

Abstract
Pulmonary fibrosis is a devastating, progressive disease and carries a prognosis worse than most cancers. Despite ongoing research, the mechanisms that underlie disease pathogenesis remain only partially understood. However, the self-perpetuating nature of pulmonary fibrosis has led several researchers to propose the existence of pathological signalling loops. According to this hypothesis, the normal wound-healing process becomes corrupted and results in the progressive accumulation of scar tissue in the lung. In addition, several negative regulators of pulmonary fibrosis are downregulated and, therefore, are no longer capable of inhibiting these feed-forward loops. The combination of pathological signalling loops and loss of a checks and balances system ultimately culminates in a process of unregulated scar formation. This review details specific signalling pathways demonstrated to play a role in the pathogenesis of pulmonary fibrosis. The evidence of detrimental signalling loops is elucidated with regard to epithelial cell injury, cellular senescence and the activation of developmental and ageing pathways. We demonstrate where these loops intersect each other, as well as common mediators that may drive these responses and how the loss of pro-resolving mediators may contribute to the propagation of disease. By focusing on the overlapping signalling mediators among the many pro-fibrotic pathways, it is our hope that the pulmonary fibrosis community will be better equipped to design future trials that incorporate the redundant nature of these pathways as we move towards finding a cure for this unrelenting disease.
Introduction
“He jests at scars that never felt a wound”
– Shakespeare
Acute and chronic lung diseases create a tremendous financial, personnel and resource burden on worldwide health systems. In this review we will focus on the pathophysiology of a chronic lung disease that is characterised by scarring of the lung tissue. Idiopathic pulmonary fibrosis (IPF) is a complex disease with only theorised potential contributory aetiologies (genetic susceptibility, environmental exposure and lifestyle habits). There is tremendous variability in the natural disease course and response to therapy. This unpredictability makes IPF difficult to diagnose, manage and study. As such, there remains an unmet need for new therapeutic targets. This review attempts to highlight the complex interplay of seemingly disparate signalling pathways to help readers identify potential new targets and consider novel approaches when designing future clinical trials.
A scar develops as part of a normal response to tissue injury. Tissue or organ fibrosis is pathological scarring that occurs when the wound-healing pathway is dysregulated, i.e. the appropriate healing response does not abate and thus becomes pathological. Wound healing involves the recruitment of fibroblasts that contribute to structural repair, homing of inflammatory cells and the release of chemokines and cytokines that induce extracellular matrix (ECM) production. When this process becomes pathological, it causes the accumulation of fibroblasts and myofibroblasts, destruction of normal tissue structure, the development of fibroblastic foci and loss of organ function. The abnormal architecture observed in pulmonary fibrosis results in restrictive lung physiology and impaired gas exchange. There are many known causes of pulmonary fibrosis, including environmental exposures, autoimmune diseases, radiation exposure, medications and respiratory infections. However, some forms of pulmonary fibrosis lack an identifiable aetiology, the most notable being IPF.
The US Food and Drug Administration (FDA) has approved only two drugs for the treatment of IPF: pirfenidone, whose precise mechanism of action is uncertain, but likely decreases production and activity of transforming growth factor-β1 (TGF-β1); and nintedanib, a triple tyrosine kinase receptor inhibitor (platelet derived growth factor (PDGF), fibroblast growth factor (FGF) and vascular endothelial growth factor (VEGF)). These medications slow the progression of the disease but do not stop or reverse the scarring and do not make patients feel less breathless [1, 2]. Currently, the only cure for IPF is lung transplantation, but this is only suitable for a small subset of patients, donated organs are a limited resource and it comes with significant potential comorbidities. Many other therapeutic targets have been studied but have failed to demonstrate clinical efficacy. We propose that the lack of efficacy may be related to the considerable redundancy in pro-fibrotic pathways involved in the complex pathogenesis of IPF.
Lung scarring develops and progresses through multiple mechanisms that involve the activation of pro-fibrotic pathways, which in turn promote their own ongoing activation [3–5]. This leads to persistent upregulation of mediators that drive fibrosis and a concurrent downregulation of negative mediators of fibrosis. Therefore, targeting any one single fibrotic pathway may be inadequate owing to the simultaneous activation of multiple pro-fibrotic, converging and cross-amplifying pathological pro-fibrotic loops. In this review we highlight the relationships between these complex wound-healing signals and explore how dysregulation of these pathways may result in pathophysiology as well as how they may be exploited for therapeutic benefit. We conceptualise these pathological loops as a parallel induction of multiple pro-fibrotic stimuli, coalescing in the synergistic and perpetual propagation of cyclical patterns of pro-fibrotic signalling. These convergent signals then work in concert to drive pathological fibrosis.
In addition, anti-fibrotic or homeostatic pathways are downregulated in pulmonary fibrosis, functionally limiting the natural “brake” on pro-fibrotic pathways. Although there are several examples of self-sustaining pro-fibrotic signalling loops, in this review we focus on 1) epithelial injury and senescence and 2) the (re)-activation of developmental pathways (figure 1). We also address how the downregulation of anti-fibrotic mediators contributes to fibrosis promotion.
FIGURE 1.
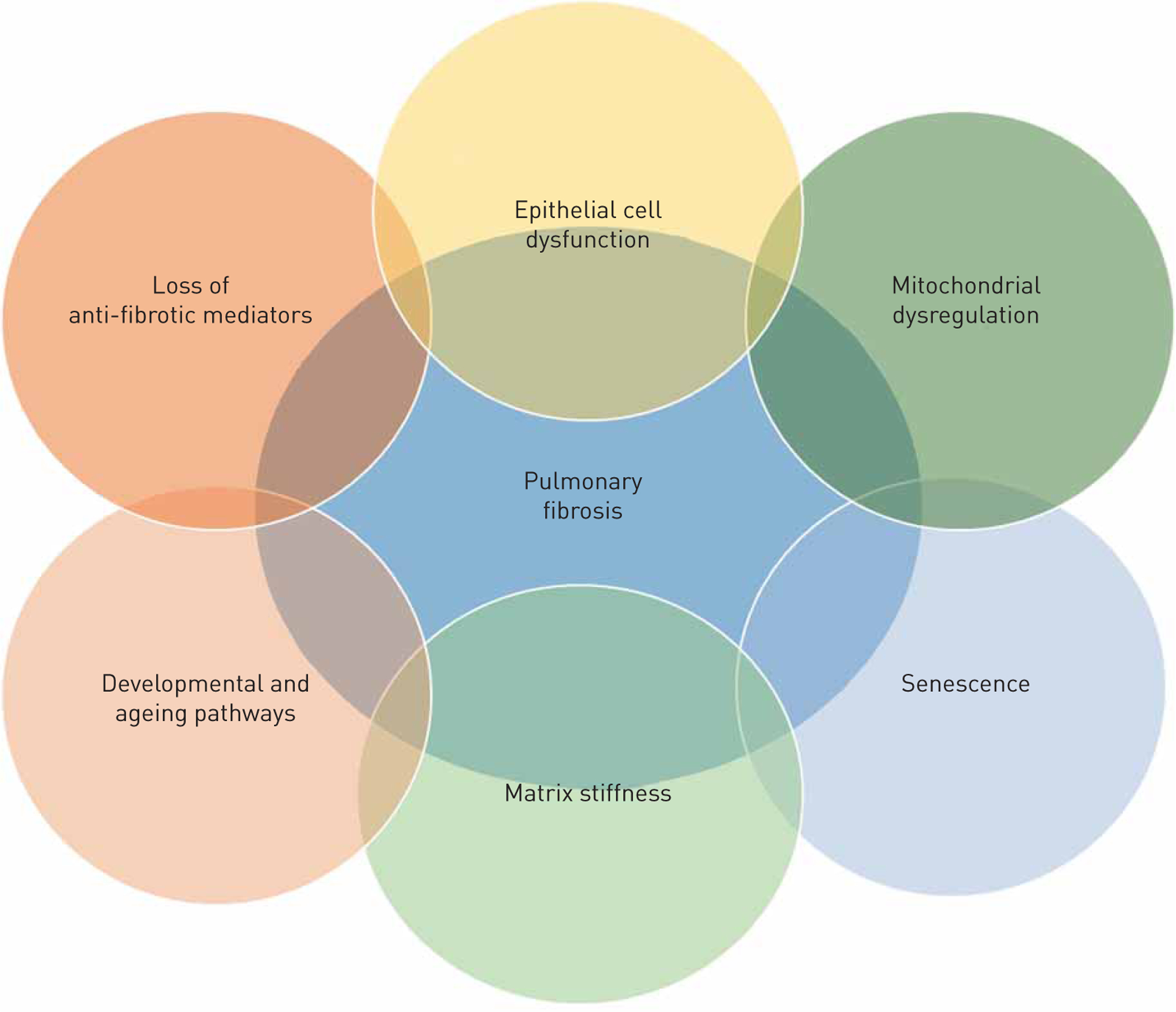
Overview of the interaction among various feed-forward loops involved in pulmonary fibrosis whereby all pathways to the development of the disease.
Epithelial cell damage, reactive oxygen species and senescence generate a pathological signalling loop
The best-supported hypothesis for the inciting event(s) in IPF is repeated injury to the alveolar epithelium [6, 7]. Sustained injury of the epithelium is hypothesised to produce an abnormal wound-healing response, resulting in the activation and differentiation of fibroblasts. This hypothesis is supported by the finding that in the alveolar epithelium of fibrotic lung tissue there are greater numbers of apoptotic type I and II cells, particularly around areas of increased myofibroblast activity [8, 9]. Interestingly, myofibroblasts have demonstrated resistance to apoptosis and senescence [8, 10]. This pattern of adjacent, phenotypically different cells exhibiting divergent behaviour highlights another aspect of the complex heterogeneity observed in IPF. Numerous in vivo studies have highlighted the importance of alveolar epithelial cell injury in the promotion of pulmonary fibrosis [11–14].
Many rodent models of pulmonary fibrosis involve the use of agents that damage the lung epithelium, resulting in epithelial cell death and subsequent development of fibrosis. These agents include bleomycin, diphtheria toxin, fluorescein isothiocyanate, silica and ionising radiation [15]. Alveolar damage has been shown to activate fibroblasts, which leads to additional epithelial injury, resulting in a self-propagating pro-fibrotic signalling loop (figure 2).
FIGURE 2.
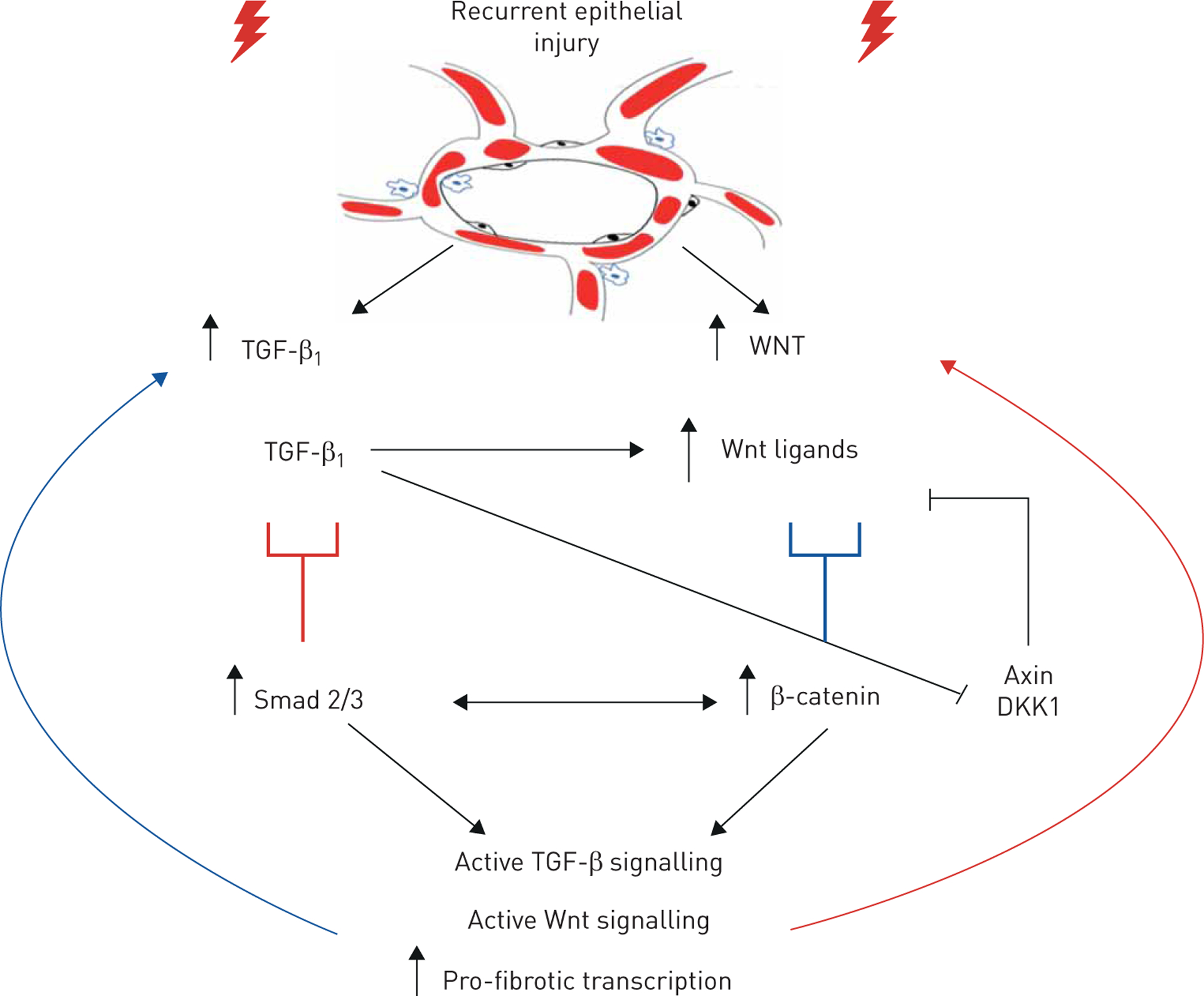
Recurrent epithelial injury leads to chronic activation of transforming growth factor-β1 (TGF-β1) and Wingless/Integrase-1 (Wnt) signalling cascades. The activation of TGF-β1 can suppress negative regulators of the Wnt pathway and ultimately activate Wnt signalling. Activation of Wnt signalling also propagates TGF-β1 signalling. This, coupled with chronic epithelial damage, further drives this cycle to result in pathological, unresolving wound healing. DKK1: Dickkopf-1
Although the specific cause of epithelial cell death remains unknown in IPF, there are genetic factors that may increase the risk of epithelial injury. The development of familial pulmonary fibrosis or sporadic IPF are associated with mutations in genes such as surfactant protein A1 (SFTPA1) [16], A2 (SFTPA2) [17] and C (SFTPC) [18, 19]; genes associated with telomere-related biology such as telomerase reverse transcriptase (TERT), telomerase RNA component (TERC) [20], regulator of telomere elongation helicase 1 (RTEL1), poly(A)-specific ribonuclease (PARN) [21] and dyskerin (DKC1) [22]; and polymorphisms in the Mucin 5B (MUC5B) [23–25] promoter. In fact, up to one third of patients with IPF have at least one common genetic variant [26].
The mechanisms by which an injured epithelium induces fibrosis remain incompletely understood, but it is hypothesised that in a healthy lung, normal alveolar epithelial cells help maintain homeostasis in neighbouring fibroblasts. However, when epithelial cells are injured, they release damage (or danger) associated molecular patterns (DAMPs) and reactive oxygen species (ROS). This disrupts the epithelial barrier through epithelial apoptosis and subsequent stimulation of local fibroblasts. For more specific information on the role of alveolar injury and the development of pulmonary fibrosis, we refer the readers to an excellent review article [27].
ROS and mitochondrial dysfunction
As organisms age there is an increase in ROS production. ROS has long been thought to play a role in pulmonary fibrosis. It is well established that damage to the epithelium results in increased ROS, which then affects neighbouring epithelial cells and fibroblasts. The capacity to regulate ROS production declines with age and this altered capacity may be one of the mechanisms through which senescent myofibroblasts and alveolar epithelial cells generate excess ROS [28, 29]. The largest endogenous source of ROS is the mitochondria, and the increased mitochondrial dysfunction that occurs with ageing is thought to be responsible for this increased production of ROS. In pulmonary fibrosis, there is increased ROS and mitochondrial dysfunction. For example, alveolar epithelial cells in patients with pulmonary fibrosis have mitochondria with morphological abnormalities and impaired enzymatic activity of the electron transport chain [30]. Furthermore, the oxidative stress generated from mitochondrial dysfunction can damage mitochondrial DNA in nearby lung epithelial cells, perpetuating a cascade of mitochondrial dysfunction and apoptosis, ultimately resulting in a pathological fibrotic response [31].
Mitochondria play a critical role in the development of lung scarring, as evidenced by the spontaneous formation of alveolar scarring and fibroblast proliferation in homozygous knockout mice of translocase on the outer mitochondrial membrane 5 (TOMM5) [32]. Increased mitochondrial ROS activates TGF-β1 and PDGF receptor (PDGFR) [33]; PDGFR expression is increased in IPF [34] and is one target of nintedanib [35]. In IPF fibroblasts, NADPH oxidase 4 (Nox-4), a marker of mitochondrial ROS, is elevated [36]. Genetic knockdown and small molecule inhibitors of Nox-4 in human lung fibroblasts inhibit TGF-β1-induced expression of myofibroblast markers and cell contractility [36, 37]. Furthermore, inhibition of Nox-4 reduces the expression of senescence markers and increases cellular susceptibility to apoptosis [28]. Both chemical inhibition of Nox-4/Nox-1 and Nox-4 RNA knockdown attenuate the development of lung fibrosis in rodents [37, 38]. TGF-β1 induces Nox-4 activity and expression, which leads to increased production of ROS [28, 36, 38]. The paracrine effect of ROS propagates an abnormal wound-healing response, which includes cell death and aberrant premature ageing [39]. Fibrosis and inflammation promote the subsequent generation of ROS, and TGF-β1 negatively regulates the antioxidant pathway [40].
Stress to mitochondria, including oxidant imbalance, causes the release of mitochondrial DNA, which acts as a DAMP recognised by the immune system [41]. Patients with IPF have increased levels of mtDNA in bronchoalveolar lavage (BAL) fluid and serum compared with healthy subjects [42]. Patients who respond to pirfenidone (response defined as a stable forced vital capacity for 1 year with treatment) experience decreased levels of circulating mtDNA in serum [42]. This suggests that treatment strategies that preserve proper mitochondrial function, reduce ROS production or decrease the levels of circulating mtDNA may serve as valuable therapeutics.
Multiple in vivo studies have shown that antioxidants, such as N-acetylcysteine (NAC), inhibit bleomycin-induced pulmonary fibrosis [43]. However, a clinical trial found that NAC did not prevent the progression of fibrosis in patients with mild-to-moderate IPF [44]. In fact, the trial was stopped prematurely because the “triple therapy” arm (prednisone, NAC and azathioprine) demonstrated a higher rate of discontinuation, adverse reactions and mortality. Yet, subsequent subgroup analyses demonstrated that a group of patients with a specific genotype of Toll interacting protein (TOLLIP), polymorphism TT, had improved or stable lung function after treatment with NAC, but those with the CC genotype had disease progression [45]. This provides supportive evidence that personalised treatment strategies could be applied clinically based on the genetic background of the patient. A clinical trial entitled PRECISIONS has recently received National Institutes of Health (NIH) funding to determine if NAC is a viable treatment for IPF patients with the TOLLIP TT genotype based on this data (NCT04300920). This is one of the first studies to provide personalised medicine within the context of IPF and represents an exciting step forwards in clinical trial development.
Senescence and senescence-associated secretory phenotype
Oxidative stress accelerates ageing and propagates signals that induce a senescent phenotype throughout the interstitium. Cellular senescence is a marker of normal ageing, but it is also more abundant in chronic lung disease (known as premature ageing). The process of senescence was initially thought to be protective from insults such as malignancy and infection because instead of inducing apoptosis or other detrimental paracrine signals, an injured cell could “hibernate” and not affect surrounding cells. However, accumulation of senescent cells has been shown to result in tissue dysfunction, age-related diseases and shortened lifespans [46]. Senescence has also been linked to telomere attrition, defective autophagy, failure of repair mechanisms, stem cell depletion and mitochondrial dysfunction [47]. Both epithelial cells and fibroblasts in the fibrotic lung experience senescence, characterised by “irreversible growth arrest” of cells. Senescence is phenotypically identified by an increase in cell size, metabolic activity, the production of inflammatory mediators and apoptosis-resistance. It is a component of several chronic lung diseases and is particularly associated with ageing. This process is implicated in the pathogenesis of lung scarring [29, 48, 49], because IPF is a disease of aged individuals.
Markers of senescence, including p16, p21 and senescence-associated β-galactosidase, are all increased in fibroblastic foci as well as in alveolar epithelial cells [28, 48]. Human lung epithelial cells treated with TGF-β1 develop a senescent phenotype. Senescent alveolar epithelial cells demonstrate a distorted morphology, have decreased barrier function and secrete an increased abundance of ROS. Importantly, senescent alveolar epithelial cells themselves are capable of inducing neighbouring fibroblasts to differentiate into myofibroblasts [48]. Similarly, fibroblasts isolated from active areas of lung fibrosis exhibit in vitro signs of enhanced cellular senescence, including abnormal cell morphology and resistance to apoptosis [29]. They also exhibit accelerated replicative senescence, are more resistant to ROS signalling and exhibit enhanced expression of myofibroblast markers [28, 29]. This creates a microenvironment in which epithelial injury, resulting in senescence, induces senescence in fibroblasts, which then develop resistance to apoptosis, continue to deposit matrix and release pro-senescent signals to perpetuate this cycle throughout the lung. Thus, senescent myofibroblasts and alveolar epithelial cells both contribute to a pro-fibrotic microenvironment, which communicates cellular stress signals and propagates a pathological wound-healing response in the lung (figure 3).
FIGURE 3.
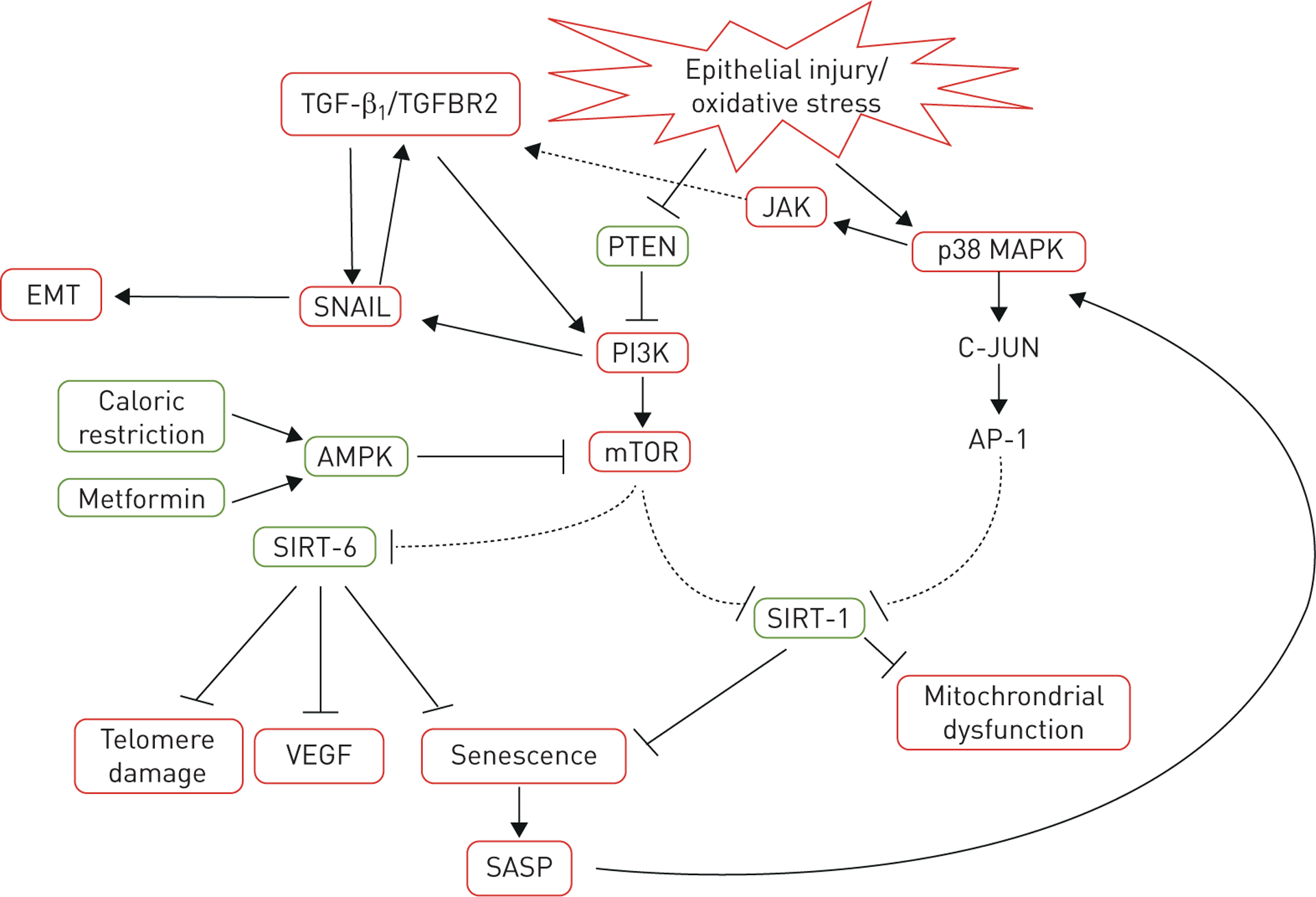
Signalling cartoon of the interaction among transforming growth factor-β1 (TGF-β1), epithelial–mesenchymal transition (EMT), epithelial injury, oxidative stress, telomere attrition and senescence. Red boxes depict pro-fibrotic pathways and green boxes indicate anti-fibrotic ones. TGFBR2: transforming growth factor-β receptor-2; PTEN: phosphatase and tensin homolog; JAK: Janus-activated kinase; p38 MAPK: p38 mitogen-activated kinase; PI3K: phosphatidylinositol-3 kinase; AMPK: 5′-AMP activated kinase; mTOR: mammalian target of rapamycin; AP-1: activator protein-1; SIRT: sirtuin protein family; VEGF: vascular endothelial growth factor; SASP: senescence-associated secretory phenotype.
Senescent cells are also metabolically active and release inflammatory signals into the microenvironment, which is termed the senescence-associated secretory phenotype (SASP) [50]. This signalling cascade is activated by p21CIP1, which then activates p38 mitogen-activated kinase (p38 MAPK) and Janus-activated kinase (JAK). Activation of JAK causes secretion of pro-inflammatory cytokines (e.g. VEGF, tumor necrosis factor-α and TGF-β1), chemokines (CXCL1 and CXCL8) and matrix metalloproteinases (MMP2 and 9). While the inflammatory consequences of SASP are established in chronic obstructive pulmonary disease and other chronic lung diseases, there is increasing evidence that supports the role of JAK activation in IPF, e.g. JAK inhibitors have been shown to attenuate bleomycin-induced pulmonary fibrosis [51]. In addition, JAK expression and activity was increased in lung tissue from patients with IPF compared to healthy controls [52]. As demonstrated in figure 3, the SASP is a downstream product of TGF-β, phosphatidylinositol-3 kinase (PI3K)/mammalian target of rapamycin (mTOR) and p38 MAPK signalling pathways. This phenotype then produces a cyclical, pro-fibrotic signalling loop that activates p38 MAPK and JAK, which induces more cellular senescence, VEGF signalling and telomere attrition, leading to more fibrosis.
Loss of negative regulation: PTEN and mTOR
The upregulation of tissue remodelling pathways causes pathological fibrosis, but the loss of anti-fibrotic signalling modalities, which serve as a homeostatic brake in the wound-healing process, also contributes to fibrosis. We present phosphatase and tensin homolog (PTEN) as a critical negative regulator of the wound-healing process and explore the consequences of the loss of such a brake on this process. We will discuss first how the loss of PTEN induces cyclical pathological signalling pathways and second how this loss causes upregulation and dysregulation of the mTOR pathway, which also drives fibrosis (figure 3).
Often examined in the context of cancer as a tumour suppressor protein, the presence of PTEN attenuates tissue fibrosis. PTEN downregulation occurs after epithelial injury (figure 3). Lung fibroblasts isolated from pulmonary fibrosis patients also display reduced protein levels and enzymatic activity of PTEN [53, 54]. Furthermore, fibroblastic foci in lung tissue from patients with pulmonary fibrosis have decreased expression of PTEN [54, 55]. Chemical inhibition or genetic knockout of PTEN in fibroblasts is necessary and sufficient to drive myofibroblast differentiation [54, 55], which supports the idea that a loss of PTEN is pro-fibrotic. In vivo, bronchoalveolar epithelial- or myeloid-specific deletion of PTEN results in more histological features of pulmonary fibrosis and collagen content in the lung in response to bleomycin [56, 57]. These deleterious effects are mitigated by restoration of PTEN. By contrast, overexpression of PTEN inhibits the development of a TGF-β1-induced fibrotic phenotype in fibroblasts [54, 55]. This underscores the importance of homeostatic PTEN signalling in the wound-healing process.
PTEN serves as a critical signalling node, limiting several pro-fibrotic pathways. At baseline levels, PTEN inhibits the phosphorylation of focal adhesion kinase (FAK), which plays a role in fibrosis through the regulation of cell migration and α-smooth muscle actin expression in fibroblasts [55]. The loss of PTEN thus allows for increased FAK phosphorylation and myofibroblast differentiation. In addition, loss of PTEN directly increases the production of TGF-β1, which then promotes the further downregulation of PTEN via transcriptional inhibition [58]. This cyclical signalling loop thus amplifies pathological signalling associated with fibrosis.
In the epithelium, PTEN expression is reduced in response to injury, including in models of fibrosis. In rodent models utilising bleomycin, there is observed PTEN depletion in epithelial cells [59]. Immunohistochemistry of lung tissue isolated from patients with IPF shows decreased PTEN expression in the alveolar epithelium as well as increased markers of senescence, p21 and β-gal [60]. In vitro, the loss of PTEN in epithelial cells induces senescence [59] and overexpression of PTEN reverses this effect [60]. There are multiple mechanisms associated with PTEN-mediated senescence in epithelial cells, including activation of NF-κB, phosphorylation of Akt and regulation of mitophagy [30, 59, 60]. This evidence suggests that PTEN regulates the response to injury of the epithelium as well as the adoption of a senescent phenotype.
An emerging area of research is interrogating how the loss of PTEN leads to the increase in other pro-fibrotic pathways such as mTOR, a developmental pathway that is dysregulated in IPF [61]. mTOR regulates a variety of essential events in embryogenesis and fetal development, from the fertilisation of oocytes [62] to the formation of complex structures in soft organ development [63]. In the lung, mTOR regulates the growth and development of branching airways [64] by coordinating expression of hypoxia-inducible factor 1α (HIF-1α) and VEGF to facilitate angiogenesis and nutrient exchange in distal regions of the lung [65]. In developed organs, mTOR regulates cellular metabolism, differentiation, apoptosis and senescence [63, 66, 67].
The reactivation of the mTOR pathway in fibrosis contributes to fibroblast proliferation, encourages cellular senescence, decreases sensitivity of fibroblasts to pro-apoptotic signals and leads to dysregulated autophagy. mTOR protein expression and that of its downstream effector p-S6 are both increased in fibroblastic foci [68, 69]. Furthermore, TGF-β1 activates mTOR signalling in human lung fibroblasts in vitro [68, 70], which in turn promotes the activation of Akt and its downstream effects, including an increase in cell proliferation, resistance to apoptosis and fibroblast migration [70]. Endogenous inhibition of mTOR is accomplished by 5′-AMP activated kinase (AMPK). This kinase is activated by low intracellular ATP concentrations, caloric restriction and metformin. Interestingly, metformin inhibits and reverses bleomycin-induced pulmonary fibrosis in mice [71]. Pharmacological inhibition of mTOR signalling by rapamycin or MLN0128 attenuates myofibroblast differentiation in vitro and bleomycin- and radiation-induced lung fibrosis in vivo [70, 72]. However, inhibition of mTOR may also increase expression of other pro-fibrotic cytokines such as connective tissue growth factor (CTGF), raising concern about the clinical utility of inhibiting mTOR [73, 74].
Mechanistically, PTEN and mTOR are connected via disinhibition of PI3K. The loss of PTEN leads to the sustained activation of PI3K and/or the FAK/Src kinase [55, 57]. Activated PI3K also generates phosphatidylinositol-3,4,5-triphosphate, which promotes the phosphorylation of Akt. mTOR, specifically mTOR complex 2, stabilises the catalytic site of Akt, which allows for maximal activation of Akt [75]. Activation of Akt induces cellular proliferation, increased glycolytic metabolism and apoptotic resistance [70, 76], all of which support pathological lung scarring. Once activated, Akt causes the activation of mTOR signalling via multiple substrates [77]. This causes a cyclical activation of Akt and mTOR, which occurs because of PTEN downregulation and subsequent PI3K activation.
Recent exciting work by the Chambers laboratory [78] and others has explored how dual inhibition of PI3K and mTOR may be effective in attenuating lung fibrosis. GSK2126458 is a dual PI3K/mTOR inhibitor originally developed for the treatment of cancer [79]. In vitro, treatment with GSK2126458 inhibits TGF-β1-induced collagen secretion in myofibroblasts. In ex vivo lung tissue from patients with IPF, GSK2126458 also attenuated collagen formation, as assessed by decreased total procollagen type 1 N-terminal propeptide (P1NP) levels and Akt phosphorylation [78]. Dual-targeting therapies such as these pave the way for more in-depth studies of how multiple, interrelated pathways drive fibrosis, and exemplify why single molecule targeting may not be efficacious in clinical practice.
Activation of mTOR signalling has also been linked with a decrease in autophagic activity. Autophagy is the process of recycling damaged organelles and cellular components, resulting in the homeostatic turnover of intracellular bodies. Markers of autophagy, including p62 and beclin 1, are decreased in the lung tissues of patients with pulmonary fibrosis, notably in fibroblasts and within active areas of fibrosis. Impaired autophagy likely promotes an ageing phenotype in epithelial cells via upregulation of senescence and mitochondrial dysfunction [80]. This increase in mTOR is correlated with increased mitochondrial biogenesis and increased ROS production, both of which can also encourage the senescent phenotype and may contribute to epithelial cell dysfunction or exhaustion in IPF.
Although mTOR may play a theoretical role in the development of pulmonary fibrosis, clinical trials studying mTOR inhibitors have been disappointing. A clinical trial utilising everolimus, a derivative of rapamycin, worsened fibrosis [81]. The reason for this outcome is not clear but may have been related to the high dose of the drug causing unanticipated detrimental off-target effects, or the fact that mTOR inhibition may increase some pro-fibrotic cytokines while inhibiting others [81]. This outcome underscores yet again the complexity of the pathogenesis of IPF and the need for a more complete understanding of the ways in which multiple intersecting pro-fibrotic pathways influence one another. Blocking a single, aberrant developmental signalling pathway may not effectively treat fibrosis owing to the overwhelming activation of various competing fibrosis-inducing signalling pathways. However, the introduction of dual-targeting therapeutics, such as GSK2126458 targeting both PI3K and mTOR, show exciting efficacy and promise in targeting signalling nodes to target multiple pro-fibrotic pathways.
Fibrotic pathogenic cascades from the activation of developmental and ageing pathways
It is estimated that 20% of genes dysregulated in IPF are related to early developmental pathways [82]. This includes signalling molecules related to cellular growth, migration, morphogenesis and differentiation, such as Wingless/Integrase-1 (Wnt)/β-catenin, bone morphogenetic protein (BMP), TGF-β1 and mTOR. Age-related DNA alterations of the lung also have demonstrated roles in the pathogenesis of IPF. These include telomere shortening, telomerase-associated mutations, mitochondrial dysfunction and generation of ROS and cellular senescence [20, 28, 67, 83].
When considering pathways associated with development, it is important to acknowledge that the timing of activation is critical to the outcome in that cell type or tissue. The research community is only beginning to understand the importance of temporal regulation of pro-fibrotic cytokines in the normal wound response of the lung. A recent single cell sequencing study by RIEMONDY et al. [84] demonstrated the importance of TGF-β1 temporal activation and subsequent downregulation in the epithelium following lipopolysaccharide injury. This highlights the need for similar research to understand the processes of resolution, thus allowing the development of novel therapeutics to attenuate fibrosis.
Wnt and TGF-β1 control critical developmental signalling pathways that act cooperatively to create a self-perpetuating, cyclical pathological signalling loop within the context of pulmonary fibrosis (reviewed in more detail in [85, 86], respectively). These pathways are activated by repeated and/or unresolved epithelial injury, resulting in the sustained activation of pro-fibrotic signals, which are then propagated and amplified throughout the mesenchyme, resulting in persistent pathological fibrosis. These master regulators of fibrosis cross-activate, or actively suppress the negative regulators, of other tissue remodelling pathways. This exacerbates the tissue remodelling response by simultaneously pressing the accelerator and removing the brakes on the wound-healing process, resulting in pathological fibrosis.
Wnt and fibrosis
Wnt/β-catenin signalling plays an important role in the developing lung through the regulation of cell fate, proliferation and determination of cellular polarity [87]. Early in development, Wnt signalling regulates airway formation and epithelial branching [88]. In the postnatal lung, Wnt is a key mediator of homeostasis through its participation in alveologenesis and progenitor cell regulation, including the maintenance and differentiation of lung stem cells [89, 90] as well as the response to injury [91, 92]. In the adult, increased Wnt activity has been demonstrated in several types of disease, including chronic lung diseases [93, 94], and it may serve to regulate the regenerative potential of the lung [95, 96]. However, aberrant reactivation of the Wnt pathway later in life could lead to exhaustion of stem and progenitor cell populations, leading to a premature ageing phenotype and reducing the capacity of the lung to respond appropriately to injurious stimuli [94, 97]. This highlights the duality of signalling proteins like TGF-β1 and Wnt; some activity is necessary for homeostasis, but prolonged activity or improperly timed signalling leads to pathology.
The Wnt family consists of 19 secreted glycoproteins that bind to transmembrane receptors, including lipoprotein receptor-related proteins (LRPs) and Frizzled 1–10 (FZD 1–10). Canonical Wnt signalling is initiated when Wnt binds to FZD and a co-receptor (LRP5/6), which allows nuclear translocation of β-catenin [98]. Once in the nucleus, β-catenin activates several transcription factors, including the main transcriptional effector, the T-cell factor/lymphoid enhancer binding factor (TCF/LEF) family. When Wnt signalling is inactive, β-catenin is phosphorylated and subsequently degraded by the ubiquitin–proteasome pathway. Detection of β-catenin protein is frequently used as an indication of active, canonical Wnt signalling. Initiation of this transcriptional programme results in the broad activation of fibrosis-associated pathways, including ECM deposition, cell cycle progression, growth factor secretion and cellular regeneration [99]. Wnt signalling also occurs independently of β-catenin, known as the non-canonical Wnt pathway. In this framework, a Wnt ligand binds an FZD receptor, which leads to the potential activation of several intracellular messengers, including protein kinase A (PKA), calcium/calmodulin, paxillin and JNK [100].
Researchers have identified increased canonical and non-canonical Wnt signalling in lung tissue of patients with IPF, indicated by the presence of Wnt ligands, receptors and/or nuclear β-catenin. Specific examples of Wnt members that are increased in IPF include the ligands Wnt1, Wnt3a and Wnt7b; the receptors Fzd1–4; and β-catenin and its downstream targets, including frizzled-related protein (FRZB), cyclin D1 and WNT1-inducible-signalling pathway protein-1 (WISP-1) [88, 101, 102]. Furthermore, Wnt/β-catenin signalling is elevated in mouse alveolar epithelial type II cells after bleomycin challenge (figure 2) [49, 103–106].
Molecules upregulated by Wnt/β-catenin signalling, such as WISP-1, fibronectin and plasminogen activator inhibitor-1 (PAI-1), induce both epithelial and myofibroblast proliferation and differentiation, resulting in the production of pro-fibrotic mediators [89, 107]. In patients with pulmonary fibrosis, the increase in Wnt ligands and downstream mediators is observed in circulating peripheral blood mononuclear cells [108] as well as in the fibrotic lung in bronchiolar lesions, damaged alveoli, fibroblastic foci [88] and type II airway epithelial cells [109]. Type II airway epithelial cells serve as one of the major progenitor cells in the lung, and prolonged Wnt expression in these cells may deplete the regenerative capacity of the lung and impair the ability of the lung to respond to injury.
The canonical Wnt/β-catenin pathway is well studied in pulmonary fibrosis, with most research focusing on the activation of β-catenin. However, it is important to note β-catenin can be activated via Wnt-independent mechanisms [110]. Within the context of pulmonary fibrosis, the upregulation of Wnt ligands, receptors and downstream mediators [109, 111, 112] indicates that the upregulation in β-catenin is at least partially dependent upon Wnt signalling. β-catenin is known to induce epithelial–mesenchymal transition and myofibroblast differentiation, increase fibroblast migration [113–115] and potentially prevent mesenchymal apoptosis [116]. In the bleomycin model of pulmonary fibrosis, β-catenin induces macrophage differentiation, which antagonises the resolution of the wound-healing process [117, 118]. In rodent models of fibrosis, β-catenin inhibitors suppress myofibroblast differentiation and induce epithelial differentiation, resulting in barrier preservation and a reduction in collagen deposition [119–122].
Dickkopf 1–4 proteins (DKK1–4) are an endogenous family of negative regulators of active Wnt signalling. The best studied is DKK1, which is thought to antagonise Wnt signalling by inducing receptor internalisation of the Wnt co-receptors LRP5/6 or by preventing the assembly of a receptor ternary complex involving LRP5/6 and FRZ receptors [123]. The DKK proteins remain relatively understudied in pulmonary fibrosis, although DKK1 is downregulated in whole lung samples from patients with pulmonary fibrosis as well as in skin from patients with systemic sclerosis [123, 124]. However, other researchers have demonstrated DKK1 is increased in whole lung tissue, with the most abundant DKK1 staining present in basal bronchial epithelial cells. In vitro, DKK1 induced dose-dependent epithelial proliferation, indicating a regulatory role for the DKK proteins in epithelial maintenance and wound repair. The concentration of DKK protein was also increased in the BAL fluid from patients with pulmonary fibrosis compared with patients without lung disease. The upregulation of DKK proteins may be a protective mechanism and could be a response to wound healing that becomes overwhelmed by other pro-fibrotic signals. Additionally, DKK proteins could have different roles in the epithelium versus the mesenchyme, which may explain these conflicting results.
TGF-β1 and fibrosis
TGF-β1 serves an essential role in development and is required for embryonic stem cell fate commitment, proliferation and tumour suppression [125]. In lung development specifically, TGF-β1 is integral to lung branching and alveolar formation [126]. In the adult lung, low levels of TGF-β1 are required to maintain homeostasis, with expression being localised to type I and II epithelial cells, mesenchymal cells, macrophages and endothelial cells [127]. TGF-β1 is one of the classic cytokines responsible for the initiation of the inflammatory response and wound healing [92]. However, when present in excess, it is critical to the development of pulmonary fibrosis. The activation of TGF-β1 in the extracellular space induces multiple downstream pro-fibrotic mediators. It is also unique in that it is able to induce its own production and subsequent activation. The self-induced activation of TGF-β1 likely plays an important physiological role in normal wound healing; however, perpetual activation may sustain a pathological environment that promotes fibrosis.
TGF-β1 signals by binding to the TGF-β receptors 1 and 2 (TGFβR1 and TGFβR2), which causes TGFβR1 to phosphorylate TGFβR2. In the canonical TGF-β1 pathway, TGFβR1 then phosphorylates Smads 2 and 3. This allows for the association of Smad4 with Smads 2/3 and this complex translocates to the nucleus to induce downstream gene transcription. There are other Smads, such as Smad7, that inhibit the association of Smad 2 and 3, as well as having several other mechanisms of TGF-β1 antagonism [126].
TGF-β1 is one of many cytokines that is significantly elevated in lung tissue and BAL fluid of patients with IPF [128–131]. Type I and type II alveolar epithelial cells, along with fibroblasts, monocytes and macrophages [128, 132, 133], produce and secrete TGF-β1 into the extracellular space, where it is maintained in an inactive form by a latent TGF-β1 complex. In vitro, TGF-β1 induces myofibroblast differentiation, activating production of ECM proteins [134]. TGF-β1 is unique in that active TGF can induce additional TGF-β1 activity [135]. In vivo, overexpression of active TGF-β1 in rodent lung tissue induces progressive and irreversible fibrosis [136, 137]. Conversely, inhibition of TGF-β1 signalling by neutralising antibodies, receptor inhibitors or via genetic silencing attenuates the development of bleomycin-induced pulmonary fibrosis in vivo [138–143]. TGF-β1 is therefore thought to be a key regulator of pulmonary fibrosis (figure 2).
Wnt and TGF-β1 cross-propagation
In development, TGF-β1 and Wnt pathways share many physiological roles, including organ formation, organisation and cellular differentiation [144]. During development these processes are tightly regulated, both spatially and temporally. After development, both pathways are then downregulated and remain relatively pedestrian in adult organisms. However, both Wnt and TGF-β1 signalling are induced in response to injury [92] to encourage tissue remodelling. In pathological fibrosis, where wound healing has become dysregulated, both Wnt and TGF-β1 signalling pathways are perpetually activated. Unfortunately, the mechanisms that govern the homeostatic activation and eventual downregulation of Wnt and TGF-β1 during normal wound healing are not well understood.
TGF-β1 is known to directly induce the expression of several Wnt ligands in bone marrow stromal cells, including Wnt2, Wnt4, Wnt5A, Wnt7A, Wnt10A and the co-receptor LRP5 [145]. In primary human lung fibroblasts, TGF-β1 induces Wnt5A and activates β-catenin [146]. Dermal fibroblasts isolated from patients with systemic sclerosis display hyper-responsiveness to Wnt ligands compared with fibroblasts isolated from patients without disease. Utilising a transgenic mouse with constitutive TGF-β1 activity in fibroblasts, researchers demonstrated reduced expression of Axin-2, which is a key negative regulator of the Wnt pathway. Reducing the levels of Axin-2 inhibits formation of the β-catenin destruction complex, allowing β-catenin to translocate into the nucleus and activate Wnt transcriptional programmes when it would otherwise be degraded [147]. This suggests that chronically high levels of TGF-β1 may prime fibroblasts to be more sensitive to Wnt ligands and to have higher basal Wnt activation, even in the absence of a ligand. This would drive activation of both pathways, contributing to a pathological, pro-fibrotic signalling loop on the receptor/ligand level. At the ligand level, TGF-β1 is able to increase Wnt ligand secretion directly and prolonged exposure to TGF-β1 sensitises fibroblasts to Wnt ligands, increasing basal Wnt activation even in the absence of stimulation (figure 2).
TGF-β1 is able to increase Wnt pathway activation while also inhibiting the negative regulators of the Wnt pathway. In a study of myocardial fibrosis by Blyszczuk et al. [148], activation of TGF-β1 led to abundant Wnt ligand secretion, which is required for both the initiation and propagation of pathological fibrosis in the heart. However, activation of the Wnt pathway while the TGF-β1 pathway was blocked was not sufficient to induce myofibroblast differentiation [148]. This suggests that some of the pro-fibrotic effects of TGF-β1 signalling may be dependent on Wnt, demonstrating the crosstalk and potential for synergism of these pathways in fibrotic disease. The pleiotropic actions of pro-fibrotic cytokines such as TGF-β1 support the concept that multiple pathological signalling loops are not only initiated in pulmonary fibrosis but also capable of perpetuating their own induction and activity across signalling cascades.
TGF-β1 suppresses DKK1 [124, 146], which is an important negative regulator of the Wnt pathway. Suppressing this negative regulator enhances Wnt/β-catenin signalling by a loss of brakes on this system. In dermal fibroblasts in vitro and an in vivo model of skin fibrosis using a TGF-β1 adenovirus, there was a TGF-β1-mediated decrease in DKK1, showing that with increasing TGF-β1 there is decreasing DKK1 protein. This imbalance increased Wnt signalling, as measured by nuclear translocation of β-catenin and activation of TCF/LEF elements [124]. Interestingly, active DKK1 attenuates active TGF-β1 secretion, indicating that inhibition of Wnt signalling also suppresses TGF-β1 signalling [149]. The redundant regulation of both pathways suggests that a loss of DKK1 may further allow both pathways to remain active without proper negative regulation (figure 2).
Shared downstream mediators of TGF-β1 and Wnt, such as Smads 2/3 and β-catenin, bind to common transcriptional elements, including Smad binding elements (SBEs) and TCF/LEF regions, resulting in dual activation of TGF-β1 and Wnt signalling at the transcriptional level. Promoter regions of many Wnt- and/or TGF-β1-responsive genes contain both SBEs and LEFs, allowing for co-transcriptional control of both signalling pathways and amplification of target genes, such as αSMA [150]. Once these genes are induced, they further signal to perpetuate Wnt/TGF-β1 signalling to cause a positive, cyclical signalling loop of fibrosis (figure 2). In addition, there is convergence of the Wnt and TGF-β1 pathways at the promoter level, where signal transducers of both pathways, such as Smads and β-catenin, form complexes that bind promoter elements to induce transcriptional activation [151, 152]. Thus, the activation of Wnt can promote sustained canonical TGF-β1 signalling and vice versa. Active TGF-β1 then suppresses the expression of negative regulators of the Wnt/β-catenin pathway, allowing for further activation of Wnt.
As an example of the delicate balance that is achieved with these interacting pathways, TGF-β1 signalling also leads to Wnt suppression. Upon TGF-β1 induction, the mediator Smad3 can form a complex with Axin and glycogen synthase kinase-3β (GSK-3β), which then induces phosphorylation of the complex, ultimately resulting in propagation of Wnt signalling without negative regulation [153]. Smad3 can also support canonical Wnt signalling by shuttling β-catenin into the nucleus [154, 155]. It is thought that, in the context of pulmonary fibrosis, this balance ultimately favours perpetuation of both signalling pathways (figure 2). However, there is likely to be some degree of temporal, spatial and cell-specific nuance to the influence that Wnt and TGF-β1 have on one another.
There are several pro-fibrotic signalling pathways that centre around developmentally activated gene programmes, specifically Wnt activation. This indicates Wnt may be a critical control point in the development of a sustained pathological fibrotic response. The pathways and mechanisms associated with aberrant Wnt activation within the context of fibrosis include the promotion of canonical TGF-β1 signalling. The convergence of the TGF-β1 and Wnt pathways is well established and essential for proper development early in life. However, reactivation of the Wnt pathway later in life, in combination with aberrant TGF-β1 signalling, is associated with several diseases that include cancer and fibrosis. We propose that recurrent epithelial injury chronically activates both TGF-β1 and Wnt pathways, leading to cross-perpetuation of both pathways because each pathway causes a loss of negative regulators in the other (figure 2).
Future directions
Several exciting frontiers of research provide hope to patients, caregivers, physicians and scientists. For example, the recently funded PRECISIONS trial demonstrates the power of collaboration between philanthropic organisations (Three Lakes Partners), a national registry and biorepository (the Pulmonary Fibrosis Foundation) and physician-scientists through the National Heart, Blood, and Lung Institute at the NIH. Precision medicine, as defined by the US National Library of Medicine, is “an emerging approach for disease treatment and prevention that takes into account individual variability in genes, environment, and lifestyle for each person”. The PRECISIONS study, as the first precision medicine trial in IPF, will establish the groundwork for future trials in the field. We also argue that innovative trial designs and data collection will lead to more ethically responsible studies with fewer participants and a more efficient arrival at clinical outcomes. For example, adaptive trials are a novel method to study the efficacy of drug(s) and allow flexibility down to the individual participant level. Because this is a relatively new approach, collaboration among biostatisticians, physicians and scientists is needed to overcome the challenging statistical methods required to assess outcomes in an adaptive trial. Harnessing the power of other non-traditional data collection methods, such as daily in-home spirometry [156], provides a rich data set and reduces the number of participants required to reach pre-determined outcomes. Clinical trials can also evolve by incorporating clinically measured (i.e. forced vital capacity) and patient-centred (e.g. efficiency and effectiveness) outcomes. From a research perspective, there are several potential therapeutic targets on the horizon, e.g. JAK 2 inhibitors and pan-FGF inhibitors. The use of big data combined with biorepositories will promote a more comprehensive understanding of individual variability and genetic differences to identify new therapeutic targets and, potentially, to discover clinically relevant biomarkers. With the explosion in publically available RNA sequencing data, basic scientists should confirm these findings at the protein and cell signalling/activity levels. Finally, using novel approaches such as organoids and co-cell culture (i.e. epithelial and fibroblasts) and creating new non-inflammatory small animal models of pulmonary fibrosis will further our understanding of the mechanisms involved in the natural progression of disease. We need to specifically determine what is involved in the normal damping down of the healing process and harness this information to reverse fibrosis. There is evidence to suggest that IPF is a subclinical disease before patients develop symptoms [157]. Therefore, dissecting the differences between wound-healing initiation, propagation and termination would add invaluable insight into pathological signalling that could be exploited to therapeutic advantage.
Conclusion
“The knot loops in upon itself; I cannot find the end”
– J.M. Coetzee
This review outlines several cyclical pathological signalling pathways of pulmonary fibrosis that stimulate the development of scar tissue, the pathological consequences of repeated and/or self-propagated epithelial damage and premature cellular ageing phenomena. In addition to the increased activation of pro-fibrotic signalling cascades in pulmonary fibrosis, there is a loss of the inhibitors of pro-fibrotic signalling.
Impaired regulators of fibrotic pathways lead to uncontrolled ECM production and scarring. Additionally, pro-fibrotic cyclical signalling pathways remain unabated in the absence of anti-fibrotic mediators, enhancing the progression of lung fibrosis.
Fibrotic signalling loops involving the loss of negative regulators of pro-fibrotic cascades contribute to the development of pulmonary fibrosis and make the discovery of single target therapeutics for lung scarring challenging. At present there are few, if any, truly effective treatments for pulmonary fibrosis. Probably owing to the significant redundancy of these pathways, many clinical trials of single-targeted therapies have failed to improve patient outcomes. Lung fibrosis is difficult to treat owing to the activation of multiple fibrotic pathways and the loss of several negative regulators of fibrogenesis that convert a normal physiological response into an irreversible pathological one.
The combination of increased pro-fibrotic and loss of anti-fibrotic proteins induces an unchecked self-fulfilling prophecy that enhances pathological lung scarring. Therefore, we propose employing the use of multi-modal therapies, in combination with precision medicine techniques and novel methodology, in future clinical trials to capture the complexity of simultaneous gain and loss of function changes that occur in IPF. Strengthening the partnerships between academia, the pharmaceutical industry, philanthropic organisations and patient advocacy organisations within the interstitial lung disease community will serve as a model as we move into the era of precision medicine.
Support statement:
This work was supported by Boehringer Ingelheim and the National Heart, Lung, and Blood Institute (grants R01HL12090804, R01HL12700104 and T32HL066988).
Footnotes
Conflict of interest: A.R. Rackow has nothing to disclose. D.J. Nagel reports grants from National Heart, Lung, and Blood Institution (NIH T32 training grant), during the conduct of the study. C. McCarthy has nothing to disclose.J. Judge has nothing to disclose. S. Lacy is employed by the US Army; the views expressed herein are those of the author and do not reflect the official policy or position of the Department of the Army, Department of Defense or the US Government. M.A.T. Freeberg has nothing to disclose. T.H. Thatcher reports grants from NIH, during the conduct of the study. R.M. Kottmann reports grants from NIH, during the conduct of the study; and has a patent for LDH inhibitors as treatment for fibrosis and fibrotic-related disorders issued. P.J. Sime reports grants from NIH, during the conduct of the study; grants from NIH, grants and personal fees for consultancy from UCB, personal fees for consultancy from Boehringer Ingelheim, personal fees from GSK, personal fees for data monitoring committee work from Intermune/Roche, personal fees for advisory board work from Prometic and Galecto, and funds for research from Guy Solimano and Greg Chandler Fund, outside the submitted work; and has a patent for methods of diagnosing and treating fibrosis issued, a patent for LDH inhibitors as treatment for fibrosis and fibrotic-related disorders issued, and a patent for method and apparatus to diagnose metastatic and progressive potential of cancer, fibrosis and other diseases pending.
References
- 1.Nathan SD, Albera C, Bradford WZ, et al. Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir Med 2017; 5: 33–41. [DOI] [PubMed] [Google Scholar]
- 2.Richeldi L, Cottin V, du Bois RM, et al. Nintedanib in patients with idiopathic pulmonary fibrosis: combined evidence from the TOMORROW and INPULSIS® trials. Respir Med 2016; 113: 74–79. [DOI] [PubMed] [Google Scholar]
- 3.Judge JL, Nagel DJ, Owens KM, et al. Prevention and treatment of bleomycin-induced pulmonary fibrosis with the lactate dehydrogenase inhibitor gossypol. PLoS One 2018; 13: e0197936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu F, Lagares D, Choi KM, et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol 2015; 308: L344–L357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nguyen XX, Muhammad L, Nietert PJ, et al. IGFBP-5 promotes fibrosis via increasing its own expression and that of other pro-fibrotic mediators. Front Endocrinol (Lausanne) 2018; 9: 601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Katzenstein AL, Myers JL. Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am J Respir Crit Care Med 1998; 157: 1301–1315. [DOI] [PubMed] [Google Scholar]
- 7.Qunn L, Takemura T, Ikushima S, et al. Hyperplastic epithelial foci in honeycomb lesions in idiopathic pulmonary fibrosis. Virchows Arch 2002; 441: 271–278. [DOI] [PubMed] [Google Scholar]
- 8.Uhal BD, Joshi I, Hughes WF, et al. Alveolar epithelial cell death adjacent to underlying myofibroblasts in advanced fibrotic human lung. Am J Physiol 1998; 275: L1192–L1199. [DOI] [PubMed] [Google Scholar]
- 9.Plataki M, Koutsopoulos AV, Darivianaki K, et al. Expression of apoptotic and antiapoptotic markers in epithelial cells in idiopathic pulmonary fibrosis. Chest 2005; 127: 266–274. [DOI] [PubMed] [Google Scholar]
- 10.Thannickal VJ, Horowitz JC. Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc Am Thorac Soc 2006; 3: 350–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Palmer KC, Snider GL, Hayes JA. An association between alveolar cell proliferation and interstitial fibrosis following acute lung injury. Chest 1976; 69: 2 Suppl., 307–309. [DOI] [PubMed] [Google Scholar]
- 12.Sisson TH, Mendez M, Choi K, et al. Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis. Am J Respir Crit Care Med 2010; 181: 254–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nureki SI, Tomer Y, Venosa A, et al. Expression of mutant Sftpc in murine alveolar epithelia drives spontaneous lung fibrosis. J Clin Invest 2018; 128: 4008–4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parimon T, Yao C, Stripp BR, et al. Alveolar epithelial type II cells as drivers of lung fibrosis in idiopathic pulmonary fibrosis. Int J Mol Sci 2020; 21: 2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moore B, Lawson WE, Oury TD, et al. Animal models of fibrotic lung disease. Am J Respir Cell Mol Biol 2013; 49: 167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nathan N, Giraud V, Picard C, et al. Germline SFTPA1 mutation in familial idiopathic interstitial pneumonia and lung cancer. Hum Mol Genet 2016; 25: 1457–1467. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, Kuan PJ, Xing C, et al. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am J Hum Genet 2009; 84: 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwartz DA. Idiopathic pulmonary fibrosis is a complex genetic disorder. Trans Am Clin Climatol Assoc 2016; 127: 34–45. [PMC free article] [PubMed] [Google Scholar]
- 19.Thomas AQ, Lane K, Phillips J 3rd, et al. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med 2002; 165: 1322–1328. [DOI] [PubMed] [Google Scholar]
- 20.Armanios MY, Chen JJ, Cogan JD, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med 2007; 356: 1317–1326. [DOI] [PubMed] [Google Scholar]
- 21.Stuart BD, Choi J, Zaidi S, et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat Genet 2015; 47: 512–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kropski JA, Mitchell DB, Markin C, et al. A novel dyskerin (DKC1) mutation is associated with familial interstitial pneumonia. Chest 2014; 146: e1–e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van der Vis JJ, Snetselaar R, Kazemier KM, et al. Effect of Muc5b promoter polymorphism on disease predisposition and survival in idiopathic interstitial pneumonias. Respirology 2016; 21: 712–717. [DOI] [PubMed] [Google Scholar]
- 24.Juge PA, Lee JS, Ebstein E, et al. MUC5B promoter variant and rheumatoid arthritis with interstitial lung disease. N Engl J Med 2018; 379: 2209–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seibold MA, Wise AL, Speer MC, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med 2011; 364: 1503–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaur A, Mathai SK, Schwartz DA. Genetics in idiopathic pulmonary fibrosis pathogenesis, prognosis, and treatment. Front Med (Lausanne) 2017; 4: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chambers RC, Mercer PF. Mechanisms of alveolar epithelial injury, repair, and fibrosis. Ann Am Thorac Soc 2015; 12: Suppl. 1, S16–S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hecker L, Logsdon NJ, Kurundkar D, et al. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci Transl Med 2014; 6: 231ra47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yanai H, Shteinberg A, Porat Z, et al. Cellular senescence-like features of lung fibroblasts derived from idiopathic pulmonary fibrosis patients. Aging (Albany NY) 2015; 7: 664–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bueno M, Lai Y-C, Romero Y, et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J Clin Invest; 125: 521–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim S-J, Cheresh P, Jablonski R, et al. The role of mitochondrial DNA in mediating alveolar epithelial cell apoptosis and pulmonary fibrosis. Int J Mol Sci 2015; 16: 21486–21519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vogel P, Read RW, Rehg JE, et al. Cryptogenic organizing pneumonia in Tomm5(−/−) mice. Vet Pathol 2013; 50: 65–75. [DOI] [PubMed] [Google Scholar]
- 33.Lei H, Kazlauskas A. A reactive oxygen species-mediated, self-perpetuating loop persistently activates platelet-derived growth factor receptor α. Mol Cell Biol 2014; 34: 110–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martinez FJ, Collard HR, Pardo A, et al. Idiopathic pulmonary fibrosis. Nat Rev Dis Primers 2017; 3: 17074. [DOI] [PubMed] [Google Scholar]
- 35.Wollin L, Wex E, Pautsch A, et al. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J 2015; 45: 1434–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amara N, Goven D, Prost F, et al. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFβ1-induced fibroblast differentiation into myofibroblasts. Thorax 2010; 65: 733–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jarman ER, Khambata VS, Cope C, et al. An inhibitor of NADPH oxidase-4 attenuates established pulmonary fibrosis in a rodent disease model. Am J Respir Cell Mol Biol 2014; 50: 158–169. [DOI] [PubMed] [Google Scholar]
- 38.Hecker L, Vittal R, Jones T, et al. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med 2009; 15: 1077–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waghray M, Cui Z, Horowitz JC, et al. Hydrogen peroxide is a diffusible paracrine signal for the induction of epithelial cell death by activated myofibroblasts. FASEB J 2005; 19: 854–856. [DOI] [PubMed] [Google Scholar]
- 40.Richter K, Kietzmann T. Reactive oxygen species and fibrosis: further evidence of a significant liaison. Cell Tissue Res 2016; 365: 591–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ellson CD, Dunmore R, Hogaboam CM, et al. Danger-associated molecular patterns and danger signals in idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol 2014; 51: 163–168. [DOI] [PubMed] [Google Scholar]
- 42.Ryu C, Sun H, Gulati M, et al. Extracellular mitochondrial DNA is generated by fibroblasts and predicts death in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2017; 196: 1571–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Day BJ. Antioxidants as potential therapeutics for lung fibrosis. Antioxid Redox Signal 2008; 10: 355–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martinez FJ, de Andrade JA, Anstrom KJ, et al. Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370: 2093–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oldham JM, Ma S-F, Martinez FJ, et al. TOLLIP, MUC5B, and the response to N-acetylcysteine among individuals with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2015; 192: 1475–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Deursen JM. The role of senescent cells in ageing. Nature 2014; 509: 439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barnes PJ, Baker J, Donnelly LE. Cellular senescence as a mechanism and target in chronic lung diseases. Am J Respir Crit Care Med 2019; 200: 556–564. [DOI] [PubMed] [Google Scholar]
- 48.Minagawa S, Araya J, Numata T, et al. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-β-induced senescence of human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol 2011; 300: L391–L401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Selman M, López-Otín C, Pardo A. Age-driven developmental drift in the pathogenesis of idiopathic pulmonary fibrosis. Eur Respir J 2016; 48: 538–552. [DOI] [PubMed] [Google Scholar]
- 50.Aoshiba K, Tsuji T, Kameyama S, et al. Senescence-associated secretory phenotype in a mouse model of bleomycin-induced lung injury. Exp Toxicol Pathol 2013; 65: 1053–1062. [DOI] [PubMed] [Google Scholar]
- 51.Eschenbrenner J, Janssen W, Kojonazarov B, et al. Role of JAK-STAT pathway in pulmonary fibrosis. Pneumologie 2013; 67: P06. [Google Scholar]
- 52.Milara J, Hernandez G, Ballester B, et al. The JAK2 pathway is activated in idiopathic pulmonary fibrosis. Respir Res 2018; 19: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.White ES, Thannickal VJ, Carskadon SL, et al. Integrin α4β1 regulates migration across basement membranes by lung fibroblasts: a role for phosphatase and tensin homologue deleted on chromosome 10. Am J Respir Crit Care Med 2003; 168: 436–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuwano K PTEN as a new agent in the fight against fibrogenesis. Am J Respir Crit Care Med 2006; 173: 5–6. [DOI] [PubMed] [Google Scholar]
- 55.White ES, Atrasz RG, Hu B, et al. Negative regulation of myofibroblast differentiation by PTEN (Phosphatase and Tensin Homolog Deleted on chromosome 10). Am J Respir Crit Care Med 2006; 173: 112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Miyoshi K, Yanagi S, Kawahara K, et al. Epithelial Pten controls acute lung injury and fibrosis by regulating alveolar epithelial cell integrity. Am J Respir Crit Care Med 2013; 187: 262–275. [DOI] [PubMed] [Google Scholar]
- 57.Kral JB, Kuttke M, Schrottmaier WC, et al. Sustained PI3K activation exacerbates BLM-induced lung fibrosis via activation of pro-inflammatory and pro-fibrotic pathways. Sci Rep 2016; 6: 23034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chow JYC, Quach KT, Cabrera BL, et al. RAS/ERK modulates TGFβ-regulated PTEN expression in human pancreatic adenocarcinoma cells. Carcinogenesis 2007; 28: 2321–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tian Y, Li H, Qiu T, et al. Loss of PTEN induces lung fibrosis via alveolar epithelial cell senescence depending on NF-κB activation. Aging Cell 2019; 18: e12858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Qiu T, Tian Y, Gao Y, et al. PTEN loss regulates alveolar epithelial cell senescence in pulmonary fibrosis depending on Akt activation. Aging (Albany NY) 2019; 11: 7492–7509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Allen RJ, Guillen-Guio B, Oldham JM, et al. Genome-wide association study of susceptibility to idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2020; 201: 564–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.O’Toole CM, Arnoult C, Darszon A, et al. Ca2+ entry through store-operated channels in mouse sperm is initiated by egg ZP3 and drives the acrosome reaction. Mol Biol Cell 2000; 11: 1571–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Land SC, Scott CL, Walker D. mTOR signalling, embryogenesis and the control of lung development. Semin Cell Dev Biol 2014; 36: 68–78. [DOI] [PubMed] [Google Scholar]
- 64.Scott CL, Walker DJ, Cwiklinski E, et al. Control of HIF-1α and vascular signaling in fetal lung involves cross talk between mTORC1 and the FGF-10/FGFR2b/Spry2 airway branching periodicity clock. Am J Physiol Lung Cell Mol Physiol 2010; 299: L455–L471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dodd KM, Yang J, Shen MH, et al. mTORC1 drives HIF-1α and VEGF-A signalling via multiple mechanisms involving 4E-BP1, S6K1 and STAT3. Oncogene 2015; 34: 2239–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell 2017; 168: 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nacarelli T, Azar A, Sell C. Aberrant mTOR activation in senescence and aging: a mitochondrial stress response? Exp Gerontol 2015; 68: 66–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gui Y-S, Wang L, Tian X, et al. mTOR overactivation and compromised autophagy in the pathogenesis of pulmonary fibrosis. PloS One 2015; 10: e0138625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Park JS, Park HJ, Park YS, et al. Clinical significance of mTOR, ZEB1, ROCK1 expression in lung tissues of pulmonary fibrosis patients. BMC Pulm Med 2014; 14: 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chang W, Wei K, Ho L, et al. A critical role for the mTORC2 pathway in lung fibrosis. PloS One 2014; 9: e106155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rangarajan S, Bone NB, Zmijewska AA, et al. Metformin reverses established lung fibrosis in a bleomycin model. Nat Med 2018; 24: 1121–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chung EJ, Sowers A, Thetford A, et al. Mammalian target of rapamycin inhibition with rapamycin mitigates radiation-induced pulmonary fibrosis in a murine model. Int J Radiat Oncol Biol Phys 2016; 96: 857–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xu X, Dai H, Geng J, et al. Rapamycin increases CCN2 expression of lung fibroblasts via phosphoinositide 3-kinase. Lab Invest 2015; 95: 846–859. [DOI] [PubMed] [Google Scholar]
- 74.Xu X, Wan X, Geng J, et al. Rapamycin regulates connective tissue growth factor expression of lung epithelial cells via phosphoinositide 3-kinase. Exp Biol Med (Maywood) 2013; 238: 1082–1094. [DOI] [PubMed] [Google Scholar]
- 75.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149: 274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sarbassov DD, Guertin DA, Ali SM, et al. Phosphorylation and regulation of Akt/PKB by the Rictor-mTOR complex. Science 2005; 307: 1098–1101. [DOI] [PubMed] [Google Scholar]
- 77.Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J 2008; 412: 179–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mercer PF, Woodcock HV, Eley JD, et al. Exploration of a potent PI3 kinase/mTOR inhibitor as a novel anti-fibrotic agent in IPF. Thorax 2016; 71: 701–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Munster P, Aggarwal R, Hong D, et al. First-in-human phase I study of GSK2126458, an oral pan-class I phosphatidylinositol–3-kinase inhibitor, in patients with advanced solid tumor malignancies. Clin Cancer Res 2016; 22: 1932–1939. [DOI] [PubMed] [Google Scholar]
- 80.Araya J, Kojima J, Takasaka N, et al. Insufficient autophagy in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 2013; 304: L56–L69. [DOI] [PubMed] [Google Scholar]
- 81.Malouf MA, Hopkins P, Snell G, et al. An investigator-driven study of everolimus in surgical lung biopsy confirmed idiopathic pulmonary fibrosis. Respirology 2011; 16: 776–783. [DOI] [PubMed] [Google Scholar]
- 82.Selman M, Pardo A, Barrera L, et al. Gene expression profiles distinguish idiopathic pulmonary fibrosis from hypersensitivity pneumonitis. Am J Respir Crit Care Med 2006; 173: 188–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zank DC, Bueno M, Mora AL, et al. Idiopathic pulmonary fibrosis: aging, mitochondrial dysfunction, and cellular bioenergetics. Front Med (Lausanne) 2018; 5: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Riemondy KA, Jansing NL, Jiang P, et al. Single cell RNA sequencing identifies TGFβ as a key regenerative cue following LPS-induced lung injury. JCI Insight 2019; 5: e123637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Burgy O, Konigshoff M. The WNT signaling pathways in wound healing and fibrosis. Matrix Biol 2018; 68–69: 67–80. [DOI] [PubMed] [Google Scholar]
- 86.Walton KL, Johnson KE, Harrison CA. Targeting TGF-β mediated SMAD signaling for the prevention of fibrosis. Front Pharmacol 2017; 8: 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Morrisey EE. Wnt signaling and pulmonary fibrosis. Am J Pathol 2003; 162: 1393–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chilosi M, Poletti V, Zamo A, et al. Aberrant Wnt/β-catenin pathway activation in idiopathic pulmonary fibrosis. Am J Pathol 2003; 162: 1495–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Königshoff M, Kramer M, Balsara N, et al. WNT1-inducible signaling protein-1 mediates pulmonary fibrosis in mice and is upregulated in humans with idiopathic pulmonary fibrosis. J Clin Invest 2009; 119: 772–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Oda K, Yatera K, Izumi H, et al. Profibrotic role of WNT10A via TGF-β signaling in idiopathic pulmonary fibrosis. Respir Res 2016; 17: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kotton DN, Morrisey EE. Lung regeneration: mechanisms, applications and emerging stem cell populations. Nat Med 2014; 20: 822–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Crosby LM, Waters CM. Epithelial repair mechanisms in the lung. Am J Physiol Lung Cell Mol Physiol 2010; 298: L715–L731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Baarsma HA, Konigshoff M. ‘WNT-er is coming’: WNT signalling in chronic lung diseases. Thorax 2017; 72: 746–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lehmann M, Baarsma HA, Konigshoff M. WNT signaling in lung aging and disease. Ann Am Thorac Soc 2016; 13: Suppl. 5, S411–S416. [DOI] [PubMed] [Google Scholar]
- 95.Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature 2005; 434: 843–850. [DOI] [PubMed] [Google Scholar]
- 96.Tammela T, Sanchez-Rivera FJ, Cetinbas NM, et al. A Wnt-producing niche drives proliferative potential and progression in lung adenocarcinoma. Nature 2017; 545: 355–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chilosi M, Doglioni C, Murer B, et al. Epithelial stem cell exhaustion in the pathogenesis of idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 2010; 27: 7–18. [PubMed] [Google Scholar]
- 98.Clevers H Wnt/β-catenin signaling in development and disease. Cell 2006; 127: 469–480. [DOI] [PubMed] [Google Scholar]
- 99.Piersma B, Bank RA, Boersema M. Signaling in fibrosis: TGF-β, WNT, and YAP/TAZ converge. Front Med (Lausanne) 2015; 2: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li C, Bellusci S, Borok Z, et al. Non-canonical WNT signalling in the lung. J Biochem 2015; 158: 355–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Selman M, Pardo A, Kaminski N. Idiopathic pulmonary fibrosis: aberrant recapitulation of developmental programs? PLoS Med 2008; 5: e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.De Langhe E, Aznar-Lopez C, De Vooght V, et al. Secreted frizzled related proteins inhibit fibrosis in vitro but appear redundant in vivo. Fibrogenesis Tissue Repair 2014; 7: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Flozak AS, Lam AP, Russell S, et al. β-Catenin/T-cell factor signaling is activated during lung injury and promotes the survival and migration of alveolar epithelial cells. J Biol Chem 2010; 285: 3157–3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Konigshoff M, Kramer M, Balsara N, et al. WNT1-inducible signaling protein-1 mediates pulmonary fibrosis in mice and is upregulated in humans with idiopathic pulmonary fibrosis. J Clin Invest 2009; 119: 772–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gottardi CJ, Königshoff M. Considerations for targeting β-catenin signaling in fibrosis. Am J Respir Crit Care Med 2013; 187: 566–568. [DOI] [PubMed] [Google Scholar]
- 106.Aumiller V, Balsara N, Wilhelm J, et al. WNT/β-catenin signaling induces IL-1β expression by alveolar epithelial cells in pulmonary fibrosis. Am J Respir Cell Mol Biol 2013; 49: 96–104. [DOI] [PubMed] [Google Scholar]
- 107.Guo Y, Xiao L, Sun L, et al. Wnt/β-catenin signaling: a promising new target for fibrosis diseases. Physiol Res 2012; 61: 337–346. [DOI] [PubMed] [Google Scholar]
- 108.Lam AP, Herazo-Maya JD, Sennello JA, et al. Wnt coreceptor Lrp5 is a driver of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2014; 190: 185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Konigshoff M, Balsara N, Pfaff EM, et al. Functional Wnt signaling is increased in idiopathic pulmonary fibrosis. PloS One 2008; 3: e2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Acebron SP, Niehrs C. β-Catenin-independent roles of Wnt/LRP6 signaling. Trends Cell Biol 2016; 26: 956–967. [DOI] [PubMed] [Google Scholar]
- 111.Sucre JMS, Deutsch GH, Jetter CS, et al. A shared pattern of β-catenin activation in bronchopulmonary dysplasia and idiopathic pulmonary fibrosis. Am J Pathol 2018; 188: 853–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Liu L, Carron B, Yee HT, et al. Wnt pathway in pulmonary fibrosis in the bleomycin mouse model. J Environ Pathol Toxicol Oncol 2009; 28: 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kim KK, Wei Y, Szekeres C, et al. Epithelial cell α3β1 integrin links β-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J Clin Invest 2009; 119: 213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chilosi M, Calio A, Rossi A, et al. Epithelial to mesenchymal transition-related proteins ZEB1, β-catenin, and β-tubulin-III in idiopathic pulmonary fibrosis. Mod Pathol 2017; 30: 26–38. [DOI] [PubMed] [Google Scholar]
- 115.Salazar KD, Lankford SM, Brody AR. Mesenchymal stem cells produce Wnt isoforms and TGF-β1 that mediate proliferation and procollagen expression by lung fibroblasts. Am J Physiol Lung Cell Mol Physiol 2009; 297: L1002–L1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chang W, Wei K, Jacobs SS, et al. SPARC suppresses apoptosis of idiopathic pulmonary fibrosis fibroblasts through constitutive activation of β-catenin. J Biol Chem 2010; 285: 8196–8206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sennello JA, Misharin AV, Flozak AS, et al. Lrp5/β-catenin signaling controls lung macrophage differentiation and inhibits resolution of fibrosis. Am J Respir Cell Mol Biol 2017; 56: 191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Redente EF. Macrophages and fibrosis resolution. Harnessing Wnt/β-catenin signaling as the way and the means. Am J Respir Cell Mol Biol 2017; 56: 150–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Henderson WR Jr, Chi EY, Ye X, et al. Inhibition of Wnt/β-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. Proc Natl Acad Sci USA 2010; 107: 14309–14314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang C, Zhu H, Sun Z, et al. Inhibition of Wnt/β-catenin signaling promotes epithelial differentiation of mesenchymal stem cells and repairs bleomycin-induced lung injury. Am J Physiol Cell Physiol 2014; 307: C234–C244. [DOI] [PubMed] [Google Scholar]
- 121.Cao H, Wang C, Chen X, et al. Inhibition of Wnt/β-catenin signaling suppresses myofibroblast differentiation of lung resident mesenchymal stem cells and pulmonary fibrosis. Sci Rep 2018; 8: 13644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chen X, Shi C, Meng X, et al. Inhibition of Wnt/β-catenin signaling suppresses bleomycin-induced pulmonary fibrosis by attenuating the expression of TGF-β1 and FGF-2. Exp Mol Pathol 2016; 101: 22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dees C, Schlottmann I, Funke R, et al. The Wnt antagonists DKK1 and SFRP1 are downregulated by promoter hypermethylation in systemic sclerosis. Ann Rheum Dis 2014; 73: 1232–1239. [DOI] [PubMed] [Google Scholar]
- 124.Akhmetshina A, Palumbo K, Dees C, et al. Activation of canonical Wnt signalling is required for TGF-β-mediated fibrosis. Nat Commun 2012; 3: 735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kitisin K, Saha T, Blake T, et al. TGF-β signaling in development. Sci STKE 2007; 2007: cm1. [DOI] [PubMed] [Google Scholar]
- 126.Bartram U, Speer CP. The role of transforming growth factor β in lung development and disease. Chest 2004; 125: 754–765. [DOI] [PubMed] [Google Scholar]
- 127.Coker RK, Laurent GJ, Shahzeidi S, et al. Diverse cellular TGF-β1 and TGF-β3 gene expression in normal human and murine lung. Eur Respir J 1996; 9: 2501–2507. [DOI] [PubMed] [Google Scholar]
- 128.Khalil N, O’Connor RN, Flanders KC, et al. TGF-β1, but not TGF-β2 or TGF-β3, is differentially present in epithelial cells of advanced pulmonary fibrosis: an immunohistochemical study. Am J Respir Cell Mol Biol 1996; 14: 131–138. [DOI] [PubMed] [Google Scholar]
- 129.Limper AH, Colby TV, Sanders MS, et al. Immunohistochemical localization of transforming growth factor-β1 in the nonnecrotizing granulomas of pulmonary sarcoidosis. Am J Respir Crit Care Med 1994; 149: 197–204. [DOI] [PubMed] [Google Scholar]
- 130.Hoyt DG, Lazo JS. Alterations in pulmonary mRNA encoding procollagens, fibronectin and transforming growth factor-β precede bleomycin-induced pulmonary fibrosis in mice. J Pharmacol Exp Ther 1988; 246: 765–771. [PubMed] [Google Scholar]
- 131.Bergeron A, Soler P, Kambouchner M, et al. Cytokine profiles in idiopathic pulmonary fibrosis suggest an important role for TGF-β and IL-10. Eur Respir J 2003; 22: 69–76. [DOI] [PubMed] [Google Scholar]
- 132.Zhang K, Flanders KC, Phan SH. Cellular localization of transforming growth factor-β expression in bleomycin-induced pulmonary fibrosis. Am J Pathol 1995; 147: 352–361. [PMC free article] [PubMed] [Google Scholar]
- 133.Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis 2010; 30: 245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Vaughan MB, Howard EW, Tomasek JJ. Transforming growth factor-β1 promotes the morphological and functional differentiation of the myofibroblast. Exp Cell Res 2000; 257: 180–189. [DOI] [PubMed] [Google Scholar]
- 135.Kelley J, Shull S, Walsh JJ, et al. Auto-induction of transforming growth factor-β in human lung fibroblasts. Am J Respir Cell Mol Biol 1993; 8: 417–424. [DOI] [PubMed] [Google Scholar]
- 136.Sime PJ, Xing Z, Graham FL, et al. Adenovector-mediated gene transfer of active transforming growth factor-β1 induces prolonged severe fibrosis in rat lung. J Clin Invest 1997; 100: 768–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lee CG, Cho SJ, Kang MJ, et al. Early growth response gene 1-mediated apoptosis is essential for transforming growth factor β1-induced pulmonary fibrosis. J Exp Med 2004; 200: 377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Giri SN, Hyde DM, Hollinger MA. Effect of antibody to transforming growth factor β on bleomycin induced accumulation of lung collagen in mice. Thorax 1993; 48: 959–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bonniaud P, Margetts PJ, Kolb M, et al. Progressive transforming growth factor β1–induced lung fibrosis is blocked by an orally active ALK5 kinase inhibitor. Am J Respir Crit Care Med 2005; 171: 889–898. [DOI] [PubMed] [Google Scholar]
- 140.Hoyles RK, Derrett-Smith EC, Khan K, et al. An essential role for resident fibroblasts in experimental lung fibrosis is defined by lineage-specific deletion of high-affinity type II transforming growth factor β receptor. Am J Respir Crit Care Med 2011; 183: 249–261. [DOI] [PubMed] [Google Scholar]
- 141.Judge JL, Owens KM, Pollock SJ, et al. Ionizing radiation induces myofibroblast differentiation via lactate dehydrogenase. Am J Physiol Lung Cell Mol Physiol 2015; 309: L879–L887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Li M, Krishnaveni MS, Li C, et al. Epithelium-specific deletion of TGF-β receptor type II protects mice from bleomycin-induced pulmonary fibrosis. J Clin Invest 2011; 121: 277–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Park SA, Kim MJ, Park SY, et al. EW-7197 inhibits hepatic, renal, and pulmonary fibrosis by blocking TGF-β/Smad and ROS signaling. Cell Mol Life Sci 2015; 72: 2023–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wang Q, Zou Y, Nowotschin S, et al. The p53 family coordinates Wnt and nodal inputs in mesendodermal differentiation of embryonic stem cells. Cell Stem Cell 2017; 20: 70–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zhou S, Eid K, Glowacki J. Cooperation between TGF-β and Wnt pathways during chondrocyte and adipocyte differentiation of human marrow stromal cells. J Bone Miner Res 2004; 19: 463–470. [DOI] [PubMed] [Google Scholar]
- 146.Baarsma HA, Engelbertink LH, van Hees LJ, et al. Glycogen synthase kinase-3 (GSK-3) regulates TGF-β1-induced differentiation of pulmonary fibroblasts. Br J Pharmacol 2013; 169: 590–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Gillespie J, Ross RL, Corinaldesi C, et al. Transforming growth factor β activation primes canonical Wnt signaling through down-regulation of Axin-2. Arthritis Rheumatol 2018; 70: 932–942. [DOI] [PubMed] [Google Scholar]
- 148.Blyszczuk P, Muller-Edenborn B, Valenta T, et al. Transforming growth factor-β-dependent Wnt secretion controls myofibroblast formation and myocardial fibrosis progression in experimental autoimmune myocarditis. Eur Heart J 2017; 38: 1413–1425. [DOI] [PubMed] [Google Scholar]
- 149.Zhuang X, Zhang H, Li X, et al. Differential effects on lung and bone metastasis of breast cancer by Wnt signalling inhibitor DKK1. Nat Cell Biol 2017; 19: 1274–1285. [DOI] [PubMed] [Google Scholar]
- 150.Kumawat K, Koopmans T, Menzen MH, et al. Cooperative signaling by TGF-β1 and WNT-11 drives sm-α-actin expression in smooth muscle via Rho kinase-actin-MRTF-A signaling. Am J Physiol Lung Cell Mol Physiol 2016; 311: L529–L537. [DOI] [PubMed] [Google Scholar]
- 151.Eger A, Stockinger A, Park J, et al. β-Catenin and TGFβ signalling cooperate to maintain a mesenchymal phenotype after FosER-induced epithelial to mesenchymal transition. Oncogene 2004; 23: 2672–2680. [DOI] [PubMed] [Google Scholar]
- 152.Cheon SS, Wei Q, Gurung A, et al. β-Catenin regulates wound size and mediates the effect of TGF-β in cutaneous healing. FASEB J 2006; 20: 692–701. [DOI] [PubMed] [Google Scholar]
- 153.Luo K Signaling cross talk between TGF-β/Smad and other signaling pathways. Cold Spring Harb Perspect Biol 2017; 9: a022137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Furuhashi M, Yagi K, Yamamoto H, et al. Axin facilitates Smad3 activation in the transforming growth factor β signaling pathway. Mol Cell Biol 2001; 21: 5132–5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Jian H, Shen X, Liu I, et al. Smad3-dependent nuclear translocation of β-catenin is required for TGF-β1-induced proliferation of bone marrow-derived adult human mesenchymal stem cells. Genes Dev 2006; 20: 666–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Russell AM, Adamali H, Molyneaux PL, et al. Daily home spirometry: an effective tool for detecting progression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2016; 194: 989–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ash SY, Washko GR. Interstitial lung abnormalities: risk and opportunity. Lancet Respir Med 2017; 5: 95–96. [DOI] [PMC free article] [PubMed] [Google Scholar]