

Abstract
Aim
The urinary glucose tetrasaccharide, Glcα1‐6Glcα1‐4Glcα1‐4Glc (Glc4), is a glycogen limit dextrin that is elevated in patients with glycogen storage disease (GSD) type III. We evaluated the potential of uncooked cornstarch therapy to interfere with Glc4 monitoring, by measuring the diurnal variability of Glc4 excretion in patients with GSD III.
Methods
Voids were collected at home over 24 hours, stored at 4°C and frozen within 48 hours. Glc4 was analyzed using liquid chromatography‐tandem mass spectrometry and normalized to creatinine.
Results
Subjects with GSD III (median age: 13.5 years, range: 3.7‐62; n = 18) completed one or more 24‐hour urine collection, and 28/36 collections were accepted for analysis. Glc4 was elevated in 16/18 subjects (median: 13 mmol/mol creatinine, range: 2‐75, reference range: <3). In collections with elevated Glc4 (23/28), two‐thirds (15/23) had low diurnal variability in Glc4 excretion (coefficient of variation [CV%] <25). The diurnal variability was significantly correlated with the Glc4 concentration (Pearson R = .644, P < .05), but not with the dose of uncooked cornstarch. High intraday variability (>25%) was not consistently observed in repeat collections by the same subject.
Conclusions
The extent and variability of Glc4 excretion relative to creatinine was not correlated with cornstarch dose. A majority of collections showed low variability over 24 hours. These findings support the use of single time‐point collections to evaluate Glc4 in patients with GSD III treated with cornstarch. However, repeat sampling over short time‐periods will provide the most accurate assessment of Glc4 excretion, as intraday variability may be increased in patients with high Glc4 excretion.
Keywords: 24‐hour urine, biomarker, Glc4, glucose tetrasaccharide, glycogen storage disease type III, Hex4, uncooked cornstarch
Abbreviations
- ALT
alanine transaminase
- AST
aspartate transaminase
- CK
creatine kinase
- CN
creatinine
- GDE
glycogen debranching enzyme
- Glc4
glucose tetrasaccharide, Glcα1‐6Glcα1‐4Glcα1‐4Glc
- GSD
glycogen storage disease
- HCC
hepatocellular carcinoma
- IS
internal standard
Synopsis.
The diurnal variability of Glc4 excretion is low for a majority of patients with glycogen storage disease III and is not correlated with cornstarch dose.
1. INTRODUCTION
Glycogen storage disease type III (GSDIII, MIM #232400) is an autosomal recessive disorder caused by a deficiency of glycogen debranching enzyme (GDE, EC 3.2.1.33, EC 2.4.1.25) encoded by AGL (MIM #610860). GDE is a cytosolic enzyme that works in combination with glycogen phosphorylase to release glucose from glycogen for energy metabolism. GDE deficiency disrupts glucose homeostasis, and results in an accumulation of abnormally structured glycogen enriched in α‐1‐6 branch points. 1 The clinical manifestations are variable and patients are classified as GSD IIIa, characterized by liver, heart, and muscle involvement, or GSD IIIb in which the liver is predominantly affected. 2 GSD III often presents in infancy or childhood with hepatomegaly and hypoglycemia due to liver disease. Ketosis, hyperlipidemia, and growth retardation are common. 3 Skeletal muscle weakness slowly progresses in GSD IIIa, becoming more prominent in the third to fourth decades of life. Liver disease becomes less apparent in adolescence and adulthood, and is associated with a decrease in liver size and serum aminotransferase levels. 4 However, long‐term hepatic complications have been reported including liver fibrosis, cirrhosis, adenomas, and hepatocellular carcinoma. 5 , 6
In addition to the general management of disease manifestations, treatment includes dietary therapy using uncooked cornstarch to minimize hypoglycemic events, and a high protein diet as an alternative energy source. 7 , 8 As new therapies are being investigated, 8 , 9 , 10 biomarkers are needed to determine the clinical severity and monitor disease progression. The glucose tetrasaccharide, Glcα1‐6Glcα1‐4Glcα1‐4Glc (Glc4) is a glycogen limit dextrin produced by circulatory amylases and neutral α1‐4‐glycosidases. 11 It is elevated in conditions associated with increased glycogen accumulation and/or release of glycogen from damaged tissues. 12 , 13 , 14 , 15 , 16 Urinary Glc4 is an established biomarker in patients with Pompe disease, correlating with skeletal muscle glycogen and disease status in these patients. 17 , 18 , 19 Glc4 is also elevated in GSD III, 16 , 20 , 21 , 22 , 23 and has potential as a biomarker in this disorder. Glc4 is usually measured in randomly collected voids (spot urines) for the convenience of patients and clinical personnel. However, it has not been determined whether Glc4 measurements in spot urines represent excretion over a 24‐hour period. Previous studies have suggested that ingestion of starch or glycogen may increase Glc4 excretion, 14 and in GSD III this could be a particular concern because of cornstarch therapy. We evaluated the diurnal variability in Glc4 excretion, to investigate whether the degree of elevation and variability in Glc4 excretion correlated with cornstarch therapy in patients with GSD III.
1.1. Materials
Acquity UPLC BEH amide 2.1 × 100 mm column, VanGuard guard column, and Sep‐Pak Vac C18 cartridges were obtained from Waters (Milford, Massachusetts), Glc4 from Toronto Research Chemicals (Toronto, Canada), glacial acetic acid, butyl‐4‐aminobenzoate, sodium cyanoborohydride from Sigma (St. Louis, MO), and methanol and acetonitrile (HPLC grade) from VWR Scientific products (Atlanta, Georgia). A stable isotope‐labeled Glc4 internal standard (IS) was synthesized as described. 24
2. METHODS
2.1. Subjects
This was a single center, prospective study of patients consented to a natural history study, approved by Duke University Health System Institutional Review Board (#Pro00047556). Patients had a confirmed diagnosis of GSD III, via AGL variant and/or enzyme analysis (Supplemental Table 1).
2.2. Glucose tetrasaccharide and creatinine analyses
Glc4 was analyzed as a butyl‐4‐aminobenzoate derivative using [13C6]Glc4 as an IS, and ultraperformance liquid chromatograph‐tandem mass spectrometry (UPLC‐MS/MS), as reported with modifications. 24 Urine (20 μL) was combined with 20 μL 50 μmol/L IS, incubated at 80°C for 1 hour with 152 mmol/L butyl‐4‐aminobenzoate, 400 mmol/L sodium cyanoborohydride, and 5.3% glacial acetic acid (vol/vol) in methanol, and excess reagent was removed using solid phase extraction. Samples were dried under nitrogen, reconstituted in 10 mmol/L ammonium acetate in 90:10 (vol:vol) acetonitrile: deionized water (diH2O), and separated with gradient elution on a UPLC BEH amide column using 10 mmol/L ammonium acetate in acetonitrile:diH2O as the mobile phase. Glc4 and the IS were detected by selected reaction monitoring (m/z 844 > 358 and m/z 850 > 364, respectively). Glc4 was normalized to creatinine (CN), analyzed as reported. 18 The Glc4 assay has acceptable intraday and interday imprecision (≤20% over the calibration range: 2‐230 μmol/L).
2.3. 24‐hour urine collections
Urine collections over 24 hours were conducted unsupervised in a residential setting. Subjects were instructed to discard the first morning void on day 1, and collect all subsequent voids separately over the next 24 hours, ending with the first morning void on the second day. Subjects were asked to reserve a small aliquot (about 1 mL) from each void in a separate container and combine the remaining urine in a single large container. Samples were stored in a cooler on cold packs and frozen within 48 hours of collection. Glc4 and creatinine were analyzed in each aliquot and pooled collections.
2.4. Statistical analyses
Descriptive statistics, Pearson correlation coefficient, linear regression, paired t test, and Bland‐Altman analyses were calculated using Microsoft Excel and GraphPad Prism V8. The diurnal variability of Glc4 excretion was calculated as the CV%. P‐values ≤.05 were considered significant.
3. RESULTS
3.1. Cohort description
Here, 18 subjects (n = 2 males, subjects 13 and 15) with GSD IIIa (n = 16) or b (n = 2, subjects 17 and 20) participated in the study. The median age was 1 year (range: 0.3‐12) at the time of diagnosis, and 13.5 years (range: 3.7‐62) at the start of the study. All were treated with various dietary regimens of cornstarch (Table 1), except the three adults (subjects #8, 13, and 29). The protein intake goal was 20% to 25% total energy consumed for all patients with GSD IIIa, achieved in seven subjects (#2, #5, #8, #10, #12, #13, and #17) using a protein supplement, and in the remaining subjects using natural sources of protein. Most subjects were ambulatory. One adult (#29) required a wheelchair and an 8‐year‐old female (#27) was considering a wheelchair for long distances. An 11‐year‐old male (#15) required ankle‐foot orthoses, an 8‐year‐old female (#7) was recommended to wear custom shoe inserts for calcaneal valgus, and one adult (#13) used assistive devices. Ten subjects had evidence of hepatomegaly on liver imaging. Of the three adults in the cohort, subject #8 showed evidence of liver fibrosis and cirrhosis, subject #13 underwent a multiorgan transplant (heart, liver, and kidney) after suffering heart failure, 25 and subject #29 had HCC treated by radioembolization. The liver disease natural history in the pediatric subjects was reported in detail elsewhere. 20
TABLE 1.
Summary of diurnal variability of Glc4 excretion in subjects with GSD III
| Subject ID‐collection # | Age (y) | Glc4 in 24‐h pooled urine (mmol/mol CN) | Diurnal variability (CV%) | Cornstarch dose and regimen | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Daily dose (g/kg/d) | Breakfast | Lunch | Dinner | Before bed | Middle of night | |||||
| <25% | >25% | g/kg | ||||||||
| 15‐1 | 10.8 | 1.7 | 16% | 3.8 | 1.1 | 1.6 | 1.1 | |||
| 15‐2 | 11.8 | 2.2 | 4% | |||||||
| 15‐3 | 12.8 | 1.7 | 39% | |||||||
| 20‐1 | 13.5 | 2.0 | 14% | 0.9 | 0.9 | |||||
| 20‐2 | 15.4 | 1.4 | 7% | |||||||
| 1‐1 | 14.6 | 6.6 | NA | 1.23 | 0.45 | 0.78 | ||||
| 08‐1 | 40.3 | 8.0 | 24% | None | ||||||
| 08‐2 | 40.7 | 10.4 | 10% | |||||||
| 08‐3 | 42.4 | 23.2 | 14% | |||||||
| 03‐1 | 15.1 | 8.8 | 7% | 0.7 | 0.7 | |||||
| 03‐2 | 16.0 | 8.6 | 20% | |||||||
| 03‐3 | 17.1 | 16.7 | 47% | |||||||
| 18‐2 | 15.4 | 12.2 | 14% | 0.73 | 0.73 | |||||
| 12‐1 | 3.7 | 11.6 | 12% | 5.57 | 1.05 | 1.05 | 1.05 | 1.21 | 1.21 | |
| 10‐1 | 4.9 | 9.5 | 25% | 2.32 | 0.93 | 0.46 | 0.93 | |||
| 10‐2 | 7.2 | 4.4 | 12% | |||||||
| 09‐1 | 6.5 | 16.5 | 21% | 1.9 | 1.1 | 0.8 | ||||
| 09‐2 | 8.3 | 11.2 | 26% | |||||||
| 05‐1 | 9.6 | 16.8 | 10% | 6 | 1.2 | 1.2 | 1.2 | 1.2 | 1.2 | |
| 05‐2 | 11.3 | 14.3 | 5% | |||||||
| 27‐1 | 7.6 | 18.1 | 26% | 3.12 | 0.78 | 0.78 | 1.56 | |||
| 02‐1 | 13.9 | 20.2 | 8% | 2.55 | 0.7 | 0.5 | 0.5 | 0.85 | ||
| 17‐1 | 5.9 | 20.6 | NA | 2.29 | 0.38 every 4 h during the day, 0.76 by continuous feed at night | |||||
| 14‐1 | 14.6 | 27.6 | 39% | 0.64 | 0.64 | |||||
| 14‐2 | 16.7 | 28.0 | 30% | |||||||
| 13‐2 | 52.2 | 28.0 | 9% | None | ||||||
| 13‐3 | 53.2 | 25.7 | 28% | |||||||
| 29‐1 | 62.4 | 29.2 | NA | None | ||||||
| 07‐1 | 8.2 | 42.2 | 39% | 0.3 | 0.3 | |||||
| 19‐1 | 17.2 | 85.8 | 41% | 0.63 | 0.63 | |||||
| 19‐2 | 19.1 | 33.6 | 12% | |||||||
Note: Collections by subjects 1, 17, and 29 were rejected for assessment of the diurnal variability, although pooled urine was used to determine Glc4 concentrations. All subjects have GSD IIIa except for subjects 17 and 20 who were diagnosed with GSD IIIb.
Abbreviations: GSD, glycogen storage disease; NA, not assessed.
3.2. 24‐hour urine collections
Thirty‐six 24‐hour urine collections were completed by 18 subjects. Six subjects completed two and another six completed three repeat collections over 1 to 3 years. Then, 28 collections by 15 subjects were accepted for diurnal variability assessments (Table 1) and 8 collections (22%) were excluded due to incomplete or inaccurate collection or recorded information, or inappropriate storage. The total urine volume was positively correlated with age (Pearson R: .758, P < .05) and weight (Pearson R: .700, P < .05; Supplemental Figure 1). The median number of voids was 6 (range: 4‐10), the median total volume of urine collected was 1265 mL (range: 262‐4000), and the median urinary output was 1.0 mL/kg/h (range: 0.53‐2.7).
3.3. Cornstarch dose and Glc4 excretion
Glc4 was elevated in all subjects except an 11 year old subject with GSD IIIa (#15) and a 13 year subject old with GSD IIIb (#20) (Table 1). Glc4 concentrations normalized to creatinine in 24‐hour collections varied widely (median: 15 mmol/mol CN, range: 2‐60, n = 18; calculated using median values for subjects with more than one collection) and were not significantly correlated with the total cornstarch dose (Figure 1). Of note, both subjects with normal Glc4 (#15 and 20) were treated with cornstarch, whereas all three subjects (#8, 13, and 29) who were not on cornstarch therapy had elevated Glc4 (Table 1). For the 15 subjects with acceptable 24‐hour urine collections, the total amount of Glc4 excreted in 24 hours was significantly correlated with weight, but not age (Supplemental Figure 2). In comparison, the total amount of creatinine excreted over 24 hours significantly increased with weight and age (Supplemental Figure 2).
FIGURE 1.
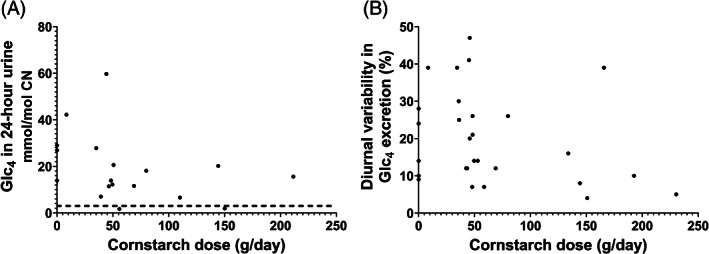
Comparison of the degree and variability of Glc4 excretion with uncooked cornstarch dose. A, Glc4 concentrations in 24‐hour urine relative to total cornstarch dose. No significant correlation was observed (Pearson R = −.349, P = .15, n = 18 patients). B, Variability (CV%) in Glc4 excretion in 24‐hour urine relative to cornstarch dose. No significant correlation was observed (Pearson R = −.278, P = .15, n = 28 collections by 15 subjects)
3.4. Variability in Glc4 excretion over 24 hours in patients with elevated Glc4
Glc4 was elevated in 23/28 of the 24‐hour urine collections, of which 15/23 had low (CV% ≤25), and 8/23 had higher (CV%: 26‐47) diurnal variability in Glc4 excretion (Table 1). Only 1/5 subjects who performed repeat collections (#14) had high variability in more than one collection. The diurnal variability was significantly correlated with the pooled 24‐hour urine Glc4 concentration normalized to creatinine (Pearson R = .644, P < .05), but not with the total dose of uncooked cornstarch (Figure 1). Furthermore, high variability was observed in one collection (#13‐2) from a subject not on cornstarch therapy. In contrast, low variability was observed in 11 collections by 8 subjects on cornstarch therapy.
No consistent trend in Glc4 excretion over 24 hours was observed in collections with high variability (Supplemental Figure 3). A closer agreement was observed between Glc4 concentrations in 24‐hour urines and the first void, compared with the last void collected (Supplemental Figure 4). However, the first and last voids did not differ significantly (paired t test, P = .41). For urines with low variability, both the first and last voids compared well with the 24‐hour urine (Supplemental Figure 5).
4. DISCUSSION
Glc4 is a promising biomarker in GSD III as it is correlated with serum transaminases in pediatric patients with GSD III 20 , 23 and a GSD III dog model, 26 and with CK in adults with GSD III. 23 These observations suggest urinary Glc4 reflects glycogen accumulation in liver more than muscle in the pediatric population, and muscle glycogen accumulation in adults. However, given the clinical variability of GSD IIIa and an increasing understanding of early muscle involvement, clinical correlation is needed in interpreting the source of Glc4 in GSD III. Patients with GSD III are treated with uncooked cornstarch which contains amylopectin, an α1‐6 branched glucose polymer that can be converted to Glc4 by amylase activity. 27 This raises a concern for the reliability of using spot urines to assess Glc4 excretion in cornstarch‐treated patients. Our results suggest that uncooked cornstarch intake does not contribute significantly to Glc4 excretion.
A previous study investigating the impact of dietary factors on Glc4 reported diurnal variation in the rate of excretion in a volunteer on a normal diet over three days. 14 There was evidence that the rate increased after meals; however, the overall daily excretion varied less than 10%. Decreased calorific intake and a low carbohydrate diet appeared to decrease the excretion rate, whereas a high carbohydrate diet consisting entirely of cooked rice (2400‐3200 kcal/24 h), resulted in a fourfold to fivefold rate increase, compared with a regular diet. 14 The authors suggested this increase might be caused by amylopectin degradation by amylase in the gastrointestinal tract. Urinary output varies over the course of the day and is impacted by fluid intake, physical activity, and sleep. 28 Thus, an increase in the Glc4 excretion rate under normal dietary conditions could be secondary, in part, to increased urinary output. The dose used to treat pediatric patients with GSD III is relatively low (typically 1 g/kg every 4 hours or longer, adjusted based on the ability of a dose to maintain euglycemia between feeds), 3 and many adults with GSD III have a minimal intake of cornstarch. In comparison, patients with GSD I generally require higher and more frequent doses (eg, 1.6‐2.5 g/kg every 3‐5 hours) due to impairment in both glycogenolysis and gluconeogenesis. 29 In our population, cornstarch intake ranged from 9 to 193 g/day, equivalent to approximately 30 to 700 kcal/day. This low dose and slower digestion of uncooked cornstarch compared with cooked starch, may explain the lack of correlation between cornstarch intake and the degree and variability of Glc4 excretion.
A higher variability in Glc4 excretion was associated with increased Glc4 concentrations, but was not consistently observed in repeat collections by the same subject. The reason for this variability is unknown. In addition to the potential for dietary influences, physical activity may be a factor. 14 Twenty‐four hour urine collections are considered a “gold standard” method for assessing urinary biomarker excretion. 30 However, 24‐hour collections impose a significant burden on subjects and a risk for collection errors, 31 as demonstrated by the high rejection rate (22%) in our study. Appropriate storage of urine is another challenge. It was proposed that Glc4 might be unstable at ambient temperature in some urine samples because of bacterial degradation, resulting in specimens with unexpectedly low concentrations. 32 Our studies indicate that Glc4 is usually stable in urine for 1 week in a climate‐controlled environment at ambient temperature (Supplemental Table 2). However, storage and transport of samples at 4°C or colder is recommended to ensure sample integrity. Several studies have demonstrated the equivalence of spot urines and 24‐hour collections for a number of analytes normalized to creatinine. 33 , 34 , 35 Our results support the use of spot urines to evaluate Glc4 in patients with GSD III.
The reliance on patients to comply with the 24‐hour urine collection protocol and the prescribed dietary cornstarch therapy in an unsupervised setting was a limitation of this study. The reliability of the 24‐hour collections accepted for analysis is supported by several lines of evidence: (a) the Glc4 excretion rate (median: 45 mg/24 hours, range: 6‐720) was comparable to published rates in patients with GSD III (9‐45 mg/24 hours) 14 ; (b) total urine volume and creatinine excreted over 24 hours significantly increased with age and weight; and (c) the voiding frequency and urine output were comparable to a previous study. 28
To conclude, spot urines are generally reliable for assessing Glc4 excretion in GSD III. Increased variability in Glc4 excretion may be observed in patients who excrete higher Glc4 concentrations. This variability does not appear to be caused by cornstarch intake. Repeat assessments in spot urines collected close together in time are advisable, to assess baseline Glc4 concentrations and monitor trends in response to therapies in GSD III.
CONFLICT OF INTEREST
S. P. Y. works for a laboratory that offers Glc4 testing, has received grant support from Sanofi Genzyme, Amicus Therapeutics, Biomarin Pharmaceutical, PTC Therapeutics, and Valerion Therapeutics, and has consulted for Amicus Therapeutics, Sanofi Genzyme, and PTC Therapeutics. P. S. K. has received research/grant support from Sanofi Genzyme, Valerion Therapeutics, Shire Pharmaceuticals, Amicus Therapeutics, Pfizer, Alexion Pharmaceuticals and Ultragenyx and consulting fees and honoraria from Sanofi Genzyme, Shire Pharmaceuticals, Alexion Pharmaceuticals, Amicus Therapeutics, Vertex Pharmaceuticals, Ultragenyx, and Asklepios Biopharmaceutical, Inc. (AskBio). P. S. K. is listed as an inventor on a licensed Duke University patent for the use of rhGAA in the treatment of GSDIII and other GSDs excluding GSDII. To date, neither Duke University nor the inventor has received any money from the commercialization of rights associated with this patent. S. A. has received consulting fees from Ultragenyx. The other authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
Sarah P. Young, Stephanie Austin, and Priya S. Kishnani: Contributed to the planning, conduct, and reporting of this work. Ela Stefanescu and Andrea M. Seifts: Contributed to the planning and conduct, and Aleena Khan and Ghada Hijazi: Contributed to the reporting of this work.
INFORMED CONSENT
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from all patients for being included in the study.
Supporting information
Appendix S1: Supplementary Information
ACKNOWLEDGMENTS
The authors would like to express their gratitude to the patient participants who volunteered to collect the 24‐hour urine collections. Without them, this study would not be possible. The authors would like to thank Dr Catherine Rehder for assistance with AGL variant nomenclature. The authors would also like to thank the study sponsor, Valerion Therapeutics, LLC, and Ultragenyx Pharmaceutical for grant support for the preparation of this manuscript.
Young SP, Khan A, Stefanescu E, et al. Diurnal variability of glucose tetrasaccharide (Glc4) excretion in patients with glycogen storage disease type III . JIMD Reports. 2021;58:37–43. 10.1002/jmd2.12181
Communicating Editor: Charles P Venditti
Funding information Ultragenyx Pharmaceutical; Valerion Therapeutics
REFERENCES
- 1. Chen Y‐T. Glycogen storage diseases. In: Scriver C, Beaudet A, Sly W, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York, NY: McGraw‐Hill; 2001:1521‐1551. [Google Scholar]
- 2. van Hoof F, Hers HG. The subgroups of type 3 glycogenosis. Eur J Biochem. 1967;2(3):265‐270. [DOI] [PubMed] [Google Scholar]
- 3. Kishnani PS, Austin SL, Arn P, et al. Glycogen storage disease type III diagnosis and management guidelines. Genet Med. 2010;12(7):446‐463. [DOI] [PubMed] [Google Scholar]
- 4. Dagli ASC, Weinstein D. Glycogen storage disease type III. In: Pago RAAM, Ardinger HH, et al., eds. Gene Reviews. Seattle, WA: University of Washington; 2010. [Google Scholar]
- 5. Haagsma EB, Smit GP, Niezen‐Koning KE, Gouw AS, Meerman L, Slooff MJ. Type IIIb glycogen storage disease associated with end‐stage cirrhosis and hepatocellular carcinoma. The liver transplant group. Hepatology. 1997;25(3):537‐540. [DOI] [PubMed] [Google Scholar]
- 6. Siciliano M, de Candia E, Ballarin S, et al. Hepatocellular carcinoma complicating liver cirrhosis in type IIIa glycogen storage disease. J Clin Gastroenterol. 2000;31(1):80‐82. [DOI] [PubMed] [Google Scholar]
- 7. Fernandes J, Leonard JV, Moses SW, et al. Glycogen storage disease: recommendations for treatment. Eur J Pediatr. 1988;147(3):226‐228. [DOI] [PubMed] [Google Scholar]
- 8. Mayorandan S, Meyer U, Hartmann H, Das AM. Glycogen storage disease type III: modified Atkins diet improves myopathy. Orphanet J Rare Dis. 2014;9:196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vidal P, Pagliarani S, Colella P, et al. Rescue of GSDIII phenotype with gene transfer requires liver‐ and muscle‐targeted GDE expression. Mol Ther. 2018;26(3):890‐901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Francini‐Pesenti F, Tresso S, Vitturi N. Modified Atkins ketogenic diet improves heart and skeletal muscle function in glycogen storage disease type III. Acta Myol. 2019;38(1):17‐20. [PMC free article] [PubMed] [Google Scholar]
- 11. Ugorski M, Seder A, Lundblad A, Zopf D. Studies on the metabolic origin of a glucose containing tetrasaccharide in human urine. Int J Exp Pathol. 1983;1(1):27‐38. [PubMed] [Google Scholar]
- 12. An Y, Young SP, Hillman SL, van Hove JL, Chen YT, Millington DS. Liquid chromatographic assay for a glucose tetrasaccharide, a putative biomarker for the diagnosis of Pompe disease. Anal Biochem. 2000;287(1):136‐143. [DOI] [PubMed] [Google Scholar]
- 13. Hallgren P, Hansson G, Henriksson KG, Hager A, Lundblad A, Svensson S. Increased excretion of a glucose‐containing tetrasaccharide in the urine of a patient with glycogen storage disease type II (Pompe's disease). Eur J Clin Invest. 1974;4:429‐433. [DOI] [PubMed] [Google Scholar]
- 14. Kumlien J, Chester MA, Lindberg BS, Pizzo P, Zopf D, Lundblad A. Urinary excretion of a glucose‐containing tetrasaccharide. A parameter for increased degradation of glycogen. Clin Chim Acta. 1988;176(1):39‐48. [DOI] [PubMed] [Google Scholar]
- 15. Oberholzer K, Sewell AC. Unique oligosaccharide (apparently glucotetrasaccharide) in urine of patients with glycogen storage diseases. Clin Chem. 1990;36(7):1381. [PubMed] [Google Scholar]
- 16. Piraud M, Pettazzoni M, de Antonio M, et al. Urine glucose tetrasaccharide: a good biomarker for glycogenoses type II and III? A study of the French cohort. Mol Genet Metabol Rep. 2020;23:100583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. An Y, Young SP, Kishnani PS, et al. Glucose tetrasaccharide as a biomarker for monitoring the therapeutic response to enzyme replacement therapy for Pompe disease. Mol Genet Metab. 2005;85(4):247‐254. [DOI] [PubMed] [Google Scholar]
- 18. Young SP, Zhang H, Corzo D, et al. Long‐term monitoring of patients with infantile‐onset Pompe disease on enzyme replacement therapy using a urinary glucose tetrasaccharide biomarker. Genet Med. 2009;11(7):536‐541. [DOI] [PubMed] [Google Scholar]
- 19. Young SP, Piraud M, Goldstein JL, et al. Assessing disease severity in Pompe disease: the roles of a urinary glucose tetrasaccharide biomarker and imaging techniques. Am J Med Genet C Semin Med Genet. 2012;160c(1):50‐58. [DOI] [PubMed] [Google Scholar]
- 20. Halaby CA, Young SP, Austin S, et al. Liver fibrosis during clinical ascertainment of glycogen storage disease type III: a need for improved and systematic monitoring. Genet Med. 2019;21(12):2686‐2694. [DOI] [PubMed] [Google Scholar]
- 21. Lennartson G, Lundblad A, Sjöblad S, Svensson S, Ockerman P. Quantitation of a urinary tetrasaccharide by gas chromatography and mass spectrometry. Biol Mass Spectrom. 1976;3(2):51‐54. [DOI] [PubMed] [Google Scholar]
- 22. Chester MA, Lundblad A, Hager A, et al. Increased urinary excretion of a glycogen‐derived tetrasaccharide in heterozygotes with glycogen storage diseases type II and III. Lancet. 1983;1(8331):994‐995. [DOI] [PubMed] [Google Scholar]
- 23. Heiner‐Fokkema MR, van der Krogt J, de Boer F, et al. The multiple faces of urinary glucose tetrasaccharide as biomarker for patients with hepatic glycogen storage diseases. Genet Med. 2020;22(11):1915‐1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Young SP, Stevens RD, An Y, Chen YT, Millington DS. Analysis of a glucose tetrasaccharide elevated in Pompe disease by stable isotope dilution‐electrospray ionization tandem mass spectrometry. Anal Biochem. 2003;316(2):175‐180. [DOI] [PubMed] [Google Scholar]
- 25. Cochrane AB, Fedson SE, Cronin DC II. Nesiritide as bridge to multi‐organ transplantation: a case report. Transplant Proc. 2007;39(1):308‐310. [DOI] [PubMed] [Google Scholar]
- 26. Brooks ED, Yi H, Austin SL, et al. Natural progression of canine glycogen storage disease type IIIa. Comp Med. 2016;66(1):41‐51. [PMC free article] [PubMed] [Google Scholar]
- 27. Nordin P, French D. 1‐phenyl‐flavazole derivatives of starch Dextrins1. J Am Chem Soc. 1958;80(6):1445‐1447. [Google Scholar]
- 28. van Hoeck K, Bael A, Lax H, Hirche H, van Gool JD. Circadian variation of voided volume in normal school‐age children. Eur J Pediatr. 2007;166(6):579‐584. [DOI] [PubMed] [Google Scholar]
- 29. Kishnani PS, Austin SL, Abdenur JE, et al. Diagnosis and management of glycogen storage disease type I: a practice guideline of the American College of Medical Genetics and Genomics. Genet Med. 2014;16(11):e1‐e1. [DOI] [PubMed] [Google Scholar]
- 30. Konig F, Andersson M, Hotz K, Aeberli I, Zimmermann MB. Ten repeat collections for urinary iodine from spot samples or 24‐hour samples are needed to reliably estimate individual iodine status in women. J Nutr. 2011;141(11):2049‐2054. [DOI] [PubMed] [Google Scholar]
- 31. Christopher‐Stine L, Petri M, Astor BC, Fine D. Urine protein‐to‐creatinine ratio is a reliable measure of proteinuria in lupus nephritis. J Rheumatol. 2004;31(8):1557‐1559. [PubMed] [Google Scholar]
- 32. Prunty H, Broomfield A, Vellodi A, et al. Update on glucose tetrasaccharide (Glc4) in urine and saliva as a potential biomarker in Pompe disease. Mol Genet. 2014;111(2):S88. [Google Scholar]
- 33. Cangemi G, Barco S, Reggiardo G, et al. Interchangeability between 24‐hour collection and single spot urines for vanillylmandelic and homovanillic acid levels in the diagnosis of neuroblastoma. Pediatr Blood Cancer. 2013;60(12):E170‐E172. [DOI] [PubMed] [Google Scholar]
- 34. Wilson T, Garcia‐Perez I, Posma JM, et al. Spot and cumulative urine samples are suitable replacements for 24‐hour urine collections for objective measures of dietary exposure in adults using metabolite biomarkers. J Nutr. 2019;149(10):1692‐1700. [DOI] [PubMed] [Google Scholar]
- 35. Teo BW, Loh PT, Wong WK, et al. Spot urine estimations are equivalent to 24‐hour urine assessments of urine protein excretion for predicting clinical outcomes. Int J Nephrol. 2015;2015:156484‐156484. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supplementary Information