

Abstract
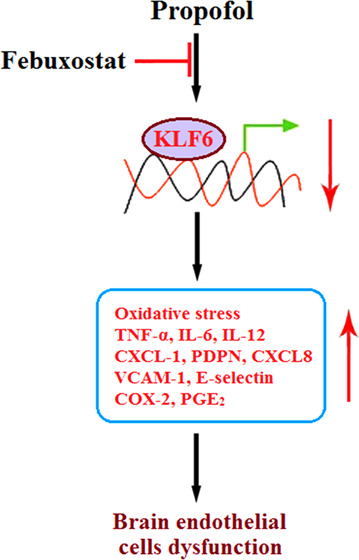
Background and purpose: A high risk of brain injury has been reported with the usage of general anesthetics such as propofol in infants. Experimental data indicated that oxidative stress and inflammation are involved in the neurotoxicity induced by propofol. Febuxostat is a novel anti-gout agent recently reported to exert an anti-inflammatory effect. The present study aims to investigate the protective property of febuxostat against the cytotoxicity of propofol in brain endothelial cells as well as the underlying preliminary mechanism. Methods: The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was utilized to screen the optimized incubation concentration of febuxostat. bEnd.3 brain endothelial cells were stimulated with 2% propofol in the presence or absence of febuxostat (10, 20 μM) for 24 h. The lactate dehydrogenase (LDH) release assay was conducted to detect cytotoxicity. The reactive oxygen species (ROS) levels were evaluated using dichloro-dihydro-fluorescein diacetate (DCFH-DA) staining, and the concentration of reduced glutathione (GSH) was determined using a commercial kit. The expressions of TNF-α, IL-6, IL-12, CXCL-1, PDPN, CXCL8, VCAM-1, and E-selectin were determined using a quantitative real-time polymerase chain reaction (qRT-PCR) and an enzyme-linked immunosorbent assay (ELISA). Western blot and qRT-PCR were utilized to determine the expressions of COX-2 and KLF6. The production of PGE2 was evaluated by ELISA. Results: First, increased LDH release induced by propofol was significantly suppressed by febuxostat. The oxidative stress (elevated ROS levels and decreased GSH level) induced by propofol was alleviated by febuxostat. Second, the upregulated inflammatory factors (TNF-α, IL-6, and IL-12), pro-inflammatory chemokines (CXCL-1, PDPN, and CXCL8), adhesion molecules (VCAM-1 and E-selectin), and inflammatory mediators (COX-2 and PGE2) induced by propofol were greatly downregulated by febuxostat. Lastly, the expression of KLF6 was significantly suppressed by propofol but greatly elevated by febuxostat. Conclusion: Febuxostat prevented the cytotoxicity of propofol in brain endothelial cells by alleviating oxidative stress and inflammatory response through KLF6.
Introduction
With the rapid development of modern medical technology, operative treatments are being performed on a number of newborns and infants with congenital defects or acquired diseases, and are inevitably accompanied by the usage of general anesthetics. However, Jevtovic-Todorovic reported that extensive degeneration of neurons and persistent learning disabilities could be induced in the brains of developing rats when immature rats were treated with general anesthetics at an early stage.1 As warned by the US Food and Drug Administration (FDA) in 2016, the development of children’s brains will be impacted under more than 3 h of anesthesia or repeated usage of general anesthetics and sedative drugs in infants or women at mid-pregnancy. Currently, multiple high-quality, polycentric, and large-sample clinical studies are being conducted to investigate the influence of general anesthetics on development and long-term cognitive and learning functions in infants.2−4 Propofol is a short-acting intravenous anesthetic widely used in clinics, especially for anesthesia in infants.5,6 Propofol was first developed by the British Imperial Chemical Industry verified by sedation function in animal experiments. It was initially reported to exert an anesthetic property in 19737,8 and was approved by the FDA in 1989. Similar to other intravenous anesthetics, propofol exerts sedative and hypnotic effects by activating γ-aminobutyric acid (GABA), which is an inhibitory neurotransmitter.9 Although approved by the FDA as an anesthetic, it has limitations, which include the maintenance of anesthesia in infants over 2 months old only and anesthesia induction for infants over 3 years old only,10 indicating the potential risk that propofol could affect the development of the brain in infants. Fundamental studies indicate that neurotoxicity in the brain of immature animals, including the rhesus monkey,11 can be induced by the application of propofol,12,13 which further triggers long-term learning and memory dysfunctions.14,15 It is reported that the overactivation of the apoptotic pathway,16 overexpression of inflammatory factors,17,18 activation of oxidative stress,19 activation of microglia, development of neural inhibition,20,21 changes of dendritic processes, and destruction of the blood–brain barrier (BBB)22 are involved in the mechanism underlying the neurotoxicity of propofol. Therefore, there is an urgent need to explore potential therapeutic methods for the safe usage of propofol in infants.
Febuxostat is an anti-gout agent. It is a non-purine selective xanthine oxidase inhibitor, which was approved for the treatment of gout in 2008 by the European Union and in 2009 by the US.23 The molecular structure of febuxostat is shown in Figure 1A. Recently, febuxostat was reported to ameliorate myocardial ischemia injury by inhibiting the production of reactive oxygen species (ROS).24 In addition, multiple pieces of research reported the anti-inflammatory and antioxidative stress effects of febuxostat.25−27 In the present study, the effects of febuxostat on inflammation and oxidative stress in brain endothelial cells induced by propofol will be investigated to explore its potential therapeutic property against clinical neurotoxicity induced by propofol.
Figure 1.

Effects of febuxostat on the cell viability of bEnd.3 brain endothelial cells. (A) Molecular structure of febuxostat. (B) Cells were stimulated with 0.1, 0.2, 1, 2, 10, 20, 100, and 200 μM febuxostat for 24 h. Cell viability was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (#P < 0.05, ##P < 0.01 vs the vehicle group, n = 6).
Results
Effects of Febuxostat on the Cell Viability of bEnd.3 Brain Endothelial Cells
To screen the optimized incubation concentration of febuxostat in bEnd.3 brain endothelial cells, the cells were stimulated with 0.1, 0.2, 1, 2, 10, 20, 100, and 200 μM febuxostat for 24 h. The cell viability of each well was evaluated using the MTT assay. As shown in Figure 1B, no significant difference in the cell viability was observed as the concentration of febuxostat increased from 0.1 to 20 μM. However, when the concentration of febuxostat exceeded 100 μM, the cell viability decreased greatly. Therefore, 10 and 20 μM febuxostat were utilized as the incubation concentrations in the subsequent experiments.
Febuxostat Prevented Propofol-Induced Release of Lactate Dehydrogenase (LDH) in bEnd.3 Brain Endothelial Cells
To evaluate the effect of febuxostat against the toxicity in bEnd.3 brain endothelial cells induced by propofol, the cells were stimulated with 2% propofol in the presence or absence of febuxostat (10, 20 μM) for 24 h, and LDH of the cells was detected. As shown in Figure 2A, irregular cell morphology was observed in the cells treated with propofol but was reversed by treatment with febuxostat. In addition, the LDH releases (Figure 2B) in the control, propofol, 10 μM febuxostat, and 20 μM febuxostat groups were 5.7, 35.2, 26.1, and 15.8%, respectively.
Figure 2.

Febuxostat prevented propofol-induced release of LDH in bEnd.3 brain endothelial cells. Cells were stimulated with 2% propofol in the presence or absence of febuxostat (10, 20 μM) for 24 h. (A) Cell morphology of bEnd.3 brain endothelial cells; scale bar, 50 μm. (B) LDH release (####P < 0.0001 vs the vehicle group; **P < 0.01, ***P < 0.001 vs the propofol group, n = 6).
Oxidative Stress in bEnd.3 Brain Endothelial Cells Induced by Propofol was Alleviated by Febuxostat
As shown in Figure 3A, the levels of ROS were significantly elevated by stimulation with propofol but were greatly suppressed by the administration of febuxostat in a dose-dependent manner. The decreased concentration of reduced glutathione (GSH) induced by the stimulation with propofol was significantly elevated by treatment with febuxostat in a dose-dependent manner. These data indicate that the activated oxidative stress induced by propofol was alleviated by febuxostat.
Figure 3.
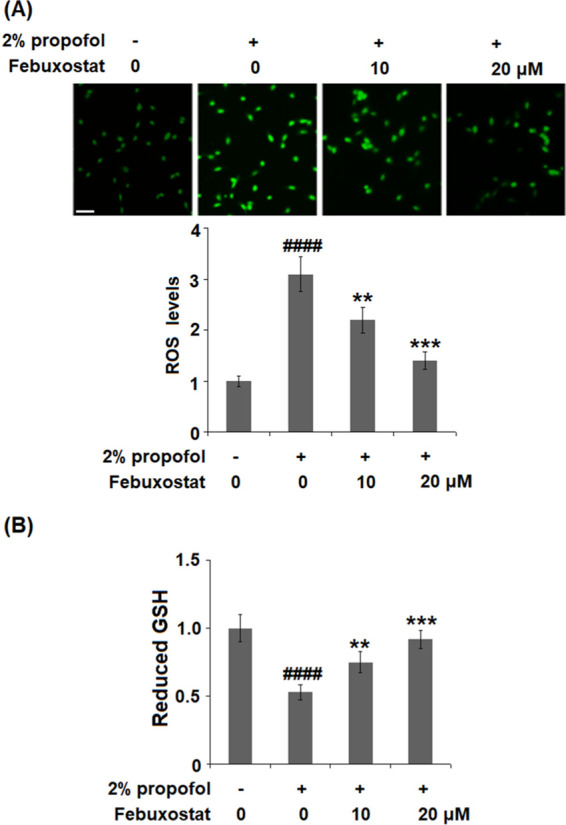
Febuxostat ameliorated propofol-induced oxidative stress in bEnd.3 brain endothelial cells. Cells were stimulated with 2% propofol in the presence or absence of febuxostat (10, 20 μM) (A) for 24 h. The levels of ROS were measured using dichloro-dihydro-fluorescein diacetate (DCFH-DA) staining; 200 μm. (B) Levels of reduced glutathione (GSH) (####P < 0.0001 vs the vehicle group; **P < 0.01, ***P < 0.001 vs the propofol group, n = 6).
Febuxostat Inhibited Propofol-Induced Expression and Production of Pro-Inflammatory Cytokines in bEnd.3 Brain Endothelial Cells
To investigate the effects of febuxostat against inflammation induced by propofol in the bEnd.3 brain endothelial cells, the concentrations of inflammatory factors released by the cells were detected following stimulation with 2% propofol in the presence or absence of febuxostat (10, 20 μM) for 24 h. As shown in Figure 4A–C, the gene expressions of TNF-α, IL-6, and IL-12 were significantly elevated by stimulation with propofol but greatly suppressed by the treatment of febuxostat in a dose-dependent manner. The concentrations of TNF-α in the control, propofol, 10 μM febuxostat, and 20 μM febuxostat groups were 76.5, 253.8, 188.1, and 143.6 pg/mL, respectively (Figure 4D). As shown in Figure 4E, approximately 103.5, 562.7, 433.2, and 311.4 pg/mL IL-6 were detected in the control, propofol, 10 μM febuxostat, and 20 μM febuxostat groups, respectively. Lastly, the concentrations of IL-12 (Figure 4F) in the control, propofol, 10 μM febuxostat, and 20 μM febuxostat groups were 91.5, 387.3, 271.9, and 212.5 pg/mL, respectively.
Figure 4.

Febuxostat inhibited propofol-induced expression and production of pro-inflammatory cytokines in bEnd.3 brain endothelial cells. Cells were stimulated with 2% propofol in the presence or absence of febuxostat (10, 20 μM) for 24 h. (A) mRNA levels of TNF-α. (B) mRNA of IL-6. (C) mRNA of IL-12. (D) Secretions of TNF-α. (E) Secretions of IL-6. (F) Secretions of IL-12 (####P < 0.0001 vs the vehicle group; **P < 0.01, ***P < 0.001 vs the propofol group, n = 6).
Febuxostat Inhibited Propofol-Induced Expression and Production of the Pro-Inflammatory Chemokines
As shown in Figure 5A–C, the gene expressions of CXCL-1, PDPN, and CXCL8 were significantly elevated by stimulation with propofol but were greatly suppressed by treatment with febuxostat in a dose-dependent manner. The concentrations of CXCL-1 in the control, propofol, 10 μM febuxostat, and 20 μM febuxostat groups were 125.6, 465.9, 376.6, and 288.5 pg/mL, respectively (Figure 5D). As shown in Figure 5E, approximately 53.5, 122.8, 89.5, and 75.3 pg/mL PDPN were detected in the control, propofol, 10 μM febuxostat, and 20 μM febuxostat groups, respectively. Lastly, the concentrations of CXCL8 (Figure 5F) in the control, propofol, 10 μM febuxostat, and 20 μM febuxostat groups were 66.4, 166.5, 121.7, and 99.8 pg/mL, respectively.
Figure 5.

Febuxostat inhibited propofol-induced expression and production of the pro-inflammatory chemokines. Cells were stimulated with 2% propofol in the presence or absence of febuxostat (10, 20 μM) for 24 h. (A) mRNA of CXCL-1. (B) mRNA of PDPN. (C) mRNA of CXCL8. (D) Secretions of CXCL-1. (E) Secretions of PDPN. (F) Secretions of CXCL8 (####P < 0.0001 vs the vehicle group; **P < 0.01, ***P < 0.001 vs the propofol group, n = 6).
Febuxostat Inhibited Propofol-Induced Expression and Production of VCAM-1 and E-Selectin
To evaluate the effects of febuxostat against the elevated expression of adhesion molecules induced by propofol, the expressions of VCAM-1 and E-selectin were detected. As shown in Figure 6A,B, the gene expression levels of VCAM-1 and E-selectin were significantly promoted by incubation with propofol but were greatly suppressed by the introduction of febuxostat in a dose-dependent manner. The concentrations of VCAM-1 and E-selectin are illustrated in Figure 6C,D. The concentrations of VCAM-1 in the control, propofol, 10 μM febuxostat, and 20 μM febuxostat groups were 156.8, 621.6, 519.9, and 422.3 pg/mL, respectively. Approximately 88.3, 375.5, 282.9, and 191.6 pg/mL E-selectin were detected in the control, propofol, 10 μM febuxostat, and 20 μM febuxostat groups, respectively.
Figure 6.

Febuxostat inhibited propofol-induced expression and production of VCAM-1 and E-selectin. Cells were stimulated with 2% propofol in the presence or absence of febuxostat (10, 20 μM) for 24 h. (A) mRNA of VCAM-1. (B) mRNA of E-selectin. (C) Secretions of VCAM-1. (D) Secretions of E-selectin (####P < 0.0001 vs the vehicle group; **P < 0.01, ***P < 0.001 vs the propofol group, n = 6).
Febuxostat Prevented Propofol-Induced Expression of Cyclooxygenase-2 (COX-2) and the Production of Prostaglandin E2 (PGE2)
To explore the effect of febuxostat on the expression of inflammatory mediators induced by propofol, the expressions of COX-2 and PGE2 were detected. As shown in Figure 7A,B, COX-2 was significantly upregulated by stimulation with propofol but greatly downregulated by the introduction of febuxostat in a dose-dependent manner. Further, the concentrations of the released PGE2 in the control, propofol, 10 μM febuxostat, and 20 μM febuxostat groups were 78.9, 235.5, 176.4, and 138.2 pg/mL, respectively.
Figure 7.

Febuxostat prevented propofol-induced expression of cyclooxygenase-2 (COX-2) and the production of prostaglandin E2 (PGE2). Cells were stimulated with 2% propofol in the presence or absence of febuxostat (10, 20 μM) for 24 h. (A) mRNA of COX-2. (B) Protein of COX-2 as measured using Western blot analysis. (C) Production of PGE2 (####P < 0.0001 vs the vehicle group; **P < 0.01, ***P < 0.001 vs the propofol group, n = 6).
Febuxostat Restored Propofol-Induced Reduction of the Transcriptional Factor KLF6
To explore the possible mechanism underlying the neuroprotective property of febuxostat, the expression of the transcriptional factor KLF6 was evaluated. As shown in Figure 8, the expression of KLF6 was significantly inhibited by the stimulation of propofol but greatly elevated by the treatment of febuxostat in a dose-dependent manner.
Figure 8.

Febuxostat restored propofol-induced reduction of the transcriptional factor KLF6. Cells were stimulated with 2% propofol in the presence or absence of febuxostat (10, 20 μM) for 24 h. (A) mRNA of KLF6. (B) Expression of KLF6 (####P < 0.0001 vs the vehicle group; **P < 0.01, ***P < 0.001 vs the propofol group, n = 6).
Discussion
As the intermediate metabolite of oxidation, ROS play an important role in conducting the cellular signal transition and maintaining the oxidant–antioxidant homeostasis.28 Under a normal physiological state, the oxidative system and the antioxidative system interact with each other to maintain the balance of production and elimination of ROS.29 When the balance is broken, direct or indirect toxicity against cells or biomolecules is triggered by the accumulated ROS, which further contributes to the excessive production of inflammatory factors, as well as irreversible injuries and apoptosis on cells.30 Oxidative stress is a biological state of cellular or tissue injuries resulting from the excessive production of ROS;31 it has been proven to be a significant inducer of the neurotoxicity caused by propofol.32 Although researchers have already reported the role of ROS-scavenging agents such as acetyl-l-carnitine, Trolox, and EUK-134 in alleviating neurotoxicity induced by propofol,33 there are still limitations for clinical applications. In the present study, significant toxicity was induced by propofol and was confirmed by the irregular morphology and elevated release of LDH. By treatment with febuxostat, the pathological state of the bEnd.3 brain endothelial cells was improved, indicating a potential protective effect of febuxostat against neurotoxicity induced by propofol. Furthermore, oxidative stress was found to be greatly activated by the stimulation of propofol, as confirmed by the elevated ROS levels and decreased reduced GSH. It was greatly reversed by the treatment with febuxostat, indicating its inhibitory effect against oxidative stress. Based on these preliminary data, we suspect that the neurotoxicity induced by propofol might be alleviated by febuxostat through inhibiting the state of oxidative stress, which, however, will be further confirmed using the animal anesthesia model in our future work.
Inflammation induces secondary injuries on the endothelial cells following direct damage from anesthesia. When the system senses the apoptosis of brain endothelial cells, the brain immune cells such as microglia and astrocytes will be recruited to the lesions by pro-inflammatory chemokines, including the CXCL family34 and podoplanin (PDPN).35 Adhesion molecules such as VCAM-1 and E-selectin promote the adhesion of astrocytes and microglia to the lesions; these are then activated to release excessive pro-inflammatory factors under the mediation of inflammatory mediators, such as COX-2 and PGE2. As a consequence, inflammation is induced in the cells, which finally contributes to the apoptosis of brain endothelial cells and dysfunction of the blood–brain barrier.36,37 In the present study, the expression levels of inflammatory factors, pro-inflammatory chemokines, adhesion molecules, and inflammatory mediators were all upregulated by stimulation with propofol, indicating an elevated inflammation state induced by propofol. By treatment with febuxostat, the inflammation state was significantly ameliorated, indicating a promising anti-inflammatory property of febuxostat. This was consistent with the reports of the effects of febuxostat in other inflammation-related diseases.38−40 The in vivo anti-inflammatory effect in the brain will be further investigated and verified in our future animal experiments.
KLF6 is reported to be an important transcriptional factor that mediates inflammation and polarization of macrophages.41,42 Also, cell proliferation ability is reported to be regulated by KLF6.43 In the present study, KLF6 was found to be significantly downregulated by stimulation with propofol, an effect reversed by treatment with febuxostat, indicating the possible mechanism by which febuxostat exerted its anti-inhibitory property by mediating the expression of KLF6. However, further verifications will be conducted on the hypothesis proposed based on the preliminary data collected in the present study, such as knocking down the expression of KLF6 using RNA interference technology, to provide evidence of the involvement of KLF6 in the anti-inflammatory effect of febuxostat.
Febuxostat has displayed clinical efficacy in reducing the levels of serum urate and its long-term use is important for improving gout flare frequency and tophus burden. However, concerns on the side effects of febuxostat have been raised. A recent cardiovascular safety study reported that febuxostat showed no difference in the primary endpoint compared to another gout medicine, allopurinol. However, administration of febuxostat led to an increased risk of heart-related deaths and death from all causes.44,45 Based on this study, the FDA issued a drug safety communication to limit the approved use of febuxostat to certain patients who are not treated effectively.46 Interestingly, natural products have increasingly received attention from both scientists and physicians due to their benefits on human health. First, natural products possess enormous structural diversity as they usually have more chiral centers and have greater molecular rigidity than synthetic chemicals. Second, natural polyphenols originating from tea or other fruits and vegetables are much safer and biologically friendlier than artificial synthetic drugs.47 Therefore, safer naturally occurring antioxidants such as dietary polyphenols from living organisms could be alternatives used as agents against oxidative stress and inflammation caused by propofol in the body.
Taken together, our results demonstrate that treatment with febuxostat mitigated the cytotoxicity of propofol in brain endothelial cells by alleviating oxidative stress and inflammatory response through KLF6.
Materials and Methods
Cell Culture and Treatments
The bEnd.3 brain endothelial cells were obtained from the cell culture collections of ATCC-LGC Standards (ATCC, Manassas), which were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7.4, gentamycin (50 mg/mL), ascorbic acid (5 mg/mL), 1% chemically defined lipid concentrate, and basic fibroblast growth factor (1 ng/mL) at 37 °C. The medium was changed every 3 days. For most experiments, cells were stimulated with 2% propofol in the presence or absence of febuxostat (10, 20 μM) for 24 h. For the MTT assay, cells were stimulated with 0.1, 0.2, 1, 2, 10, 20, 100, and 200 μM febuxostat for 24 h.
MTT Assay
After the treatments, the bEnd.3 brain endothelial cells were incubated with a medium containing 10 μL of MTT solution (5 mg/mL, Sigma-Aldrich) at 37 °C for 4 h, followed by the addition of 150 μL of dimethyl sulfoxide (DMSO) to terminate the reaction. After shaking the plates for 15 min, the absorbance of each well at 490 nm was measured with a microplate reader (Bio-Tek Instruments, Winooski). The OD values were used to calculate cell viability.
LDH Release Assay
Briefly, the treated bEnd.3 brain endothelial cells were seeded on 96-well plates at a density of 2 × 104 cells/mL, followed by centrifugation to collect the supernatants. Subsequently, the supernatants were mixed with 20 μL of 2,4-dinitrophenylhydrazine at 37 °C for 15 min, followed by the addition of 250 μL of 0.4 M NaOH, and incubated at 37 °C for another 15 min. Lastly, the absorbance at 450 nm was measured with a microplate reader (Bio-Tek Instruments, Winooski).
DCFH-DA Assay
The cells were seeded on wells at a density of 1 × 105 cells/well for 24 h, and 1 mL of DCFH-DA (Sigma-Aldrich, MO) solution was added following the removal of the culture medium; it was diluted with serum-free medium at a ratio of 1:1000. Subsequently, the treated bEnd.3 brain endothelial cells were washed using phosphate-buffered saline (PBS) buffer to clear the residual DCFH-DA solution following incubation at 37 °C for 20 min. Lastly, fluorescence was measured at 488 nm (excitation) and 525 nm (emission) using an inverted fluorescence microscope (Olympus, Tokyo, Japan) and quantified with a fluorescent microplate reader (Thermo Fisher, MA).
Measurement of Intracellular GSH
The concentration of the intracellular GSH was detected using a reduced GSH quantification kit (Dojindo, MD). Briefly, the treated bEnd.3 brain endothelial cells were lysed, followed by pretreatment with a coenzyme working solution. Subsequently, the enzyme working solution was added to the supernatants, followed by incubation for 2 h at room temperature. The substrate working solution was added to the plates, followed by incubation at room temperature for 2 h. Lastly, the absorbance at 405 nm was measured using a microplate reader (Bio-Tek Instruments, Winooski).
Real-Time Polymerase Chain Reaction (PCR) Analysis
The trizol reagents (Thermo Fisher Scientific, MA) were used to isolate the total RNA from the treated bEnd.3 brain endothelial cells, which were reverse-transcribed to cDNA using the RT Master Mix kit (Takara, Tokyo, Japan). The SYBR Master Mix kit (Takara, Tokyo, Japan) with the StepOne-Plus system (Takara, Tokyo, Japan) was used to perform the PCR by denaturing at 95 °C for 30 s, annealing at 60 °C for 1 min, and extending at 95 °C for 5 s. The relative gene expressions of related proteins were quantified using the 2–ΔΔCt method, with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as the internal negative control.
Enzyme-Linked Immunosorbent Assay (ELISA)
The production of TNF-α, IL-6, IL-12, CXCL-1, PDPN, CXCL8, VCAM-1, E-selectin, and PGE2 was measured using ELISA kits (Thermo Fisher Scientific, MA). First, the samples were incubated with 1% bovine serum albumin (BSA) for 1 h to remove nonspecific binding proteins, followed by mixing with the primary antibodies against each protein at room temperature for 1 h. Subsequently, the samples were incubated with streptavidin–horseradish peroxidase (HRP)-conjugated secondary antibodies for 20 min at room temperature. Lastly, the absorbance at 450 nm was measured using a microplate spectrophotometer (Thermo Fisher, MA).
Western Blot Assay
The radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime, Shanghai, China) was used to lyse the treated bEnd.3 brain endothelial cells for 15 min on ice and the proteins were quantified using a bicinchoninic acid (BCA) protein quantitative kit (Beyotime, Shanghai, China). Subsequently, approximately 30 μg samples were loaded and separated using 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The samples were then transferred to a poly(vinylidene difluoride) (PVDF) membrane, followed by blotting with antibodies against KLF6 (1:1000, Cat#sc-365633, Santa Cruz Biotechnology) and COX-2 (1:1000, Cat#12282, Cell Signaling Technologies) for 2 h at room temperature. After washing with Tris-buffered saline with Tween 20 (TBST) buffer, the membranes were incubated with the secondary anti-rabbit (1:1000, #7074, Cell Signaling Technologies) and anti-mouse antibodies (1:1000, #7076, Cell Signaling Technologies) for 1.5 h at room temperature. β-Actin (1:10 000, Cat#4970, Cell Signaling Technologies) was used as the negative control. Images were taken and analyzed with the software Image J.
Statistical Analysis
All procedures were repeated with three biological replicates to verify the results. GraphPad Prism 7.0 (GraphPad Software) was employed to perform statistical analysis. Data are presented as mean ± SD. Results were statistically analyzed using one-way analysis of variance (ANOVA) with Tukey’s post hoc test for multigroup comparisons. Data with P values < 0.05 were considered statistically significant.
Acknowledgments
This study was supported by the Cancer Hospital of China Medical University.
The authors declare no competing financial interest.
References
- Jevtovic-Todorovic V.; Hartman R. E.; Izumi Y.; Benshoff N. D.; Dikranian K.; Zorumski C. F.; Olney J. W.; Wozniak D. F. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J. Neurosci. 2003, 23, 876–882. 10.1523/JNEUROSCI.23-03-00876.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andropoulos D. B.; Greene M. F. Anesthesia and Developing Brains - Implications of the FDA Warning. N. Engl. J. Med. 2017, 376, 905–907. 10.1056/NEJMp1700196. [DOI] [PubMed] [Google Scholar]
- Gleich S. J.; Flick R.; Hu D.; Zaccariello M. J.; Colligan R. C.; Katusic S. K.; Schroeder D. R.; Hanson A.; Buenvenida S.; Wilder R. T.; Sprung J.; Voigt R. G.; Paule M. G.; Chelonis J. J.; Warner D. O. Neurodevelopment of children exposed to anesthesia: design of the Mayo Anesthesia Safety in Kids (MASK) study. Contemp. Clin. Trials 2015, 41, 45–54. 10.1016/j.cct.2014.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson A. J.; Disma N.; de Graaff J. C.; Withington D. E.; Dorris L.; Bell G.; Stargatt R.; Bellinger D. C.; Schuster T.; Arnup S. J.; Hardy P.; Hunt R. W.; Takagi M. J.; Giribaldi G.; Hartmann P. L.; Salvo I.; Morton N. S.; von Ungern Sternberg B. S.; Locatelli B. G.; Wilton N.; Lynn A.; Thomas J. J.; Polaner D.; Bagshaw O.; Szmuk P.; Absalom A. R.; Frawley G.; Berde C.; Ormond G. D.; Marmor J.; McCann M. E. Neurodevelopmental outcome at 2 years of age after general anaesthesia and awake-regional anaesthesia in infancy (GAS): an international multicentre, randomised controlled trial. Lancet 2016, 387, 239–250. 10.1016/S0140-6736(15)00608-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chidambaran V.; Costandi A.; D’Mello A. Propofol: a review of its role in pediatric anesthesia and sedation. CNS Drugs 2015, 29, 543–563. 10.1007/s40263-015-0259-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbas M. S.; El-Hakeem E. E. A.; Kamel H. E. Three minutes propofol after sevoflurane anesthesia to prevent emergence agitation following inguinal hernia repair in children: a randomized controlled trial. Korean J. Anesthesiol. 2019, 72, 253–259. 10.4097/kja.d.18.00345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson K. A.; Goodale D. B. The recent development of propofol (DIPRIVAN). Intensive Care Med. 2000, 26, S400–S404. 10.1007/PL00003783. [DOI] [PubMed] [Google Scholar]
- James R.; Glen J. B. Synthesis, biological evaluation, and preliminary structure-activity considerations of a series of alkylphenols as intravenous anesthetic agents. J. Med. Chem. 1980, 23, 1350–1357. 10.1021/jm00186a013. [DOI] [PubMed] [Google Scholar]
- Sanna E.; Mascia M. P.; Klein R. L.; Whiting P. J.; Biggio G.; Harris R. A. Actions of the general anesthetic propofol on recombinant human GABAA receptors: influence of receptor subunits. J. Pharmacol. Exp. Ther. 1995, 274, 353–360. [PubMed] [Google Scholar]
- Smith M. C.; Williamson J.; Yaster M.; Boyd G. J.; Heitmiller E. S. Off-label use of medications in children undergoing sedation and anesthesia. Anesth. Analg. 2012, 115, 1148–1154. 10.1213/ANE.0b013e3182501b04. [DOI] [PubMed] [Google Scholar]
- Creeley C.; Dikranian K.; Dissen G.; Martin L.; Olney J.; Brambrink A. Propofol-induced apoptosis of neurones and oligodendrocytes in fetal and neonatal rhesus macaque brain. Br. J. Anaesth. 2013, 110, i29–i38. 10.1093/bja/aet173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briner A.; Nikonenko I.; De Roo M.; Dayer A.; Muller D.; Vutskits L. Developmental Stage-dependent persistent impact of propofol anesthesia on dendritic spines in the rat medial prefrontal cortex. Anesthesiology 2011, 115, 282–293. 10.1097/ALN.0b013e318221fbbd. [DOI] [PubMed] [Google Scholar]
- Zhong Y.; Liang Y.; Chen J.; Li L.; Qin Y.; Guan E.; He D.; Wei Y.; Xie Y.; Xiao Q. Propofol inhibits proliferation and induces neuroapoptosis of hippocampal neurons in vitro via downregulation of NF-kappaB p65 and Bcl-2 and upregulation of caspase-3. Cell Biochem. Funct. 2014, 32, 720–729. 10.1002/cbf.3077. [DOI] [PubMed] [Google Scholar]
- Gao J.; Peng S.; Xiang S.; Huang J.; Chen P. Repeated exposure to propofol impairs spatial learning, inhibits LTP and reduces CaMKIIalpha in young rats. Neurosci. Lett. 2014, 560, 62–66. 10.1016/j.neulet.2013.11.061. [DOI] [PubMed] [Google Scholar]
- Yu D.; Jiang Y.; Gao J.; Liu B.; Chen P. Repeated exposure to propofol potentiates neuroapoptosis and long-term behavioral deficits in neonatal rats. Neurosci. Lett. 2013, 534, 41–46. 10.1016/j.neulet.2012.12.033. [DOI] [PubMed] [Google Scholar]
- Fredriksson A.; Ponten E.; Gordh T.; Eriksson P. Neonatal exposure to a combination of N-methyl-D-aspartate and gamma-aminobutyric acid type A receptor anesthetic agents potentiates apoptotic neurodegeneration and persistent behavioral deficits. Anesthesiology 2007, 107, 427–436. 10.1097/01.anes.0000278892.62305.9c. [DOI] [PubMed] [Google Scholar]
- Milanović D.; Pesic V.; Popic J.; Tanic N.; Kanazir S.; Jevtovic-Todorovic V.; Ruzdijic S. Propofol anesthesia induces proapoptotic tumor necrosis factor-alpha and pro-nerve growth factor signaling and prosurvival Akt and XIAP expression in neonatal rat brain. J. Neurosci. Res. 2014, 92, 1362–1373. 10.1002/jnr.23409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popic J.; Pesic V.; Milanovic D.; Todorovic S.; Kanazir S.; Jevtovic-Todorovic V.; Ruzdijic S. Propofol-induced changes in neurotrophic signaling in the developing nervous system in vivo. PLoS One 2012, 7, e34396 10.1371/journal.pone.0034396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C.; Du F.; Cang J.; Xue Z. Pink1 attenuates propofol-induced apoptosis and oxidative stress in developing neurons. J. Anesth. 2018, 32, 62–69. 10.1007/s00540-017-2431-2. [DOI] [PubMed] [Google Scholar]
- Al-Jahdari W. S.; Saito S.; Nakano T.; Goto F. Propofol induces growth cone collapse and neurite retractions in chick explant culture. Can. J. Anaesth. 2006, 53, 1078 10.1007/BF03022874. [DOI] [PubMed] [Google Scholar]
- Wang H.; Luo Z.; Xue Z. G.; Cang J. Propofol inhibits neuronal differentiation of mouse embryonic stem cells in vitro. Chin. Med. J. 2013, 126, 4186–4188. 10.3760/cma.j.issn.0366-6999.20130856. [DOI] [PubMed] [Google Scholar]
- Sharma H. S.; Ponten E.; Gordh T.; Eriksson P.; Fredriksson A.; Sharma A. Propofol promotes blood-brain barrier breakdown and heat shock protein (HSP 72 kd) activation in the developing mouse brain. CNS Neurol. Disord. Drug Targets 2014, 13, 1595–1603. 10.2174/1871527313666140806122906. [DOI] [PubMed] [Google Scholar]
- Takano Y.; Hase-Aoki K.; Horiuchi H.; Zhao L.; Kasahara Y.; Kondo S.; Becker M. A. Selectivity of febuxostat, a novel non-purine inhibitor of xanthine oxidase/xanthine dehydrogenase. Life Sci. 2005, 76, 1835–1847. 10.1016/j.lfs.2004.10.031. [DOI] [PubMed] [Google Scholar]
- Shafik A. N. Febuxostat improves the local and remote organ changes induced by intestinal ischemia/reperfusion in rats. Dig. Dis. Sci. 2013, 58, 650–659. 10.1007/s10620-012-2391-1. [DOI] [PubMed] [Google Scholar]
- Amirshahrokhi K. Febuxostat attenuates ulcerative colitis by the inhibition of NF-kappaB, proinflammatory cytokines, and oxidative stress in mice. Int. Immunopharmacol. 2019, 76, 105884 10.1016/j.intimp.2019.105884. [DOI] [PubMed] [Google Scholar]
- Khan S. I.; Malhotra R. K.; Rani N.; Sahu A. K.; Tomar A.; Garg S.; Nag T. C.; Ray R.; Ojha S.; Arya D. S.; Bhatia J. Febuxostat Modulates MAPK/NF-kappaBp65/TNF-alpha Signaling in Cardiac Ischemia-Reperfusion Injury. Oxid. Med. Cell. Longevity 2017, 2017, 8095825 10.1155/2017/8095825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahmi A. N.; Shehatou G. S.; Shebl A. M.; Salem H. A. Febuxostat protects rats against lipopolysaccharide-induced lung inflammation in a dose-dependent manner. Naunyn Schmiedebergs Arch. Pharmacol. 2016, 389, 269–278. 10.1007/s00210-015-1202-6. [DOI] [PubMed] [Google Scholar]
- Devasagayam T. P.; Tilak J. C.; Boloor K. K.; Sane K. S.; Ghaskadbi S. S.; Lele R. D. Free radicals and antioxidants in human health: current status and future prospects. J. Assoc. Physicians India 2004, 52, 794–804. [PubMed] [Google Scholar]
- Li J.; Zhou K.; Meng X.; Wu Q.; Li S.; Liu Y.; Wang J. Increased ROS generation and SOD activity in heteroplasmic tissues of transmitochondrial mice with A3243G mitochondrial DNA mutation. Genet. Mol. Res. 2008, 7, 1054–1062. 10.4238/vol7-4gmr480. [DOI] [PubMed] [Google Scholar]
- Taşman F.; Atac A.; Er N.; Dagdeviren A.; Kendir B. Expression of beta 1 integrins in human dental pulp in vivo: a comparative immunohistochemical study on healthy and chronic marginal periodontitis samples. Int. Endod. J. 2001, 34, 11–15. 10.1046/j.1365-2591.2001.00324.x. [DOI] [PubMed] [Google Scholar]
- Maritim A. C.; Sanders R. A.; Watkins J. B. 3rd Diabetes, oxidative stress, and antioxidants: a review. J. Biochem. Mol. Toxicol. 2003, 17, 24–38. 10.1002/jbt.10058. [DOI] [PubMed] [Google Scholar]
- Tang F.; Zhao L.; Yu Q.; Liu T.; Gong H.; Liu Z.; Li Q. Upregulation of miR-215 attenuates propofol-induced apoptosis and oxidative stress in developing neurons by targeting LATS2. Mol. Med. 2020, 26, 38 10.1186/s10020-020-00170-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.; Rainosek S. W.; Sadovova N.; Fogle C. M.; Patterson T. A.; Hanig J. P.; Paule M. G.; Slikker W. Jr.; Wang C. Protective effect of acetyl-L-carnitine on propofol-induced toxicity in embryonic neural stem cells. Neurotoxicology 2014, 42, 49–57. 10.1016/j.neuro.2014.03.011. [DOI] [PubMed] [Google Scholar]
- Antonelli A.; Ferrari S. M.; Giuggioli D.; Ferrannini E.; Ferri C.; Fallahi P. Chemokine (C-X-C motif) ligand (CXCL)10 in autoimmune diseases. Autoimmun. Rev. 2014, 13, 272–280. 10.1016/j.autrev.2013.10.010. [DOI] [PubMed] [Google Scholar]
- Tejchman A.; Lamerant-Fayel N.; Jacquinet J. C.; Bielawska-Pohl A.; Mleczko-Sanecka K.; Grillon C.; Chouaib S.; Ugorski M.; Kieda C. Tumor hypoxia modulates podoplanin/CCL21 interactions in CCR7+ NK cell recruitment and CCR7+ tumor cell mobilization. Oncotarget 2017, 8, 31876–31887. 10.18632/oncotarget.16311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K.; Fan J.; Han J. Ameliorating effects of traditional Chinese medicine preparation, Chinese materia medica and active compounds on ischemia/reperfusion-induced cerebral microcirculatory disturbances and neuron damage. Acta Pharm. Sin. B 2015, 5, 8–24. 10.1016/j.apsb.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi B.; Angiari S.; Zenaro E.; Budui S. L.; Constantin G. Vascular inflammation in central nervous system diseases: adhesion receptors controlling leukocyte-endothelial interactions. J. Leukocyte Biol. 2011, 89, 539–556. 10.1189/jlb.0710432. [DOI] [PubMed] [Google Scholar]
- Heikal M. M.; Shaaban A. A.; Elkashef W. F.; Ibrahim T. M. Effect of febuxostat on biochemical parameters of hyperlipidemia induced by a high-fat diet in rabbits. Can. J. Physiol. Pharmacol. 2019, 97, 611–622. 10.1139/cjpp-2018-0731. [DOI] [PubMed] [Google Scholar]
- Nomura J.; Kobayashi T.; So A.; Busso N. Febuxostat, a Xanthine Oxidoreductase Inhibitor, Decreases NLRP3-dependent Inflammation in Macrophages by Activating the Purine Salvage Pathway and Restoring Cellular Bioenergetics. Sci. Rep. 2019, 9, 17314 10.1038/s41598-019-53965-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno Y.; Yamamotoya T.; Nakatsu Y.; Ueda K.; Matsunaga Y.; Inoue M. K.; Sakoda H.; Fujishiro M.; Ono H.; Kikuchi T.; Takahashi M.; Morii K.; Sasaki K.; Masaki T.; Asano T.; Kushiyama A. Xanthine Oxidase Inhibitor Febuxostat Exerts an Anti-Inflammatory Action and Protects against Diabetic Nephropathy Development in KK-Ay Obese Diabetic Mice. Int. J. Mol. Sci. 2019, 20, 4680 10.3390/ijms20194680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho E. A.; Piquette-Miller M. KLF6 and HSF4 transcriptionally regulate multidrug resistance transporters during inflammation. Biochem. Biophys. Res. Commun. 2007, 353, 679–685. 10.1016/j.bbrc.2006.12.090. [DOI] [PubMed] [Google Scholar]
- Kim G. D.; Ng H. P.; Chan E. R.; Mahabeleshwar G. H. Kruppel-like factor 6 promotes macrophage inflammatory and hypoxia response. FASEB J. 2020, 34, 3209–3223. 10.1096/fj.201902221R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T.; Aizawa K.; Matsumura T.; Nagai R. Vascular implications of the Kruppel-like family of transcription factors. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1135–1141. 10.1161/01.ATV.0000165656.65359.23. [DOI] [PubMed] [Google Scholar]
- White W. B.; Saag K. G.; Becker M. A.; et al. Cardiovascular safety of febuxostat or allopurinol in patients with gout. N. Engl. J. Med. 2018, 378, 1200–1210. 10.1056/NEJMoa1710895. [DOI] [PubMed] [Google Scholar]
- Robinson P. C.; Dalbeth N. Febuxostat for the treatment of hyperuricaemia in gout. Expert Opin. Pharmacother. 2018, 19, 1289–1299. 10.1080/14656566.2018.1498842. [DOI] [PubMed] [Google Scholar]
- https://www.fda.gov/drugs/drug-safety-and-availability/fda-adds-boxed-warning-increased-risk-death-gout-medicine-uloric-febuxostat.
- Angeloni C.; Pirola L.; Vauzour D.; Maraldi T. Dietary polyphenols and their effects on cell biochemistry and pathophysiology.. Oxid. Med. Cell. Longevity 2012, 2012, 583901 10.1155/2012/583901. [DOI] [PMC free article] [PubMed] [Google Scholar]