

Abstract
Alport syndrome (AS) is a severe inherited glomerulopathy caused by mutations in the genes encoding the α-chains of type-IV collagen, the most abundant component of the extracellular glomerular basement membrane (GBM). Currently most AS mouse models are knockout models for one of the collagen-IV genes. In contrast, about half of AS patients have missense mutations, with single aminoacid substitutions of glycine being the most common. The only mouse model for AS with a homozygous knockin missense mutation, Col4a3-p.Gly1332Glu, was partly described before by our group. Here, a detailed in-depth description of the same mouse is presented, along with another compound heterozygous mouse that carries the glycine substitution in trans with a knockout allele. Both mice recapitulate essential features of AS, including shorten lifespan by 30–35%, increased proteinuria, increased serum urea and creatinine, pathognomonic alternate GBM thinning and thickening, and podocyte foot process effacement. Notably, glomeruli and tubuli respond differently to mutant collagen-IV protomers, with reduced expression in tubules but apparently normal in glomeruli. However, equally important is the fact that in the glomeruli the mutant α3-chain as well as the normal α4/α5 chains seem to undergo a cleavage at, or near the point of the mutation, possibly by the metalloproteinase MMP-9, producing a 35 kDa C-terminal fragment. These mouse models represent a good tool for better understanding the spectrum of molecular mechanisms governing collagen-IV nephropathies and could be used for pre-clinical studies aimed at better treatments for AS.
Keywords: Collagen-IV, Glomerular basement membrane, Alport syndrome, Glycine missense mutation, Kidney disease, Mouse model
Abbreviations: AS, alport syndrome; ARAS, autosomal recessive alport syndrome; BSA, bovine serum albumin; EM, electron microscopy; ESRD, end stage renal disease; GBM, glomerular basement membrane; PAS, periodic acid schiff; TGF-b1, transforming growth factor beta1; UPR, unfolded protein response; TBM, tubular basement membrane
Highlights
-
•
Two mouse models were generated that recapitulate essential features of AS patients.
-
•
Glomeruli and tubuli respond differently to mutant collagen IV protomers.
-
•
The mutant colIV protomers in glomeruli probably undergo a cleavage process by MMP9.
-
•
The two AS mouse models represent a good tool for studying collagen-IV nephropathies.
-
•
These models could be used for pre-clinical studies aimed at better treatments.
Introduction
Alport syndrome (AS) is a hereditary glomerulopathy characterized by progressive kidney disease and abnormalities of the ears and eyes. It is caused by mutations in genes encoding the α3, α4 or α5 chains of collagen-IV, a major structural component of the extracellular basement membranes [[1], [2], [3], [4]]. Collagen-IV is the most abundant molecule in the GBM, along with laminin, perlecan and other proteoglycans [5]. The GBM is one of the three crucial components of the glomerular filtration barrier, along with the fenestrated capillary endothelium on the inside and the podocytes with the slit diaphragm on the outside, and it represents a specialized sheet-like extracellular matrix (ECM), which maintains barrier function. There are six alpha chains that participate in collagen IV trimer formation, α1-α6. Only three trimer-combinations are biochemically permissible and found in nature, α1α1α2, α3α4α5 & α5α5α6, with distinct tissue distribution. Collagens function as triple helical structures in their mature form and each α-chain contains repeating aminoacid triplets of Gly-X-Y, where X and Y are frequently prolines and 4-hydroxyprolines which contribute to the thermal stability [6]. Glycine, as the smallest aminoacid, is the only one that can fit at the centre of the helix where the chains meet. In mature human kidneys collagen-IV heterotrimers in the GBM switch from the α1α1α2 configuration to α3α4α5 protomers, produced by the podocytes [7]. While in humans, the α3α4α5 trimer is present in the GBM and some distal tubular BMs, in mice the same trimer exists also in all tubular basement membranes [8].
Three different modes of AS inheritance are known, the X-linked AS, caused by mutations in the COL4A5 gene (~80–85% of patients) [9], and the autosomal recessive AS (ARAS, ~10–15%), resulting from compound heterozygous or homozygous mutations in the COL4A3 or COL4A4 [4,10]. About 1% of AS is described as autosomal dominant (ADAS), caused by COL4A3 or COL4A4 heterozygous mutations, with later age at onset of end-stage renal disease (ESRD) in the 40–60s and rarely with extra-renal manifestations [11]. AS has a prevalence of 1/5000–10,000 for X-linked AS (XLAS) and 1/50000 for autosomal recessive AS (ARAS). Heterozygous patients present with hematuria due to thin basement membrane nephropathy with variable severity and predisposition to later age-at-onset of end-stage renal disease [12,13].
Clinical presentation includes hematuria, progressive nephritis with proteinuria and declining renal function, which inevitably leads to ESRD at early age, if untreated [[14], [15], [16], [17]]. Histologically, the major pathognomonic features on electron microscopy (EM) are alternate thinning and thickening of the GBM, in the presence of splitting and lamellation, accompanied by podocyte foot processes effacement. Additionally, patients develop extra renal manifestations, hearing impairment (~70%) and ocular defects (~40% of patients) [18]. A genotype-phenotype correlation supports that patients with missense glycine (Gly) substitutions demonstrate a later age-at-onset of ESRD compared to patients with non-missense defects [19,20].
Two mouse models for ARAS have been created by knocking out the Col4a3 gene [21,22] and a third was developed by random insertional mutagenesis of the Col4a3-Col4a4 genes [23]. An X-linked AS model has a Gly5>STOP mutation in the Col4a5 gene [24]. The best studied model, created by insertional mutagenesis of Col4a3, exhibited a very severe phenotype with microhematuria and proteinuria and reached ESRD at age ~14-weeks. Transmission EM revealed focal multi-lamellation and thinning of the GBM, which at the age of 12–15 weeks, became thick and heavily rarefied. Heterozygous Col4a3 knock-out mice, also showed shorter lifespan, persistent hematuria, proteinuria, and significantly thin GBM coinciding with tubulointerstitial fibrosis [25]. Despite the fact that Gly-substitution represents the most common type of mutation in human Alport there is only one knockin mouse model of Alport with a Gly-substitution (Gly1332Glu) in Col4a3, created recently by our group, initially seeking to recapitulate the phenotype of heterozygous Gly1334Glu mutation in humans, which causes thin basement membrane nephropathy, with FSGS-like features. Heterozygous patients of this mutation present with a wide phenotypic spectrum, ranging from isolated microscopic hematuria to ESRD, whereas compound heterozygous patients for Col4a3-p.Gly1334Glu and Col4a3-p.Gly871Cys develop severe ARAS [26,27]. The mutant mice showed activation of the unfolded protein response (UPR) due to ER-stress in glomerular isolates, but the effect on the kidney phenotype awaits detailed characterization [28]. Here, we characterized in detail the kidney phenotype of two mouse models for AS, one that carries the Col4a3-p.Gly1332Glu in homozygosity and one that carries the Col4a3-p.Gly1332Glu substitution in compound heterozygosity with a Col4a3 knocked-out allele.The latter were generated by crossing the knockin mouse with the well-studied knockout model [21].
Results
We studied mice homozygous for the missense mutation Col4α3-Gly1332Glu (knockin) and mice with compound heterozygosity that carry the Col4α3-Gly1332Glu mutation in the one allele and is knock-out for the other (compound heterozygous). The latter were generated by crossing the knockin mouse with the well-studied knockout model [21]. In some experiments where necessary, the following mice were used as controls: heterozygous mice for the Col4α3-Gly1332Glu, homozygous mice for the knockout allele and wild type mice.
Brief description:
Col4a3 +/+ mice: Wild type mice
Col4a3 mut/mut mice: Homozygous for the missense mutation Col4α3-Gly1332Glu
Col4a3 mut/+ mice: Heterozygous for the missense mutation Col4α3-Gly1332Glu
Col4a3 mut/− mice: Compound heterozygous for the missense mutation Col4α3-Gly1332Glu and the Col4a3-knock-out allele
Col4a3 −/− mice: Homozygous for the Col4α3-knockout allele
GBM thickening and splitting in Col4a3 mut/mut and Col4a3 mut/− mice
EM studies showed that Col4a3 mut/mut and Col4a3 mut/− mice exhibited the characteristic alternate thinning-thickening and splitting of the GBM. The heterozygous Col4a3 mut/+ mice displayed a thinner GBM while the control Col4a3 +/+ mice showed normal thickness, the characteristic trilaminar appearance with regular podocyte foot processes and normal fenestrations of the endothelium (Fig. 1A, B).
Fig. 1.

Ultrastructural pathology of the compound heterozygous and knockin mice is consistent with AS nephritis. (A) Wild type mice (+/+) display regular GBM, with a thickness of 280–300 nm. The Col4a3 mut/mut mice and the Col4a3 mut/− mice demonstrate thinner GBMs, ranging between 140 and 160 nm (white arrows), with areas of severe irregular thickening (black arrows), consistent with AS nephritis. The heterozygous Col4a3 mut/+ mice display a relatively thinner basement membrane compared to controls. The age of all mice is 20 weeks. (B) Mean values of the thin areas in the GBM of homozygous Col4a3 mut/mut, compound heterozygous Col4a3 mut/− mice, heterozygous Col4a3 mut/+ and Col4a3 +/+ wild type mice. The GBM thickness histograms were derived from 25 to 40 glomeruli from three mice of each group. Results were analyzed using one-way ANOVA with Tukey post-testing (***p=0.0009, ****p≤0.0001).
Col4a3 mutant mice show albuminuria, which progresses over time
We monitored the urine albumin concentration per month of Col4a3 mut/mut mice (n=8), Col4a3 mut/− compound heterozygous mice (n=8) and wild-type mice (n=4), for a period of 13 months. Albuminuria of >0.1 g/L was noted after 3-months in the compound heterozygous mice and increased to >1 g/L after 12-months (Fig. 2A-B). The finding of albuminuria in Col4α3 mut/mut mice was inconsistent during early stages of the disease, for reasons we do not fully understand. Perhaps it is attributed to the general heterogeneity observed due to the mice being incipient congenic instead of a perfect inbred strain.
Fig. 2.
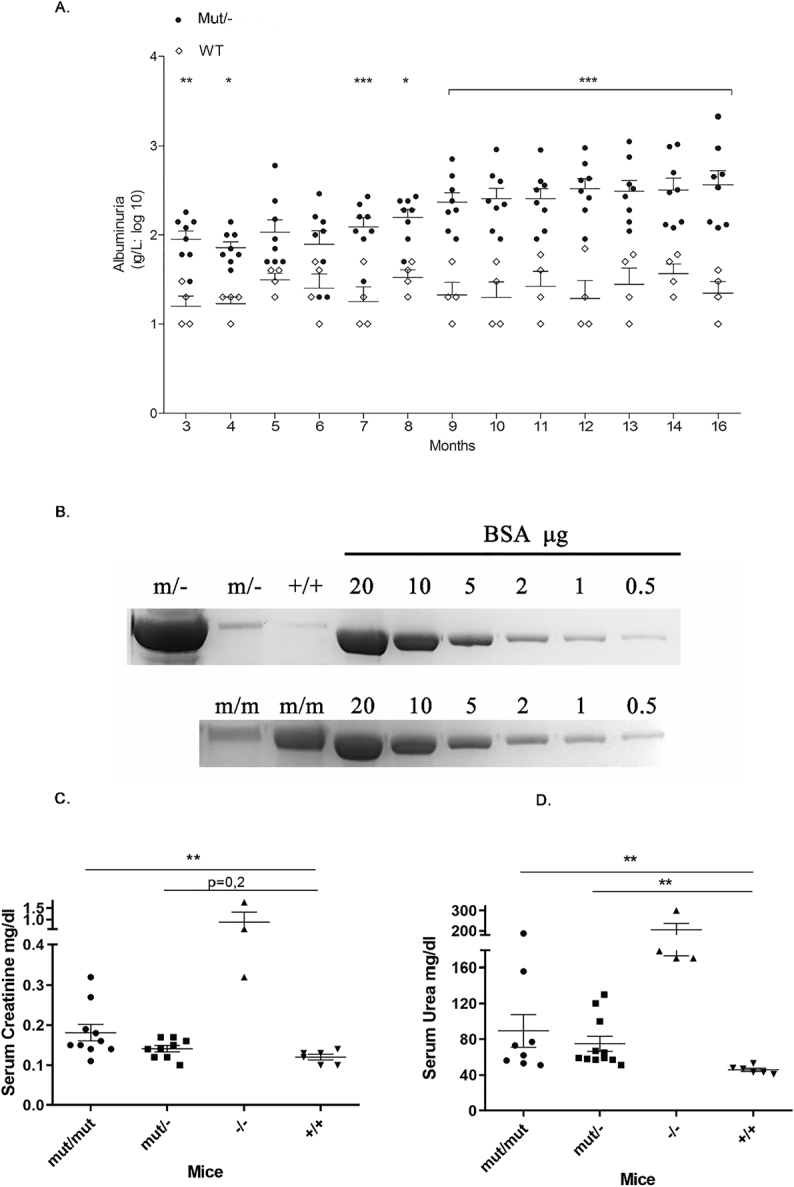
Increased albuminuria in Col4a3 mut/− mice (A&B). Increased serum urea and creatinine levels in some Col4a3 mut/− and Col4a3 mut/mut mice (C&D).
(A) The time course of urine albumin concentration(g/L) in eight compound heterozygous mice (black circles) and four wild type mice (white rhombus) for a period of 13 months. Albuminuria of >0.1 g/L was noted after 3-mo of life and increased to >1 g/L after 12-mo for most Col4a3 mut/− mice. Results were analyzed using two-way ANOVA on log10-transformed values (P<0.001). * Significant difference of P<0.05, ** P<0.01, *** P<0.001.
(B) An illustrative example of urine fractionation by SDS-PAGE. Twenty μL of urine from two mut/− mice (upper panel), two mut/mut (lower panel) and one wild type mouse (+/+) of the same age, were fractionated together with standard concentrations of Bovine serum albumin on a 7.5% SDS-PAGE and stained with Coomassie blue. Note the high albumin levels in one of the two mutants. (C&D) Mean concentration (mg/dL) of creatinine(C) and urea (D) in serum from Col4a3 mut/− (n=11), Col4a3 mut/mut (n=8), Col4a3 −/− knockout (n=4) and wild type (n=6) mice (+/+). Serum urea levels of five mut/mut and seven mut/− mice were higher than those of normal mice. It is noteworthy that for some mice, the urea levels have reached very high levels, similar to those observed in the homozygous Col4a3 knockout mice (−/−), which die around the third month of life. Creatinine levels, on the other hand, have generally shown a small increase in the total mut/mut and mut/− mice. Note that the mut/mut mice which had very high levels of urea also showed high levels of creatinine that reached the levels of Col4a3 knockout mice. For the analysis, serum was collected from 15 to 22 months old knockin mutant,compound heterozygous mice and wild type mice, and from 2,5 months old knockout mice. Results were analyzed using one-way ANOVA with Tukey multiple comparison adjustment (* p≤0.05, ** p≤0.01, *** p≤0.001, **** p≤0.0001). Individual comparison p-values for urea are as follows: +/+ vs mut/mut: 0,0054 and +/+ vs mut/−: 0,0023, and for creatinine: +/+ vs mut/mut: 0,0088. mut/−, compound heterozygous; mut/mut, homozygous knockin; −/−, homozygous knockout; +/+, WT mice.
We then examined two separate cohorts of mice at an advanced age, >15-months old. In Col4α3 mut/mut mice (n=32), the high levels of albuminuria (>1 g/L) were observed in 62.5%, while 65% of Col4α3 mut/− mice (n=20) had high albuminuria (Fig. 2B). Additionally, microscopic analysis showed that a 70% or 75% of mutant mice presented intermittent microscopic hematuria after their 3rd month of life, indicating that the Gly-substitution mutation, with or without the combination of total allele loss, leads to erythrocytes loss in the urine (not shown).
Serum creatinine and urea reached high values in a subset of Col4α3 mutant mice
The serum urea and creatinine of 62,5% Col4a3 mut/mut and 63% Col4a3 mut/− mice were higher compared to normal controls, indicating impaired renal function. It is noteworthy that urea in 5 of the 19 total mutant mice tested, and creatinine in 2 of the 10 total Col4a3 mut/mut mice reached high values, which approached those from Col4a3 double-knockout mice that die around the third month of life (Fig. 2C-D).
Renal fibrosis in Col4a3 mutant mice
Both AS models exhibited degenerative changes in their kidneys (Fig. 3A). Specifically, renal sections from mutant mice exhibited moderate to severe diffuse periglomerular and interstitial fibrosis that in most cases were accompanied by mild to moderate infiltration by lymphocytes. Segmental or global glomerulosclerosis is also present.
Fig. 3.

Moderate to severe fibrosis in kidney samples of Col4a3 mutants. (A) Commonly used stains on kidney sections of the following mouse models to assess renal fibrosis. Collagen deposition in both periglomerular and interstitial locations are indicated with arrowheads and arrows respectively. Each staining was performed on 6 μm paraffin kidney sections from three mice of each group. The magnification of all images in panel A was 20×.
Wild-type mice (20 months old),
H&E, exhibiting minimal alterations, Masson's trichrome exhibiting Bowman's capsules and renal tubuli lined by thin regular line of collagen. PAS, outlining a fine basement membrane in both renal glomeruli and renal tubules colored bright purple. Sirious red, with collagen fibers indicated as fine red lines.
Col4a3 mut/− mice (20-months-old).
H&E, exhibiting moderate diffuse periglomerular and interstitial fibrosis. Masson's trichrome reveals collagen deposition in both periglomerular and interstitial locations. PAS staining reveals mild deposition of PAS-positive material. Sirius red staining confirms the findings of H&E and Masson's trichrome.
Col4a3 mut/mut mice (20 months old).
H&E, Masson's trichrome exhibiting moderate multifocal interstitial fibrosis. PAS exhibiting moderate diffuse deposition of PAS-positive material both in the Bowman's capsule as well as in the glomerular tufts. Sirius red staining reveals deposition of collagen fibers both in interstitial and periglomerular locations confirming the findings of H&E and Masson's trichrome.
Col4a3−/− knockout mice (6 weeks old).
H&E, staining exhibiting severe glomerular and interstitial fibrosis. Masson's trichrome staining reveals collagen deposition in both periglomerular and interstitial locations. PAS exhibiting severe deposition of PAS-positive material. Sirius red staining, note the opalescent glomerulus at the centre of the field exhibiting severe fibrosis of the Bowman's capsule and glomerular tufts.
(B)Western blot analysis of profibrotic markers TGF-β1 and Acta2 displaying increased levels in glomeruli isolates from 20-month-old homozygous Col4a3 mut/mut and Col4a3 mut/− mice. This result in mutant mice was reproducible in multiple repetitions, compared to the wild type counterparts. β-Tubulin expression in the same samples was used as an equal loading control.
(C-D) Quantification of representative blots as in B is shown in graphic form. The expression levels of TGF-β1 and Acta2 in either glomeruli (C) or whole kidney (D) isolates were normalized to β-tubulin levels. Data are means±SEM (n≥3). Results were analyzed using one-way ANOVA with Tukey post-testing (*p≤0.0164, **p≤0.004,***p≤0.0009, ****p≤0.0001). mut/−, compound heterozygous; mut/mut, homozygous knockin; −/−, homozygous knockout; +/+, wild type mice; Acta2, alpha-smooth muscle actin.
Among 12 mutant mice that were tested for fibrosis, a 67% of them, had <25% degree of tubular injury, while a 33% of mutant mice had 75% degree of injury. The degree of interstitial fibrosis was either <25% or 26–45%. Mice with the more severe phenotype had a 20–23% degree of segmentally or globally sclerosed glomeruli. None of the lesions described were observed in wild-type mice of same age. Notably, the mice exhibiting the most severe histological lesions were those with the most severe biochemical alterations, as previously shown [22].
Moreover, transforming growth factor beta-1 (TGF-β1) and α-smooth muscle actin (α-SMA or Acta2) were upregulated in glomerular isolates from Col4a3 mut/mut and Col4a3 mut/− mice compared with wild-type littermates (Fig. 3B-D). Similarly, upregulation of these markers was evident in whole kidney isolates from old-aged mutant mice. TGF-β1 as the major pro-fibrotic cytokine mediates many of the central processes involved in fibrosis. In CKD, TGF-β over-expression induces renal fibrosis [29]. Myofibroblasts that represent key players in the cellular pathology underlying renal fibrosis, are often identified by the expression of Acta2 [30,31].
Lifespan of Col4α3 mutant mice is significantly shorter compared to controls
From all mice that were monitored for their lifespan, Col4a3 mut/mut mice (n=31) and Col4a3 mut/− mice (n=33), lived on average for 15.1 and 16.07 months respectively (Fig. 4). This lifespan was 30–35% shorter than in wild-type controls (n=23) that lived for 22.7 months. The difference was significant using log-rank (Mantel–Cox) test (P<0.05). The average lifespan of the heterozygous mice for the glycine substitution, Col4a3 mut/+ (n=25), was also shorter than normal, at 17.9 months.
Fig. 4.
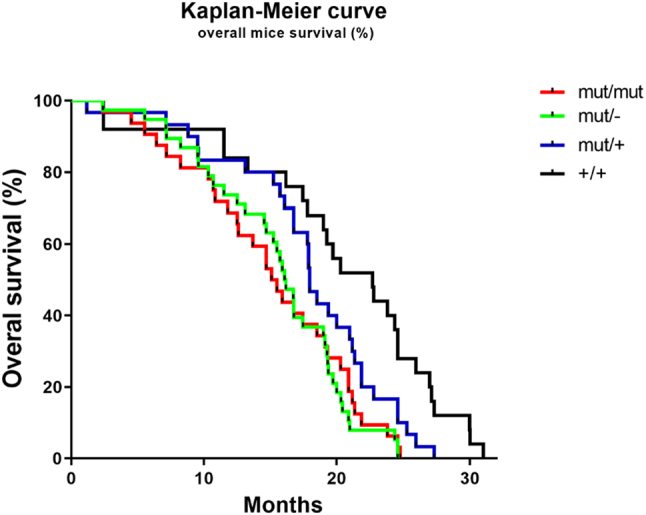
Significantly shortened lifespan of Col4α3 mutant mice compared to controls. Survival diagram showing reduced life span of Col4a3 mut/mut and Col4a3 mut/− mice in comparison with heterozygous mice and wild-type controls. The Col4a3 mut/mut mice lived 15.1 mo (n=31) while the Col4a3 mut/− lived 16.07 mo (n=33). This was significantly less than in 25 Col4a3 mut/+ mice or 23 wild-type mice Col4a3 +/+ in which mean lifespan was 17.9 and 22.7 mo, respectively. Kaplan-Meier survival analysis was used to assess mouse lifespan and data were compared using log-rank (Mantel–Cox) testing; with a P value= 0.016 the survival curves were significantly different.
Reduced expression of Col4α3-α5 chains in tubules of Col4α3 mut/mut and mut/- kidneys
The distribution of collagen-IV alpha-chains was examined by immunofluorescence staining with antibodies that recognize the NC1 domain of each collagen-IV chain. In wild-type and in heterozygous Col4a3 mut/+ kidneys, there is normal positive staining for the α3/α4/α5 chains, both in the GBM and the tubular basement membrane (TBM). However, in kidneys from mutant Col4α3 mut/mut or Col4α3 mut/−, the expression of alpha-chains is reduced in tubules but normal in glomeruli (Fig. 5A, B and Supp. Fig. 3). Importantly, the expression of the α1 and α2 chains in mutant kidneys was distributed normally throughout the GBM and TBM (Fig. 5A, C).
Fig. 5.

Expression of the three collagen chains a3, a4 and a5, is reduced in the tubules of Col4a3 mut/mut and Col4a3 mut/− mice. Immunofluorescence on 6 μm kidney sections of 20-week-old Col4a3 mut/−, Col4a3+/+ (wild type) and 3-month old Col4a3−/− mice (A) and of 20-week- old Col4a3 mut/mut mice, Col4a3mut/+ and Col4a3+/+ mice (B) for each collagen-IV alpha chain (a3, a4, a5) of the a3a4a5 protomer and for the chains a1 and a2 (C), corresponding to the α1α1α2 protomer. In kidneys from both mutant mice (mut/mut and mut/−), the expression of the a3, a4 and a5 chains is reduced in tubules but appears normal in glomeruli. Overall expression of type IV collagen was evaluated by staining with mAb JK2 which recognizes a common epitope in the NC1 domain of α1, α3 and α5(IV) collagen (C). In normal controls and heterozygous mut/+ mice, the a3, a4, and a5 chains are localized to the GBM and in the TBM. No signal was detected on kidneys from 3mo old Col4a3−/− (negative controls). The expression of the collagen-IV chains a1 (A and C) and a2 (C) in the tubules and glomeruli of both mutant mice is normal. Therefore, the mutation in Col4a3 affects only the expression pattern of itself and of its partner chains in the a3a4a5 heterotrimer. Scale bar was 150 μm for a3 (low magnification), 50 μm for a3 (high magnification), a4 and a5 (IV) staining in A and 25 μm for a1 (IV). In B the original magnification was 200×. Images of 400× magnification are also included. Col4a3 mut/mut, knockin; Col4a3 mut/−, compound heterozygous; Col4a3 mut/+, heterozygous knockin; Col4a3−/−, knockout; Col4a3 +/+, wild-type mice.
Podocytes secrete effectively mutant collagen-IV
To examine whether the mutant Col4α3 protein is secreted normally from the podocytes to the GBM, double immunofluorescence staining was performed on Col4a3 mut/mut and mut/− kidney sections, for Col4α3 and laminin, both localized in the GBM and the TBM. Col4a3 and laminin appear to co-localize in both mutant mice, in the GBM, with no perceptible difference compared to wild-type mice (Fig. 6). Double immunofluorescence was also performed for the Col4α3 protein and synaptopodin, a protein marker for podocyte cytoplasm. The mutated Col4α3 protein is not co-localized with synaptopodin inside the podocytes of mutant kidneys, demonstrating a staining pattern similar to that of wild-type mice (Fig. 7). Results suggest that the presence of the glycine substitution in Col4α3 protein does not prevent the secretion of at least a fraction of the trimer from the podocytes to the GBM.
Fig. 6.

Colocalization of Col4a3 and laminin in the GBM of Col4a3 mutants shown by double immunofluorescence staining. Double immunofluorescence for Col4a3 (red) and of the GBM marker laminin (green), on 6 μm paraffin kidney sections of 20 mo Col4a3 mut/− mice, 20 mo Col4a3 mut/mut mice, 20 mo wild type (+/+) mice and 2,5 mo Col4a3 (−/−) mice. Co-localization of Col4a3 and laminin corresponds to the yellow areas in merged images. The expression of Col4a3 in either mut/mut or mut/− mice is co-localized with that of laminin in the GBM as it is normally seen in wild type mice (+/+). This result indicates that Col4a3 protein is secreted effectively into the GBM. Scale bar, 50 μm.
Fig. 7.

No colocalization of Col4a3 and synaptopodin in glomeruli of Col4a3 mutants shown by double immunofluorescence staining. Double immunofluorescence for Col4a3 (green) and the podocyte marker synaptopodin (red) on 6 μm kidney paraffin sections of 20 mo Col4a3 mut/−, Col4a3 mut/mut mice, wild type (+/+) mice and 2,5 mo Col4a3 −/− mice. Images at the right represent overlays of Col4a3 with synaptopodin staining. As it is normally observed in wild type mice, the expression of Col4a3 in either mut/mut or mut/− mice is not co-localized with the expression of synaptopodin, a podocyte marker, suggesting effective secretion of normal and mutant collagen trimers. Scale bar, 50 μm.
Decreased alpha-chain levels in mutant whole kidneys. In glomeruli, the collagen-IV chains are normally expressed but are probably cleaved
Col4α3 expression was assessed by Western Blot (WB) in lysates from glomerular and whole kidney isolates. In glomerular isolates of both mutant mice, the expression of Col4α3 chain is reduced at 160 kDa (MW of Collagen-IV chains); accordingly, a de novo 35 kDa protein fragment was detected by the same antibody against Col4α3, only in mutant mice. The same was observed for the non-mutated chains Col4α4 and Col4α5 (Fig. 8A, B). Importantly, the small de novo protein fragment (35 kDa) of each α-chain was detected mainly in glomeruli from mutant mice but not in whole kidney isolates, in which the presence of the three chains at 160–200 kDa, was decreased (Fig. 8C, D). Moreover, in mutant glomeruli, the truncated amino-terminal collagen-IV fragments, which consist of the 7S and part of the collagenous domain (CD), were not detectable with any of the two antibodies used that recognize epitopes in the N-terminus or middle region (Supp. Fig. 1). The most likely explanation is that the amino-terminal truncated fragments are physicochemically unstable and rapidly degraded extracellularly. In support of this, the expression of the intact full-length alpha-chains was reduced at 160 kDa in glomeruli and in whole kidneys, compared to wild-type mice (Fig. 8A-D). In order to validate our method and confirm that the protein used was isolated exclusively from glomeruli, we performed WBs and tested for the expression of synaptopodin and podocin as podocyte markers and MUC1 as a tubular marker. The results showed none or negligible presence of MUC1 protein (Supp. Fig. 2).
Fig. 8.

Downregulation of α3, α4 and α5 chains in whole kidney lysates and occurrence of a small Col4 fragment in glomerular isolates of Col4a3 mutants.
(A) Representative WB to demonstrate protein expression level change in glomeruli from 20-month-old Col4a3 wild type (+/+), Col4a3 mut/mut and Col4a3 mut/− mice. The glomeruli were harvested using Dynabeads perfusion and then equal amounts of isolated protein were resolved by SDS-PAGE gel followed by WB. Specific antibodies that either recognize the collagenous domain (CD) of Col4a3 or the NC1 domain of the ColIV a3, a4 and a5 chains were used to identify their expression. Although the a3, a4 and a5 chains are barely detected at 160 kDa, a 35 kDa protein fragment is detected by the antibodies recognizing the NC1 domain of Col4a3, a4 and a5 in mutants respectively. (C) Representative WB to demonstrate protein expression level change in homogenates of whole kidney lysates from 15-month-old Col4a3 mut/−, mut/mut and wild type mice. Specific antibodies were used to detect the levels of the three Col4 chains: a3, a4 and a5. The expression of Col4a3, a4 and a5 is drastically reduced in whole kidneys of Col4a3 mut/mut and mut/− mice. Specific antibodies that either recognize the NH2-terminous (NH2-end) domain or the NC1 domain of ColIV chains were used to identify the expression of the a3, a4 and a5 chains. (B, D) Quantification of representative blots as in A and C is shown in graphic form. The expression levels of the alpha chains in either glomeruli (B) or whole kidney (D) isolates were normalized to β-tubulin levels. β-Tubulin expression in the same samples was used as an equal loading control. Data are means±SEM (n≥3). Results were analyzed using one-way ANOVA with Tukey post-testing (**p≤0.0043***p≤0.0003, ****p≤0.0001).
Possible association of the MMP-9 protein activation with the cleavage of the mutant collagen IV protomer
Glomerular protein was assayed at several time points over a knockin mouse's life in order to examine whether the 35kda fragment of Col4a3 starts out as cleaved or is cleaved at a specific point in disease progression. The 35kda fragment appears at early ages albeit at low levels and reaches high levels at a later stage, probably around 15-months of life (Fig. 9A). Furthermore, glomerular protein from the same timepoints as above was assayed for the expression or activation of several MMPs (MMP-2, MMP-3, MMP-9 and MMP-12) in order to examine whether the time specific cleavage of the mutant collagen IV protomer has any correlation with a metalloproteinase. The expression of MMP-2, MMP-12 (data not shown) and MMP-3 (Fig. 9B) was not altered in mutant mice, however the activated form of MMP-9 (63 kDa) is detected near the 6th month of a knockin mouse's life and remains stable at advanced age (Fig. 9B). The fact that the activated form of MMP-9 is detected during the month that the levels of the 35 kDa Col4a3 fragment begin to elevate, suggests that MMP-9 could be the enzyme that mediates the cleavage of the mutant Col-IV protomer. Note that the activated form of MMP-9 was also slightly detected in some WT mice after the 17 month of life, probably due to its role for the generation of tumstatin, a 28kda Col4a3 fragment that is also detected under normal conditions.
Fig. 9.

Representative WB to demonstrate protein expression level change in glomeruli from 4, 6 and 15-month-old Col4a3 mut/mut and 6 and 15-month-old Col4a3 wild type (+/+) mice. (A) Specific antibody that recognizes the NC1 domain of Col4a3 chain was used to detect the 35 kDa fragment of a3 (Col-IV) chain. The expression levels of the α3 chains in glomeruli isolates were normalized to β-tubulin levels. The level of the 35 kDa fragment of Col4a3 progressively increases with aging of Col4a3 mutants. (B) A specific antibody that recognizes the 82 kda and the 63 kDa active forms of MMP-9 and one that recognizes the MMP3 were used. The 63 kDa active form of MMP-9 was detected from 6th month of knockin mice and maintained until the 15th month. The expression levels of the 63 kDa fragment of MMP-9 in glomeruli isolates from 4, 6, 10, 12 and 15-month-old Col4a3 mut/mut and 4, 10 and 15-month-old wild type (+/+) mice were normalized to actin levels. Data are means±SEM (n≥3). Results were analyzed using one-way ANOVA with Tukey post-testing (**p≤0.007, ***p≤0.0008 ****p≤0.0001). NC1, non-collagenous domain; CD, collagenous domain.
Discussion
It is well-known that glycine, the smallest aminoacid, is indispensable at every third position of collagenous sequences and its substitution leads to triple-helix misfolding and destabilization. Around 51% of AS patients inherit single aminoacid substitutions in Col4a3 or Col4a4 or Col4a5, 73% of which are glycines [16,32].
Therefore, we created the first knockin mouse, homozygous for mutation Col4a3-p.Gly1332Glu, and a compound heterozygous mutant carrying this mutation in trans with a knocked-out allele. Both mice developed AS, with a later age-at-onset of ESRD, compared to homozygous knockout mice. Later age-at-onset of ESRD is also the case for humans with missense mutations compared to those with non-missense mutations [17,19,33,34].
Col4a3 mut/mut mice provide a model to study the biochemical and pathophysiological consequences in AS, the same way we study patients with Gly-substitutions with either ARAS or XLAS. The Gly1334Glu mutation introduces a break in a stretch of 33 Gly-X-Y repeats, which form the penultimate collagenous domain of α345(IV) collagen; similar to nearby pathogenic COL4A5 mutations Gly1333Ser, Gly1342Arg, Gly1354Cys [[35], [36], [37]]. Interestingly, a study that looked at triple helical glycine substitutions suggested that mutations near the carboxy terminal region are more severe that those near the amino terminal region of COL4A5 [19]. Additionally, there are reports showing that Gly-substitutions nearer the carboxy terminus in fibrillar collagens of type I [38,39], type II [40,41] or type III [42] are more often associated with severe forms of the relevant diseases.
Additionally, the compound heterozygous mouse could be an exemplar study model for AS patients who inherit analogous mutations in compound heterozygosity in collagen-IV genes [27,[43], [44], [45]].
Clinical features and pathology
Mice of both models demonstrate a similar 30–35% reduction in their lifespan compared to normal mice, most probably due to impaired kidney function. A similar age at onset of ESRD was also reported before, in human patients who inherit either homozygous or compound heterozygous mutations in COL4A3 and COL4A4 mutations [43]. Decreased glomerular filtration resulted in uremia and elevated creatinine, which despite its variability, in some mice reached the high levels observed in Col4a3−/− mice, which die early (Fig. 2). Notably, variability on the levels of serum urea and creatinine between mutants has been also observed before in other AS mouse models [24,46,47]. Importantly, mice with the most severe urea and creatinine levels displayed the most severe histological abnormalities, which agrees with findings in another Col4a3−/− mouse [22].
Proteinuria and haematuria are hallmark features of collagen-IV nephropathies. Both mice showed impaired glomerular permeability with high levels of albuminuria, although of variable degree, indicating that the Gly1332Glu substitution, with or without the combination of the null allele, leads to defective blood filtration. Hematuria was not a consistent finding and was demonstrated intermittently. Notably, hematuria was also not evident in one [21] of the two Col4a3−/− mice previously described [21,22]. Overall, the variability in symptoms described in mutant mice here has been also reported in AS patients [11,[48], [49], [50]], highlighting the heterogeneity of the severity of this disease, the drivers of which are not absolutely clear. A glycine substitution in the collagenous domain of Col4a3 in mice recapitulates late onset Alport syndrome. Glomerular and interstitial fibrosis is a common later-onset event in these mice, perhaps of dual etiology, attributed to the chronic glomerulonephritis and to the reduced expression of collagen α3α4α5 protomers in the tubules. Evidence of focal and segmental glomerulosclerosis was also present while there were multiple histological abnormalities accompanied by increased profibrotic markers, TGF-β1 and Acta2 (Fig. 3). Overall, the observed variability in the rate of appearance of renal failure and disease severity, may be attributed to the mice being incipient congenic. Ultrastructurally, the presence of typical GBM abnormalities in both models, including podocyte foot process effacement, is evidence that they recapitulate credibly the AS GBM phenotype (Fig. 1) [51].
Podocytes vs tubular epithelial cells
All three alpha-chains are drastically decreased in the tubules of mutant mice, as confirmed by immunofluorescence studies on kidney sections and WB analysis on whole kidneys specimens. Importantly, immunofluorescence studies showed that the expression of alpha-chains in the glomeruli was not detectably altered compared to wild-type animals. GBM positivity for collagen-IV chains was reported before in a mouse model for AS with a mutation in the conserved GT-splice donor of Col4a4, intron 30 [52]. Positive GBM staining for collagen-IV chains was not totally unexpected, as it has been reported in many patients with Gly-substitutions [53,54] although some Gly-substitutions result in negative α3/4/5 staining [17].
Collagen quality control explains also the podocyte ER stress reported previously. Based on previous work, some or most mutant α345(IV) is likely degraded, possibly by ER-phagy [55], but a variable proportion of mutant trimers may escape degradation and be secreted in GBM, owing to activation of the UPR. The continuous presence of α345(IV) may prevent activation of the DDR1 receptor, thus resulting in delayed and milder renal fibrosis and longer lifespan [56].
WB of glomerular proteins showed something remarkable: there is a possibility that the alpha-chains making the α345(IV) trimer, undergo a specific cleavage, releasing COOH-terminal protein fragments of 35 kDa. It is hypothesised that this targeted cleavage is exerted in the region of Gly1332Glu substitution, perhaps due to local imperfect folding of the protomers. Notably, the 35 kDa fragment was observed only in mutant glomerular lysates, not from whole kidney lysates in which the isolated protein corresponds predominantly to tubules. This suggests that the two kidney compartments, respond differently with mutant trimers. These different molecular mechanisms are under further investigation. The observed distribution of Col4α3 protein in the GBM (Fig. 6, Fig. 7), is probably due to the presence of the 35 kDa fragment (Fig. 8A, 9A), since the anti-Col4α3 antibody used in immunofluorescence studies recognizes the NC1 domain of α3-chain, which is part of the 35 kDa fragment and is located shortly after the Gly1332Glu substitution (Supp. Fig. 1). If α345(IV) is proteolytically cleaved at or around position 1332, then the carboxyl-terminal 35 kDa fragment may remain attached to the GBM via a disulfide bonding between Cys1354 or 1367 or others around there in Col4a4, and with other α345(IV) trimers [α4(IV)-chain has numerous unpaired cysteines in the collagenous domain which can only form inter-molecular disulfide bonds].
The cleavage of the Col4a3-35 kDa fragment is performed after the assembly of α345(IV) protomers, and most probably after secretion in the GBM, as a similar 35 kDa fragment is detected by different antibodies that recognize the NC1-domain of both Col4α4 and Col4α5 in mutant glomerular isolates. Interestingly, the progressive increase of the levels of α3(IV) 35 kDa fragment during aging (Fig. 9A), could be correlated with the late onset of the clinical symptoms in mutant mice. A possible scenario could be that the accumulation of cleaved collagen-IV fragments leads to weakening of the GBM meshwork, thus contributing further to the worsening of the phenotype.
Moreover, as the Col4α3 antibody against the anti-collagenous domain (CD) in WB (Fig. 8A and Supp. Fig. 1), did not recognize any protein fragment at 125 kDa (estimated size of the large truncated NH2-terminal fragment) but only reduced levels of the full-length Col4α3 protein, it is hypothesised that the large truncated fragment of Col4α3 is rapidly degraded in the GBM in a targeted enzymatic digestion most probably by a metalloproteinase. The specific antibody was functional only in WB assays. It is possible that the Gly1332Glu substitution delays the trimer-folding towards the amino-terminus, while also creating a triple-helix imperfection and exposing a cleavage site to a metalloproteinase.
It is expected that the 35 kDa fragment is resistant to degradation due to high density of cysteine residues in the NC1 domains of α3, α4 and α5(IV) chains [57], which provides a tighter configuration, as it remains attached to the final mature protomer, in contrast to fibrillar collagens [58,59].
Importantly a recent study showed that AS-related Gly missense mutations in COL4A5, one of which resembled the one we study here with a Gly to Glu substitution at position 400, significantly reduced the structural stability of the collagen, decreased the melting temperature and accelerated the rate of degradation of the collagen by trypsin and chymotrypsin, based on experimental studies and computational dynamics simulations [60].
Studies of COL1A1 and COL1A2 mutations causing osteogenesis imperfecta show that Gly substitutions (for example COL1A1-p.Gly904Cys), introduce breaks/interruptions in the collagenous triple-helical structure, with the effects of (a) dramatically lowering the melting temperature [61,62] and (b) increasing susceptibility to proteases in in vitro assays [63]. Similarly, a COL3A1-p.Gly790Ser substitution in a patient with Ehlers-Danlos syndrome makes the procollagen molecule sensitive to proteases [[64], [65], [66]]. A similar cleavage may explain the 35 kDa fragment in mutant mice here.
The main group of enzymes responsible for collagen and other protein turnover in extracellular matrix (ECM) are matrix metalloproteinases (MMPs) [67,68]. The spatial expression of MMPs in the kidney is complex and has not been fully characterized yet [[69], [70], [71]]. The expression of MMP-9 appears to be mainly detected to the glomerulus [72,73]. MMPs can be controlled at the protein level by their endogenous activators or inhibitors and by factors that trigger their secretion.
All MMPs are initially synthesized as inactive proenzymes and are processed into active isoforms by proteolytic removal of a pro-peptide [74]. MMP-9 is found as active isoforms at both 82 and 63 kDa. The activation of pro-MMP-9, occurs by exclusion of the NH2-terminal pro-peptide, yielding the 82 kDa that is inhibited by tissue inhibitor of metalloproteinase-1 (TIMP-1), its natural inhibitor. On the other hand, the 63-kDa active isoform which is yielded by a COOH-terminal cleavage, is not inhibited by TIMP-1 [75,76]. MMP-9 has different activation mechanisms and substrate specificity. Here, we showed that the 63-kDa active isoform of MMP-9 is being detected in the glomeruli of knockin mice around the month where the 35 kDa Col4a3 fragment starts to accumulate in glomeruli, suggesting that the cleavage is mediated possibly by MMP-9. One explanation for the fact that the MMP-9 levels remain stable while α3-NCI increases as the mutants grow, is a possible progressive accumulation of the α3-NCI fragments in the GBM due to the long lasting half-life of GBM Collagen IV, estimated to be greater than 100 days [77]. Importantly, among the numerous MMPs that are expressed in kidneys [69], MMP-9 was previously proved to have a closer affinity to Col4a3, being the most effective enzyme in the generation of tumstatin domain from type IV collagen [78]. Tumstatin is a 28-kDa fragment that corresponds to the bioactive NC1 domain of Col4α3, and is being cleaved by MMP-9 acting either as an angiogenesis inhibitor of endothelial cell proliferation and blood vessel formation or as a proapoptotic factor [78,79]. Interestingly, the expression of MMP-2 or MMP-3 and of the MMP-12, a metalloproteinase that was shown previously to be induced in glomeruli from Col4a3 knockout mice [80] was not altered in knockin mice.
Conclusion
We described two mouse models of ARAS that can serve as tools for better understanding of the mechanisms governing collagen-IV nephropathies and for the development of better future therapies. The Col4a3 mut/mut mouse represents the first that carries a substitution mutation and recapitulates clinical and pathologic features of the disease. In accordance with human studies, these mice have a later-onset phenotype compared to mice with a double knockout of the α3-chain. This milder phenotype supports that even though the mutant trimers might disrupt the GBM meshwork, they are better tolerated than null secretion, partly rescuing the phenotype. This may justify using synthetic chaperones for facilitating better trimer folding and secretion.
Experimental procedures
Animals
We used the homozygous knockin (Col4a3 mut/mut) mouse that carries the mutation Col4a3-Gly1332Glu, previously generated in our lab initially on a mixed background [28]. The mouse line was backcrossed for five generations in order to maintain the mutation in homozygosity on incipient congenic 129×1/SvJ genetic background. The compound heterozygous (Col4a3 mut/−) mouse carries the mutant allele Col4a3-Gly1332Glu and the Col4a3 knockout (−) allele. This mouse derived through breeding of the heterozygous knockout mice [21] with the homozygous Col4a3 knockin mice.
Cross-breeding and PCR-based genotyping of homozygous Col4a3−/−, heterozygous Col4a3+/− and wild-type control mice on 129×1/SvJ genetic background were performed as described previously [81,82]. Genotyping for detecting the knockin substitution mutation Col4a3-Gly1332Glu, was as described in [24]. In comparisons between different mouse models and controls, an effort was made to use age-matched animals. However, due to the fact that knockout mice die at around the 3rd month of their life, we had to collect samples of earlier ages. All animal protocols were approved by the Veterinary Services of the Ministry of Agriculture, Rural Development and Environment (approval code CY/EXP/PR.L5).
Electron microscopy
For EM studies, kidney sections were processed and examined under a transmission electron microscope (JEM2100HR; JEOL, Inc., Tokyo, Japan) equipped with an ES500W Erlangshen CCD camera (Gatan GmbH,Munchen, Germany). GBM thickness was measured in open capillary loops using Digital-Micrograph software (Gatan). The GBM thickness histograms were derived from 25 to 40 glomeruli from three mice of each group. The number of capillary loops that were used for getting the mean values of the thin areas in the GBM from each group was: 44 from homozygous Col4a3 mut/mut, 65 from compound heterozygous Col4a3 mut/− mice, 38 from heterozygous Col4a3 mut/+ and 22 from Col4a3 +/+ wild type mice.
Biochemical studies and proteinuria/hematuria
Twenty-four-hour urine was collected from knockin (Col4a3 mut/mut), compound heterozygous (Col4a3 mut/−) and wild-type (WT) mice (Col4a3+/+) every 4-weeks over 14 months. Urine collected in metabolic cages for 24-h, was evaluated for proteinuria and hematuria. Protein was tested qualitatively, by microelectrophoresis of 20 μL urine on SDS-polyacrylamide gel and Coomassie Blue staining followed by densitometry, using bovine serum albumin as molecular-weight standard. Hematuria was tested by using a dipstick test (Roche) and by calculation of red blood cell number/ml urine, under optic microscopy. Serum parameters (urea and creatinine) were analyzed in blood collected from the heart of sacrificed animals.
Trichrome/PAS/H-E staining
Kidneys were collected from homozygous Col4a3 mut/mut (n=6), compound heterozygous Col4a3 mut/− (n=6), homozygous Col4a3−/− (n=3), and wild-type (n=4) control mice. We used mice of both sexes. Mouse kidneys were fixed in 4% paraformaldehyde following standard protocols. Four micrometer-thick sections were either stained with Hematoxylin and Eosin (H/E) or with a Masson's trichrome staining kit (Sigma -Aldrich, UK) or periodic acid-Schiff (PAS) stain (Sigma -Aldrich, UK) or Sirius Red (Chondrex, USA) following manufacturer's instructions. The staining procedure was repeated on different sections (n>3) from each sample. To evaluate tubular injury (defined as tubular atrophy, dilatation, and thickening of the basement membrane), and interstitial fibrosis (blue stain on Trichome-stained sections and red stain on Sirius red stain) ten cortical fields from each animal were examined at ×200 magnification and the percentage of tubules demonstrating injury was assessed by the method described before [83]. To quantitate the degree of glomerular injury, 100 glomeruli were examined, and the number of segmental or globally sclerosed glomeruli was expressed as a percentage. The sections were evaluated by two renal pathologists in a blinded fashion.
Isolation of glomeruli
Glomeruli were isolated following the protocol by Takemoto et al. [84], which involves the use of Dynabeads (Dynal, Oslo, Norway) through the heart of anesthetized mice. Glomeruli total RNA and protein were isolated using a commercially available kit (Macherey-Nagel), according to manufacturer's instructions.
Immunofluorescence
Immunofluorescense experiments were performed either using cryosections or paraffin sections of kidney samples following standard procedures.For cryosections, kidneys were embedded in OCT. 6-μm sections were post-fixed in acetone, penetrated with 0.2% triton X. For antigen retrieval, 6 M urea-0.1 M glycine, pH 3.5, for 30-min at 4 °C was used. The primary antibodies (Supp. Table 1) were diluted in 5% serum in 1% BSA/PBS and applied overnight at 4 °C. Alexa 488 or Cy3 conjugated secondary antibodies were used.
For paraffin sections, kidneys were fixed in 4% paraformaldehyde, dehydrated and embedded in paraffin wax using standard protocols. For double immunofluorescence experiments, 6 μm kidney sections were penetrated with 0.2% triton X in PBS and antigen retrieval was carried out by microwave heating into a 0.2 N HCL solution for 10 min and treated with 6 M urea-0.1 M glycine, pH 3.5, for 30 min at 4 °C. The next steps were as described above for cryosections. Slides were viewed by a TCSL confocal microscope (Leica, Germany).
Western blot
15 μg of soluble protein extracted from isolated glomeruli or whole kidney lysates were prepared for electrophoresis on 10% SDS-PAGE. After transfer to nitrocellulose paper and blocking, immunostaining was performed in either 5% milk or BSA in TBST buffer or ChonBlock (Chondrex, USA). Primary and secondary antibodies are listed in Supp. Table1. Secondary antibodies, either goat anti-mouse or donkey anti-rabbit (Santa Cruz Biotechnology) were labeled with peroxidase. Proteins were detected using the Enhanced ChemiLuminescence Plus Blotting Detection system (Amersham Biosciences, UK) and were visualized through the ChemiDoc™ XRS+ System (BioRad, CA, USA). All transblots were reprobed with anti–β-tubulin antibody (Santa Cruz Biotechnology) (1:500), for normalization. Band density was defined with the ImageJ Software (http://imagej.nih.gov/ij).
Statistical analyses
GBM thickness was evaluated in EM images and results from different groups of animals were statistically evaluated using one-way ANOVA followed by Tukey post-testing. Survival of animals was evaluated using a standard Kaplan-Meier analysis paired to a log-rank Mantel-Cox testing. Albuminuria analysis was performed using two-way ANOVA on log10-transformed values. GraphPad Prism-5 software was used for all analyses. Differences of p-values ≤0.05 were considered as significant.
CRediT authorship contribution statement
C.D., C.O., I.S. designed the study; C.O., I.S. and P.I. carried out most experiments and analyzed the data; P.P. and M.P. had designed the knockin mouse and contributed to interpretation of results. D-B.B. assisted with experimentation and contributed to drafting the manuscript; K.A and C.S carried out a part of experiments; C.O., I.S., M.P., D-B.B., C.D. drafted & revised the manuscript; G.P. and N.M. assisted with molecular & cell biology experimentation; K.S. carried out the electron microscopy imaging and analyzed the data; G.N. and D.G.L analyzed histological slides; O.G. assisted in the research plan, interpretation of the results and the discussion; all authors contributed to the writing and approved the final version of the manuscript.
Acknowledgments
Acknowledgements
We acknowledge microscopy support by Dr. Pantelis Georgiades at University of Cyprus. We also thank Christos Karaiskos for technical support at The Cyprus Institute of Neurology & Genetics.
Funding
This work was funded partly by the Alport Syndrome Foundation Inc. (ASF), Pedersen Family and the Kidney Foundation of Canada (KFOC) Funding Program and the University of Cyprus, to C.D., and partly by a POST-DOC/0916/0190 grant, through the Cyprus Research and Innovation Foundation, to C.O. DBB gratefully acknowledges support by funds from the Meharry Medical College and the Meharry Translational Research Center grant U54 MD007593 from the National Institute on Minority Health and Health Disparities of the National Institutes of Health. O.G. is supported by the German Research Foundation DFG (GR 1852/6-1) and the “Clincial Trials” Programme of the German Ministry of Education and Research BMBF (01KG1104). Part of this work was presented as a poster at the 56th ERA-EDTA Congress, June 13–16, 2019, Budapest, Hungary.
Declaration of competing interest
The authors whose names are listed immediately below certify that they have NO known competing financial interests or personal relation-ships that could have appeared to influence the work reported in this paper.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.mbplus.2020.100053.
Appendix A. Supplementary data
Supplementary material
References
- 1.Alport A.C. Hereditary familial congenital haemorrhagic nephritis. Br. Med. J. 1927;1(3454):504–506. doi: 10.1136/bmj.1.3454.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barker D.F., Hostikka S.L., Zhou J. Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science. 1990;248(4960):1224–1227. doi: 10.1126/science.2349482. [DOI] [PubMed] [Google Scholar]
- 3.Hudson B.G. The molecular basis of Goodpasture and Alport syndromes: beacons for the discovery of the collagen IV family. J. Am. Soc. Nephrol. 2004;15(10):2514–2527. doi: 10.1097/01.ASN.0000141462.00630.76. [DOI] [PubMed] [Google Scholar]
- 4.Mochizuki T., Lemmink H.H., Mariyama M. Identification of mutations in the alpha 3(IV) and alpha 4(IV) collagen genes in autosomal recessive Alport syndrome. Nat. Genet. 1994;8(1):77–81. doi: 10.1038/ng0994-77. [DOI] [PubMed] [Google Scholar]
- 5.Miner J.H. Glomerular basement membrane composition and the filtration barrier. Pediatr. Nephrol. 2011;26(9):1413–1417. doi: 10.1007/s00467-011-1785-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hudson BG, Reeders ST, Tryggvason K. Type IV collagen: structure, gene organization, and role in human diseases. Molecular basis of Goodpasture and Alport syndromes and diffuse leiomyomatosis. J. Biol. Chem. 1993;268(35):26033–26036. [PubMed]
- 7.Abrahamson D.R., Hudson B.G., Stroganova L., Borza D.B., St John P.L. Cellular origins of type IV collagen networks in developing glomeruli. J. Am. Soc. Nephrol. 2009;20(7):1471–1479. doi: 10.1681/ASN.2008101086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ninomiya Y., Kagawa M., Iyama K. Differential expression of two basement membrane collagen genes, COL4A6 and COL4A5, demonstrated by immunofluorescence staining using peptide-specific monoclonal antibodies. J. Cell Biol. 1995;130(5):1219–1229. doi: 10.1083/jcb.130.5.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martin P., Heiskari N., Zhou J. High mutation detection rate in the COL4A5 collagen gene in suspected Alport syndrome using PCR and direct DNA sequencing. J. Am. Soc. Nephrol. 1998;9(12):2291–2301. doi: 10.1681/ASN.V9122291. [DOI] [PubMed] [Google Scholar]
- 10.Lemmink H.H., Mochizuki T., van den Heuvel L.P. Mutations in the type IV collagen alpha 3 (COL4A3) gene in autosomal recessive Alport syndrome. Hum. Mol. Genet. 1994;3(8):1269–1273. doi: 10.1093/hmg/3.8.1269. [DOI] [PubMed] [Google Scholar]
- 11.Pescucci C., Mari F., Longo I. Autosomal-dominant Alport syndrome: natural history of a disease due to COL4A3 or COL4A4 gene. Kidney Int. 2004;65(5):1598–1603. doi: 10.1111/j.1523-1755.2004.00560.x. [DOI] [PubMed] [Google Scholar]
- 12.Deltas C., Pierides A., Voskarides K. Molecular genetics of familial hematuric diseases. Nephrol. Dial. Transplant. 2013;28(12):2946–2960. doi: 10.1093/ndt/gft253. [DOI] [PubMed] [Google Scholar]
- 13.Deltas C., Savva I., Voskarides K., Papazachariou L., Pierides A. Carriers of autosomal recessive Alport syndrome with thin basement membrane nephropathy presenting as focal segmental glomerulosclerosis in later life. Nephron. 2015;130(4):271–280. doi: 10.1159/000435789. [DOI] [PubMed] [Google Scholar]
- 14.Ciccarese M., Casu D., Ki Wong F. Identification of a new mutation in the alpha4(IV) collagen gene in a family with autosomal dominant Alport syndrome and hypercholesterolaemia. Nephrol. Dial. Transplant. 2001;16(10):2008–2012. doi: 10.1093/ndt/16.10.2008. [DOI] [PubMed] [Google Scholar]
- 15.Demosthenous P., Voskarides K., Stylianou K. X-linked Alport syndrome in Hellenic families: phenotypic heterogeneity and mutations near interruptions of the collagen domain in COL4A5. Clin. Genet. 2012;81(3):240–248. doi: 10.1111/j.1399-0004.2011.01647.x. [DOI] [PubMed] [Google Scholar]
- 16.Heidet L., Arrondel C., Forestier L. Structure of the human type IV collagen gene COL4A3 and mutations in autosomal Alport syndrome. J. Am. Soc. Nephrol. 2001;12(1):97–106. doi: 10.1681/ASN.V12197. [DOI] [PubMed] [Google Scholar]
- 17.Jais J.P., Knebelmann B., Giatras I. X-linked Alport syndrome: natural history in 195 families and genotype- phenotype correlations in males. J. Am. Soc. Nephrol. 2000;11(4):649–657. doi: 10.1681/ASN.V114649. [DOI] [PubMed] [Google Scholar]
- 18.Jais J.P., Knebelmann B., Giatras I. X-linked Alport syndrome: natural history and genotype-phenotype correlations in girls and women belonging to 195 families: a “European Community Alport syndrome concerted action” study. J. Am. Soc. Nephrol. 2003;14(10):2603–2610. doi: 10.1097/01.asn.0000090034.71205.74. [DOI] [PubMed] [Google Scholar]
- 19.Gross O., Netzer K.O., Lambrecht R., Seibold S., Weber M. Meta-analysis of genotype-phenotype correlation in X-linked Alport syndrome: impact on clinical counselling. Nephrol. Dial. Transplant. 2002;17(7):1218–1227. doi: 10.1093/ndt/17.7.1218. [DOI] [PubMed] [Google Scholar]
- 20.Tsiakkis D., Pieri M., Koupepidou P., Demosthenous P., Panayidou K., Deltas C. Genotype-phenotype correlation in X-linked Alport syndrome patients carrying missense mutations in the collagenous domain of COL4A5. Clin. Genet. 2012;82(3):297–299. doi: 10.1111/j.1399-0004.2012.01849.x. [DOI] [PubMed] [Google Scholar]
- 21.Cosgrove D., Meehan D.T., Grunkemeyer J.A. Collagen COL4A3 knockout: a mouse model for autosomal Alport syndrome. Genes Dev. 1996;10(23):2981–2992. doi: 10.1101/gad.10.23.2981. [DOI] [PubMed] [Google Scholar]
- 22.Miner J.H., Sanes J.R. Molecular and functional defects in kidneys of mice lacking collagen alpha 3(IV): implications for Alport syndrome. J. Cell Biol. 1996;135(5):1403–1413. doi: 10.1083/jcb.135.5.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu W., Phillips C.L., Killen P.D. Insertional mutation of the collagen genes Col4a3 and Col4a4 in a mouse model of Alport syndrome. Genomics. 1999;61(2):113–124. doi: 10.1006/geno.1999.5943. [DOI] [PubMed] [Google Scholar]
- 24.Rheault M.N., Kren S.M., Thielen B.K. Mouse model of X-linked Alport syndrome. J. Am. Soc. Nephrol. 2004;15(6):1466–1474. doi: 10.1097/01.asn.0000130562.90255.8f. [DOI] [PubMed] [Google Scholar]
- 25.Beirowski B., Weber M., Gross O. Chronic renal failure and shortened lifespan in COL4A3+/− mice: an animal model for thin basement membrane nephropathy. J. Am. Soc. Nephrol. 2006;17(7):1986–1994. doi: 10.1681/ASN.2005101044. [DOI] [PubMed] [Google Scholar]
- 26.Pierides A., Voskarides K., Athanasiou Y. Clinico-pathological correlations in 127 patients in 11 large pedigrees, segregating one of three heterozygous mutations in the COL4A3/ COL4A4 genes associated with familial haematuria and significant late progression to proteinuria and chronic kidney disease from focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2009;24(9):2721–2729. doi: 10.1093/ndt/gfp158. [DOI] [PubMed] [Google Scholar]
- 27.Voskarides K., Damianou L., Neocleous V. COL4A3/COL4A4 mutations producing focal segmental glomerulosclerosis and renal failure in thin basement membrane nephropathy. J. Am. Soc. Nephrol. 2007;18(11):3004–3016. doi: 10.1681/ASN.2007040444. [DOI] [PubMed] [Google Scholar]
- 28.Pieri M., Stefanou C., Zaravinos A. Evidence for activation of the unfolded protein response in collagen IV nephropathies. J. Am. Soc. Nephrol. 2014;25(2):260–275. doi: 10.1681/ASN.2012121217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schnaper HW, Jandeska S, Runyan CE, et al. TGF-beta signal transduction in chronic kidney disease. Front Biosci (Landmark Ed). 2009;14:2448–2465. [DOI] [PMC free article] [PubMed]
- 30.Ina K., Kitamura H., Tatsukawa S., Fujikura Y. Significance of alpha-SMA in myofibroblasts emerging in renal tubulointerstitial fibrosis. Histol. Histopathol. 2011;26(7):855–866. doi: 10.14670/HH-26.855. [DOI] [PubMed] [Google Scholar]
- 31.Zhang G., Moorhead P.J., el Nahas A.M. Myofibroblasts and the progression of experimental glomerulonephritis. Exp. Nephrol. 1995;3(5):308–318. [PubMed] [Google Scholar]
- 32.http://www.hgmd.cf.ac.uk/ac/all.php
- 33.Matthaiou. Prevalence of clinical, pathological and molecular features of glomerular basement membrane nephropathy caused by COL4A3 or COL4A4 mutations: a systematic review. Clinical Kidney Journal. 2020;In press. [DOI] [PMC free article] [PubMed]
- 34.Storey H., Savige J., Sivakumar V., Abbs S., Flinter F.A. COL4A3/COL4A4 mutations and features in individuals with autosomal recessive Alport syndrome. J. Am. Soc. Nephrol. 2013;24(12):1945–1954. doi: 10.1681/ASN.2012100985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Plant K.E., Green P.M., Vetrie D., Flinter F.A. Detection of mutations in COL4A5 in patients with Alport syndrome. Hum. Mutat. 1999;13(2):124–132. doi: 10.1002/(SICI)1098-1004(1999)13:2<124::AID-HUMU4>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 36.Weber S., Strasser K., Rath S. Identification of 47 novel mutations in patients with Alport syndrome and thin basement membrane nephropathy. Pediatr. Nephrol. 2016;31(6):941–955. doi: 10.1007/s00467-015-3302-4. [DOI] [PubMed] [Google Scholar]
- 37.Zhang X., Zhang Y., Zhang Y. X-linked Alport syndrome: pathogenic variant features and further auditory genotype-phenotype correlations in males. Orphanet J Rare Dis. 2018;13(1):229. doi: 10.1186/s13023-018-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Byers P.H., Wallis G.A., Willing M.C. Osteogenesis imperfecta: translation of mutation to phenotype. J. Med. Genet. 1991;28(7):433–442. doi: 10.1136/jmg.28.7.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marini J.C., Forlino A., Cabral W.A. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum. Mutat. 2007;28(3):209–221. doi: 10.1002/humu.20429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barat-Houari M., Dumont B., Fabre A. The expanding spectrum of COL2A1 gene variants IN 136 patients with a skeletal dysplasia phenotype. Eur. J. Hum. Genet. 2016;24(7):992–1000. doi: 10.1038/ejhg.2015.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoornaert K.P., Vereecke I., Dewinter C. Stickler syndrome caused by COL2A1 mutations: genotype-phenotype correlation in a series of 100 patients. Eur. J. Hum. Genet. 2010;18(8):872–880. doi: 10.1038/ejhg.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pope F.M., Narcisi P., Nicholls A.C., Germaine D., Pals G., Richards A.J. COL3A1 mutations cause variable clinical phenotypes including acrogeria and vascular rupture. Br. J. Dermatol. 1996;135(2):163–181. [PubMed] [Google Scholar]
- 43.Savige J., Storey H., Il Cheong H. X-linked and autosomal recessive Alport syndrome: pathogenic variant features and further genotype-phenotype correlations. PLoS One. 2016;11(9) doi: 10.1371/journal.pone.0161802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y., Ding J., Zhang H. Effect of heterozygous pathogenic COL4A3 or COL4A4 variants on patients with X-linked Alport syndrome. Mol Genet Genomic Med. 2019;7(5) doi: 10.1002/mgg3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao X., Chen C., Wei Y. Novel mutations of COL4A3, COL4A4, and COL4A5 genes in Chinese patients with Alport syndrome using next generation sequence technique. Mol Genet Genomic Med. 2019;7(6) doi: 10.1002/mgg3.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Falcone S., Wisby L., Nicol T. Modification of an aggressive model of Alport syndrome reveals early differences in disease pathogenesis due to genetic background. Sci. Rep. 2019;9(1):20398. doi: 10.1038/s41598-019-56837-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hashikami K., Asahina M., Nozu K., Iijima K., Nagata M., Takeyama M. Establishment of X-linked Alport syndrome model mice with a Col4a5 R471X mutation. Biochem Biophys Rep. 2019;17:81–86. doi: 10.1016/j.bbrep.2018.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cervera-Acedo C., Coloma A., Huarte-Loza E., Sierra-Carpio M., Dominguez-Garrido E. Phenotype variability in a large Spanish family with Alport syndrome associated with novel mutations in COL4A3 gene. BMC Nephrol. 2017;18(1):325. doi: 10.1186/s12882-017-0735-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Funk S.D., Bayer R.H., Malone A.F., McKee K.K., Yurchenco P.D., Miner J.H. Pathogenicity of a human laminin beta2 mutation revealed in models of Alport syndrome. J. Am. Soc. Nephrol. 2018;29(3):949–960. doi: 10.1681/ASN.2017090997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Langsford D, Tang M, Djurdjev O, Er L, Levin A. The variability of estimated glomerular filtration rate decline in Alport syndrome. Can J Kidney Health Dis. 2016;3:2054358116679129. [DOI] [PMC free article] [PubMed]
- 51.Okada S., Inaga S., Kitamoto K. Morphological diagnosis of Alport syndrome and thin basement membrane nephropathy by low vacuum scanning electron microscopy. Biomed. Res. 2014;35(5):345–350. doi: 10.2220/biomedres.35.345. [DOI] [PubMed] [Google Scholar]
- 52.Korstanje R., Caputo C.R., Doty R.A. A mouse Col4a4 mutation causing Alport glomerulosclerosis with abnormal collagen alpha3alpha4alpha5(IV) trimers. Kidney Int. 2014;85(6):1461–1468. doi: 10.1038/ki.2013.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gubler M.C. Inherited diseases of the glomerular basement membrane. Nat. Clin. Pract. Nephrol. 2008;4(1):24–37. doi: 10.1038/ncpneph0671. [DOI] [PubMed] [Google Scholar]
- 54.Gubler M.C., Knebelmann B., Beziau A. Autosomal recessive Alport syndrome: immunohistochemical study of type IV collagen chain distribution. Kidney Int. 1995;47(4):1142–1147. doi: 10.1038/ki.1995.163. [DOI] [PubMed] [Google Scholar]
- 55.Forrester A, De Leonibus C, Grumati P, et al. A selective ER-phagy exerts procollagen quality control via a Calnexin-FAM134B complex. EMBO J. 2019;38(2). [DOI] [PMC free article] [PubMed]
- 56.Gross O., Girgert R., Beirowski B. Loss of collagen-receptor DDR1 delays renal fibrosis in hereditary type IV collagen disease. Matrix Biol. 2010;29(5):346–356. doi: 10.1016/j.matbio.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 57.Khoshnoodi J., Pedchenko V., Hudson B.G. Mammalian collagen IV. Microsc. Res. Tech. 2008;71(5):357–370. doi: 10.1002/jemt.20564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stynes G.D., Kiroff G.K., Page R.S., Morrison W.A., Kirkland M.A. Surface-bound collagen 4 is significantly more stable than collagen 1. J. Biomed. Mater. Res. A. 2017;105(5):1364–1373. doi: 10.1002/jbm.a.36019. [DOI] [PubMed] [Google Scholar]
- 59.Sundaramoorthy M., Meiyappan M., Todd P., Hudson B.G. Crystal structure of NC1 domains. Structural basis for type IV collagen assembly in basement membranes. J. Biol. Chem. 2002;277(34):31142–31153. doi: 10.1074/jbc.M201740200. [DOI] [PubMed] [Google Scholar]
- 60.Yeo J., Qiu Y., Jung G.S., Zhang Y.W., Buehler M.J., Kaplan D.L. Adverse effects of Alport syndrome-related Gly missense mutations on collagen type IV: insights from molecular simulations and experiments. Biomaterials. 2020;240:119857. doi: 10.1016/j.biomaterials.2020.119857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Witecka J., Augusciak-Duma A.M., Kruczek A. Two novel COL1A1 mutations in patients with osteogenesis imperfecta (OI) affect the stability of the collagen type I triple-helix. J. Appl. Genet. 2008;49(3):283–295. doi: 10.1007/BF03195625. [DOI] [PubMed] [Google Scholar]
- 62.Constantinou C.D., Nielsen K.B., Prockop D.J. A lethal variant of osteogenesis imperfecta has a single base mutation that substitutes cysteine for glycine 904 of the alpha 1(I) chain of type I procollagen. The asymptomatic mother has an unidentified mutation producing an overmodified and unstable type I procollagen. J. Clin. Invest. 1989;83(2):574–584. doi: 10.1172/JCI113920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Valli M., Zolezzi F., Mottes M. Gly85 to Val substitution in pro alpha 1(I) chain causes mild osteogenesis imperfecta and introduces a susceptibility to protease digestion. Eur. J. Biochem. 1993;217(1):77–82. doi: 10.1111/j.1432-1033.1993.tb18220.x. [DOI] [PubMed] [Google Scholar]
- 64.Stolle C.A., Pyeritz R.E., Myers J.C., Prockop D.J. Synthesis of an altered type III procollagen in a patient with type IV Ehlers-Danlos syndrome. A structural change in the alpha 1(III) chain which makes the protein more susceptible to proteinases. J. Biol. Chem. 1985;260(3):1937–1944. [PubMed] [Google Scholar]
- 65.Tromp G., Kuivaniemi H., Shikata H., Prockop D.J. A single base mutation that substitutes serine for glycine 790 of the alpha 1 (III) chain of type III procollagen exposes an arginine and causes Ehlers-Danlos syndrome IV. J. Biol. Chem. 1989;264(3):1349–1352. [PubMed] [Google Scholar]
- 66.Westerhausen A., Kishi J., Prockop D.J. Mutations that substitute serine for glycine alpha 1-598 and glycine alpha 1-631 in type I procollagen. The effects on thermal unfolding of the triple helix are position-specific and demonstrate that the protein unfolds through a series of cooperative blocks. J. Biol. Chem. 1990;265(23):13995–14000. [PubMed] [Google Scholar]
- 67.Iozzo RV, Gubbiotti MA. Extracellular matrix: the driving force of mammalian diseases. Matrix Biol. 2018;71–72:1–9. [DOI] [PMC free article] [PubMed]
- 68.Karamanos NK, Theocharis AD, Neill T, Iozzo RV. Matrix modeling and remodeling: a biological interplay regulating tissue homeostasis and diseases. Matrix Biol. 2019;75–76:1–11. [DOI] [PMC free article] [PubMed]
- 69.Catania J.M., Chen G., Parrish A.R. Role of matrix metalloproteinases in renal pathophysiologies. Am. J. Physiol. Ren. Physiol. 2007;292(3):F905–F911. doi: 10.1152/ajprenal.00421.2006. [DOI] [PubMed] [Google Scholar]
- 70.Tan R.J., Liu Y. Matrix metalloproteinases in kidney homeostasis and diseases. Am. J. Physiol. Ren. Physiol. 2012;302(11):F1351–F1361. doi: 10.1152/ajprenal.00037.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zakiyanov O., Kalousova M., Zima T., Tesar V. Matrix metalloproteinases in renal diseases: a critical appraisal. Kidney Blood Press. Res. 2019;44(3):298–330. doi: 10.1159/000499876. [DOI] [PubMed] [Google Scholar]
- 72.Kuroda T., Yoshida Y., Kamiie J. Expression of MMP-9 in mesangial cells and its changes in anti-GBM glomerulonephritis in WKY rats. Clin. Exp. Nephrol. 2004;8(3):206–215. doi: 10.1007/s10157-004-0289-8. [DOI] [PubMed] [Google Scholar]
- 73.McMillan J.I., Riordan J.W., Couser W.G., Pollock A.S., Lovett D.H. Characterization of a glomerular epithelial cell metalloproteinase as matrix metalloproteinase-9 with enhanced expression in a model of membranous nephropathy. J. Clin. Invest. 1996;97(4):1094–1101. doi: 10.1172/JCI118502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Springman E.B., Angleton E.L., Birkedal-Hansen H., Van Wart H.E. Multiple modes of activation of latent human fibroblast collagenase: evidence for the role of a Cys73 active-site zinc complex in latency and a “cysteine switch” mechanism for activation. Proc. Natl. Acad. Sci. U. S. A. 1990;87(1):364–368. doi: 10.1073/pnas.87.1.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bellini T., Trentini A., Manfrinato M.C. Matrix metalloproteinase-9 activity detected in body fluids is the result of two different enzyme forms. J. Biochem. 2012;151(5):493–499. doi: 10.1093/jb/mvs014. [DOI] [PubMed] [Google Scholar]
- 76.Vempati P., Karagiannis E.D., Popel A.S. A biochemical model of matrix metalloproteinase 9 activation and inhibition. J. Biol. Chem. 2007;282(52):37585–37596. doi: 10.1074/jbc.M611500200. [DOI] [PubMed] [Google Scholar]
- 77.Price R.G., Spiro R.G. Studies on the metabolism of the renal glomerular basement membrane. Turnover measurements in the rat with the use of radiolabeled amino acids. J. Biol. Chem. 1977;252(23):8597–8602. [PubMed] [Google Scholar]
- 78.Hamano Y., Zeisberg M., Sugimoto H. Physiological levels of tumstatin, a fragment of collagen IV alpha3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via alphaV beta3 integrin. Cancer Cell. 2003;3(6):589–601. doi: 10.1016/s1535-6108(03)00133-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Petitclerc E., Boutaud A., Prestayko A. New functions for non-collagenous domains of human collagen type IV. Novel integrin ligands inhibiting angiogenesis and tumor growth in vivo. J. Biol. Chem. 2000;275(11):8051–8061. doi: 10.1074/jbc.275.11.8051. [DOI] [PubMed] [Google Scholar]
- 80.Rao V.H., Meehan D.T., Delimont D. Role for macrophage metalloelastase in glomerular basement membrane damage associated with alport syndrome. Am. J. Pathol. 2006;169(1):32–46. doi: 10.2353/ajpath.2006.050896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gross O., Beirowski B., Koepke M.L. Preemptive ramipril therapy delays renal failure and reduces renal fibrosis in COL4A3-knockout mice with Alport syndrome. Kidney Int. 2003;63(2):438–446. doi: 10.1046/j.1523-1755.2003.00779.x. [DOI] [PubMed] [Google Scholar]
- 82.Gross O., Schulze-Lohoff E., Koepke M.L. Antifibrotic, nephroprotective potential of ACE inhibitor vs AT1 antagonist in a murine model of renal fibrosis. Nephrol. Dial. Transplant. 2004;19(7):1716–1723. doi: 10.1093/ndt/gfh219. [DOI] [PubMed] [Google Scholar]
- 83.Rangan G.K., Pippin J.W., Coombes J.D., Couser W.G. C5b-9 does not mediate chronic tubulointerstitial disease in the absence of proteinuria. Kidney Int. 2005;67(2):492–503. doi: 10.1111/j.1523-1755.2005.67106.x. [DOI] [PubMed] [Google Scholar]
- 84.Takemoto M., Asker N., Gerhardt H. A new method for large scale isolation of kidney glomeruli from mice. Am. J. Pathol. 2002;161(3):799–805. doi: 10.1016/S0002-9440(10)64239-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material