

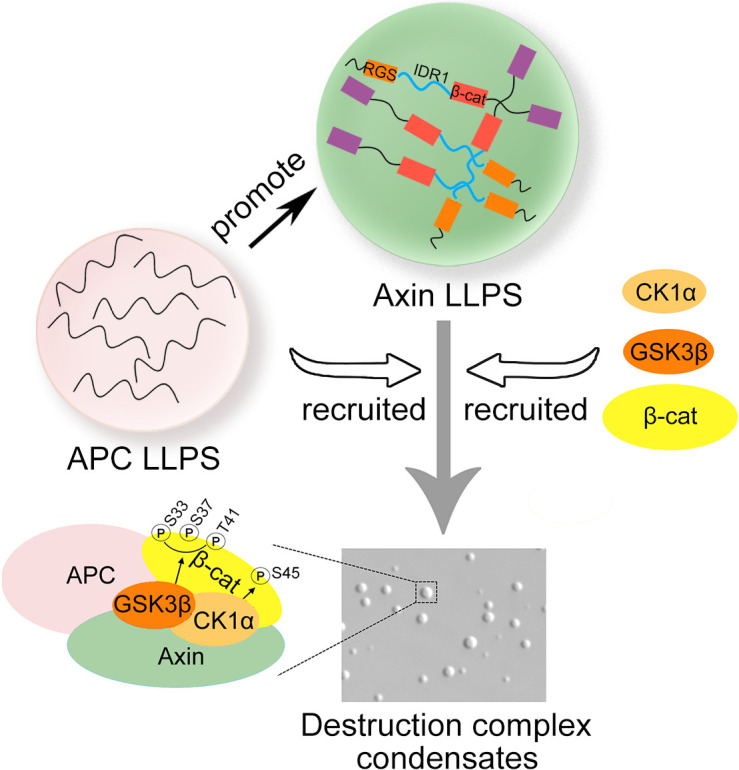
Assembly of the destruction complex is critical for Wnt/β-catenin signaling. Nong et al. find that Axin undergoes liquid–liquid phase separation, which is promoted by APC. Phase-separated Axin provides a scaffold for recruitment of GSK3β, CK1α, and β-catenin and facilitates GSK3β-mediated β-catenin phosphorylation and thus degradation.
Abstract
In Wnt/β-catenin signaling, the β-catenin protein level is deliberately controlled by the assembly of the multiprotein β-catenin destruction complex composed of Axin, adenomatous polyposis coli (APC), glycogen synthase kinase 3β (GSK3β), casein kinase 1α (CK1α), and others. Here we provide compelling evidence that formation of the destruction complex is driven by protein liquid–liquid phase separation (LLPS) of Axin. An intrinsically disordered region in Axin plays an important role in driving its LLPS. Phase-separated Axin provides a scaffold for recruiting GSK3β, CK1α, and β-catenin. APC also undergoes LLPS in vitro and enhances the size and dynamics of Axin phase droplets. The LLPS-driven assembly of the destruction complex facilitates β-catenin phosphorylation by GSK3β and is critical for the regulation of β-catenin protein stability and thus Wnt/β-catenin signaling.
Graphical Abstract
Introduction
The level of the key mediator β-catenin in Wnt/β-catenin signaling is tightly regulated to ensure appropriate expression of target genes (Clevers and Nusse, 2012; Nusse and Clevers, 2017). In the absence of Wnt ligands, the soluble β-catenin protein level is low due to its constant phosphorylation-dependent ubiquitination and subsequent proteasome degradation. β-catenin phosphorylation is mediated by the sequential actions of CK1α and GSK3β (MacDonald et al., 2009) in a protein destruction complex assembled by the scaffolding protein Axin and adenomatous polyposis coli (APC; Gammons and Bienz, 2018; Schaefer et al., 2018; Stamos and Weis, 2013; Wu and Pan, 2010). Despite extensive efforts in investigations, the mechanism underlying the assembly and regulation of the β-catenin destruction complex is still largely unclear.
There are two Axin genes (Axin1 and Axin2) in vertebrate genomes. Both Axin1 and Axin2 proteins harbor multiple protein-binding domains, including the N-terminal Regulators of G protein Signaling (RGS) domain that binds APC, the C-terminal DAX domain that mediates homo- (Axin–Axin) and hetero-polymerization (Axin–Dvl), and the binding sites for GSK3β and β-catenin, which are located in the middle region (Bienz, 2014; Pronobis et al., 2017). The tumor suppressor APC has a conserved region mediating self-oligomerization and Armadillo repeats in the N-terminal region and the multiple binding sites for β-catenin and Axin in the middle region (Kunttas-Tatli et al., 2014; Stamos and Weis, 2013). Axin and APC share features with known components of biomolecular condensates, including multivalency, the presence of intrinsically disordered regions (IDRs), and the ability to form puncta. Phase separation concentrates molecules in spatially confined compartments, facilitating biochemical reactions and signaling events (Alberti et al., 2019; Banani et al., 2017). Therefore, it has been proposed that APC and Axin may undergo phase separation in living cells (Schaefer and Peifer, 2019; Sear, 2007), but no experimental evidence has been provided. As Axin2 expression is at a low level at the physiological condition and is elevated upon Wnt signaling, and in turn it controls the intensity and duration of Wnt signaling in a negative feedback manner (Jho et al., 2002), we chose Axin1 as the focus in this study. We showed that Axin1 undergoes protein liquid–liquid phase separation (LLPS). Phase-separated Axin condensates recruit other components of the destruction complex, GSK3β, CK1α, and β-catenin, facilitating β-catenin phosphorylation by GSK3β.
Results and discussion
Axin1 undergoes phase separation
Axin1 is the critical player in organizing the β-catenin destruction complex (Faux et al., 2008; Lee et al., 2003; Li et al., 2012), and it formed membraneless spherical punctate structures in cells (Cliffe et al., 2003; Faux et al., 2008; Fig. 1 A). As Axin2 was expressed at a low level in HEK293T cells (Fig. S1 A), we generated Axin1 knockout (KO) HEK293T cells (Fig. S1 B). The reporter assay showed that Axin1 KO yielded a higher basal Wnt signaling, which could be blocked by reexpression of Axin1 (Fig. S1 C). As shown previously (Schwarz-Romond et al., 2007), EGFP-tagged Axin1 formed punctate structures in Axin1 KO HEK293T cells (Fig. 1 A) when it was expressed at a level close to the endogenous protein level in WT HEK293T cells (Fig. S1 D). The fluorescence signal of Axin1 protein in puncta was quickly recovered after photobleaching (FRAP), reaching ∼60% within 60 s (Fig. 1 B). Furthermore, the two encountering Axin puncta fused and coalesced into a larger sphere (Fig. 1 C), indicating that Axin1 puncta in living cells are highly dynamic and possess liquid-like properties.
Figure 1.

Axin1 undergoes LLPS. (A) Confocal images showing Axin1-EGFP puncta in Axin1 KO HEK293T cells. The nuclei were counterstained by DAPI (blue). (B) FRAP showing the recovery of the fluorescent signal in Axin1-EGFP puncta in Axin1 KO cells. (C) Droplet fusion of Axin1-EGFP puncta in an Axin1 KO cells. (D) Droplet formation of 2 µM Axin1 protein with 10% PEG8000. (E) Axin1 LLPS with 2.5% PEG8000. Left: Differential interference contrast (DIC) imaging. Right: turbidity measurement. (F) FRAP analysis of green-labeled Axin1 droplets. (G) Axin1 droplet fusion. (H) Quantification of colocalized LLPS droplets of green- and red-labeled Axin1 protein. ***, P < 0.001. n = 300. (I) Schematic diagram of Axin1 and its mutants. (J) Confocal images of EGFP-tagged proteins in Axin1 KO cells. (K) FRAP analysis of EGFP-tagged protein puncta in Axin1 KO cells. (L) Droplet formation of 2 µM Axin1 and its mutant proteins with 10% PEG8000. The droplet number was counted from three independent DIC fields. The size of droplets was measured from 200 droplets for each group. **, P < 0.01; ***, P < 0.001. (M) FRAP analysis of red-labeled Axin1 and its mutant protein droplets. Data in H, L, and M are shown as mean ± SD (n = 3). Scale bars in A and J, 10 µm; in B–D, F, G, and K–M, 2 µm. β-cat, β-catenin; del, deletion.
Figure S1.

Axin1 LLPS depends on protein concentration and salt concentration. (A) The mRNA levels of Axin1 and Axin2 in HEK293T cells were examined by quantitative RT-PCR. Data are shown as mean ± SD (n = 3). (B) The CRISPR/Cas9 system was applied to generate two Axin1 KO cell lines from HEK293T cells using single-guide RNA 5′-TTCGCTGTACCGTCTACTGG-3′ targeting the exon1 region of Axin1. The efficiency of Axin1 KO was examined by immunoblotting. KO1 and KO2 are the two KO clones used in this study. The genomic DNA sequencing results depicting Axin1 mutations in two Axin1 KO clones were shown above. (C) WT and Axin1 KO HEK293T cells were transfected with TopFlash-luciferase reporter and HA-Axin1-EGFP, then treated with or without Wnt3a conditional medium for 24 h before harvesting for luciferase determination. The asterisks indicate significant differences between the relative luciferase activity of each group in Axin1 KO HEK293T cells with that in WT HEK293T cells. Data are shown as mean ± SD (n = 3). (D) Axin1 KO HEK293T cells were transfected with the plasmids as indicated. Cell lysates were subjected to immunoblotting. The expression level of endogenous Axin1 protein in WT HEK293T cells was compared with the transfected HA-Axin1-GFP protein in Axin1 KO HEK293T cells. (E) Differential interference contrast (DIC) images of the droplets formed by different concentrations of purified Axin1 protein after induction by 10% PEG8000. The lower panel shows the quantification of the droplet size (n = 300). Data are shown as mean ± SD (n = 3). (F) DIC images of the Axin1 droplet formation in buffer containing different NaCl concentrations after LLPS induction with 2.5% PEG8000. (G) DIC images of Axin1 droplets treated with or without 1,6-hexanediol (1,6-Hex). (H) FRAP analysis of Sulfo-Cyanine3 maleimide–labeled Axin1 and FUS. A defined region of a droplet or aggregate structure (box) was bleached, and the recovery of the fluorescent signal in the bleached area was monitored. Quantification is shown below, and data are shown as mean ± SD (n = 3). (I and J) Fluorescence images of Axin1 droplet fusion (I) or colocalization (J) after LLPS induction, with the proteins labeled with either Alexa 488 NHS ester (green) or Sulfo-Cyanine3 maleimide (red). (K) DIC images of Axin1 (1 µM, 2.5 µM, 5 µM, and 12.5 µM) droplets in buffers containing the physiological salt concentration after incubation with PreScission protease. (L) FRAP analysis of Sulfo-Cyanine3 maleimide–labeled Axin1 (12.5 µM) droplets. Quantification of the FRAP data are shown in the right panel. Data are shown as mean ± SD (n = 3). (M) DIC images of Axin (15 µM) clusters formed in 10% PEG8000. (N) FRAP analysis of AD2-EGFP, AD4-EGFP, and AD6-EGFP puncta formed after 12 h with or without Wnt3a conditional medium stimulation in Axin1 KO HEK293T cells. Quantification of the FRAP data are shown as mean ± SD (n = 3). (O) Confocal images of EGFP-tagged proteins in Axin1 KO cells. (P) LLPS of AD1, AD2, AD3, AD4, AD5, and AD6 at different NaCl concentrations after induction by 2.5% PEG8000 as assessed by DIC imaging. Turbidity of purified Axin1 mutants after induced phase separation in different protein and salt concentrations. (Q) DIC images of droplets formed by 2 µM purified AD8, AD9, and M1 after induction by 10% PEG8000. The droplet number was counted from three independent DIC fields. The size of droplets was measured from 200 droplets for each group. Data are shown as mean ± SD. ***, P < 0.001. (R) Turbidity of purified 2 µM Axin1, AD7, AD7hnRNPA1, AD8, AD9, AD10, M1, and M2 after induced phase separation with 10% PEG8000. (S) DIC images of Axin1 ΔDAX (0.5 µM, 1 µM, and 2 µM) droplets induced by 10% PEG8000. Statistical analysis was performed with a two-tailed unpaired t test (*, P < 0.05; **, P < 0.01; ***, P < 0.001) in A, C, E, Q, and R. Scale bar in O, 10 µm; in E–N, Q, and S, 2 µm. Luc, luciferase.
To determine whether Axin1 undergoes LLPS, purified maltose-binding protein (MBP)-tagged Axin1 protein was incubated in a buffer containing 150 mM NaCl and 20 mM Hepes, pH 7.4. Spherical droplets were formed within a few seconds after adding 10% polyethylene glycol (PEG) 8000 (Fig. 1 D), and their size was bigger when the protein concentration was increased (Fig. S1 E). Axin1 LLPS in 2.5% PEG8000 was sensitive to protein concentration and salt concentration (Fig. 1 E and Fig. S1 F). Formation of Axin1 droplets in 10% PEG8000 was insensitive to 20% 1,6-hexanediol (Fig. S1 G). FRAP analysis with green-labeled Axin1 proteins showed that the fluorescent signal recovered to 30% within 3 min after photobleaching in droplets (Fig. 1 F), while aggregates formed by the RNA-binding protein fused in sarcoma (FUS) exhibited no recovery after bleaching (Fig. S1 H), indicating that Axin1 droplets are in a dynamic liquid-like phase. Consistently, two encountering Axin droplets were able to undergo fusion (Fig. 1, G and H; and Fig. S1, I and J). After cleavage of the MBP tag, Axin1 could still form droplets at the concentration of 2.5 µM at the physiological salt condition (Fig. S1 K), indicating that Axin1 can undergo LLPS without a crowder. However, PEG8000 apparently promoted Axin1 LLPS as the critical concentration of Axin1 for droplet formation was reduced to 1 µM with 2.5% PEG8000 (Fig. 1 E and Fig. S1 F), even to 0.5 µM with 10% PEG8000 (Fig. S1 E). The latter Axin1 concentration was close to the endogenous Axin1 protein level in HEK293T cells (by synthesizing three peptides of Axin1 as standards for mass-spectrometry quantification, the endogenous Axin1 protein concentration was assessed as 0.59 µM). The internal mobility of Axin1 droplets was also slow without PEG8000 as the fluorescent signal recovered to 10% within 4 min after photobleaching (Fig. S1 L). When the droplets reached a certain size, the droplets formed clusters (Fig. S1 M), a property that has been found for other condensates formed by LLPS (Zhang et al., 2015). These results indicate that the Axin1 protein forms droplets with liquid-like properties.
IDR1 of Axin1 is necessary for LLPS
To map the regions that are essential for LLPS, a series of Axin1 deletion mutants was expressed in Axin1 KO HEK293T cells (Fig. 1 I). Mutants lacking the IDR1 (aa 209–464; AD1, AD3, and AD5) failed to form puncta, while the IDR1-retaining mutants (AD2, AD4, and AD6) still formed puncta in cells (Fig. 1 J), indicating that IDR1 is critical for puncta formation. However, the mutant containing IDR1 alone was diffusely distributed. FRAP confirmed that the puncta formed by mutant proteins exhibited high internal mobility (Fig. 1 K and Fig. S1 N). Furthermore, replacement of the aa 209–376 region with the LLPS-driving low-complexity domains (aa 186–320) of hnRNPA1 (AD7hnRNPA1) or the C-terminal domain (aa 267–414) of TDP43 (AD7TDP43; Conicella et al., 2016; Molliex et al., 2015) could restore punta formation (Fig. 1 J and Fig. S1 O). The detailed mutagenesis analyses revealed that the aa 281–306 region, especially the five positively charged arginine residues, was essential for Axin1 LLPS.
Purified AD2, AD4, AD6, and IDR1 formed visible droplets, while the droplet-forming ability of IDR1-lacking AD1, AD3, and AD5 was much attenuated with smaller numbers and smaller size (Fig. 1 L and Fig. S1 P), which was supported by the turbidity measurement at OD600 (Wang et al., 2018; Fig. 1 E and Fig. S1 P). Droplets formed by AD2 and AD4 showed a similar internal mobility, while AD6 showed faster recovery than WT Axin1 (Fig. 1 M). These results indicate that IDR1 makes a major contribution to Axin LLPS, consistent with the requirement for IDR1 in formation of Axin puncta in cells. The droplet-forming ability of purified AD7, AD8, AD9, AD10, M1, and M2 was also impaired with smaller numbers and smaller size of droplets induced by 10% PEG8000 (Fig. 1 L; and Fig. S1, Q and R), while AD7hnRNPA1 recovered droplet-forming ability, which agreed with the in vivo puncta formation (Fig. 1 J).
Previous studies suggest that the DAX domain–mediated reversible polymerization of Axin is necessary for puncta formation and signalosome formation in cells (Bienz, 2014; Schwarz-Romond et al., 2007). However, the ΔDAX Axin1 mutant (aa 746–826 deletion) still formed droplets in vitro (Fig. S1 S), indicating that the DAX domain is not essential for LLPS in vitro. These results indicate that different domains of Axin may play distinct roles in regulating Wnt signaling at different steps.
APC promotes Axin LLPS
Ectopically expressed Axin1-EGFP formed punctate structures in both SW480 cells harboring mutant APC and HCT116 cells expressing WT APC (Fig. 2 A). However, the recovery of the fluorescent signal in Axin1 puncta after photobleaching was much slower in SW480 cells (Fig. 2 B), suggesting that APC may enhance the dynamics of Axin condensates. As it was difficult to express WT human APC in mammalian cells due to its huge size, we used Drosophila APC2 (dAPC2), which is functional in suppressing Wnt/β-catenin signaling in mammalian cells (Roberts et al., 2012; Fig. S2 A). dAPC2-EGFP and Axin1-mCherry formed puncta in SW480 cells, and the fluorescent signals were overlapped when coexpressed (Fig. S2 B). dAPC2 elevated the internal mobility of Axin puncta (Fig. 2 C and Fig. S2 C). dAPC2 formed puncta in both WT and Axin1 KO HEK293T cells, and the puncta were dynamic (Fig. 2, D–F).
Figure 2.

APC undergoes LLPS and promotes Axin1 condensate formation. (A and B) Confocal images (A) and FRAP analysis (B) of Axin1 puncta in HCT116 and SW480 cells. (C) FRAP analysis of Axin1 puncta in SW480 cells coexpressing dAPC2. (D) dAPC2-EGFP puncta Rel.in WT and Axin1 KO HEK293T cells. (E and F) FRAP analysis (E) and confocal images showing fusion (F) of dAPC2-EGFP puncta in Axin1 KO HEK293T cells. (G) dAPC2 protein droplets with 10% PEG8000. (H) dAPC2 droplet formation with 3.5% PEG8000. (I) Overlapping of Axin1 (green) and dAPC2 (red) in droplets. (J) Axin1 droplets with or without dAPC2. Data are shown as mean ± SD of droplets in six fields (220 × 166 mm) for each condition. (K) FRAP analysis of Axin1 droplets with or without dAPC2. Data are shown as mean ± SD (n = 3). Scale bars in A and D, 10 µm; in B, E–G, I, and J, 2 µm. ***, P < 0.001.
Figure S2.

APC protein undergoes LLPS. (A) Axin1 KO HEK293T cells (KO1) transfected with TopFlash-luciferase reporter and dAPC2 were treated with or without Wnt3a conditional medium for 24 h before harvesting for luciferase determination. The data are shown as mean ± SD (n = 3). Statistical analysis was performed with a two-tailed unpaired t test (***, P < 0.001). (B) Confocal fluorescence images showing colocalization of Axin1 and dAPC2 puncta in SW480 cells. (C) FRAP analysis of Axin1-mCherry protein puncta in SW480 cells with or without dAPC2-EGFP protein. (D) Differential interference contrast (DIC) images of droplets formed by different concentrations of dAPC2 after adding 10% PEG8000. (E) DIC images of the droplets formed by dAPC2 at different NaCl concentrations after adding 3.5% PEG8000. (F) DIC images of Axin1 droplets with or without the same concentration of dAPC2 protein (1 µM, 2.5 µM, and 5 µM) at 150 mM NaCl without a crowder. (G) Axin1 LLPS with or without dAPC2 in different concentrations without a crowder, as assessed by DIC imaging. (H) Turbidity of purified Axin1 with or without the same concentration of dAPC2 protein (0.5 µM, 1 µM, 2.5 µM, and 5 µM). Scale bar in B, 10 µm; in C–F, 2 µm. Vec, vector.
dAPC2 protein formed droplets in the presence of 10% PEG8000 (Fig. 2 G). Larger droplets formed as the protein concentration increased (Fig. S2 D), and dAPC2 droplet formation was sensitive to salt concentration (Fig. 2 H and Fig. S2 E). Axin1 and dAPC2 were present in the same droplet (Fig. 2 I), and the droplets formed by the dAPC2 and Axin1 mixture were larger than the ones formed by either individual protein (Fig. 2 J). Further, dAPC2 increased the internal mobility of Axin1 droplets (Fig. 2 K). dAPC2 also enhanced Axin1 LLPS without a crowder by reducing the critical concentration to undergo LLPS from 2.5 µM to 1 µM (Fig. S2, F and G), which was also supported by the turbidity measurement (Fig. S2 H). These data together indicate that APC promotes LLPS of Axin and increases the dynamics of Axin condensates.
Axin1 acts as a scaffold to assemble the destruction complex
In Axin1 KO HEK293T cells, dAPC2 existed as puncta, and GSK3β was mainly localized in the cytosol, while CK1α and β-catenin were mainly found in the nucleus (Fig. S3 A). Interestingly, when these proteins were coexpressed with Axin1, they colocalized in puncta, except CK1α, which was still mostly found in the nucleus. Wnt3a had no apparent effect on the colocalization of Axin1 with dAPC2 or GSK3β, while inducing β-catenin entering into the nucleus (Fig. S3, B and C). GSK3β, CK1α, and β-catenin proteins did not form droplets in vitro (Fig. S3 D). To determine whether Axin was able to recruit other components of the destruction complex into its droplets in vitro, green-labeled Axin1 and the red-labeled other proteins were premixed. Upon LLPS induction with 10% PEG8000, APC, GSK3β, CK1α, and β-catenin proteins showed colocalization with Axin1 droplets (Fig. 3 A). When one of the dye-labeled proteins was mixed with the other four unlabeled proteins, labeled protein was found in the droplets (Fig. 3 B). These data suggest that Axin1 condensates can recruit β-catenin, CK1α, and GSK3β into droplets.
Figure S3.

Colocalization of Axin with APC, GSK3β, and β-catenin in HEK293T cells and interaction of Axin1 or its deletion mutants with APC, GSK3β, CK1α, or β-catenin. (A) Confocal fluorescence images showing the subcellular localization of dAPC2-mCherry, β-catenin-mCherry, GSK3β-mCherry, and CK1α-mCherry in Axin1 KO HEK293T cells. The nuclei were counterstained by DAPI (blue). (B) Confocal fluorescence images showing colocalization of Axin1-EGFP with dAPC2-mCherry, β-catenin–mCherry, CK1α-mCherry, or GSK3β-mCherry in Axin1 KO HEK293T cells. Graphs show the fluorescence intensity profiles along the line indicated by the arrow in the merge panels. The distance along the arrow is shown on the x axis, and the corresponding fluorescence intensities are shown on the y axis. The overlapping EGFP and mCherry signals indicate colocalization. (C) Colocalization of Axin1-EGFP with dAPC2-mCherry, β-catenin–mCherry, or GSK3β-mCherry. (B) After 12 h with or without Wnt3a conditional medium, stimulation in Axin1 KO HEK293T cells was quantified. n = 100, with three biological replicates. (D) No droplets were observed with purified β-catenin, GSK3β, or CK1α protein in 20 mM Hepes, pH 7.4, 1 M NaCl, and 10% PEG8000. (E) Differential interference contrast (DIC) images of droplets formed by 1 µM Axin1 protein and the turbidity of Axin1 LLPS were measured in the solution for the in vitro phosphorylation assay, containing 2.5 µM CK1α, 50 nM GSK3β, and 1.5 µM β-catenin and Axin1 (0.5 µM, 1 µM, and 2.5 µM) with or without 2.5% PEG8000. The droplet number was counted from three independent DIC fields, and the quantification data are shown as mean ± SD (n = 3). Statistical analysis was performed with a two-tailed unpaired t test (***, P < 0.001). (F) Confocal fluorescence images showing colocalization of Axin1-mCherry, AD1-mCherry, AD2-mCherry, and AD3-mCherry with dAPC2-EGFP, and Axin1-EGFP, AD1-EGFP, AD2-EGFP, and AD3-EGFP with GSK3β-mCherry or β-catenin–mCherry in Axin1 KO HEK293T cells. (G) FRAP analysis of dAPC2-EGFP, GSK3β-mCherry, and β-catenin–mCherry puncta when Axin1-mCherry, AD2-mCherry or Axin1-EGFP, AD2-EGFP were coexpressed. Quantification data are shown as mean ± SD (n = 3). (H) Axin1 KO HEK293T cells transfected with Axin1-EGFP were treated with or without Wnt3a conditional medium for 24 h, and then harvested for anti-GFP immunoprecipitation and anti-GSK3β, anti-CK1α, anti–β-catenin, or anti-APC immunoblotting. (I) Axin1 KO HEK293T cells (KO1, a different KO line from the one shown in Fig. 4 A) transfected with TopFlash-luciferase and Axin1-EGFP WT or mutants were treated with or without Wnt3a conditional medium for 24 h and then harvested for luciferase determination. The expression level of Axin1 WT and mutant proteins in the reporter assay was detected with immunoblotting. Data are shown as mean ± SD (n = 3). Statistical analysis was performed with a two-tailed unpaired t test (*, P < 0.05; **, P < 0.01; ***, P < 0.001). (J) The expression level of the HA-tagged Axin1 WT and mutants in the fertilized Xenopus eggs (Fig. 4 C). The expressed proteins are indicated with an asterisk. Scale bars in A, B, and F, 10 µm; in D, E, and G, 2 µm. IB, immunoblot; IP, immunoprecipitation; Vec, vector; TCL, total cell lysate.
Figure 3.

Axin1 recruits GSK3β, CK1α, and β-catenin to its droplets and promotes GSK3β-mediated β-catenin phosphorylation. (A) Green-labeled Axin1 protein and the indicated red-labeled proteins were mixed and subjected to LLPS assay. Relative levels of the proteins recruited into the Axin1 droplets were obtained by fitting to the standard calibration curve with the MBP level set to one. Statistical analyses were performed with a two-tailed unpaired t test (***, P < 0.001). Data are shown as mean ± SD (n = 3). (B) Axin1, dAPC2, GSK3β, CK1α, and β-catenin proteins were mixed (2 µM each) for LLPS assay. Only the indicated protein was labeled by red. (C) Axin1 protein (2.5 µM) was incubated with 1.5 µM β-catenin and different concentrations of GSK3β or CK1α for phosphorylation assay and analyzed by immunoblotting. (D) Axin1 protein (2.5 µM) was incubated with 1.5 µM β-catenin, 0.5 µM GSK3β, or 2.5 µM CK1α for phosphorylation assay. (E) Axin1 protein (1 µM) was incubated with 1.5 µM β-catenin, 2.5 µM CK1α, or GSK3β at indicated concentrations (0.05–1 µM) for phosphorylation assay. (F) Axin1 protein (1 µM) was incubated with 50 nM GSK3β or 2.5 µM CK1α with or without addition of 2.5% PEG8000 for phosphorylation assay. (G) Axin1 protein or its mutants (1 µM) were incubated with 50 nM GSK3β and/or 2.5 µM CK1α for phosphorylation assay. (H) Axin1 KO HEK293T cells transfected with indicated plasmids were treated with 2.5 µg/ml MG-132 for 12 h before harvesting for immunoblotting. (I) β-Catenin (6 µM), GSK3β (0.25 µM), and CK1α (2.5 µM) were incubated with or without 1.5 µM Axin1 or dAPC2 for phosphorylation assay. The purified proteins are shown with asterisks. (C–G) The band intensity of phosphorylated β-catenin was normalized to total β-catenin protein. Scale bars in A and B, 2 µm. Vec, vector; Rel., relative.
Axin LLPS promotes GSK3β-mediated β-catenin phosphorylation
To investigate the role of LLPS of the destruction complex in regulating β-catenin stability, we performed in vitro phosphorylation assays. A low concentration of GSK3β protein failed to phosphorylate β-catenin at S33, S37, and T41, while the GSK3β-mediated phosphorylation was significantly increased in the presence of CK1α that phosphorylates β-catenin at S45 (Fig. 3, C and D) and further enhanced by Axin1 (Fig. 3 E). In the presence of PEG8000, the phosphorylation of β-catenin was dramatically facilitated by GSK3β and also by the presence of both GSK3β and CK1α, indicating the enzyme activity was significantly promoted by LLPS (Fig. 3 F). Consistently, PEG8000 enhanced Axin1 LLPS in the same phosphorylation assay condition (Fig. S3 E). All these data indicated that the LLPS-driven assembly of the destruction complex facilitates β-catenin phosphorylation by GSK3β.
Axin1 and AD2, but not AD1, AD3, and AD5, increased β-catenin phosphorylation by GSK3β, but not by CK1α (Fig. 3 G), indicating that the IDR1-mediated LLPS of Axin is critical to facilitate β-catenin phosphorylation by GSK3β. Also, the GSK3β-mediated phosphorylation required the ability of Axin1 to bind with GSK3β and β-catenin, as AD4 and AD6 did not enhance phosphorylation. Furthermore, AD2, like WT Axin1, was well colocalized with dAPC2, GSK3β, and β-catenin when they were coexpressed in Axin1 KO HEK293T cells (Fig. S3 F), and a fast recovery dynamic characteristic of dAPC2, GSK3β, and β-catenin in the puncta was observed (Fig. S3 G).
The importance of Axin LLPS in the GSK3β-mediated phosphorylation of β-catenin was confirmed in Axin1 KO HEK293T cells. WT Axin1 and AD2 increased β-catenin phosphorylation by GSK3β and reduced β-catenin protein level much more strongly than AD1, AD3, AD4, and AD5 (Fig. 3 H). A pulldown assay confirmed that the GSK3β-binding motif (aa 376–406) within IDR1 was essential for the GSK3β interaction (Fig. S3 H). IDR2 was critical for the interaction with APC and CK1α as both AD2 and AD3 showed no interaction. Intriguingly, AD3 did not bind β-catenin, indicating that both IDR1 and IDR2 contribute to β-catenin binding in addition to the known β-catenin–binding region (aa 464–520). dAPC2 also synergized with Axin1 to enhance β-catenin phosphorylation by GSK3β, but not by CK1α, in vitro (Fig. 3 I), consistent with the promoting effect of dAPC2 on Axin1 LLPS.
Axin LLPS is critical for Wnt/β-catenin signaling
Axin1 and AD2 significantly inhibited Wnt3a-induced luciferase expression in Axin1 KO HEK293T cells, while AD1 and AD3 failed to inhibit Wnt signaling and to promote β-catenin degradation (Fig. 4, A and B; and Fig. S3 I). Interestingly, both the expression of AD1 and AD3 led to a higher basal Wnt signaling, suggesting that they acted in a dominant negative manner, which is also consistent with the note that they caused a diffused distribution of dAPC2 (Fig. S3 F). AD4, AD5, AD6, and AD7 still retained some ability to inhibit Wnt-induced reporter expression, while AD7hnRNPA1 and AD7TDP43 exhibited a similar inhibitory effect to WT Axin1 (Fig. 4 A and Fig. S3 I). Wnt/β-catenin signaling regulates early embryogenesis and vertebrate dorsal axis formation, and its deregulation leads to abnormal development of Xenopus embryos (Clevers, 2006). Inhibition of Wnt/β-catenin signaling results in loss of axis (a ventralization phenotype) in Xenopus embryos (Kao and Elinson, 1989). The expression of Axin1, AD2, and AD5 in one-cell-stage Xenopus embryos resulted in morphologically abnormal embryos at the tail bud stage, by exhibiting a severe ventralized phenotype (partial axis and no axis; absence of axis structures such as cement gland and eye; Fig. 4 C and Fig. S3 J). AD1 and AD4 had the opposite effect and caused severe dorsalization (most of the embryos exhibit multiple or radial cement gland pigment), suggesting they may function in a dominant negative manner in the embryos. AD3 had a minimal effect, showing a loss-of-function phenotype. AD6 that lacks β-catenin binding domain caused a range of phenotypes, from normal to partial ventralized phenotype or partial dorsalized embryos, suggesting its partial loss-of-function.
Figure 4.

The LLPS property of Axin is needed to inhibit Wnt signaling. (A) Axin1 KO HEK293T cells were transfected with TopFlash-luciferase reporter and WT Axin1-EGFP or its mutants, then treated with or without Wnt3a conditional medium for 12 h and harvested for luciferase determination. Statistical analyses were performed with two-tailed unpaired t test. Data are shown as mean ± SD (n = 3). The stars (*) indicate the significant differences between each mutant group with the WT Axin1 group, while the hash symbols (#) indicate the significant differences between AD7TDP43 or AD7hnRNPA1 with the AD7 group. **, P < 0.01; ***, P < 0.001; ###, P < 0.0001. (B) Axin1 KO HEK293T cells transfected with WT Axin1-EGFP or its mutants were treated with 2.5 µg/ml cycloheximide (CHX) for the indicated time before harvesting for immunoblotting. (C) The Xenopus embryos were dejellied 30 min after fertilization and injected with mRNA encoding WT Axin1 or mutants. Embryos were cultured to the tailbud stage (stage 32) for phenotyping. Embryos with different morphologies and their percentages are shown. The histogram summarizes results from two independent experiments. β-cat, β-catenin; Vec, vector; Rel., relative.
These data together indicate that the LLPS property of Axin1 is important for it to inhibit Wnt signaling in Xenopus embryos. However, we realize that the in vivo function of Axin1 is more complex. In addition to regulating Wnt signaling, Axin1 is known to modulate other signaling pathways, such as JNK signaling and TGF-β signaling by interacting with MEKK, I-MFA, DCAP, Grb4, and Smad3 (Luo and Lin, 2004), and these signaling pathways are important to regulating the development of Xenopus embryos. Moreover, different from Axin1 KO HEK293T cells, the existence of endogenous Axin1 protein in embryos may also affect the phenotypic readout. Therefore, Axin1 deletion mutants may produce more complex effects in Xenopus embryos, leading to some differences between the Xenopus embryo phenotypes and the Wnt reporter expression.
Axin has been shown to recruit destruction complex components and β-catenin into its puncta in cells (Faux et al., 2008; Pronobis et al., 2015; Schaefer et al., 2018; Schaefer and Peifer, 2019). Here, we demonstrated that the IDR1-mediated Axin LLPS enhanced by APC recruits and concentrates GSK3β, CK1α, and β-catenin in condensates, greatly facilitating GSK3β-mediated β-catenin phosphorylation and thus degradation, which is critical for Wnt/β-catenin signaling. Compared with a diffuse degradation complex, Axin1-mediated phase separation can enhance the phosphorylation and degradation of β-catenin by enhancing the local concentration of destruction complex component, and reducing the concentration of GSK3β required for phosphorylation of β-catenin. Moreover, formation of LLPS-confined compartments could also ensure the spatially controlled signaling transduction and its quick response to stimuli.
Materials and methods
Antibodies
Mouse monoclonal anti-β-catenin (SC7963; Santa Cruz Technology), rabbit monoclonal anti-CK1α (ab108296; Abcam), rabbit monoclonal anti-GSK3β (12456; Cell Signaling Technology), rabbit monoclonal anti-β-catenin (S45; 9564; Cell Signaling Technology), rabbit monoclonal anti-β-catenin (S33,37,T41; 2009; Cell Signaling Technology), rabbit monoclonal anti-Axin1 (2074; Cell Signaling Technology), rabbit monoclonal anti-APC (2504; Cell Signaling Technology), mouse monoclonal anti-GFP (scc8334; Santa Cruz Technology), mouse monoclonal anti-MBP (M6295; Sigma-Aldrich), and rabbit monoclonal anti-His-probe (H-15; sc803; Santa Cruz Technology) antibodies, mouse IgG (12-371B; Millipore), and rabbit IgG (12–370; Cell Signaling Technology) were purchased from the indicated suppliers.
Cell culture, plasmids, and transfection
Human embryonic kidney HEK293T cells (CRL-3216) and human colorectal adenocarcinoma SW480 (CCL-228) and HCT116 cells (CCL-247) were obtained from American Type Culture Collection. HEK293T cells were maintained in DMEM (Corning) supplemented with 10% FBS (Gibco) at 37°C in a humidified, 5% CO2 incubator. HCT116 cells were maintained in McCoy’s 5a Modified Medium (Corning) supplemented with 10% FBS, while SW480 cells were maintained in Leibovitz’s L-15 Medium (Corning) supplemented with 10% FBS without CO2. Cell transfection was conducted with VigoFect (Vigorous Biotechnology) or Lipofectamine 2000 (Invitrogen).
All plasmids used in this study were generated by subcloning corresponding cDNAs into HA-pcDNA3.1 (for mammalian cell expression), pET32M.3C, pETMBP.3C (for bacteria expression), or pCS107-HA (for in vitro transcription) vectors. The sequences encoding human Axin1 (NCBI RefSeq no. NM_181050.3), human APC (NM_000038.6), human GSK3β (NM_001146156.2), β-catenin (NM_001098209.2), and human CK1α (NM_001025105.3) were amplified using standard PCR procedures with cDNA from HEK293T cells and subcloned into HA-pcDNA3.1, pETMBP.3C, and pCS107-HA vectors. Drosophila melanogaster dAPC2 (NM_001347814.1) was amplified and subcloned into pETMBP.3C vectors. Plasmids encoding Axin1 deletions or mutations were generated by PCR using primers with appropriate nucleotide substitutions.
Gene editing with CRISPR/Cas9
HEK293T cells were transfected with plasmids encoding Cas9 and a single-guide RNA 5′-TTCGCTGTACCGTCTACTGG-3′ targeting the exon1 region of Axin1. Axin1 KO clones were screened by immunoblotting and confirmed by genomic DNA sequencing. Two clones (KO1 and KO2) were obtained and used in this study.
Immunoblotting, coimmunoprecipitation, immunofluorescence, pulldown, and reporter assays
All genes were PCR-amplified and cloned into the HA-pcDNA3.1 vector (for mammalian cell expression) to produce HA-tag–fused recombinant proteins. To express EGFP- or mCherry-fused proteins, the EGFP or mCherry cDNA was fused to the 3′ end of target cDNAs, and therefore the expressed proteins were tagged with HA at the N terminus and EGFP or mCherry at the C terminus. Point mutations and deletions were introduced by site-directed mutagenesis.
For immunoblotting, the cells were lysed on ice with the lysis solution (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.5% NP-40, 2 mM EDTA, 1 mM NaVO3, 10 mM NaF, and protease inhibitors) and rotated for 30 min at 4°C. The lysates were subjected to SDS-PAGE, and immunoblotting was performed with primary antibodies as indicated and secondary anti-rabbit or anti-mouse antibodies conjugated to horseradish peroxidase, followed by detection with enhanced chemiluminescent substrate (Pierce) according to the manufacturer’s instructions. For coimmunoprecipitation, the cells were lysed as above. After an aliquot was taken for total protein expression analysis, immunoprecipitation was performed by adding precleared GFP-Nanoab-Agarose (LABLEAD) to the remaining cell lysate followed by incubation at 4°C overnight with gentle rotation. Then the immune complex was isolated by centrifugation, washed with lysis solution, and analyzed by SDS-PAGE and immunoblotting. For immunofluorescence, cells transfected with the indicated plasmids were washed two times with PBS, fixed with 4% formaldehyde solution for 20 min, permeabilized with 0.1% Triton X-100 in PBS for 5 min, and blocked in 5% BSA in PBS for 30 min at room temperature. Then, DAPI was added for 1 h, and images were obtained with Nikon A1 RMP Multiphoton microscope. Images were analyzed with NIS-Elements AR Analysis. For pull-down assays, cells were lysed with the lysis solution. The lysates were incubated with GFP-Nanoab-Agarose (LABLEAD) at 4°C for 2 h. The beads were then collected, washed three times with lysis buffer, and subjected to immunoblotting. For in vitro pulldown assays, proteins were incubated with GFP-Nanoab-Agarose (LABLEAD) in the buffer containing 20 mM Hepes, pH 7.4, and 1 M NaCl, and incubated at 4°C for 2 h. The beads were then collected, washed three times with buffer, and subjected to immunoblotting. For reporter assays, cells were transfected with TopFlash-luciferase reporter and Renilla as an internal control and treated with or without Wnt3a conditional medium for 24 h, and luciferase activity was measured after 36 h with the dual reporter assay system (Promega) following the manufacturer’s protocol. The experiments were repeated in triplicate, and the data are presented as the mean ± SD after normalization to Renilla luciferase activity.
Protein expression and purification
All genes were PCR-amplified and cloned into the pET32M.3C vector to produce His6-tag–fused recombinant proteins or pETMBP.3C vector to produce MBP-tag–fused recombinant proteins. To express EGFP- or mCherry-fused proteins, the EGFP or mCherry cDNA was fused to the 3′ end of target cDNAs. Point mutations and deletions were introduced by the site-directed mutagenesis approach. All recombinant proteins used in this study were expressed in Escherichia coli BL21-CodonPlus (DE3) with induction by 1 M IPTG for 16 h at 18°C. To purify the MBP-His6-Axin, MBP-His6-Axin1-GFP, MBP-His6-dAPC2, MBP-His6-GSK3β, MBP-His6-CK1α, MBP-His6-β-catenin, MBP-His6-AD1, MBP-His6-AD2, MBP-His6-AD3, MBP-His6-AD4, MBP-His6-AD5, or MBP-His6-AD6 proteins expressed with pETMBP.3C, E. coli cells were resuspended in binding buffer (50 mM Tris-Cl, pH 7.9, 2 M NaCl, and 10 mM imidazole), lysed with a high-pressure homogenizer, and sedimented at 18,000 rpm for 30 min. The supernatant lysates were purified in Amylose Resin (NEB). After extensive washing with binding buffer, proteins were eluted with MBP elution buffer (50 mM Tris-Cl, pH 7.9, 2 M NaCl, and 10 mM imidazole, for MBP-tagged proteins), then purified with a HiPrep 26/60 Sephacryl S-200 HR column (17–1195-01; GE Healthcare) on an AKTA purifier (GE Healthcare), and eluted with a buffer containing 20 mM Hepes, pH 7.4, and 1 M NaCl. To purify the His6-FUS proteins expressed with pET32M.3C, E. coli cells were resuspended in binding buffer (50 mM Tris-Cl, pH 7.9, 2 M NaCl, and 10 mM imidazole), lysed with a high-pressure homogenizer, and sedimented at 18,000 rpm for 30 min. The supernatant lysates were purified on Ni-NTA agarose beads (for His6-tagged proteins; QIAGEN). After extensive washing with binding buffer, the proteins were eluted with elution buffer (50 mM Tris-Cl, pH 7.9, 2 M NaCl, and 2 M imidazole, for His6-tagged proteins), then loaded onto a desalting column (17–0851-01; GE Healthcare), and finally eluted with a buffer containing 20 mM Hepes, pH 7.4, and 1 M NaCl. All the purified proteins were concentrated by centrifugal filtration (Millipore) All the purified proteins were concentrated by centrifugal filtration (Millipore) and stored in aliquots at −80°C.
In vitro phase separation assay
Proteins dissolved in the buffer containing 20 mM Hepes, pH 7.4, and 1 M NaCl were mixed, and the concentration of NaCl was adjusted to 150 mM with buffer containing 20 mM Hepes, pH 7.4. The mixture was treated with PEG8000 or PreScission protease immediately, the concentration of NaCl was further adjusted to 150 mM NaCl, and then droplet formation was examined. FUS formed aggregates by induction of phase separation and agitated (middle and bottom) for 1 d at 25°C in the presence of RNA (0.4:1 FUS, by mass). For imaging, droplets were observed either on a glass slide or in a glass-bottom cell culture dish for differential interference contrast or fluorescence imaging (Imager M2 and Zeiss LSM 880) with a 63 × 1.40 oil-immersion objective lens (Plan-Apochromatlan; Zeiss) and a camera (Axiocam HRm; Zeiss) at room temperature. To examine the effect of 1,6-hexanediol (an aliphatic alcohol that disrupts weak hydrophobic interactions) on droplets, the phase-separated droplet solution (0.5 ml) was dispensed into plastic cuvettes, and then 1,6-hexanediol was added for further incubation at room temperature. Zeiss LSM software (Version 4.2 SP1) and ZEN software 2012 (Zeiss) were used to analyze these images.
Turbidity measurement
Turbidity was quantified by absorbance of 600-nm-wavelength light for proteins diluted to 20 mM Hepes, pH 7.4, and different concentrations of NaCl. Phase separation was initiated by addition of PreScission protease or PEG8000. Turbidity was monitored by VARIOSKAN FLASH (Thermo Fisher Scientific). All measurements were performed in triplicate with three repeats of each group, and similar results were obtained.
Protein fluorescence labeling
Alexa Fluor-488 NHS ester (green; Thermo Fisher Scientific) and Sulfo-Cyanine3 maleimide (red; Lumiprobe) were dissolved in DMSO and incubated with the corresponding protein at room temperature for 1 h (fluorophore to protein molar ratio was 1:1). The reaction was quenched by 20 mM Hepes, pH 7.4, and 1 M NaCl. The fluorophores and other small molecules were removed from the proteins by passing the reaction mixture through a desalting column (17–0851-01; GE Healthcare) with buffer containing 20 mM Hepes, pH 7.4, and 1 M NaCl. In imaging assays, fluorescence-labeled proteins were further diluted with the corresponding unlabeled proteins in the same buffer.
FRAP analysis
FRAP was performed with a confocal microscope (LSM 880 Meta plus Zeiss Axiovert zoom; Zeiss) at room temperature. Fluorescence-labeled protein was adjusted to 1% by diluting the labeled protein into the unlabeled one. Defined regions were photobleached at a specific wavelength using the 561-nm or 488-nm laser, and the fluorescence intensities in these regions were collected every 3 s (for in vitro droplets) and normalized to the initial intensity before bleaching. Image intensity was measured by mean mean region of interest and further analyzed by Prism (GraphPad).
Quantification of protein concentration in the condensed phase droplets
Only one component was labeled with Sulfo-Cyanine3 maleimide (red). To generate a standard calibration curve, the fluorescence intensity of a protein at different concentrations was measured by confocal microscopy (LSM 880 Meta plus Zeiss Axiovert zoom; Zeiss) and analyzed by the ImageJ software. The protein concentration in the measured droplets was obtained by fitting the measured fluorescence intensity into the standard calibration curve.
In vitro phosphorylation assay
The purified proteins were mixed in 40 µl of reaction buffer containing 25 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10 mM MgCl2, 1 mM DTT, and 5 mM ATP for 30 min at room temperature. The reaction was stopped by adding SDS loading buffer, and proteins were analyzed by immunoblotting.
Quantification of endogenous protein concentrations by liquid chromatography–tandem mass spectrometry
For the quantitative analysis of human Axin1 and APC protein, cells were lysed by lysis buffer containing 8 M urea and protease inhibitor (Roche) in PBS. 100 µg protein was reduced, alkylated, and digested by trypsin, followed by being desalted. The tryptic peptides and standard synthetic peptides (Axin1: LEPCDSNEEK, EQVEAEATR, and FAEELIHR; APC: GAATDEK, QLEYEAR, YSDEQLNSGR) were labeled by TMT reagent and mixed together. The mixture was desalted using C18 stage-tips.
For liquid chromatography–tandem mass spectrometry analysis, the peptides were separated by a C18 column (75 µm inner diameter, 150 mm length, 5 µm, 300 Å) with a Thermo-Dionex Ultimate 3000 HPLC system, which was directly connected with a Thermo Fisher Scientific Q-Exactive HF-X Hybrid Quadrupole-Orbitrap mass spectrometer. A series of adjusted linear gradients according to the hydrophobicity of fractions with a flow rate of 300 nl/min was applied. The mass spectrometer was programmed to acquire in the data-dependent acquisition mode. The survey scan was from m/z 300 to 1,800 with resolution of 60,000 at m/z 400. After one microscan, the top 40 most intense peaks with charge state 2 and above were dissociated by normalized collision energy of 32%. The isolation window was set at 0.7 D width, and the dynamic exclusion time was 15 s. The MS2 spectra were acquired with a resolution of 15,000, an automatic gain control target of 1e5, and a maximum IT of 50 ms.
The generated tandem mass spectrometry spectra were searched against the database containing sequences of target proteins by using the SEQUEST search engine in Proteome Discoverer 2.2 software. The search criteria were as follows: full tryptic specificity was required, one missed cleavage was allowed, carbamidomethylation on cysteine and TMT-6plex on lysine/peptide N terminus were set as the fixed modifications, oxidation on methionine was set as the variable modification, precursor ion mass tolerances were set at 20 ppm for all mass spectrometry acquired in an Orbitrap mass analyzer, and the fragment ion mass tolerance was set to 20 mmu for all MS2 spectra acquired. Peptide spectral matches were validated using Fixed Value PSM Validator provided by Proteome Discoverer software based on q-values at a 1% false discovery rate.
Xenopus embryo injection
Xenopus laevis (J strain) was obtained from Nasco, Inc. and maintained according to the Institutional Animal Care and Use Committee protocol. The animal manipulation protocol was approved by the Tsinghua University Animal Care and Use Committee. Fertilized eggs were dejellied using 2% L-cysteine (pH 8.0) in 0.1× Marc’s Modified Ringer (MMR), then thoroughly washed with 0.1× MMR and injected with mRNA encoding WT or deletion mutants of Axin1 (AD1–AD6). The dose for mRNA injection was 1 ng per embryo. For microinjection after fertilization, eggs were cultured in 0.5× MMR plus 2% Ficoll400 (GE Healthcare). Microinjected embryos were transferred into fresh 0.1× MMR at stage 8 and cultured to the tailbud stages (stage 32) for phenotyping and recording.
Quantification and statistical analysis
Statistical analyses were performed with a two-tailed unpaired t test. *, P < 0.05; **, P < 0.01; and ***, P < 0.001 were considered statistically significant. For the bar charts, data were plotted as mean ± SD from at least three independent experiments. For immunoblot gel quantification, the gels were scanned, and the band intensities were quantified with ImageJ. The band intensity of total proteins was normalized to the loading control (GAPDH), and the band intensity of phosphorylated proteins was normalized to the total proteins. Graphs were generated in GraphPad Prism 5.
Online supplemental material
Fig. S1 shows that LLPS of Axin1 protein depends on protein concentration and salt concentration. Fig. S2 shows that APC protein undergoes LLPS. Fig. S3 shows the colocalization of Axin with APC, GSK3β, and β-catenin in HEK293T cells and interaction of Axin1 or its deletion mutants with APC, GSK3β, CK1α, or β-catenin.
Acknowledgments
We thank the Protein Chemistry and Proteomics Facility at the Technology Center for Protein Sciences of Tsinghua University for sample analysis and Dr. Pilong Li for insightful suggestions.
This work was supported by grants from the National Natural Science Foundation of China (31988101 and 31730056) and the National Key Research and Development Program of China (2017YFA0103601) to Y.-G. Chen, and the National Natural Science Foundation of China (31571531) to Q. Tao.
The authors declare no competing financial interests.
Author contributions: J. Nong, K. Kang, Q. Shi, Q. Tao, and Y.-G. Chen designed the experiments; J. Nong, K. Kang, Q. Shi, and X. Zhu performed the experiments; J. Nong, K. Kang, Q. Shi, Q. Tao, and Y.-G. Chen analyzed the data; and J. Nong, K. Kang, Q. Shi, and Y.-G. Chen wrote the paper.
References
- Alberti, S., Gladfelter A., and Mittag T.. 2019. Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell. 176:419–434. 10.1016/j.cell.2018.12.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banani, S.F., Lee H.O., Hyman A.A., and Rosen M.K.. 2017. Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 18:285–298. 10.1038/nrm.2017.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienz, M. 2014. Signalosome assembly by domains undergoing dynamic head-to-tail polymerization. Trends Biochem. Sci. 39:487–495. 10.1016/j.tibs.2014.08.006 [DOI] [PubMed] [Google Scholar]
- Clevers, H. 2006. Wnt/beta-catenin signaling in development and disease. Cell. 127:469–480. 10.1016/j.cell.2006.10.018 [DOI] [PubMed] [Google Scholar]
- Clevers, H., and Nusse R.. 2012. Wnt/β-catenin signaling and disease. Cell. 149:1192–1205. 10.1016/j.cell.2012.05.012 [DOI] [PubMed] [Google Scholar]
- Cliffe, A., Hamada F., and Bienz M.. 2003. A role of Dishevelled in relocating Axin to the plasma membrane during wingless signaling. Curr. Biol. 13:960–966. 10.1016/S0960-9822(03)00370-1 [DOI] [PubMed] [Google Scholar]
- Conicella, A.E., Zerze G.H., Mittal J., and Fawzi N.L.. 2016. ALS Mutations Disrupt Phase Separation Mediated by α-Helical Structure in the TDP-43 Low-Complexity C-Terminal Domain. Structure. 24:1537–1549. 10.1016/j.str.2016.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faux, M.C., Coates J.L., Catimel B., Cody S., Clayton A.H., Layton M.J., and Burgess A.W.. 2008. Recruitment of adenomatous polyposis coli and beta-catenin to axin-puncta. Oncogene. 27:5808–5820. 10.1038/onc.2008.205 [DOI] [PubMed] [Google Scholar]
- Gammons, M., and Bienz M.. 2018. Multiprotein complexes governing Wnt signal transduction. Curr. Opin. Cell Biol. 51:42–49. 10.1016/j.ceb.2017.10.008 [DOI] [PubMed] [Google Scholar]
- Jho, E.H., Zhang T., Domon C., Joo C.K., Freund J.N., and Costantini F.. 2002. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol. Cell. Biol. 22:1172–1183. 10.1128/MCB.22.4.1172-1183.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao, K.R., and Elinson R.P.. 1989. Dorsalization of mesoderm induction by lithium. Dev. Biol. 132:81–90. 10.1016/0012-1606(89)90207-8 [DOI] [PubMed] [Google Scholar]
- Kunttas-Tatli, E., Roberts D.M., and McCartney B.M.. 2014. Self-association of the APC tumor suppressor is required for the assembly, stability, and activity of the Wnt signaling destruction complex. Mol. Biol. Cell. 25:3424–3436. 10.1091/mbc.e14-04-0885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, E., Salic A., Krüger R., Heinrich R., and Kirschner M.W.. 2003. The roles of APC and Axin derived from experimental and theoretical analysis of the Wnt pathway. PLoS Biol. 1:E10. 10.1371/journal.pbio.0000010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, V.S., Ng S.S., Boersema P.J., Low T.Y., Karthaus W.R., Gerlach J.P., Mohammed S., Heck A.J., Maurice M.M., Mahmoudi T., and Clevers H.. 2012. Wnt signaling through inhibition of β-catenin degradation in an intact Axin1 complex. Cell. 149:1245–1256. 10.1016/j.cell.2012.05.002 [DOI] [PubMed] [Google Scholar]
- Luo, W., and Lin S.C.. 2004. Axin: a master scaffold for multiple signaling pathways. Neurosignals. 13:99–113. 10.1159/000076563 [DOI] [PubMed] [Google Scholar]
- MacDonald, B.T., Tamai K., and He X.. 2009. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell. 17:9–26. 10.1016/j.devcel.2009.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molliex, A., Temirov J., Lee J., Coughlin M., Kanagaraj A.P., Kim H.J., Mittag T., and Taylor J.P.. 2015. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell. 163:123–133. 10.1016/j.cell.2015.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusse, R., and Clevers H.. 2017. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell. 169:985–999. 10.1016/j.cell.2017.05.016 [DOI] [PubMed] [Google Scholar]
- Pronobis, M.I., Rusan N.M., and Peifer M.. 2015. A novel GSK3-regulated APC:Axin interaction regulates Wnt signaling by driving a catalytic cycle of efficient βcatenin destruction. eLife. 4:e08022. 10.7554/eLife.08022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronobis, M.I., Deuitch N., Posham V., Mimori-Kiyosue Y., and Peifer M.. 2017. Reconstituting regulation of the canonical Wnt pathway by engineering a minimal β-catenin destruction machine. Mol. Biol. Cell. 28:41–53. 10.1091/mbc.e16-07-0557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, D.M., Pronobis M.I., Poulton J.S., Kane E.G., and Peifer M.. 2012. Regulation of Wnt signaling by the tumor suppressor adenomatous polyposis coli does not require the ability to enter the nucleus or a particular cytoplasmic localization. Mol. Biol. Cell. 23:2041–2056. 10.1091/mbc.e11-11-0965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer, K.N., and Peifer M.. 2019. Wnt/Beta-Catenin Signaling Regulation and a Role for Biomolecular Condensates. Dev. Cell. 48:429–444. 10.1016/j.devcel.2019.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer, K.N., Bonello T.T., Zhang S., Williams C.E., Roberts D.M., McKay D.J., and Peifer M.. 2018. Supramolecular assembly of the beta-catenin destruction complex and the effect of Wnt signaling on its localization, molecular size, and activity in vivo. PLoS Genet. 14:e1007339. 10.1371/journal.pgen.1007339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz-Romond, T., Metcalfe C., and Bienz M.. 2007. Dynamic recruitment of axin by Dishevelled protein assemblies. J. Cell Sci. 120:2402–2412. 10.1242/jcs.002956 [DOI] [PubMed] [Google Scholar]
- Sear, R.P. 2007. Dishevelled: a protein that functions in living cells by phase separating. Soft Matter. 3:680–684. 10.1039/b618126k [DOI] [PubMed] [Google Scholar]
- Stamos, J.L., and Weis W.I.. 2013. The β-catenin destruction complex. Cold Spring Harb. Perspect. Biol. 5:a007898. 10.1101/cshperspect.a007898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, A., Conicella A.E., Schmidt H.B., Martin E.W., Rhoads S.N., Reeb A.N., Nourse A., Ramirez Montero D., Ryan V.H., Rohatgi R., et al. 2018. A single N-terminal phosphomimic disrupts TDP-43 polymerization, phase separation, and RNA splicing. EMBO J. 37:e97452. 10.15252/embj.201797452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, D., and Pan W.. 2010. GSK3: a multifaceted kinase in Wnt signaling. Trends Biochem. Sci. 35:161–168. 10.1016/j.tibs.2009.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H., Elbaum-Garfinkle S., Langdon E.M., Taylor N., Occhipinti P., Bridges A.A., Brangwynne C.P., and Gladfelter A.S.. 2015. RNA Controls PolyQ Protein Phase Transitions. Mol. Cell. 60:220–230. 10.1016/j.molcel.2015.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]