

Abstract
Renal tubular acidosis is comprised of a diverse group of congenital or acquired disease with the common dominator of defective renal acid excretion with protean manifestation but in adults, recurrent kidney stones and nephrocalcinosis are main modes of presentation. Calcium phosphate stones and nephrocalcinosis are frequently encountered in distal hypokalemic RTA type 1. Alkaline urinary pH, hypocitraturia, and less frequently hypercalciuria, are the tripartite lithogenic factors in dRTA predisposing to calcium phosphate stone formation; the latter two are also commonly encountered in other causes of urolithiasis. While the full blown syndrome is easily diagnosed by conventional clinical criteria, an attenuated forme fruste called incomplete dRTA typically evades clinical testing and only uncovered by provocative acid loading challenges. Stone formers that cannot acidify urine pH <5.3 during acid loading are considered to have incomplete dRTA. However, urinary acidification capacity is not a dichotomous but rather a continuous trait so incomplete dRTA is not a distinct entity but maybe one end of a spectrum. Recent findings suggest that incomplete dRTA can be attributed to heterozygous carriers of hypofunctional V-ATPase. The value of incomplete dRTA diagnosis by provocative testing and genotyping candidate genes are valuable research tools but remains unclear at the moment whether they alter clinical practice, and need further clarification. No randomized control trials have been performed in SF with dRTA or calcium phosphate stones and until such data is available, treatment of calcium phosphate stones are centered on reversing the biochemical abnormalities encountered in the metabolic work-up. SF with type I dRTA should receive alkali therapy, preferentially in the form of K-citrate delivered judiciously to treat the chronic acid retention which drives both stone formation and bone disease.
Keywords: Kidney stones, renal tubular acidosis, acidification test, alkali therapy
Introduction
Homeostasis or fixité du milieu intérieur, as first described by the pioneering French physiologist Claude Bernard, is a prerequisite for multicellular life and ensured by multiple organs, including the kidney. In addition to numerous other homeostatic tasks, the kidney plays a pivotal role in maintaining body acid-base balance and body fluid pH by the reclamation of bicarbonate from the glomerular filtrate and excretion of net acid. Renal net acid excretion regenerates bicarbonate decomposed by non-volatile acids. If renal excretion is defective or overwhelmed as in the case of non-volatile acid overproduction, metabolic acidosis ensues. In contrast, renal tubular acidosis (RTA) is a group of congenital or acquired disorders characterized not by non-volatile acid overproduction, but defective renal acid excretion in the setting of preserved glomerular filtration rate (GFR). Traditionally, RTA is classified as “proximal” or “distal”, depending on the site of the tubular lesion. There is also a popular but less informative numeric classification separating RTA to types I–IV (see Table 1). Clinically, all forms of RTAs – with the exception of the incomplete form of distal RTA-present as normal anion gap metabolic acidosis. To secure the diagnosis of RTA, other forms of normal anion gap acidosis need to be excluded; which includes non-renal alkali-losing states and overproduction acidosis with successful renal excretion of the conjugate anion of the acid. Depending on the type of RTA, serum K and urinary pH are either high or low and allow further separation (see Table 1)1. Low urine ammonium and its surrogate of highly positive urinary anion gap, are present in distal RTAs (dRTAs) but typically not in in proximal RTA (pRTA).
Table 1.
Classification of renal tubular acidosis by nephron segment and phenotypic features
| Tubular segment | Numeric type | Synonym(s) | Plasma chemistry | Urine chemistry | Clinical features |
|---|---|---|---|---|---|
| Proximal | II |
|
|
Maximally acidic urine in the absence of bicarbonaturia. Alkalinuria when plasma [HCO3−] is raised to normal. β2-microglobulinuria in the absence of albuminuria. | No nephrolithiasis |
| Distal | I |
|
|
Alkaline urine, urine pH > 6 Hypocitraturia | Calcium nephrolithiasis, Nephrocalcinosis (frequent) |
| IV |
|
|
Urine pH <6 Low urinary ammonium |
Increased risk of uric acid nephrolithiasis if urinary pH <5.3, especially if type II DM, high BMI. | |
| Combined proximal and distal | III |
|
|
Alkaline urine, urine pH > 6 Hypocitraturia | Calcium nephrolithiasis, Nephrocalcinosis (unusual) |
The clinical presentation of RTA ranges from completely asymptomatic to increasingly dire outcomes such as recurrent kidney stones, nephrocalcinosis, end stage renal disease, and sudden death from hypokalemic dysrhythmias. The main focus of this review lies on kidney stone formation in incomplete dRTA (idRTA), but we provide a briefly account of the association of all RTA forms with renal stone disease.
Proximal RTA (Type II)
Impaired tubular bicarbonate reclamation, either isolated or generalized in the form of a Fanconi syndrome, is the hallmark of pRTA2. Isolated forms of pRTA are mostly congenital with either autosomal-recessive or autosomal-dominant inheritance. While the former is due to bi-allelic mutations in the basolateral sodium/bicarbonate cotransporter NBCe1, encoded by the SLC4A4 gene, the cause of the latter remains elusive2, 3. Although patients with pRTA are acidemic (low blood pH), daily accruing non-volatile acid equivalents can be eliminated by the distal nephron and no net acid retention occurs under steady-state conditions3, 4. As a result, the classical promoters of calcium phosphate (CaP) stone formation encountered in type I dRTA and idRTA (hypercalciuria, hypocitraturia and alkalinuria) are not present in pRTA. Thus, with the exception of some specific rare scenarios (e.g. Fanconi syndrome associated with Dent’s disease), nephrocalcinosis and nephrolithiasis are not common features of pRTA.
Hyperkalemic forms of RTA: low urinary pH and uric acid stones
In contrast to pRTA, all forms of dRTA may be associated with recurrent kidney stones. The pathogenic mechanisms of stone formation are similar for hypokalemic type I, combined type III and incomplete dRTA, stone formation. In hyperkalemic forms of dRTA, however, the mechanism is different. Hyperkalemic dRTA associated with aldosterone deficiency, also known as Type IV RTA, is the most frequent form of RTA encountered clinically and is characterized by low urine pH and decreased acid excretion. This is in contrast to other forms of hyperkalemic dRTA where aldosterone is not decreased and urine pH always fails to decrease as discussed elsewhere in this issue. Calcareous stone formation is uncommon in patients with hyperkalemic forms of dRTA5–8. A main reason for that is that patients with hyperkalemic RTA have typically some degree of CKD with marked reduction in urinary calcium excretion. Type IV dRTA can potentially be a cause of uric acid calculi, especially if associated with low urine pH (<5.5), type II diabetes or high body mass index (BMI)9–11. Note that the majority of classic uric acid stone formers (SF) have unduly aciduria but rarely have hyperkalemic RTA12. The fundamental pathophysiology in idiopathic uric acid nephrolithiasis is an increased acid load to the kidney and inadequate ammonia production/excretion13. The solubility of undissociated uric acid is low; ~0.5 mM in human urine at 37 °C. Thus, at a urine pH of 5.35 (pKa of uric acid), only ~1 mM uric acid (sum of dissociated and undissociated forms of uric acid) can be solubilized. Physiological concentrations of uric acid in the urine are typically > 1 mM, and as such uric acid stone formation is a simple consequence of a low urinary pH. Low urinary pH in type IV RTA is due to a shortage of the urinary buffer ammonium, caused by hyperkalemia-induced impaired ammoniagenesis in the proximal tubule9, 11, 14. Treatment of uric acid calculi associated with hyperkalemic type IV RTA should be targeted at eliminating the underlying cause (e.g. cessation of offending drugs, treatment of obstructive uropathy). If this is not feasible, increase of urinary pH by alkali supplementation effectively prevents stone formation.
Hypokalemic distal RTA (Type I): High urinary pH and calcium phosphate stones
The first description of type I dRTA in an autopsy series with six children was presented by Lightwood in 193515. Albright and associates recognized the tubular origin of the entity in 1946 and the term “renal tubular acidosis” was coined by Pines and Mudge in 195116, 17. Type I dRTA can be acquired or inherited (Table 2). A myriad of acquired causes are known to cause type I dRTA, the most classical one being Sjögren’s syndrome with autoantibodies directed at α–intercalated cells18. For familial cases, autosomal-recessive and autosomal-dominant mutations in the anion exchanger 1 (AE1, encoded by SLC4A1 gene), autosomal-recessive mutations in the B1 and a4 subunits of the V-ATPase (encoded by ATP6V1B1 and ATP6V0A4 genes, respectively) and recently autosomal-recessive mutations in the transcription factor Foxi1 (encoded by the FOXI1 gene) have been identified as the underlying monogenic causes19–22. Overall, type I dRTA is considered a rare cause of calcareous nephrolithiasis23, 24. Mechanistically, rate- (or capacity-) limited distal tubular H+ secretion is the reason for reduced urinary net acid excretion and alkaline urinary pH25. Unlike in pRTA, there is systemic H+ retention in patients with type I dRTA3, 4. As a consequence of H+ retention, intestinal calcium absorption and release of calcium from bone increase and renal calcium reabsorption decreases, resulting in hypercalciuria.26–28. Hypocitraturia due to avid reclamation of citrate by proximal tubular cells in the setting of systemic acidosis is another hallmark of type I dRTA.
Table 2.
Etiology of hypokalemic type I distal tubular acidosis
| Type | Etiology | Clinical features |
|---|---|---|
| Congenital: monogenic mutations | B1 | Autosomal recessive. Early onset sensorineural hearing loss. |
| a4 | Autosomal recessive. Late onset sensorineural hearing loss. | |
| AE1 | Autosomal recessive or dominant forms. Hemolytic anemia. | |
| Foxi1 | Autosomal recessive. Early onset sensorineural hearing loss. | |
| CA | Autosomal recessive. Mixed pRTA and dRTA | |
| Acquired conditions | Autoimmune diseases | Sjögren syndrome. Systemic lupus erythematosus, Cryoglubulinemia. Primary biliary cirrhosis. Thyroiditis |
| Dysproteinemic states | Hyperglobulinemia, Paraprotein states. | |
| Endocrine hypercalciuria | Hyperparathyroidism. Vitamin D intoxication. Medullary sponge kidney. Idiopathic hypercalciuria | |
| Drugs and toxins | Amphotericin B. Lithium. Vanadate. Cyclamate. Carbonic anhydrase inhibitors (topiramate, acetazolamide) | |
| Tubulointerstitial disease | Interstitial nephritis. Chronic pyelonephritis. Obstructive uropathy. Balkan nephropathy. Renal transplantation, bariatric surgery with hyperoxaluria | |
| Liver disease | Autoimmune hepatitis. Cirrhosis. |
The sequelae of type I dRTA are recurrent nephrolithiasis, nephrocalcinosis and bone disease. The three key prolithogenic factors in type I dRTA (hypercalciuria, hypocitraturia and relatively alkaline to very alkaline urinary pH) favor CaP precipitation (Fig. 1). The typical calculus in type I dRTA consists of carbonate apatite (95.7%) with only minute brushite admixture (1.4%) and has a characteristic morphology with a smooth aspect and a glazed brown-yellow appearance with tiny cracks29, 30. Stone composition similar to dRTA is observed in patients with carbonic anhydrase inhibitor treatment (acetazolamide, topiramate). In contrast, patients with primary hyperparathyroidism and renal phosphate leak typically have CaP stones that contain significantly more brushite (29.1 and 23.9% respectively)29. The degree of CaP admixture is accurately reflected by the magnitude of brushite supersaturation31. As the stone composition ranges from CaOx to mixed CaOx-CaP, to CaP, the prevalence of a urinary acidification deficit increases from 5 to 40%32, 33. The concurrent existence of type I dRTA and nephrocalcinosis has been known since 1950’s and the order of causality is likely bidirectional34; i.e. nephrocalcinosis can be the result of dRTA or vice versa. The pathogenesis of nephrocalcinosis in type I dRTA is not known but possibly involves the same lithogenic factors that also foster the development of stones. The increase in basolateral uptake of citrate by the proximal tubule from the peritubular capillaries can lower interstitial citrate concentration as the vasa recta traverse the deep medulla. Counter to this effect is the reduced pumping of H+ into the tubular lumen which will decrease basolateral bicarbonate exit from the collecting duct and decrease interstitial pH, hence reducing the risk of CaP precipitation. Interstitial ectopic calcifications may also be a variant of Randall’s plaques which occurs without acidification defects35. In familial cases, the prevalence of nephrocalcinosis typically depends on the severity of the disease with late occurrence in autosomal-dominant AE1 mutations but early and severe in autosomal-recessive AE1 and V-ATPase mutations.
Figure 1.
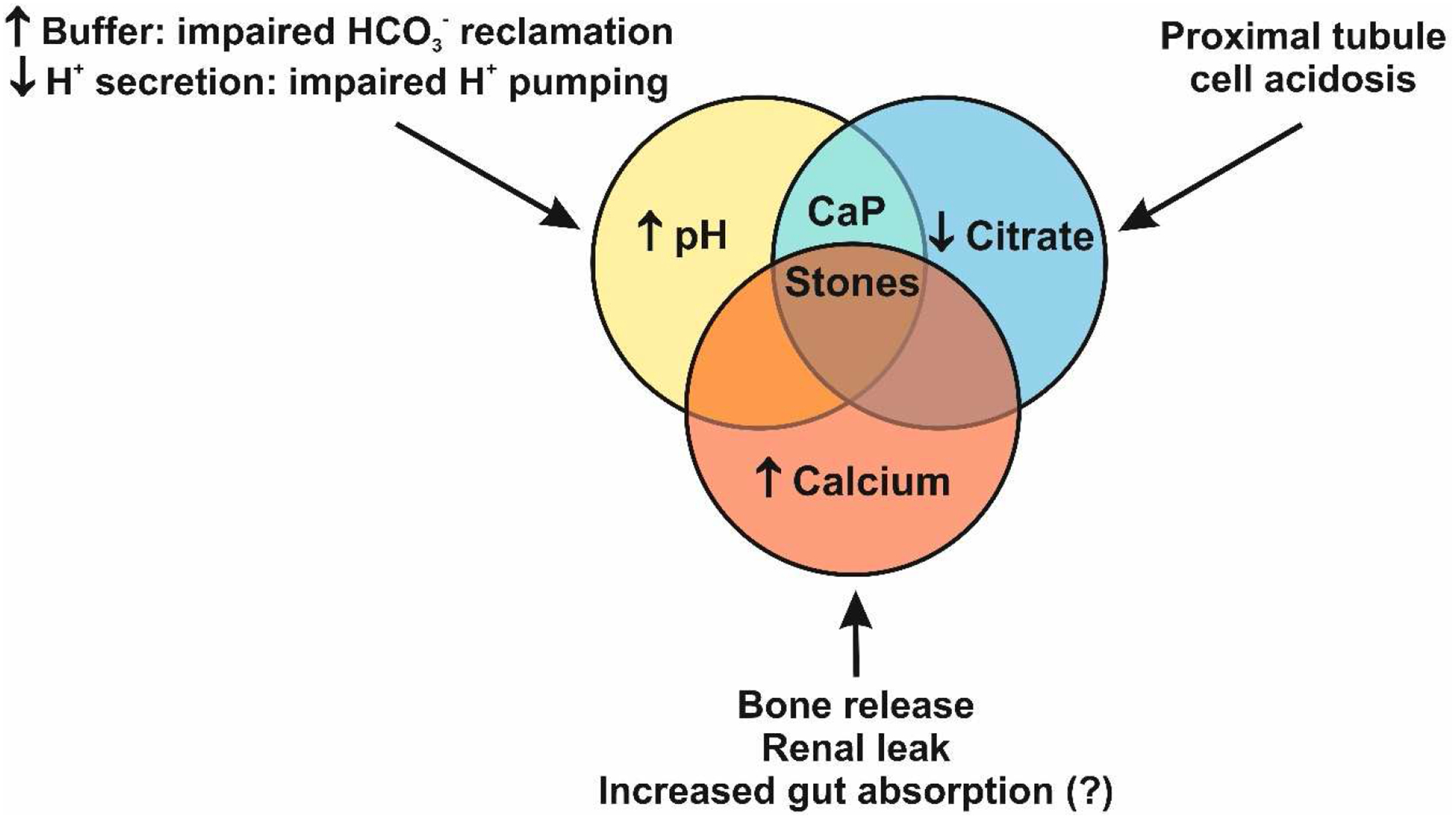
The three key prolithogenic urinary abnormalities in type I dRTA. Urinary abnormalities are similar in type I dRTA and idRTA but typically less severe in the latter. In contrast do dRTA, hypercalciuria is not a common finding in idRTA. The proposed underlying mechanisms are shown.
Unlike pRTA, patients with type I dRTA typically present with low bone mass primarily due to reduced bone formation and turnover rates, and to some extent defective mineralization and reduced non-collagenous proteins36–38. Overt osteomalacia is rare in dRTA36, but dRTA from Sjögren’s syndrome can be associated with proximal tubule phosphate wasting and secondary osteomalacia39.
Incomplete dRTA
The entity “incomplete dRTA“ (idRTA) was first described by Wrong and Davies in 195940. They reported three patients with bilateral nephrocalcinosis without systemic metabolic acidosis (defined by hypobicarbonatemia) that were unable to maximally acidify the urine by a one day ammonium chloride acid loading test (0.1g NH4Cl/Kg body weight). Trough urinary pH reached were 5.73, 5.91 and 6.5, respectively. Trough urinary pH of 10 healthy subjects tested in the same study ranged from 4.59 to 5.24. Only one of the three patients with idRTA described was hypercalciuric, no patient had kidney stones. This entity is not commonly diagnosed because the routine clinical test are all normal. Since this original publication, the one day ammonium chloride loading test was considered the gold standard for the diagnosis of idRTA. Typically, a pH < 5.3 has been accepted as threshold to rule out idRTA but there has never been a clear consensus on this threshold pH and various other definitions have been employed in the past24, 41–46. Using non-uniform definitions and provocative test procedures, a wide range of prevalence of idRTA from 2 to 21 % have been reported in recurrent SF24, 47, 48. Much higher prevalence rates of idRTA were reported in subgroup of patients with recurrent CaP stones, chronic pyelonephritis, nephrocalcinosis or medullary sponge kidney.
Unfortunately, gastrointestinal side effects occur frequently during ammonium chloride loading. Thus, in the decades following the first description, several alternative tests that impose an acute acid load with better tolerability or safety profile were introduced (furosemide, furosemide/fludrocortisone, reduced dose 3 day ammonium chloride test, arginine-HCl or calcium chloride loading test), but rigorous comparative studies of test procedures were lacking49, 50. A recent prospective study in an unselected cohort of 170 SF compared the performance of the frequently used furosemide/fludrocortisone test or of non-provocative parameters against the gold standard one day ammonium chloride loading test. The study showed that the two non-provocative parameters, second morning fasting urinary pH <5.3 and plasma K+ >3.8 mM/l, or the furosemide/fludrocortisone test had excellent negative predictive but low positive predictive values for the diagnosis of idRTA51. Thus, idRTA can be reliably excluded in SF by either the use of the two non-provocative parameters of second morning fasting urinary pH and plasma K+ or the furosemide/fludrocortisone test. If idRTA cannot be excluded by either approach, the diagnosis can be sought by the ammonium chloride loading test. The study also revealed that there was a negative association of nadir urinary pH in the ammonium chloride test with 24 h urinary citrate excretion, SF with idRTA had significantly lower urinary citrate than SF without idRTA. With respect to 24 h urinary calcium excretion, no differences were found between SF with or without idRTA.
IdRTA shares many features of overt type I dRTA, including urinary traits (hypercalciuria, hypocitraturia and alkaline pH) and the association with nephrolithiasis and nephrocalcinosis. The degree of biochemical urinary abnormalities is typically less severe than in type I dRTA and especially hypercalciuria is often not seen in idRTA. An important but often neglected difference between type I dRTA and idRTA is the fact that ammonium excretion is typically normal or occasionally increased in idRTA while it is significantly reduced in the case of type I dRTA44, 52. Thus, although no rigorous balance studies have been performed as in type I dRTA or pRTA, idRTA patients are classically considered to be in acid-base steady-state (i.e. stable serum bicarbonate concentration).
Low bone mass is a frequent finding in recurrent SF, vertebral fracture risk is increased fourfold compared to non-SF53, 54. idRTA may also be associated with low bone mass, but it is not as established as in type I dRTA. Osther et al. first reported increased markers of bone formation and bone resorption in SF with idRTA compared to SF without idRTA55. In patients with primary osteoporosis, Weger et al. discovered a high prevalence of idRTA56, 57 and children with idRTA exhibit significantly reduced height58. However, in a population-based study in an area of endemic tubular acidosis, idRTA was not associated with lower bone mass59. Furthermore, a recent study in 150 recurrent SF found no difference in bone mineral density between patients with and without idRTA60. Thus, additional longitudinal studies are needed to examine if a urinary acidification defect in the absence of frank systemic acidosis is associated with reduced bone mass and/or increased fracture risk.
IdRTA is typically considered an acquired condition (e.g. Sjögren’s syndrome, lithium therapy), but familial associations have been described (e.g. medullary sponge kidney). Rarely, idRTA may represent a “pre-acidotic” forme fruste version of type I dRTA, patients with transition of idRTA to overt type I dRTA have been described and causes of idRTA and type I dRTA overlap (e.g. nephrocalcinosis, Sjögren’s syndrome)40, 52, 61. Unfortunately, longitudinal studies in patients with idRTA are lacking and we do not know which patients with idRTA will eventually progress to type I dRTA. Certainly, given the large prevalence difference of overt type I dRTA and idRTA in recurrent SF, transition from idRTA to type I dRTA must be a rare event.
In most SF with idRTA, the cause of the urinary acidification deficit remains obscure. Since we do not usually obtain tissue, some form of unrecognized interstitial nephritis may be present in some patients. An important recent finding is that heterozygous carriers in a large family with an autosomal-recessive V-ATPase B1 subunit truncation mutation (p.F468fsX487) or SF heterozygous for the non-synonymous polymorphism p.E161K in the V-ATPase B1 subunit are not normal but exhibit a urinary acidification deficit, compatible with idRTA62, 63. These findings are compatible with haploinsufficiency of B1 in humans. If other known V-ATPase B1 or a4 subunit missense mutations also cause a detectable deficit in urinary acidification in a heterozygous state is currently unknown but is worthy of investigation. Thus, it is conceivable that allelic variants of genes involved in H+ secretion in α–intercalated cells or in ammonia synthesis in proximal tubular cells are associated with a urinary acidification deficit and thus the development of idRTA. It is noteworthy that the heterozygous carriers look indistinguishable from the regular idiopathic calcium SF and can be missed. The clinical features of the p.E161K heterozygotes include younger age of onset, more likely to have a positive family history, higher incidence of CaP stones, and an abnormally high trough urine pH upon NH4Cl challenge62, 63.
Treatment of nephrolithiasis in type I dRTA and incomplete dRTA
The management of dRTA needs to be directed at the treatment of the underlying cause if one can be identified and is amenable to therapy. Interstitial nephritis and dRTA of Sjögren’s syndrome typically respond to immunosuppressive therapy64. Alkali is the cornerstone of therapy if definitive elimination of the underlying causes is not possible. The situation is clear in children with congenital forms of type I dRTA with metabolic acidosis which leads to reduced GFR, treatment with alkali improves bone and somatic growth65, 66. Sodium bicarbonate supplementation normalizes height in children with idRTA and preserved GFR67.
In adults, treatment indication and modality are less well established. There are no true RCTs for stones prevention in adults with type I dRTA or idRTA48. Even RCTs for calcareous stones in the absence of dRTA have not addressed specifically the outcomes of patients with CaP stones. In small studies, treatment with alkali in adults with either dRTA or idRTA decreased skeletal Ca mobilization and hypercalciuria, increased bone density, increased citraturia and reduced stone formation26, 38, 68, 69. K-citrate is preferable over Na-citrate because K-citrate tends to reduce calciuria in addition to increasing citraturia and improves CaOx saturation. In contrast, Na-citrate at equimolar doses can increase calciuria and saturation for brushite and Na-urate while leaving that of CaOx unchanged68. In patients with dRTA associated with medullary sponge kidney which may represent a form of idRTA, alkali therapy also led to a decrease in stone passage and improvement of the associated bone disease70–72.
The amount of alkali needed to increase citraturia in SF with dRTA is hard to predict and even more challenging to adjust. Although compliance and gut absorption can be assured by 24 hr urine K excretion, it is not unusual that doses > 60 meq/d of potassium alkali are needed to significantly raise citraturia by only a few meq/d. The reason for the discrepancy between the kaliuresis and citraturia is not known. Apart from increasing citraturia, alkali therapy also raises urinary pH, potentially worsening brushite supersaturation and thus promote CaP stone formation. Unfortunately, due to the lack of RCT data we currently do not know at which point benefit ends and harm starts with alkali therapy. Computer-based programs (Equil-2, JESS) can aid the clinician in estimating urinary brushite saturation and guide therapy73–75. In contrast to saturation index (SI) calculated by JESS, relative supersaturation ratio (RSR) determined by Equil-2 seems to overestimate the pH effect on brushite saturation when compared to the empirically determined concentration-to-product ratio (CPR) (Figure 2)73. Clinically it is important to consider both urinary pH and citrate when treating a patient with alkali. It is possible to raise urinary citrate by small amounts (e.g. 2 mmoles/d) which may not be accompanied with alkalinuria but adequate to chelate urinary calcium. If the increase in urinary pH is not paralleled by a concomitant rise in urinary citrate, alkali therapy will be harmful. Alternatively, thiazide diuretics can be employed to reduce calciuria which will reduce urinary brushite saturation by decreasing urinary calcium and pH76.
Figure 2.
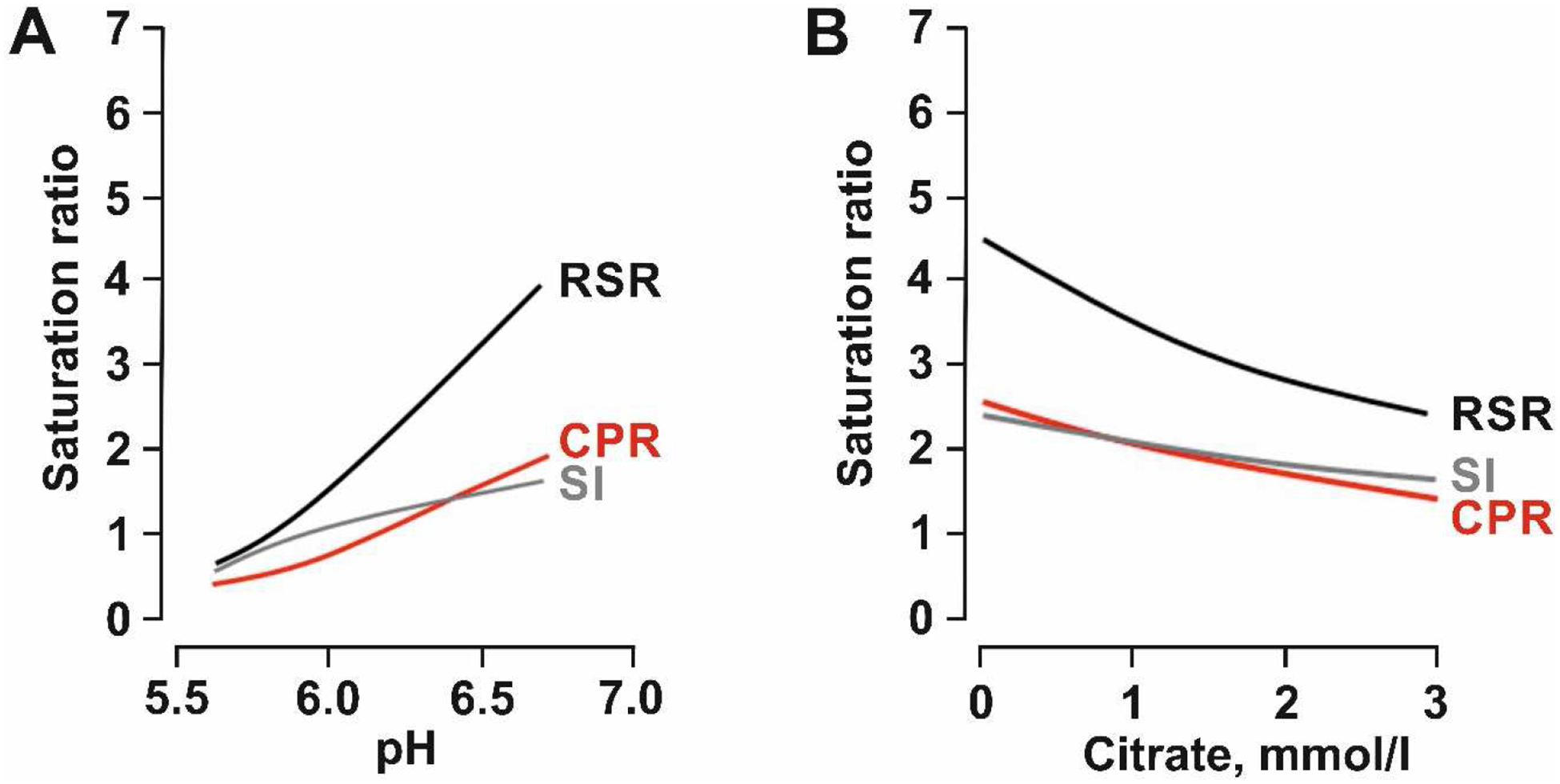
Brushite saturation in relation to urinary pH (A) und urinary citrate concentration (B). RSR (relative supersaturation ratio) and SI (saturation index) are calculated by the two programs EQUIL-2 (RSR) and JESS (SI). CPR (concentration-to-product ratio) is based on empirical physicochemical determination of brushite saturation in urine. Data are taken from Pak et al.73, 96.
Provocative thoughts on provocative diagnostic testing for incomplete dRTA
Since the initial description of the unmasking procedure by provocative testing, idRTA was always considered a separate entity and patients suffering from it a distinct group of SF. Traditionally, only selected SF (e.g. alkaline fasting urinary pH, CaP-containing stones or unexplained hypocitraturia) were subjected to provocative testing in clinical practice. Prospective studies in which large cohorts of unselected SF underwent provocative testing are lacking. Recent data challenges the dogma of idRTA as a separate entity51, 77. In an unselected cohort of 170 SF, urinary acidification capacity was found to be not a dichotomous but a continuous trait (Figure 3). Hypercalciuria, hypocitraturia and low bone mass are variable characteristics encountered in all calcareous SF and equally variable for idRTA.
Figure 3.
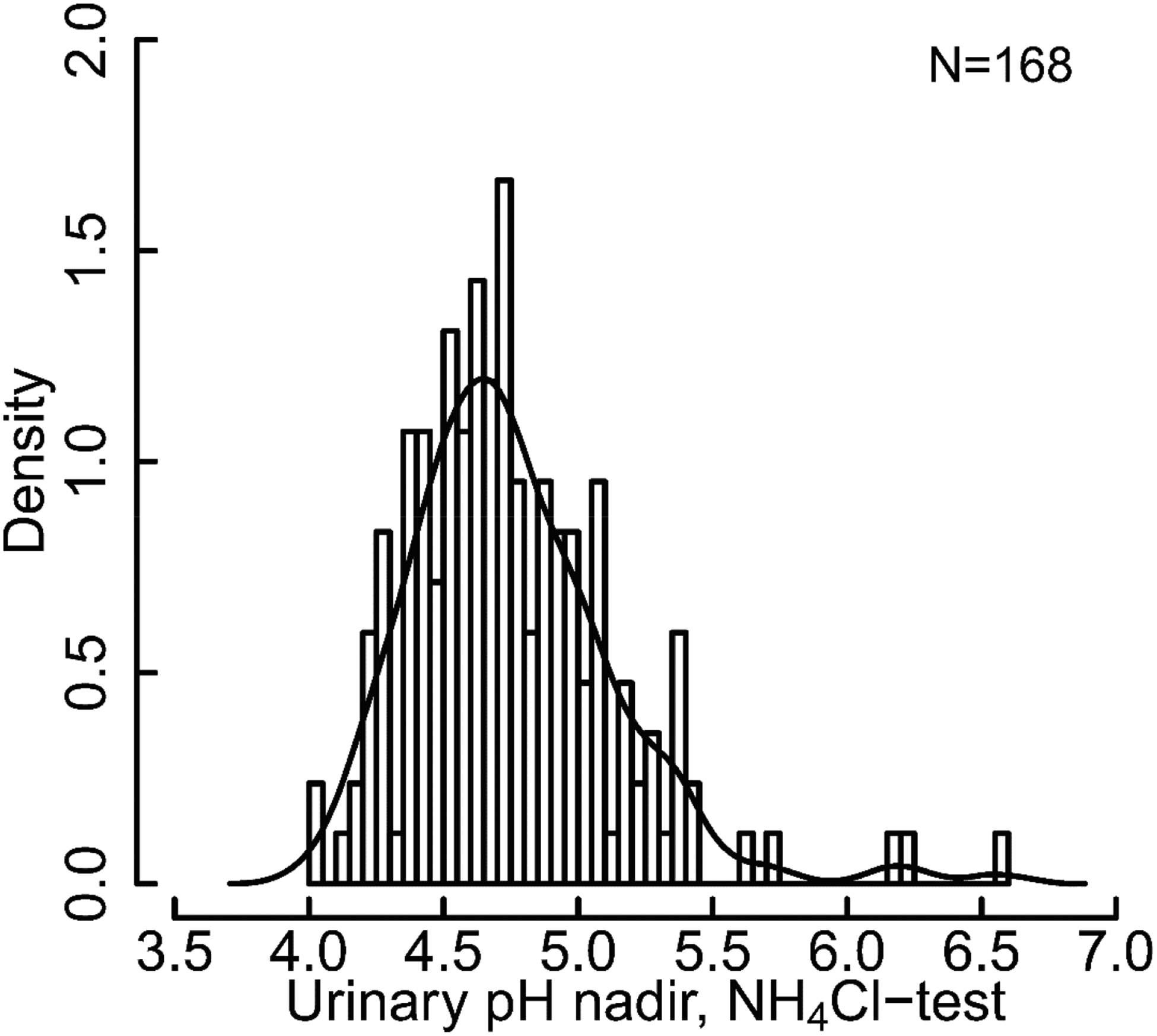
Histogram of urinary pH nadir achieved in ammonium chloride test in unselected cohort of 168 SF with Kernel density plot. Data are taken from Dhayat et al.51.
In addition to the classical read-out of urinary pH and acid excretion one can also prepare membranes from the urine that contains various fractions such as exosomes or ectosomes that can serves as a ‘window” permitting a glimpse at the apical membrane of the renal tubules in humans78. While normal subjects showed an increase in the B1 subunits in urine pellets in response to an acid load, it is was not observed in patients with dRTA79, 80.
As outlined above, there are no randomized clinical trials in SF with idRTA. In small studies, treatment with alkali in adult SF with idRTA decreased hypercalciuria, increased citraturia, reduced stone formation and improved associated bone disease26, 68, 70–72, 81. Results of these uncontrolled studies suggested that SF with idRTA indeed constitute a unique subset of patients that may benefit from alkali treatment. However, alkali treatment was also found to be effective in the recurrence prevention in unselected cohorts of calcareous SF82. Thus, longitudinal and interventional trials are needed to further explore the prognostic and therapeutic relevance of diagnosing idRTA in SF. Until the advent of such studies, provocative testing for idRTA diagnosis should, in our opinion, be prescribed only conservatively in clinical routine and be mainly viewed as a research tool.
Combined proximal-distal RTA (Type III)
Combined proximal-distal RTA (Type 3) is a rare entity which was originally described as a transient condition in children83. The genetic condition with osteopetrosis and RTA is due to congenital type II carbonic anhydrase deficiency84, 85. A more common form of combined pdRTA is from carbonic anhydrase inhibition. In a small study of 27 elderly patients receiving acetazolamide (250 to 1,000 mg/day) for glaucoma, 4 patients had mild acidosis (pH 7.29 – 7.31), 10 had moderate acidosis (pH 7.20 – 7.29), and 1 severe acidosis (pH 7.15)86. Another drug that has multiple properties with one being carbonic anhydrase inhibition is topiramate which is used to treat seizures, migraine headache, weight loss, and many other off-label use.
The chemical lithogenicity is identical to that of dRTA composed of alkalinuria, hypocitraturia, and modest hypercalciuria giving rise to CaP stones. When normal individuals were given topiramate, all of them developed alkalinuria, hypocitraturia and double the relative saturation ratio of brushite87. While biochemical stone risks are increased in every subject on topiramate, not all subjects gets kidney stones. The prevalence of kidney stones are dependent on dose and duration of therapy (Table 3). In some, though not all of the studies, imaging was used to detect asymptomatic stones. Once stones start forming, it is very unlikely that they will subside. The best countermeasure is a discussion with the neurologist to explore alternative therapy if possible. If cessation is not possible and topiramte has to be continued, alkali in the form of potassium citrate can be administered88.
Table 3.
Kidney stone incidence in patients treated with topiramate
| n | Mean Age | Dose (mg/day) | Rx Duration (Months) | Stone Incidence | References89–95 |
|---|---|---|---|---|---|
| ? | ? | 250–450 mg | ? | 1.5 % | Product monograph |
| 170 | 47 | 250–450 mg | 6 | 1.8 % | Stephen et al. Epilepsia 2000 |
| 45 | 16 | 4 mg/Kg | 15.8 | 4.5 % | Coppola et al. Epilepsy Res 2002a |
| 18 | 9 | 5 mg/Kg | 11.9 | 5.6 % | Coppola et al. Epilepsy Res 2002b |
| 197 | 58 | 400–800 mg | 12 | 9.1 % | Cudkowicz et al. Neurology 2003 |
| 24 | 21 | 7.9 mg/kg | 34 | 54 % | Goyal et al. Ped Neurology 2009 |
| 96 | 7 | 5 mg/kg | 12 | 5.2 % | Mahmoud et al. Epilepsia 2011 |
| 54 | 12 | 4–5 mg/kg | 24 | 12 % | Corbin Bush et al. Ped Urol 2012 |
Conclusions
RTA’s encompass a broad group of congenital or acquired disorders, characterized by defective renal acid excretion, which can lead to recurrent kidney stones and nephrocalcinosis. Hyperkalemic RTA can be occasionally associated with uric acid nephrolithiasis, in the setting of type II diabetes or the metabolic syndrome but it is not a main cause of uric acid stones. Proximal type II RTA is very rarely associated with urolithiasis or nephrocalcinosis. In contrast, CaP stones and nephrocalcinosis are frequently encountered in patients with hypokalemic type I dRTA or idRTA. While the former can be diagnosed by conventional clinical criteria, diagnosis of idRTA is classically only achieved with provocative acid loading tests. SF that cannot acidify urine pH <5.3 during provocative testing are considered to have idRTA. In addition to alkaline urinary pH, hypercalciuria and hypocitraturia are the two other prolithogenic urinary abnormalities encountered in SF with idRTA which are also encountered in SF without idRTA.
Urinary acidification capacity is a continuous trait, questioning the view of idRTA as a separate entity. Alkaline urinary pH > 6 is an important lithogenic factor for CaP stone formation and can easily be detected by spot fasting or 24 h urine pH measurements. The value of idRTA diagnosis by provocative testing and genotyping the candidate genes, are important research tools but remains unclear at the moment whether they alter clinical practice, and need further clarification. Similarly, no randomized clinical trials have been performed in SF with dRTA or CaP stones. Until the availability of such data, one should focus treatment of SF with CaP stones on the biochemical abnormalities encountered in the metabolic work-up. SF with type I dRTA should receive alkali therapy, preferentially in the form of K-citrate delivered cautiously, to treat the chronic acid retention which drives both stone formation and bone disease.
Clinical Summary:
Distal renal tubular acidosis, which can be caused by a variety of congenital or acquired conditions, predisposes to calcium phosphate stones via pathophysiologic intermediates of alkalinuria, hypocitraturia, and to a lesser extent hypercalciuria.
Incomplete distal renal tubular acidosis usually escapes routine clinical detection and its diagnosis requires the demonstration of inadequate urinary acidification from provocative acid loading testing but such traits are not “normal vs. abnormal” but in fact is a continuum, and at the present momentum it is unclear whether these tests should extend from the human research lab into clinical practice.
The mainstream therapy of complete and incomplete dRTA is still alkali supplement such as potassium citrate, to render the urine less lithogenic by raising citrate and to prevent bone loss, but the citraturic effect can be offset by worsening alkalinuria so cautious dose titration is of critical importance.
ACKNOWLEDGEMENTS
DF was supported by the Swiss National Centre of Competence in Research NCCR TransCure and the Swiss National Science Foundation (grants # 31003A_135503, 31003A_152829 and 33IC30_166785/1). OWM was supported by the National Institutes of Health (P30 DK-079328, R01 DK081423, and T32DK007257), the American Heart Foundation, and the Charles Pak Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES
DF has served as a consultant for Otsuka Pharmaceuticals. DF has received unrestricted research funding from Novartis, Abbvie and Otsuka Pharmaceuticals. OM served on the Advisory Boards for AbbVie, Allena, Ardelyx, Genzyme-Sanofi, and Triceda.
REFERENCES
- 1.Batlle DC, Hizon M, Cohen E, Gutterman C, Gupta R. The use of the urinary anion gap in the diagnosis of hyperchloremic metabolic acidosis. N Engl J Med. 1988;318:594–599. [DOI] [PubMed] [Google Scholar]
- 2.Igarashi T, Inatomi J, Sekine T, et al. Mutations in SLC4A4 cause permanent isolated proximal renal tubular acidosis with ocular abnormalities. Nat Genet. 1999;23:264–266. [DOI] [PubMed] [Google Scholar]
- 3.Lemann J Jr., Adams ND, Wilz DR, Brenes LG. Acid and mineral balances and bone in familial proximal renal tubular acidosis. Kidney Int. 2000;58:1267–1277. [DOI] [PubMed] [Google Scholar]
- 4.Goodman AD, Lemann J Jr., Lennon EJ, Relman AS. Production, Excretion, and Net Balance of Fixed Acid in Patients with Renal Acidosis. J Clin Invest. 1965;44:495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Batlle DC, Arruda JA, Kurtzman NA. Hyperkalemic distal renal tubular acidosis associated with obstructive uropathy. N Engl J Med. 1981;304:373–380. [DOI] [PubMed] [Google Scholar]
- 6.Arruda JA, Batlle DC, Sehy JT, Roseman MK, Baronowski RL, Kurtzman NA. Hyperkalemia and renal insufficiency: role of selective aldosterone deficiency and tubular unresponsiveness to aldosterone. Am J Nephrol. 1981;1:160–167. [DOI] [PubMed] [Google Scholar]
- 7.Uribarri J, Oh MS, Pak CY. Renal stone risk factors in patients with type IV renal tubular acidosis. Am J Kidney Dis. 1994;23:784–787. [DOI] [PubMed] [Google Scholar]
- 8.Batlle DC. Hyperkalemic hyperchloremic metabolic acidosis associated with selective aldosterone deficiency and distal renal tubular acidosis. Seminars in Nephrology. Vol 1: Elsevier; 1981:260–274. [Google Scholar]
- 9.Cameron MA, Maalouf NM, Adams-Huet B, Moe OW, Sakhaee K. Urine composition in type 2 diabetes: predisposition to uric acid nephrolithiasis. J Am Soc Nephrol. 2006;17:1422–1428. [DOI] [PubMed] [Google Scholar]
- 10.Maalouf NM, Cameron MA, Moe OW, Adams-Huet B, Sakhaee K. Low urine pH: a novel feature of the metabolic syndrome. Clin J Am Soc Nephrol. 2007;2:883–888. [DOI] [PubMed] [Google Scholar]
- 11.Maalouf NM, Cameron MA, Moe OW, Sakhaee K. Metabolic basis for low urine pH in type 2 diabetes. Clin J Am Soc Nephrol. 2010;5:1277–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sakhaee K, Adams-Huet B, Moe OW, Pak CY. Pathophysiologic basis for normouricosuric uric acid nephrolithiasis. Kidney Int. 2002;62:971–979. [DOI] [PubMed] [Google Scholar]
- 13.Wiederkehr MR, Moe OW. Uric Acid Nephrolithiasis: A Systemic Metabolic Disorder. Clin Rev Bone Miner Metab. 2011;9:207–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maalouf NM, Cameron MA, Moe OW, Sakhaee K. Novel insights into the pathogenesis of uric acid nephrolithiasis. Curr Opin Nephrol Hypertens. 2004;13:181–189. [DOI] [PubMed] [Google Scholar]
- 15.Lightwood R Communication no. 1. Arch Dis Child. 1935;10:205. [Google Scholar]
- 16.Albright F, Burnett CH, Parson W, Reifenstein EC, Roos A. THE VARIOUS ETIOLOGIES MET IN THE UNITED STATES WITH EMPHASIS ON THAT RESULTING FROM A SPECIFIC FORM OF RENAL ACIDOSIS, THE THERAPEUTIC INDICATIONS FOR EACH ETIOLOGICAL SUB-GROUP, AND THE RELATIONSHIP BETWEEN OSTEOMALACIA AND MILKMAN’S SYNDROME. Medicine. 1946;25:399–479 %R. [DOI] [PubMed] [Google Scholar]
- 17.Pines KL, Mudge GH. Renal tubular acidosis with osteomalacia: Report of three cases. The American journal of medicine. 1951;11:302–311. [DOI] [PubMed] [Google Scholar]
- 18.Cohen EP, Bastani B, Cohen MR, Kolner S, Hemken P, Gluck SL. Absence of H(+)-ATPase in cortical collecting tubules of a patient with Sjogren’s syndrome and distal renal tubular acidosis. J Am Soc Nephrol. 1992;3:264–271. [DOI] [PubMed] [Google Scholar]
- 19.Karet FE, Finberg KE, Nelson RD, et al. Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet. 1999;21:84–90. [DOI] [PubMed] [Google Scholar]
- 20.Smith AN, Skaug J, Choate KA, et al. Mutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116-kD subunit, cause recessive distal renal tubular acidosis with preserved hearing. Nat Genet. 2000;26:71–75. [DOI] [PubMed] [Google Scholar]
- 21.Enerback S, Nilsson D, Edwards N, et al. Acidosis and Deafness in Patients with Recessive Mutations in FOXI1. J Am Soc Nephrol. 2018;29:1041–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karet FE, Gainza FJ, Gyory AZ, et al. Mutations in the chloride-bicarbonate exchanger gene AE1 cause autosomal dominant but not autosomal recessive distal renal tubular acidosis. Proc Natl Acad Sci U S A. 1998;95:6337–6342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pak CY, Britton F, Peterson R, et al. Ambulatory evaluation of nephrolithiasis. Classification, clinical presentation and diagnostic criteria. Am J Med. 1980;69:19–30. [DOI] [PubMed] [Google Scholar]
- 24.Ito H, Kotake T, Suzuki F. Incidence and clinical features of renal tubular acidosis-1 in urolithiasis. Urol Int. 1993;50:82–85. [DOI] [PubMed] [Google Scholar]
- 25.Donnelly S, Kamel KS, Vasuvattakul S, Narins RG, Halperin ML. Might distal renal tubular acidosis be a proximal tubular cell disorder? Am J Kidney Dis. 1992;19:272–281. [DOI] [PubMed] [Google Scholar]
- 26.Preminger GM, Sakhaee K, Pak CY. Hypercalciuria and altered intestinal calcium absorption occurring independently of vitamin D in incomplete distal renal tubular acidosis. Metabolism. 1987;36:176–179. [DOI] [PubMed] [Google Scholar]
- 27.Bushinsky DA, Frick KK. The effects of acid on bone. Curr Opin Nephrol Hypertens. 2000;9:369–379. [DOI] [PubMed] [Google Scholar]
- 28.Alexander RT, Cordat E, Chambrey R, Dimke H, Eladari D. Acidosis and Urinary Calcium Excretion: Insights from Genetic Disorders. J Am Soc Nephrol. 2016;27:3511–3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Daudon M, Bouzidi H, Bazin D. Composition and morphology of phosphate stones and their relation with etiology. Urol Res. 2010;38:459–467. [DOI] [PubMed] [Google Scholar]
- 30.Dessombz A, Letavernier E, Haymann JP, Bazin D, Daudon M. Calcium Phosphate Stone Morphology Can Reliably Predict Distal Renal Tubular Acidosis. J Urol. 2014. [DOI] [PubMed] [Google Scholar]
- 31.Parks JH, Coward M, Coe FL. Correspondence between stone composition and urine supersaturation in nephrolithiasis. Kidney Int. 1997;51:894–900. [DOI] [PubMed] [Google Scholar]
- 32.Pak CY, Poindexter JR, Adams-Huet B, Pearle MS. Predictive value of kidney stone composition in the detection of metabolic abnormalities. Am J Med. 2003;115:26–32. [DOI] [PubMed] [Google Scholar]
- 33.Pak CY, Poindexter JR, Peterson RD, Heller HJ. Biochemical and physicochemical presentations of patients with brushite stones. J Urol. 2004;171:1046–1049. [DOI] [PubMed] [Google Scholar]
- 34.Drosos AA, Pennec YL, Elisaf M, et al. Sjogren’s syndrome in patients with the CREST variant of progressive systemic scleroderma. J Rheumatol. 1991;18:1685–1688. [PubMed] [Google Scholar]
- 35.Gambaro G, Abaterusso C, Fabris A, et al. The origin of nephrocalcinosis, Randall’s plaque and renal stones: a cell biology viewpoint. Arch Ital Urol Androl. 2009;81:166–170. [PubMed] [Google Scholar]
- 36.Domrongkitchaiporn S, Pongsakul C, Stitchantrakul W, et al. Bone mineral density and histology in distal renal tubular acidosis. Kidney Int. 2001;59:1086–1093. [DOI] [PubMed] [Google Scholar]
- 37.Disthabanchong S, Domrongkitchaiporn S, Sirikulchayanonta V, Stitchantrakul W, Karnsombut P, Rajatanavin R. Alteration of noncollagenous bone matrix proteins in distal renal tubular acidosis. Bone. 2004;35:604–613. [DOI] [PubMed] [Google Scholar]
- 38.Domrongkitchaiporn S, Pongskul C, Sirikulchayanonta V, et al. Bone histology and bone mineral density after correction of acidosis in distal renal tubular acidosis. Kidney Int. 2002;62:2160–2166. [DOI] [PubMed] [Google Scholar]
- 39.Fulop M, Mackay M. Renal tubular acidosis, Sjogren syndrome, and bone disease. Arch Intern Med. 2004;164:905–909. [DOI] [PubMed] [Google Scholar]
- 40.Wrong O, Davies HE. The excretion of acid in renal disease. Q J Med. 1959;28:259–313. [PubMed] [Google Scholar]
- 41.Joshi A, Gupta SK, Srivastava A. Metabolic evaluation in first-time renal stone formers in North India: a single center study. Saudi J Kidney Dis Transpl. 2013;24:838–843. [DOI] [PubMed] [Google Scholar]
- 42.Osther PJ, Hansen AB, Rohl HF. Screening renal stone formers for distal renal tubular acidosis. Br J Urol. 1989;63:581–583. [DOI] [PubMed] [Google Scholar]
- 43.Gault MH, Chafe LL, Morgan JM, et al. Comparison of patients with idiopathic calcium phosphate and calcium oxalate stones. Medicine (Baltimore). 1991;70:345–359. [DOI] [PubMed] [Google Scholar]
- 44.Tannen RL, Falls WF Jr., Brackett NC Jr., Incomplete renal tubular acidosis: some clinical and physiological features. Nephron. 1975;15:111–123. [DOI] [PubMed] [Google Scholar]
- 45.Backman U, Danielson BG, Johansson G, Ljunghall S, Wikstrom B. Incidence and clinical importance of renal tubular defects in recurrent renal stone formers. Nephron. 1980;25:96–101. [DOI] [PubMed] [Google Scholar]
- 46.Stitchantrakul W, Kochakarn W, Ruangraksa C, Domrongkitchaiporn S. Urinary risk factors for recurrent calcium stone formation in Thai stone formers. J Med Assoc Thai. 2007;90:688–698. [PubMed] [Google Scholar]
- 47.Wikstrom B, Backman U, Danielson BG, Fellstrom B, Johansson G, Ljunghall S. Ambulatory diagnostic evaluation of 389 recurrent renal stone formers. A proposal for clinical classification and investigation. Klin Wochenschr. 1983;61:85–90. [DOI] [PubMed] [Google Scholar]
- 48.Gambaro G, Croppi E, Coe F, et al. Metabolic diagnosis and medical prevention of calcium nephrolithiasis and its systemic manifestations: a consensus statement. J Nephrol. 2016;29:715–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Walsh SB, Shirley DG, Wrong OM, Unwin RJ. Urinary acidification assessed by simultaneous furosemide and fludrocortisone treatment: an alternative to ammonium chloride. Kidney Int. 2007;71:1310–1316. [DOI] [PubMed] [Google Scholar]
- 50.Batlle DC. Segmental characterization of defects in collecting tubule acidification. Kidney Int. 1986;30:546–554. [DOI] [PubMed] [Google Scholar]
- 51.Dhayat NA, Gradwell MW, Pathare G, et al. Furosemide/Fludrocortisone Test and Clinical Parameters to Diagnose Incomplete Distal Renal Tubular Acidosis in Kidney Stone Formers. Clin J Am Soc Nephrol. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Buckalew VM Jr., McCurdy DK, Ludwig GD, Chaykin LB, Elkinton JR Incomplete renal tubular acidosis. Physiologic studies in three patients with a defect in lowering urine pH. Am J Med. 1968;45:32–42. [DOI] [PubMed] [Google Scholar]
- 53.Sakhaee K, Maalouf NM, Kumar R, Pasch A, Moe OW. Nephrolithiasis-associated bone disease: pathogenesis and treatment options. Kidney Int. 2011;79:393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Melton LJ 3rd, Crowson CS, Khosla S, Wilson DM, O’Fallon WM. Fracture risk among patients with urolithiasis: a population-based cohort study. Kidney Int. 1998;53:459–464. [DOI] [PubMed] [Google Scholar]
- 55.Osther PJ, Bollerslev J, Hansen AB, Engel K, Kildeberg P. Pathophysiology of incomplete renal tubular acidosis in recurrent renal stone formers: evidence of disturbed calcium, bone and citrate metabolism. Urol Res. 1993;21:169–173. [DOI] [PubMed] [Google Scholar]
- 56.Weger M, Deutschmann H, Weger W, Kotanko P, Skrabal F. Incomplete renal tubular acidosis in ‘primary’ osteoporosis. Osteoporos Int. 1999;10:325–329. [DOI] [PubMed] [Google Scholar]
- 57.Weger W, Kotanko P, Weger M, Deutschmann H, Skrabal F. Prevalence and characterization of renal tubular acidosis in patients with osteopenia and osteoporosis and in non-porotic controls. Nephrol Dial Transplant. 2000;15:975–980. [DOI] [PubMed] [Google Scholar]
- 58.Sharma AP, Sharma RK, Kapoor R, Kornecki A, Sural S, Filler G. Incomplete distal renal tubular acidosis affects growth in children. Nephrol Dial Transplant. 2007;22:2879–2885. [DOI] [PubMed] [Google Scholar]
- 59.Pongchaiyakul C, Domrongkitchaiporn S, Stitchantrakul W, Chailurkit LO, Rajatanavin R. Incomplete renal tubular acidosis and bone mineral density: a population survey in an area of endemic renal tubular acidosis. Nephrol Dial Transplant. 2004;19:3029–3033. [DOI] [PubMed] [Google Scholar]
- 60.Arampatzis S, Ropke-Rieben B, Lippuner K, Hess B. Prevalence and densitometric characteristics of incomplete distal renal tubular acidosis in men with recurrent calcium nephrolithiasis. Urol Res. 2012;40:53–59. [DOI] [PubMed] [Google Scholar]
- 61.Norman ME, Feldman NI, Cohn RM, Roth KS, McCurdy DK. Urinary citrate excretion in the diagnosis of distal renal tubular acidosis. J Pediatr. 1978;92:394–400. [DOI] [PubMed] [Google Scholar]
- 62.Zhang J, Fuster DG, Cameron MA, et al. Incomplete distal renal tubular acidosis from a heterozygous mutation of the V-ATPase B1 subunit. Am J Physiol Renal Physiol. 2014;307:F1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dhayat NA, Schaller A, Albano G, et al. The Vacuolar H+-ATPase B1 Subunit Polymorphism p.E161K Associates with Impaired Urinary Acidification in Recurrent Stone Formers. J Am Soc Nephrol. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ren H, Wang WM, Chen XN, et al. Renal involvement and followup of 130 patients with primary Sjogren’s syndrome. J Rheumatol. 2008;35:278–284. [PubMed] [Google Scholar]
- 65.Nash MA, Torrado AD, Greifer I, Spitzer A, Edelmann CM Jr., Renal tubular acidosis in infants and children. Clinical course, response to treatment, and prognosis. J Pediatr. 1972;80:738–748. [DOI] [PubMed] [Google Scholar]
- 66.McSherry E, Morris RC Jr., Attainment and maintenance of normal stature with alkali therapy in infants and children with classic renal tubular acidosis. J Clin Invest. 1978;61:509–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sharma AP, Singh RN, Yang C, Sharma RK, Kapoor R, Filler G. Bicarbonate therapy improves growth in children with incomplete distal renal tubular acidosis. Pediatr Nephrol. 2009;24:1509–1516. [DOI] [PubMed] [Google Scholar]
- 68.Preminger GM, Sakhaee K, Pak CY. Alkali action on the urinary crystallization of calcium salts: contrasting responses to sodium citrate and potassium citrate. J Urol. 1988;139:240–242. [DOI] [PubMed] [Google Scholar]
- 69.Greenberg AJ, McNamara H, McCrory WW. Metabolic balance studies in primary renal tubular acidosis: effects of acidosis on external calcium and phosphorus balances. J Pediatr. 1966;69:610–618. [DOI] [PubMed] [Google Scholar]
- 70.Fabris A, Bernich P, Abaterusso C, et al. Bone disease in medullary sponge kidney and effect of potassium citrate treatment. Clin J Am Soc Nephrol. 2009;4:1974–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fabris A, Lupo A, Bernich P, et al. Long-term treatment with potassium citrate and renal stones in medullary sponge kidney. Clin J Am Soc Nephrol. 2010;5:1663–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Higashihara E, Nutahara K, Niijima T. Renal hypercalciuria and metabolic acidosis associated with medullary sponge kidney: effect of alkali therapy. Urol Res. 1988;16:95–100. [DOI] [PubMed] [Google Scholar]
- 73.Pak CY, Moe OW, Maalouf NM, Zerwekh JE, Poindexter JR, Adams-Huet B. Comparison of semi-empirical and computer derived methods for estimating urinary saturation of brushite. J Urol. 2009;181:1423–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rodgers A, Allie-Hamdulay S, Jackson G. Therapeutic action of citrate in urolithiasis explained by chemical speciation: increase in pH is the determinant factor. Nephrol Dial Transplant. 2006;21:361–369. [DOI] [PubMed] [Google Scholar]
- 75.Werness PG, Brown CM, Smith LH, Finlayson B. EQUIL2: a BASIC computer program for the calculation of urinary saturation. J Urol. 1985;134:1242–1244. [DOI] [PubMed] [Google Scholar]
- 76.Bergsland KJ, Worcester EM, Coe FL. Role of proximal tubule in the hypocalciuric response to thiazide of patients with idiopathic hypercalciuria. Am J Physiol Renal Physiol. 2013;305:F592–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Goldfarb DS. Refining Diagnostic Approaches in Nephrolithiasis: Incomplete Distal Renal Tubular Acidosis. Clin J Am Soc Nephrol. 2017;12:1380–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.van Balkom BW, Pisitkun T, Verhaar MC, Knepper MA. Exosomes and the kidney: prospects for diagnosis and therapy of renal diseases. Kidney Int. 2011;80:1138–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pathare G, Dhayat NA, Mohebbi N, et al. Changes in V-ATPase subunits of human urinary exosomes reflect the renal response to acute acid/alkali loading and the defects in distal renal tubular acidosis. Kidney Int. 2018;93:871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pathare G, Dhayat N, Mohebbi N, et al. Acute regulated expression of pendrin in human urinary exosomes. Pflugers Arch. 2018;470:427–438. [DOI] [PubMed] [Google Scholar]
- 81.Preminger GM, Sakhaee K, Skurla C, Pak CY. Prevention of recurrent calcium stone formation with potassium citrate therapy in patients with distal renal tubular acidosis. J Urol. 1985;134:20–23. [DOI] [PubMed] [Google Scholar]
- 82.Fink HA, Wilt TJ, Eidman KE, et al. Medical management to prevent recurrent nephrolithiasis in adults: a systematic review for an American College of Physicians Clinical Guideline. Ann Intern Med. 2013;158:535–543. [DOI] [PubMed] [Google Scholar]
- 83.Donckerwolcke RA, Valk C, van Wijngaarden-Penterman MJ, van Stekelenburg GJ. A case of transient renal tubular acidosis type 1,4 hybrid RTA: a study of the pathophysiologic characteristics of the acidification defect. Pediatr Res. 1979;13:1177–1178. [DOI] [PubMed] [Google Scholar]
- 84.Sly WS, Hewett-Emmett D, Whyte MP, Yu YS, Tashian RE. Carbonic anhydrase II deficiency identified as the primary defect in the autosomal recessive syndrome of osteopetrosis with renal tubular acidosis and cerebral calcification. Proc Natl Acad Sci U S A. 1983;80:2752–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sly WS, Whyte MP, Sundaram V, et al. Carbonic anhydrase II deficiency in 12 families with the autosomal recessive syndrome of osteopetrosis with renal tubular acidosis and cerebral calcification. N Engl J Med. 1985;313:139–145. [DOI] [PubMed] [Google Scholar]
- 86.Heller I, Halevy J, Cohen S, Theodor E. Significant metabolic acidosis induced by acetazolamide. Not a rare complication. Arch Intern Med. 1985;145:1815–1817. [PubMed] [Google Scholar]
- 87.Welch BJ, Graybeal D, Moe OW, Maalouf NM, Sakhaee K. Biochemical and stone-risk profiles with topiramate treatment. Am J Kidney Dis. 2006;48:555–563. [DOI] [PubMed] [Google Scholar]
- 88.Jhagroo RA, Wertheim ML, Penniston KL. Alkali replacement raises urinary citrate excretion in patients with topiramate-induced hypocitraturia. Br J Clin Pharmacol. 2016;81:131–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Stephen LJ, Sills GJ, Brodie MJ. Topiramate in refractory epilepsy: a prospective observational study. Epilepsia. 2000;41:977–980. [DOI] [PubMed] [Google Scholar]
- 90.Coppola G, Caliendo G, Veggiotti P, et al. Topiramate as add-on drug in children, adolescents and young adults with Lennox-Gastaut syndrome: an Italian multicentric study. Epilepsy Res. 2002;51:147–153. [DOI] [PubMed] [Google Scholar]
- 91.Coppola G, Capovilla G, Montagnini A, et al. Topiramate as add-on drug in severe myoclonic epilepsy in infancy: an Italian multicenter open trial. Epilepsy Res. 2002;49:45–48. [DOI] [PubMed] [Google Scholar]
- 92.Cudkowicz ME, Shefner JM, Schoenfeld DA, et al. A randomized, placebo-controlled trial of topiramate in amyotrophic lateral sclerosis. Neurology. 2003;61:456–464. [DOI] [PubMed] [Google Scholar]
- 93.Goyal M, Grossberg RI, O’Riordan MA, Davis ID. Urolithiasis with topiramate in nonambulatory children and young adults. Pediatr Neurol. 2009;40:289–294. [DOI] [PubMed] [Google Scholar]
- 94.Mahmoud AA, Rizk T, El-Bakri NK, Riaz M, Dannawi S, Al Tannir M. Incidence of kidney stones with topiramate treatment in pediatric patients. Epilepsia. 2011;52:1890–1893. [DOI] [PubMed] [Google Scholar]
- 95.Corbin Bush N, Twombley K, Ahn J, et al. Prevalence and spot urine risk factors for renal stones in children taking topiramate. J Pediatr Urol. 2013;9:884–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pak CY, Rodgers K, Poindexter JR, Sakhaee K. New methods of assessing crystal growth and saturation of brushite in whole urine: effect of pH, calcium and citrate. J Urol. 2008;180:1532–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]