA extracted from each sample were quantified as described above. The quantification of CD3-DNA copy numbers allowed us to determine the number of cells per sample, thus allowing the normalization of HIV-RNA copy numbers per 106 cells (2 CD3-DNA copies/cell). Finally, the ratio between HIV-RNA/106 cells and HIV-DNA/106 cells was calculated, indicative of HIV transcription.
2.7. TILDA
The frequency of CD4+ T-cells with inducible multiply spliced HIV-RNA was determined using a modified version of TILDA, as we previously described [66]. Briefly, enriched CD4+ T-cells were rested for 3-5 h at 37°C, 5% CO2, in the presence of antiretroviral drugs (200 nM raltegravir and 180 nM AZT). CD4+ T-cells were stimulated for 18 h with PMA (100 ng/mL) and Ionomycin (1 μg/mL) (Sigma). After stimulation, cells were washed with PBS 10% FBS, counted and serially diluted to 18 × 106 cells/mL, 9 × 106 cells/mL, 3 × 106 cells/mL and 1 × 106 cells/mL in PBS 10% FBS. Primers for tat/rev pre-amplification were Tat1.4 and Rev; the Tat/Rev primers for the real-time PCR were Tat2 and Rev; and the probe used in these reactions was msHIV FamZen (Supplemental Table 3). Positive wells at each dilution were counted and the maximum likelihood method was used to calculate the frequency of cells with inducible HIV msRNA (http://bioinf.wehi.edu.au/software/elda) [66].
2.8. Statistics
Statistical analyses were performed using the GraphPad Prism 8 Software (GraphPad Software, Inc.). The non-parametric Wilcoxon test was used to determine statistical significance for differences between matched groups. The use of non-parametric tests is justified by the fact that data sets did not pass the normal distribution test Kolmogorov-Smirnov (data not shown), calculated using GrapPad Prism Software. Based on Martin Bland's recommendations relative to p-value interpretation (https://www-users.york.ac.uk/~mb55/msc/applbio/week4/signif_compact.pdf) p-values are indicated on the Figures and interpreted, as follows: little/no evidence (p>0.1), weak evidence (p=0.1-0.05), evidence (0.01-0.05), strong evidence (<0.01), and very strong evidence (<0.001). Finally, pairwise correlations between measurements and corresponding "CA HIV RNA/DNA" values were performed using Spearman's method, as implemented in R stats package (version 4.0.3) [R Core Team (2020). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria (https://www.R-project.org)]. The outliers were identified, as reflected in the Q-Q plots (Extended data in support of Fig. 2, Fig. 3, Fig. 4, Fig. 5, Fig. 6, Fig. 7 and Supplemental Figures 1-5), but they were not excluded from the analysis because there were no technical or clinical justification.
Fig. 2.
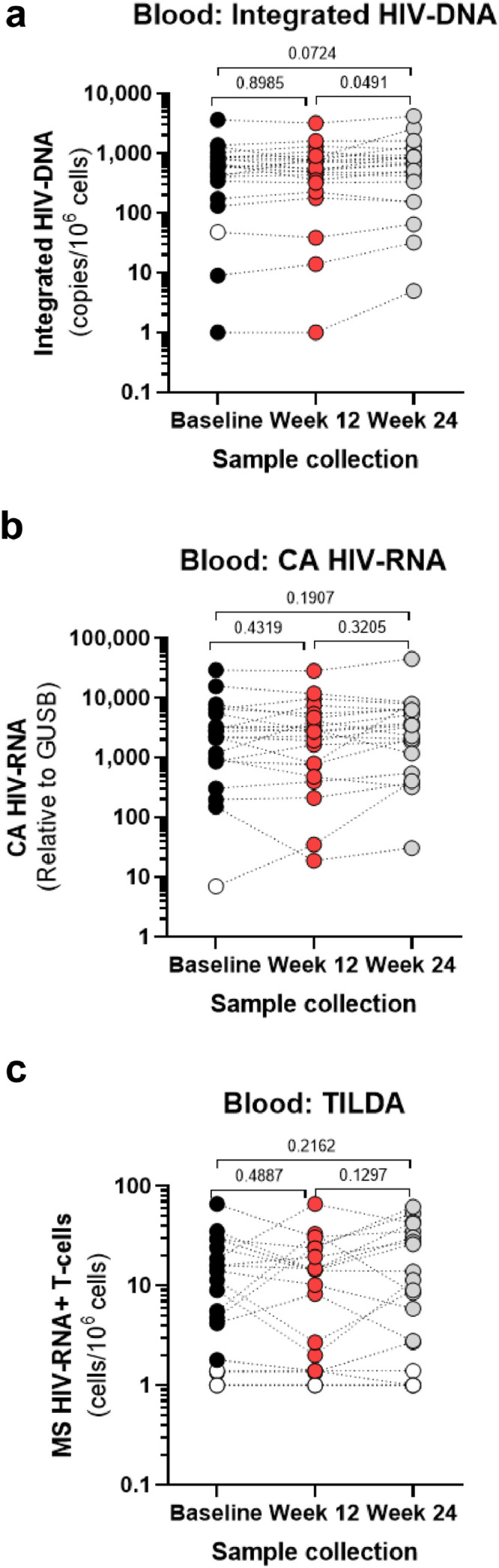
Effects of metformin treatment on viral reservoirs in peripheral blood CD4+ T-cells. Total CD4+ T-cells were enriched from fresh PBMCs isolated at Baseline (back circles), Week 12 (red circles) and Week 24 (grey circles) from the blood of study participants (n=22, Supplemental Table 1) by negative selection. (a) Levels of integrated HIV-DNA (Alu/HIV-LTR primers) were quantified by real-time nested PCR in cell lysates; shown are HIV-DNA copy numbers per 106 cells (two CD3-DNA copies/cell). (b) Total RNA was extracted from total CD4+ T-cells and CA HIV-RNA (LTR/Gag primers) levels were quantified by real-time RT-PCR. Shown are CA HIV-RNA copy numbers relative to GUSB mRNA (housekeeping gene). (c) The frequency of inducible MS HIV-RNA in CD4+ T cells was quantified by TILDA; shown are values normalized per 106 cells. (a-c) Grey bars represent mean values of different virological measurements. Values under the limit of detection are represented with open circles. Wilcoxon p-values for matched paired comparisons are depicted on the graphs.
Fig. 3.

Effects of metformin treatment on peripheral blood and colon CD4+ T-cell frequency and expression of activation markers. Matched blood and colon (SCB) samples were collected from n=13 participants (Supplemental Table 1) at Baseline (black circles) and after 12 weeks of metformin treatment (red circles). Cells were extracted and stained with a cocktail of specific fluorochrome-conjugated Abs. A viability dye was used to exclude dead cells from the analysis. Lineage markers included CD8 (CD8+ T-cells), CD19 (B cells), and CD66b (granulocytes) Abs. (a) Shown is the gating strategy for flow cytometry identification of total (Lineage−CD326−CD3+CD4+) and memory (CD45−) CD4+ T-cells in blood (upper panels) and colon (lower panels). CD8+ T-cells were identified as Lineage+CD326−CD3+CD4− (data not shown). (b-c) Shown are statistical analyses of the frequency of total and memory CD4+ T-cells within live Lineage−CD326− cells. Memory CD8+ T-cells were identified as CD326− Lin+CD3+CD45− cells (gating strategy not shown). (d-e) Shown are statistical analyses of the frequency of HLA-DR+CD38+ within the memory CD4+ (d) and CD8+ (e) T-cells from blood and colon at Baseline and Week 12. Grey bars represent the mean values. Wilcoxon p-values for matched paired comparisons are depicted on the graphs.
Fig. 4.

Metformin reduces mTOR phosphorylation in colon-infiltrating CCR6+ CD4+ T-cells. The gating strategy for flow cytometry identification of memory CD4+ and CD8+ T-cells in matched blood and colon samples (SCB) of n=13 study participants (Supplemental Table 1) is explained in the legend of Fig. 3. (a) Shown are statistical analyses of intracellular expression of phosphorylated mTOR (p-mTOR) in memory CD4+ and CD8+ T-cells from blood and colon at Baseline (black circles) and Week 12 of metformin treatment (red circles). Wilcoxon p-values for matched comparisons between Baseline and Week 12 are depicted on the graphs. (b)-c Shown are dot plots from a representative study participant (LILAC #2) of intracellular p-mTOR and surface CCR6 expression in memory CD4+ T-cells from the blood and the colon at Baseline (black dot plots) and Week 12 (red dot plots). (d) Shown are statistical analyses of the frequency of CCR6+ and CCR6−p-mTOR+ in matched blood and colon memory CD4+ T-cells before (black circles) and after 12 weeks of metformin (red circles). Grey bars represent the mean values. Wilcoxon p-values for matched paired comparisons are depicted on the graphs. For graphical simplicity only p-values <0.2 are depicted.
Fig. 5.

Effects of metformin treatment on markers of gut-homing, HIV permissiveness, and cell survival in colon-infiltrating CCR6+/CCR6− CD4+ T-cells. The gating strategy for flow cytometry identification of memory CCR6+/CCR6− CD4+ T-cells from matched blood and SCB is depicted in Fig. 4A. (a-d) Shown are the statistical analyses of the differential surface expression of integrin β7 (a), and CCR5 (b), as well as the of intracellular SAMHD1 (c) and Bcl-2 expression (d) in blood and colon memory CCR6+ and CCR6− CD4+ T-cells at Baseline (black circles) and Week 12 of metformin treatment (red circles). Grey bars represent the mean values. Wilcoxon p-values for matched paired comparisons are depicted on the graphs.
Fig. 6.

Metformin-mediated effects on residual HIV transcription in colon-infiltrating memory CD4+ T-cells of a fraction of ART-treated PLWH. Memory CD4+ T-cells were sorted by FACS from SCB of n=13 study participants (Supplemental Table 1), using the gating strategy depicted in Fig. 3A. The total CA DNA and RNA were dually extracted from FACS-sorted colon memory CD4+ T-cells. Total CA HIV-DNA (Gag primers) and HIV-RNA (Gag primers) levels were quantified by real time nested PCR and RT-PCR, respectively. Shown are changes in (a) absolute CA HIV-DNA levels, as well as (b) CA HIV-RNA/HIV-DNA ratios at Baseline (black bars) and Week 12 (red bars) are depicted on the graphs. Wilcoxon p-values for matched paired comparisons are depicted on the graphs.
Fig. 7.
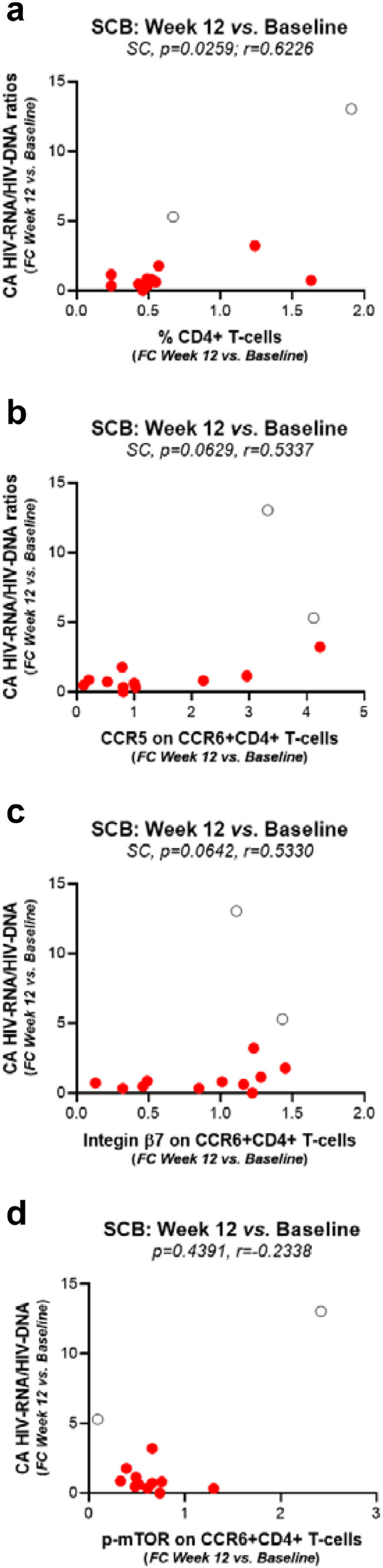
Metformin-mediated changes in HIV-RNA/HIV-DNA ratios in relationship with the phenotype and frequency of colon-infiltrating CD4+ T-cells. The Spearman correlation (SC) model was first applied to determine metformin-induced changes (Week 12 versus Baseline) in HIV-RNA/HIV-DNA ratios in relationship with changes in the frequency of total CD4+ T-cells infiltrating the colon (a) and that of CCR6+CD4+ T-cells expressing CCR5 (b), integrin β7 (c), or p-mTOR (d) in n=13 study participants. Although two outliers were identified, as depicted on the graphs (open black circles), there were no technical or clinical justifications for the outliers to be excluded from the statistical analysis. The SC p and r values are indicated on the graphs.
2.9. Samples size calculation
In view of a future randomized clinical trial (RCT), we used data from the pilot study to determine means, standard deviations, and used the Wilcoxon matched pairs ranked test to calculate the sample size with the help of the G*Power software.
2.10. Role of the funding source
The Funders had no role in study design, data collection, data analyses, interpretation, or writing of report.
3. Results
3.1. Clinical outcomes
The study design is depicted in Fig. 1. Briefly, blood samples were collected from participants enrolled in Montreal (n=13) and in Ottawa (n=9) at Baseline, 12 weeks of metformin treatment (Week 12), and 12 weeks after metformin discontinuation (Week 24). Matched sigmoid colon biopsies (SCB) were collected from participants enrolled in Montreal (n=13) at Baseline and Week 12. Detailed study objectives, inclusion criteria for study participants, and safety measures in the Lilac pilot clinical trial were recently reported by our group [61,62]. The absolute CD4+ and CD8+ T-cell counts, as well as the CD4+/CD8+ T-cell ratios, remained stable between Baseline, Week 12, and Week 24 (Supplemental Figure 1), as we previously reported [61,62].
3.2. Effects of metformin on plasma inflammation and gut-barrier dysfunction markers
Multiple plasma markers indicative of systemic inflammation [soluble CD14 (sCD14), lipopolysaccharide-binding protein (LBP)] and mucosal barrier dysfunction [intestinal fatty acid-binding protein (I-FABP)] were previously linked to HIV disease progression and the development of non-AIDS co-morbidities [17,18,67]. To gain insights into the potential benefits of metformin treatment, a set of soluble factors were quantified in the plasma. Quantifications by ELISA revealed no changes in LBP and I-FABP levels after metformin treatment, while a minor decrease in sCD14 levels were observed between Week 12 and Baseline (median: 1,818 versus 1,778; p=0.0595; Wilcoxon) and Baseline versus Week 24 (median: 1,760 versus 1,818; p=0.0158; Wilcoxon) (Supplemental Figure 2a-c). Further, quantifications by Luminex Multiplex Assay of 45 plasma markers associated to inflammation and immune activation revealed changes that, in majority, did not reach statistical significance, with high inter-donor variations (Supplemental Figure 2d). Among the most modulated factors at Week 12 versus Baseline, levels the chemokine CCL11/Eotaxin-1 [binds to CCR3, a reported minor HIV receptor [68]] were significantly up-regulated (median: 270 versus 207; p<0.0001; Wilcoxon), while levels of CCL20/MIP-3α (median: 105 versus 118; p=0.0209; Wilcoxon) [CCR6 ligand overexpressed during HIV infection [69]] were significantly down-regulated (Supplemental Figure 2e). For CXCL10/IP-10, a chemokine previously associated with disease progression [70], changes between time points did not reach statistical significance (Supplemental Figure 2g); nevertheless, the exclusion of the 2 outliers revealed a minor decrease in CXCL10 levels at Week 12 versus Baseline (median: 1,148 versus 1,350; p=0.0539; Wilcoxon), followed by a significant increase at Week 24 versus Week 12 (median: 1,538 versus 1,148; p=0.0107; Wilcoxon) (data not shown). In conclusion, metformin prompted a modest changes in the expression of plasma markers associated with systemic inflammation and gut barrier dysfunction.
3.3. Effects of metformin on HIV reservoir markers in blood CD4+ T-cells
To determine the effect of metformin on markers of HIV persistence, we performed virologic measurements in blood CD4+ T-cells at Baseline, Week 12 and Week 24. The PCR-based quantification of integrated HIV-DNA levels [43] demonstrated a minor increase in the pool of cells carrying integrated HIV reservoirs after metformin treatment, between Week 24 and Baseline (median: 682 versus 624; p=0.0724; Wilcoxon) and between Week 24 and Week 12 (median 682 versus 543; p=0.0491; Wilcoxon) (Fig. 2a). The size of transcriptionally-competent HIV reservoirs, quantified by RT-PCR as cell-associated (CA) unspliced (US) HIV-RNA ex vivo [65], revealed no statistically significant changes in response to metformin (Fig. 2b). The frequency of CD4+ T-cells carrying inducible multiply spliced (MS) HIV-RNA, quantified by the Tat/Rev induced limiting dilution assay (TILDA) after stimulation in vitro [66], revealed detectable HIV transcription in the majority of donors, with important inter-donor variations but no significant changes in response to metformin (Figs. 2c). In conclusion, these results reveal a minor increase in integrated HIV-DNA levels at 12 weeks after metformin discontinuation (Week 24) compared to Baseline, while no significant changes in transcription-competent HIV reservoirs were observed between Baseline, Week 12 and Week 24 in blood CD4+ T-cells.
3.4. Effects of metformin on the frequency and phenotype of blood T-cell subsets
To further explore the immunological effects of metformin treatment on CD4+ T-cells, polychromatic flow cytometry analysis was performed on frozen PBMCs. Changes in the frequency of naive (CD45RA+CCR7+CD27+), central memory (CM, CD45RA–CCR7+CD27+), transitional memory (TM, CD45RA−CCR7−CD27+), effector memory (EM, CD45RA–CCR7–CD27–), and terminally differentiated (TD, CD45RA+CCR7–CD27–) CD4+ and CD8+ T-cell subsets (Supplemental Figure 3a) revealed no major changes between Baseline, Week 12 and Week 24 (Supplemental Figure 3b-c). Nevertheless, some differences that reached statistical significance were noted for N, EM, and TD CD4+ T-cells (Supplemental Figure 3b) and for CM and TD CD8+ T-cells (Supplemental Figure 3c). In addition, the expression levels of the HIV co-receptor CCR5 and the immune checkpoint molecules PD-1, LAG3, and TIGIT on total and CD4+ and CD8+ T-cell subsets was relatively stable between Baseline, Week 12, and Week 24, despite some minor reductions in expression that reached statistical significance, especially for PD-1 and LAG3 (Supplemental Figure 4). Finally, the expression of tissue-specific homing integrins α4β7, α4β1, αEβ7, and αEβ1 on naive and memory CD4+ T-cell subsets were also relatively stable between Baseline, Week 12, and Week 24 (Supplemental Figure 5). In conclusion, 12 weeks of metformin treatment did not promote major and sustained changes in the frequency of naive and memory CD4+ and CD8+ T-cell subsets in the blood, nor in their expression levels of markers linked to HIV permissiveness, T-cell exhaustion, and tissue-specific homing.
3.5. Effects of metformin on the frequency and phenotype of colon CD4+ T-cells
Considering the importance of gut-associated lymphoid tissues (GALT) in HIV pathogenesis [22] and the fact that the intestine is the principal organ targeted by metformin [58], subsequent investigations were performed using matched blood and SCB samples collected at Baseline and Week 12 (LILAC #1-13; Supplemental Table 1). As expected, in contrast to blood, the pool of CD4+ and CD8+ T-cells infiltrating the colon exhibited in majority a memory phenotype (Fig. 1a-c; data not shown). While no changes were observed in the blood, the frequency of total and memory CD4+ T-cells significantly decreased in the colon at Week 12 compared to Baseline (Fig. 3a-c). The frequency of memory CD4+ and CD8+ T-cells displaying an activated phenotype (HLA-DR+CD38+) was higher in the colon compared to the blood at both Baseline and Week 12 (Fig. 3d-e). Metformin-mediated changes in the expression of HLA-DR/CD38 on blood and colon-infiltrating CD4+ and CD8+ T-cells did not reach statistical significance (Fig. 3d-e). Nevertheless, marked inter-donor variations were observed, with the frequency of colon-infiltrating HLA-DR+CD38+ CD4+ and CD8+ T-cells robustly decreasing in a fraction of 8/13 and 9/13 participants, respectively, at Week 12 versus Baseline (Fig. 3d-e). In conclusion, metformin was associated with a significant decrease in the frequency of colon-infiltrating CD4+ T-cells, with a reduction in the frequency of activated T-cells being observed only in a fraction of participants.
3.6. Metformin decreases mTOR phosphorylation in colon memory CCR6+CD4+ T-cells
Metformin is documented to inhibit the mTOR signaling pathway [55], [56], [57]. To determine the effectiveness of metformin treatment, the expression of the active phosphorylated form of mTOR (p-mTOR) was analyzed in CD4+ and CD8+ T-cells from matched blood and SCB. Compared to blood, at Baseline p-mTOR expression was considerably higher in the colon CD4+ T-cells (median: 3.9% versus 20.4%; p=0.0002; Wilcoxon and CD8+ T-cells (median: 4% versus 17.6%; p=0.0081; Wilcoxon) (Fig. 4a). In the blood, no changes in mTOR activation in CD4+ and CD8+ T-cells were observed in response to metformin (Fig. 4a). In contrast, in the colon, a decrease in p-mTOR expression was observed for both CD4+ T-cells (median: 20.4% vs. 15.5%; p=0.064; Wilcoxon) and CD8+ T-cells (median: 17.6% vs. 8.27%, Baseline vs. Week 12; p=0.039; Wilcoxon) (Fig. 4a).
Considering our previous findings that p-mTOR is preferentially expressed in colon-infiltrating memory CCR6+ compared to CCR6− CD4+ T-cells [43], we investigated the effect of metformin treatment on p-mTOR expression in these subsets (Fig. 4b-c). Indeed, as we previously reported [43], at Baseline p-mTOR expression was significantly higher in the colon compared to the blood for both CCR6+ (1.75% versus 13.35%; p=0.0002; Wilcoxon) and CCR6- (1.12% versus 3.8%; p=0.0012; Wilcoxon) CD4+ T-cells, with the highest levels of expression in the colon on CCR6+ versus CCR6− T-cells (13.35% versus 3.8%; p=0.0002; Wilcoxon) (Fig. 4d). Metformin treatment resulted in a significant decrease in p-mTOR expression in colon CCR6+ CD4+ T-cells (median: 13% vs. 7.87%, Baseline vs. Week 12; p=0.0129; Wilcoxon) (Fig. 4d). Notably, this decrease was observed in the majority of participants with the exception of two: LILAC#6 (6.08% vs. 14.7%) and LILAC#8 (4.15% vs. 5.38%Baseline versus Week 12). Although p-mTOR expression levels were much lower in CCR6−CD4+ compared to CCR6+CD4+ T-cells at Baseline, metformin significantly reduced the frequency of p-mTOR+ cells in this subset as well (median: 4.07% vs. 1.44%, Baseline vs. Week 12; p=0.0139; Wilcoxon) (Fig. 4d). These results confirm the superior p-mTOR expression in CCR6+ versus CCR6−CD4+ T-cells [43] and demonstrate the capacity of metformin to decrease mTOR phosphorylation in colon-infiltrating CD4+ T-cells.
3.6.1. Effects of metformin on the expression of integrin β7, CCR5, SAMHD1, and Bcl-2 in colon memory CCR6+CD4+ T-cells
We further investigated changes in the expression of functional markers, such as the gut-homing and HIV binding molecule integrin β7 (ITGB7) [71], the HIV co-receptor CCR5 [1], the HIV restriction factor SAMHD1 [72], and the survival molecule Bcl-2 [73] on CCR6+ and CCR6− CD4+ T-cells from blood and matched colon biopsies. At Baseline, colon-infiltrating CCR6+ T-cells expressed higher levels of integrin β7 (p=0.0005; Wilcoxon) and CCR5 (p=0.0005; Wilcoxon) compared to their blood counterparts (Fig. 5a-b). Similarly, within the colon, CCR6+ compared to CCR6− T-cells expressed higher levels of integrin β7 (p=0.0002; Wilcoxon) and CCR5 (p=0.0479; Wilcoxon) (Fig. 5a-b). Metformin-induced changes in integrin β7 and CCR5 expression on CCR6+/CCR6− T-cells from blood and colon were subject to tremendous donor-to-donor variations and did not reach statistical significance (Fig. 5a-b). In terms of SAMHD1 expression, at Baseline, colon-infiltrating CCR6+ T-cells expressed higher levels compared with blood CCR6+ T-cells (p=0.0547; Wilcoxon) and colon-infiltrating CCR6− T-cells (p=0.0078; Wilcoxon) (Fig. 5c). Metformin significantly increased the frequency of CCR6+ T-cells expressing SAMHD1 in blood (p=0.007; Wilcoxon) (Fig. 5c). A similar increase that did not reach statistical significance was observed in the colon, with 6/8 participants showing an increase in the frequency of colon SAMHD1+CCR6+ T-cells upon metformin treatment (median: 24.7% vs. 38.3%) (Fig. 5c). Finally, the expression of Bcl-2 was the lowest in colon-infiltrating CCR6− T-cells compared to blood CCR6− T-cells (p=0.0078; Wilcoxon) and colon-infiltrating CCR6+ T-cells (p=0.0078; Wilcoxon) at Baseline (Fig. 5d). Metformin mediated a significant increase in Bcl-2 expression in colon-infiltrating CCR6+ T-cells (p=0.0078; Wilcoxon), with Bcl-2 levels at Week 12 being significantly higher in CCR6+ compared to CCR6- T-cells in the colon (Fig. 5d). Overall, these results reveal metformin-mediated changes, notably a statistically significant increase in the expression of SAMHD1 and Bcl-2 in CCR6+CD4+ T-cells from blood and colon, respectively.
3.6.2. Metformin reduces residual HIV transcription in colon memory CD4+ T-cells in a fraction of ART-treated PLWH
We have shown that metformin treatment results in a considerable decrease in p-mTOR in colon-infiltrating memory CCR6+CD4+ T-cells (Fig. 4), a subsets enriched in HIV-DNA in the colon of ART-treated PLWH [31,33]. As mTOR positively regulates different HIV replication steps, including transcription [49,50], we further investigated the effects of metformin on HIV reservoirs in the colon. Specifically, CA total HIV-DNA and HIV-RNA levels were quantified using DNA/RNA dually extracted from memory CD4+ T-cells of SCB collected at Baseline and Week 12. Consistent with results in the blood (Fig. 2), levels of total HIV-DNA were stable in colon memory CD4+ T-cells between Baseline and Week 12 (Fig. 6a). The HIV-RNA/HIV-DNA ratios, indicative of HIV transcription [65], are depicted in Fig. 6b. Although differences between Baseline and Week 12 did not reach statistical significance, 8/13 participants showed a decrease in HIV transcription ranging from 15% to 92% decrease (LILAC #1, #2, #3, #4, #5, #7, #8, #12), while HIV transcription increased in 4/13 (LILAC #6, #9, #11, #13) and remained stable in 1/13 participants (LILAC #10, Supplemental Table 1; Fig. 6b). Thus, metformin reduced the HIV-RNA/HIV/DNA ratios, indicative of decreased residual HIV transcription, in the colon of 61.5% study participants.
Finally, pairwise correlations between phenotypic measurements in colon-infiltrating CD4+ T-cells, in terms of frequency (the % of total CD4+ T-cells) and phenotype (the % of CD4+ T-cells with a p-mTOR+CCR6+, CCR5+CCR6+ and ITGB7+CCR6+ phenotype), and the corresponding CA HIV RNA/DNA ratios were performed using the Spearman's method in the n=13 LILAC participants. Results in Fig. 7 demonstrate that metformin-mediated changes at Week 12 versus Baseline in the CA HIV-RNA/HIV-DNA ratios positively correlated with changes in the frequency of total CD4+ T-cells infiltrating the colon (p=0.0259, r=0.6226; Spearman correlation; Fig. 7a) and that of CCR6+CD4+ T-cells expressing CCR5 (p=0.0629, r=0.5337; Spearman correlation; Fig. 7b) and integrin β7 (p=0.0642, r=0.5330; Spearman correlation; Fig. 7c). A positive correlation between the decrease in CA HIV RNA/DNA ratios and the decrease in mTOR phosphorylation in CCR6+CD4+ T-cells was not observed (Fig. 7d), likely because changes in the frequencies of p-mTOR+CCR6+CD4+ T-cells were strongly decreased after 12 weeks of metformin treatment (Fig. 4d), while this was not the case for CA HIV RNA/DNA ratios (Fig. 6).
Together, these results are in line with the knowledge that CCR6+CD4+ T-cells expressing the HIV co-receptor CCR5, are major sites for HIV replication/transcription [21,22], and point to the beneficial effects of metformin in reducing T-cell activation and residual HIV transcription in the colon of ART-treated PLWH.
3.7. Novel sample size calculations
In view of a future RCT, we used the data presented in Fig. 3d to determine means and standard deviations, and used the Wilcoxon matched pairs ranked test to calculate the sample size needed to observe a significant reduction in the frequency of activated CD4+ T cells (defined as HLADR+CD38+) in the colon after 12 weeks of metformin supplementation of ART in PLWH. The data from this pilot study performed on n=13 SCB participants indicated that the frequency of colon-infiltrating HLADR+CD38+CD4+ T cells was 5.78% (SD: 3.45%) at Baseline and 4.60% (SD: 3.00%) after 12 weeks of metformin treatment. Based on this, we estimated that a sample size of n=58 participants will have 80% power to observe a difference at least as great as the one reported in the pilot study, with two-sided alpha=0.05 (data not shown). Thus, future randomized clinical trials (RCT) should be performed on least n=58 participants in an effort to improve the likelihood of reaching statistical significance for multiple parameters, including metformin-mediated changes in the residual HIV transcription in the colon of ART-treated PLWH.
4. Discussion
Here, we report results from the Lilac pilot clinical trial (NCT02659306), in which metformin was administered for 12 weeks to n=22 non-diabetic ART-treated PLWH with CD4/CD8 ratios below 0.8. Our investigations revealed metformin-mediated functional changes in the sigmoid colon, including i) decreased infiltration of memory CD4+ T-cells; ii) reduced mTOR phosphorylation/activation in CCR6+CD4+ T-cells, and iii) decreased residual HIV transcription in memory CD4+ T-cells 61.5% study participants. At Baseline, the HIV-RNA/HIV-DNA ratios, a surrogate marker for residual HIV transcription, were positively correlated with the frequency of CCR6+CD4+ T-cells expressing CCR5 and phosphorylated mTOR. At Week 12 vs. Baseline, changes in residual HIV transcription in the colon positively correlated with a decreased infiltration of memory CD4+ T-cells in the colon. Our findings support a model in which metformin exerts beneficial effects in attenuating inflammation and residual HIV transcription in the colon of ART-treated PLWH via mechanisms involving a reduction in mTOR phosphorylation/activation (Fig. 7). These findings add to recent publications by our group demonstrating that metformin supplementation of ART was well tolerated and exerted positive effects on the body weight and gut microbiota [61,62].
The current knowledge on metformin acting as an mTOR inhibitor [55], [56], [57], together with the documented role of mTOR in regulating cell metabolism [[44], [45], [46], [47],52] and HIV transcription [49,50], mainly in Th17 cells [22,43,59], prompted us to evaluate the effects of metformin supplementation of ART on multiple immunological and virological markers. The peripheral blood is the most accessible compartment for clinical investigations. In our study, metformin showed negligible efficacy in changing plasma levels of markers of inflammation/activation and gut-barrier dysfunction, as well as CD4+ T-cell markers linked to HIV permissiveness (CCR5), T-cell exhaustion (PD-1, LAG3, and TIGIT), and tissue-specific homing (α4β7, α4β1, αEβ7, and αEβ1). Similarly, metformin prompted minor changes in the size of HIV reservoirs in blood CD4+ T-cells. Nested real-time PCR quantifications [43]), showed a minor but statistically significant increase in HIV-DNA integration between week 12 and Week 24; this may be explained by the recirculation of the T cells from tissues, consistent with the metformin-mediated decrease in the frequency of colon-infiltrating CD4+ T-cells and in the plasma levels of CCL20, a chemokine involved in CCR6+ T cell trafficking [74]. In contrast, changes in the frequency of cells carrying inducible MS HIV-RNA (measured by TILDA [66]), and replication-competent HIV reservoirs (measured by QVOA [75]) did not reach statistical significance between Baseline, Week 12, and Week 24, consistent with the low expression of phosphorylated mTOR in blood cells. These results are in line with the well-described stability of HIV reservoirs under ART and the difficulty to perturb HIV latency upon short time interventions [2], [3], [4]. This may be explained by the relatively short time of metformin treatment, which was 12 weeks. Indeed, a recently published study, demonstrated that metformin supplementation of ART for 48 weeks decreased T-cell exhaustion as revealed by the measurement of PD-1, TIGIT and Tim3 in the blood [76]. Therefore, whether metformin administration for 48 weeks or longer time periods will decrease viral reservoirs in the blood requires future investigations.
During the course of HIV infection, immunological and virological changes were reported to occur in the GALT before any differences could be observed in the blood [14]. Indeed, the GALT is the most important site of T-cell activation, the portal site of HIV entry, and an important anatomic HIV reservoir [22,77]. Therefore, we investigated the effect of metformin treatment in the colon, which is also a major site of metformin action [58]. While the quantification of total HIV-DNA in matched blood and colon memory CD4+ T-cells confirmed the stability of HIV reservoirs [2], [3], [4], the HIV-RNA/DNA ratios, indicative of residual HIV transcription, in colon memory CD4+ T-cells were markedly decreased in 8/13 study participants. This effect could be the direct consequence of a reduced mTOR phosphorylation, consistent with the role of mTOR in the positive regulation of HIV transcription/latency in CD4+ T-cells [49,50].
Previous studies by our group demonstrated that gut-homing Th17-polarized CCR6+CD4+ T-cells distinguishing from non-Th17 CCR6−CD4+ T-cells by their transcriptional program prone to HIV replication/persistence and exhibit a CCR5+integrinβ7+p-mTOR+ phenotype in the colon of ART-treated PLWH [22,31]. Consistently, in this study, we confirmed the predominant expression of the HIV co-receptor CCR5, the gut-homing integrin β7, and the metabolic regulator mTOR by colon-infiltrating CCR6+ compared to CCR6−CD4+ T-cells at Baseline. Our results show that metformin-mediated changes in mTOR phosphorylation in total memory and CCR6+CD4+ T-cells correlated positively with changes in residual HIV transcription in the colon. These findings are in line with reports by other groups describing an mTOR-dependent increase in HIV transcriptions via CDK9 phophorylation, a component of the p-TEFb complex involved in Tat-mediated HIV elongation [9,49,50]. Metformin also increased expression of Bcl-2, a marker of cell survival, in colon-infiltrating CCR6+CD4+ T-cells. This is consistent with the fact that mTOR inhibits Bcl-2 expression [73] thus, additionally supporting the efficacy of metformin treatment on colon-infiltrating T-cells.
Another interesting finding of our study included the metformin-mediated increase in total SAMHD1 expression in CCR6+CD4+ T-cells observed in all participants in the blood and in a subset of participants in the colon. The active (non-phosphorylated) form of SAMHD1 restricts HIV replication at the level of viral reverse transcription [72]. Previous studies reported that, in the blood, CCR6- compared to CCR6+CD4+ T-cells express higher levels of SAMHD1 [78], thus providing an explanation for the relative resistance of the CCR6- T-cell subset to HIV infection compared to the CCR6+ T-cells [22]. In our study, SAMHD1 expression was surprisingly higher in colon-infiltrating CCR6+ versus CCR6−CD4+ T-cells. Of note, the Abs we used recognizes total rather than the phosphorylated inactive form of SAMHD1 [72] thus, preventing any conclusions on SAMHD1 activity in these cells. A direct link between mTOR and SAMHD1 is not described in the literature. Nevertheless, SAMHD1 is known to regulate cell metabolism by modulating the pool of intracellular dNTPs [72]. Metabolically active cells express low levels of SAMHD1, creating an environment prone to RNA synthesis and favorable to HIV reverse transcription [79]. Additionally, it was recently demonstrated that SAMHD1 suppresses HIV-LTR transcription [80]. Thus, its increased expression in colon CCR6+ T-cells may result in an attenuated HIV transcription. Hence, elevated SAMHD1 expression after metformin administration may reflect a reduction of the metabolic activity at the cellular level, in line with the decreased expression of phosphorylated mTOR in colon-infiltrating CCR6+CD4+ T-cells. Such conclusions require though larger sample size studies.
In line with the possibility to use mTOR inhibitors for HIV cure/remission, a pilot clinical trial performed on ART-treated PLWH receiving sirolimus for 20 weeks demonstrated a decrease in CCR5 expression and markers of cell cycling and immune exhaustion (Henrich et al., Abstract, CROI 2019). The effect of sirolimus and other mTOR inhibitors on the normalization of immune activation and the control of residual HIV transcription during ART deserves investigations.
Fig. 8.

Proposed model of metformin-mediated decrease in HIV transcription in ART-treated PLWH. Shown is the graphical representation of the current knowledge on the effect of metformin on mTOR activation via two mechanisms: 1) an AMP-Activated Protein Kinase (AMPK)-dependent manner by inhibiting mitochondria function and increasing the adenosine monophosphate (AMP):adenosine triphosphate (ATP) ratio, thus leading to AMP-dependent AMPK activation and AMPK-mediated mTORC1 inhibition and 2) an AMPK-independent mechanism via the inhibition of Rag GTPase (RAG) activity, which activates mTORC1 [55], [56], [57]. Subsequently, mTOR pathway activation, as a consequence of its phosphorylation (P), facilitates HIV transcription via the regulation of multiple factors including CDK9 phosphorylation (P), a component of the p-TEFb complex [9], involved in Tat-mediated HIV transcription [49]. Together, our results, reveal the capacity of metformin treatment to reduce residual HIV transcription in ART-treated PLWH by reducing mTOR activation/phosphorylation.
In conclusion, our investigations performed in the framework of the LILAC pilot clinical study reveal that metformin diminishes residual HIV transcription in the colon CD4+ T-cells of more than half of study participants, likely via mTOR-dependent mechanisms (Fig. 7). Other beneficial effect of metformin include a reduced infiltration of CD4+ T cells in the colon, in line with the decrease in plasma levels of CCL20, a chemokine involved in CCR6+ T-cell trafficking [26,74], and a reduction in mTOR phosphorylation, mainly in colon CCR6+ T-cells, a subset documented to carry HIV reservoirs in ART-treated PLWH [31], [32], [33]. Considering the promising results observed upon only 12 weeks of metformin treatment, the long-term metformin supplementation of ART may represent a possible new strategy to be exploited in view of achieving the goal of immunological restoration and/or HIV reservoir purging in PLWH. Future randomized clinical trials with long-term metformin supplementation of ART should test this possibility. Finally, our findings highlight the importance of investigating the GALT, and in particular the gut-homing CCR6+ Th17 cells, during clinical intervention aimed at HIV remission/cure.
Declaration of Competing Interest
EAC is a member of the Scientific Advisory Board of Theratechnologies. JBA performed contract research and/or served on Advisory Boards for Gilead Sciences Canada Inc., Merck Canada Inc., Abbvie Corp., ViiV Healthcare, Bristol Myers Squibb, Janssen Inc., and Argos Pharmaceuticals. NC received research funding from EMD Serono and served on Advisory Boards for Gilead Sciences. JPR performed contract research and/or served on Advisory Boards for Gilead Sciences Canada Inc., Merck Canada Inc., Abbvie Corp., ViiV Healthcare, Bristol Myers Squibb, Janssen Inc., Argos Pharmaceuticals from InnaVirVax, and Theravectys. PA's laboratory receives research funding from Glaxo Smith Klein/NeoMed for projects not related to the present study. PA served as a Consultant at Merck Canada Inc. relative to research projects different from the present study. The other co-authors (DP, AP, RP, AF, LRM, MM, AG, VM, FPD, JPG, SI, SL, and MPG) have no financial or non-financial competing interests to disclose.
Acknowledgments
Contributors
DP designed and performed research, analyzed data, prepared figures, and wrote the manuscript. AP performed research and analyzed data related to HIV reservoir measurements. RP, AF, LRM, MM, AG, VM, FPD, and SI performed research related to clinical study design, sample collection/preparation and/or HIV reservoir measurements. JPG performed statistical analysis. SL and EAC provided protocols and contributed to research design. MPG provided access to matched blood and colon biopsies. JBA contributed to clinical study design and recruited participants. NC provided protocols and reagents, contributed to research design and manuscript writing. JPR conceived the clinical study hypothesis and design, set up clinical research protocols and recruited participants, provided access to biological samples and clinical information, and contributed to manuscript writing. PA conceived the research study hypothesis, designed research, analyzed data, prepared figures, performed statistical analysis, and wrote the manuscript. All authors read and approved the final version of the manuscript, and have had access to the raw data.
Acknowledgements
This study was funded by grants from the Canadian Institutes of Health Research (CIHR; #IBC-154053 to PA; #HOP-103230, PTJ-166049 to JPR); the Vaccines & Immunotherapies Core of the CIHR Canadian HIV Trials Network (grant CTN PT247 to JPR); the Fonds de Recherche du Québec-Santé (FRQ-S)/AIDS and Infectious Diseases Network (to PA and JPR); the FRQ‐S Cell Therapy Network (to JPR); the Canadian HIV Cure Enterprise Team Grant (CanCURE 1.0) funded by the CIHR in partnership with CANFAR and IAS (CanCURE 1.0; # HIG-133050 to PA, EAC, JBA, and JPR); and The Canadian HIV Cure Enterprise Team Grant (CanCURE 2.0) funded by the CIHR (#HB2–164064 to PA, EAC, JBA, NC, and JPR). Core facilities and HIV-infected patients’ cohorts were supported by the Fondation du CHUM and the FRQ-S Network. The funding institutions played no role in the design, collection, analysis, and interpretation of data. DP received a Doctoral award from the Université de Montréal and the FRQ-S. VM received a FRQ-S post-doctoral fellowship award. SI was supported by a postdoctoral fellowship from the Canadian Institutes of Health, HIV Clinical trials network (CTN). SL is an FRQ-S Research Scholars Emeritus awardee. JPR holds a Louis Lowenstein Chair in Hematology and Oncology, McGill University.
The authors thank Dr. Dominique Gauchat and Philippe St Onge (Flow Cytometry Core Facility, CHUM-Research Center, Montréal, QC, Canada) for expert technical support with polychromatic flow cytometry sorting. The authors thank Olfa Debbeche (Biosafety Level 3 Core Facility CHUM-Research Center, Montréal, QC, Canada); Dr. Olivier Schwartz (Institut Pasteur, Paris, France) for providing the anti-SAMHD1 antibody; Mario Legault for help with ethical approvals and informed consents, and Josée Girouard and Angie Massicotte for their key contribution to LILAC study participant recruitment and access to matched blood and sigmoid biopsies and clinical information from HIV-infected and uninfected donors. The authors address a special thanks to all Lilac pilot clinical trial participants from Montréal and Ottawa for their gift of blood and colon biopsies and key contribution to this work.
Data sharing
Data from this manuscript is available from the corresponding author upon reasonable request
Footnotes
Supplementary material associated with this article can be found in the online version at doi:10.1016/j.ebiom.2021.103270.
Contributor Information
Delphine Planas, Email: delphine.planas@pasteur.fr.
Amélie Pagliuzza, Email: amelie.pagliuzza.chum@ssss.gouv.qc.ca.
Rosalie Ponte, Email: ponte.rosalie@gmail.com.
Augustine Fert, Email: augustine.fert@umontreal.ca.
Laurence Raymond Marchand, Email: laurence.raymond-marchand.chum@ssss.gouv.qc.ca.
Marta Massanella, Email: mmassanella@irsicaixa.es.
Annie Gosselin, Email: annie.gosselin.chum@gmail.com.
Vikram Mehraj, Email: vikram.mehraj@mail.mcgill.ca.
Franck P Dupuy, Email: dura007@hotmail.fr.
Stéphane Isnard, Email: stephane.isnard@mail.mcgill.ca.
Jean-Philippe Goulet, Email: gouletjp@icloud.com.
Sylvie Lesage, Email: sylvie.lesage@umontreal.ca.
Eric A. Cohen, Email: Eric.Cohen@ircm.qc.ca.
Mager Peter Ghali, Email: ghali98@hotmail.com.
Jonathan B. Angel, Email: jangel@ohri.ca.
Nicolas Chomont, Email: nicolas.chomont@umontreal.ca.
Jean-Pierre Routy, Email: jean-pierre.routy@mcgill.ca.
Petronela Ancuta, Email: petronela.ancuta@umontreal.ca.
Appendix. Supplementary materials
References
- 1.Barre-Sinoussi F, Ross AL, Delfraissy JF. Past, present and future: 30 years of HIV research. Nat Rev Microbiol. 2013;11(12):877–883. doi: 10.1038/nrmicro3132. [DOI] [PubMed] [Google Scholar]
- 2.Sengupta S, Siliciano RF. Targeting the latent reservoir for HIV-1. Immunity. 2018;48(5):872–895. doi: 10.1016/j.immuni.2018.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davenport MP, Khoury DS, Cromer D, Lewin SR, Kelleher AD, Kent SJ. Functional cure of HIV: the scale of the challenge. Nat Rev Immunol. 2019;19(1):45–54. doi: 10.1038/s41577-018-0085-4. [DOI] [PubMed] [Google Scholar]
- 4.Cohn LB, Chomont N, Deeks SG. The Biology of the HIV-1 Latent Reservoir and Implications for Cure Strategies. Cell Host Microbe. 2020;27(4):519–530. doi: 10.1016/j.chom.2020.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hunt PW, Lee SA, Siedner MJ. Immunologic biomarkers, morbidity, and mortality in treated HIV infection. J Infect Dis. 2016;214(Suppl 2):S44–S50. doi: 10.1093/infdis/jiw275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zicari S, Sessa L, Cotugno N, Ruggiero A, Morrocchi E, Concato C. Immune activation, inflammation, and non-AIDS Co-Morbidities in HIV-infected patients under long-term ART. Viruses. 2019;11(3) doi: 10.3390/v11030200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Massanella M, Fromentin R, Chomont N. Residual inflammation and viral reservoirs: alliance against an HIV cure. Curr Opin HIV AIDS. 2016;11(2):234–241. doi: 10.1097/COH.0000000000000230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thenin-Houssier S, Valente ST. HIV-1 capsid inhibitors as antiretroviral agents. Curr HIV Res. 2016;14(3):270–282. doi: 10.2174/1570162x14999160224103555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bouchat S, Van Lint C. Viral latency of HIV-1. Virologie (Montrouge) 2019;23(4):195–210. doi: 10.1684/vir.2019.0782. [DOI] [PubMed] [Google Scholar]
- 10.Kearney MF, Wiegand A, Shao W, Coffin JM, Mellors JW, Lederman M. Origin of rebound plasma HIV includes cells with identical proviruses that are transcriptionally active before stopping of antiretroviral therapy. J Virol. 2016;90(3):1369–1376. doi: 10.1128/JVI.02139-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siliciano JD, Siliciano RF. Nonsuppressible HIV-1 viremia: a reflection of how the reservoir persists. J Clin Invest. 2020;130(11):5665–5667. doi: 10.1172/JCI141497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Halvas EK, Joseph KW, Brandt LD, Guo S, Sobolewski MD, Jacobs JL. HIV-1 viremia not suppressible by antiretroviral therapy can originate from large T cell clones producing infectious virus. J Clin Invest. 2020;130(11):5847–5857. doi: 10.1172/JCI138099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Veazey RS, DeMaria M, Chalifoux LV, Shvetz DE, Pauley DR, Knight HL. Gastrointestinal tract as a major site of CD4+ T cell depletion and viral replication in SIV infection. Science. 1998;280(5362):427–431. doi: 10.1126/science.280.5362.427. [DOI] [PubMed] [Google Scholar]
- 14.Veazey RS, Lackner AA. Getting to the guts of HIV pathogenesis. J Exp Med. 2004;200(6):697–700. doi: 10.1084/jem.20041464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brenchley JM, Schacker TW, Ruff LE, Price DA, Taylor JH, Beilman GJ. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med. 2004;200(6):749–759. doi: 10.1084/jem.20040874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mehandru S, Poles MA, Tenner-Racz K, Horowitz A, Hurley A, Hogan C. Primary HIV-1 infection is associated with preferential depletion of CD4+ T lymphocytes from effector sites in the gastrointestinal tract. J Exp Med. 2004;200(6):761–770. doi: 10.1084/jem.20041196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12(12):1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 18.Ancuta P, Kamat A, Kunstman KJ, Kim EY, Autissier P, Wurcel A. Microbial translocation is associated with increased monocyte activation and dementia in AIDS patients. PLoS One. 2008;3(6):e2516. doi: 10.1371/journal.pone.0002516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Douek DC, Roederer M, Koup RA. Emerging concepts in the immunopathogenesis of AIDS. Annu Rev Med. 2009;60:471–484. doi: 10.1146/annurev.med.60.041807.123549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ancuta P, Monteiro P, Sekaly RP. Th17 lineage commitment and HIV-1 pathogenesis. Curr Opin HIV AIDS. 2010;5(2):158–165. doi: 10.1097/COH.0b013e3283364733. [DOI] [PubMed] [Google Scholar]
- 21.Wacleche VS, Landay A, Routy JP, Ancuta P. The Th17 lineage: from barrier surfaces homeostasis to autoimmunity, cancer, and HIV-1 pathogenesis. Viruses. 2017;9(10) doi: 10.3390/v9100303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Planas D, Routy JP, Ancuta P. New Th17-specific therapeutic strategies for HIV remission. Curr Opin HIV AIDS. 2019;14(2):85–92. doi: 10.1097/COH.0000000000000522. [DOI] [PubMed] [Google Scholar]
- 23.Dong C. TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol. 2008;8(5):337–348. doi: 10.1038/nri2295. [DOI] [PubMed] [Google Scholar]
- 24.Kelley CF, Kraft CS, de Man TJ, Duphare C, Lee HW, Yang J. The rectal mucosa and condomless receptive anal intercourse in HIV-negative MSM: implications for HIV transmission and prevention. Mucosal Immunol. 2017;10(4):996–1007. doi: 10.1038/mi.2016.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stieh DJ, Matias E, Xu H, Fought AJ, Blanchard JL, Marx PA. Th17 cells are preferentially infected very early after vaginal transmission of SIV in macaques. Cell Host Microbe. 2016;19(4):529–540. doi: 10.1016/j.chom.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McGary CS, Alvarez X, Harrington S, Cervasi B, Ryan ES, Iriele RI. The loss of CCR6(+) and CD161(+) CD4(+) T-cell homeostasis contributes to disease progression in SIV-infected rhesus macaques. Mucosal Immunol. 2017;10(4):1082–1096. doi: 10.1038/mi.2016.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schuetz A, Deleage C, Sereti I, Rerknimitr R, Phanuphak N, Phuang-Ngern Y. Initiation of ART during early acute HIV infection preserves mucosal Th17 function and reverses HIV-related immune activation. PLoS Pathog. 2014;10(12) doi: 10.1371/journal.ppat.1004543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muranski P, Borman ZA, Kerkar SP, Klebanoff CA, Ji Y, Sanchez-Perez L. Th17 cells are long lived and retain a stem cell-like molecular signature. Immunity. 2011;35(6):972–985. doi: 10.1016/j.immuni.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun H, Kim D, Li X, Kiselinova M, Ouyang Z, Vandekerckhove L. Th1/17 Polarization of CD4 T Cells Supports HIV-1 Persistence during Antiretroviral Therapy. J Virol. 2015;89(22):11284–11293. doi: 10.1128/JVI.01595-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wacleche VS, Goulet JP, Gosselin A, Monteiro P, Soudeyns H, Fromentin R. New insights into the heterogeneity of Th17 subsets contributing to HIV-1 persistence during antiretroviral therapy. Retrovirology. 2016;13(1):59. doi: 10.1186/s12977-016-0293-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gosselin A, Wiche Salinas TR, Planas D, Wacleche VS, Zhang Y, Fromentin R. HIV persists in CCR6+CD4+ T cells from colon and blood during antiretroviral therapy. AIDS. 2017;31(1):35–48. doi: 10.1097/QAD.0000000000001309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khoury G, Anderson JL, Fromentin R, Hartogenesis W, Smith MZ, Bacchetti P. Persistence of integrated HIV DNA in CXCR3 + CCR6 + memory CD4+ T cells in HIV-infected individuals on antiretroviral therapy. AIDS. 2016;30(10):1511–1520. doi: 10.1097/QAD.0000000000001029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anderson JL, Khoury G, Fromentin R, Solomon A, Chomont N, Sinclair E. Human immunodeficiency virus (HIV)-Infected CCR6+ Rectal CD4+ T Cells and HIV persistence on antiretroviral therapy. J Infect Dis. 2020;221(5):744–755. doi: 10.1093/infdis/jiz509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramesh R, Kozhaya L, McKevitt K, Djuretic IM, Carlson TJ, Quintero MA. Pro-inflammatory human Th17 cells selectively express P-glycoprotein and are refractory to glucocorticoids. J Exp Med. 2014;211(1):89–104. doi: 10.1084/jem.20130301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu L, Hollinshead KER, Hao Y, Au C, Kroehling L, Ng C. Niche-selective inhibition of pathogenic Th17 cells by targeting metabolic redundancy. Cell. 2020;182(3):641–654. doi: 10.1016/j.cell.2020.06.014. e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.El Hed A, Khaitan A, Kozhaya L, Manel N, Daskalakis D, Borkowsky W. Susceptibility of human Th17 cells to human immunodeficiency virus and their perturbation during infection. J Infect Dis. 2010;201(6):843–854. doi: 10.1086/651021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gosselin A, Monteiro P, Chomont N, Diaz-Griffero F, Said EA, Fonseca S. Peripheral blood CCR4+CCR6+ and CXCR3+CCR6+CD4+ T cells are highly permissive to HIV-1 infection. J Immunol. 2010;184(3):1604–1616. doi: 10.4049/jimmunol.0903058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bernier A, Cleret-Buhot A, Zhang Y, Goulet JP, Monteiro P, Gosselin A. Transcriptional profiling reveals molecular signatures associated with HIV permissiveness in Th1Th17 cells and identifies peroxisome proliferator-activated receptor gamma as an intrinsic negative regulator of viral replication. Retrovirology. 2013;10:160. doi: 10.1186/1742-4690-10-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cleret-Buhot A, Zhang Y, Planas D, Goulet JP, Monteiro P, Gosselin A. Identification of novel HIV-1 dependency factors in primary CCR4(+)CCR6(+)Th17 cells via a genome-wide transcriptional approach. Retrovirology. 2015;12:102. doi: 10.1186/s12977-015-0226-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Christensen-Quick A, Lafferty M, Sun L, Marchionni L, DeVico A, Garzino-Demo A. Human Th17 cells lack HIV-inhibitory RNases and are highly permissive to productive HIV infection. J Virol. 2016;90(17):7833–7847. doi: 10.1128/JVI.02869-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kurebayashi Y, Nagai S, Ikejiri A, Ohtani M, Ichiyama K, Baba Y. PI3K-Akt-mTORC1-S6K1/2 axis controls Th17 differentiation by regulating Gfi1 expression and nuclear translocation of RORgamma. Cell Rep. 2012;1(4):360–373. doi: 10.1016/j.celrep.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 42.Kastirr I, Crosti M, Maglie S, Paroni M, Steckel B, Moro M. Signal Strength and Metabolic Requirements Control Cytokine-Induced Th17 Differentiation of Uncommitted Human T Cells. J Immunol. 2015;195(8):3617–3627. doi: 10.4049/jimmunol.1501016. [DOI] [PubMed] [Google Scholar]
- 43.Planas D, Zhang Y, Monteiro P, Goulet JP, Gosselin A, Grandvaux N. HIV-1 selectively targets gut-homing CCR6+CD4+ T cells via mTOR-dependent mechanisms. JCI Insight. 2017;2(15) doi: 10.1172/jci.insight.93230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Waickman AT, Powell JD. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol Rev. 2012;249(1):43–58. doi: 10.1111/j.1600-065X.2012.01152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Linke M, Fritsch SD, Sukhbaatar N, Hengstschlager M, Weichhart T. mTORC1 and mTORC2 as regulators of cell metabolism in immunity. FEBS Lett. 2017;591(19):3089–3103. doi: 10.1002/1873-3468.12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Myers DR, Wheeler B, Roose JP. mTOR and other effector kinase signals that impact T cell function and activity. Immunol Rev. 2019;291(1):134–153. doi: 10.1111/imr.12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168(6):960–976. doi: 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heredia A, Le N, Gartenhaus RB, Sausville E, Medina-Moreno S, Zapata JC. Targeting of mTOR catalytic site inhibits multiple steps of the HIV-1 lifecycle and suppresses HIV-1 viremia in humanized mice. Proc Natl Acad Sci U S A. 2015;112(30):9412–9417. doi: 10.1073/pnas.1511144112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Besnard E, Hakre S, Kampmann M, Lim HW, Hosmane NN, Martin A. The mTOR Complex Controls HIV Latency. Cell Host Microbe. 2016;20(6):785–797. doi: 10.1016/j.chom.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jin S, Liao Q, Chen J, Zhang L, He Q, Zhu H. TSC1 and DEPDC5 regulate HIV-1 latency through the mTOR signaling pathway. Emerg Microbes Infect. 2018;7(1):138. doi: 10.1038/s41426-018-0139-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Campbell GR, Bruckman RS, Herns SD, Joshi S, Durden DL, Spector SA. Induction of autophagy by PI3K/MTOR and PI3K/MTOR/BRD4 inhibitors suppresses HIV-1 replication. J Biol Chem. 2018;293(16):5808–5820. doi: 10.1074/jbc.RA118.002353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Taylor HE, Calantone N, Lichon D, Hudson H, Clerc I, Campbell EM. mTOR overcomes multiple metabolic restrictions to enable HIV-1 reverse transcription and intracellular transport. Cell Rep. 2020;31(12) doi: 10.1016/j.celrep.2020.107810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bailey CJ, Turner RC. Metformin. N Engl J Med. 1996;334(9):574–579. doi: 10.1056/NEJM199602293340906. [DOI] [PubMed] [Google Scholar]
- 54.Foretz M, Guigas B, Viollet B. Understanding the glucoregulatory mechanisms of metformin in type 2 diabetes mellitus. Nat Rev Endocrinol. 2019;15(10):569–589. doi: 10.1038/s41574-019-0242-2. [DOI] [PubMed] [Google Scholar]
- 55.Kalender A, Selvaraj A, Kim SY, Gulati P, Brule S, Viollet B. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010;11(5):390–401. doi: 10.1016/j.cmet.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol. 2020;21(4):183–203. doi: 10.1038/s41580-019-0199-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia. 2017;60(9):1577–1585. doi: 10.1007/s00125-017-4342-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ouyang J, Isnard S, Lin J, Fombuena B, Marette A, Routy B. Metformin effect on gut microbiota: insights for HIV-related inflammation. AIDS Res Ther. 2020;17(1):10. doi: 10.1186/s12981-020-00267-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Limagne E, Thibaudin M, Euvrard R, Berger H, Chalons P, Vegan F. Sirtuin-1 Activation Controls Tumor Growth by Impeding Th17 Differentiation via STAT3 Deacetylation. Cell Rep. 2017;19(4):746–759. doi: 10.1016/j.celrep.2017.04.004. [DOI] [PubMed] [Google Scholar]
- 60.Nath N, Khan M, Paintlia MK, Singh I, Hoda MN, Giri S. Metformin attenuated the autoimmune disease of the central nervous system in animal models of multiple sclerosis. J Immunol. 2009;182(12):8005–8014. doi: 10.4049/jimmunol.0803563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Routy JP, Isnard S, Mehraj V, Ostrowski M, Chomont N, Ancuta P. Effect of metformin on the size of the HIV reservoir in non-diabetic ART-treated individuals: single-arm non-randomised Lilac pilot study protocol. BMJ Open. 2019;9(4) doi: 10.1136/bmjopen-2018-028444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Isnard S, Lin J, Fombuena B, Ouyang J, Varin TV, Richard C. Repurposing metformin in nondiabetic people with HIV: influence on weight and gut microbiota. Open Forum Infect Dis. 2020;7(9):ofaa338. doi: 10.1093/ofid/ofaa338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Victor VM, Rovira-Llopis S, Banuls C, Diaz-Morales N, Lopez-Domenech S, Escribano-Lopez I. Metformin modulates human leukocyte/endothelial cell interactions and proinflammatory cytokines in polycystic ovary syndrome patients. Atherosclerosis. 2015;242(1):167–173. doi: 10.1016/j.atherosclerosis.2015.07.017. [DOI] [PubMed] [Google Scholar]
- 64.Cattaneo D, Resnati C, Rizzardini G, Gervasoni C. Dolutegravir and metformin: a clinically relevant or just a pharmacokinetic interaction? AIDS. 2018;32(4):532–533. doi: 10.1097/QAD.0000000000001720. [DOI] [PubMed] [Google Scholar]
- 65.Planas D, Fert A, Zhang Y, Goulet JP, Richard J, Finzi A. Pharmacological inhibition of PPARy boosts HIV reactivation and Th17 effector functions, while preventing progeny virion release and de novo infection. Pathog Immun. 2020;5(1):177–239. doi: 10.20411/pai.v5i1.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Procopio FA, Fromentin R, Kulpa DA, Brehm JH, Bebin AG, Strain MC. A novel assay to measure the magnitude of the inducible viral reservoir in HIV-infected individuals. EBioMedicine. 2015;2(8):874–883. doi: 10.1016/j.ebiom.2015.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.El-Far M, Tremblay CL. Gut microbial diversity in HIV infection post combined antiretroviral therapy: a key target for prevention of cardiovascular disease. Curr Opin HIV AIDS. 2018;13(1):38–44. doi: 10.1097/COH.0000000000000426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.He J, Chen Y, Farzan M, Choe H, Ohagen A, Gartner S. CCR3 and CCR5 are co-receptors for HIV-1 infection of microglia. Nature. 1997;385(6617):645–649. doi: 10.1038/385645a0. [DOI] [PubMed] [Google Scholar]
- 69.Fontaine J, Poudrier J, Roger M. Short communication: persistence of high blood levels of the chemokines CCL2, CCL19, and CCL20 during the course of HIV infection. AIDS Res Hum Retroviruses. 2011;27(6):655–657. doi: 10.1089/aid.2010.0261. [DOI] [PubMed] [Google Scholar]
- 70.Ploquin MJ, Madec Y, Casrouge A, Huot N, Passaes C, Lecuroux C. Elevated basal pre-infection CXCL10 in plasma and in the small intestine after infection are associated with more rapid HIV/SIV disease onset. PLoS Pathog. 2016;12(8) doi: 10.1371/journal.ppat.1005774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Arthos J, Cicala C, Nawaz F, Byrareddy SN, Villinger F, Santangelo PJ. The Role of Integrin alpha4beta7 in HIV Pathogenesis and Treatment. Curr HIV/AIDS Rep. 2018;15(2):127–135. doi: 10.1007/s11904-018-0382-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Simon V, Bloch N, Landau NR. Intrinsic host restrictions to HIV-1 and mechanisms of viral escape. Nat Immunol. 2015;16(6):546–553. doi: 10.1038/ni.3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Faivre S, Kroemer G, Raymond E. Current development of mTOR inhibitors as anticancer agents. Nat Rev Drug Discov. 2006;5(8):671–688. doi: 10.1038/nrd2062. [DOI] [PubMed] [Google Scholar]
- 74.Wang C, Kang SG, Lee J, Sun Z, Kim CH. The roles of CCR6 in migration of Th17 cells and regulation of effector T-cell balance in the gut. Mucosal Immunol. 2009;2(2):173–183. doi: 10.1038/mi.2008.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang Y, Planas D, Raymond Marchand L, Massanella M, Chen H, Wacleche VS. Improving HIV outgrowth by optimizing cell-culture conditions and supplementing with all-trans. Retinoic Acid. Front Microbiol. 2020;11:902. doi: 10.3389/fmicb.2020.00902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shikuma CM, Chew GM, Kohorn L, Souza SA, Chow D, SahBandar IN. Short communication: metformin reduces CD4 T cell exhaustion in HIV-infected adults on suppressive antiretroviral therapy. AIDS Res Hum Retroviruses. 2020;36(4):303–305. doi: 10.1089/aid.2019.0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wong JK, Yukl SA. Tissue reservoirs of HIV. Curr Opin HIV AIDS. 2016;11(4):362–370. doi: 10.1097/COH.0000000000000293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ruffin N, Brezar V, Ayinde D, Lefebvre C, Schulze Zur Wiesch J, van Lunzen J. Low SAMHD1 expression following T-cell activation and proliferation renders CD4+ T cells susceptible to HIV-1. AIDS. 2015;29(5):519–530. doi: 10.1097/QAD.0000000000000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Valle-Casuso JC, Angin M, Volant S, Passaes C, Monceaux V, Mikhailova A. Cellular metabolism is a major determinant of HIV-1 reservoir seeding in CD4(+) T cells and offers an opportunity to tackle infection. Cell Metab. 2019;29(3):611–626. doi: 10.1016/j.cmet.2018.11.015. e5. [DOI] [PubMed] [Google Scholar]
- 80.Antonucci JM, Kim SH, St Gelais C, Bonifati S, Li TW, Buzovetsky O. SAMHD1 impairs HIV-1 gene expression and negatively modulates reactivation of viral latency in CD4(+) T cells. J Virol. 2018;92(15) doi: 10.1128/JVI.00292-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.