

Abstract
A novel series of anthracenyl-isoxazole amide (AIM) antitumor agents containing N-heterocycles in the 10 position (N-het) were synthesized using palladium cross-coupling. The unique steric environment of the N-het-AIMs required individual optimization in each case. Lanthanide mediated double activation was used to couple the dimethylamino pyrrole moiety, required for antitumor action. Robust antitumor activity was observed against breast and brain cancer cell lines. The compounds were docked with the c-myc oncogene promoter sequence, which adopts a G4 quadruplex DNA conformation, and represents the working hypothesis for biological action. The N-het-AIMs have useful fluorescence properties, allowing for observation of their distribution within tumor cells.
1. Introduction
G-quadruplexes (G4s) are comprised of multiple planar decks or G-quartets which result from Hoogsteen base-pairing between four guanines. The cavity formed in the center of the quadruplex is usually occupied by cations such as K+ or Na+; these ions aid in stabilizing the G4 structure. G4s are formed from guanine rich sequences of DNA or RNA including sequences found in telomeres and oncogene promotor regions. It is thought G4s play a role in regulating transcription and their stabilization has been shown to suppress gene-expression. Thus, G-quadruplexes represent a promising target for small molecule anti-tumor agents. Small molecules which bind G-quadruplex DNA are of continuing interest for potential applications as antitumor agents.1-3 Analogous to B-DNA albeit on a larger molecular scale, small molecules have been reported which interact with Quadruplex DNA by stacking with the G-deck (Telomestatin),4 binding in the groove (Distamycin),5 and external stacking with extrahelical bases and electrostatic interactions with the sugar phosphate backbone (TMPyP4)6 (Fig. 1).7 More recently, research groups have endeavored to develop and understand the molecular basis for selectivity among the numerous putative quadruplex forming sequences found in the human genome.8-10 We have previously reported quadruplex binding anthracenyl isoxazole amides (AIMs) with IC50 values in the sub-micromolar range against a human glioma cell line (SNB-19).11 With the attainment of potent activity, we sought to address potential pharmacokinetic issues, namely bioavailability (see Fig 2).
Fig. 1.
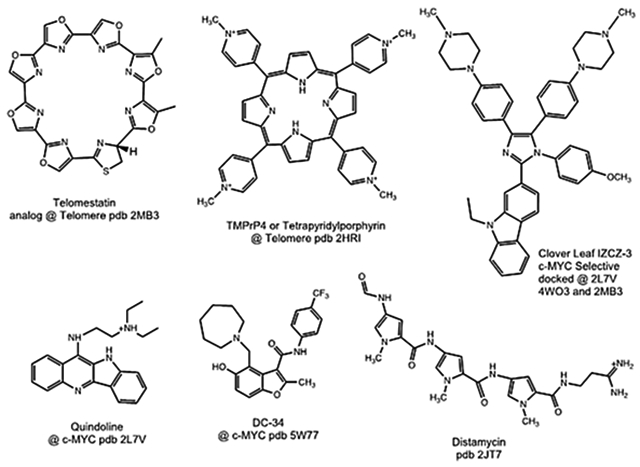
Molecules which bind quadruplex DNA, and PDB accession numbers.
Fig. 2.
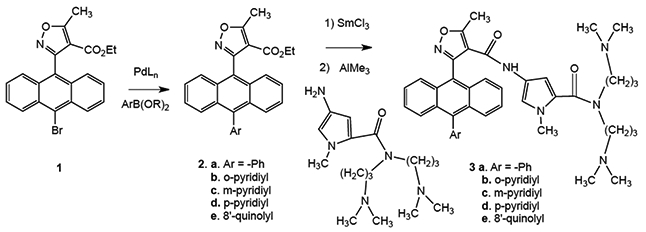
Synthesis of N-heterocyclic AIMs used in the study.
2. Chemistry
2.1. Boronate cross-coupling
Containing an anthracene moiety, the AIMs are quite lipophilic; however, introducing a pyridine or quinoline into the C10 position of the anthracene would be expected to reduce the clogD significantly. The C10 position of anthracene ring systems is a notoriously difficult partner in Suzuki couplings owing to its steric bulk.12 Our previous AIM cross-coupling methodology proved inadequate for the nitrogen heterocycles, in some cases giving 0% yield (Table 1, Entries 1 and 2).12 Through a systematic study of reaction conditions, optimized conditions were developed, only a select number of experiments are summarized here from a total effort of almost one hundred attempts (Table 1). The rate of oxidative addition and transmetalation is greatly reduced for substrates containing di-ortho substituents and para electron withdrawing groups. These difficulties can be addressed by increasing L1Pd(0) concentration or by using a strong base, such as NaOH. Boronic acids transmetalate slowly owing to the C-B bond being relatively inert which slows interaction with the Pd center. The base’s role is to convert the stable boronic acid into a more reactive boronate species which transmetalates rapidly. The stronger the base, the higher the boronate concentration, however the use of strong bases can be problematic in the presence of base-sensitive functional groups, such as the ester in AIM precursors (Fig. 3).
Table 1.
Palladium catalyzed cross coupling preparation of N-heterocyclic isoxazole anthryl esters, 2a–2e.
| Entry | Palladium reagent | Boron reagent (Product) |
Phosphine | Base | Temperature (°C) solvent | Time | Yield |
|---|---|---|---|---|---|---|---|
| 1 | Pd2(dba)3 | 4-PyrB(TMP) (2d) | (t-Bu)3P | KF | RT THF | 5 d | 0 |
| 2 | PdL4 | 4-PyrB(OH)2 (2d) | Ph3P | KF | RT | 72 h | 0 |
| 3 | PdL4 | 4-PyrB(OH)2 (2d) | Ph3P | KF | 40 | 24 h | 10 |
| 4 | XPhos Pd G3 | 4-PyrB(OH)2 (2d) | XPhos | Na2CO3 (aq) | 90 | 24 h | 40 |
| 5 | PdCl2(dppf) | 4-PyrB(TMP) (2d) | Dppf | KOAc | CH3CN reflux | 3 d | 50 |
| 6 | XPhos Pd G3 | 4-PyrB(OH)2 (2d) | XPhos | Na2CO3 (aq) | 120, microwave | 20 m | 68 |
| 7 | PdL4 | 3-PyrB(OH)2 (2c) | Ph3P | Na2CO3 | 120, microwave | 1 h | 81 |
| 8 | XPhos Pd G3 CuOAc2 | 2-PyrMIDA (2b) | XPhos | K3PO4 | 100 | 24 h | 20 |
| 9 | XPhos Pd G3 | 8′-QuinB(OH)2 (2e) | XPhos | Na2CO3 | 40 | 72 h | 57 |
Abbreviations: dba dibenzylidene acetone, dppf bisdiphenylphosphineferrocene, TMP tetramethylpicolinate, RT room temperature.
Fig. 3.
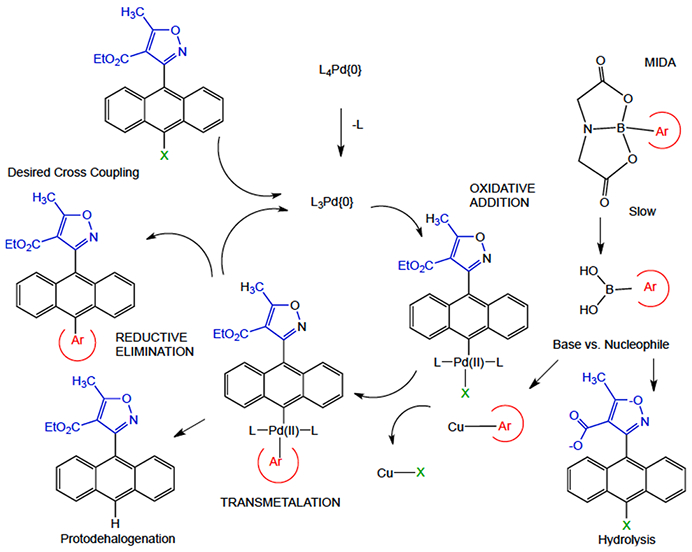
Role of MIDA in the boronate in cross coupling.
It has been shown that monoligated Pd species afford higher catalyst turnover rates in comparison to L2Pd(0). Buchwald’s group has developed a library of phosphine ligands which preferentiate monoligated Pd species.13 2-pyridyl boronic acids require rapid reaction rates as they are prone to protodeborylation. It is possible to circumvent this problem by employing 2-pyridyl N-methyl-iminodiacetic acid (MIDA) boronate developed by Burke’s group.14 The MIDA acts as a protecting group and allows for base promoted slow-release of the boronic acid reactive species (Fig. 3) (see Fig 4).
Fig. 4.

Sc-xrd characterization of the N-heterocyclic isoxazole esters A. m-pyridyl B. p-pyridyl C. 8-quinolinyl.
For the quinoline, m-pyridyl, and p-pyridyl boronic acids utilization of XPhos Pd G3 precatalyst in a biphasic system proved optimal. The 2-pyridyl derivative proved exceptionally difficult to prepare. This was due to the lability of the 2-pyridyl boron bond as well as previously mentioned properties of the AIM structure. Modest success was finally achieved through the implementation of the MIDA boronate method. The MIDA group allows for release of the 2-pyridyl boronic acid at a rate slower than the catalyst turnover. Typically, boronic acids are converted to the boronate species in situ which undergo transmetalation with Pd in the catalytic cycle. The 2-pyridyl boronic acid is too unstable, even under slow release conditions, to be a viable coupling partner. For this reason, the MIDA method includes the addition of Cu(II) acetate and diethanolamine (DEA). The copper combines with DEA forming Cu (DEA)2 and this complex transmetalates with the boronic acid to produce a 2-pyridyl copper intermediate species. The 2-pyridyl copper species then enters the catalytic cycle through transmetalation with the Pd (Fig. 3). In addition, under the MIDA conditions, ring opening of the isoxazole was evidenced, which contributed to the lower yield, however, sufficient material was obtained to proceed with our SAR study.15
2.2. Lanthanide catalyzed double activation
The N-het-isoxazole esters 2 were then coupled with the bis-dimethylamino pyrrole using our previously described lanthanide double activation (Fig. 2), which is necessitated in this case because of the side reactions expected with basic N-heterocycles under usual Shotten-Bauman amide formation.16,17 While the conversions of coupling products 3 were modest, the balance of material was recovered ester.
2.3. Small molecule crystallography
The AIMs previously reported have a characteristic topology wherein the 9-anthryl moiety enforces an approximate orthogonal relationship with the isoxazole. The addition of the N-heterocycles in the 10 position gives rise to the possibility of diastereomers with respect to the two axes. As a starting point to understand the potential solution dynamics of AIMs, we examined the single crystal x-ray diffractometry of three of the intermediate isoxazole esters. The experimental parameters for the crystals, data collection and refinement are shown in Table 2. The accuracy of the structures was quite good as reflected in R values of 0.040 to 0.047 (Table 2). The dihedral angle between the mean planes described by the isoxazole and anthracene were more shallow than most of those previously observed, but still closer to orthogonal than conjugated or planar. The p-pyridyl exhibits the smallest isoxazole to anthracene dihedral to date at 68°, compared to the previous range of 72–88°. In the two cases where diastereomers were possible with respect to the relationship between the heterocyclic rings, in both cases they were observed as a single cis- diastereomer in the solid state. The dihedral angle between the N-heterocyclic ring and the anthracene is within a few degrees of the corresponding dihedral with the isoxazole, and thus the relationship of the heterocycles is that the heteroatoms roughly point in the same direction (dihedral angle of approximately 5–25°).
Table 2.
Crystal Data for N-heterocyclic isoxazole esters 2c-e. Crystals 2c-e were mono-clinic, experiments were carried out at 100 K. The BA was triclinic and run at 293 K.
| Compound | m-Pyr 2c | p-Pyr 2d | 8-Quin 2e |
|---|---|---|---|
| Crystal Data | |||
| Empirical Formula | C26H20N2O3 | C26H20N2O3 | C30H22N2O3 |
| Formula Weight | 408.44 | 408.44 | 458.49 |
| Space group | P21/c | C2/c | P21/c |
| a/Å | 8.2571 (7) | 30.3595 (6) | 11.1995(12) |
| b/Å | 11.5857 (9) | 7.2167 (1) | 14.4696(16) |
| c/Å | 20.9084 (17) | 22.2409 (5) | 14.3170(16) |
| α/° | 90 | 90 | 90 |
| β/° | 90.242 (3) | 125.2256 (9) | 101.662(4) |
| γ/° | 90 | 90 | 90 |
| Volume/Å3 | 2000.2 (3) | 3980.59 (14) | 2272.2(4) |
| Z | 4 | 8 | 4 |
| Dx mg/m3 | 1.356 | 1.363 | 1.340 |
| μ/mm1 | 0.72 | 0.09 | 0.087 |
| F(000) | 856 | 1712 | 960.0 |
| Crystal appearance | Yellow Cube | Yellow Prism | Colorless Prism |
| Crystal size/mm3 | 0.4 × 0.25 × 0.2 | 0.49 × 0.24 × 0.16 | 0.24 × 0.16 × 0.08 |
| Data Collection | |||
| Radiation (λ) | Cu Kα (1.54178) | Mo Kα (0.71073) | Mo Kα (0.71073) |
| Θ /° | 4.2–72.3 | 2.7–28.7 | 2.9 – 28.7 |
| Index ranges | −10 ≤ h ≤ 10 | −40 ≤ h ≤ 40 | −15 ≤ h ≤ 15 |
| −14 ≤ k ≤ 13 | −9 ≤ k ≤ 9 | −19 ≤ k ≤ 19 | |
| −24 ≤ l ≤ 25 | −30 ≤ l ≤ 29 | −19 ≤ l ≤ 19 | |
| Measured Reflections | 36,456 | 25,023 | 105,583 |
| Independent reflections | 3955 | 5141 | 5905 |
| Reflections with I > 2σ(I) | 3627 | 3707 | 4752 |
| Refinement | |||
| Parameters | 282 | 282 | 318 |
| Goodness-of-fit on F2 | 1.08 | 1.02 | 1.031 |
| R[F2 > 2σ(F2)] | 0.040 | 0.043 | 0.047 |
| wR(F2) | 0.105 | 0.106 | 0.1200 |
| Largest diff. peak/hole / e Å−3 | 0.31/−0.30 | 0.31/−0.21 | 0.47/−0.19 |
| Calculated features | |||
| Isoxazole/Anthracene dihedral/° | 77.25 | 68.09 | 73.74 |
| Anthracene/N-heterocycle dihedral/° | 79.21 | 74.46 | 72.05 |
| Isoxazole/N-heterocycle angle/° | 25.28 | 6.81 | 4.76 |
During chromatographic separation, the stereoisomers were observed to elute and were separable and isolable as disastereomers, however, on standing in solution at room temperature the diastereomers equilibrated to 50:50 mixtures over several days. This allows us to estimate the rotation barrier at <26 kcal/mol. Previously we had examined rotational barriers of isoxazole esters and AIMs using quantum mechanical methods at the B3LYP/6-31G* level of theory and compared those to experimental barriers determined on chiral amides. The barrier predictions were found to be in good agreement with computation, with the isoxazolyl 4-carbonyl substituent (Ester verses (S)-sec-butyl amide verses pyrrole amide within 1.2 kcal/mole) having only a small effect on the anthryl rotational barrier, with the predicted ΔG‡ by computation of 23.2 kcal/mole for the pyrrole amide, which is approximately the barrier at which the half-life of the rotation would be observable. This is also consistent with the equilibration of the AIM under physiological temperature to accommodate the biomolecular target.
Another stereochemical feature observed in the solid state was the conformation of the ethyl ester. All of the ester dihedral angles are in the range of conjugation with the isoxazole, with the m-pyridyl having the highest dihedral angle at 19.32°, and the quinolyl the most planar at 5.13°. The ethyl group of the ethyl ester was observed in the m- and p-pyridyl examples to be oriented endo- over the anthracene, but in the quinoline case facing away from that ring in an exo- relationship. As expected for a solution average populated by the endo- conformer, significant anisotropy is consistently observed for the ethyl group in the proton NMR, and this analogous effect gives rise to observed anisotropy during interaction with oligonucleotides (Figure 6).
Fig. 6.
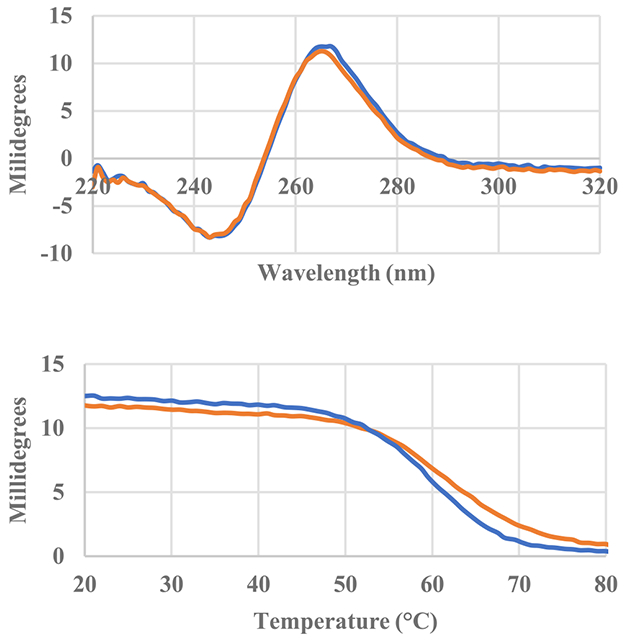
Circular dichroism spectra of Pu22 oligonucleotide with and without two equivalents of AIM compound in solution. Pu22 only (blue), 10-Quin (orange). Circular dichroism thermal melting curves of Pu22 oligonucleotide taken at 265 nm with and without two equivalents of the AIM compound.
3. Computational chemistry
3.1. Computational docking and scoring prediction
The N-heterocyclic AIMs were initially docked with coordinates from solution studies (PDBID: 2L7V), using the Discovery Studio platform v4.0. The oligonucleotide structure was typed with the CHARMm forcefield and energy minimized with the smart minimizer protocol within Discovery Studio using the Generalized-Born with simple switching implicit solvent model to a root mean square gradient (RMS) convergence <0.001 kcal/mol prior to use in the docking studies.18,19 Docking was performed using the flexible docking protocol, which allows for flexibility in both the ligand and the binding site residues.20,21 The final ranking of the docked poses was performed via consensus scoring, combining the predicted binding energy with the Jain, PLP2, and Ludi3 scoring functions.22,23 The poses were scored using Cdocker Interaction Energies, a proprietary weighting of the energy of ligand and protein, subtracted from the complex energy. The initial training set of 12 discreet known AIMs, and the four 10-N-heterocycles was examined using Discovery Studio v4.0. During the Ligand Preparation step imidate tautomers were generated at the secondary amide (listed in Supplementary Material table as structure 2 of each corresponding AIM). Two conclusions stood out: (1) the quindoline ligands studied with 2L7V had their best –Cdocker scores of 47.1 (Site 1) and 42.2 (Site 2) compared to best poses in the range of 75.5 to 78.5 for the N-heterocyclic AIM series,18 and (2) the best N-heterocycle score was observed for the 10- quinoline AIM (Quin-AIM). Furthermore, several of the top poses for the Quin-AIM possessed similar orientations with respect to Pu22 in site 1, that is the dimethylamino tails oriented toward the single strand 5′-TGA- end of the oligonucleotide, which tends to increase confidence in the veracity of the pose. The top three scoring poses for the Quin-AIM at site 1 in our initial survey of the training set is shown in supplementary data, with the important intermolecular interactions summarized for the top score in a LigPlot. Similarly, the top 6 of 10 scoring poses at site 2 exhibited similar binding modes.
Given the observation that the Quin-AIM possessed the highest cytotoxicity of the series, we then further examined the docking of the Quin-AIM using Discovery Studio v2017, which allowed us to begin with the ligand in the cis- diastereomer (akin to the x-ray of the intermediate isoxazole ester), and allow the ligand to accommodate in its optimal binding to the receptor by rotating at both anthracene to heterocycle rings junctures. The top 7 scores obtained robust Cdocker interaction energies of over 80 kcal/mole, with the highest at 83.72 (Fig. 5A). These grouped into two sets of poses. The highest scoring 3 (out of 7) anchor the double tail (DT) towards the extrahelical G5 and point the isoxazole C5 towards an aromatic and largely lipophilic gulley (Fig. 5A), while the other 4 of 7 orient in an opposite sense, and points the C3 of the anthracene into this same gulley.
Fig. 5.
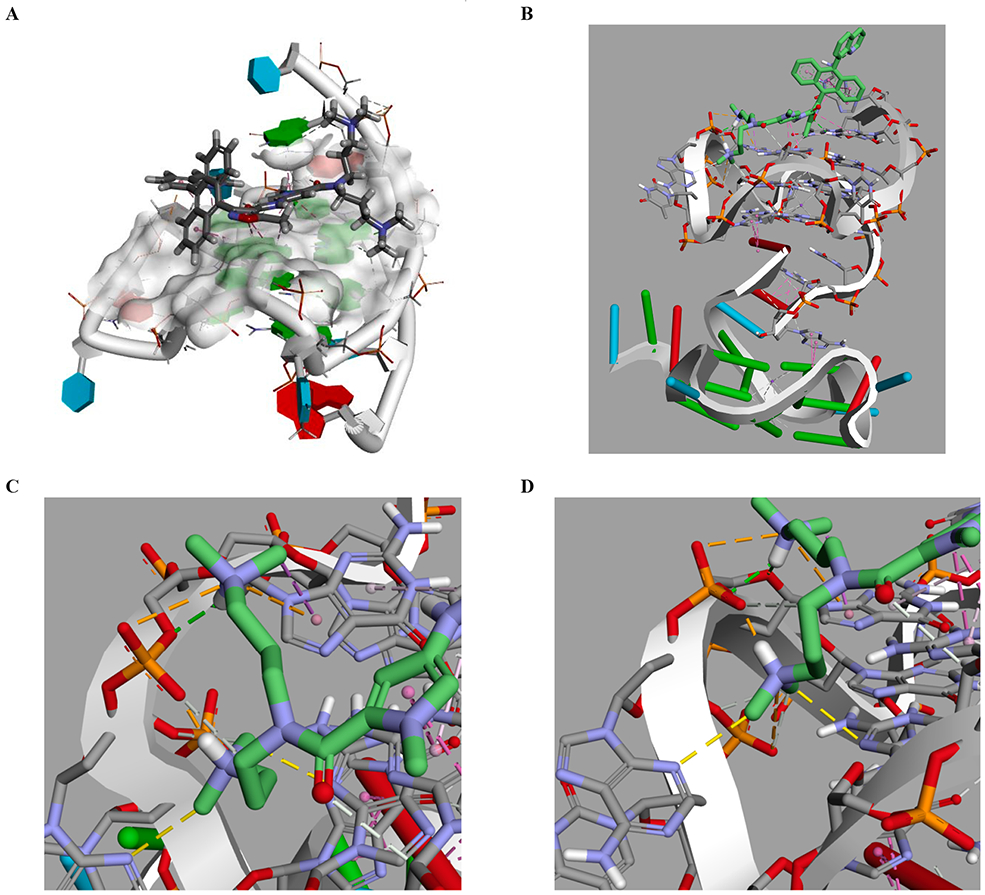
Discovery Studio docking of Quin-AIM at c-myc PDB accession number 2L7V. A. Side view of the highest score. B c-myc at 6 ns, then simulated for an additional 25 ns with Quin-AIM 3e. C. the dimethyl amino tail shows unusual interactions (dotted yellow lines) with both extrahelical A16 (lower left) and G-deck base G12. D. the other dimethylamino tail exhibits both an expected electrostatic interaction with the phosphate and an unexpected π-cation interaction with G16 (upper right, dotted orange lines).
We find the Discovery Studio calculation useful also for what it suggests as working hypotheses. Both the pyrrole N-methyl and the isoxazole C-5 methyl play roles in the binding energy, however, there is a large predominantly aromatic gulley adjacent to the isoxazole C-5 methyl, as well as adjacent to the 2- and/or 3- position of the anthryl, suggesting that hydrophobic or aromatic substituents located precisely in this position have additional binding possibilities. This and other opportunities suggested by using computation as a working hypothesis will be pursued and described in due course.
The observation that computation produced ensembles of ligand-receptor interactions that were similar, yet not identical, lead us to consider a molecular dynamics study to determine whether the poses represented a kinetic local minimum or a global thermodynamic equilibrium.
3.2. Molecular dynamics
Molecular Dynamics (MD) simulations were performed using YASARA, which utilizes the Amber forcefield, using the c-myc coordinates from our own work (PDBID: 6AU4).24,25 Over the course of 25 ns dynamics the oligonucleotide initially showed a partial unfolding, and then a refolding motion. The Quin-AIM was docked with the MD at specific timeframes: 0, 6, 14.5, 18.5, and 25 ns which were chosen to afford a high diversity in the ensemble of starting structures. The minimum energy was observed with the oligonucleotide conformer at 6 ns, which was then further simulated for an additional 25 ns. The best scoring pose is subtly distinct from the Discovery Studio docking at 2L7V in that the N-heterocyclic moiety has “walked” out slightly towards the G5 (Fig. 5B), a guanine not contained with a G-tetrad. The intermolecular interactions are a combination of expected and unexpected. Part of the expected effect of introduction of N-heterocycles, was the anticipation of interactions of the Lewis basic nitrogen with proximal Lewis and Bronsted acid partners. The nitrogen in the heterocycle of Quin-AIM has a hydrogen bond with the amino group of the guanine (Fig. 5B). The anthryl moiety has extensive face-to-face π-stacking with the G5 guanine, totaling five interactions from each of the anthryl rings to both base rings. The isoxazole group acts as a bridge between the TGA terminus and the top G-tetrad, by anchoring with a hydrogen bond between the ring O of the isoxazole with the amino group of A6. The isoxazole also shows T-shaped π-π interactions with both G5 and A6. The isoxazole C5 methyl bridges from A6 to G11 and G16 of the top G-tetrad. The pyrrole group has π-π face-to-face interactions with both rings of G11. The double dimethylamino tail group is expected to interact with the sugar phosphate backbone, and in fact, this interaction with the protonated dimethyl amino group has been considered essential for DNA binding small molecules to overcome the negative charge surrounding the DNA perimeter. Thus, electrostatic interactions between both protonated dimethylamino groups was expected. What is unusual is interaction between the dimethyl amino group and extrahelical (N3 of A15) and G-tetrad bases (N3 of G12, which is contained in the middle G-tetrad) as illustrated in Fig. 6C. Finally, the second dimethylamino tail has a π-cation interaction with G16 of the top G-deck (Fig. 5D). This suggests the possibility that the dimethylamino groups not only initiate sequence reading by the small molecule as expected, but also that the group can contribute to additional interactions with both extrahelical and G-tetrad bases.
3.3. Limitations of the computation
All computer aided drug design is subject to the logical fallacies of correlation and confirmation bias.26 Thus, further experimental studies to verify the binding details suggested by computation are always warranted. One especially troublesome temptation of confirmation bias is the tendency to rule out alternate explanations once an alluring hypothesis appears supported, and given the complexity of the Warburg phenotype, the number of possibilities are daunting.
3.4. Circular dichroism NMR spectroscopy
The Pu22 oligonucleotide exhibits a CD Spectrum characteristic of the parallel quadruplex conformation in solution (Fig. 7). Addition of the Quin-AIM to the Pu22 in solution confirms that the G4 conformer is maintained. Thermal melting curves indicate that the Quin-AIM also increases the melting temperature of the G4 under these conditions (Tm = 58.4 °C for Pu22 only and 61.7 °C with 2 equivalents Quin-AIM).
Figure 7.
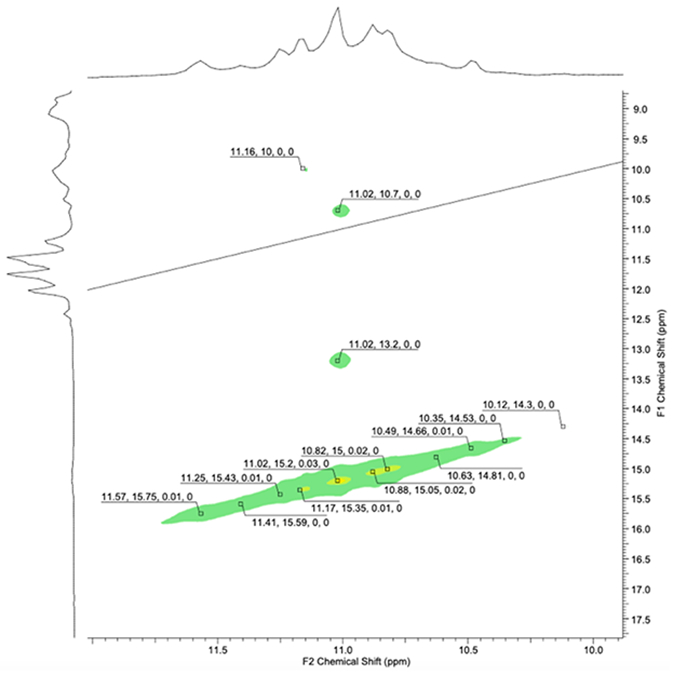
NMR of Pu22 in saturated solution of Quin-AIM 3e (approximately 2 equivalents).
NMR was examined for the Ph-AIM with the Pu22 oligomer, in a similar manner to our study of AIMs with a telomeric oligomer described previously.11 The G-deck hydrogen bonding region of Pu22 (10.95–11.6 ppm) produced a clean and relatively sharp spectrum, and titration with Ph-AIM gave rise to subtle but measurable and reproducible anisotropic shifts (the spectrum and assignments for c-myc Pu22 are shown in the Supporting Data) and this behavior corresponds with a stabilization of the G4 as determined experimentally by CD melting studies. The up-field shifts were observed at both site 1 and site 2 of c-myc, and the binding interactions suggested by computation were examined in detail to determine whether those interactions were consistent with the experiment. Similarly, titration of the Pu22 oligonucleotide with Quin-AIM 3e lead to signal broadening and upfield shifts of G residues at both binding sites 1 and 2 (Fig. 7, Table 3).
Table 3.
NMR c-myc oligonucleotide interactions with 10Ph-AIM. Blue 5′-Site 1; Green S′-Site 2, NMR assignments according to (Dai, 2011), NMR c-myc oligonucleotide interactions with 10-Quin-AIM 3e.
| Chemical shift (δ) | Guanine Assign. | Chemical shift (δ) | (Δδ) | Chemical shift (δ) | (Δδ) | Chemical shift (δ) | (Δδ) |
|---|---|---|---|---|---|---|---|
| c-myc (Pu22) | (Dai, 2011) | +Ph-AIM (2 eq.) | +Ph-AIM (2 eq.) | +Quin-AIM (1 eq.) | +Quin-AIM (1 eq.) | +Quin-AIM (2 eq.) | +Quin-AIM (2 eq.) |
| 11.79 | G16 | 11.8 | +0.01 | 11.79 | 0 | 11.67 | −0.12 |
| 11.67 | G7 | 11.65 | −0.02 | 11.65 | −0.02 | 11.64 | −0.13 |
| 11.60 | G11 | 11.58 | −0.02 | 11.60 | 0 | 11.48 | −0.12 |
| 11.41 | G12 | 11.40 | −0.01 | 11.40 | −0.01 | 11.32 | −0.09 |
| 11.28 | G21 | 11.28 | 0 | 11.27 | −0.01 | 11.25 | −0.03 |
| 11.18 | G20 | 11.21 | +0.03 | unres. | 11.09 | −0.09 | |
| 11.17 | G8 | 11.16 | +0.01 | unres. | 11.09 | −0.08 | |
| 11.16 | G17 | 11.14 | −0.02 | 11.16 | 0 | 11.09 | −0.07 |
| 10.99 | G13 | 10.98 | −0.01 | 10.97 | −0.02 | 10.95 | −0.03 |
| 10.98 | G22 | 10.95 | −0.03 | unres. | 10.90 | −0.05 | |
| 10.95 | G18 | 10.92 | −0.03 | 10.92 | −0.03 | 10.70 | −0.25 |
| 10.56 | G9 | 10.56 | 0 | 10.56 | 0 | 10.56 | 0 |
In general, face-to-face or edge-to-face π-π interactions were predicted for each signal shifted in the NMR titration, however, the computation predicted additional anisotropic interactions that were not observed (a table of specific predicted interactions by computation and those observed by NMR is shown in Supplementary Information).
The change in chemical shift is greater in magnitude for 10-Quin-AIM 3e verses the C-10 chloro and, the 10-Ph-AIM analogs. We conclude based on these studies that quadruplex binding is a plausible contributor to the anti-tumor activity observed, although additional or alternative mechanisms are not ruled out.
3.5. Antitumor activity
The N-heterocyclic AIMs exhibit robust antitumor activity against both brain and breast cancer cell lines. In both cases the quinoline was the most active compound (Table 4). Against the breast cancer cell line MDA-468 all of the N-heterocycles exhibited single digit micro molar cytotoxicity, with the m-pyridyl AIM roughly equipotent to the quinoline. In the brain tumor cell line SNB-19, the quinoline was considerably more active than the other heterocycles, at 2 μM verses 7 for the m- and p-pyridyl analogs.
Table 4.
Antitumor activity against breast cancer cell line MDA-468, and two brain tumor cell lines, human Glioblastoma multiforme SNB-19, and C6 rat glioma. The last column is a summary of initial computational docking of the N-heterocyclic AIMs at the Pu22 quadruplex of the c-myc oncogene, PDB accession number 2L7V, using the Discovery Studio suite version 4.0, unless otherwise noted. §Discovery Studio version 2017.
| SNB-19 |
MDA-468 |
C6 |
|||||
|---|---|---|---|---|---|---|---|
| 10-Ar AIM |
IC50 (μM) |
95% CI (±) |
IC50 (μM) |
95% CI (±) |
IC50 (μM) |
95% CI (±) |
Cdocker Interaction Energy (kcal/ mole) conformation |
| o-Pyridyl | >25 | 6.34 | 0.67 | >25 | 75.6 | ||
| m-Pyridyl | 7.76 | 0.42 | 1.29 | 0.3 | >25 | 72.0 | |
| p-Pyridyl | 7.08 | 0.26 | 3.1 | 0.27 | 16.9 | 0.5 | 77.6 |
| Quinoline | 2.03 | 0.09 | 1.17 | 0.25 | 6.71 | 0.12 | 78.5 Anti 76.7 Syn (locked) 83.7 Syn (eq)§ |
In addition we examined a rat glioma cell line (C6 in Table 4), against which the N-heterocyclic AIMs were somewhat less active, however, the Quin-AIM did retain single digit micromolar potency at 6.71 μM, which argues that this cell line could be potentially used in an animal model for pre-clinical characterization of the AIMs.
3.6. Fluorescent properties for tumor cell imaging
The N-heterocyclic AIMs were notably more fluorescent than analogs we have previously synthesized. This is a useful property when examining the localization of the antitumor agents in cancer cells, which can be critical during surgical resection, referred to as tumor paint. The tumor cells were observed to exhibit some of the expected hallmarks of apoptosis, such as punctate localization of the antitumor agents, and membrane blebbing and packaging characteristic of organized cell death (Fig. 8). One consistent observation is that the AIMs localize rapidly in the tumor cells, far more efficiently than would be expected for passive diffusion. There was also a notable concentration of the AIMs inside the cytoplasm. However, it also appears that there is little or no fluorescence inside the nucleus. This could be the result of the AIMs not permeating the nucleus, or the result of efflux by MDR-1 or related efflux pumps. However, we have also reported that AIMs bound to G4 forming oligonucleotides exhibit a pronounced hypochromic effect, therefore, it is also plausible that AIMs bound to G4 could escape detection by this method.17 Our working hypothesis has been that the AIMs exert their antitumor effect by G4 quadruplex binding, and in addition to G4 found in telomeres and oncogenes, there are also reports of G4 quadruplex DNA located in the mitochondria. Alternative molecular mechanisms of action will be considered in detail in future reports.
Fig. 8.
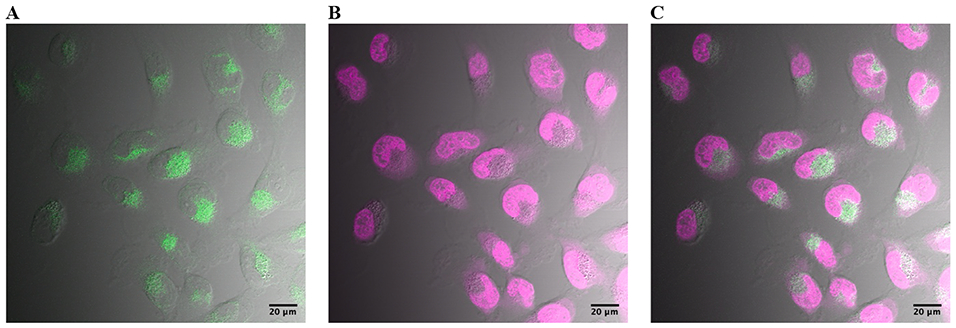
SNB-19 cells treated with 500 nM of the 10-Quinoline AIM (green, A), 1x HCS NuclearMask (magenta, B), and overlay (white, C). Scale bar shown is 20 μM.
We have in other studies reported on the preparation of relatively robust MDR-1 inhibitors, and the possibility of increasing the cytotoxicity of the AIM agents to tumor cells using efflux transporter inhibitors in combination appears to be a worthy pursuit.27
Another issue which is surprising, centers on the question of how compounds with such apparent high lipophilicity cross any membrane with such facility. It has been previously reported that intramolecular hydrogen bonding can mask the apparent surface area and lipophilicity of a molecule, and result in improved membrane permeability and absorption, expanding what would be expected based on Lipinski’s rule of five for the corresponding unfolded conformation.28 In the case of the AIMs, an analogous intramolecular π-cation interaction would plausibly give rise to such a folded conformer, which obviously reduces both the surface area and effective lipophilicity of the AIM (Fig. 9). This could in turn give rise to more rapid passive diffusion through a membrane. Alternatively, there is the possibility of facilitated or active transport through an organic cation transporter. We intend to test these hypotheses in future studies.
Fig. 9.
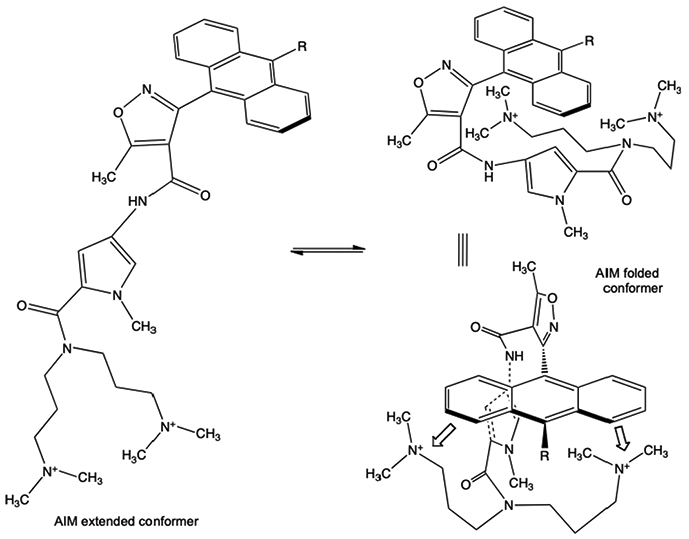
The apparent high lipophilicity of the extended AIM conformer (left), could be dramatically reduced via intramolecular interactions leading to folded conformers (right).
4. Experimental
4.1. Cell viability assay
Growth inhibition of SNB-19 human glioblastoma cells was determined using the MTT colorimetric assay as previously described.11 IC50 values were calculated using the ‘R’ statistical computing software and the ‘ic50′ package.29,30 This package fits a logistic model to the dose-response data collected using the MTT assay and approximates the concentration of the compound required to inhibit the growth of the cells by 50% versus the DMSO control (IC50).
4.2. Confocal imaging
Cells were diluted to 100,000 cells/mL and plated on an 8-well Lab-Tek II chambered coverglass slide at a density of 20,000 cells/well in 0.2 mL growth medium. Cells were grown in optimal growth conditions and allowed to adhere overnight. The following day, treatment medium was made by dilution of 5 mM DMSO stock solutions of AIM compounds in growth medium. Culture medium was removed by aspiration and treatment medium containing the desired compound was added. The cells were then incubated at growth conditions 10 min prior to removing treatment medium by aspiration. Each well was washed twice with 0.2 mL of PBS and then cells were then fixed in 4% PFA for 10 min on ice prior to imaging. Imaging was completed on an Olympus FV-1000 confocal laser scanning microscope immediately following fixation. Images were captured using a 60x oil-immersion objective lens (NA 1.42). Compound fluorescence was visualized using selective excitation at 405 nm and emission collected at 422 nm. HCS NuclearMask staining was performed according to manufacturer protocol.
4.3. Circular dichroism
DNA oligonucleotide (Pu22: 5′-TGAGGGTGGGTAGGGTGGGTAA-3′) was dissolved in nuclease-free water to make a 1 mM stock solution. The compound sample was prepared using previously made 5 mM solutions of AIM compounds in DMSO. Samples were made at 5 μM strand concentration in 1 mM potassium phosphate buffer (pH 6) for both the oligonucleotide alone and oligonucleotide plus 2 equivalents compound. Samples were annealed by heating to 90 °C for 10 min and then gradually cooling to 4 °C overnight prior to data collection.
CD spectra and thermal melting curves were acquired using Applied Photophysics Chirascan CD Spectrophotometer. CD spectra were gathered from 200 nm to 400 nm with a 1 nm bandwidth and 0.2 nm step size at 5°C and 0.5 s time-per-point. CD thermal melting curves were gathered at 265 nm from 20 – 90 °C at a rate of 1 °C/min in 0.5 °C steps. The CD thermal melting curves were acquired at 24 s time-per-point with 1 nm bandwidth.
4.4. Circular dichroism data analysis
Following data collection, thermal melting curves were fit to a six-parameter logistic curve using the qpcR package for the R statistical computing software environment.29,31 The six-parameter logistic fit was chosen to minimize the number fitting parameters while maximizing goodness-of-fit criteria such as AIC, BIC and R2. Thermal melting values (Tm) for each condition were assigned by computing the minimum value of the first derivative of the fitted curve for each sample.
4.5. General procedure for cross-coupling reactions
The crystalline isoxazole ester was ground into a fine powder and liquid reagents were degassed before use. All solid reagents were added to a reaction vessel under an inert atmosphere. The vessel was then degassed and back-filled with argon three times before adding liquid reagents.
Ethyl 5-methyl-3-(10-(pyridin-2-yl)anthracen-9-yl)isoxazole-4-carboxylate, 2b.
To ground 10-Br-ester 1 (1 mmol), 2-Pyridyl MIDA (1.5 mmol), and XPhos Pd G3 (0.05 mmol), K3PO4 (5 mmol), Cu(OAc)2 (0.5 mmol), diethanolamine (1 mmol), was added DMF 8 mL. and the mixture was allowed to react for 24 hrs at 100 °C. The reaction mixture was cooled to room temperature and transferred to a separatory funnel using 20 mL of ether. 10 mL of 2 N HCl was added to the separatory funnel which was carefully shaken. To the separatory funnel was then added 2 N NaOH and it was carefully shaken. The organic layer was removed, and the aqueous phase was extracted 3 times with 10 mL of ether. The combined organic fractions were washed with 10 mL of brine and dried over anhydrous sodium sulfate. After filtration and concentration, the product 2b was obtained as a solid in 20% yield after isolation and purification. The majority of the material balance was debrominated anthracene.
1H NMR (400 MHz, CHCl3-d) δ 8.98 (br. s., 1H), 8.01 (d, J = 8.2 Hz, 1H), 7.69 (d, J = 8.4 Hz, 2H), 7.64–7.48 (m, 5H), 7.47–7.33 (m, 5H), 3.81–3.66 (m, 2H), 2.94 (d, J = 4.3 Hz, 3H), 0.45 (t, J = 7.1 Hz, 1H), 0.38 (t, J = 7.1 Hz, 2H).
The x-ray quality crystal was obtained by repeated slow evaporation from ethyl acetate and hexane and was solved in cooperation with the SCrALS program (and will be reported elsewhere).
4.5.1. Ethyl 5-methyl-3-(10-(pyridin-3-yl)anthracen-9-yl)isoxazole-4-carboxylate, 2c.
Using the procedure described above, 10-Br-ester 1 (3 mmol), 3-Pyridyl boronic acid (4.5 mmol), and tetrakis (0.15 mmol), Na2CO3 (9 mmol, 3 mL of 3 M aqueous solution), dimethyl acetamide (30 mL). 1 hr 120 °C microwave irradiation.
The reaction mixture was cooled to room temperature and transferred to a separatory funnel using 20 mL of ether. The organic layer was then washed 3 times with 10 mL of water, once with 10 mL of brine, then dried over sodium sulfate. Filtration and concentration produced the product which was purified by silica gel chromatography, the yield after isolation and purification was 81%.
Similarly, 10-Br-ester 1 (1 mmol), 3-pyridyl boronic acid (1.5 mmol), and XPhos Pd G3 (0.05 mmol), Na2CO3 (3 mmol, 1 mL of 3 M aqueous solution), dimethyl acetamide (10 mL). 1 hr 120°C microwave irradiation. The reaction mixture was cooled to room temperature and transferred to a separatory funnel using 20 mL of ether. The organic layer was then washed 3 times with 10 mL of water, once with 10 mL of brine, then dried over sodium sulfate. Gave 76% isolated and purified yield.
1H NMR (400 MHz, CHCl3-d) δ = 8.88–8.78 (m, 1H), 8.71 (d, J = 10.5 Hz, 1H), 7.86–7.76 (m, 1H), 7.70 (d, J = 8.6 Hz, 2H), 7.64–7.51 (m, 3H), 7.49–7.32 (m, 4H), 3.83–3.67 (m, 2H), 2.94 (s, 3H), 0.47 (t, J = 7.1 Hz, 1H), 0.40 (t, J = 7.2 Hz, 2H).
4.5.2. Ethyl 5-methyl-3-(10-(pyridin-4-yl)anthracen-9-yl)isoxazole-4-carboxylate, 2d
Using the procedure described above, 10-Br-ester 1 (1 mmol), 4-Pyridyl boronic acid (1.5 mmol), and tetrakis (0.02 mmol), Na2CO3 (3 mmol, 1 mL of 1 M aqueous solution), dimethyl acetamide (10 mL) were combined for 1 hr and subject to 120 °C microwave irradiation. The reaction mixture was cooled to room temperature and transferred to a separatory funnel using 20 mL of ether. The organic layer was then washed 3 times with 10 mL of water, once with 10 mL of brine, then dried over sodium sulfate. After filtration, concentration and silica gel chromatography the isolated purified yield was 50%
This compound was obtained as a yellow crystal, which was recrystallized from hexane-EtOAc (6:1) solution, after chromatography: mp 158–160 °C; 1H -NMR (CHCl3-d): δ(ppm) 8.90(d, J = 4.8 Hz, 2H), 7.72(d, J = 8.4 Hz, 2H), 7.60 (d, J = 8.4 Hz, 2H), 7.43(m, 6H), 3.80(q, J = 7.2 Hz, 2H), 2.97(s, 3H), 0.47(t, J = 7.2 Hz, 3H); 13C -NMR (CDCl3): δ(ppm) 176.24, 161.37, 160.54, 150.12, 150.03, 147.28, 135.81, 130.35, 128.85, 126.42, 126.31, 126.21, 126.16, 125.85, 125.76, 123.99, 111.42, 60.15, 13.51, 12.94; MS (EI): m/z 408.15 (100.00, M+), 320.10 (30.59, M─COOC2H5─CH3), 296.15 (20.60), 292.15 (18.15), 254.15 (17.08), 226.10 (17.61); Anal. Calc’d for C26H20N2O3: C, 76.46; H, 4.94; N, 6.86. Found: C, 76.31; H, 5.10; N, 7.15.
4.5.3. Ethyl 5-methyl-3-(10-(quinolin-8-yl)anthracen-9-yl)isoxazole-4-carboxylate, 2e
Using the general procedure described above, 10-Br-Ester 1 (3 mmol), 8-Quinoline boronic acid (4.5 mmol), and XPhos Pd G3 (0.15 mmol), Na2CO3 (6 mmol, 12 mL of 0.5 M aqueous solution), were combined and THF (6 mL)was added. The reaction mixture was warmed for 72 hr at 40 °C. and then was cooled to room temperature and transferred to a separatory funnel with 10 mL of water and DCM. The aqueous layer was extracted 3 times with 10 mL of DCM. The combined organic fractions were then washed with 10 mL of brine. Filtration and concentration was followed by silica gel chromatography, and the 10-Quin-ester 2e was obtained in 57% yield.
1H NMR (400 MHz, CHCl3-d) δ = 8.72 (dd, J = 1.8, 4.2 Hz, 1H), 8.33–8.25 (m, 1H), 8.09–8.03 (m, 1H), 7.81–7.75 (m, 1H), 7.75–7.70 (m, 3H), 7.43–7.33 (m, 5H), 7.25–7.17 (m, 2H), 3.81–3.70 (m, 2H), 2.99–2.91 (m, 3H), 0.47–0.35 (m, 3H).
13C NMR (101 MHz,CHCl3-d) δ = 176.20, 176.10, 161.78, 161.69, 160.86, 160.77, 150.78, 150.13, 147.77, 138.33, 137.33, 136.38, 132.73, 130.65, 130.56, 130.48, 130.39, 128.56, 128.50, 128.44, 128.36, 127.21, 127.16, 126.50, 126.27, 125.84, 125.77, 125.71, 125.65, 125.06, 123.33, 121.39, 121.31, 111.67, 111.53, 60.51, 60.03, 13.48, 13.40, 12.89, 12.50.
HRMS Calc’d for C30H23N2O3 459.1709 ([M + 1]+), found 459.1691, 3.9 ppm.
4.6. General experimental procedure for double activation
THF was freshly distilled from sodium and benzophenone. Methanol was dried over calcium hydride. The esters and SmCl3 were added to a dry reaction vessel under an inert atmosphere and the vessel was degassed three times·THF was added and the solution was allowed to stir for 6 h.
The nitro-pyrrole double-tail, palladium on carbon, and MeOH were added to the hydrogenation flask under an inert atmosphere. The flask was evacuated and back-filled three times with hydrogen. The flask was stirred for 4 h under hydrogen at 40 psi. The flask was then evacuated and back-filled with argon. The mixture was filtered through a glass fiber filter disc and the filter and was washed three times with MeOH. The solution was then concentrated and placed under high vacuum.
The flask containing the dry amino-pyrrole double tail was evacuated and back-filled with argon three times before THF was added. The solution was placed in an ice bath and AlMe3 was added dropwise to the cold stirring solution. The flask was removed from the ice bath and allowed to stir for 1 h. The flask was then returned to an ice bath and the solution containing the ester and SmCl3 was added dropwise to the amine solution over 30 min via a cannula. The solution was removed from the ice bath and allowed to warm to room temperature before being brought to reflux for 18 h.
The reaction was then cooled to room temperature and sodium sulfate decahydrate was added in small portions until no more bubbles formed. The solution was then diluted with MeOH and filtered through Celite. Three portions of MeOH containing 10% ammonium hydroxide were used to wash the Celite.
The filtrate was then concentrated and purified on a silica column equilibrated with DCM half- saturated with ammonia. The column was eluted with 20% MeOH in DCM half-saturated with ammonia. We attribute the low yields after isolation and purification to the extreme steric hindrance of the N-het-AIM systems, however, most of the remaining material balance was recovered starting material.
4.6.1. N-(5-(bis(3-(dimethylamino)propyl)carbamoyl)-1-methyl-1H-pyrrol-3-yl)-5-methyl-3-(10-(pyridin-2-yl)anthracen-9-yl)isoxazole-4-carboxamide, 3b.
As described above, o-Pyridyl ester (0.67 mmol) SmCl3 (0.67 mmol), were combined and THF (28 mL) was added. The nitro-pyrrole double-tail (1.35 mmol), was reduced using Pd on carbon (0.47 g), MeOH (30 mL), H2 (40 psi), over 4 hr. The amino-pyrrole double-tail so obtained was treated with, AlMe3 (2.02 mmol), THF (14 mL). The solutions were combined via cannula, and refluxed. The yield after isolation and purification by silica gel chromatography was 10%.
1H NMR (400 MHz, MeOH-d4) d = 8.86 (br. s., 1H), 8.17 (s, 1H), 7.84 (d, J = 8.2 Hz, 2 H), 7.69 (br. s., 2 H), 7.57–7.49 (m, 4 H), 7.46 (s, 3H), 6.78 (s, 1H), 5.13 (s, 1H), 3.58–3.45 (m, 8 H), 3.37–3.33 (m, 2 H), 3.03–2.78 (m, 17H), 2.78–2.64 (m, 11H), 2.09 (br. s., 5 H)
MW calculated for 721, HPLC-MS observed m/Z 776 ([M + CH3CN + 2 MeOH]+, 100% Rel. intensity).
4.6.2. N-(5-(bis(3-(dimethylamino)propyl)carbamoyl)-1-methyl-1H-pyrrol-3-yl)-5-methyl-3-(10-(pyridin-3-yl)anthracen-9-yl)isoxazole-4-carboxamide, 3c
As describe above m-Pyridyl ester 2c (2.02 mmol) SmCl3 (2.02 mmol), were combined in THF (84 mL) solution. Nitro-pyrrole double-tail (4.04 mmol), was reduced using Pd on carbon (1.4 g), MeOH (91 mL), H2 (40 psi), over 4 hr. The resulting amino-pyrrole double-tail, was treated with AlMe3 (6.06 mmol), THF (42 mL). The yield after purification by silica gel chromatography was 22%
1H NMR 8.785 (d, J = 3.9 Hz, 1H), 8.625 (d, J = 3.5 Hz, 1H), 7.99–7.91 (m, 1H), 7.84 (d, J = 8.7 Hz, 2 H), 7.74 (dd, J = 5.0, 7.7 Hz, 1H), 7.58 (d, J = 8.6 Hz, 2 H), 7.54–7.38 (m, 4 H), 6.83–6.76 (m, 1H), 6.09 (dd, J = 1.7, 11.4 Hz, 1H), 3.50 (s, 3 H), 3.42 (t, J = 7.5 Hz, 4 H), 3.36–3.29 (m, 1H), 2.86 (d, J = 2.1 Hz, 3 H), 2.25 (br. s., 7 H), 2.16 (br. s., 11H), 1.73 (br. s., 4 H)
4.6.3. N-(5-(bis(3-(dimethylamino)propyl)carbamoyl)-1-methyl-1H-pyrrol-3-yl)-5-methyl-3-(10-(pyridin-4-yl)anthracen-9-yl)isoxazole-4-carboxamide, 3d
Prepared as described above from Nitro-pyrrole double-tail (1.4 mmol), Pd on carbon (0.47 g), MeOH (30 mL), H2 (40 psi), 4 hr., the resulting. Amino-pyrrole double-tail, was combined with AlMe3 (4.07 mmol), THF (14 mL). The p-Pyridyl ester 2d (0.68 mmol) SmCl3 (0.68 mmol), were combined and dissolved in THF (28 mL). The yield after purification by silica gel chromatography was 24%.
1H NMR (400 MHz, MeOH-d4) d = 8.81 (d, J = 4.8 Hz, 2H), 7.82 (d, J = 8.7 Hz, 2H), 7.58–7.50 (m, 5H), 7.48 (d, J = 8.6 Hz, 2H), 7.45–7.38 (m, 2H), 6.79 (d, J = 1.5 Hz, 1H), 6.14 (d, J = 1.6 Hz, 1H), 3.49 (s, 3H), 3.42 (t, J = 7.4 Hz, 4H), 2.84 (s, 3H), 2.29 (dd, J = 3.4, 7.5 Hz, 8H), 2.20 (br. s., 11H), 1.80–1.66 (m, 5H).
4.6.4. N-(5-(bis(3-(dimethylamino)propyl)carbamoyl)-1-methyl-1H-pyrrol-3-yl)-5-methyl-3-(10-(quinolin-8-yl)anthracen-9-yl)isoxazole-4-carboxamide, 3e
Prepared as above, the nitro-pyrrole double-tail (0.7 mmol), was initilly reduced with hydrogen and Pd on carbon (0.24 g), in MeOH as solvent (15 mL), H2 (40 psi), 4 hr, filtered and concentrated. The resulting amino-pyrrole double-tail, was treated with AlMe3 (1 mmol), in THF (7 mL), to which was added 10-Quinoline ester 2e (0.35 mmol) and SmCl3 (0.35 mmol), THF (14 mL). The yield after purification by silica gel chromatography was 14%.
1H NMR (400 MHz, MeOH-d4) d = 8.65–8.54 (m, 2H), 8.27 (d, J = 8.6 Hz, 1H), 7.94–7.91 (m, 1H), 7.91–7.78 (m, 4H), 7.64–7.53 (m, 1H), 7.51–7.44 (m, 2H), 7.38 (t, J = 9.4 Hz, 2H), 7.32 (dd, J = 6.7, 7.7 Hz, 2H), 6.67 (d, J = 1.6 Hz, 1H), 6.63 (d, J = 1.6 Hz, 1H), 3.63 (s, 3H), 3.57–3.47 (m, 5H), 3.06 (br. s., 4H), 2.95 (s, 2H), 2.90 (s, 2H), 2.81–2.58 (m, 11H), 2.10–1.98 (m, 4H).
13C NMR (101 MHz, MeOH-d4) δ = 171.98, 170.45, 165.04, 164.98, 160.17, 159.61, 159.07, 159.03, 149.97, 149.89, 138.05, 136.71, 133.24, 133.02, 130.78, 130.68, 130.54, 130.49, 129.01, 128.88, 126.83, 126.70, 126.27, 126.05, 125.51, 125.42, 125.38, 122.51, 122.21, 122.12, 121.56, 121.51, 121.38, 121.30, 117.39, 117.01, 115.98, 115.37, 105.58, 104.82, 54.70, 42.04, 34.56, 34.56, 34.47, 23.31, 11.80, 11.54.
Supplementary Material
Acknowledgements
The authors thank the ALSAM Foundation for support of this work. We thank Dan DeCato for small molecule crystallography of 2c and 2e performed by the small molecule x-ray core, supported by National Science Foundation (NSF)-MRI (CHE - 1337908), and National Institutes of Health (CoBRE NIGMS P20GM103546). We thank Professor Bruce Bowler for use of his CD. We thank Dr. Kevin Rider for helpful discussions on isoxazole poisoning of palladium cross-couplings, and Christina Gates for help in HPLC-MS.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmc.2020.115781.
References
- 1.Neidle S Therapeutic Applications of Quadruplex Nucleic Acids. Academic Press; 2012. [Google Scholar]
- 2.Neidle S Quadruplex nucleic acids as novel therapeutic targets. J Med Chem. 2016;59(13):5987–6011. 10.1021/acs.jmedchem.5b01835. [DOI] [PubMed] [Google Scholar]
- 3.Neidle S Quadruplex nucleic acids as targets for anticancer therapeutics. Nature Reviews Chemistry. 2017;1(5):s41570-017–0041-017. doi: 10.1038/s41570-017-0041. [DOI] [Google Scholar]
- 4.Chung WJ, Heddi B, Tera M, Iida K, Nagasawa K, Phan AT. Solution structure of an intramolecular (3 + 1) human telomeric G-quadruplex bound to a telomestatin derivative. J Am Chem Soc. 2013;135(36):13495–13501. 10.1021/ja405843r. [DOI] [PubMed] [Google Scholar]
- 5.Martino L, Virno A, Pagano B, et al. Structural and thermodynamic studies of the interaction of distamycin A with the parallel quadruplex structure [d(TGGGGT)]4. J Am Chem Soc. 2007;129(51):16048–16056. 10.1021/ja075710k. [DOI] [PubMed] [Google Scholar]
- 6.Parkinson GN, Ghosh R, Neidle S. Structural basis for binding of porphyrin to human telomeres. Biochemistry. 2007;46(9):2390–2397. 10.1021/bi062244n. [DOI] [PubMed] [Google Scholar]
- 7.Silverman RB, Holladay MW. The Organic Chemistry of Drug Design and Drug Action. Academic Press; 2014. [Google Scholar]
- 8.Calabrese DR, Chen X, Leon EC, et al. Chemical and structural studies provide a mechanistic basis for recognition of the MYC G-quadruplex. Nat Commun. 2018;9(1):4229. 10.1038/s41467-018-06315-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu M-H, Wang Y-Q, Yu Z-Y, et al. Discovery of a new four-leaf clover-like ligand as a potent c-MYC transcription inhibitor specifically targeting the promoter G-quadruplex. J Med Chem. 2018. 10.1021/acs.jmedchem.7b01697. [DOI] [PubMed] [Google Scholar]
- 10.Todd AK, Johnston M, Neidle S. Highly prevalent putative quadruplex sequence motifs in human DNA. Nucleic Acids Res. 2005;33(9):2901–2907. 10.1093/nar/gki553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weaver MJ, Kearns AK, Stump S, et al. AIMing towards improved antitumor efficacy. Bioorg Med Chem Lett. 2015;25(8):1765–1770. 10.1016/j.bmcl.2015.02.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han X, Li C, Rider KC, Blumenfeld A, Twamley B, Natale NR. The isoxazole as a linchpin for molecules that target folded DNA conformations: selective lateral lithiation and palladation. Tetrahedron Lett. 2002;43(43):7673–7677. 10.1016/S0040-4039(02)01845-2. [DOI] [Google Scholar]
- 13.Martin R, Buchwald SL. Palladium-catalyzed Suzuki–Miyaura cross-coupling reactions employing dialkylbiaryl phosphine ligands. Acc Chem Res. 2008;41(11): 1461–1473. 10.1021/ar800036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dick GR, Woerly EM, Burke MD. A general solution for the 2-pyridyl problem. Angew Chem Int Ed. 2012;51(11):2667–2672. 10.1002/anie.201108608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahneman DT, Estrada JG, Lin S, Dreher SD, Doyle AG. Predicting reaction performance in C-N cross-coupling using machine learning. Science. 2018;360(6385):186–190. 10.1126/science.aar5169. [DOI] [PubMed] [Google Scholar]
- 16.Zhou P, Mosher MD, Taylor WD, Crawford GA, Natale NR. Double activation preparation of an acridinyl-isoxazolyl-lexitropsin. Bioorg Med Chem Lett. 1997;7(19):2455–2456. 10.1016/S0960-894X(97)10011-7. [DOI] [Google Scholar]
- 17.Han X, Li C, Mosher MD, et al. Design, synthesis and biological evaluation of a novel class of anticancer agents: anthracenylisoxazole lexitropsin conjugates. Bioorg Med Chem. 2009;17(4):1671–1680. 10.1016/j.bmc.2008.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dai J, Carver M, Hurley LH, Yang D. Solution structure of a 2:1 quindoline-c-MYC G-quadruplex: insights into G-quadruplex-interactive small molecule drug design. J Am Chem Soc. 2011;133(44):17673–17680. 10.1021/ja205646q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brooks BR, Brooks CL, MacKerell AD, et al. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30(10):1545–1614. 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koska J, Spassov VZ, Maynard AJ, et al. Fully automated molecular mechanics based induced fit protein-ligand docking method. J Chem Inf Model. 2008;48(10): 1965–1973. 10.1021/ci800081s. [DOI] [PubMed] [Google Scholar]
- 21.Parrill AL. Rational Drug Design. In: Rational Drug Design. Vol 719. ACS Symposium Series. American Chemical Society; 1999. doi: 10.1021/bk-1999-0719.fw001. [DOI] [Google Scholar]
- 22.Jain AN. Scoring noncovalent protein-ligand interactions: A continuous differentiable function tuned to compute binding affinities. J Computer-Aided Mol Des. 1996;10(5):427–440. 10.1007/BF00124474. [DOI] [PubMed] [Google Scholar]
- 23.Böhm HJ. On the use of LUDI to search the fine chemicals directory for ligands of proteins of known three-dimensional structure. J Comput Aided Mol Des. 1994;8(5): 623–632. 10.1007/BF00123669. [DOI] [PubMed] [Google Scholar]
- 24.Krieger E, Vriend G. YASARA view – molecular graphics for all devices – from smartphones to workstations. Bioinformatics. 2014;30(20):2981–2982. 10.1093/bioinformatics/btu426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stump S, Mou T-C, Sprang SR, Natale NR, Beall HD. Crystal structure of the major quadruplex formed in the promoter region of the human c-MYC oncogene. PLoS ONE. 2018;13(10), e0205584. 10.1371/journal.pone.0205584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jain AN, Cleves AE. Does your model weigh the same as a duck? J Comput Aided Mol Des. 2012;26(1):57–67. 10.1007/s10822-011-9530-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steiger SA, Li C, Backos DS, Reigan P, Natale NR. Dimeric isoxazolyl-1,4-dihydropyridines have enhanced binding at the multi-drug resistance transporter. Bioorg Med Chem. 2017;25(12):3223–3234. 10.1016/j.bmc.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alex A, Millan DS, Perez M, Wakenhut F, Whitlock GA. Intramolecular hydrogen bonding to improve membrane permeability and absorption in beyond rule of five chemical space. Med Chem Commun. 2011;2(7):669–674. 10.1039/G1MD00093D. [DOI] [Google Scholar]
- 29.R Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 30.P F, Rk T. Standardized high-throughput evaluation of cell-based compound screens. BMC Bioinformatics. 2008;9:475–475. doi: 10.1186/1471-2105-9-475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ritz C, Spiess A-N. qpcR: an R package for sigmoidal model selection in quantitative real-time polymerase chain reaction analysis. Bioinformatics. 2008;24(13):1549–1551. 10.1093/bioinformatics/btn227. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.