

Abstract
Introduction
This study characterizes retinal capillary blood flow in subjects with autosomal dominant Alzheimer's disease (ADAD)‐causing mutations.
Methods
Carriers of PSEN1 or APP mutations were prospectively recruited and split into early‐stage (ES) and late‐stage (LS) groups. Controls were normal subjects and non‐carriers from the at‐risk group. Capillary blood flow was quantified using an optical coherence tomography angiography‐based measure of erythrocyte flux through capillary segments. Statistical analyses were adjusted for correlation between two eyes of the same subject.
Results
ES carriers had significantly greater capillary blood flow than controls and LS carriers. ES and LS carriers had significantly greater capillary blood flow heterogeneity than controls. There was no difference between capillary blood flow of LS carriers and controls.
Discussion
ES ADAD carriers demonstrate increased retinal capillary blood flow and flow heterogeneity compared to controls. These findings support the hypothesis that increased perfusion is a pathophysiologic feature of presymptomatic stages of ADAD.
Keywords: autosomal dominant Alzheimer's disease, biomarkers, capillary blood flow, Latino, optical coherence tomography angiography, retina
1. INTRODUCTION
Pathologic changes have been found in the retina in association with late onset Alzheimer's disease (LOAD). Eye histopathology in human subjects affected by LOAD has demonstrated optic nerve atrophy 1 and thinning of retinal layers. 2 Retinal thinning in vivo, particularly of the retinal nerve fiber layer, has also been confirmed in numerous human clinical studies. 3 Various forms of amyloid beta (Aβ) protein have been demonstrated in the eyes of at least some persons with LOAD. 4 , 5 , 6 , 7 Pathologic changes in capillaries may be some of the earliest vascular changes associated with LOAD. 8 In vivo studies of retinal blood flow in human subjects have demonstrated either decreased capillary density, impaired arteriolar vessel reactivity, or reduced foveal avascular zone size among subjects with LOAD. 9 , 10 , 11 Collectively these findings suggest that vascular changes in the retina may parallel changes in the brain and be related to the underlying pathological process in LOAD. However, the specific contribution of LOAD as opposed to confounding factors such as age, hypertension, diabetes, and other vascular disease is difficult to ascertain in aged subjects. 4 , 5 , 6 , 7 , 12
There is a small subset of patients, estimated to be around 1%, for whom Alzheimer's disease (AD) is inherited in an autosomal dominant fashion (autosomal dominant AD or ADAD). 13 For those that carry ADAD mutations, development of disease is essentially certain and the age of symptom onset is both young and to some extent predictable. 14 , 15 , 16 Though differences exist, 4 , 5 , 6 , 7 , 12 the clinical and pathological phenotype of ADAD is similar to that of LOAD, and therefore ADAD is a valuable model for studying LOAD biomarkers. This is particularly true because subjects with ADAD are young and lack confounding comorbid vascular disease that is inherent in subjects with LOAD. To date, there are no studies evaluating retinal vascular changes in ADAD subjects.
In this study we use a noninvasive, Food and Drug Administration–approved imaging modality, optical coherence tomography angiography (OCTA), to quantify changes in carriers of ADAD mutations (A431E and F388S substitutions in PSEN1 or V717I substitution in APP) at the capillary level. We also implement a novel method of assessing erythrocyte flux within capillaries to determine whether subclinical changes in capillary blood flow occur before the onset of measurable changes in capillary density.
2. METHODS
This was a cross‐sectional, observational study of ADAD mutation carriers and healthy controls recruited from an ongoing National Institutes of Health–funded study of families with ADAD. This study was approved by the institutional review board of the University of Southern California (USC) and adhered to the tenets of the Declaration of Helsinki. Informed consent was obtained from each subject or their appropriate surrogate.
2.1. Subjects
All ADAD subjects were persons of Latino origin known to be affected by or at risk for inheriting the F388S or A431E substitutions in PSEN1 17 or V717I substitution in APP 18 mutations. Numerous families at risk for the A431E mutation have been identified, representing a founder effect centered in Jalisco State in Mexico. 17 , 19 Non‐carriers from the at‐risk group were included in the control group.
RESEARCH IN CONTEXT
Systematic Review: The authors reviewed the literature using traditional (e.g., PubMed) sources. There has been much research on retinal biomarkers for late onset Alzheimer's disease (LOAD) but not for autosomal dominant AD (ADAD).
Interpretation: Capillary blood flow as assessed by optical coherence tomography angiography (OCTA) is increased in preclinical carriers of ADAD causing mutations. This is consistent with findings from other studies on subjects at risk for LOAD demonstrating increased cerebral blood flow in carriers of the apolipoprotein E ε4 allele using arterial spin labeling and positron emission tomography.
Future directions: This article provides evidence that retinal capillary blood flow, as assessed by OCTA, may play a useful role in detection of vascular abnormalities before onset of ADAD. Larger prospective studies are needed to confirm and extend these findings.
Subjects from ADAD families were recruited based on their “at‐risk” status. As the genetic tests performed in this study were not performed in a Clinical Laboratory Improvement Act (CLIA) laboratory, participants were not provided with these specific results and they signed consents acknowledging this. However, all participants were given the option of undergoing clinical presymptomatic testing through a genetic counselor outside of the study at no cost to them. Some subjects were already overtly affected with dementia and/or spastic paraparesis, a feature atypical for LOAD but that is seen in association with some PSEN1 mutations. 20 The age of dementia diagnosis can be fairly consistent based on the mutation variant; ≈42 years for the A431E PSEN1, 17 25 years for the F388S PSEN1, and 52 years for the V717I APP mutation. 16 , 19 , 21 All participants had study partners who provided collateral information.
The medical and symptom history of participants at risk for ADAD was obtained using the Uniform Data Set 3 of the Alzheimer's Disease Center protocol, 22 which includes the Clinical Dementia Rating (CDR) scale. 23 The CDR is a structured interview of the participant and study partner in which participants are categorized as either being cognitively normal (CDR = 0); having mild or questionable impairment (CDR = 0.5); or having mild, moderate, or severe dementia (CDR scores of 1, 2, and 3, respectively). ADAD and control subjects with major psychiatric illness or known history of any non‐AD neurological disorder (judged to possibly affect study measures, such as cysticercosis or major cerebrovascular accident) were excluded. Subjects were also excluded if they had any past ocular history of glaucoma, hypertensive retinopathy, retinal surgery, or any past medical history of significant vascular disease. Controls consisted of age‐matched healthy controls recruited at the USC Roski Eye Institute and four non‐mutation carrying persons from the ADAD families.
All subjects underwent comprehensive ophthalmic examination including assessment of visual acuity, intraocular pressure, slit‐lamp examination, and dilated fundus examination by a board‐certified ophthalmologist (AHK). Participants were excluded if they had any past medical history of vascular diseases including diabetes, heart disease, or vasculitis. All subjects with significant media opacity such as cataracts or any other obscuration of the fundus were also excluded.
2.2. Genetic testing
The presence of the APP V717I and PSEN1 A431E and F388S substitutions were assessed at the USC Alzheimer's Disease Research Center using the Sanger sequencing method. Briefly, DNA was extracted from an aliquot of whole blood using the Quick‐DNATM Miniprep Kit (Cat. No. D3024, Zymo Research). Approximately 50 ng DNA was amplified in a 25 μL reaction mixture with AccuPrimeTM Taq DNA Polymerase (Thermo Fisher Scientific) and following respective primers. The forward and reverse primers for APP were 5′‐CAACCAGTTGGGCAGAGAAT‐3′ and 5′‐AGCATCATGGAAGCACACTG‐3′, respectively. The forward and reverse primers for PSEN1 were 5′‐TTCCAGATTGAATGAACGTCT‐3′ and 5′‐CGCAGTGTCAGTGAAATCGT‐3′, respectively. The polymerase chain reaction (PCR) products were purified using a one‐step ExoSAP‐ITTM PCR product cleanup reagent (Cat. No. 78200, Thermo Fisher Scientific) before sequencing.
2.3. OCTA imaging protocol
The 3 × 3 mm scans centered on the fovea were acquired by a commercially available spectral domain OCTA system (AngioPlex, Carl Zeiss Meditec), with a central wavelength of 840 nm and a scan speed of 68,000 A‐scans per second. Each OCTA volume is formed by 350 horizontal scans (B‐scans) with each B‐scan formed by 300 A‐scans. The device uses an optical microangiography (OMAG) algorithm 24 , 25 to determine the decorrelation signal, which represents the movement of erythrocytes. OCTA capture was blinded to genetic data. Subjects who were already overtly affected due to their clinical status were apparent to the imaging technicians. However, clinical status was not known to those performing image analyses.
Images with poor decentration of the foveal avascular zone (FAZ), signal strength less than seven (as assessed by the manufacturer's image quality designation), significant motion artifacts, or vitreous floaters were excluded. The OCTA volumes were automatically segmented by the manufacturer's software to construct en face superficial retinal layer (SRL) images. The SRL extends from the inner limiting membrane (ILM) through the inner plexiform layer (IPL), which is approximated as 70% of the thickness between the ILM and outer plexiform layer (OPL). This segmentation methodology is commercially available and has been described before. 26 Segmentation for all scans was manually reviewed to ensure accuracy. In no case was it necessary to modify the automated segmentation results.
2.4. Quantitative capillary flow and morphometric measures
Capillary density and caliber were calculated as described previously 26 , 27 , 28 with modifications to allow for exclusion of non‐capillary vessels such as arterioles and venules. Specifically, capillary density was measured as vessel skeleton density (VSD), which represents the total length of capillaries. It is calculated by skeletonizing the diameter of all the vessels from a binarized OCTA image to 1 pixel width, and then dividing the number of skeletonized vessel pixels by the total area of the image that is not occupied by large vessels. Vessel diameter index (VDI) provides a relative measurement of vessel caliber in the OCTA image. It is calculated by dividing the binarized OCTA image by the skeletonized image to yield an index of vessel caliber. 26 , 27 , 28 The FAZ and non‐capillary vessels (arterioles and venules) were excluded from both VSD and VDI measures using a manual threshold set empirically to exclude vessels that appear larger than individual capillaries (Figure 1A‐D).
FIGURE 1.
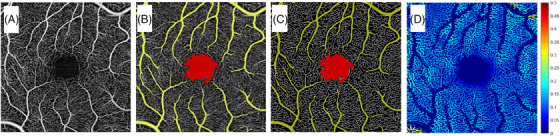
Segmented en face superficial retinal layer (SRL) 3 × 3 mm optical coherence tomography angiography (OCTA) scan centered around the fovea of a control subject. A, Original image of retinal perfusion. B, Image after exclusion of large vessels in yellow and foveal avascular zone (FAZ) in red. C, Skeletonized image of retinal perfusion after exclusion of large vessels in yellow and FAZ in red. D, Pseudo‐colored image of OCTA flux, representing capillary blood flow, with warmer colors corresponding to higher flow
Unlike larger vessels, capillary blood flow occurs by passage of individual erythrocytes through thin‐walled vessels that are approximately the diameter of an erythrocyte. Therefore, the raw decorrelation intensity values from the capillaries in an OCTA image correspond to movement of erythrocytes and the magnitude of the values is correlated with the number of erythrocytes passing through a capillary segment (also called capillary flux). Measurement of capillary flux is effectively a measure of total capillary blood flow through a segment of capillary. Non‐capillary vessels (arterioles and venules) do not behave in a similar manner and were therefore excluded from the blood flow measurements. Total capillary blood flow for a given OCTA image was calculated by averaging all the flux intensity values of the pixels occupied by capillaries and exclusive of arterioles or venules (Figure 1D).
The standard deviation for the flux intensity values for each image was also calculated and represents capillary blood flow heterogeneity based on similar methods used in animal models to calculate capillary velocity heterogeneity using OCTA. 29 Capillary blood flow heterogeneity quantifies the variability in blood flow through all the capillaries within a single image.
2.5. Grouping and statistical analysis
Adjusted age, a measure of estimated years until or since diagnosis of dementia, was calculated for all carriers by the following method based on a methodology used in prior studies on ADAD. 14 , 15 , 30 For those already meeting criteria for dementia, adjusted age was the age on the date of imaging minus the age of initial diagnosis of dementia, when available, or its best estimate based on clinical information. Therefore, all subjects meeting criteria for dementia at the time of imaging had an adjusted age of greater than or equal to zero. For subjects not yet meeting the criteria for dementia, the adjusted age was age at the time of imaging minus the median age at which persons with the mutation for which they were at risk are diagnosed with dementia. Therefore, all subjects not meeting criteria for dementia at the time of imaging had an adjusted age of less than zero. Mutation carriers were divided into early‐stage (ES) and late‐stage (LS) groups based on an age‐adjusted cutoff of –4 (4 years before predicted age of dementia diagnosis based on mutation variant); –4 was chosen as the adjusted age cutoff to best separate clinical from preclinical carriers based on evidence that cognition among ADAD carriers becomes significantly different from non‐carriers 2 or 3 years before symptom onset. 30
Pearson's χ2 analysis was used to test whether there were significant between‐group differences for sex, age, and ethnicity. A generalized estimating equations model adjusting for correlated data due to inclusion of both eyes from the same subject was used to determine associations between OCTA measures (capillary density, caliber, blood flow, and blood flow heterogeneity) and carrier status (ES carrier, LS carrier, and non‐carrier controls). Our model did not adjust for sex because it was not significantly different between groups in our univariate analysis. We also conducted sensitivity analyses using data from only one randomly selected eye from each subject, and using data excluding non‐Latino controls, current smokers, or those with hypertension. We did not include age as a covariate, because the independent variable (carrier stage) was defined based on adjusted age, the difference between age and predicted/historical age of diagnosis of dementia. Each OCTA measure was analyzed separately from the other measures to avoid inflation of parameter estimate standard errors due to collinearity. A P < .05 was accepted as statistically significant. SAS® (version 9.4; SAS Institute Inc.) was used for all statistical analyses.
3. RESULTS
Our study included 39 control eyes from 21 subjects (mean age 37.6 ± 10.3), 9 ES carrier eyes from 5 subjects (mean age 30 ± 8.4), and 11 LS carrier eyes from 8 subjects (mean age 41 ± 7.1). Among the five ES mutation carriers, four had the A431E PSEN1 mutation and one had the V717I APP mutation. Among the eight LS carriers, seven had the A431E PSEN1 mutation and one had the F388S PSEN1 mutation. Among the five ES mutation carriers, four had CDR scores of 0 and one had a CDR score of 0.5. Among the LS carriers, two had CDR scores of 0.5, three had CDR scores of 1, two had CDR scores of 2, and one had a CDR score of 3.0. Table 1 summarizes the subject demographics. None of the subjects were taking antihypertensives or antiplatelets.
TABLE 1.
Subject demographics
| Carriers | ||||
|---|---|---|---|---|
| Controls | Early‐stage | Late‐stage | P for between‐group difference | |
| Mutations | N/A |
V717I APP, n = 1 A431E PSEN1, n = 4 |
F388S PSEN1, n = 1 A431E PSEN1, n = 7 |
– |
| Number of eyes | 39 | 9 | 11 | – |
| Number of subjects | 21 | 5 | 8 | – |
| Male sex, N (%) a | 14 (66.7%) | 2 (60%) | 6 (75%) | 0.175 |
| Age, mean ± SD | 37.6 ± 10.3 | 30 ± 8.4 | 41 ± 7.1 | 0.00008 |
| CDR‐SOB, mean ± SD | N/A | 0.3 ± 0.4 | 8 ± 4.9 | – |
| CDR total score | N/A |
CDR = 0, n = 4, CDR = 0.5, n = 1,. |
CDR = 0.5. n = 2 CDR = 1, n = 3 CDR = 2. n = 2 CDR = 3, n = 1 |
– |
| Hypertension, N (%) | 1 (4.8%) | 0 (0%) | 0 (0%) | – |
| Current smoker, N (%) | 2 (9.5%) | 2 (40%) | 0 (0%) | – |
| Ethnicity | 9 Latino, 7 Asian, 3 White, 1 Other, 1 Refused | All Latino | All Latino | 0.003 |
The mutation variant, number, sex, age, adjusted age, Clinical Dementia Rating Scale Sum of Boxes scores (CDR‐SOB), CDR total score, hypertension status, smoking status and ethnicity of subjects is summarized.
Abbreviations: CDR, Clinical Dementia Rating; SD, standard deviation.
No significant differences between groups.
Figure 2 shows examples of the capillary blood flow in an ES carrier, LS carrier, and control. Capillary flow appears qualitatively greater in the ES carrier than in both the LS and the control images. Figure 3A‐B illustrates histograms of capillary blood flow value (flux) distributions for all the OCTA scan pixels from one representative subject in each group confirming the right‐ward shift in the blood flow values among ES and LS carriers compared to controls. Quantitative analysis of the whole cohort confirmed significantly higher capillary flow among the ES carriers compared to both the LS carriers (0.134 ± 0.002 vs. 0.120 ± 0.003, P < .0001) and controls (0.134 ± 0.002 vs. 0.119 ± 0.002, P < .0001; Figure 4A). There was no significant difference in capillary flow between controls and LS carriers (0.119 ± 0.002 vs. 0.120 ± 0.003, P = .7 Figure 4A).
FIGURE 2.

Segmented en face pseudo‐colored images representing magnitude of capillary flow, in early‐stage (ES) carrier on left, late‐stage (LS) carrier in middle, and control on right. Colder colors correspond to lower flow and warmer colors correspond to higher flow. Flow appears qualitatively greatest in ES carrier
FIGURE 3.
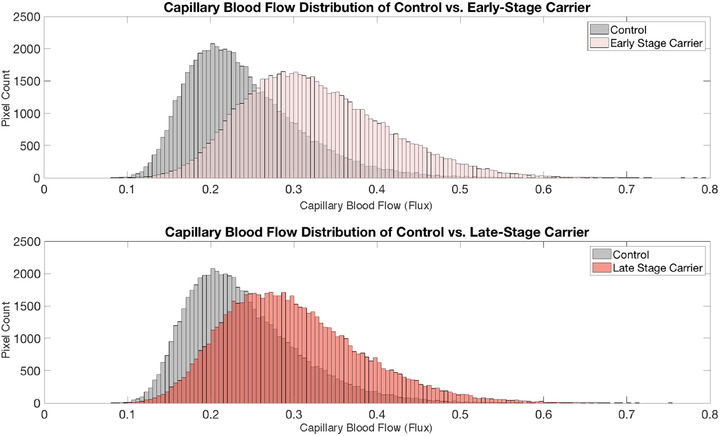
Histogram distributions of capillary flow values (flux) from all the pixels in an optical coherence tomography angiography scan from a representative control subject and early‐stage carrier (A), and a representative control subject and late‐stage carrier (B). Flow heterogeneity, that is, standard deviation of all pixel flow values, is greater in both the early and late stage carriers than the control subject
FIGURE 4.
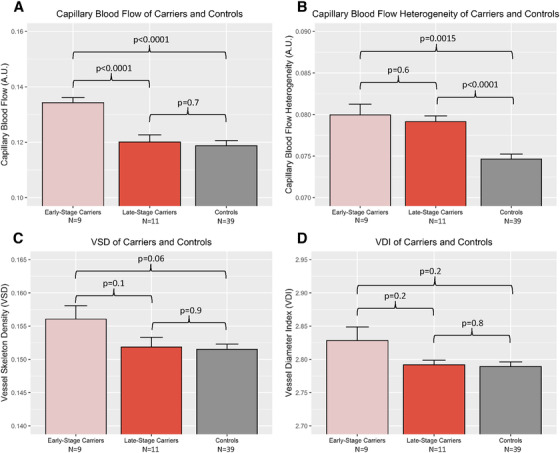
Capillary blood flow and flow heterogeneity among early‐stage (ES) carriers, late‐stage (LS) carriers, and controls. Capillary blood flow is significantly greater in ES carriers than either LS carriers or controls (A), capillary blood flow heterogeneity is significantly greater in ES carriers and LS carriers than in controls (B), capillary density is not significantly different among the three groups in the cohort (C), and vessel caliber is not significantly different among the three groups in the cohort (D). Error bars indicate standard error
Capillary blood flow heterogeneity, as measured by the standard deviation of the blood flow values for each group, was significantly higher in both the ES carriers (0.0800 ± 0.001 vs. 0.0747 ± 0.001, P = .0015) and in the LS carriers (0.0792 ± 0.001 vs. 0.0747 ± 0.001, P < .0001) compared to the controls (Figure 4B). There was no difference in blood flow heterogeneity between ES and LS carriers (0.0800 ± 0.001 vs. 0.0792 ± 0.001, P = .6, and Figure 4B).
Capillary density of the ES carriers was not significantly different than that of LS carriers (0.156 ± 0.002 vs. 0.152 ± 0.001, P = .1) or controls (0.156 ± 0.002 vs. 0.152 ± 0.0008, P = .06 and Figure 4C). There was also no difference between capillary density of LS carriers and controls (0.152 ± 0.001 vs. 0.152 ± 0.0008, P = .9 and Figure 4C). Vessel caliber of the ES carriers was not significantly different than that of the LS carriers (2.83 ± 0.020 vs. 2.79 ± 0.007, P = .2 ) and that of the controls (2.83 ± 0.020 vs. 2.78 ± 0.007, P = .2 and Figure 4C). There was no difference between vessel caliber of LS carriers and controls (2.79 ± 0.007 vs. 2.78 ± 0.007, P = .8; Figure 4D). Repeating the above analyses using data from only one randomly selected eye from each subject, instead of both eyes, did not significantly change the results or conclusions. Nor did repeating the above analyses excluding non‐Latino controls, subjects with hypertension, or current smokers change the results of conclusions.
All of our ES carriers (i.e., those > 4 years younger than predicted age of dementia diagnosis based on mutation variant) were asymptomatic, except for one. This subject was an outlier because he/she developed symptoms many years earlier than was predicted by his/her mutation variant but he/she had not yet reached the threshold for dementia diagnosis. To be conservative, we repeated our analysis with this subject reclassified into the late‐stage category (i.e., comparing all asymptomatic to symptomatic carriers) and our results were unchanged. Specifically, capillary blood flow among ES carriers was still significantly higher than that of LS carriers (0.134 ± 0.006 vs. 0.122 ± 0.01, P < .0001) and controls (0.134 ± 0.006 vs. 0.119 ± 0.01, P < .001). There was no significant difference in capillary blood flow between LS carrier and controls (0.122 ± 0.01 vs. 0.119 ± 0.01, P = .3). Capillary blood flow heterogeneity was significantly higher in both ES carriers (0.080 ± 0.004 vs. 0.075 ± 0.004, P = .015) and LS carriers (0.079 ± 0.002 vs. 0.075 ± 0.004, P < .0001) than controls. There was no difference in capillary blood flow heterogeneity between ES and LS carriers (0.080 ± 0.004 vs. 0.079 ± 0.02, P = .6). Capillary density of ES carriers was significantly greater than controls (0.158 ± 0.005 vs. 0.152 ± 0.005, P = .0004) but not LS carriers (0.158 ± 0.005 vs. 0.151 ± 0.005, P = .1). There was no difference in capillary density between controls and LS carriers (0.152 ± 0.005 vs. 0.151 ± 0.005, P = .9). Vessel caliber was not significantly different between ES carriers and LS carriers (2.80 ± 0.04 vs. 2.81 ± 0.05, P = .2), ES carriers and controls (2.80 ± 0.04 vs. 2.79 ± 0.04, P = .4), or LS carriers and controls (2.81 ± 0.05 vs. 2.79 ± 0.04, P = .3). Figure S1 in supporting information shows the capillary blood flow and adjusted age (estimated age to dementia diagnosis) of our outlier in the context of the rest of our subjects.
4. DISCUSSION
This was a pilot cross‐sectional study investigating retinal capillary blood flow, caliber, and density in presymptomatic and symptomatic carriers of the ADAD‐causing mutations, A431E and F388S in PSEN1 and V717I in APP, which confer a predictable age of symptom onset and dementia diagnosis. We show that ES carriers of these ADAD mutations demonstrate greater capillary blood flow without any concomitant measurable change in capillary density or caliber. This finding was accompanied by greater capillary blood flow heterogeneity in both ES and LS carriers. Collectively these findings suggest that there are measurable abnormalities in blood flow during early stages of AD pathophysiology, before there is attenuation of capillary density.
These observed differences in retinal capillary blood flow may parallel those previously described in the brain of preclinical LOAD subjects. Specifically, studies using arterial spin labeling, positron emission tomography, and functional magnetic resonance imaging have shown that apolipoprotein E (APOE) ε4 carriers display greater cerebral blood flow (CBF) in the presymptomatic state. 31 , 32 , 33 , 34 , 35 , 36 A model has been established to explain this observation of cerebral hyperperfusion in early stages of AD. 37 It argues that AD causes damage to capillaries resulting in increasing capillary flow and flow heterogeneity. A similar phenomenon has been demonstrated using OCTA in aging mice. 29 Namely, older mice with age‐related capillary damage were found to have increased capillary velocities as well as increased capillary velocity heterogeneity compared to younger mice.
Our results and their implications in ADAD are consistent with studies implicating vascular changes in LOAD. Specifically, similar to our data, Querques et al. recently demonstrated that subjects with mild cognitive impairment (MCI) or LOAD also have no measurable change in macular capillary density using OCTA but do have impaired retinal arteriolar reactivity in response to physiologic retinal stimulation. 9 Abnormal arteriolar dilation could be a possible explanation for the increased capillary flow heterogeneity that we observe in our data. Additionally, it is feasible that the increased flow demonstrated in ES carriers is compensatory for any potentially impaired arteriolar dilation that may be occurring in our population. At least one other study demonstrates impaired retinal arteriolar blood flow among subjects with AD and MCI. 38 These observations seem to support similar pathophysiological mechanisms underlying LOAD and ADAD.
It is noteworthy that there are several published findings of decreased retinal capillary density in subjects with LOAD. 10 , 39 , 40 , 41 , 42 These studies generally describe lower retinal capillary density measures among symptomatic LOAD subjects 39 , 41 , 43 but not subjects with earlier stages of disease or MCI. 43 In addition, most of these studies only find significant differences in FAZ area 11 or foveal capillary density 39 and not global density measures. These studies tend to support the idea that changes in capillary density, if present, occur later in the course of LOAD.
Our study has some limitations. As this was a pilot study and these ADAD mutations are rare, our sample size of ES and LS carriers is small. Some subjects had potential confounders of retinal vascular flow, such as smoking and hypertension, but excluding those subjects did not materially change our results. Our controls included Latinos and healthy subjects from other racial/ethnic groups. There may be racial/ethnic differences in retinal capillary blood flow that may confound the observed differences between controls and carrier groups; however, when excluding non‐Latino controls our results were materially unchanged. Because this was a cross‐sectional rather than longitudinal study, our observed differences between our ES and LS carriers may be influenced by intersubject variability and not reflect chronic changes associated with disease progression in carriers. Nevertheless, these findings need to be assessed further in larger prospective studies and in studies that follow ADAD patients longitudinally.
CONFLICTS OF INTEREST
John M. Ringman is a DMC member of Renew Group Private Limited and a consultant for InnoSense. Carl Zeiss Meditec has provided grant funding, equipment, and financial support to Amir H Kashani. Ruikang K. Wang holds patents with Carl Zeiss Meditec and Kowa Inc and is a consultant for Carl Zeiss Meditec and Insight Phototonic Solutions.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
The authors would like to thank Berislav Zlokovic, Abhay Sagare, and Melanie D. Sweeney for their help with genotyping of the subjects for the study. This study was supported by the Tai Family Research Scholars Fund (MBS) and by grants from the NIH: K08EY027006, R01EY030564, UH2NS100614, UH3NS100614, P50AG05142, R01AG062007; Bright Focus Foundation CA2020004; unrestricted departmental funding from Research to Prevent Blindness (New York, NY, USA); and research grants from Carl Zeiss Meditec Inc (Dublin, CA, USA). Carl Zeiss Meditec, Inc was not, however, consulted in the design, implementation, or analysis of the study data.
Singer MB, Ringman JM, Chu Z, et al. Abnormal retinal capillary blood flow in autosomal dominant Alzheimer's disease. Alzheimer's Dement. 2021;13:e12162. 10.1002/dad2.12162
REFERENCES
- 1. Hinton DR, Sadun AA, Blanks JC, Miller CA. Optic‐nerve degeneration in Alzheimer's disease. N Engl J Med. 1986;315:485‐487. [DOI] [PubMed] [Google Scholar]
- 2. Blanks JC, Hinton DR, Sadun AA, Miller CA. Retinal ganglion cell degeneration in Alzheimer's disease. Brain Res. 1989;501:364‐372. [DOI] [PubMed] [Google Scholar]
- 3. Chan VTT, Sun Z, Tang S, et al. Spectral‐domain OCT measurements in Alzheimer's disease: a systematic review and meta‐analysis. Ophthalmology. 2019;126:497‐510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. den Haan J, Morrema THJ, Verbraak FD, et al. Amyloid‐beta and phosphorylated tau in post‐mortem Alzheimer's disease retinas. Acta Neuropathol Commun. 2018;6:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Koronyo Y, Biggs D, Barron E, et al. Retinal amyloid pathology and proof‐of‐concept imaging trial in Alzheimer's disease. JCI Insight. 2017;2:e93621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. La Morgia C, Ross‐Cisneros FN, Koronyo Y, et al. Melanopsin retinal ganglion cell loss in Alzheimer's disease. Ann Neurol. 2016;79:90‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Koronyo‐Hamaoui M, Koronyo Y, Ljubimov AV, et al. Identification of amyloid plaques in retinas from Alzheimer's patients and noninvasive in vivo optical imaging of retinal plaques in a mouse model. NeuroImage. 2011;54 Suppl 1:S204‐217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bell RD, Zlokovic BV. Neurovascular mechanisms and blood‐brain barrier disorder in Alzheimer's disease. Acta Neuropathol (Berl). 2009;118:103‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Querques G, Borrelli E, Sacconi R, et al. Functional and morphological changes of the retinal vessels in Alzheimer's disease and mild cognitive impairment. Sci Rep. 2019;9:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lahme L, Esser EL, Mihailovic N, et al. Evaluation of ocular perfusion in Alzheimer's disease using optical coherence tomography angiography. J Alzheimers Dis JAD. 2018;66:1745‐1752. [DOI] [PubMed] [Google Scholar]
- 11. O'Bryhim BE, Apte RS, Kung N, Coble D, Van Stavern GP. Association of preclinical Alzheimer's disease with optical coherence tomographic angiography findings. JAMA Ophthalmol. 2018;136:1242‐1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ringman JM, Goate A, Masters CL, et al. Genetic heterogeneity in Alzheimer's disease and implications for treatment strategies. Curr Neurol Neurosci Rep. 2014;14:499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moulder KL, Snider BJ, Mills SL, et al. Dominantly inherited Alzheimer's Network: facilitating research and clinical trials. Alzheimers Res Ther. 2013;5:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bateman RJ, Xiong C, Benzinger TLS, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. 2012;367:795‐804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ringman JM, Younkin SG, Pratico D, et al. Biochemical markers in persons with preclinical familial Alzheimer's disease. Neurology. 2008;71:85‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ryman DC, Acosta‐Baena N, Aisen PS, et al. Symptom onset in autosomal dominant Alzheimer's disease: a systematic review and meta‐analysis. Neurology. 2014;83:253‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Murrell J, Ghetti B, Cochran E, et al. The A431E mutation in PSEN1 causing Familial Alzheimer's Disease originating in Jalisco State, Mexico: an additional fifteen families. Neurogenetics. 2006;7:277‐279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Goate A, Chartier‐Harlin M‐C, Mullan M, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349:704‐706. [DOI] [PubMed] [Google Scholar]
- 19. Yescas P, Huertas‐Vazquez A, Villarreal‐Molina MT, et al. Founder effect for the Ala431Glu mutation of the presenilin 1 gene causing early‐onset Alzheimer's disease in Mexican families. Neurogenetics. 2006;7:195‐200. [DOI] [PubMed] [Google Scholar]
- 20. Soosman SK, Joseph‐Mathurin N, Braskie MN, et al. Widespread white matter and conduction defects in PSEN1‐related spastic paraparesis. Neurobiol Aging. 2016;47:201‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mullan M, Tsuji S, Miki T, et al. Clinical comparison of Alzheimer's disease in pedigrees with the codon 717 Val→Ile mutation in the amyloid precursor protein gene. Neurobiol Aging. 1993;14:407‐419. [DOI] [PubMed] [Google Scholar]
- 22. Besser L, Kukull W, Knopman DS, et al. Version 3 of the National Alzheimer's Coordinating Center's Uniform Data Set. Alzheimer Dis Assoc Disord. 2018;32:351‐358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Burke WJ, Miller JP, Rubin EH, et al. Reliability of the Washington University Clinical Dementia Rating. Arch Neurol. 1988;45:31‐32. [DOI] [PubMed] [Google Scholar]
- 24. Wang RK, An L, Francis P, Wilson DJ. Depth‐resolved imaging of capillary networks in retina and choroid using ultrahigh sensitive optical microangiography. Opt Lett. 2010;35:1467‐1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. An L, Qin J, Wang RK. Ultrahigh sensitive optical microangiography for in vivo imaging of microcirculations within human skin tissue beds. Opt Express. 2010;18:8220‐8228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim AY, Chu Z, Shahidzadeh A, Wang RK, Puliafito CA, Kashani AH. Quantifying microvascular density and morphology in diabetic retinopathy using spectral‐domain optical coherence tomography angiography. Invest Ophthalmol Vis Sci. 2016;57:OCT362‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reif R, Qin J, An L, Zhi Z, Dziennis S, Wang R. Quantifying optical microangiography images obtained from a spectral domain optical coherence tomography system. Int J Biomed Imaging. 2012;2012:509783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chu Z, Lin J, Gao C, et al. Quantitative assessment of the retinal microvasculature using optical coherence tomography angiography. J Biomed Opt. 2016;21:66008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li Y, Choi WJ, Wei W, et al. Aging‐associated changes in cerebral vasculature and blood flow as determined by quantitative optical coherence tomography angiography. Neurobiol Aging. 2018;70:148‐159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McDade E, Wang G, Gordon BA, et al. Longitudinal cognitive and biomarker changes in dominantly inherited Alzheimer's disease. Neurology. 2018;91:e1295‐306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fleisher AS, Podraza KM, Bangen KJ, et al. Cerebral perfusion and oxygenation differences in Alzheimer's disease risk. Neurobiol Aging. 2009;30:1737‐1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Scarmeas N, Habeck CG, Stern Y, Anderson KE. APOE genotype and cerebral blood flow in healthy young individuals. JAMA. 2003;290:1581‐1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thambisetty M, Beason‐Held L, An Y, Kraut MA, Resnick SM. APOE epsilon4 genotype and longitudinal changes in cerebral blood flow in normal aging. Arch Neurol. 2010;67:93‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wierenga CE, Clark LR, Dev SI, et al. Interaction of Age and APOE genotype on cerebral blood flow at rest. J Alzheimers Dis. 2013;34:921‐935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bookheimer SY, Strojwas MH, Cohen MS, et al. Patterns of brain activation in people at risk for Alzheimer's disease. N Engl J Med. 2000;343:450‐456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ringman JM, Medina LD, Braskie M, et al. Effects of Risk genes on BOLD activation in presymptomatic carriers of Familial Alzheimer's Disease Mutations during a novelty encoding task. Cereb Cortex N Y NY. 2011;21:877‐883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Østergaard L, Aamand R, Gutiérrez‐Jiménez E, et al. The capillary dysfunction hypothesis of Alzheimer's disease. Neurobiol Aging. 2013;34:1018‐1031. [DOI] [PubMed] [Google Scholar]
- 38. Jiang H, Liu Y, Wei Y, et al. Impaired retinal microcirculation in patients with Alzheimer's disease. PloS One. 2018;13:e0192154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zabel P, Kaluzny JJ, Wilkosc‐Debczynska M, et al. Comparison of retinal microvasculature in patients with Alzheimer's Disease and primary open‐angle glaucoma by optical coherence tomography angiography. Invest Ophthalmol Vis Sci. 2019;60:3447‐3455.. [DOI] [PubMed] [Google Scholar]
- 40. Zhang YS, Zhou N, Knoll BM, et al. Parafoveal vessel loss and correlation between peripapillary vessel density and cognitive performance in amnestic mild cognitive impairment and early Alzheimer's Disease on optical coherence tomography angiography. PloS One. 2019;14:e0214685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bulut M, Kurtuluş F, Gözkaya O, et al. Evaluation of optical coherence tomography angiographic findings in Alzheimer's type dementia. Br J Ophthalmol. 2018;102:233‐237. [DOI] [PubMed] [Google Scholar]
- 42. Jiang H, Wei Y, Shi Y, et al. Altered macular microvasculature in mild cognitive impairment and Alzheimer Disease. J Neuroophthalmol. 2018;38:292‐298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yoon SP, Grewal DS, Thompson AC, et al. Retinal microvascular and neurodegenerative changes in Alzheimer's Disease and mild cognitive impairment compared with control participants. Ophthalmol Retina. 2019;3:489‐499. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information