

Summary
The gastrointestinal tract (GI) is the primary site of lymphoproliferative lesions, spanning from reactive lymphoid hyperplasia to overt lymphoma. The diagnosis of these diseases is challenging and an integrated approach based on clinical, morphological, immunohistochemical and molecular data is needed. To reach to confident conclusions, a stepwise approach is highly recommended. Histological evaluation should first assess the benign versus neoplastic nature of a given lymphoid infiltrate. Morphological and phenotypic analyses should then be applied to get to a definite diagnosis.
This review addresses the key histological features and diagnostic workup of the most common GI non-Hodgkin lymphomas (NHLs). Differential diagnoses and possible pitfalls are discussed by considering distinct groups of lesions (i.e. small to medium B-cell NHLs; medium to large B-cell NHLs; T-cell NHLs; and mimickers of Hodgkin lymphoma). The key clinical and epidemiological features of each entity are also described.
Key words: lymphoma, gastrointestinal lymphoma, Hodgkin lymphoma, B-cell lymphoma, T-cell lymphoma
Introduction and clinical relevance
The gastrointestinal (GI) tract plays several immunologic functions, including fine-tuning the intestinal microbiome, mediating tolerance toward food antigens and mounting immune responses against enteric pathogens 1. Such functions rely on dedicated anatomical structures, widely spread along the stomach, the small and large bowel (i.e. Peyer’s patches and rectal tonsil) 1,2. The physiologic and/or neoplastic expansion of such structures gives rise to a broad spectrum of lesions, spanning from benign lymphoid hyperplasia to malignant B and T cell lymphomas 3.
Gastrointestinal lymphoid proliferations are commonly found in the surgical pathology practice and their diagnosis can be challenging even for trained pathologists. This is due to their disguising and overlapping histological features, the small amount of diagnostic material and sampling artifacts. Despite such limitations, histology is the gold standard for the diagnosis of GI lymphoproliferative disorders (LPDs) and guides the management of patients 4. For these reasons, when dealing with lymphoid infiltrates in the GI tract, every effort should be taken to get to a diagnosis as accurate and informative as possible.
This review will discuss the histological features of GI LPDs and non-Hodgkin lymphomas (NHLs), specifically addressing the key diagnostic features and pitfalls of each entity. A practical approach will be followed, by grouping GI LPDs into major diagnostic categories, discussing their general features and considering their differential diagnosis. This work is intended to support the surgical pathology practice. For a detailed discussion of the clinical and pathophysiological features of each entity, the reader is referred to the many excellent reviews published on this topic 5-10.
The diagnosis of GI LPDs: benign versus malignant lesions
The first most compelling issue concerning GI LPDs is to define whether they are benign or malignant in nature. Clues favoring a diagnosis of lymphoma over reactive lymphoid hyperplasia include: (i) tissue effacement by confluent sheets of lymphoid cells (even with polyp formation), (ii) infiltration and disruption of glandular units (i.e. “lymphoepithelial lesions”; LELs), (iii) atypical follicles, follicular colonization or expanded mantle zones, (iv) the monomorphic composition of the lymphoid infiltrate, and (v) the documentation of cytologically or phenotypically atypical elements, singly or in aggregate. None of these findings is pathognomonic of lymphoma per se, nor all lymphomas disclose each of these features. It is rather the integration of multiple parameters that leads to a correct diagnosis (Fig. 1).
Figure 1.
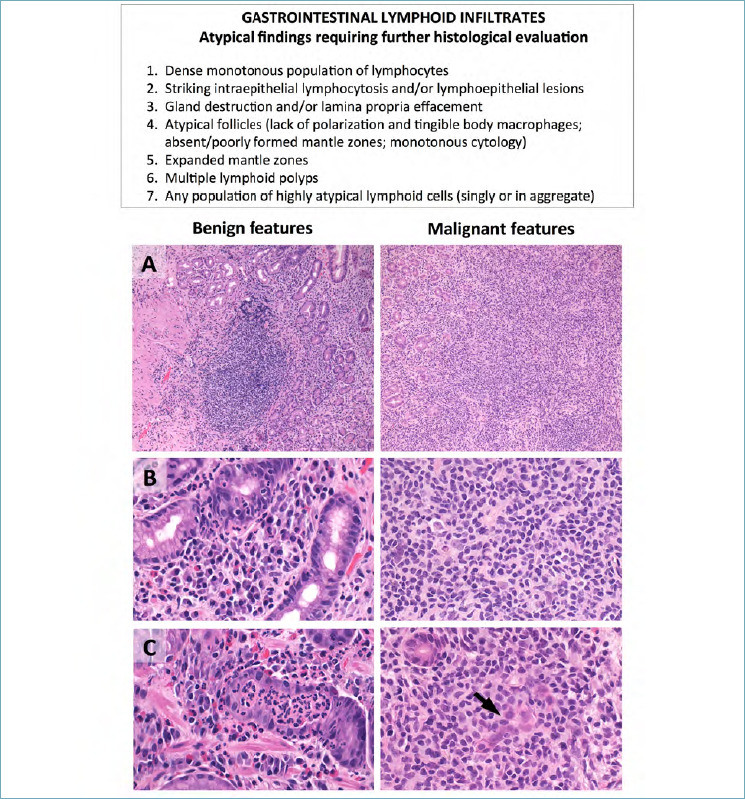
Differential diagnosis between benign and malignant GI lymphoid proliferations. The table summarizes the key morphological features favoring a diagnosis of neoplastic over reactive lymphoid infiltrate. (A) Reactive infiltrates are non-destructive and encompass well-demarcated lymphoid nodules (left figure); non-Hodgkin lymphomas are instead characterized by dense infiltrates occupying the entire lamina propria (right figure). (B) Reactive infiltrates are polymorphic and include variable numbers of small lymphocytes, granulocytes, histiocytes and plasma cells (left figure). Non-Hodgkin lymphomas mostly consist of monomorphic, atypical lymphocytes (right figure). (C) In reactive conditions, intra-glandular cells are mainly granulocytes and/or scattered, small lymphocytes (left figure); in non-Hodgkin lymphomas, native glands are infiltrated/effaced by neoplastic lymphocytes (“lymphoepithelial lesions”) (right figure; arrow). (H&E stain; original magnification, 10x and 20x).
Molecular biology may also support the differential diagnosis between reactive hyperplasia and lymphoma, the former being associated with polyclonal rearrangements of the immunoglobulin and T cell receptor genes, the latter showing monoclonal gene patterns. Exceptions nonetheless exist to this rule. In fact, antigen-driven monoclonal populations are commonly found in inflammatory, infectious or autoimmune responses 11 and polyclonal patterns may characterize highly mutant NHLs, as a result of limited primer annealing and poor amplification of the monoclonal sequences 12. Thus, the finding of a monoclonal peak does not necessarily imply a diagnosis of lymphoma and the results of molecular biology should always be integrated with the clinical-pathological findings.
Despite extensive phenotypic and molecular characterization, in a minority of cases a definite diagnosis cannot be made. In such instances, the degree of uncertainty should be stated in the pathology report and provisional statements of “atypical lymphoid hyperplasia/infiltrates”, “lymphoid infiltrate of uncertain potential/significance”, or “lymphoid infiltrate suspicious but not certain for lymphoma” may be proposed 13. The management of such lesions will depend on the clinical picture, the endoscopic findings and the severity of histological atypia.
Primary GI lymphomas versus secondary involvement by systemic disease
Once the neoplastic nature of a lymphoid infiltrate has been established, another important issue is defining its origin. The GI tract can indeed host both primary NHLs and secondary localizations of systemic disease. The clinical criteria distinguishing these two groups of neoplasms were first proposed by Dawson in 1961. A NHL is considered of primary GI origin if, at presentation: (i) peripheral and/or mediastinal lymphadenopathies are lacking; (ii) blood cell counts are normal; (iii) the lesion is predominantly localized within the GI tract and the only affected lymph nodes are adjacent to such lesion; and (iv) the liver and spleen are spared 14.
By applying Dawson’s criteria, primary GI NHLs account for about 30-40% of all extra-nodal lymphomas and for 1-4% of GI malignancies 15. Secondary GI involvement is by far more frequent, being reported in 5-20% of NHLs 16. The anatomic distribution of GI NHLs varies across geographical areas: the stomach is primarily involved in Western countries 17, while the small bowel is mostly affected in Middle East regions 18. In all, gastric and small intestinal NHLs account for 80-85% of all GI cases.
Primary GI NHLs are mostly of B-cell lineage (90% of cases), spanning from indolent to very aggressive neoplasms. Peripheral T-cell lymphomas are less common and typically associated with poor prognosis. In contrast to patients with known classic Hodgkin lymphoma (cHL), great caution is required before making a diagnosis of primary cHL in extranodal sites (stage IE is extremely rare: 0.25-1% of cases). Mimickers of cHL are well documented and will be addressed in a separate section of this review. Finally, the GI tract is frequently site of post-transplant LPDs (PTLDs). These lesions are frequently driven by EBV infection and include benign, non-destructive lymphoid or plasma cell proliferations (i.e. follicular hyperplasia, plasma cell hyperplasia, infectious mononucleosis-like LPDs), polymorphic and monomorphic B and T/NK-cell PTLDs (Tab. I).
Table I.
Classification of GI lymphomas
| Histologic subtype | Predilection site |
|---|---|
| B Cell Origin (90% of cases) | |
| Extranodal marginal zone lymphoma, MALT type | Stomach, small intestine |
| Alpha heavy chain disease (IPSID) | Jejunum, Ileum |
| Diffuse large B cell lymphoma, NOS | Stomach, Ileocecal |
| Burkitt lymphoma | Ileocecal, stomach, small intestine |
| Plasmablastic lymphoma | Anorectum |
| Mantle cell lymphoma | Any sites |
| Follicular lymphoma | Duodenum, large intestine |
| Posttransplant lymphoprolipherative disorder | Any sites |
| T/NK cell origin (5-9% of cases) | |
| Enteropathy associates T cell lymphoma (EALT) | Small intestine |
| Monomorphic epitheliotropic intestinal T cell lymphoma (MEILT) | Small intestine |
| NK/T-cell lymphoma, nasal type | Small intestine, large intestine |
| Periphetal T cell lymphoma, NOS | Large intestine, small intestine |
| Indolent T/NK lymphoprolipherative disorder | Small intestine, large intestine |
| Posttransplant lymphoprolipherative disorders | Any sites |
| Classical Hodgkin Lymphoma (< 5% of cases) | Rectum |
NON-HODGKIN B CELL LYMPHOMAS OF THE GI TRACT
For diagnostic purposes, B-cell NHLs can be grouped in two broad disease categories: (i) tumors consisting of small to medium size B-cells; and (ii) tumors consisting of medium to large size B-cells. The first group encompasses extra-nodal marginal zone lymphoma (ENMZL), mantle cell lymphoma (MCL), follicular lymphoma (FL) and chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL). The second group mainly includes Burkitt lymphoma (BL), diffuse large B-cell lymphoma (DLBCL), high-grade B-cell lymphoma (HGBL) not otherwise specified (HGBL NOS), HGBL with MYC and BCL2 and/or BCL6 rearrangements (i.e. double/triple-hit lymphoma, DHL/THL), large B-cell lymphoma (LBCL) with IRF4 rearrangements, and plasmablastic lymphoma (PBL).
SMALL TO MEDIUM SIZE B-CELL NHLs
This group includes entities primarily arising in the GI tract or secondarily spreading to this site. A stepwise immunohistochemical approach supporting their diagnosis is proposed in Figure 2.
Figure 2.
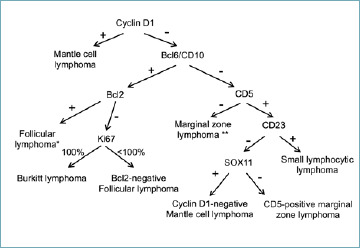
Immunohistochemical algorithm for the diagnosis of small to medium-sized B-cell NHLs. Notes: *CD10-negative follicular lymphomas can be encountered; **A similar phenotype is shared by lymphoplasmacytic lymphoma, which is not included in this review due to its exceptional occurrence in the GI tract.
Extra-nodal marginal zone lymphomas (ENZML) are indolent B-cell NHLs, mostly arising in the mucosa-associated lymphoid tissue (MALT) of the stomach and small bowel. A consistent number of cases develop after chronic antigen stimulation by H. pylori (gastric ENZML) or C. jejuni (small intestinal ENMZL) infection 5. Alpha heavy chain disease (also known as “immunoproliferative small intestinal disease; IPSID) is a clinical-pathological variant of small bowel ENMZL, characterized by monoclonal alpha heavy chain secretion with no immunoglobulin light chains. IPSID affects adolescents and young adults of Mediterranean origin and presents with malabsorption, diarrhea, fever, and weight loss 19.
Histologically, ENMZLs disclose sheets of B-cells, expanding the lamina propria with possible extension to the muscolaris mucosae and submucosa. Reactive germinal centers (GCs) are scattered throughout the tumor, often with features of GC colonization (i.e. effacement of GCs and disruption of follicular dendritic cell [FDC] meshworks). Neoplastic cells also infiltrate the surrounding glands, producing LELs (Fig. 3A). These are defined as intra-epithelial aggregates of ≥ 3 marginal zone cells that disrupt the gland architecture. Epithelial cells of LELs frequently undergo eosinophilic degeneration (i.e. oxyphilic change). The neoplastic population consists of small to medium-sized mature B cells with variable amounts of plasma cells and scattered large B blasts. The mature cell component has variable cytological features, spanning from marginal zone to monocytoid, centrocyte-like, small-cell and plasmacytic morphology. Marginal zone cells are medium-sized, have pale cytoplasm, slightly irregular nuclear contours, moderately dispersed chromatin and inconspicuous nucleoli. Monocytoid cells have larger amounts of paler cytoplasm, while centrocyte-like cells are small to medium-sized with irregular/cleaved nuclei and dense chromatin. Small cell morphology consists of mature B-cells with round nuclear contours and clumped chromatin, resembling SLL/CLL. Plasmacytic differentiation occurs in one third of gastric ENMZL and may include plasma cells with intra-nuclear immunoglobulin inclusion (i.e. Dutcher bodies). Such histological features are even more prominent in IPSID, where they occur together with villous widening and pseudo-atrophy. LELs are limited to residual B-cell areas.
Figure 3.
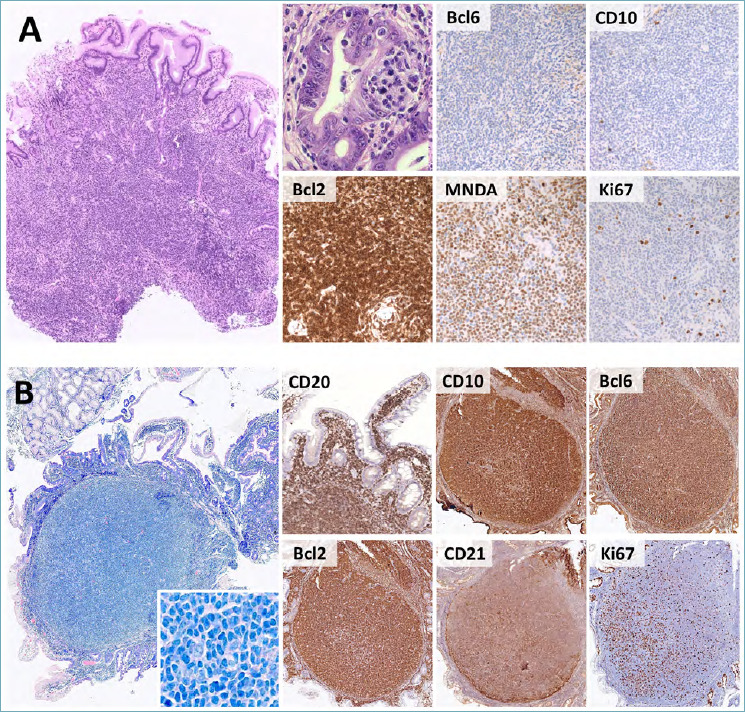
Histological features of ENMZL and primary duodenal FL. (A) ENMZL presents as a dense infiltrate of small to medium B-cells, with frequent LELs (H&E, high power picture). Phenotypically, CD10 and Bcl6 are negative, while Bcl2 is diffusely positive. MNDA expression supports the diagnosis, while the Ki67 proliferation index is low. (B) Primary duodenal FL presents as superficially located lymphoid nodules with villous widening and lamina propria expansion. The neoplastic population includes numerous centrocytes with sparse centroblasts (large box, insert). CD20, CD10, Bcl6 and Bcl2 are diffusely positive. CD21 immunostain highlights peripheral ring-shaped FDC networks, while the Ki67 index is low-to-moderate and lacks polarization. (H&E, Giemsa and immunoperoxidase stain; original magnification 7x, 20x and 80x).
On immunohistochemistry (IHC), ENMZLs express the pan-B cell antigen CD20 and are negative for CD10, Bcl6, CD23, Cyclin D1 and CD5. IgM are usually positive in ENMZL, while IgA heavy chains are characteristically expressed in IPSID. Positivity for IRTA1 has been reported in subsets of cases (mostly with monocytoid morphology) and supports the diagnosis. MNDA and CD43 are also variably expressed (Fig. 3A). In case of plasma cell differentiation, light chain restriction can be documented by kappa and lambda immunostains 3.
The diagnosis of ENMZL may prove challenging on occasion. This holds particularly true for chronic inflammatory conditions (e.g. H. pylori-related gastritis) or post-treatment assessments, in which the border between reactive lymphoid hyperplasia and ENMZL is blurred. In such instances, uncertainty should be reported through validated scoring systems, such as the Wotherspoon score (for treatment-naïve cases) 20 and GELA score (for post-treatment evaluations) 21 (Tab. II).
Table II.
Scoring systems for treatment-naïve and post-treatment ENMZL
| Wotherspoon scoring system for treatment-naïve gastric ENMZL* | ||
|---|---|---|
| Score | Diagnosis | Histological features |
| 0 | Normal mucosa | Scattered lymphocytes and plasma cells in the lamina propria; no lymphoepithelial lesions |
| 1 | Active gastritis | Inflammatory lymphoid infiltrate in the lamina propria; no lymphoepithelial lesions |
| 2 | Active gastritis with lymphoid follicles | Inflammatory lymphoid infiltrate with follicles; no lymphoepithelial lesions |
| 3 | Suspicious lymphoid infiltrate, probably reactive | Dense lymphoid infiltrate with follicles surrounded by small lymphocytes that infiltrate diffusely the lamina propria with occasional lymphoepithelial lesions |
| 4 | Suspicious lymphoid infiltrate, probably lymphoma | Dense lymphoid infiltrate with follicles surrounded by marginal zone lymphocytes that infiltrate diffusely the lamina propria and the glandular epithelium with focal lymphoepithelial lesions |
| 5 | ENMZL | Dense lymphoid infiltrate effacing gastric mucosa with diffuse lymphoepithelial lesions |
| GELA scoring system for post-treatment gastric ENMZL** | ||
| Score | Diagnosis | Histological features |
| CR | Complete histological remission | Scattered lymphocytes and plasma cells in the lamina propria; no lymphoepithelial lesions; empty or fibrotic lamina propria |
| pMRD | Probable minimal residual disease | Lymphoid aggregates in the lamina propria, muscularis mucosae or submucosa; no lymphoepithelial lesions; empty or fibrotic lamina propria |
| rRD | Responding residual disease | Dense periglandular lymphoid infiltrate with/without lymphoepithelial lesions; focal empty or fibrotic lamina propria |
| NC | No change (persistent ENMZL) | Dense lymphoid infiltrate effacing gastric mucosa, mostly with lymphoepithelial lesions |
Notes: *modified form ref. 20;
**modified from ref. 21.
Note that molecular biology tests are not included in either scoring system. Even in post-treatment settings, monoclonal peaks per se cannot be assumed as evidence of residual disease. Nonetheless, the documentation of identical peaks in pre- and post-treatment samples is highly suggestive of pMRD/rRd.
The differential diagnosis of ENMZL includes a number of entities. First, ENZML must be distinguished from other small-to-medium B-cell NHLs. As marginal zone markers (e.g. MNDA and IRTA1) have only limited sensitivity or specificity, the diagnosis of ENMZL is often one of exclusion and requires thorough immunophenotyping (Fig. 2). In the pediatric population, ENMZL must also be distinguished from “atypical marginal zone hyperplasia” (AMZH) 22. This lesion occurs in the appendix and tonsil of children and adolescents, as a consequence of unknown inflammatory triggers. Histologically, AMZH recapitulates the morphological features of ENMZL with frequent lambda-chain restriction on immunophenotyping, being nonetheless characterized by polyclonal rearrangements of the immunoglobulin genes and never progressing to locally-spread or systemic disease 22. When the plasmacytic differentiation is so prominent to occupy most of the tissue sample, the risk of a misdiagnosis of extramedullary plasmacytoma does exist. The clinical features and search for an associated B-cell component in subsequent sections usually allows for a reliable diagnosis. Finally, ENMZL with increased large cell fractions must be distinguished from DLBCL. By definition, any sheet-like proliferation of large cells (i.e. aggregates > 20 large cells and/or areas with > 10% of cohesive large cells) excludes ENMZL and prompts a diagnosis of DLBCL. If both high and low-grade areas are present, a diagnosis of DLBCL with accompanying ENMZL is advised and the percentage amount of each component should be reported 3.
In the GI tract, follicular lymphoma (FL) occurs as either primary disease or (more frequently) as secondary involvement by systemic cases 23. Primary FL of the GI tract usually arises in the duodenum, with possible synchronous seeding to the jejunum and ileum 23,24. Primary-duodenal FL is a disease variant with excellent prognosis and unique clinical-pathological features 3,23. It presents as small polypoid lesions, incidentally found during upper GI endoscopy. Histologically, the lymphoid infiltrate is confined to the lamina propria of duodenal villi with minimal (if any) extension to the submucosa. The duodenal wall and regional lymph nodes are characteristically spared. Neoplastic follicles are non-polarized, lack tingible-body macrophages and are devoid of mantle zones. The tumor population consists of small centrocytes (coarse chromatin and cleaved nuclei) and scattered centroblasts (open chromatin and multiple nucleoli) (Fig. 3B). Centrocytes and centroblasts disclose the classic FL phenotype (positivity for CD10, Bcl6 and Bcl2; negativity for CD5, Cyclin D1, CD23, MNDA and IRTA1), while FDC meshworks are characteristically pushed at the periphery of the neoplastic follicles. This feature is readily highlighted by CD21 and/or CD23 immunostain. The Ki-67 proliferation index is low and non-polarized (Fig. 3B). Like systemic FLs, primary-duodenal cases bear the t(14;18)(q32;q21), which juxtaposes the BCL2 and IGH genes 3,23. Given its excellent outcome, primary-duodenal FL must be distinguished from duodenal involvement by systemic FL. The latter should be favored in case of deep infiltration of the duodenal wall, the mesentery and/or regional lymph nodes, with preserved expanded FDC meshworks and in cases lacking Bcl2 protein expression and/or BCL2 gene translocations. Moreover, primary-duodenal FL is usually a low-grade neoplasm (i.e. G1/G2: < 15 centroblasts/high-power field [HPF]). The documentation of high-grade morphology (i.e. G3A/G3B: > 15 centroblasts/HPF) should prompt investigation for secondary disease 23.
In rare instances, primary GI FL occurs in non-duodenal sites. These tumors account for < 4% of all GI NHLs, usually involve the small and large bowel and present with abdominal pain, GI obstruction or intussusception. Unlike primary duodenal FL, the intestinal wall is deeply infiltrated by neoplastic follicles, variably associated with interstitial fibrosis. Most cases are low grade (G1/G2), express CD10, Bcl6 and Bcl2 and disclose expanded FDC meshworks 24. The differential diagnosis between such cases and secondary GI involvement by systemic FL relies on Dawson’s criteria (see above).
Mantle cell lymphoma (MCL) is a biologically aggressive neoplasm, accounting for about 3-10% of NHLs 3. It affects middle-aged to elderly patients and presents as nodal/extra-nodal disease, with frequent GI involvement (15-30% of cases). In the large bowel, it may occasionally appear as multiple polypoid lesions, known as lymphomatous polyposis. Histologically, classic MCL consists of diffuse, vaguely nodular or (rarely) perifollicular lymphoid infiltrates localized in the lamina propria and submucosa. Unlike ENMZL, native glands are spared, LELs are rarely found and the infiltrate is more deeply seated with limited effacement of the superficial lamina propria. The neoplastic infiltrate consists of small to medium-sized cells with irregular nuclear contours (Fig. 4A). Blastoid, pleomorphic and (more rarely) small or marginal zone cell variants are reported. The immunophenotype of MCL is highly characteristic, with strong positivity for CD5, Cyclin D1, and SOX11. Negativity for one of such markers is nonetheless possible and correlates with specific clinical-prognostic features. In particular, CD5-negative MCLs typically present as extra-nodal disease (also in the GI tract) and bear more favorable prognosis (Fig. 4A) 25. CD10, Bcl6 and CD23 are usually (yet not invariably) negative 3,26. The proliferation index varies and should always be assessed, as values > 30% correlate with worse prognosis 27. Over 95% of MCLs bear the t(11;14)(q13;32), which juxtaposes the IGH with the Cyclin D1-coding CCND1 gene. In selected cases, Fluorescence In Situ Hybridization (FISH) analysis for this translocation supports the diagnosis 3 (Fig. 4A). Small subsets of Cyclin D1-positive cells properly located in the mantle zone of otherwise reactive follicles can be discovered in inflammatory infiltrates of patients with Cyclin D1-positive monoclonal B-cell lymphocytosis. In the absence of manifest lymphoma, this picture should not be diagnosed as clinically overt MCL 28.
Figure 4.
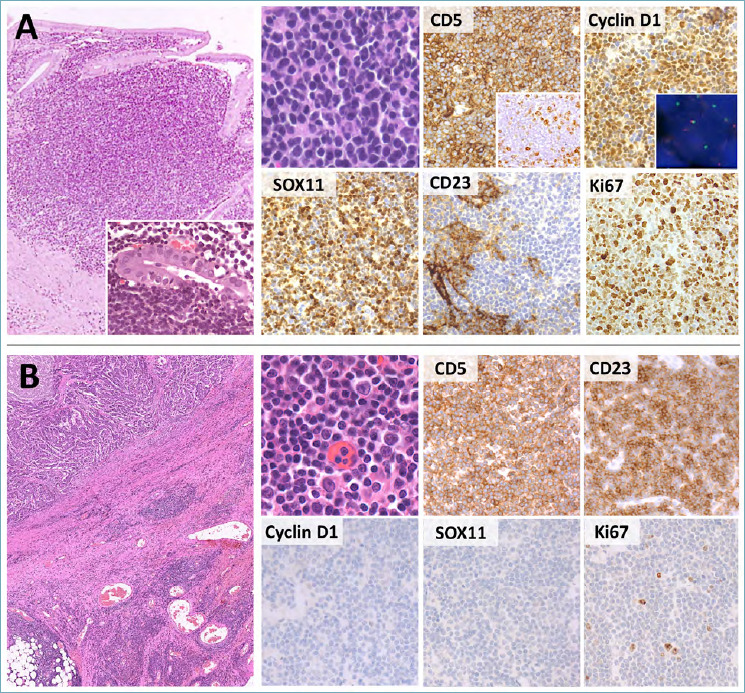
Histological features of MCL and CLL/SLL. (A) MCL presents as a diffuse or vaguely nodular proliferation of monomorphic small-sized B-cells. Rarely, LELs can be documented (large box, insert). The tumor cells express CD5, Cyclin D1 and SOX11. Rare cases with aberrant phenotypes, such as CD5-negativity (CD5 box, insert), are nonetheless encountered. Cyclin D1 over-expression is caused by CCND1 translocations, which can be documented by FISH (Cyclin D1, insert; break apart probes). CD23 is negative in tumor cells and positive in residual FDC meshworks. The Ki-67 index is usually low to moderate, despite occasional cases (mostly blastoid or pleomorphic variant, as in this figure) are highly proliferating. (B) CLL/SLL is usually an incidental finding in surgical specimens removed for other causes (e.g. colon adenocarcinoma; large box, upper left). Neoplastic lymphocytes are small, with scattered paraimmunoblasts (H&E, high power picture). They are positive for CD5 and CD23, but negative for Cyclin D1 and SOX11. The Ki67 index is low and limited to the paraimmunoblast component. (H&E, immunoperoxidase and DAPI stain; original magnification 5x, 20x and 40x, 80x).
The major differential diagnosis of MCL is chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL). This is an indolent B-cell neoplasm, usually affecting elderly people with peripheral blood and/or nodal involvement 3. Despite CLL/SLL never occurs as primary GI lymphoma, subtle interstitial and intravascular infiltrates may be encountered in the stomach, bowel and appendix as a result of systemic dissemination. This is also true for specimens removed for complicated diverticulitis or carcinoma: in this setting, CLL/SLL cells are likely recruited by the inflammatory milieu and appear as multiple, discrete and monotonous lymphoid nodules (Pizzi M, Bellan A; unpublished data) (Fig. 4B). Histologically, CLL/SLL is characterized by an interstitial, diffuse or vaguely nodular infiltrate, made of small lymphocytes with round nuclei and dark chromatin. Paler pseudo-follicles enriched in medium-sized nucleolated paraimmunoblasts are commonly seen. On IHC, CLL/SLL is positive for CD20, CD23 and CD5, but (unlike MCL) it stains negative for Cyclin D1 and SOX11 3 (Fig. 4B). CD200 and LEF1 are also usually positive 29.
MEDIUM TO LARGE SIZE B-CELL NHLs
This group includes a variety of clinically aggressive entities (BL, DLBCL, HGBL NOS, DHL/THL, LBCL with IRF4 rearrangements and PBL), presenting with bulky lesions, intestinal occlusion, GI bleeding and/or perforation. They occur in immunocompetent as well as immunocompromised patients 3.
Burkitt lymphoma (BL) is an aggressive NHL, typically occurring in extra-nodal sites or as leukemic disease. Three epidemiological variants are described: (i) endemic BL, (ii) sporadic BL, and (iii) immunodeficiency-associated BL. Endemic BL occurs in equatorial Africa, affects children and young adolescents and presents as bulky lesions of the jaws and facial bones. Sporadic and immunodeficiency (mostly HIV)-associated cases are reported throughout the world, have a broad age distribution and account for about 1-2% of adult lymphomas in Western countries 30. Immunodeficiency-associated BL usually presents with nodal or leukemic disease, while sporadic BL mostly occurs in the GI tract and abdominal cavity 3.
The histological and cytogenetic features of BL are highly characteristic. In prototypical cases, the tumor shows a diffuse, cohesive and monotonous proliferation of medium-sized lymphocytes with squared-off borders, round nuclei, finely clumped chromatin, inconspicuous nucleoli and scant basophilic cytoplasm with lipid vacuoles. The latter are more readily appreciable in cytological preparations or by adipophilin immunostain 31. Rare (usually immunodeficiency-related) cases disclose a more pleomorphic cytology and/or plasmacytoid differentiation. Irrespective of the morphological variants, tingible body macrophages are scattered throughout the lesion, imparting the classic “starry sky” pattern (Fig. 5A). Phenotypically, tumor cells are almost invariably positive for CD10 and Bcl6 and negative (or only weakly positive) for Bcl2. The Ki-67 proliferation index is equal (or close) to 100%. Myc protein is diffusely and strongly expressed in > 80% of neoplastic cells. Negative staining may nonetheless occur in a minority of cases and does not prevent the diagnosis, if all other BL features are present 32. CD38, MUM1, CD43 and SOX11 expression is variable, while TdT, Cyclin D1, CD23, and CD5 are consistently negative (Fig. 5A). Positivity for EBER in situ hybridization is documented in most endemic and immunodeficiency-associated BLs, while it is less common in sporadic cases. Most BLs bear translocations juxtaposing MYC to one of the immunoglobulin genes (IGH, IGK and/or IGL): the resulting t(8;14)(q24;q32) or alternative t(2;8)(p12;q24) and t(8;22)(q24;q11) play a key pathogenic role and help to confirm the diagnosis (Figure 5A). By definition, BCL6 and/or BCL2 rearrangements are not present 3. Of note, about 10% of otherwise classical BLs lacks MYC rearrangements 33,34, indicating that alternative pathogenic mechanisms may occur. In such cases, strict clinical, morphological and phenotypic criteria should be applied to rule out possible BL mimickers.
Figure 5.
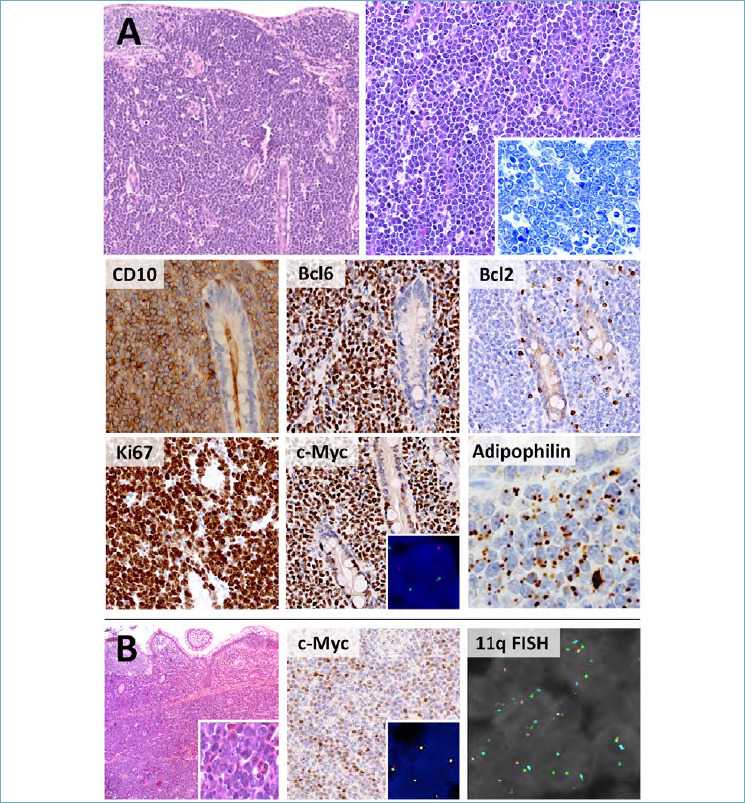
Histological features of BL and Burkitt-like lymphoma with 11q aberrations. (A) BL presents with bulky GI lesions, featuring a diffuse proliferation of monomorphic, medium-sized lymphocytes with squared-off borders and inconspicuous nucleoli. A starry sky pattern is usually present. Phenotypically, BL is positive for GC markers (CD10, Bcl6) and negative/weakly positive for Bcl2. The Ki-67 index approximates 100% and c-Myc is strongly and diffusely expressed. MYC translocations are present in >90% of cases (c-Myc box, insert; break apart probes). Tumor cells are also positive for adipophilin, a marker of deranged lipid metabolism. (B) Burkitt-like lymphoma with 11q aberrations discloses more cytological heterogeneity (H&E, insert) and weaker c-Myc expression than conventional BL. MYC translocations are not documented (c-Myc box, insert; break apart probes), while 11q aberrations (gain/loss signals at FISH) are present by definition. (H&E, Giemsa, immunoperoxidase and DAPI stain; original magnification 10x, 20x and 80x).
The diagnosis of BL is usually straightforward. Small or poorly representative biopsy samples may nonetheless pose the differential diagnosis with other high-grade B-cell NHLs with medium size or blastoid morphology (i.e. blastoid MCL, B-cell lymphoblastic leukemia/lymphoma [B-ALL], HGBL or, rarely, DLBCL, and Burkitt-like lymphoma with 11q aberration). Negativity for CD5 and Cyclin D1 excludes blastoid MCL, while negativity for TdT (and other immaturity markers, such as CD34) argues against B-ALL 35. HGBLs (NOS or DHL/THL) and DLBCL have features that more commonly diverge from the classical morphological, phenotypic and cytogenetic presentation of BL (e.g. pleomorphic or large-sized cells, negativity for CD10 and/or Bcl6; diffuse, strong positivity for Bcl2; lack of MYC translocations; occurrence of BCL2 and/or BCL6 translocations) 3. Helpful immunohistochemical algorithms have been reported in challenging cases 36. Finally, Burkitt-like lymphoma with 11q aberration is a rare, aggressive B-cell NHL, mostly occurring at nodal sites in children, young adults and in the post-transplant setting. The differential diagnosis with BL relies on a greater degree of cytological pleomorphism, a vaguely nodular (or even follicular) growth pattern and on the less intense positivity for Myc protein. MYC rearrangements are typically lacking, while chromosome 11q alterations (proximal gains and telomeric losses) are present by definition (Figs. 5B, 6) 3,37.
Figure 6.
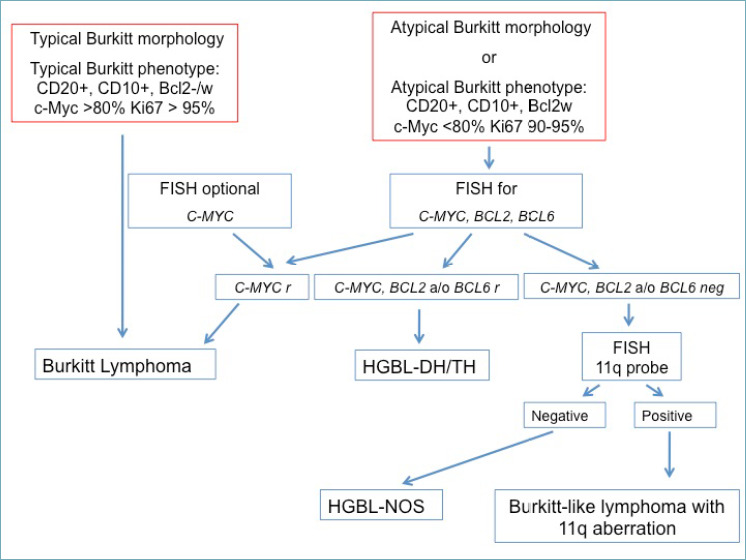
Diagnostic flow chart of BL and its mimickers.
Diffuse large B-cell lymphomas (DLBCLs) are clinically and biologically heterogeneous neoplasms, defined by purely morphological criteria (i.e. diffuse growth pattern of large B cells). DLBCLs account for about 45-50% of all primary GI NHLs and develop as either primary disease or secondary evolution of a prior low-grade lymphoma (ENMZL; FL or CLL/SLL) or MCL 38. The identification of secondary cases is based on the clinical history, disease presentation, and on the histological documentation of a clonally related (prior or coexisting) indolent component.
Despite several DLBCL variants are described, the NOS subtype is by far the most common. The tumor typically presents as a bulky, ulcerated lesion with extensive infiltration of the affected organ. Tumor cells resemble centroblasts (medium to large-sized blasts with multiple membrane-bound nucleoli), immunoblasts (large-sized blasts with a single, centrally located nucleolus) or have anaplastic morphology. Spindle or signet-ring cytology is rarely found 3,38. In the GI tract, immunoblastic morphology is by far the most frequent (Fig. 7A).
Figure 7.
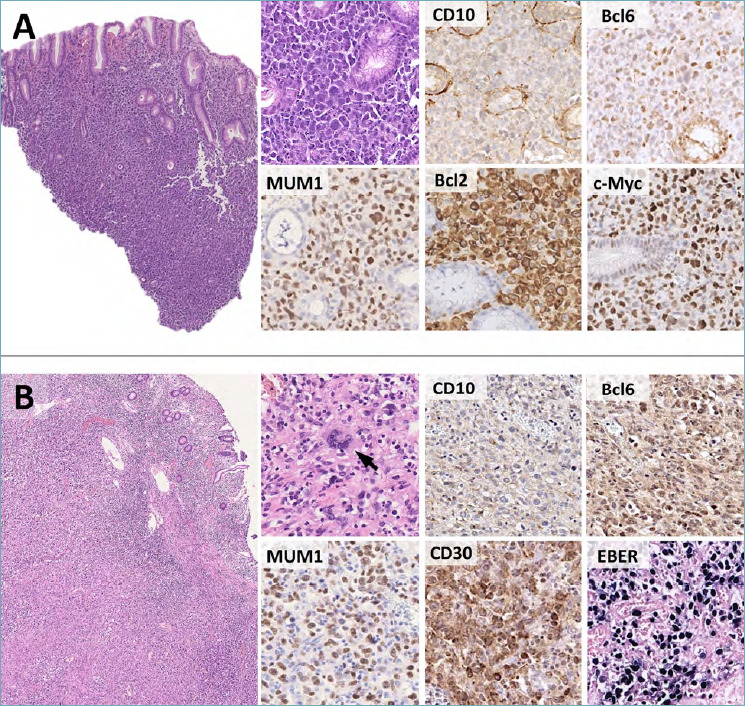
Histological features of DLBCL. (A) In the GI tract, DLBCLs present as sheets of large, atypical cells (H&E, high power picture). Most cases disclose a non-GCB phenotype (e.g. negativity for CD10, weak positivity for Bcl6, strong and diffuse expression of MUM1). Bcl2 and c-Myc are variably positive and their joint expression (i.e. double expressor phenotype) is associated with worse prognosis. (B) Rare cases of DLBCLs are EBV-positive. These tumors are more polymorphic than EBV-negative DLBCLs and may contain sternbergoid cells (H&E, high power picture; arrow). They are usually negative for CD10 and positive for MUM1 and CD30. Bcl6 is variable (positive in this case). (H&E and immunoperoxidase stain; original magnification 5x and 40x).
Given their biological heterogeneity, DLBCLs have highly variable phenotypes. Blast cells are strongly positive for panB-cell markers (CD20, CD19, PAX5, CD79a), though some of them may exceptionally be lacking, with variable expression of CD10, Bcl6, MUM1, Bcl2 and Myc (Fig. 7A). CD30, CD23 and CD5 can be occasionally positive, while Cyclin D1, SOX11 and TdT are consistently negative. The Ki-67 proliferation index is variable, but typically exceeds 25-30%. EBER in situ hybridization is positive in a minority of cases and is usually documented in elderly patients. These lymphomas fall within the WHO category of “EBV-positive, DLBCL NOS” and should be diagnosed as such (Fig. 7B). EBV positivity may also be documented in immunodeficiency-related cases and in the post-transplant setting 3.
For prognostic purposes, newly diagnosed DLBCLs should be stratified according to the putative cell of origin into germinal center-derived (GCB) or non-GCB types 39. In the routine practice, this is possible by applying IHC algorithms, including the Visco-Young, Choi and/or Hans algorithm40-42. The latter is most widely used and is based on the sequential assessment of CD10, Bcl6 and MUM1 expression. All DLBCL cases should also be stained for Myc and Bcl2, as double expressor cases bear significantly worse prognosis and should be reported as such (cutoffs for Myc and Bcl2 positivity: 40% and 50% of neoplastic cells, respectively) 43 (Fig 7A). Of note, Myc/Bcl2 double expression is not a surrogate marker of MYC and BCL2 gene rearrangements, although Myc protein positivity in > 70% of the neoplastic cells is more likely sustained by MYC translocations 44. As such, in defined clinical and pathological settings, Myc and Bcl2 joint positivity should prompt FISH analyses for DHL/THL, as recently stated by a consensus paper from the Italian Hematopathology Group 45.
A subset of high-grade B-cell NHLs discloses ambiguous features, not perfectly fitting within the BL and/or DLBCL categories. These cases (formerly known as “large B-cell lymphomas with features intermediate between DLBCL and BL”) are currently referred to as high-grade B-cell lymphomas (HGBL) and further sub-classified into: (i) HGBL NOS and (ii) HGBL with rearrangements of MYC and BCL2 and/or BCL6 (i.e. DHL/THL) 3,46. Most often, these tumors have large-cell or blastoid morphology (i.e. medium-sized blasts with finely dispersed chromatin and inconspicuous nucleoli). Their phenotype is variable and the proliferation index is usually high, reflecting the heterogeneous (yet very aggressive) nature of these neoplasms. CD10, Bcl6, Bcl2 and Myc are variably expressed in HGBL NOS, while they are typically positive in DHL/THL 3,47. TdT and Cyclin D1 are invariably negative, thus excluding a diagnosis of B-ALL and blastoid/pleomorphic MCL 35. By definition, DHL/THL harbor MYC translocations together with BCL2 and/or BCL6 rearrangements. Instead, HGBLs NOS are not translocated or feature rearrangements in only one of such genes.
Large B-cell lymphoma (LBCL) with IRF4 rearrangement is a recently described lymphoid neoplasm, included as a provisional entity in the revised 4th edition of the WHO Classification 3. The tumor primarily occurs in children and young adults, accounting for < 1% of all high-grade B-cell NHLs. Despite Waldeyer ring and cervical lymph nodes are most commonly affected, primary GI involvement is reported in up to one third of cases 48. Histologically, LBCL with IRF4 rearrangement consists of a monotonous proliferation of medium to large-sized lymphocytes with open chromatin. The tumor discloses a diffuse, nodular and diffuse or purely nodular growth pattern. In the latter case, neoplastic nodules are large, round and back-to-back with absent or attenuated mantle zones, resembling G3 FL. A starry-sky pattern is typically absent. On IHC, MUM1 is strongly and diffusely expressed. CD20 and Bcl6 are positive, while CD10 and Bcl2 are variable. The Ki67 proliferation index is high (> 30% of neoplastic cells). Cytogenetic analyses disclose cryptic rearrangements of IRF4, more often with the immunoglobulin genes. MYC and/or BCL2 translocations are never documented 42.
The differential diagnosis of LBCL with IRF4 rearrangement includes FL (for cases with nodular growth pattern) and DLBCL NOS (for cases with diffuse or mixed growth pattern). In the appropriate context (i.e. GI B-cell NHL with high-grade morphology in children or young adults), the striking positivity for MUM1 should prompt FISH analysis for IRF4 translocations. Their documentation is virtually diagnostic of LBCL with IRF4 rearrangement and excludes other entities 3,48.
Plasmablastic lymphoma (PBL) is an aggressive NHL with plasmacytic differentiation, most commonly arising in immunocompromised or elderly patients. The tumor typically develops in extra-nodal locations (mostly the oral cavity and head and neck region). Primary PBL of the GI tract accounts for about 20% of cases and occurs in the stomach, terminal ileum, large bowel, rectum and anal canal 49. In these locations, the tumor presents with large, ulcerated lesions causing abdominal pain, bleeding, diarrhea and/or constipation. Monoclonal components in the peripheral blood are only rarely documented. Histologically, PBL is characterized by sheets of large cells, spanning from immunoblasts to plasmablasts (i.e. blasts with eccentrically located nuclei, evident nucleoli and abundant cytoplasm). Immunoblast morphology frequently occurs in the oral and nasal cavity of HIV-positive patients, while plasmablastic differentiation is more characteristic of other anatomic sites. A starry sky pattern and abundant mitotic figures are common. A plasma cell component at various stages of maturation is not a feature of PBL and should prompt consideration of alternative diagnoses (e.g. anaplastic plasmacytoma/multiple myeloma). On IHC, PBL discloses sharp plasmacytic differentiation with positivity for CD38, CD138, MUM1, IgG and either kappa or lambda chains. The Ki-67 proliferation index is very high (> 80%). CD30 and EMA are frequently expressed, while CD79a, CD56 and c-Myc are positive in subsets of cases. CD45, CD20 and PAX5 are negative or only focally positive, thus ruling out DLBCL NOS and other high-grade B-cell NHLs. Most PBLs (75% of cases) are positive for EBER in situ hybridization (Fig. 8A). This feature and the overall clinical presentation are extremely helpful in differentiating PBL from extramedullary anaplastic plasmacytoma and multiple myeloma, which are rarely associated with EBV infection. In some instances, however, the distinction is not feasible and a descriptive diagnosis of “plasmablastic neoplasm, consisting with PBL or anaplastic plasmacytoma” is advisable 3,50. In PBL, negativity for ALK1 and HHV8 rules out other CD20-negative NHLs with plasmablastic morphology, such as ALK1-positive large B-cell lymphoma and extra-cavitary primary effusion lymphoma (Fig. 8B).
Figure 8.
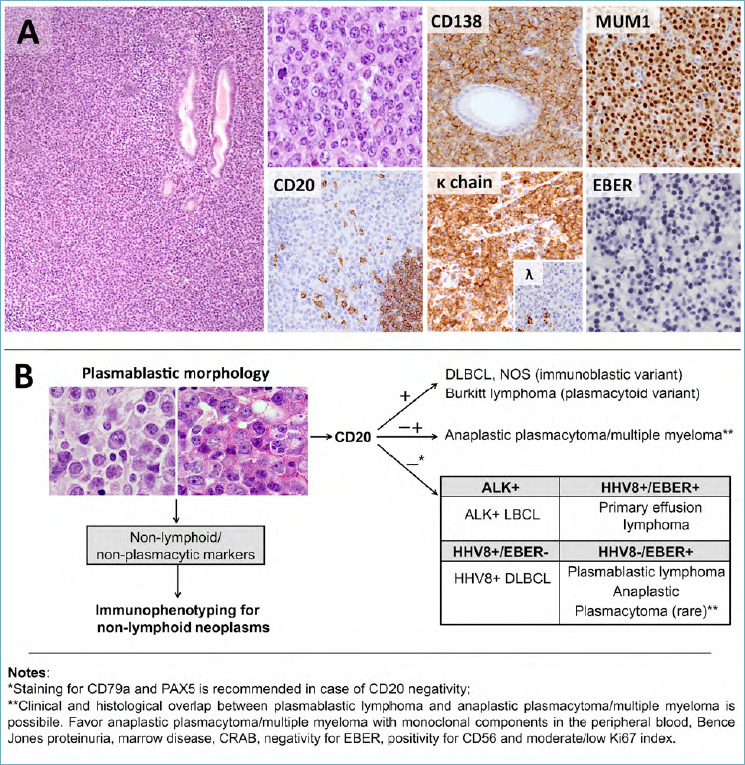
Histological features and differential diagnosis of PBL. (A) PBL consists of sheets of large cells with plasmablastic/plasmacytoid features. Tumor cells are characteristically negative for CD20 and express plasma cell markers, such as CD138 and MUM1. Light chain restriction is documented by kappa and lambda immunostains. The vast majority of cases is driven by EBV infection (positive EBER in situ hybridization). (H&E and immunoperoxidase stain; original magnification 10x, 20x and 40x). (B) Diagnostic flow chart for GI neoplasms with plasmablastic morphology.
NON-HODGKIN T CELL LYMPHOMAS OF THE GI TRACT
T-cell NHLs represent an absolute minority of GI lymphoid neoplasms. Despite any T-cell neoplasm may virtually involve the GI tract, some entities localize electively to the small intestine. These include: (i) enteropathy-associated T-cell lymphoma (EATL); (ii) monomorphic epitheliotropic intestinal T-cell lymphomas (MEITL); (iii) some cases of extra-nodal NK/T-cell lymphoma (ENKTL), nasal type; (iv) rare intestinal T-cell lymphomas, NOS; and (v) the so-called “indolent T-cell LPDs of the GI tract”.
Enteropathy-associated T-cell lymphoma (formerly known as type 1 EATL) is a rare and very aggressive T-cell neoplasm, affecting patients with prior or concomitant history of coeliac disease (CD). It accounts for < 5% of all GI NHLs and mostly occurs in Western countries. In Italy, it constitutes about two thirds of all primary GI T-cell neoplasms, with an estimated incidence among CD patients of 0.2-2/100.000/year 51,52. The disease typically occurs in the jejunum or ileum sometimes with multifocal lesions, and presents with ulcerated, perforated or stenotic masses that cause abdominal pain, bleeding, intestinal occlusion and systemic symptoms. Patients also complain of CD-related symptoms 53.
Histologically, EATL is characterized by a diffuse proliferation of polymorphic atypical cells of medium to large size that colonize the surface epithelium and deeply infiltrate the intestinal wall. Anaplastic features are documented in about 40% of cases, while angioinvasion, angiocentricity and necrotic areas are commonly observed. The neoplastic population is accompanied by a rich inflammatory infiltrate, consisting of histiocytes, eosinophils and small lymphocytes (Fig. 9A). Spreading to regional lymph nodes, liver, skin and other extra-abdominal organs is frequent. The uninvolved small intestinal mucosa discloses the classical histological features of CD (variable degrees of villous atrophy with increased intra-epithelial CD3-positive T lymphocytes) (Fig. 9A). On IHC, EATL expresses the panT cell markers CD3 and CD7, with variable loss of CD2 and CD5. Neoplastic cells disclose an activated cytotoxic phenotype (positivity for TIA1, granzyme B and perforin) and are almost invariably positive for CD30. The latter is expressed at higher intensity in cases with anaplastic cytology. EATL is usually negative for CD4, CD8 and CD56 and does not express T-cell receptor (TCR) antigens (Fig. 9A). EBER in situ hybridization is typically negative and its documentation should prompt consideration of an immunodeficiency-related LPD or other NK/T-cell entities (see below) 3.
Figure 9.
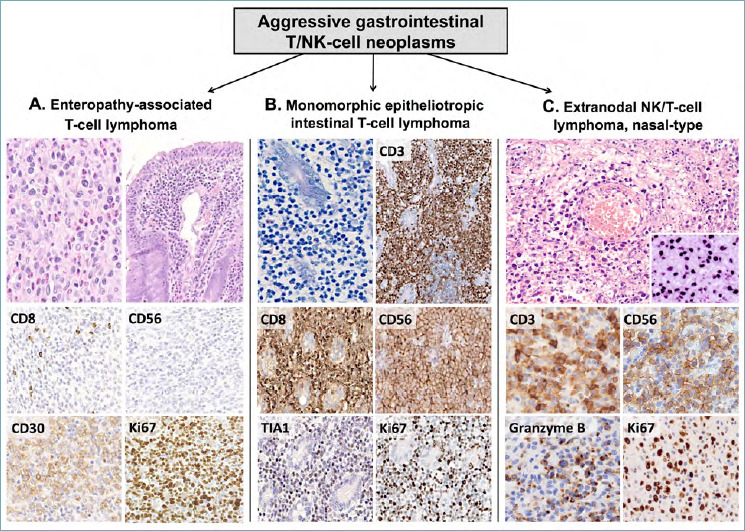
Differential diagnosis of GI aggressive T/NK-cells NHLs. EATL, MEITL and ENKTL, nasal type are aggressive T/NK lymphoid neoplasms of the GI tract. (A) EATL affects celiac patients and presents as polymorphous sheets of atypical T-cells with a rich inflammatory background (eosinophils, histiocytes and small lymphocytes). The uninvolved intestinal mucosa shows signs of enteropathy (H&E, upper right picture). Tumor cells are positive for various panT-cell markers and CD30, but lack CD8 and CD56. The ki67 index is high. (B) MEITL is an aggressive epitheliotropic NHL made of monomorphic atypical cells. CD3 is positive and highlights the striking epitheliotropism of tumor cells. CD8, CD56 and TIA1 are diffusely expressed, CD30 is negative (not shown) and the Ki-67 index is high. (C) ENKTL, nasal type is characterized by prominent angiotropism and diffuse positivity for EBER (large box, insert). It typically expresses cytoplasmic CD3, CD56 and cytotoxic markers (e.g. Granzyme B). The Ki67 index is high. (H&E, Giemsa and immunoperoxidase stain; original magnification 5x, 20x and 40x).
The main differential diagnoses of EATL include refractory CD and secondary GI involvement by systemic T-cell NHLs (mostly anaplastic large cell lymphoma; ALCL). Refractory CD (RCD) is defined as CD not responding to > 6-12 months of strictly gluten-free diet. Two forms of RCD are described: (i) type 1 RCD, which is histologically indistinguishable from classic CD; and (ii) type 2 RCD, which is characterized by > 50% atypical intraepithelial T-cells, closely resembling EATL (i.e. positivity for CD3; double negativity for CD8 and CD4) 54. Several lines of evidence suggest that type 2 RCD is the precursor lesion of EATL 55. The differential diagnosis between the two entities relies on RCD being an intra-epithelial disease of mostly small atypical cells, whereas EATL is a large cell lymphoma and massively infiltrates the intestinal wall.
Like EATL, ALCL is characterized by large atypical blasts with a defective T-cell phenotype, diffuse expression of CD30 and positivity for T-cell cytotoxic markers 56. In ALCL, however, there is no history of CD, the uninvolved intestinal mucosa lacks features of enteropathy and the positivity for CD30 is much more intense and diffuse than in EATL. However, if CD features are not seen on the tissue samples and there is no history of CD, the differentiation can be challenging and CD clinical investigation should be suggested.
Monomorphic epitheliotropic T-cell lymphoma (MEITL; formerly known as type 2 EATL) is another aggressive T-cell NHL, typically arising in the small bowel. Unlike EATL, it is not associated with CD and accounts for the vast majority of primary GI T-cell NHLs in Asia 7. Histologically, intestinal villi are distorted and widened by sheets of monotonous medium-sized cells with pale cytoplasm, sparse chromatin and small nucleoli. Epitheliotropism is striking, while angiotropism, necrotic areas and/or accompanying inflammatory cells are characteristically absent (Fig. 9B). The phenotype of MEITL differs from EATL in that the neoplastic cells are positive for CD8 and CD56 with negativity for CD30. MATK, TCRγδ chains and TIA1 are usually positive, while perforin and granzyme B are more variable 3,57 (Fig. 9B). About 25% of cases aberrantly express CD20, but negativity for PAX5, CD19, CD79a and other B-cell markers supports the diagnosis of a T-cell neoplasm 58. EBER in situ hybridization is consistently negative, thus excluding ENKTL, nasal type 3.
Extra nodal NK/T-cell lymphoma (ENKTL) is an EBV-driven aggressive lymphoma, most commonly occurring in the upper respiratory tract. The GI is the primary site of disease in 2-7% of cases, but secondary involvement by extra-intestinal neoplasms is reported. ENKTL, nasal type usually arises in the jejunum and ileum of middle-aged males, presenting as large ulcerated lesions with bleeding and/or perforation. Key histological features are the prominent angiotropism, the presence of large necrotic areas and/or ulceration without epitheliotropism and the diffuse positivity of neoplastic cells for EBER (Fig. 9C). Tumor cells are cytologically variable, spanning from relatively small to large atypical blasts with “sternbergoid” features in a mixed inflammatory background. Most cases disclose a NK phenotype, with positivity for CD56, cytoplasmic CD3 and cytotoxic markers. Surface CD3, CD5, CD4 and CD8 are negative, while CD2, CD7 and CD30 are variably expressed 59 (Fig. 9C). Rare cases with T-cell (either TCRγδ or TCRαβ) phenotype do also occur 60. Irrespective of the cell lineage, EBER positivity is always documented and its negativity should prompt consideration of other entities (e.g. MEITL, PTCL NOS and indolent T-cell LPDs of the GI tract; see below). Molecular biology supports the differential diagnosis, as monoclonal TCR gene rearrangements are not documented in most ENKTL, nasal type (i.e. cases of NK derivation), while are usually present in other T-cell entities.
Rare intestinal T-cell NHLs do not fulfill the diagnostic criteria of EATL, MEITL and ENKTL, nasal type. These tumors, currently referred to as intestinal T-cell lymphomas NOS, are not a specific disease entity, but a diagnostic category to be used when other more common neoplasms are ruled out or not assessable (i.e. small biopsy samples lacking surface epithelium or inadequate material for immunophenotyping) 3. These cases are not associated with CD, usually arise in the large bowel or the stomach and have a very aggressive clinical course. The morphological and immunohistochemical features are highly variable, yet most cases disclose a cytotoxic phenotype and lack TCR expression. The differential diagnosis with secondary GI involvement by peripheral T-cell lymphoma NOS is based on clinical data and imaging studies 61.
In recent years, indolent T-cell LPDs of the GI tract have also been described 62. These cases mostly occur in the stomach, small intestine and large bowel and present with abdominal pain, weight loss, diarrhea or dyspepsia. Endoscopically, the mucosa of affected sites is thickened, polypoid or hyperemic with superficial erosions. Histology discloses a dense, lamina propria-limited, non-angioinvasive lymphoid infiltrate, displacing (yet not infiltrating) the mucosal glands or surface epithelium 62. Epithelioid granulomas and occasional eosinophils may be present, yielding the differential diagnosis with Crohn disease. Cytologically, neoplastic cells are monomorphic and small to medium-sized. Cases with NK phenotype disclose brightly eosinophilic cytoplasmic granules. On IHC, most indolent T-cell LPDs have a non-activated cytotoxic phenotype (i.e. positivity for CD3, CD8 and TIA1; negativity for perforin and granzyme B), but positivity for CD4 or even NK cell markers has been reported 63,64. Irrespective of the cell lineage, these lesions have very low proliferation index (usually < 10% of neoplastic cells) and a protracted clinical course, with limited response to conventional chemotherapy. Molecular biology shows monoclonal TCR rearrangements in all cases 3.
HODGKIN LYMPHOMA MIMICKERS IN THE GI TRACT
Primary classic Hodgkin lymphoma (cHL) is extremely rare in the GI tract. Occasional GI LPDs may nonetheless feature neoplastic elements that closely resemble Hodgkin and Reed-Sternberg (HRS) cells. EBV-positive muco-cutaneous ulcer (EBVMCU) is the most challenging of such entities.
This recently described disease affects immunosuppressed and elderly males and presents as large ulcerated lesions of the skin, oral cavity, and GI tract 3,65. In the latter site, the rectum and sigmoid colon are most commonly involved. The clinical course is usually benign, with nearly all cases responding to reduction of immunosuppressive therapy 65. Radiation and chemotherapy are also effective. Systemic spread is very rare, but relapse and local progression have been reported 3.
Histologically, EBVMCU appears as a well-demarcated ulcer with a dense, polymorphic sub-epithelial lymphoid infiltrate. The latter includes small lymphocytes, plasma cells, histiocytes and eosinophils. Variable numbers of large atypical cells are also present, resembling either immunoblasts or HRS cells (i.e. very large cells with multi-lobated nuclei, eosinophilic nucleoli and abundant cytoplasm). The deepest margin of the lesion is sharp and consists of a band-like infiltrate of reactive lymphocytes (Fig. 10A). Necrotic areas and angioinvasion are frequently documented. On IHC, the atypical cells are positive for CD30, MUM1, PAX5, CD79a and OCT2. Expression of CD20 is variable and CD15 is found in about 50% of cases. CD10 and Bcl6 are usually negative. A key diagnostic feature is the strong and diffuse positivity for EBER in the large atypical blasts as well as in small to medium-sized B cells. The background lymphoid infiltrate and deep margin of the lesion mainly consists of CD8-positive T-cells (Fig. 10A). Molecular biology shows monoclonal rearrangements of the immunoglobulin genes in about 50% of cases 65.
Figure 10.
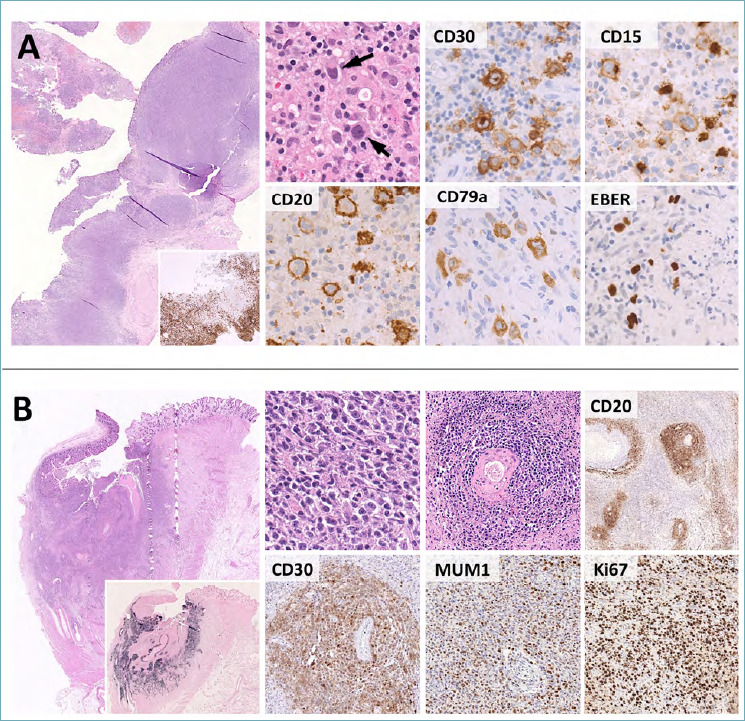
Histological features of EBVMCU and monomorphic PTLD. (A) EBVMCU is an ulcerated, sharply demarcated lymphoid lesion with a band-like CD3-positive T-cell infiltrate at the base (large box, insert). Hodgkin/Reed Sternberg-like cells are characteristically present (high power picture, arrow) and express CD30, multiple B-cell markers (e.g. CD20, CD79a), MUM1 and EBER. CD15 is present in subsets of cases. These features need to be distinguished from cHL, whose primary involvement of the GI tract is extremely rare. (B). The differential diagnosis of EBVMCU encompasses various EBV-positive LPDs, including monomorphic PTLD. This consists of sheets of atypical blasts with variable histological features (the case reported here has lymphomatoid granulomatosis-like features with marked angiotropism). CD20, CD30 and MUM1 are typically expressed, EBER positivity is strong and diffuse (large box, insert) and the Ki67 index is high. (H&E, immunoperoxidase stain; original magnification 2x, 10x, 20x and 40x).
Depending on the amount and morphological features of the large atypical cells, EBVMCU enters the differential diagnosis with cHL, monomorphic/polymorphic PTLD and EBV-positive DLBCL NOS. Distinction from cHL is based on the clinical presentation and on the sharp circumscription of the lesion (deep band-like margin of T cells). The positivity for EBER even in small lymphocytes and the B-cell phenotype of the atypical cells are also not features of cHL. The differential diagnosis with monomorphic/polymorphic PTLD and EBV-positive DLBCL NOS is more challenging and largely depends on the clinical findings and the localized, non-infiltrative nature of EBVMCU 66 (Fig. 10B). Finally, small and superficial biopsies of EBVMCU may lead to the wrong impression of florid granulation tissue associated with benign mucous ulcers. In such instances, only a high degree of suspicion and careful histological examination leads to the correct diagnosis. Immunophenotyping for CD30, MUM1 and EBER help highlighting the atypical cell component, thus favoring EBVMCU over reactive inflammation.
Conclusions
Non-Hodgkin lymphomas of the GI tract constitute a broad spectrum of neoplasms with variable clinical and biological features. The management of such entities is based on an integrated diagnostic framework, considering morphological, immunophenotypic and molecular/cytogenetic data. As discussed in this review, a step-wise approach is highly recommended to reduce the spectrum of differential diagnoses and to get to confident conclusions. Key features for any diagnostic evaluation are the assessment of tumor cell cytology (typical vs atypical; small vs large size) and lineage of differentiation (B versus T cell proliferations). This first evaluation will guide any subsequent ancillary test, reducing the number of unnecessary (or even confounding) analyses. Several factors may limit the diagnostic reliability of the biopsy findings (sampling artifacts; little amount of diagnostic material; overlapping features among different entities). Pathologists should be aware of such limitations, stating all of them in the final report. This will likely limit the over-interpretation of the histological findings, favoring the teamwork between pathologists and clinicians and the proper management of patients.
Figures and tables
Footnotes
ABBREVATIONS:
AMZH: atypical marginal zone hyperplasia; B-ALL: B-cell lymphoblastic leukemia/lymphoma; BL: Burkitt lymphoma; CD: coeliac disease; cHL: classic Hodgkin lymphoma; CLL/SLL: chronic lymphocytic leukemia/small lymphocytic lymphoma; DLBCL: diffuse large B-cell lymphoma; EALT: enteropathy-associated T-cell lymphoma; EBVMCU: EBV-positive muco-cutaneous ulcer; ENKTL: extra-nodal NK/T-cell lymphoma; ENMZL: extra-nodal marginal zone lymphoma; FDC: follicular dendritic cell; FISH: Fluorescence In Situ; FL: follicular lymphoma; GC: germinal centers; GCD: germinal center-derived; GI: gastrointestinal tract; HGBL: high-grade B-cell lymphoma; HGBL NOS: high-grade B-cell lymphoma not otherwise specified; HPF: high-power field; HRS: Hodgkin and Reed-Sternberg cells; IHC: immunohistochemistry;
IPSID: immunoproliferative small intestinal disease; LBCL: large B-cell lymphoma;
LELs: lymphoepithelial lesions; LPDs: lymphoproliferative disorders; MALT: mucosa-associated lymphoid tissue MALT; MCL: mantle cell lymphoma; MEITL: Monomorphic epitheliotropic T-cell lymphoma; NHLs: non-Hodgkin lymphomas;
PBL: plasmablastic lymphoma; PTLDs: post-transplant LPDs; RCD: Refractory CD;
TCR: T-cell receptor.
References
- 1.Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol 2014;14:667-85. https://doi.org/10.1038/nri3738 10.1038/nri3738 [DOI] [PubMed] [Google Scholar]
- 2.Brandtzaeg P, Kiyono H, Pabst R, et al. Terminology: nomenclature of mucosa-associated lymphoid tissue. Mucosal Immunol 2008;1:31-7. https://doi.org/10.1038/mi.2007.9 10.1038/mi.2007.9 [DOI] [PubMed] [Google Scholar]
- 3.Swerdlow SH, World Health Organization, International Agency for Research on Cancer . WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon: International Agency for Research on Cancer; 2017. [Google Scholar]
- 4.Ghimire P, Wu GY, Zhu L. Primary gastrointestinal lymphoma. World J Gastroenterol 2011;17:697-707. https://doi.org/10.3748/wjg.v17.i6.697 10.3748/wjg.v17.i6.697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakamura S, Ponzoni M. Marginal zone B-cell lymphoma: lessons from Western and Eastern diagnostic approaches. Pathology 2020:52:15-29. https://doi.org/10.1016/j.pathol.2019.08.012 10.1016/j.pathol.2019.08.012 [DOI] [PubMed] [Google Scholar]
- 6.Duffles Amarante G, Collins G, Rocha V. What do we know about duodenal-type follicular lymphoma? From pathological definition to treatment options. Br J Haematol 2020:188:831-7. https://doi.org/10.1111/bjh.16348 10.1111/bjh.16348 [DOI] [PubMed] [Google Scholar]
- 7.van Vliet C, Spagnolo DV. T- and NK-cell lymphoproliferative disorders of the gastrointestinal tract: review and update. Pathology 2020:52:128-41. https://doi.org/10.1016/j.pathol.2019.10.001 10.1016/j.pathol.2019.10.001 [DOI] [PubMed] [Google Scholar]
- 8.Ott G. Aggressive B-cell lymphomas in the update of the 4th edition of the World Health Organization classification of haematopoietic and lymphatic tissues: refinements of the classification, new entities and genetic findings. Br J Haematol 2017:178:871-87. https://doi.org/10.1111/bjh.14744 10.1111/bjh.14744 [DOI] [PubMed] [Google Scholar]
- 9.Jain P, Wang M. Mantle cell lymphoma: 2019 update on the diagnosis, pathogenesis, prognostication, and management. Am J Hematol 2019:94:710-25. https://doi.org/10.1002/ajh.25487. 10.1002/ajh.25487 [DOI] [PubMed] [Google Scholar]
- 10.Montes-Moreno S, King RL, Oschlies I, et al. Update on lymphoproliferative disorders of the gastrointestinal tract: disease spectrum from indolent lymphoproliferations to aggressive lymphomas. Virchows Arch 2020:476:667-81. https://doi.org/10.1007/s00428-019-02704-8 10.1007/s00428-019-02704-8 [DOI] [PubMed] [Google Scholar]
- 11.Lo WY, Li JY, Chan YK, et al. Instability of clonality in gastric lymphoid infiltrates: a study with emphasis on serial biopsies. Am J Surg Pathol 2005:29:1582-92. https://doi.org/10.1097/01.pas.0000188031.40836.00 10.1097/01.pas.0000188031.40836.00 [DOI] [PubMed] [Google Scholar]
- 12.Theriault C, Galoin S, Valmary S, et al. PCR analysis of immunoglobulin heavy chain (IgH) and TcR-gamma chain gene rearrangements in the diagnosis of lymphoproliferative disorders: results of a study of 525 cases. Mod Pathol 2000:13:1269-79. https://doi.org/10.1038/modpathol.3880232 10.1038/modpathol.3880232 [DOI] [PubMed] [Google Scholar]
- 13.Hussein MR. Atypical lymphoid proliferations: the pathologist’s viewpoint. Expert Rev Hematol 2013:6:139-53. https://doi.org/10.1586/ehm.13.4 10.1586/ehm.13.4 [DOI] [PubMed] [Google Scholar]
- 14.Dawson IM, Cornes JS, Morson BC. Primary malignant lymphoid tumours of the intestinal tract. Report of 37 cases with a study of factors influencing prognosis. Br J Surg 1961:49:80-9. https://doi.org/10.1002/bjs.18004921319 10.1002/bjs.18004921319 [DOI] [PubMed] [Google Scholar]
- 15.Loehr WJ, Mujahed Z, Zahn FD, et al. Primary lymphoma of the gastrointestinal tract: a review of 100 cases. Ann Surg 1969:170:232-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freeman C, Berg JW, Cutler SJ. Occurrence and prognosis of extranodal lymphomas. Cancer 1972:29:252-60. https://doi.org/10.1097/00000658-196908000-00011 10.1097/00000658-196908000-00011 [DOI] [PubMed] [Google Scholar]
- 17.Koch P, del Valle F, Berdel WE, et al. Primary gastrointestinal non-Hodgkin’s lymphoma: I. Anatomic and histologic distribution, clinical features, and survival data of 371 patients registered in the German Multicenter Study GIT NHL 01/92. J Clin Oncol 2001:19:3861-73. https://doi.org/10.1200/JCO.2001.19.18.3861 10.1200/JCO.2001.19.18.3861 [DOI] [PubMed] [Google Scholar]
- 18.Salem P, el-Hashimi L, Anaissie E, et al. Primary small intestinal lymphoma in adults. A comparative study of IPSID versus non-IPSID in the Middle East. Cancer 1987:59:1670-6. doi:10.1002/1097-0142(19870501)59:9<1670::aid-cncr2820590925>3.0.co;2-d [DOI] [PubMed] [Google Scholar]
- 19.Al-Saleem T, Al-Mondhiry H. Immunoproliferative small intestinal disease (IPSID): a model for mature B-cell neoplasms. Blood 2005:105:2274-80. https://doi.org/10.1182/blood-2004-07-2755 10.1182/blood-2004-07-2755 [DOI] [PubMed] [Google Scholar]
- 20.Wotherspoon AC, Doglioni C, Diss TC, et al. Regression of primary low-grade B-cell gastric lymphoma of mucosa-associated lymphoid tissue type after eradication of Helicobacter pylori. Lancet 1993:342(8871):575-7. https://doi.org/10.1016/0140-6736(93)91409-f 10.1016/0140-6736(93)91409-f [DOI] [PubMed] [Google Scholar]
- 21.Copie-Bergman C, Gaulard P, Lavergne-Slove A, et al. Proposal for a new histological grading system for post-treatment evaluation of gastric MALT lymphoma. Gut 2003:52:1656. https://doi.org/10.1136/gut.52.11.1656 10.1136/gut.52.11.1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Attygalle AD, Liu H, Shirali S, et al. Atypical marginal zone hyperplasia of mucosa-associated lymphoid tissue: a reactive condition of childhood showing immunoglobulin lambda light-chain restriction. Blood 2004:104:3343-8. https://doi.org/10.1182/blood-2004-01-0385 10.1182/blood-2004-01-0385 [DOI] [PubMed] [Google Scholar]
- 23.Marks E, Shi Y. Duodenal-type follicular lymphoma: a clinicopathologic review. Arch Pathol Lab Med 2018:142:542-7. https://doi.org/10.5858/arpa.2016-0519-RS 10.5858/arpa.2016-0519-RS [DOI] [PubMed] [Google Scholar]
- 24.Misdraji J, Harris NL, Hasserjian RP, et al. Primary follicular lymphoma of the gastrointestinal tract. Am J Surg Pathol 2011:35:1255-63. https://doi.org/10.1097/PAS.0b013e318224e661 10.1097/PAS.0b013e318224e661 [DOI] [PubMed] [Google Scholar]
- 25.Miao Y, Lin P, Saksena A, et al. CD5-negative mantle cell lymphoma: clinicopathologic correlations and outcome in 58 patients. Am J Surg Pathol 2019:43:1052-60. https://doi.org/10.1097/PAS.0000000000001278 10.1097/PAS.0000000000001278 [DOI] [PubMed] [Google Scholar]
- 26.Pizzi M, Agostinelli C, Righi S, et al. Aberrant expression of CD10 and BCL6 in mantle cell lymphoma. Histopathology 2017:71:769-77. https://doi.org/10.1111/his.13286 10.1111/his.13286 [DOI] [PubMed] [Google Scholar]
- 27.Ye H, Desai A, Zeng D, et al. Smoldering mantle cell lymphoma. J Exp Clin Cancer Res 2017:36:185. https://doi.org/10.1186/s13046-017-0652-8 10.1186/s13046-017-0652-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Espinet B, Ferrer A, Bellosillo B, et al. Distinction between asymptomatic monoclonal B-cell lymphocytosis with cyclin D1 overexpression and mantle cell lymphoma: from molecular profiling to flow cytometry. Clin Cancer Res 2014:20:1007-19. https://doi.org/10.1158/1078-0432.CCR-13-1077 10.1158/1078-0432.CCR-13-1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang XM, Aguilera N. New immunohistochemistry for B-cell lymphoma and Hodgkin lymphoma. Arch Pathol Lab Med 2014:138:1666-72. https://doi.org/10.5858/arpa.2014-0058-RA 10.5858/arpa.2014-0058-RA [DOI] [PubMed] [Google Scholar]
- 30.Piccaluga PP, De Falco G, Kustagi M, et al. Gene expression analysis uncovers similarity and differences among Burkitt lymphoma subtypes. Blood 2011:117:3596-3608. https://doi.org/10.1182/blood-2010-08-301556 10.1182/blood-2010-08-301556 [DOI] [PubMed] [Google Scholar]
- 31.Ambrosio MR, Piccaluga PP, Ponzoni M, et al. The alteration of lipid metabolism in Burkitt lymphoma identifies a novel marker: adipophilin. PLoS One 2012:7:e44315. https://doi.org/10.1371/journal.pone.0044315 10.1371/journal.pone.0044315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mundo L, Ambrosio MR, Raimondi F, et al. Molecular switch from MYC to MYCN expression in MYC protein negative Burkitt lymphoma cases. Blood Cancer J 2019:9:91. https://doi.org/10.1038/s41408-019-0252-2 10.1038/s41408-019-0252-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hummel M, Bentink S, Berger H, et al. A biologic definition of Burkitt’s lymphoma from transcriptional and genomic profiling. N Engl J Med 2006:354:2419-30. https://doi.org/10.1056/NEJMoa055351 10.1056/NEJMoa055351 [DOI] [PubMed] [Google Scholar]
- 34.Leucci E, Cocco M, Onnis A, et al. MYC translocation-negative classical Burkitt lymphoma cases: an alternative pathogenetic mechanism involving miRNA deregulation. J Pathol 2008:216:440-50. https://doi.org/10.1002/path.2410 10.1002/path.2410 [DOI] [PubMed] [Google Scholar]
- 35.Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016:127:2375-90. https://doi.org/10.1182/blood-2016-01-643569 10.1182/blood-2016-01-643569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Naresh KN, Ibrahim HA, Lazzi S, et al. Diagnosis of Burkitt lymphoma using an algorithmic approach--applicable in both resource-poor and resource-rich countries. Br J Haematol 2011:154:770-6. https://doi.org/10.1111/j.1365-2141.2011.08771.x 10.1111/j.1365-2141.2011.08771.x [DOI] [PubMed] [Google Scholar]
- 37.Wagener R, Seufert J, Raimondi F, et al. The mutational landscape of Burkitt-like lymphoma with 11q aberration is distinct from that of Burkitt lymphoma. Blood 2019:133:962-6. https://doi.org/10.1182/blood-2018-07-864025 10.1182/blood-2018-07-864025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xie Y, Pittaluga S, Jaffe ES. The histological classification of diffuse large B-cell lymphomas. Semin Hematol 2015:52:57-66. https://doi.org/10.1053/j.seminhematol.2015.01.006 10.1053/j.seminhematol.2015.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000:403(6769):503-11. https://doi.org/10.1038/35000501 10.1038/35000501 [DOI] [PubMed] [Google Scholar]
- 40.Hans CP, Weisenburger DD, Greiner TC, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004:103:275-82. https://doi.org/10.1182/blood-2003-05-1545 10.1182/blood-2003-05-1545 [DOI] [PubMed] [Google Scholar]
- 41.Visco C, Li Y, Xu-Monette ZY, et al. Comprehensive gene expression profiling and immunohistochemical studies support application of immunophenotypic algorithm for molecular subtype classification in diffuse large B-cell lymphoma: a report from the International DLBCL Rituximab-CHOP Consortium Program Study. Leukemia 2012:26:2103-13. https://doi.org/10.1038/leu.2012.83 10.1038/leu.2012.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Choi WW, Weisenburger DD, Greiner TC, et al. A new immunostain algorithm classifies diffuse large B-cell lymphoma into molecular subtypes with high accuracy. Clin Cancer Res 2009:15:5494-5502. https://doi.org/10.1158/1078-0432.CCR-09-0113 10.1158/1078-0432.CCR-09-0113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li L, Li Y, Que X, et al. Prognostic significances of overexpression MYC and/or BCL2 in R-CHOP-treated diffuse large B-cell lymphoma: a systematic review and meta-analysis. Sci Rep 2018:8:6267. https://doi.org/10.1038/s41598-018-24631-5 10.1038/s41598-018-24631-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ziepert M, Lazzi S, Santi R, et al. A 70% cut-off for MYC protein expression in diffuse large B cell lymphoma identifies a high-risk group of patients. Haematologica 2020. https://doi.org/10.3324/haematol.2019.235556. Online ahead of print. 10.3324/haematol.2019.235556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Di Napoli A, Remotti D, Agostinelli C, et al. A practical algorithmic approach to mature aggressive B cell lymphoma diagnosis in the double/triple hit era: selecting cases, matching clinical benefit: a position paper from the Italian Group of Haematopathology (G.I.E.). Virchows Arch 2019:475:513-18. https://doi.org/10.1007/s00428-019-02637-2 10.1007/s00428-019-02637-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rimsza L, Pittaluga S, Dirnhofer S, et al. The clinicopathologic spectrum of mature aggressive B cell lymphomas. Virchows Arch 2017:471:453-66. https://doi.org/10.1007/s00428-017-2199-7 10.1007/s00428-017-2199-7 [DOI] [PubMed] [Google Scholar]
- 47.Li S, Desai P, Lin P, et al. MYC/BCL6 double-hit lymphoma (DHL): a tumour associated with an aggressive clinical course and poor prognosis. Histopathology 2016:68:1090-8. https://doi.org/10.1111/his.12884 10.1111/his.12884 [DOI] [PubMed] [Google Scholar]
- 48.Salaverria I, Philipp C, Oschlies I, et al. Translocations activating IRF4 identify a subtype of germinal center-derived B-cell lymphoma affecting predominantly children and young adults. Blood 2011:118:139-47. https://doi.org/10.1182/blood-2011-01-330795 10.1182/blood-2011-01-330795 [DOI] [PubMed] [Google Scholar]
- 49.Luria L, Nguyen J, Zhou J, et al. Manifestations of gastrointestinal plasmablastic lymphoma: a case series with literature review. World J Gastroenterol 2014:20:11894-11903. https://doi.org/10.3748/wjg.v20.i33.11894 10.3748/wjg.v20.i33.11894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lorsbach RB, Hsi ED, Dogan A, et al. Plasma cell myeloma and related neoplasms. Am J Clin Pathol 2011:136:168-82. https://doi.org/10.1309/AJCPENJ68FFBRIYB 10.1309/AJCPENJ68FFBRIYB [DOI] [PubMed] [Google Scholar]
- 51.Gao Y, Kristinsson SY, Goldin LR, et al. Increased risk for non-Hodgkin lymphoma in individuals with celiac disease and a potential familial association. Gastroenterology 2009:136:91-8. https://doi.org/10.1053/j.gastro.2008.09.031. 10.1053/j.gastro.2008.09.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Corrao G, Corazza GR, Bagnardi V, et al. Mortality in patients with coeliac disease and their relatives: a cohort study. Lancet 2001:358(9279):356-61. https://doi.org/10.1016/s0140-6736(01)05554-4 10.1016/s0140-6736(01)05554-4 [DOI] [PubMed] [Google Scholar]
- 53.Delabie J, Holte H, Vose JM, et al. Enteropathy-associated T-cell lymphoma: clinical and histological findings from the international peripheral T-cell lymphoma project. Blood 2011:118:148-55. https://doi.org/10.1182/blood-2011-02-335216 10.1182/blood-2011-02-335216 [DOI] [PubMed] [Google Scholar]
- 54.van Gils T, Nijeboer P, van Wanrooij RL, et al. Mechanisms and management of refractory coeliac disease. Nat Rev Gastroenterol Hepatol 2015:12:572-579. https://doi.org/10.1038/nrgastro.2015.155 10.1038/nrgastro.2015.155 [DOI] [PubMed] [Google Scholar]
- 55.Chander U, Leeman-Neill RJ, Bhagat G. Pathogenesis of Enteropathy-Associated T Cell Lymphoma. Curr Hematol Malig Rep 2018:13:308-17. https://doi.org/10.1007/s11899-018-0459-5 10.1007/s11899-018-0459-5 [DOI] [PubMed] [Google Scholar]
- 56.Inghirami G, Pileri SA, European TCLSG. Anaplastic large-cell lymphoma. Semin Diagn Pathol 2011:28:190-201. https://doi.org/10.1053/j.semdp.2011.03.002 10.1053/j.semdp.2011.03.002 [DOI] [PubMed] [Google Scholar]
- 57.Tan SY, Ooi AS, Ang MK, et al. Nuclear expression of MATK is a novel marker of type II enteropathy-associated T-cell lymphoma. Leukemia 2011:25:555-7. https://doi.org/10.1038/leu.2010.295 10.1038/leu.2010.295 [DOI] [PubMed] [Google Scholar]
- 58.Tan SY, Chuang SS, Tang T, et al. Type II EATL (epitheliotropic intestinal T-cell lymphoma): a neoplasm of intra-epithelial T-cells with predominant CD8alphaalpha phenotype. Leukemia 2013:27:1688-96. https://doi.org/10.1038/leu.2013.41 10.1038/leu.2013.41 [DOI] [PubMed] [Google Scholar]
- 59.Yu BH, Shui RH, Sheng WQ, et al. Primary intestinal extranodal natural killer/t-cell lymphoma, nasal type: a comprehensive clinicopathological analysis of 55 cases. PLoS One 2016:11:e0161831. https://doi.org/10.1371/journal.pone.0161831 10.1371/journal.pone.0161831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pongpruttipan T, Sukpanichnant S, Assanasen T, et al. Extranodal NK/T-cell lymphoma, nasal type, includes cases of natural killer cell and alphabeta, gammadelta, and alphabeta/gammadelta T-cell origin: a comprehensive clinicopathologic and phenotypic study. Am J Surg Pathol 2012:36:481-99. https://doi.org/10.1097/PAS.0b013e31824433d8 10.1097/PAS.0b013e31824433d8 [DOI] [PubMed] [Google Scholar]
- 61.Attygalle AD, Cabecadas J, Gaulard P, et al. Peripheral T-cell and NK-cell lymphomas and their mimics; taking a step forward - report on the lymphoma workshop of the XVIth meeting of the European Association for Haematopathology and the Society for Hematopathology. Histopathology 2014:64:171-99. https://doi.org/10.1111/his.12251 10.1111/his.12251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Perry AM, Warnke RA, Hu Q, et al. Indolent T-cell lymphoproliferative disease of the gastrointestinal tract. Blood 2013:122:3599-3606. https://doi.org/10.1182/blood-2013-07-512830 10.1182/blood-2013-07-512830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Soderquist CR, Patel N, Murty VV, et al. Genetic and phenotypic characterization of indolent T-cell lymphoproliferative disorders of the gastrointestinal tract. Haematologica 2019. https://doi.org/10.3324/haematol.2019.230961. Online ahead of print. 10.3324/haematol.2019.230961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Takeuchi K, Yokoyama M, Ishizawa S, et al. Lymphomatoid gastropathy: a distinct clinicopathologic entity of self-limited pseudomalignant NK-cell proliferation. Blood 2010:116:5631-7. https://doi.org/10.1182/blood-2010-06-290650 10.1182/blood-2010-06-290650 [DOI] [PubMed] [Google Scholar]
- 65.Dojcinov SD, Venkataraman G, Raffeld M, et al. EBV positive mucocutaneous ulcer--a study of 26 cases associated with various sources of immunosuppression. Am J Surg Pathol 2010:34:405-17. https://doi.org/10.1097/PAS.0b013e3181cf8622 10.1097/PAS.0b013e3181cf8622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Natkunam Y, Goodlad JR, Chadburn A, et al. EBV-Positive B-Cell proliferations of varied malignant potential: 2015 SH/EAHP workshop report-part 1. Am J Clin Pathol 2017:147:129-52. https://doi.org/10.1093/ajcp/aqw214 10.1093/ajcp/aqw214 [DOI] [PMC free article] [PubMed] [Google Scholar]