

Abstract
Background
Orthopedic disease progresses in mucopolysaccharidosis type I (MPS I), even with approved therapies and remains a major factor in persistent suffering and disability. Novel therapies and accurate predictors of response are needed. The primary objective of this study was to identify surrogate biomarkers of future change in orthopedic disease.
Methods
As part of a 9‐year observational study of MPS I, range‐of‐motion (ROM), height, pelvic radiographs were measured annually. Biomarkers in year 1 were compared to healthy controls. Linear regression tested for associations of change in biomarkers over the first year with change in long‐term outcomes.
Results
MPS I participants (N = 19) were age 5 to 16 years and on average 6.9 ± 2.9 years post treatment initiation. Healthy controls (N = 51) were age 9 to 17 years. Plasma IL‐1β, TNF‐α, osteocalcin, pyridinolines, and deoxypyridinolines were higher in MPS than controls. Within MPS, progression of hip dysplasia was present in 46% to 77%. A 1 pg/mL increase in IL‐6 was associated with −22°/year change in ROM (−28 to −15; P < .001), a 20 nmol/mmol creatinine/year increase in urine PYD was associated with a −0.024 Z‐score/year change in height Z‐score (−0.043 to −0.005; P = .016), and a 20 nmol/mmol creatinine/year increase in urine PYD was associated with a −2.0%/year change in hip dysplasia measured by Reimers migration index (−3.8 to −0.1; P = .037).
Conclusions
Inflammatory cytokines are high in MPS I. IL‐6 and PYD were associated with progression in joint contracture, short stature, and hip dysplasia over time. Once validated, these biomarkers may prove useful for predicting response to treatment of skeletal disease in MPS I.
Keywords: biomarker, bone, cytokines, Hurler, inflammation, mucopolysaccharidosis, Scheie
SYNOPSIS.
Change in plasma and urine biomarkers over 1 year are associated with skeletal disease progression over 9 years in individuals with mucopolysaccharidosis type I.
1. INTRODUCTION
Mucopolysaccharidosis type I (MPS I) is a lysosomal storage disease caused by a mutation in the IDUA gene that manifests as central nervous system (cognitive impairment) and somatic (eg, bone, cartilage, heart, liver) abnormalities that vary broadly in severity. 1 The severe form of MPS I, involving rapid neurocognitive deterioration and early death before 10 years of age, is designated MPS IH (for Hurler syndrome; OMIM 607014), whereas the phenotypes with less severe neurocognitive symptoms and longer lifespan are called “attenuated” (MPS IA; including the phenotypic spectrum from Hurler‐Scheie syndrome (OMIM 607015) to Scheie syndrome (OMIM 607016)). These distinct forms of MPS I are treated very differently. Children with MPS IH require hematopoietic stem cell transplantation (HSCT), sometimes supplemented with peri‐HSCT enzyme replacement therapy (ERT). It is well recognized that HSCT can alter the course of neurocognitive deterioration whereas ERT alone does not. 2 , 3 , 4 , 5 , 6 , 7 In contrast, children with MPS IA are treated with ERT alone which attenuates somatic disease without the inherent risks of HSCT. 8 , 9
HSCT and ERT therapies have considerably extended the life expectancy for patients, but significant skeletal and joint disease continues to cause severe physical disability and a negative impact on quality of life for individuals with MPS I. 3 , 8 , 9 , 10 Otherwise successfully treated patients with MPS I show one or more progressive orthopedic complications (eg, hip dysplasia, kyphosis, scoliosis, osteoarthritis) that causes chronic pain, decreased physical function, and poor quality of life. 3 , 11 , 12 , 13 , 14 Thus, therapies to alleviate the significant debilitating disease are needed and are critical to improve the health, function and quality of life of individuals with MPS I.
Based upon animal studies, the crippling skeletal and joint disease manifestations of MPS appear to be, at least in part, inflammatory in nature. 15 , 16 , 17 , 18 , 19 , 20 For example, individuals with MPS I, II, and VI have high circulating levels of the inflammatory cytokine TNF‐α and these high TNF‐α levels are associated with more pain and physical disability. 12 In animal models of MPS, elevated levels of TNF‐α, IL‐1β, and RANKL have all been implicated in MPS‐related joint disease. 16 , 18 , 20
Currently, design of therapeutic clinical trials is limited by the lack of control data on the natural history of skeletal disease progression and minimal information regarding early biomarkers that predict disease progression. Without this information, the time required to demonstrate a significant clinical effect based on disease progression is protracted to the point of being impractical. Thus, identifying predictive biomarkers of disease response to treatment is critical in promoting the efficient advancement of treatments for MPS.
The goals of this prospective observational study are to measure the change in skeletal and joint disease over time in MPS I, evaluate pathways of inflammation and bone modeling and remodeling, and identify potential biomarkers that are predictive of change in bone and joint disease in MPS I. These could be tested in future clinical trials for their ability to predict MPS disease response to treatment.
2. MATERIALS AND METHODS
2.1. Study design
Plasma and urine samples from 19 participants with MPS I (14 MPS IH; 5 MPS IA), age 5 to 16 years, who were enrolled in a 9‐year, multicentered, longitudinal observational study of children and young adults with MPS (Lysosomal Disease Network and Clinicaltrials.gov NCT01521429) and had participated in the study for a minimum of 5 years were included in this analysis. Inclusion criteria were diagnosis of MPS I, age 5 to 33 years of age, and ability to travel to the study center. All participants with MPS IA were being treated with ERT for more than 1 year at study initiation. Exclusion criteria were pregnancy, participation in any other study within the past 6 months that would increase radiation exposure above 500 mrem for the calendar year, and inability to comply with study procedures.
Healthy control subjects were recruited for a separate study of the association between bone and energy metabolism. 21 Inclusion criteria included otherwise healthy adolescents between 8 and 17 years of age. Exclusion criteria included diagnosis of diabetes mellitus, polycystic ovarian syndrome, treatment with a medication that alters insulin sensitivity, secretion, or beta‐cell mass, participation in a concurrent intervention trial, and pregnancy.
2.2. Biomarkers
Blood and urine samples were collected from both populations the morning after a minimum 8‐hour fast in 4 mL EDTA tubes, immediately processed, frozen, and stored at −80°C. All MPS samples were run in one batch. All control samples were run in another batch.
Markers of inflammation (interleukin‐1 β [IL‐1β], interleukin‐6 [IL‐6], tumor necrosis factor‐α [TNF‐α]), bone formation (osteocalcin [OCN]), bone resorption (deoxypyridinolines [DPD], pyridinolines [PYD]), and cartilage degradation (PYD) were measured. The bone biomarkers were chosen based on their prior use in pediatric studies of bone 22 , 23 and availability of well‐established assays described below. IL‐1β was measured in plasma using the Quantikine high sensitivity Human IL‐1β Immunoassay from R & D Systems (Minneapolis, MN): intra‐assay coefficient of variation (CV) was <8.2% and inter‐assay CV was <16.1%. IL‐6 was measured in plasma using the Quantikine high sensitivity Human IL‐6 Immunoassay (R & D Systems): intra‐assay CV < 6.0% and inter‐assay CV <10.5%. TNF‐α was measured in plasma using the Quantikine high sensitivity Human TNF‐α Immunoassay (R & D Systems): intra‐assay CV <6.8% and inter‐assay CV <10.7%. OCN was measured in serum using the MicroVue Osteocalcin EIA kit from Quidel Corporation (San Diego, California): intra‐assay %CV <5.0 and inter‐assay %CV <12.0. DPD was measured in urine using the MicroVue DPD EIA kit (Quidel Corporation): intra‐assay CV <8.4% and inter‐assay CV <4.8%. PYD was measured in urine using the MicroVue PYD EIA kit (Quidel Corporation): intra‐assay CV <9.9% and inter‐assay CV <11.2%.
2.3. Outcome measures
Shoulder flexion, elbow extension, and knee extension were measured in each joint three times and the average taken. Measurement of range‐of‐motion (ROM) was standardized at all three sites with a training manual provided for reference at each site. In addition, the same one to two individuals performed the measurements at each visit. A joint summary score was calculated as the summary of all six joints (higher number = better ROM). Height was measured with a wall‐mounted stadiometer, three times standing without shoes, and the average recorded. Each subject was repositioned between each measurement. Height Z‐score (ie, adjusted for age and sex) was calculated based on the National Center for Health Statistics 2000 data as provided by the Center for Disease Control. Tonnis angle, Reimers migration index (RMI), and Tonnis Grade were measured from anterior‐posterior pelvic radiographs by a single individual (Dr K. K. W., Pediatric Orthopedic Surgery). Tonnis angle and RMI are measurements of hip dysplasia. Tonnis Grade is a measurement of hip osteoarthritis.
2.4. Statistical analysis
Descriptive statistics are presented as mean ± SD for continuous variables, and as frequency and percent for nominal variables. Linear regression models were used to compare biomarkers at baseline in MPS vs controls with adjustment for BMI Z‐score for inflammatory biomarkers (TNF‐α, IL‐6, IL‐1β) and adjustment for age for bone and cartilage biomarkers (OCN, DPD, PYD). Furthermore, linear regression was used to examine the association of annual rate of change in biomarker over study years 1 to 2 with the annual rate of change in ROM over years 5 to 9 and height Z‐score over years 2 to 9 in participants with MPS, adjusted for age. Outcomes of annual rate of change for ROM and height Z‐score were calculated using linear regression to fit a slope for change in each outcome over time, per individual. Outliers of change in biomarkers were removed based on being greater than three SD from the mean and by visual inspection. Annualized rate of change of Tonnis angle and RMI were calculated for each participant and for right and left hips over study visits 7 to 9. Rather than average across hips for each participant, the hip that showed the most severe disease progression in Tonnis angle and RMI was used as the outcome in linear regression models with each of the change in biomarkers during the first year of study as independent variables. These models were further adjusted for age (months) and the first recorded value of the Tonnis angle or RMI. Statistical analysis of change in ROM, height Z‐score, Tonnis angle, and RMI over time in MPS IH vs MPS IA is not included due to low sample size within MPS groups.
Analyses and graphing were performed using R, version 3.5.3, STATA 14, Adobe Photoshop, and Prism.
3. RESULTS
Population characteristics are described in detail in Table 1. Of note, the study included 51 healthy controls and 19 participants with MPS I (14 MPS IH; 5 MPS IA). Controls were, on average, older than MPS participants. Controls and MPS participants were similar in BMI Z‐scores. All participants with MPS IH were treated with HSCT ≥2.9 years prior to study enrollment, and all participants with MPS IA were currently treated with ERT (laronidase). None were wheelchair dependent. Within the MPS I group, baseline patient records were examined for evidence of infection due to the potential influence on cytokine analysis; there was one participant with mild nasal congestion assumed related to seasonal allergies given normal physical exam, and one participant with tinea corporis.
TABLE 1.
Population characteristics
| Controls | All MPS I | MPS IH | MPS IA | |
|---|---|---|---|---|
| N = 51 | N = 19 | N = 14 | N = 5 | |
| Age at visit, y | 14.6 ± 2.0 (9.2‐17.9) | 9.7 ± 4.2 (5.0‐16.5) | 8.0 ± 2.9 (5.0‐12.4) | 14.3 ± 3.9 (7.4‐16.5) |
| Treated with HSCT, y | NA | 14 (74) | 14 (100) | 0 (0) |
| Age at HSCT initiation, y | NA | 1.3 ± 0.7 (0.2‐2.5) | 1.3 ± 0.7 (0.2‐2.5) | NA |
| Treated with ERT, yes | NA | 13 (68) | 8 (57) a | 5 (100) |
| Age at ERT initiation, y | NA | 3.8 ± 4.0 (0.5‐13.3) | 1.3 ± 0.6 (0.5‐2.3) | 7.8 ± 4.0 (2.5‐13.3) |
| Time since HSCT, y | NA | 6.7 ± 3.0 (2.9‐11.2) | 6.7 ± 3.0 (2.9‐11.2) | NA |
| Time since ERT initiation, y | NA | 5.5 ± 2.1 (3.1‐10.7) | 4.5 ± 1.0 (3.1‐6.3) | 6.9 ± 2.7 (3.6‐10.7) |
| Sex, female | 27 (53) | 8 (42) | 7 (50) | 1 (20) |
| Race | ||||
| White b | 38 (75) | 19 (100) | 14 (100) | 5 (100) |
| Black | 5 (10) | 0 (0) | 0 (0) | 0 (0) |
| American Indian | 1 (2) | 0 (0) | 0 (0) | 0 (0) |
| Mixed | 7 (13) | 0 (0) | 0 (0) | 0 (0) |
| Ethnicity | ||||
| Hispanic | 5 (10) | 1 (0.05) | 0 (0) | 1 (20) |
| Non‐Hispanic | 46 (90) | 18 (95) | 14 (100) | 4 (80) |
| BMI, Z‐score | 1.1 ± 1.2 (−1.4 to 2.8) | 0.8 ± 0.9 (−0.5 to 2.4) | 0.7 ± 0.9 (−0.5 to 1.9) | 1.1 ± 1.0 (0.2‐2.4) |
| Bone age, y | 15.1 ± 2.1 (10.0‐19.0) | 9.4 ± 5.0 (3.0‐19.0) | 7.6 ± 3.6 (3.0‐13.5) | 14.5 ± 5.0 (7.0‐19.0) |
| Joint Summary Score | NA | 215 ± 84 | 249 ± 67 | 118 ± 41 |
| Height Z‐score | 0.3 ± 1.0 (−2.1 to 1.9) | −2.3 ± 1.1 (−4.2 to −0.7) | −2.4 ± 1.2 (−4.2 to −0.7) | −2.0 ± 0.7 (−3.2 to −1.5) |
| Tonnis grade (highest hip) | NA | |||
| 0 | 9 (50)1 | 7 (50) | 2 (50)1 | |
| 1 | 1 (6) | 1 (7) | 0 | |
| 2 | 2 (11) | 2 (14) | 0 | |
| 3 | 5 (28) | 4 (29) | 1 (25) | |
| 5 | 1 (6) | 0 | 1 (25) | |
| Tonnis angle right | NA | 17 ± 12(1) (1‐40) | 18 ± 13 (1‐40) | 15 ± 10(1) (8‐30) |
| Tonnis angle left | NA | 20 ± 14(1) (−2 to 44) | 23 ± 14 (−2 to 44) | 9 ± 4(1) (6‐15) |
| RMI right | NA | 36 ± 22(3) (6‐80) | 36 ± 22(2) (6‐80) | 35 ± 22(1) (15‐66) |
| RMI Left | NA | 38 ± 18(2) (11‐75) | 40 ± 19(1) (11‐75) | 30 ± 7(1) (22‐38) |
| IL‐1β, pg/mL | 0.17 ± 0.04(24) | 0.34 ± 0.35 | 0.36 ± 0.41 | 0.25 ± 0.05 |
| TNF‐α, pg/mL | 1.2 ± 0.4 | 5.7 ± 2.8 | 6.3 ± 3.0 | 4.2 ± 1.2 |
| IL‐6, pg/mL | 1.7 ± 1.3 | 1.9 ± 5.6(1) | 2.3 ± 6.3 | 0.2 ± 0.2(1) |
| OCN, pg/mL | 17.9 ± 8.3 | 27.1 ± 9.8 | 30.4 ± 9.0 | 17.9 ± 4.6 |
| DPD, nmol/mmol creatinine | 9.1 ± 5.0 | 26.0 ± 15.4 | 29.5 ± 15.6 | 16.1 ± 10.1 |
| PYD, nmol/mmol creatinine | 111.1 ± 60.1 | 146.2 ± 81.4 | 159.8 ± 80.6 | 108.0 ± 79.0 |
Note: Superscript numbers indicate number of missing data. Data are presented as mean ± SD (min‐max) or N (%).
Abbreviations: BMI, body mass index; DPD, urine deoxypyridinolines; ERT, enzyme replacement therapy; HSCT, hematopoietic stem cell transplantation; IL‐1β, interleukin‐1β; IL‐6, plasma interleukin‐6; OCN, plasma osteocalcin; PYD, urine pyridinolines; RMI, Reimers migration index; TNF‐α, tumor necrosis factor‐α.
Peri‐transplant ERT only.
Includes Hispanic.
Growth failure and joint contractures were highly prevalent in the MPS I cohort, despite treatment with HSCT and/or ERT. All MPS IH participants and 60% with MPS IA participants had short stature by last follow up. All MPS I participants had joint contracture of at least one joint. Participants treated with only ERT (ie, MPS IA participants) compared to those treated with HSCT (ie, MPS IH participants) had more severe joint contractures based on the ROM summary score; however, they were significantly older than the HSCT treated cohort (Table 1; Figure 1). One MPS participant had undergone hip replacement due to severe osteoarthritis along with hip dysplasia, and 50% of MPS participants had some degree of hip osteoarthritis by Tonnis grade (Table 1).
FIGURE 1.
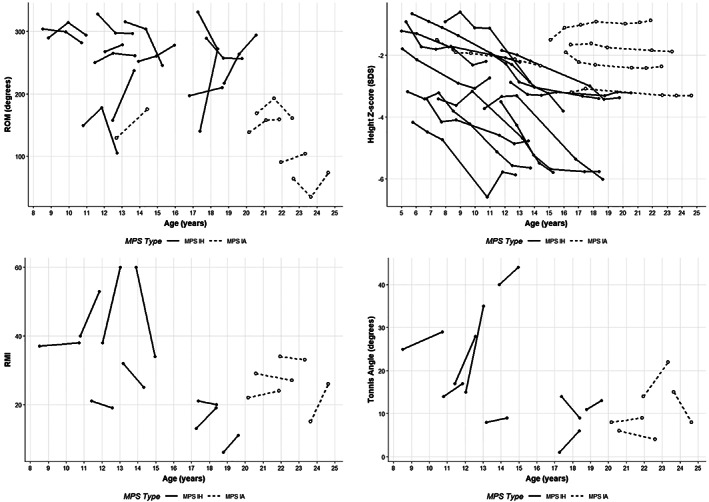
Change in joint range of motion (ROM) summary score (sum of bilateral shoulder flexion, elbow, and knee extension), height Z‐score, Reimers migration index (RMI), and Tonnis angle over time. Filled circles and solid lines = MPS IH; open circles and dotted lines = MPS IA
Joint contracture worsened with age at entry into this observational study in the MPS IA but not in the MPS IH cohort (Figure 1). Height Z‐score decreased gradually over time in MPS IH (Figure 1). Progression of hip dysplasia was present in 46% (N = 6) by RMI and 77% (N = 10) by Tonnis angle (Figure 1).
TNF‐α, IL‐1β, and DPD were higher in MPS vs controls (all P‐values <.05), and there was no significant difference in IL‐6, OCN, or PYD values between groups (Figure 2). Regression analysis in MPS I revealed that the within‐individual change in IL‐6 over the first 2 years of the study was significantly associated with the change in joint ROM over years 5 to 9 (−22°/year change in ROM for every 1 pg/mL increase in IL‐6 (95%CI −29 to −15; P < .001); this association remained significant when repeating the analysis with only MPS IH participants (P < .001) confirming that the association is not due to differences between MPS IH and MPS IA.
FIGURE 2.
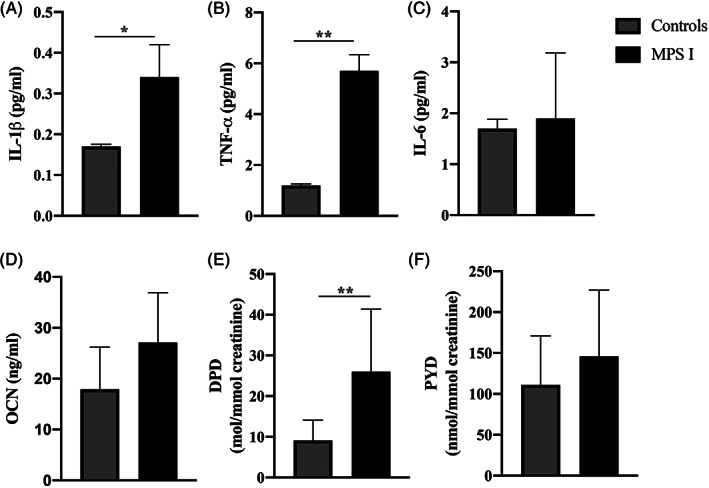
Inflammatory, A‐C, and bone, D‐F, biomarker concentrations in mucopolysaccharidosis type I (MPS I) compared to healthy controls. Mean ± SEM is shown. P ‐ values adjusted for BMI Z‐score, A‐C, or age, D‐F. *P < .05; **P < .001. DPD, urine deoxypyridinolines; IL‐1β, interleukin‐1β; IL‐6, plasma interleukin‐6; OCN, plasma osteocalcin; PYD, urine pyridinolines; TNF‐α, tumor necrosis factor‐α
There was also a within‐individual change of −0.024 SDS/year in height Z‐score for every 20 nmol/mmol creatinine/year change in urine PYD (95% CI −0.043 to −0.005; P = .016) and a −2.0°/year in RMI for every 20 nmol/mmol creatinine/year change in urine PYD (95% CI −3.8 to −0.1; P = 0.037). There were no other statistically significant associations between biomarkers and joint ROM, height Z‐score, RMI, or Tonnis angle (Table 2).
TABLE 2.
Associations between change in biomarkers over first year of study with annualized change in joint ROM over years 5 to 9, annualized height Z‐score over years 2 to 9, annualized Tonnis angle, and RMI over years 7 to 9
| Estimated annual rate of change (95% CI) | P‐value | ||
|---|---|---|---|
| Joint ROM over years 5‐9 | IL‐1β (per 0.1 pg/mL) | −11.0 (−30.6, 8.5) | .246 |
| TNF‐α (per 10 pg/mL) | −43.7 (−129.8, 42.5) | .295 | |
| IL‐6 (per 1 pg/mL) | −22.0 (−28.7, −15.3) | <.001 | |
| OCN (per 10 pg/mL) | 4.1 (−12.7, 20.8) | .614 | |
| DPD (per 20 nmol/mmol) | 18.2 (−22.9, 59.2) | .358 | |
| PYD (per 20 nmol/mmol) | 0.4 (−4.5, 5.4) | .857 | |
| Height Z‐score over years 2‐9 | IL‐1β (per 0.1 pg/mL) | −0.005 (−0.10, 0.089) | .908 |
| TNF‐α (per 10 pg/mL) | −0.06 (−0.48, 0.35) | .746 | |
| IL‐6 (per 1 pg/mL) | −0.03 (−0.10, 0.04) | .365 | |
| OCN (per 10 pg/mL) | −0.04 (−0.12, 0.04) | .291 | |
| DPD (per 20 nmol/mmol) | −0.01 (−0.16, 0.14) | .849 | |
| PYD (per 20 nmol/mmol) | −0.024 (−0.043, −0.005) | .016 | |
| Tonnis angle over years 7‐9 | IL‐1β (per 0.1 pg/mL) | −0.91 (−5.10, 3.27) | .634 |
| TNF‐α (per 10 pg/mL) | −5.03 (−23.61, 13.55) | .550 | |
| IL‐6 (per 1 pg/mL) | 0.08 (−0.70, 0.86) | .826 | |
| OCN (per 10 pg/mL) | −0.92 (−5.54, 3.69) | .662 | |
| DPD (per 20 nmol/mmol) | −5.22 (−16.14, 5.71) | .303 | |
| PYD (per 20 nmol/mmol) | 0.28 (−0.79, 1.36) | .564 | |
| RMI over years 7‐9 | IL‐1β (per 0.1 pg/mL) | −0.01 (−9.33, 9.31) | .998 |
| TNF‐α (per 10 pg/mL) | 11.47 (−23.12, 46.06) | .467 | |
| IL‐6 (per 1 pg/mL) | −0.75 (−2.27, 0.76) | .286 | |
| OCN (per 10 pg/mL) | 1.22 (−8.03, 10.47) | .772 | |
| DPD (per 20 nmol/mmol) | −8.11 (−30.03, 13.81) | .418 | |
| PYD (per 20 nmol/mmol) | −1.99 (−3.84, −0.14) | .037 |
Note: All estimates adjusted for age in months and the first recorded value of Tonnis angle or RMI for these measures.
Abbreviations: DPD, urine deoxypyridinolines; IL‐1β, interleukin‐1β; IL‐6, plasma interleukin‐6; OCN, plasma osteocalcin; PYD, urine pyridinolines; RMI, Reimers migration index; ROM, range of motion summary score (sum of bilateral shoulder flexion, elbow, and knee extension); TNF‐α, tumor necrosis factor‐α.
4. DISCUSSION
In this study, we report several important findings. First, although the presence of inflammation has been found in animal models of MPS I and treatment naïve patients with MPS IH, 24 , 25 our study identified elevated levels of biomarkers of inflammation in treated MPS I individuals compared to healthy controls. Second, we present data showing that despite current therapies joint contractures, short stature, hip dysplasia, and osteoarthritis persists, and may even worsen over time. Third, we identified PYD as a prognostic biomarker of future growth and hip dysplasia and IL‐6 of future rate of change in joint ROM. Given that joint contractures and skeletal disease progression are among the most significant, impactful contributors to persistent suffering and disability in individuals with MPS I, these findings of a link between biomarker change and future change in a clinical outcome have important implications for understanding disease pathology and potentially predicting therapeutic impact in future clinical trials.
Biomarkers obtained prior to the onset of disease that are prognostic of the disease‐related severity or predictive of treatment response are used in other, non‐MPS, populations. In the skeletal system, biomarkers of bone turnover are prognostic of the risk of fracture in adult populations 26 , 27 , 28 and osteoporosis treatment efficacy in bisphosphonate clinical trials. 29 , 30 , 31 The predictability of inflammatory and cartilage turnover markers for disease response to treatment has been evaluated in individuals with inflammatory joint disease treated with TNF‐α inhibitors as well. These studies have found associations between baseline C‐reactive protein, TNF‐α, or markers of cartilage turnover and response to treatment. 32 Additional biomarkers of inflammation and cartilage turnover (eg, cartilage oligomeric matrix protein, soluble E‐selectin, and intercellular adhesion molecule‐1) decrease with treatment and have variable predictability of disease response to therapy in other inflammatory skeletal diseases such as juvenile idiopathic arthritis, rheumatoid arthritis, and ankylosing spondylitis. 32 , 33 , 34 Similar inflammatory and cartilage biomarkers may be useful in predicting disease response to treatments over time in individuals with MPS.
In MPS, prior studies have shown a change in a biomarker in response to therapy, but not an association with future change in bone or cartilage outcomes. One study found that heparan sulfate (HS) and dermatan sulfate (DS), two glycosaminoglycans (GAGs) that accumulate in MPS I, decrease after initiation of ERT, but HS or DS did not correlate with clinical outcomes in patients with MPS I treated with ERT. 35 Another potential biomarker is heparin cofactor II‐thrombin complex, which also decreases in response to treatment with ERT and HSCT, and increases with the clinical response of enzyme infusion reaction of flushing. 36 , 37 However, we know of no studies attempting to use these markers to predict future bone or cartilage disease using clinical outcomes.
We found significant systemic inflammation in MPS I, demonstrated by the elevated IL‐1β and TNF‐α cytokine levels compared to healthy controls. Elevated IL‐1β cytokine levels are synonymous with NLRP3 inflammasome activation. 38 Our findings are consistent with preclinical reports of the importance of TNF‐α and inflammasome activation in MPS disorders. CNS and somatic disease in MPS animal models have been shown to be driven, in part, by the GAG‐induced inflammatory response through the TLR4/MyD88 signaling pathways, resulting in increased secretion of TNF‐α from cells such as microglia and macrophage, along with NLRP3 inflammasome activation. 15 , 16 , 17 , 18 , 39 , 40 , 41 , 42 , 43 , 44 , 45 , 46 , 47 , 48 , 49 , 50 , 51 This may be most consistent with the elevated IL‐1β and TNF‐α levels we found in children and adolescents with MPS I, but at present, this is speculative.
Surprisingly, although we found an association between PYD and growth, we did not find a similar association for DPD, a similar biomarker of bone and cartilage turnover, and growth. We speculate that this is because PYD originates from both bone and cartilage, but DPD is from only bone, 52 , 53 and would suggest that chondrocyte abnormalities leading to abnormal endochondral growth and increased cartilage turnover, more so than abnormal bone modeling/remodeling, play a significant role in the development of short stature in children with MPS I.
At first glance, the fact that there was not a statistically significant within‐individual change in ROM over time in our MPS I cohort may seem to detract from our findings of a significant association IL‐6 with ROM summary score. However, we believe the lack of statistically significant within‐individual change in ROM summary score may be due to the relative short duration of measurement, small sample size, and the variability of measurement inherent in the ROM outcome. Furthermore, the estimated change in ROM (−22°/y) that is associated with a 1 pg/mL change in IL‐6 is far greater than what would be considered clinically significantly. This, along with the lack of associations between IL‐1β and TNF‐α cytokine levels with joint contracture changes over time, may also simply be due to the same limitations which are common problems with studies of rare diseases.
A variety of factors can influence bone and inflammatory biomarkers. Our samples were collected in the morning after a minimum 8‐hours fast to avoid variability related to hormonal changes throughout the day and diet. Although inflammatory biomarkers are increased in adolescents with obesity, 54 we have previously shown that obesity does not influence levels of the bone biomarkers osteocalcin and bone specific alkaline phosphatase in MPS I. 21 However, bone biomarkers do change with age and pubertal stage. 55 For this reason, we adjusted our analyses using bone biomarkers for age but not BMI Z‐score. Inflammatory biomarkers are not influenced by age in our cohort of individuals with MPS I (data not shown), but as already mentioned, they are influenced by obesity. Thus, we included BMI Z‐score in the analysis of the inflammatory biomarkers.
This study is limited by the difference in mean age between our MPS and control cohorts, as well as a small sample of MPS IA participants making comparisons between the MPS I groups not statistically feasible. To address this difference in age between groups, we adjusted for age in our analyses of bone biomarkers which are known to change with age and pubertal stage.
5. CONCLUSIONS
In conclusion, inflammasome disease is a prominent feature, or perhaps the driving pathology, of skeletal system disease as demonstrated by increased levels of cytokines and is a possible therapeutic target for treatments of MPS I in addition to HSCT and ERT. Biomarkers that are associated with long‐term clinical outcomes, such as we report here, are needed to most effectively and efficiently test potential therapies due to the rarity of the disease and relatively slow rate of disease progression. We have identified two biomarkers that were prognostic of long‐term change in growth, joint contractures, and hip dysplasia. Once validated as predictive of long‐term response to a therapy aimed at treating the joint and skeletal manifestations of MPS I, these biomarkers could be used for predicting outcomes in both clinical practice and clinical trials.
CONFLICT OF INTERESTS
Dr T. C. L. receives research support from Sanofi‐Genzyme. Dr J. B. E. has received honoraria, consulting fees, and/or research support from ArmaGen, JCR pharmaceuticals, Orchard Therapeutics, Bluebird Bio, Shire/Takeda, and Sanofi‐Genzyme. Ms R. L. F. declares that she has no conflict of interest. Dr K. D. R. declares that he has no conflict of interest. Dr E. B. F. is a consultant for BioMarin Pharmaceuticals and Ascendis. Dr B. S. M. is a consultant for AbbVie, Ascendis, Ferring, Novo Nordisk, Pfizer, Sandoz, and Versartis and has received research support from Alexion, Ascendis, Endo Pharmaceuticals, Genentech, Genzyme, Novo Nordisk, Opko, Sandoz, Sangamo, Shire, Tolmar, and Versartis. Dr K. K. W. is a consultant for BioMarin Pharmaceuticals, has received honoraria from BioMarin Pharmaceuticals, Sanofi‐Genzyme, has grant funding from BioMarin Pharmaceuticals and Ultragenyx, and receives royalties from UpToDate.com. Dr C. B. W. is a consultant for Sanofi‐Genzyme. Dr P. J. O. receives research and clinical trial support from Sanofi‐Genzyme, Horizon, Magenta, and Bluebird Bio. Dr L. E. P. is a speaker and consultant for Sanofi‐Genzyme, has received research support from Sanofi‐Genzyme, Shire, BioMarin, and Pfizer‐Therachon, and is a consultant for Sangamo, Immusoft, Pfizer‐Therachon, and BioMarin.
AUTHOR CONTRIBUTIONS
Troy C. Lund and Terence M. Doherty: Wrote the first draft of the manuscript and are thus co‐first authors. Rebecca L. Freese and Kyle D. Rudser: Performed statistical analysis. Ellen B. Fung, Bradley S. Miller, Chester B. Whitley, and Lynda E. Polgreen: Designed the research studies. Ellen B. Fung, Bradley S. Miller, and Lynda E. Polgreen: Conducted experiments and acquired data. Klane K. White: Interpreted radiographs. Finally, all authors provided critical feedback and helped shape the analysis and manuscript.
ETHICS APPROVAL AND PATIENT CONSENT
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from all patients for being included in the study.
ACKNOWLEDGMENTS
The authors thank the patients and their families for their participation in these studies, and the study personnel at participating centers, including Jane Kennedy, Eva Villa‐Lopez, Angel Zozobrado, and Ashley Weisenburger. National Institute of Arthritis and Musculoskeletal and Skin Diseases K23AR057789; National Institute of Neurological Disorders and Stroke U54NS065768; National Center for Advancing Translational Sciences of the National Institutes of Health (NIH) UL1TR001881, UL1TR000114, UL1TR002484, and UL1RR024131; Sanofi‐Genzyme 21493‐01. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH National Center for Advancing Translational Sciences.
Lund TC, Doherty TM, Eisengart JB, et al. Biomarkers for prediction of skeletal disease progression in mucopolysaccharidosis type I. JIMD Reports. 2021;58:89–99. 10.1002/jmd2.12190
Troy C. Lund and Terence M. Doherty are co‐first authors.
Communicating Editor: Carla E. Hollak
Funding information National Center for Advancing Translational Sciences, Grant/Award Numbers: UL1TR001881, UL1TR000114, UL1TR002484, UL1RR024131; National Institute of Arthritis and Musculoskeletal and Skin Diseases, Grant/Award Number: K23AR057789; National Institute of Neurological Disorders and Stroke, Grant/Award Number: U54NS065768; Sanofi Genzyme, Grant/Award Number: 21493‐01
REFERENCES
- 1. Pastores GM, Arn P, Beck M, et al. The MPS I registry: design, methodology, and early findings of a global disease registry for monitoring patients with mucopolysaccharidosis type I. Mol Genet Metab. 2007;91(1):37‐47. [DOI] [PubMed] [Google Scholar]
- 2. Whitley CB, Belani KG, Chang PN, et al. Long‐term outcome of Hurler syndrome following bone marrow transplantation. Am J Med Genet. 1993;46(2):209‐218. 10.1002/ajmg.1320460222. [DOI] [PubMed] [Google Scholar]
- 3. Aldenhoven M, Wynn RF, Orchard PJ, et al. Long‐term outcome of Hurler syndrome patients after hematopoietic cell transplantation: an international multicenter study. Blood. 2015;125(13):2164‐2172. 10.1182/blood-2014-11-608075. [DOI] [PubMed] [Google Scholar]
- 4. Bjoraker KJ, Delaney K, Peters C, Krivit W, Shapiro EG. Long‐term outcomes of adaptive functions for children with mucopolysaccharidosis I (Hurler syndrome) treated with hematopoietic stem cell transplantation. J Dev Behav Pediatr. 2006;27(4):290‐296. [DOI] [PubMed] [Google Scholar]
- 5. Eisengart JB, Rudser KD, Tolar J, et al. Enzyme replacement is associated with better cognitive outcomes after transplant in Hurler syndrome. J Pediatr. 2013;162(2):375‐380.e1. 10.1016/j.jpeds.2012.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Eisengart JB, Rudser KD, Xue Y, et al. Long‐term outcomes of systemic therapies for Hurler syndrome: an international multicenter comparison. Genet Med. 2018;20(11):1423‐1429. 10.1038/gim.2018.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Eisengart JB, Jarnes J, Ahmed A, et al. Long‐term cognitive and somatic outcomes of enzyme replacement therapy in untransplanted Hurler syndrome. Mol Genet Metab Rep. 2017;13:64‐68. 10.1016/j.ymgmr.2017.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Clarke LA, Wraith JE, Beck M, et al. Long‐term efficacy and safety of laronidase in the treatment of mucopolysaccharidosis I. Pediatrics. 2009;123(1):229‐240. [DOI] [PubMed] [Google Scholar]
- 9. Kakkis ED, Muenzer J, Tiller GE, et al. Enzyme‐replacement therapy in mucopolysaccharidosis I. N Engl J Med. 2001;344(3):182‐188. [DOI] [PubMed] [Google Scholar]
- 10. Hobbs JR, Hugh‐Jones K, Barrett AJ, et al. Reversal of clinical features of Hurler's disease and biochemical improvement after treatment by bone‐marrow transplantation. Lancet. 1981;2(8249):709‐712. [DOI] [PubMed] [Google Scholar]
- 11. Polgreen LE, Tolar J, Plog M, et al. Growth and endocrine function in patients with Hurler syndrome after hematopoietic stem cell transplantation. Bone Marrow Transpl. 2008;41(12):1005‐1011. [DOI] [PubMed] [Google Scholar]
- 12. Polgreen LE, Vehe RK, Rudser K, et al. Elevated TNF‐α is associated with pain and physical disability in mucopolysaccharidosis types I, II, and VI. Mol Genet Metab. 2016;117(4):427‐430. 10.1016/j.ymgme.2016.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Langereis EJ, den Os MM, Breen C, et al. Progression of hip dysplasia in mucopolysaccharidosis type I Hurler after successful hematopoietic stem cell transplantation. J Bone Joint Surg Am. 2016;98(5):386‐395. 10.2106/JBJS.O.00601. [DOI] [PubMed] [Google Scholar]
- 14. Thawrani DP, Walker K, Polgreen LE, Tolar J, Orchard PJ. Hip dysplasia in patients with Hurler syndrome (mucopolysaccharidosis type 1H). J Pediatr Orthop. 2013;33(6):635‐643. 10.1097/BPO.0b013e31829abfe0. [DOI] [PubMed] [Google Scholar]
- 15. Simonaro CM, Ge Y, Eliyahu E, He X, Jepsen KJ, Schuchman EH. Involvement of the toll‐like receptor 4 pathway and use of TNF‐α antagonists for treatment of the mucopolysaccharidoses. Proc Natl Acad Sci U S A. 2010;107(1):222‐227. 10.1073/pnas.0912937107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Simonaro CM, D'Angelo M, Haskins ME, Schuchman EH. Joint and bone disease in mucopolysaccharidoses VI and VII: identification of new therapeutic targets and biomarkers using animal models. Pediatr Res. 2005;57(5 Pt 1):701‐707. [DOI] [PubMed] [Google Scholar]
- 17. Eliyahu E, Wolfson T, Ge Y, Jepsen KJ, Schuchman EH, Simonaro CM. Anti‐TNF‐alpha therapy enhances the effects of enzyme replacement therapy in rats with mucopolysaccharidosis type VI. PLoS One. 2011;6(8):e22447. 10.1371/journal.pone.0022447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Simonaro CM, Haskins ME, Schuchman EH. Articular chondrocytes from animals with a dermatan sulfate storage disease undergo a high rate of apoptosis and release nitric oxide and inflammatory cytokines: a possible mechanism underlying degenerative joint disease in the mucopolysaccharidoses. Lab Investig J Tech Methods Pathol. 2001;81(9):1319‐1328. [DOI] [PubMed] [Google Scholar]
- 19. Schuchman EH, Ge Y, Lai A, et al. Pentosan polysulfate: a novel therapy for the mucopolysaccharidoses. PLoS One. 2013;8(1):e54459. 10.1371/journal.pone.0054459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. de Oliveira PG, Baldo G, Mayer FQ, et al. Characterization of joint disease in mucopolysaccharidosis type I mice. Int J Exp Pathol. 2013;94(5):305‐311. 10.1111/iep.12033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chahla SE, Frohnert BI, Thomas W, Kelly AS, Nathan BM, Polgreen LE. Higher daily physical activity is associated with higher osteocalcin levels in adolescents. Prev Med Rep. 2015;2:568‐571. 10.1016/j.pmedr.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang L, Grey V. Pediatric reference intervals for bone markers. Clin Biochem. 2006;39(6):561‐568. 10.1016/j.clinbiochem.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 23. Stevenson DA, Rudser K, Kunin‐Batson A, et al. Biomarkers of bone remodeling in children with mucopolysaccharidosis types I, II, and VI. J Pediatr Rehabil Med. 2014;7(2):159‐165. 10.3233/PRM-140285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Raymond GV, Pasquali M, Polgreen LE, et al. Elevated cerebral spinal fluid biomarkers in children with mucopolysaccharidosis I‐H. Sci Rep. 2016;6:38305. 10.1038/srep38305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Simonaro CM, Tomatsu S, Sikora T, et al. Pentosan polysulfate: oral versus subcutaneous injection in mucopolysaccharidosis type I dogs. PLoS One. 2016;11(4):e0153136. 10.1371/journal.pone.0153136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Miller PD. Bone density and markers of bone turnover in predicting fracture risk and how changes in these measures predict fracture risk reduction. Curr Osteoporos Rep. 2005;3(3):103‐110. [DOI] [PubMed] [Google Scholar]
- 27. Chapurlat RD, Garnero P, Bréart G, Meunier PJ, Delmas PD. Serum type I collagen breakdown product (serum CTX) predicts hip fracture risk in elderly women: the EPIDOS study. Bone. 2000;27(2):283‐286. [DOI] [PubMed] [Google Scholar]
- 28. Garnero P, Hausherr E, Chapuy MC, et al. Markers of bone resorption predict hip fracture in elderly women: the EPIDOS prospective study. J Bone Miner Res. 1996;11(10):1531‐1538. 10.1002/jbmr.5650111021. [DOI] [PubMed] [Google Scholar]
- 29. Bonnick SL, Shulman L. Monitoring osteoporosis therapy: bone mineral density, bone turnover markers, or both? Am J Med. 2006;119(4 suppl 1):S25‐S31. [DOI] [PubMed] [Google Scholar]
- 30. Eastell R, Barton I, Hannon RA, Chines A, Garnero P, Delmas PD. Relationship of early changes in bone resorption to the reduction in fracture risk with risedronate. J Bone Miner Res. 2003;18(6):1051‐1056. 10.1359/jbmr.2003.18.6.1051. [DOI] [PubMed] [Google Scholar]
- 31. Reginster J‐Y, Adami S, Lakatos P, et al. Efficacy and tolerability of once‐monthly oral ibandronate in postmenopausal osteoporosis: 2 year results from the MOBILE study. Ann Rheum Dis. 2006;65(5):654‐661. 10.1136/ard.2005.044958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Emery P, Dörner T. Optimising treatment in rheumatoid arthritis: a review of potential biological markers of response. Ann Rheum Dis. 2011;70(12):2063‐2070. 10.1136/ard.2010.148015. [DOI] [PubMed] [Google Scholar]
- 33. Horneff G, Fitter S, Foeldvari I, et al. Double‐blind, placebo‐controlled randomized trial with adalimumab for treatment of juvenile onset ankylosing spondylitis (JoAS): significant short term improvement. Arthritis Res Ther. 2012;14(5):R230. 10.1186/ar4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. den Broeder AA, Joosten LAB, Saxne T, et al. Long term anti‐tumour necrosis factor alpha monotherapy in rheumatoid arthritis: effect on radiological course and prognostic value of markers of cartilage turnover and endothelial activation. Ann Rheum Dis. 2002;61(4):311‐318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Langereis EJ, van Vlies N, Church HJ, et al. Biomarker responses correlate with antibody status in mucopolysaccharidosis type I patients on long‐term enzyme replacement therapy. Mol Genet Metab. 2015;114(2):129‐137. 10.1016/j.ymgme.2014.10.012. [DOI] [PubMed] [Google Scholar]
- 36. Clarke LA, Hemmelgarn H, Colobong K, et al. Longitudinal observations of serum heparin cofactor II‐thrombin complex in treated mucopolysaccharidosis I and II patients. J Inherit Metab Dis. 2012;35(2):355‐362. 10.1007/s10545-011-9369-6. [DOI] [PubMed] [Google Scholar]
- 37. Langford‐Smith KJ, Mercer J, Petty J, et al. Heparin cofactor II‐thrombin complex and dermatan sulphate:chondroitin sulphate ratio are biomarkers of short‐ and long‐term treatment effects in mucopolysaccharide diseases. J Inherit Metab Dis. 2011;34(2):499‐508. 10.1007/s10545-010-9254-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jo E‐K, Kim JK, Shin D‐M, Sasakawa C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol. 2016;13(2):148‐159. 10.1038/cmi.2015.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wilkinson FL, Holley RJ, Langford‐Smith KJ, et al. Neuropathology in mouse models of mucopolysaccharidosis type I, IIIA and IIIB. PLoS One. 2012;7(4):e35787. 10.1371/journal.pone.0035787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ausseil J, Desmaris N, Bigou S, et al. Early neurodegeneration progresses independently of microglial activation by heparan sulfate in the brain of mucopolysaccharidosis IIIB mice. PLoS One. 2008;3(5):e2296. 10.1371/journal.pone.0002296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Martins C, Hůlková H, Dridi L, et al. Neuroinflammation, mitochondrial defects and neurodegeneration in mucopolysaccharidosis III type C mouse model. Brain J Neurol. 2015;138(Pt 2):336‐355. 10.1093/brain/awu355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Arfi A, Richard M, Gandolphe C, Bonnefont‐Rousselot D, Thérond P, Scherman D. Neuroinflammatory and oxidative stress phenomena in MPS IIIA mouse model: the positive effect of long‐term aspirin treatment. Mol Genet Metab. 2011;103(1):18‐25. 10.1016/j.ymgme.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 43. Holley RJ, Ellison SM, Fil D, et al. Macrophage enzyme and reduced inflammation drive brain correction of mucopolysaccharidosis IIIB by stem cell gene therapy. Brain J Neurol. 2018;141(1):99‐116. 10.1093/brain/awx311. [DOI] [PubMed] [Google Scholar]
- 44. Simonaro CM. Cartilage and chondrocyte pathology in the mucopolysaccharidoses: the role of glycosaminoglycan‐mediated inflammation. J Pediatr Rehabil Med. 2010;3(2):85‐88. 10.3233/PRM-2010-0120. [DOI] [PubMed] [Google Scholar]
- 45. Simonaro CM, D'Angelo M, He X, et al. Mechanism of glycosaminoglycan‐mediated bone and joint disease: implications for the mucopolysaccharidoses and other connective tissue diseases. Am J Pathol. 2008;172(1):112‐122. 10.2353/ajpath.2008.070564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Polgreen LE, Vehe RK, Rudser K, et al. TNF‐alpha levels are increased in children with mucopolysaccharidosis types I, II and VI treated with ERT. Mol Genet Metab. 2014;111(2):S86‐S87. [Google Scholar]
- 47. Polgreen LE, Menk J, Rudser K, et al. Exploring surrogate biomarkers of skeletal and joint disease progression in mucopolysaccharidosis type I. Mol Genet Metab. 2019;126(2):S118‐S119. 10.1016/j.ymgme.2018.12.302. [DOI] [Google Scholar]
- 48. Bigger BW, Holley R, Ellison S, et al. Interleukin‐1 plays a central role in behaviour abnormalities in mucopolysaccharidosis type III (MPS III). Mol Genet Metab. 2018;123(2):S24‐S25. 10.1016/j.ymgme.2017.12.038. [DOI] [Google Scholar]
- 49. Parker H, Bigger BW. The role of innate immunity in mucopolysaccharide diseases. J Neurochem. 2019;148(5):639‐651. 10.1111/jnc.14632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Parker H. Identifying the role of IL‐1 in neuroinflammation of MPSIIIA. 2018. https://www.research.manchester.ac.uk/portal/files/93531836/FULL_TEXT.PDF
- 51. Metcalf JA, Zhang Y, Hilton MJ, Long F, Ponder KP. Mechanism of shortened bones in mucopolysaccharidosis VII. Mol Genet Metab. 2009;97(3):202‐211. 10.1016/j.ymgme.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Szulc P, Seeman E, Delmas PD. Biochemical measurements of bone turnover in children and adolescents. Osteoporos Int. 2000;11(4):281‐294. [DOI] [PubMed] [Google Scholar]
- 53. Civitelli R, Armamento‐Villareal R, Napoli N. Bone turnover markers: understanding their value in clinical trials and clinical practice. Osteoporos Int. 2009;20(6):843‐851. 10.1007/s00198-009-0838-9. [DOI] [PubMed] [Google Scholar]
- 54. Al‐Shorman A, Al‐Domi H, Faqih A. Markers of subclinical atherosclerosis in schoolchildren with obesity and metabolic syndrome. Swiss Med Wkly. 2017;147:w14446. 10.4414/smw.2017.14446. [DOI] [PubMed] [Google Scholar]
- 55. Fares JE, Choucair M, Nabulsi M, Salamoun M, Shahine CH, Fuleihan GE‐H. Effect of gender, puberty, and vitamin D status on biochemical markers of bone remodedeling. Bone. 2003;33(2):242‐247. 10.1016/s8756-3282(03)00160-1. [DOI] [PubMed] [Google Scholar]