

Abstract
Nobiletin was found to protect against acute myocardial infarction (AMI)‐induced cardiac function decline and myocardial remodeling, although the dose–effect relationship and underlying pathways remained unclear. In the current research, different doses of Nobiletin (7.5, 15 and 30 mg/kg/day) were administered to AMI rat model for 21 days. Survival rate, echocardiography, and histological analysis were assessed in vivo. In addition, MTT assay, flow cytometry, and Western blotting were conducted to explore Nobiletin's cytotoxicity and antiapoptotic effect on H9C2 cells. Mechanistically, the activation of MAPK effectors and p38 in vivo was studied. The results showed medium‐ and high‐dose Nobiletin could significantly improve survival rate and cardiac function and reduce the area of infarction and cardiac fibrosis. Medium dose showed the best protection on cardiac functions, whereas high dose showed the best protective effect on cellular apoptosis and histological changes. JNK activation was significantly inhibited by Nobiletin in vivo, which could help to explain the partial contribution of autophagy to AMI‐induced apoptosis and the discrepancy on dose–effect relationships. Together, our study suggested that JNK inhibition plays an important role in Nobiletin‐induced antiapoptotic effect in myocardial infarction, and medium‐dose Nobiletin demonstrated the strongest effect in vivo.
Keywords: acute myocardial infarction, apoptosis, cardiac remodeling, hypertrophy, nobiletin
Abbreviations
- AMI
acute myocardial infarction
- AMPK
AMP‐activated protein kinase
- CO
cardiac output
- ERK
extracellular signal‐regulated kinases
- JNK
c‐Jun N‐terminal kinase
- LAD
left anterior descending
- LVAWd
left ventricular end‐diastolic anterior wall thickness
- LVAWs
left ventricular end‐systolic anterior wall thickness
- LVEF
left ventricular ejection fraction
- LVFS
left ventricular fractional shortening
- LVIDd
left ventricular end‐diastolic diameter
- LVIDs
left ventricular end‐systolic diameter
- LVM
left ventricular mass
- LVM/BM
left ventricular mass/body mass
- LVPWd
left ventricular end‐diastolic posterior wall thickness
- LVPWs
left ventricular end‐systolic posterior wall thickness
- MAPK
mitogen‐activated protein kinase
- mTOR
mammalian target of rapamycin
- MTT
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide
- OGD
oxygen‐glucose deprivation
- PI3K
Phosphoinositide 3‐kinase
- TTC
2, 3, 5‐triphenyl tetrazolium chloride
1. INTRODUCTION
Acute myocardial infarction (AMI) is an imminent threat to public health, as it is one of the leading causes of death globally. 1 Previous studies on animal and cell models suggested that myocardial cell death is modulated by the interplay between oxidative stress, inflammation, apoptosis, and autophagy, which contribute to the progression and prognosis of cardiac ischemic diseases. 2 Loss of cardiomyocyte induced by ischemia injury is a key factor that results in cardiac remodeling, pathological hypertrophy and life‐threatening heart failure. 3 , 4 Thus finding novel agents to modulate cell death during ischemia injury is an important therapeutic target that draws constant research attention.
Nobiletin is a polymethoxy flavonoid enriched in citrus, which was reported to have anti‐inflammatory, 5 , 6 anticancer, 7 and antiapoptotic effects. 5 , 6 , 7 In the past few years, the protective effects of Nobiletin against ischemic injury were reported in hepatic, renal, and cerebral ischemic diseases. 8 , 9 , 10 Studies have shown that Nobiletin could alleviate hippocampal injury following cerebral ischemia and improve learning and memory. 11 , 12 , 13 While there are few studies of Nobiletin on myocardial infarction, our previous study found that Nobiletin could protect against myocardial ischemic injury through restoration of autophagic flux, alleviation of apoptosis, and reduction in cardiac remodeling. 14 Interestingly, neither chloroquine (inhibitor of autophagic flux) nor 3‐MA (inhibitor of autophagosome formation) could fully reverse the antiapoptotic effect of Nobiletin (previous study, Figure 4A and B). Moreover, the study showed AMI‐induced activation of classical autophagy upstream signaling pathways mTOR, AMPK, and PI3K were unaffected by Nobiletin treatment. Therefore, it is speculated that diverse cellular signaling pathways contribute to the protective effect of NOB.
FIGURE 4.
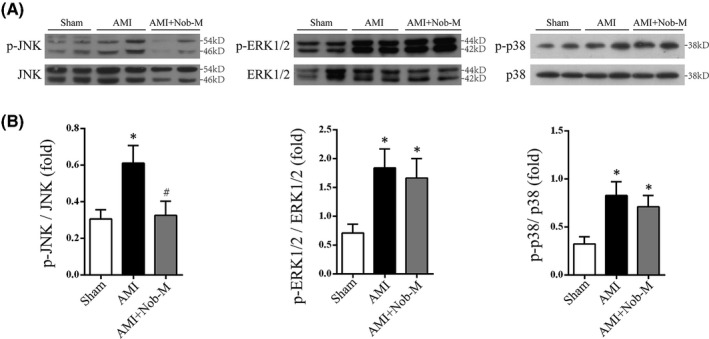
Nobiletin exerts antiapoptotic effect via modulation of JNK pathway in AMI rat model. (A) Representative immunoblots of p‐JNK, JNK, p‐ERK1/2, ERK1/2, p‐p38, and p38 levels in tissue lysate from Sham, AMI, and AMI+Nob‐M. (B) Statistical analysis of Western blot in (A). Data were represented as mean±SEM. (*p < .05 vs. Sham, # p < 0.05 vs. AMI; n = 5)
JNK (c‐Jun N‐terminal kinase) is an important third‐tier kinase in the MAPK pathway and it is at the crosstalk of a wide range of stimuli and regulates a multitude of downstream activities ranging from transcription regulation, cytoskeletal dynamics to apoptosis induction. 15 , 16 It plays an important part in apoptosis induction, including extrinsic, mitochondrial, and ER‐stress‐induced apoptotic pathways, by regulating apoptosis‐related gene expression and protein modification. 17 , 18 , 19 , 20 , 21 For example, JNK could activate the apoptotic pathway by increasing apoptotic gene expression via c‐Jun activation, inhibiting Bcl‐2 directly via phosphorylation, or releasing proapoptotic Bad and Bax. 16 Therefore, JNK has received a lot of attention as a research target to develop potential therapy against myocardial ischemic injury. 22 The relationship between JNK and autophagy is more complex and maybe context‐dependent. Studies have found that JNK could upregulate ATG gene expression, 23 and activated JNK could release Beclin1 from Bcl‐2/Beclin1 complex via phosphorylation, 24 which induces detrimental autophagy. 25 , 26 In this study, the modulatory effect on major MAPK family members JNK, ERK1/2, and p38 MAPK were explored to elucidate the regulation on the upstream pathway by Nobiletin. Furthermore, the dose–effect relationship of the protective effect of Nobiletin was explored by assays.
2. MATERIALS AND METHODS
2.1. Animals
All experiments conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication no. 85–23, revised 1996), and the protocol was approved by the Animal Research Committee of Guangzhou Medical University. Adult male Sprague–Dawley rats (7–8 weeks of age, 200–220 g) were purchased from the Medical Experimental Animal Center of Guangdong Province and kept on a 12‐h light/dark cycle under temperature‐ and humidity‐controlled environment for 7 days. All rats received food and water available ad libitum.
All rats were randomly divided into six groups (n = 54 per group): (1) Sham group (Sham); (2) AMI group (AMI); (3) AMI+Nobiletin low‐dose group (AMI+Nob‐L, Nobiletin 7.5 mg/kg/day); (4) AMI+Nobiletin medium‐dose group (AMI+Nob‐M, Nobiletin 15 mg/kg/day); (5) AMI+Nobiletin high‐dose group (AMI+Nob‐H, Nobiletin 30 mg/kg/day); and (6) AMI +Enalapril (AMI+Ena, Enalapril 10 mg/kg/day). Rats in Nobiletin and Enalapril groups received Nobiletin (#962600, Sigma‐Aldrich) or Enalapril (Merck Sharp & Dohme), respectively, via intraperitoneal injection once every 24 h for 21 days. Rats in the Sham and AMI group were administered with equal volume of saline solution.
2.2. Rat model of acute myocardial infarction
Animal model of myocardial infarction was induced by left anterior descending (LAD) coronary artery ligation as we previously described. 14 , 27 In brief, rats were anesthetized with 2% (v/v) isoflurane in O2 and connected to a respirator for mechanical ventilation. Body temperature was maintained with a heating pad. After exposure of the heart by thoracotomy and pericardiotomy between the fourth and fifth rib on the left, LAD coronary artery was ligated with a 6‐0 silk suture 2 mm below the tip of the left atrial appendage. Proper LAD ligation was confirmed by the whitening of the myocardial tissue distal to the suture and by electrocardiography. Sham animals underwent thoracotomy and pericardiotomy, but not LAD ligation.
2.3. Measurement of infarction size and fibrosis
After 21 days of LAD ligation, the rats were euthanized via intraperitoneal injection of pentobarbital sodium (200 mg/kg), and the hearts were rapidly removed. The left ventricle including the septum was dissected free and weighed. The myocardial tissue was sectioned for H&E staining and histology as we described previously. 14 Myocardial infarct size was measured by 2, 3, 5‐triphenyl tetrazolium chloride (TTC) staining, whereas fibrosis was measured by fixing the cardiac tissue in 4% paraformaldehyde for Masson's staining as previously described. 14 For TTC staining, the left ventricle was sliced transversely into 2‐mm‐thick slices perpendicular to the axis of the LAD immediately and incubated in TTC (pH 7.4) for 20 min at 37°C. The tissue from area‐at‐risk (identified as red tissue staining) was isolated from the infarct area (absence of staining) and the nonrisk region (purple tissue staining) for use in molecular studies. The area of infarction and area of fibrosis were analyzed and quantified by Image‐Pro Plus v.5.0 (Media Cybernetics Inc.).
2.4. H9C2 cell culture
The H9C2 rat cardiac myoblast cell line was acquired from American Type Culture Collection (ATCC), and cultured as previously described. 28 Cells were cultured in complete high‐glucose DMEM medium supplemented with 10% fetal bovine serum and 100 U/ml penicillin‐streptomycin, maintained under an atmosphere of 95% humidified air and 5% (v/v) CO2 at 37°C. Cells were passed when the monolayer reached 90% confluence. Passages 3–9 were used for study.
2.5. Establishment of oxygen‐glucose deprivation model
As we previously described, 14 cells were subjected to serum‐free, glucose‐free, and sodium pyruvate‐free DMEM (#D5030, Sigma‐Aldrich) after rinsing twice with the same medium, and were subjected to hypoxia treatment for 12 h in an anoxia chamber (InVivo 500, Ruskinn Life Science) (94%N2/5%CO2/1%O2) at 37°C. The Nobiletin‐treated group was exposed to 20 mM Nobiletin 2 h before oxygen‐glucose deprivation (OGD) treatment.
2.6. Cell viability assay
The cells were seeded in 96‐well plates and allowed to recover for more than 24 h before OGD treatment as described earlier. The viability of cells was determined by MTT (3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide) assay and the absorbance at 570 and 620 nm was determined with a Biorad plate reader.
2.7. Detection of cell apoptosis by flow cytometry
The Annexin V‐FITC/PI Apoptosis Detection Kit (#556547, BD Pharmingen) was used to measure apoptotic cell population according to the manufacturer's instruction. In brief, after Nobiletin pretreatment at different concentrations and OGD exposure, cells were trypsinized, washed twice with cold PBS and re‐suspended in binding buffer. Then the cells were incubated with Annexin V‐FITC and PI working solution in the dark at room temperature for 15 min. Cellular fluorescence was measured by flow cytometer (FACSAria; Beckman‐Coulter Inc.) and the data were analyzed by FlowJo v.7.6 (TreeStar Inc.). Each experiment was repeated at least three times.
2.8. Western blot analysis
The tissue from area‐at‐risk was homogenized with cold RIPA lysis buffer with protease inhibitor cocktail (Sigma) before centrifugation and separation of supernatant. Protein concentration was determined by BCA assay (Pierce). An equal amount of protein extracts (20 µg) from each sample was electrophoretically separated by 10% SDS polyacrylamide gels and electro‐transferred to polyvinylidene difluoride (PVDF) membrane (Roche). The membranes were blocked with 5% BSA and incubated overnight at 4°C with primary antibodies against p‐JNK (#4668, Cell Signaling Technology), JNK (#9252, Cell Signaling Technology), p‐p38 MAPK (#4511, Cell Signaling Technology), p38 (#8690, Cell Signaling Technology), p‐ERK1/2 (#4370, Cell Signaling Technology), ERK1/2 (#4695, Cell Signaling Technology), and GAPDH (SC‐48166, Santa Cruz Biotechnology) at a working dilution of 1:1000. Membranes were washed three times with TBST and incubated with secondary antibody at a dilution of 1:5000 for 1 h at room temperature. After sufficient washing, blots were developed by 20X LumiGLO Reagent (Cell Signaling Technology). The band intensity was recorded by densitometry and analyzed with ImageJ (NIH). All the experiments were conducted at least three times.
2.9. Statistical analysis
All data in this study were analyzed using GraphPad Prism v.5.0 (GraphPad Software Inc.). Quantitative data were represented as Mean±SEM. Statistical comparisons were analyzed using one‐way ANOVA with Tukey's post hoc test. Differences with p < .05 were considered statistically significant.
2.10. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY, 29 and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20. 30
3. RESULTS
3.1. Nobiletin improves survival rate and ameliorates cardiac function deterioration in AMI rats
Male Sprague–Dawley rats were subjected to LAD ligation to establish AMI model. 24 h after the procedure, low, medium, or high dose of Nobiletin (7.5, 15 or 30 mg/kg/day) was administered intraperitoneally once per day for 21 days. Enalapril is an angiotensin‐converting enzyme inhibitor (ACE‐I) that was found to have cardioprotective and antihypertrophic effect in clinical use, 31 , 32 , 33 and was administered as a positive drug control (10 mg/kg/day, i.p.). The Kaplan–Meier survival analysis suggested that AMI+Nob‐M (p = .0424), AMI+Nob‐H (p = .2890), and AMI+Ena (p = .0458) groups have better prognosis compared to AMI group (Figure 1A). Cardiac functions measured by transthoracic echocardiography (Figure 1B and C) also suggested that in AMI+Nob‐M, AMI+Nob‐H, and AMI+Ena groups, left ventricular internal diameter in systole (LVIDs), left ventricular anterior wall thickness at end‐systole (LVAWs) were significantly improved compared to AMI group. Both end‐systolic volume (ESV) and end‐diastolic volume (EDV) were increased in AMI rats (Figure S1), although both Nobiletin and Enalapril exerted a protective effect on EDV, the overall results were statistically insignificant (Table S1). On the other hand, medium‐ and high‐dose Nobiletin significantly alleviated ESV increase, leading to improved left ventricular fractional shortening (LVFS), left ventricular ejection fraction (LVEF) and cardiac output (Figure 1C), although LVPWd, LVPWs, and LVAWd showed no significant improvement (Figure S1). In addition to the improved left ventricular systolic function, Nobiletin treatment also significantly reduced AMI‐induced left ventricular mass (LVM) increase (Figure 1C), suggesting alleviation of cardiac hypertrophy. In terms of the dose‐dependency of Nobiletin, medium dose exhibited better protective effect on survival rate, LVEF, LVFS, and cardiac output than high‐dose treatment, although the result is statistically insignificant.
FIGURE 1.
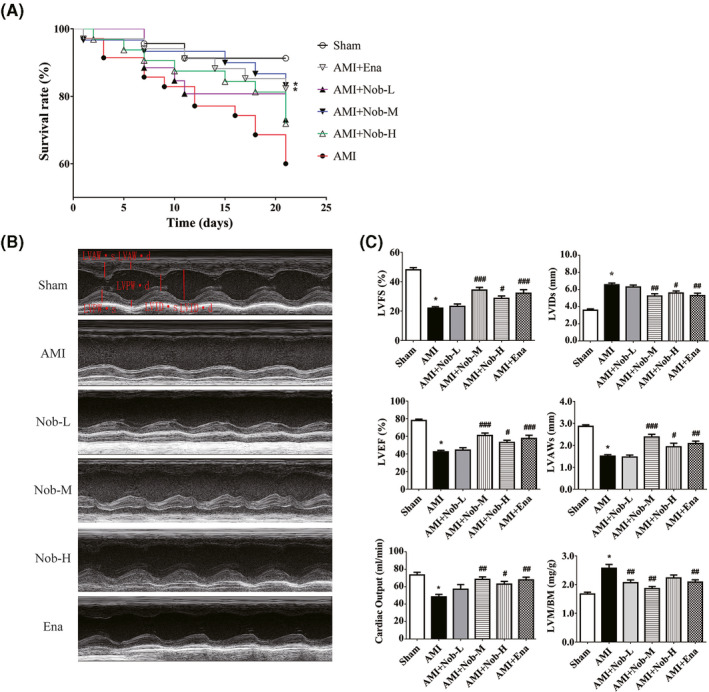
Nobiletin's effect on survival rates and cardiac function deterioration in AMI rats. (A) Survival curve for rats 21 day after LAD ligation (n = 15 per group). *Kaplan–Meier analysis performed shows p value <.05. (B) Representative M‐mode echocardiogram recordings of each group 21 days after LAD ligation. (C) Left ventricular mass/body weight (LVM/BM), left ventricular end‐systolic anterior wall thickness (LVAWs), left ventricular ejection fraction (LVEF), left ventricular fractional shortening (LVFS), left ventricular end‐systolic diameter (LVIDs) and cardiac output (CO) were calculated according to M‐mode echocardiography in (B). (*p < .05 vs. Sham; # p < .01 vs. AMI; ## p < .01 vs. AMI; ### p < .001 vs. AMI; n = 15)
These results were consistent with our previous study that Nobiletin is protective against acute cardiac infarction in rat model. Furthermore, the findings suggested that medium‐dose Nobiletin exhibited better protective effect on animal survival, cardiac function preservation, and alleviation of hypertrophy.
3.2. Nobiletin alleviates myocardial infarction and fibrosis in AMI rats
To investigate the protective effect of Nobiletin on cardiac infarction and fibrosis after ischemic injury, myocardial tissues were stained by HE staining, TTC staining, and Masson's staining, respectively, at the end of the 21‐day treatment (Figure 2A and B). HE staining showed regularly arranged myocardial fibers in the Sham group, which was replaced by fibrous tissue formation, inflammatory cell infiltration, and evidence of hemorrhage in AMI group. Nobiletin treatment could significantly reduce the disruption of myocardial fiber arrangement, fibrosis formation, and neutrophil infiltration (Figure 2A, left panel). Masson's trichrome staining could define the degree of fibrogenesis, as the collagen fibers were stained blue and the muscle fibers stained red in the histological sections. The result further confirmed reduced fibrotic area in AMI+Nob‐M, AMI+Nob‐H, and AMI+Ena groups compared with AMI (Figure 2A, right panel). TTC‐stained sections could delineate the infarct myocardium as the unstained area and the intact myocardium as red area (Figure 2A). Compared to AMI group, treatment of medium‐ and high‐dose Nobiletin and Enalapril could significantly reduce the infarction area (Figure 2B), and intriguingly, the protective effect follows a dose‐dependent manner.
FIGURE 2.
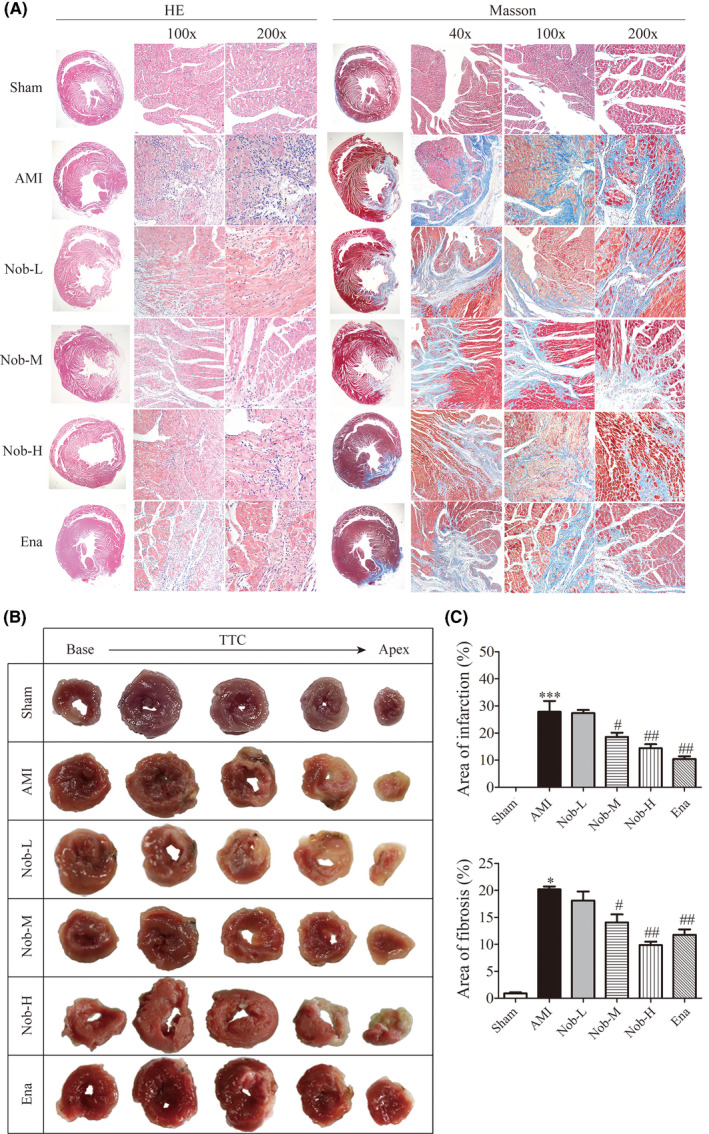
Nobiletin's effect on myocardial infarction and fibrosis in AMI rats. (A) Representative images of HE staining and Masson staining with different magnifications. (B) Representative sections of TTC staining. (C) Statistical analysis of area of infarction and fibrosis, from analysis on TTC staining sections and Masson staining sections, respectively. (*p < .05 vs. Sham; # p < .05 vs. AMI, ## p < .01 vs. AMI; n = 15)
These data suggested that Nobiletin‐induced cardiac protection against ischemic injury was related to reduction in infarction area and tissue fibrosis. In addition, high Nobiletin concentration (30 mg/kg/day) conferred better cardiac protection on in vivo histological assays.
3.3. The cytotoxicity and antiapoptotic effect of Nobiletin on H2C9 cardiomyocytes against OGD treatment
The cytotoxic effect of Nobiletin alone was determined by MTT assay and its protective effect on H9C2 cells after 12‐h oxygen‐glucose deprivation (OGD) treatment was determined by Western blotting and flow cytometry. In terms of cytotoxicity, at concentrations up to 40 μM, Nobiletin exerted no apparent cytotoxic effect in vitro after 24 h (Figure 3A). As for antiapoptotic protection, cleaved caspase 3, the major effector of apoptosis, increased after 12 h OGD treatment and was alleviated by 2 h Nobiletin pretreatment at 20, 40, and 80 μM (Figure 3B) in a dose‐dependent manner. Flow cytometry with FITC‐Annexin V/PI double staining confirmed that apoptotic cell population increased after OGD treatment from 7.54 ± 0.94% to 33.36 ± 4.13%, which was reduced by Nobiletin pretreatment to 16.35 ± 1.41% at 80 μM (Figure 3C). Moreover, higher dosage (20, 40 and 80 μM) of Nobiletin conferred better protection against apoptosis compared to lower dosages (5 and 10 μM).
FIGURE 3.
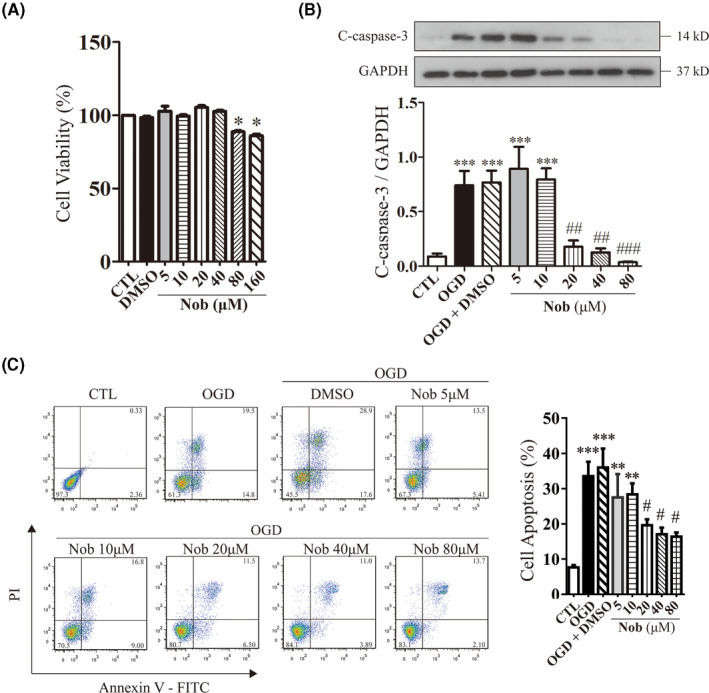
The cytotoxicity and antiapoptotic effect of Nobiletin on H2C9 cardiomyocytes against OGD treatment. (A) Cell viability was analyzed by MTT assay on H9C2 cardiomyocytes pretreated with various concentrations of Nobiletin for 24 h. (*p < .05 vs. CTL; n = 6). (B) Representative immunoblots and analysis of cleaved caspase‐3 levels in H9C2 cardiomyocytes after 12 h OGD treatment in the presence or absence of 2 h Nobiletin pretreatments. The data were represented as the mean ± SEM. (*p < .05 vs. CTL, # p < .05 vs. OGD; n = 6). (C) Representative images and analysis of FITC‐Annexin V/PI double stain of H9c2 cardiomyocytes by flow cytometry. (**p < .01 vs. CTL, ***p < .001 vs. CTL, # p < .05 vs. OGD; n = 6)
These findings confirmed our previous study, that the protective effect of Nobiletin against ischemic injury originates from its modulatory effect on caspase‐3 cleavage and cell apoptosis, which follow a dose‐dependent manner. However, it could inhibit cell viability at high dosage (80 and 160 μM).
3.4. Nobiletin exerts antiapoptotic effect via modulation of JNK pathway in AMI rat model
MAPK is a family of kinases that regulate cell proliferation, differentiation and survival through a sequential three‐tiered kinase phosphorylation, 34 , 35 which was shown to be modulated by Nobiletin in cerebral ischemia injury. 9 Western blotting was conducted to identify the effect of Nobiletin on JNK, p38 MAPK, and ERK1/2, three important MAPK members that transduce respective signals to downstream pathways in AMI tissue. JNK and p38 MAPK, two pro‐death MAPK responsive to stress stimuli were significantly activated, as shown by increased phosphorylation. Similarly, ERK1/2, a prosurvival MAPK molecule was also activated in AMI model (Figure 4A and B). Pretreatment with Nobiletin (15 mg/kg/day) exerted differential modulatory effects on MAPK molecules, as the activation of JNK in AMI group was significantly reversed (from 0.61 ± 0.09 fold to 0.32 ± 0.08 fold, p < .01), whereas the phosphorylation of ERK1/2 and p38 MAPK remained unchanged (Figure 4C).
4. DISCUSSION
Loss of cardiomyocyte after myocardial infarction is a critical event that involves necrosis, apoptosis and autophagic cell death, which leads to impairment in left ventricular function, pathological cardiac remodeling, and heart failure. 36 , 37 The regulated cell death mechanisms such as apoptosis and autophagic cell death have been under investigation as important therapeutic targets in the treatment of AMI. 38 , 39 , 40
Consistent with our previous research, Nobiletin can significantly reduce the myocardial infarction area, improve cardiac function, and reduce left ventricular remodeling in vivo (Figure 1). In addition, the study on dose–effect relationship showed medium‐dose Nobiletin treatment (15 mg/kg/day) showed better protective effect than high‐dose treatment (30 mg/kg/day), although the difference is statistically insignificant. Interestingly, in vivo and in vitro assays (HE, TTC and Masson staining; and cell viability assay) suggested better protection by high‐dose Nobiletin (Figures 2 and 3) against histological changes and cell apoptosis induced by AMI. The antiapoptotic effect also seemed to be dose‐dependent by Nobiletin treatment on both in vitro and in vivo levels, as apoptotic population and caspase 3 cleavage were reduced by Nobiletin preconditioning in OGD‐treated cells (Figure 3B and C). On the other hand, Nobiletin at a higher dosage (80 μM) showed an inhibitory effect on cell viability (Figure 3A) while maintaining its antiapoptotic effect (Figure 3B and C). This may help to explain the discrepancy in the dose–effect relationship in functional study result (Figure 1).
Our previous study showed that by restoring autophagic flux, Nobiletin could partially alleviate AMI‐induced apoptosis, and classical autophagy signaling pathways were unaffected. Our current research took a step further to investigate the roles of major MAPK pathway effectors in the protective effect of Nobiletin against AMI. All three MAPK effectors, including proapoptotic JNK and p38 MAPK and antiapoptotic ERK1/2 were activated in ischemia injury (Figure 4). Our study showed that only JNK was suppressed by Nobiletin treatment.
The results suggested that inhibition of JNK is the key mechanism of action. JNK activation by ROS production was found to mediate autophagy initiation, 41 , 42 which leads to caspase‐dependent apoptosis. 25 , 43 , 44 , 45 In addition, a previous study suggested that JNK mediates autophagic cell death in Bax/Bak double‐knockout cells, a pathway independent of apoptosis, 46 which is in line with the other studies suggesting caspase‐independent autophagic cell death mediated by JNK. 26 , 47 , 48 On the other hand, the activation of JNK could be induced by blockage of autophagic flux, which leads to increased ER stress, 49 , 50 activation of PERK and IRE1, and ROS production. 18 , 51 Studies also found enhanced autophagic flux could reverse JNK activation, leading to reduced apoptotic cell death. 52 , 53 Therefore, we hypothesize a feedforward loop of ROS production, JNK activation, and autophagic flux blockage, which is interrupted by Nobiletin treatment (Figure 5). As autophagic cell death and apoptosis are two separate downstream death pathways in this process, this is in agreement with our previous results, that inhibition of autophagy initiation (by 3‐MA) and more importantly the inhibition of autophagic flux (by CQ) could partially reverse the antiapoptotic effect of Nobiletin. 14
FIGURE 5.
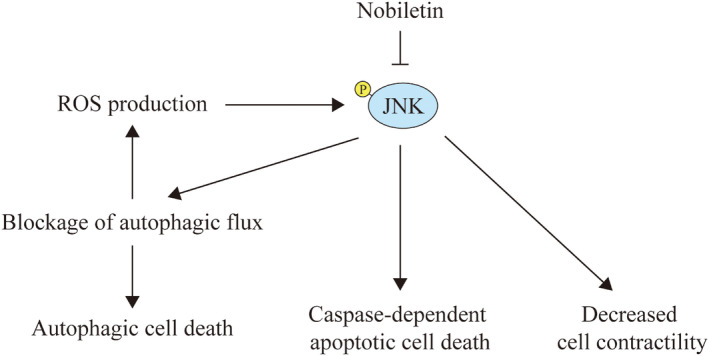
Schematic representation of JNK interaction with autophagic flux and its biological functions in Nobiletin‐modulated AMI protection. The activated JNK by ROS accumulation could mediate the blockage of autophagic flux, which could exacerbate the production of ROS, leading to a feedforward loop of JNK activation. Nobiletin treatment could alleviate the activation of JNK, which help to curb the ensuing autophagic cell death and caspase‐dependent apoptosis. A recent study also suggest a possible role of increasing cardiomyocyte contractility by JNK inhibition 54
In addition, JNK inhibition was reported to play a protective effect on cardiomyocyte contractility, 54 thus inhibition of JNK activation by Nobiletin might also help to explain the discrepancy that medium dose (15 mg/kg/d) exhibited better cardiac function and overall animal survival, whereas high dose showed better protective effect for cell survival and structural preservation. However, this warrants further study for confirmation.
Collectively, our study confirmed that natural product Nobiletin exerts protection against AMI by inhibiting myocardial cell death both in vitro and in vivo, whereas the dose–effect relationship remains differential between functional and histological studies. We also proposed that inhibition of JNK activation by Nobiletin could disrupt the vicious feedforward loop of ROS production, autophagic flux blockage, and JNK activation, leading to the rescue of both noncanonical autophagic death and caspase‐dependent apoptosis. This finding could tie up the loose ends in our previous study and explain the findings in our study. Since the modulation of JNK is increasingly investigated in the treatment of myocardial ischemic injury, Nobiletin could be a potential therapeutic option in the treatment of AMI patients.
CONFLICT OF INTEREST
We declared that the authors have no conflicts of interest.
Supporting information
Figure S1
Table S1
ACKNOWLEDGEMENTS
This work was supported by the National Natural Science Foundation of China [grant No. 81573429 to Dr. X Wu].
Zumei Liu, Zhimin Gao and Lihuan Zeng authors are contributed equally to this work.
DATA AVAILABILITY STATEMENT
All data generated and analyzed in the study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Roth GA, Johnson C, Abajobir A, et al. Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. J Am Coll Cardiol. 2017;70(1):1‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang X, Guo Z, Ding Z, Mehta JL. Inflammation, autophagy, and apoptosis after myocardial infarction. J Am Heart Assoc. 2018;7(9):e008024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Engel D, Peshock R, Armstong RC, Sivasubramanian N, Mann DL. Cardiac myocyte apoptosis provokes adverse cardiac remodeling in transgenic mice with targeted TNF overexpression. Am J Physiol‐Heart Circ Physiol. 2004;287(3):H1303‐H1311. [DOI] [PubMed] [Google Scholar]
- 4. Haudek SB, Taffet GE, Schneider MD, Mann DL. TNF provokes cardiomyocyte apoptosis and cardiac remodeling through activation of multiple cell death pathways. J Clin Investig. 2007;117(9):2692‐2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang L, Zhang X, Zhang C, et al. Nobiletin promotes antioxidant and anti‐inflammatory responses and elicits protection against ischemic stroke in vivo. Brain Res. 2016;1636:130‐141. [DOI] [PubMed] [Google Scholar]
- 6. Malik S, Bhatia J, Suchal K, et al. Nobiletin ameliorates cisplatin‐induced acute kidney injury due to its anti‐oxidant, anti‐inflammatory and anti‐apoptotic effects. Exp Toxicol Pathol. 2015;67(7–8):427‐433. [DOI] [PubMed] [Google Scholar]
- 7. Chen J, Chen AY, Huang H, et al. The flavonoid nobiletin inhibits tumor growth and angiogenesis of ovarian cancers via the Akt pathway. Int J Oncol. 2015;46(6):2629‐2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Güvenç M, Cellat M, Uyar A, et al. Nobiletin protects from renal ischemia‐reperfusion injury in rats by suppressing inflammatory cytokines and regulating iNOS‐eNOS expressions. Inflammation. 2020;43(1):336‐346. [DOI] [PubMed] [Google Scholar]
- 9. Wang T, Wang F, Yu L, Li Z. Nobiletin alleviates cerebral ischemic‐reperfusion injury via MAPK signaling pathway. Am J Transl Res. 2019;11(9):5967. [PMC free article] [PubMed] [Google Scholar]
- 10. Dusabimana T, Kim SR, Kim HJ, Park SW, Kim H. Nobiletin ameliorates hepatic ischemia and reperfusion injury through the activation of SIRT‐1/FOXO3a‐mediated autophagy and mitochondrial biogenesis. Exp Mol Med. 2019;51(4):1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zheng Y, Bu J, Yu L, Chen J, Liu H. Nobiletin improves propofol‐induced neuroprotection via regulating Akt/mTOR and TLR 4/NF‐κB signaling in ischemic brain injury in rats. Biomed Pharmacother. 2017;91:494‐503. [DOI] [PubMed] [Google Scholar]
- 12. Yamamoto Y, Shioda N, Han F, et al. Nobiletin improves brain ischemia‐induced learning and memory deficits through stimulation of CaMKII and CREB phosphorylation. Brain Res. 2009;1295:218‐229. [DOI] [PubMed] [Google Scholar]
- 13. Zhang L, Zhao H, Zhang X, et al. Nobiletin protects against cerebral ischemia via activating the p‐Akt, p‐CREB, BDNF and Bcl‐2 pathway and ameliorating BBB permeability in rat. Brain Res Bull. 2013;96:45‐53. [DOI] [PubMed] [Google Scholar]
- 14. Wu X, Zheng D, Qin Y, et al. Nobiletin attenuates adverse cardiac remodeling after acute myocardial infarction in rats via restoring autophagy flux. Biochem Biophys Res Comm. 2017;492(2):262‐268. [DOI] [PubMed] [Google Scholar]
- 15. Zeke A, Misheva M, Reményi A, Bogoyevitch MA. JNK signaling: regulation and functions based on complex protein‐protein partnerships. Microbiol Mol Biol Rev. 2016;80(3):793‐835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dhanasekaran DN, Reddy EP. JNK‐signaling: a multiplexing hub in programmed cell death. Genes Cancer. 2017;8(9‐10):682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dhanasekaran DN, Reddy EP. JNK signaling in apoptosis. Oncogene. 2008;27(48):6245‐6251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dong Y, Chen H, Gao J, Liu Y, Li J, Wang J. Molecular machinery and interplay of apoptosis and autophagy in coronary heart disease. J Mol Cell Cardiol. 2019;136:27‐41. [DOI] [PubMed] [Google Scholar]
- 19. Wang X‐T, Pei D‐S, Xu J, et al. Opposing effects of Bad phosphorylation at two distinct sites by Akt1 and JNK1/2 on ischemic brain injury. Cell Signal. 2007;19(9):1844‐1856. [DOI] [PubMed] [Google Scholar]
- 20. Donovan N, Becker EB, Konishi Y, Bonni A. JNK phosphorylation and activation of BAD couples the stress‐activated signaling pathway to the cell death machinery. J Biol Chem. 2002;277(43):40944‐40949. [DOI] [PubMed] [Google Scholar]
- 21. Papadakis ES, Finegan KG, Wang X, et al. The regulation of Bax by c‐Jun N‐terminal protein kinase (JNK) is a prerequisite to the mitochondrial‐induced apoptotic pathway. FEBS Lett. 2006;580(5):1320‐1326. [DOI] [PubMed] [Google Scholar]
- 22. Shvedova M, Anfinogenova Y, Atochina‐Vasserman EN, Schepetkin IA, Atochin DN. c‐Jun N‐terminal kinases (JNKs) in myocardial and cerebral ischemia/reperfusion injury. Front Pharmacol. 2018;9:715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhou Y‐Y, Li Y, Jiang W‐Q, Zhou L‐F. MAPK/JNK signalling: a potential autophagy regulation pathway. Biosci Rep. 2015;35(3):e00199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1‐mediated phosphorylation of Bcl‐2 regulates starvation‐induced autophagy. Mol Cell. 2008;30(6):678‐688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhu Z, Huang Y, Lv L, et al. Acute ethanol exposure‐induced autophagy‐mediated cardiac injury via activation of the ROS‐JNK‐Bcl‐2 pathway. J Cell Physiol. 2018;233(2):924‐935. [DOI] [PubMed] [Google Scholar]
- 26. Trenti A, Grumati P, Cusinato F, Orso G, Bonaldo P, Trevisi L. Cardiac glycoside ouabain induces autophagic cell death in non‐small cell lung cancer cells via a JNK‐dependent decrease of Bcl‐2. Biochem Pharmacol. 2014;89(2):197‐209. [DOI] [PubMed] [Google Scholar]
- 27. Wu X, He L, Cai Y, et al. Induction of autophagy contributes to the myocardial protection of valsartan against ischemia‐reperfusion injury. Mol Med Rep. 2013;8(6):1824‐1830. [DOI] [PubMed] [Google Scholar]
- 28. Zheng D, Liu Z, Zhou Y, et al. Urolithin B, a gut microbiota metabolite, protects against myocardial ischemia/reperfusion injury via p62/Keap1/Nrf2 signaling pathway. Pharmacol Res. 2020;153:104655. [DOI] [PubMed] [Google Scholar]
- 29. Harding S, Sharman J, Faccenda E, et al. NC‐IUPHAR (2018). The IUPHAR/BPS guide to pharmacology in. 2018.
- 30. Alexander SP, Fabbro D, Kelly E, et al. The concise guide to pharmacology 2019/20: enzymes. Br J Pharmacol. 2019;176:S297‐S396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tani S, Nagao K, Anazawa T, et al. Effects of enalapril and losartan in left ventricular remodeling after acute myocardial infarction: a possible mechanism of prevention of cardiac events by angiotensin‐converting enzyme inhibitors and angiotensin receptor blockers in high‐risk myocardial infarction. Intern Med. 2009;48(11):877‐882. [DOI] [PubMed] [Google Scholar]
- 32. Wang J, Guo X, Dhalla NS. Modification of myosin protein and gene expression in failing hearts due to myocardial infarction by enalapril or losartan. Biochim et Biophys Acta (BBA)‐Mol Basis Dis. 2004;1690(2):177‐184. [DOI] [PubMed] [Google Scholar]
- 33. González GE, Wilensky L, Cassaglia P, Morales C, Gelpi RJ. Early administration of Enalapril prevents diastolic dysfunction and ventricular remodeling in rabbits with myocardial infarction. Cardiovasc Pathol. 2016;25(3):208‐213. [DOI] [PubMed] [Google Scholar]
- 34. Roux PP, Blenis J. ERK and p38 MAPK‐activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004;68(2):320‐344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rose BA, Force T, Wang Y. Mitogen‐activated protein kinase signaling in the heart: angels versus demons in a heart‐breaking tale. Physiol Rev. 2010;90(4):1507‐1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wencker D, Chandra M, Nguyen K, et al. A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Investig. 2003;111(10):1497‐1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. van Empel VP, Bertrand AT, Hofstra L, Crijns HJ, Doevendans PA, De Windt LJ. Myocyte apoptosis in heart failure. Cardiovasc Res. 2005;67(1):21‐29. [DOI] [PubMed] [Google Scholar]
- 38. Teringova E, Tousek P. Apoptosis in ischemic heart disease. J Transl Med. 2017;15(1):87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ma S, Wang Y, Chen Y, Cao F. The role of the autophagy in myocardial ischemia/reperfusion injury. Biochim et Biophys Acta (BBA)‐Mol Basis Dis. 2015;1852(2):271‐276. [DOI] [PubMed] [Google Scholar]
- 40. Aghaei M, Motallebnezhad M, Ghorghanlu S, et al. Targeting autophagy in cardiac ischemia/reperfusion injury: a novel therapeutic strategy. J Cell Physiol. 2019;234(10):16768‐16778. [DOI] [PubMed] [Google Scholar]
- 41. Xu P, Das M, Reilly J, Davis RJ. JNK regulates FoxO‐dependent autophagy in neurons. Genes Dev. 2011;25(4):310‐322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lorin S, Pierron G, Ryan KM, Codogno P, Djavaheri‐Mergny M. Evidence for the interplay between JNK and p53‐DRAM signaling pathways in the regulation of autophagy. Autophagy. 2010;6(1):153‐154. [DOI] [PubMed] [Google Scholar]
- 43. Zhao Y‐S, An J‐R, Yang S, et al. Hydrogen and oxygen mixture to improve cardiac dysfunction and myocardial pathological changes induced by intermittent hypoxia in rats. Oxid Med Cell Longev. 2019;2019:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Guo W, Liu X, Li J, et al. Prdx1 alleviates cardiomyocyte apoptosis through ROS‐activated MAPK pathway during myocardial ischemia/reperfusion injury. Int J Biol Macromol. 2018;112:608‐615. [DOI] [PubMed] [Google Scholar]
- 45. Li M, Zhao H, Wu J, et al. Nobiletin protects against acute liver injury via targeting c‐Jun N‐Terminal Kinase (JNK)‐induced apoptosis of hepatocytes. J Agric Food Chem. 2020;68(27):7112‐7120. 10.1021/acs.jafc.0c01722 [DOI] [PubMed] [Google Scholar]
- 46. Shimizu S, Konishi A, Nishida Y, et al. Involvement of JNK in the regulation of autophagic cell death. Oncogene. 2010;29(14):2070‐2082. [DOI] [PubMed] [Google Scholar]
- 47. Wong CH, Iskandar KB, Yadav SK, Hirpara JL, Loh T, Pervaiz S. Simultaneous induction of non‐canonical autophagy and apoptosis in cancer cells by ROS‐dependent ERK and JNK activation. PLoS One. 2010;5(4):e9996. 10.1371/journal.pone.0009996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen M‐C, Lee N‐H, Hsu H‐H, et al. Thymoquinone induces caspase‐independent, autophagic cell death in CPT‐11‐resistant LoVo colon cancer via mitochondrial dysfunction and activation of JNK and p38. J Agric Food Chem. 2015;63(5):1540‐1546. 10.1021/jf5054063 [DOI] [PubMed] [Google Scholar]
- 49. Gonzalez‐Rodriguez A, Mayoral R, Agra N, et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014;5(4):e1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xiang X‐Y, Yang X‐C, Su J, et al. Inhibition of autophagic flux by ROS promotes apoptosis during DTT‐induced ER/oxidative stress in HeLa cells. Oncol Rep. 2016;35(6):3471‐3479. [DOI] [PubMed] [Google Scholar]
- 51. Ornatowski W, Lu Q, Yegambaram M, et al. Complex interplay between autophagy and oxidative stress in the development of pulmonary disease. Redox Biol. 2020;36:101679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ogata M, Hino S‐I, Saito A, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26(24):9220‐9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cheng Z, Zhang M, Hu J, et al. Mst1 knockout enhances cardiomyocyte autophagic flux to alleviate angiotensin II‐induced cardiac injury independent of angiotensin II receptors. J Mol Cell Cardiol. 2018;125:117‐128. [DOI] [PubMed] [Google Scholar]
- 54. Bozi LH, Takano AP, Campos JC, et al. Endoplasmic reticulum stress impairs cardiomyocyte contractility through JNK‐dependent upregulation of BNIP3. Int J Cardiol. 2018;272:194‐201. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Table S1
Data Availability Statement
All data generated and analyzed in the study are available from the corresponding author upon reasonable request.