

Abstract
Circulating tumor cells (CTCs) are shed by cancer into the bloodstream, where a viable subset overcomes oxidative stress to initiate metastasis. We show that single CTCs from melanoma patients coordinately upregulate lipogenesis and iron homeostasis pathways. These are correlated with both intrinsic and acquired resistance to BRAF inhibitors across clonal cultures of BRAF-mutant CTCs. The lipogenesis regulator SREBF2 directly induces transcription of the iron carrier Transferrin (TF), reducing intracellular iron pools, reactive oxygen species (ROS) and lipid peroxidation, thereby conferring resistance to inducers of ferroptosis. Knockdown of endogenous TF impairs tumor formation by melanoma CTCs, and their tumorigenic defects are partially rescued by the lipophilic anti-oxidants Ferrostatin-1 and Vitamin E. In a prospective melanoma cohort, presence of CTCs with high lipogenic and iron metabolic RNA signatures is correlated with adverse clinical outcome, irrespective of treatment regimen. Thus, SREBF2-driven iron homeostatic pathways contribute to cancer progression, drug resistance and metastasis.
Keywords: Circulating tumor cells, Melanoma, SREBP, lipogenesis, Transferrin, Metastasis, Ferroptosis
INTRODUCTION
Metastatic melanoma remains a highly lethal malignancy, despite breakthroughs in the targeted treatment of BRAF-mutant cases (1–3) and the application of immune checkpoint therapies irrespective of genotype (4–8). Melanomas with BRAFV600E and associated oncogenic variants typically undergo rapid tumor shrinkage following treatment with combination BRAF and MEK inhibitors, although there is considerable variation in the degree of initial response, and virtually all cases recur with drug-resistant disease. The effectiveness of immune-targeted therapies appears to be determined in part by total mutational burden in the tumor and the associated presentation of neoantigens, which are typically high in melanoma (9–14), but only a third of patients have sustained responses (15,16). For both targeted and immune-based therapies, innate and acquired resistance is compounded by tumor heterogeneity in signaling pathways, expression of cell surface epitopes and intrinsic cell survival pathways.
The transition from primary melanoma to metastatic disease is characterized by blood-based dissemination of circulating tumor cells (CTCs) to diverse metastatic sites, as is the progression from oligometastatic to widespread metastatic disease. While melanoma cells are notoriously invasive and resistant to any therapeutic interventions other than immunological and BRAF-targeted treatments, the exceptional environment in the bloodstream may provide new therapeutic opportunities to suppress metastasis.
Specifically, the high oxygen content of blood has been shown to render primary melanoma cells vulnerable to reactive oxygen species (ROS) (17). Identifying innate mechanisms that confer resistance to ROS, and thereby enable a subset of melanoma cells to remain viable as they circulate in the blood, may thus provide new therapeutic targets to suppress metastatic precursors.
The small number of CTCs present within a blood specimen, admixed to billions of normal blood cells, presents significant technical challenges to their isolation and functional analysis (18). Neural crest-derived melanoma cells do not express the epithelial cell surface protein EpCAM, which is traditionally used to capture CTC using magnetically-conjugated antibody enrichment (19,20). Furthermore, melanoma cells display a high degree of heterogeneity in cell surface epitope expression, which complicates their direct capture using lineage or tumor-specific antibody panels (21). In designing a tumor agnostic strategy for isolating CTCs within blood specimens, we developed a platform (CTC-iChip) for efficient depletion of normal blood cells, thereby enriching for cancer cells irrespective of cell surface markers (22,23). Taking advantage of microfluidic flow kinetics, this CTC-iChip achieves initial size-based exclusion of RBCs and platelets, followed by inertial focusing of all nucleated blood cells into a single streamline, through which antibody-tagged leukocytes are magnetically separated from unlabeled CTCs. Across multiple tumor types, this platform achieves 104 enrichment for CTCs, and we have applied a digital RNA-based readout to allow highly sensitive measurement of melanoma tumor cell burden in the blood (24). The enrichment of untagged and unmanipulated CTCs not only enhances their RNA quality for such diagnostic purposes, but it also helps preserve their cell viability for functional studies.
We have previously reported the establishment of long-term CTC cultures from blood specimens of women with hormone-receptor positive breast cancer, which are often oligoclonal in origin, and which recapitulate somatic mutations acquired during the course of therapy, and their epigenetic cell states (25–27). Here, we established CTC cultures from blood samples of patients with metastatic melanoma, comparing their transcriptomes across different cases, as well as in multiple single CTC-derived isogenic lines from the same patient. In these cultured melanoma CTCs and in single CTCs freshly isolated from blood specimens, we find striking upregulation of Sterol Regulatory Element-Binding Protein (SREBP)-driven lipogenic pathways, together with iron homeostatic pathways, including the iron transport protein Transferrin (TF). The master regulator of cholesterol homeostasis SREBF2 directly induces expression of TF, which in turn suppresses ROS and drug-induced ferroptotic cell death. Modulating these pathways in mouse models has a dramatic effect on metastasis, while their overexpression within patient-derived CTCs is strongly correlated with an adverse clinical outcome. Thus, the coordinated overexpression of SREBP target genes and TF in melanoma CTCs points to a regulatory pathway linking lipogenesis with iron homeostasis, contributing to the ability of viable CTCs to overcome oxidative damage in the circulation.
RESULTS
Melanoma patient-derived tumorigenic CTC cultures overexpress lipogenic and iron homeostatic pathways
We generated patient-derived CTC cultures following microfluidic enrichment from blood samples of patients with metastatic melanoma. Five independent cultures were established from four patients (out of a cohort of 37 patients undergoing treatment on IRB-approved protocols at Massachusetts General Hospital Cancer Center). The CTC-iChip was used to deplete normal hematopoietic cells from 10ml of whole blood, and the enriched, unlabeled CTCs were maintained in anchorage-independent media, under hypoxic conditions and in the presence of heparin and Rho kinase inhibitor (22,23,25) (see MATERIALS AND METHODS). To confirm their identity as melanoma cells, the cultured CTCs were tested for expression of characteristic melanoma markers (24) (Figures 1A, S1A, S1B), and shown to have shared mutational profiles with their respective parental tumors, including the canonical drug-sensitizing BRAFV600E or NRASQ61K mutations, as well as displaying predicted BRAF and MEK inhibitor sensitivity patterns (Figures 1A, S1C, S1D). The melanoma CTC lines were tested for tumorigenesis potential in immunosuppressed NSG mice. All five cultured lines are highly tumorigenic following subcutaneous inoculation and four of them (with the exception of PEM-78) generated metastases upon direct intravascular injection, either by tail-vein or intracardiac inoculation (Figures 1B, 1C; S1E–J). Both primary tumors and metastases generated from GFP-luciferase-tagged CTCs show preservation of characteristic melanosome markers (Figures 1C, S1F, S1H, S1K). We selected two of these CTC lines, Mel-167 and Mel-182–2, for detailed functional studies.
Figure 1. Lipogenic and iron homeostasis signatures are elevated in melanoma patient-derived CTC lines.
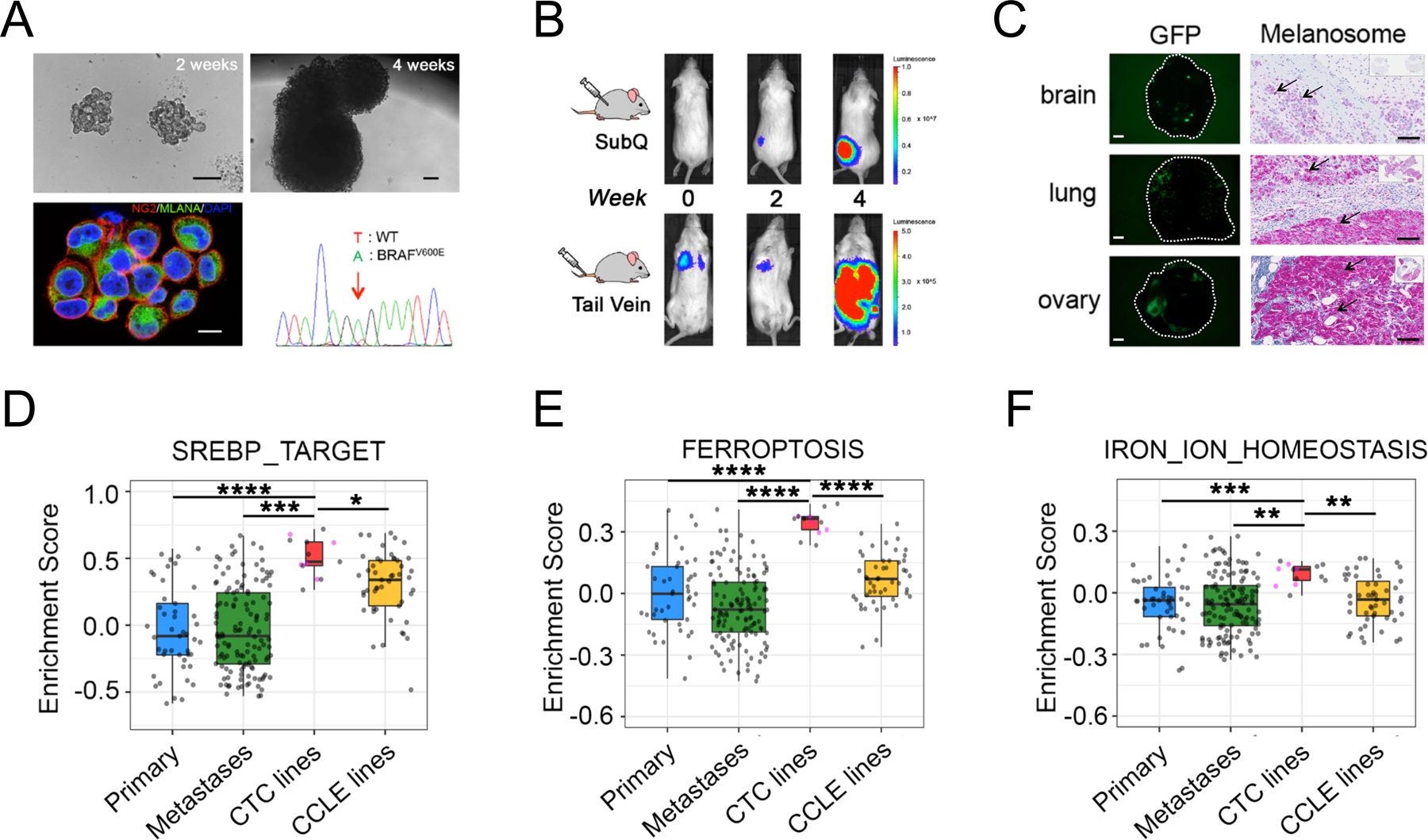
(A) Characterization of ex vivo-cultures of melanoma Mel-167 CTCs. Upper panels: bright field images of suspension cultures at 2 weeks (left, scale bar: 100 μm) and 4 weeks (right, scale bar: 100 μm); Lower left: Representative immunofluorescence image of cultured CTCs co-stained for melanoma markers CSPG4 (red) and MLANA (green), with nuclear DAPI (blue). Scale bar: 10μm. Lower right: DNA sequencing identifies the heterozygous BRAFV600E mutation (T-> A at nucleotide 1799) in the cultured melanoma Mel-167 CTCs.
(B) Melanoma Mel-167 CTCs are tumorigenic in mice. Primary tumors (Upper panel) and metastases (Lower panel) following subcutaneous and tail vein injection in NSG mice, respectively, of GFP-luciferase-tagged Mel-167 CTCs, monitored over 4 weeks using in vivo imaging (IVIS).
(C) Intra-cardiac inoculation of GFP-luciferase-tagged Mel-167 CTCs into NSG mice leading to metastases in brain, lung and ovary. Representative images of GFP expressing tumor cells in tissue sections (Left panels, hatched circles, scale bar: 2mm) and immunohistochemical staining for melanosomes (Right panels, purple stain of individual melanoma cells marked by black arrows, with whole tissue section shown on the top right box; scale bar: 100 μm).
(D-F) GSVA pathway enrichment box plots of (D) SREBP_TARGET, (E) FERROPTOSIS and (F) IRON_ION_HOMEOSTASIS, comparing melanoma CTC cell line samples (n = 13, including 5 distinct lines colored in pink and 8 sample repeats of these 5 lines in grey) with TCGA high purity primary melanomas (n = 45) (Primary), TCGA high purity metastatic melanomas (n = 129) (Metastases) and CCLE melanoma cell lines (n = 49). The mean GSVA enrichment scores were calculated for replicates of each CTC line. Pairwise comparisons between CTC lines and the other three categories were performed and statistical significance was assessed by two-sided Welch’s t-test. ****P < 0.0001; ***P < 0.001; ** P < 0.01; * P < 0.05. Curated pathway signature gene lists are found in Supplementary Table S2.
Having established robust tumorigenic and metastatic competent CTC cultures, we compared their transcriptomic profiles with those of standard melanoma tumor-derived cell lines (Cancer Cell Line Encyclopedia; CCLE), and primary and metastatic melanoma tumors (The Cancer Genome Atlas; TCGA) (28,29). By gene set variation analysis (GSVA)(30), we found the most significantly enriched pathways in the CTC cell lines were those involved in lipogenesis (“SREBP_TARGET”, “FATTY_ACID_METABOLISM”, “ADIPOGENESIS” and “CHOLESTEROL_HOMEOSTASIS”), along with iron-related pathways, including “OXIDATIVE_PHOSPHORYLATION”, “FERROPTOSIS”, and “IRON_ION_HOMEOSTASIS”, after correction for batch effect (Figures 1D–F, S2A–B). The dramatic enrichment of multiple pathways in CTC lines appears to be independent of tumor site of origin, or differences in culture media and hypoxic conditions in vitro. A modest increase in SREBP_TARGET signatures is also observed in CCLE cell lines when compared to primary tumor samples (Figure 1D, Tables S1–S3). Thus, transcriptional profiling of cultured melanoma CTC lines from multiple patients, compared with traditional melanoma cell lines and with primary melanoma specimens reveals concurrent upregulation of lipogenesis and iron-dependent metabolic signatures.
Single cell-derived CTC cultures exhibit innate resistance to BRAF inhibition through enhanced lipogenesis
Mel-167 CTC cultures, established prior to any treatment administration from a patient who was newly diagnosed with metastatic melanoma, are multi-clonal in origin. Despite having the characteristic BRAFV600E drug-sensitizing mutation, the patient had only a transient (<3 months) clinical response to the standard combined BRAF/MEK inhibition regimen, suggesting the presence of intrinsically drug resistant cancer cells at the time of diagnosis. Upon generating 13 single CTC-derived clones from the pre-treatment specimen, we indeed observed a range of sensitivity to the BRAF inhibitor vemurafenib among these untreated cultured CTC clones (Figures 2A, B). Defining the resistant versus sensitive clones as those with an IC50 for vemurafenib that is either above or below that of the bulk CTC population, we first tested for mutational heterogeneity using whole-exome sequencing (WES) of four resistant clonal lines, versus two sensitive clones, the bulk parental Mel167 culture, and the matched primary melanoma tumor (Figure S3A). No new driver mutations were detected across these clonally-related lines that could account for the differential drug sensitivity. However, RNA-seq profiles of all 13 isogenic CTC lines, followed by GSVA gene set analysis, identified lipogenesis pathways as being highly enriched in the drug-resistant CTC clones, compared with sensitive lines (CHOLESTEROL_HOMEOSTATIS, P=0.0098; SREBF1_TARGET, P=0.0135; SREBF2_TARGET, P=0.0212 and SREBP_TARGET, P=0.0201) (Figures 2C, S3B). The individual clones were heterogeneous with respect to tumorigenic phenotypes (Figure S3C). Thus, upregulation of SREBP-dependent lipogenesis is correlated with innate resistance to BRAF inhibition, within a subset of untreated patient-derived isogenic clonal CTC lines.
Figure 2. Clonal heterogeneity in SREBP-dependent lipogenesis as a mediator of intrinsic resistance to BRAF inhibitor among melanoma CTCs.
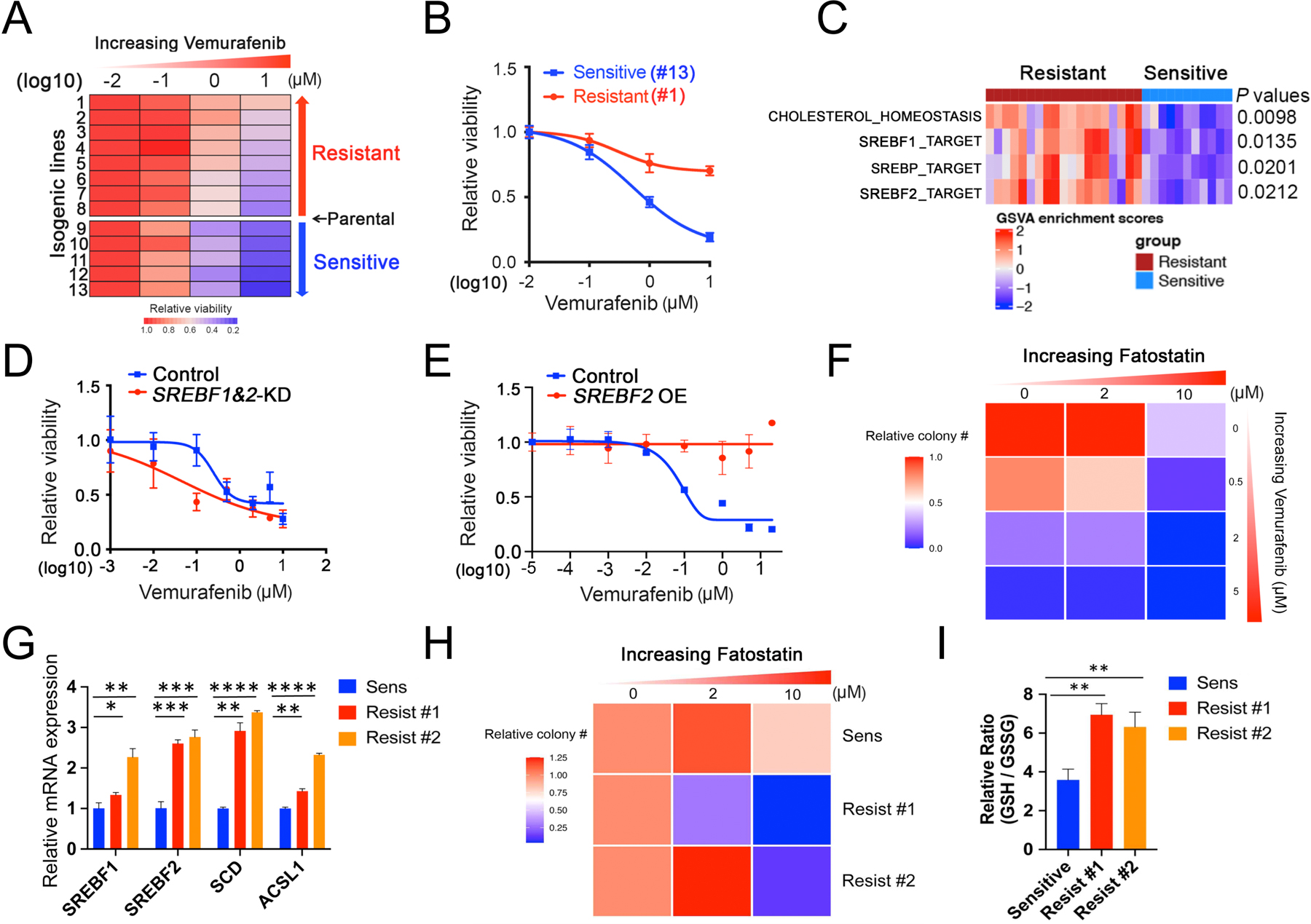
(A) Heatmap of BRAF inhibitor (BRAFi, vemurafenib) drug sensitivity performed on 13 single CTC-derived isogenic clones derived from one pretreatment blood sample from patient Mel-167 (tumor is BRAFV600E positive). Rows: left to right, relative cell viability with increasing vemurafenib concentrations: 0 (DMSO control), 0.1, 1 and 10 μM. Columns: top to bottom, isogenic CTC lines ranked from lowest to highest sensitivity to BRAFi. The 13 isogenic lines are divided into sensitive (n = 5) versus resistant (n = 8) groups based on their BRAFi sensitivity compared to the uncloned parental CTC culture. The isogenic lines have virtually identical mutational profiles compared to the primary tumor specimen, the parental Mel-167 CTC culture and with each other (refer to Figure S3A).
(B) Vemurafenib sensitivity curves of two representative isogenic melanoma CTC lines, #1 (sensitive, blue line) and #13 (resistant, red line). Y-axis, relative cell viability; X-axis, drug concentrations in log(10) scale. Statistical significance is assessed by two-sided T test with unequal variance when comparing differences in cell viabilities between line #1 and line #13 treated with Vemurafenib at different concentrations: P = 0.0009 (1 μM) and P = 7.56 × 10−7 (10 μM).
(C) Heatmap showing GSVA enrichment scores of pathways that were significantly different between BRAF inhibitor-sensitive (blue) and resistant (red) isogenic CTC lines. The statistical significance was defined as the gene sets with FDR-adjusted P values < 0.05 and absolute mean difference in GSVA scores between two groups > 0.25. P values were assessed by two-sided Welch’s t-test. The heatmap for all pathways is shown in Fig S3B.
(D) Vemurafenib sensitivity of the BRAFV600E mutant CTCs following depletion SREBF1 and SREBF2 using siRNAs, compared with cells treated with control. Y-axis represents relative viability; X-axis represents drug concentration in μM, log10 scale. The knockdown efficiency against SREBF1 and SREBF2 are shown in Figure S3C. Statistical significance is assessed by two-sided T test with unequal variance. P = 0.0121 when comparing differences in cell viabilities between control and SREBF1&2-KD groups treated with 0.1 μM Vemurafenib.
(E) Vemurafenib sensitivity of the BRAFV600E mutant CTCs with ectopic expression of a mature and activated form of SREBF2. Vector transfected cells are shown as controls. Y-axis represents relative viability; X-axis represents drug concentration in μM, log10 scale. The mRNA and protein level of SREBF2 overexpression is shown in Figures 3H and 3I, respectively. Statistical significance is assessed by two-sided T test with unequal variance and P values are generated when comparing cell viabilities between control and SREBF2 OE groups treated with Vemurafenib at different concentrations: P = 0.0022 (0.1 μM); P = 0.0417 (1 μM ); P = 0.0126 (5 μM); P = 2.11 ×10−6 (20 μM).
(F) Heatmap representing soft agar colony numbers following treatment of Mel-167 CTCs with increasing concentrations of the BRAFi Vemurafenib and SREBP inhibitor Fatostatin, which show cooperative cell toxicity. The drug effect on colony size is shown in Figure S3E.
(G) Elevated expression of SREBF1, SREBF2 and their downstream targets SCD and ACSL1 in parental Mel-167 CTC cultures (sensitive) and in two clones with acquired resistance to Vemurafenib (clones #1, #2). Y-axis: relative fold change of mRNAs (real time qPCR) shown between sensitive parental and the resistant lines (Actin internal control). Data was obtained from three biological repeats. Statistical significance is assessed by two-sided T test with unequal variance. P = 0.0001 for SREBF1; P = 0.0008 for SREBF2; P = 0.0009 for SCD; P = 0.0005 for ACSL1. **P < 0.001.
(H) Heatmap representing soft agar clonogenic ability of the parental (sensitive) Mel-167 CTCs, compared with two Vemurafenib-resistant derivative clones (#1, #2), following treatment with increasing concentrations of the SREBP inhibitor Fatostatin. The Vemurafenib-resistant CTCs show increased sensitivity to Fatostatin. Statistical significance was assessed by two-sided T test with Welch’s correction (P = 0.0168, comparing resistant clone #1 to sensitive line; P = 0.0139 comparing resistant clone #2 to sensitive line). The drug effect on colony size is shown in Figure S3F.
(I) Increased ratio of reduced to oxidized glutathione (GSH/GSSG), indicative of enhanced reductive capacity, in two Mel-167-CTC clonal lines with acquired resistance to Vemurafenib compared with the control sensitive cells. Statistical significance was assessed by two-sided T test with Welch’s correction. P = 0.0020, comparing Resist #1 to Sensitive and P = 0.0092 comparing Resist #2 to Sensitive line. ** P < 0.01.
To test the functional consequences of modulating SREBP activity in these untreated cultured melanoma CTCs, we achieved double knockdown of SREBF1 and SREBF2 using siRNA combinations in parental Mel-167 CTCs, showing significant enhancement of sensitivity to the BRAF inhibitor vemurafenib (Figures 2D, S3D). Conversely, doxycycline-inducible expression of a mature isoform of SREBF2, which bypasses normal cellular processing and directly translocates to the nucleus to activate lipogenic signaling (31), results in a high degree of resistance to vemurafenib (Figures 2E). We also tested the effect of the SREBP inhibitor Fatostatin: Mel-167 CTCs display strong cooperative sensitivity to vemurafenib and Fatostatin in suppressing soft agar colony number (Figure 2F) and soft agar colony size (Figure S3E), established in vitro correlates of tumorigenicity.
To test whether the effect of SREBP-dependent lipogenesis extends from innate resistance to vemurafinib in untreated melanoma cells to acquired drug-induced resistance, we cultured Mel-167 CTCs in the presence of 1μM vemurafenib for 3 months, generating two independent resistant CTC lines. In both resistant lines, we find a significant increase in expression of endogenous SREBF1 and SREBF2, along with increased expression of the SREBP target genes SCD and ACSL1 (Figure 2G). Both Mel-167 CTC lines with acquired vemurafenib resistance display enhanced sensitivity to the SREBP inhibitor Fatostatin, as measured by reduced number and size of soft agar colonies (Figures 2H, S3F). Finally, consistent with an effect of lipogenic regulation on cellular redox potential, both SREBP-high, vemurafenib-resistant Mel-167 clones show an increased GSH/GSSG ratio, indicative of enhanced ROS neutralizing capacity (Figure 2I, total GSH and GSSG levels are shown in Figures S3G–H).
Taken all together, resistance by BRAF-mutant CTCs to vemurafenib is correlated with SREBP activity, both among heterogeneous clonal cultures drawn from an untreated patient, as well as following in vitro drug selection to generate acquired resistance. Suppression of SREBP activity by knocking down both SREBF1 and SREBF2 or treatment with Fatostatin abrogates this drug resistance phenotype, while ectopic SREBF2 expression alone is sufficient to dramatically enhance vemurafenib resistance.
SREBF2 induces Transferrin Transcription
SREBF1 and SREBF2 are central players of lipogenesis, with SREBF1 primarily linked to lipid metabolism and SREBF2 being the master regulator of cholesterol synthesis. Both SREBP paralogs are known to be aberrantly expressed in some cancers, although their functional significance in carcinogenesis remains poorly understood (32,33). To determine which SREBP genes are functionally important in mediating the melanoma CTC tumorigenic and drug resistance phenotypes, we first compared SREBF1 and SREBF2 mRNA expression levels in CTC lines using RNA-seq. All 5 CTC lines show higher abundance of SREBF2 mRNA, compared SREBF1 (Figure S4A). Consistent with this finding, higher activity of SREBF2, compared to SREBF1, is detected by GSVA enrichment scores of SREBF1/2 target gene expression in these CTC lines (Figure S4B, Table S2). We used specific antisense oligos (ASOs) to effectively suppress SREBF1 or SREBF2 individually, or both together (SREBF1-KD, SREBF2-KD and SREBF1&2-KD, respectively) in the Mel-167 CTC line (Figure 3A), quantifying clonogenic growth in soft agar, a well-established correlate of tumorigenesis. SREBF2-KD significantly suppresses soft agar growth, whereas SREBF1-KD has no effect (Figure 3B). The combination of the two ASOs (SREBF1&2-KD) demonstrates modestly increased colony suppression, compared with SREBF2-KD alone (Figure 3B, P = 0.0480 for SREBF2-KD vs Control and P = 0.0277 for SREBF1&2-KD vs Control). Interestingly, mTOR signaling has been implicated in lipogenic signaling (34), and while its inhibitor Torin 1 suppresses expression of SREBF1 in Mel-167 CTCs, it does not affect levels of SREBF2 (Figures S4C, S4D). Thus, while SREBF1 may enhance the effect of SREBF2, the latter is most relevant to Mel-167 CTC tumorigenesis.
Figure 3. Transferrin is a transcriptional target of SREBF2.

(A) Knockdown of SREBF1 (SREBF1-KD), SREBF2 (SREBF2-KD) or both (SREBF1&2-KD) in Mel-167 cultured CTCs, using antisense oligonucleotides (ASO), demonstrating both specificity for each individual gene and effective dual targeting. Y-axis, relative mRNA fold change normalized to Actin. Statistical significance was assessed by two-sided Welch’s t-tests. ** P < 0.01; *** P < 0.001; *** P < 0.0001.
(B) Quantification of soft agar colony numbers formed by Mel-167 CTCs transfected with control, SREBF1-KD, SREBF2-KD or SREBF1&2-KD ASO sequences. Y-axis, relative colony number normalized to control. Statistical significance was assessed by two-sided Welch’s t-test. * P < 0.05; n.s, not significant.
(C) Heatmap representation of SREBF2 ChIP-seq in Mel-167 CTCs, showing enrichment of SREBF2 binding sites within the transcriptional start sites (TSS) of genes comprising three pathways: SREBP_TARGET, FERROPTOSIS and IRON_ION_HOMEOSTASIS. For each pathway, the first two columns represent replicate experiments, and the third shows the input reads. The GSEA pathway enrichment plots and assessment of statistical significance of SREBF2 ChIP-seq are shown in Table S4. The heatmap color scale (Y-axis) represents the read intensities in bins per million mapped reads (BPM: set the maximum value at 3).
(D) Venn diagram showing the top 10 genes at the intersection of SREBF2 bound promoters in CTCs and genes with increased expression in CTCs, compared with primary and metastatic melanoma (TCGA) and standard tumor-derived melanoma lines (CCLE). The mean fold change in individual gene expression is listed below the Venn diagram, with transferrin (TF) as the top hit.
(E) Integrative Genomic Viewer (IGV) plot showing SREBF2 ChIP-seq peaks in the TF gene promoter region (framed in red). Two experimental repeats (rep 1, 2) are shown. Input genomic DNA serves as control. The scales in bins per million mapped reads (BPM) of peak window for each sample are shown in brackets, and the genomic structure of the TF gene is shown below the IGV plot.
(F) In vivo binding of SREBF2 to the TF gene promoter, as shown by ChIP-qPCR analysis in Mel-167 melanoma CTCs. Top: Schematic representation of expected qPCR products (#1, 2, 3, 4) spanning regions of the TF gene promoter including those containing the two predicted SREBP binding sites (#2, 3), which are shown in blue and red (35). Bottom: ChIP-qPCR performed using anti-FLAG antibody to precipitate FLAG-SREBF2-DNA complexes. Y axis shows relative fold enrichment of TF gene promoter fragments (normalized to control IgG antibody), with strong in vivo binding of SREBF2 to fragments #2 and #3 that contain the SREBP consensus sequences, but not to neighboring fragments (#1 and #4) or to unrelated sequences (a, b). Data are normalized to 2% of total genomic DNA input.
(G) Suppression of TF mRNA expression in Mel-167 CTCs, following treatment with ASOs targeting SREBF1 and SREBF2 alone or together, compared with control (see Figure 3A for knockdown efficiency and specificity), demonstrating that SREBF2 is the primary regulator of TF expression, with modest enhancement by combined SREBF1&2 KD.
(H) Induction of TF mRNA expression by SREBF2 in Mel-167 CTCs, demonstrated by real-time q-PCR analysis, 48 hours following doxycycline-mediated inducible expression of SREBF2. SREBF2 also mediates a modest increase in SREBF1 mRNA. Data are normalized to actin. Y-axis shows relative fold change in SREBF2-expressing cells compared with uninduced controls. Statistical significance was assessed by two-sided Welch’s t-test. P = 0.003 for TF; P = 0.0002 for SREBF2; P = 0.0009 for SREBF1. **P < 0.01; ***P < 0.001.
(I) Induction of TF protein by doxycycline-inducible FLAG-tagged SREBF2, quantified by Western blot analysis in Mel-167 CTCs. Actin is shown as loading control.
To identify SREBF2-regulated genes mediating these tumor-enhancing effects, we undertook chromatin immunoprecipitation followed by Next Generation Sequencing (ChIP-Seq), defining direct SREBF2 transcriptional targets in cultured melanoma CTCs. Well established SREBP targets implicated in lipogenesis are readily identified by ChIP-Seq (Figure 3C and Table S4). However, additional in vivo SREBP binding sites are also identified in the promoters of multiple genes involved in ferroptosis and iron homeostasis (Figure 3C and Table S4), supporting the coordinated expression of these pathways, previously observed in multiple CTC lines (Figures 1D–F). Key to the intersection between these pathways appears to be the iron binding protein Transferrin (TF): Comparing the set of genes commonly upregulated across the cultured melanoma CTCs versus primary tumors, metastases and CCLE melanoma lines (N=586), with the set of genes whose promoters are bound by SREBF2 in CTCs (N=3813), we identified 258 candidate targets, of which the most highly upregulated hit is TF (Mean fold change = 54.19, Figures 3D, S5A). In an independent analysis, we compared RNA-seq transcriptional profiles of two melanoma CTC lines versus their individually matched archival tumor specimens: Mel-167 (CTCs versus matched primary tumor) and PEM-22 (CTCs versus 6 patient-matched independent metastatic lesions). Among ten differentially expressed transcripts shared in these two pairwise comparisons (genes upregulated > 32 fold in CTCs), TF was the most highly differentially expressed (1st hit in PEM-22, 4th in Mel-167) (Figure S5B). Interestingly, endogenous TF expression is also significantly upregulated in the clonally-derived Mel-167 CTC lines with intrinsic vemurafenib resistance, compared with the sensitive isogenic clones (Figure S5C). We therefore selected the iron binding protein TF as a novel candidate SREBF2 target, with potential relevance to lipogenic regulation of iron metabolism and tumorigenesis.
Direct visualization of the ChIP-Seq reads confirms a sharp peak of SREBP binding at the site of the TF gene promoter (Figure 3E). The promoter contains two adjacent predicted SREBP binding sites (Catalog of Inferred Sequence Binding Preferences; CIS-BP) (35): motifs M2388_1.02, ATGAGGTGAT, and M6488_1.02, GGGTTGGGAGAGG (Figures 3F, S5D). Direct in vivo binding of SREBF2 to TF promoter fragments containing the two predicted sites is evident using chromatin immunoprecipitation followed by PCR amplification of the DNA sequences (ChIP-qPCR) (Figures 3F, S5D). We note that ASOs targeting SREBF2 suppress TF expression effectively, while knockdown of SREBF1 has a mild effect on TF expression in Mel-167 CTCs (Figure 3G). Soft agar growth suppression is only observed in SREBF2-KD but not in SREBF1-KD Mel-167 CTCs (Figure 3B), and a modest enhancement of TF suppression is evident with the combined knockdown (Figure 3G). mTOR inhibition, which suppresses SREBF1 but not SREBF2 expression, also does not affect TF expression (Figures S4C, S4D). Finally, to confirm the functional consequence of these chromatin binding studies, we used doxycycline-inducible SREBF2, demonstrating robust induction of TF mRNA and TF protein expression in cultured melanoma CTCs (Figures 3H, 3I). Taken all together, TF is a novel SREBF2 target gene in melanoma CTCs, raising the possibility that may contribute to the lipogenic tumorigenesis phenotype through modulation of iron homeostasis and ferroptosis.
Transferrin enhances tumorigenesis by melanoma CTC cultures
TF plays a critical role in regulating iron trafficking and metabolism. It is normally expressed only in the liver, from which it is secreted into the bloodstream where it binds tightly to iron, ultimately interacting with the transferrin receptor expressed by multiple cell types and internalized (36). While expression of the Transferrin Receptor (TFRC) gene is common in cancer (37), TF transcripts are rarely detected except in liver and in some cell types in the brain (38,39). To exclude the possibility that aberrant TF expression in CTCs results from in vitro culture conditions, we undertook single cell RNA-seq of individually picked melanoma CTCs following microfluidic enrichment from primary blood specimens. Among 76 individual CTCs freshly isolated from 22 patients and validated to be melanoma cells by their expression of melanoma lineage markers (24), 7 (9.2%) express TF mRNA (mean expression = 17.64, Transcripts Per Million (TPM)) (Figure 4A). Such fresh CTCs include both viable and pre-apoptotic cells with variable RNA quality. However, TF expression is detectable in as many as 9/20 (45%) single CTCs that have been incubated in culture medium for 4–8 weeks, a condition in which only viable CTCs persist, but before they initiate in vitro proliferation (mean expression = 90.14 TPM) (Figure 4A). No TF expression is evident in individually selected leukocytes nor is it detectable in similarly isolated prostate or hormone receptor-positive breast CTCs, suggesting a melanoma-specific pathway (Figure S5E)(25,40,41).
Figure 4. Depletion of Transferrin impairs tumor formation.
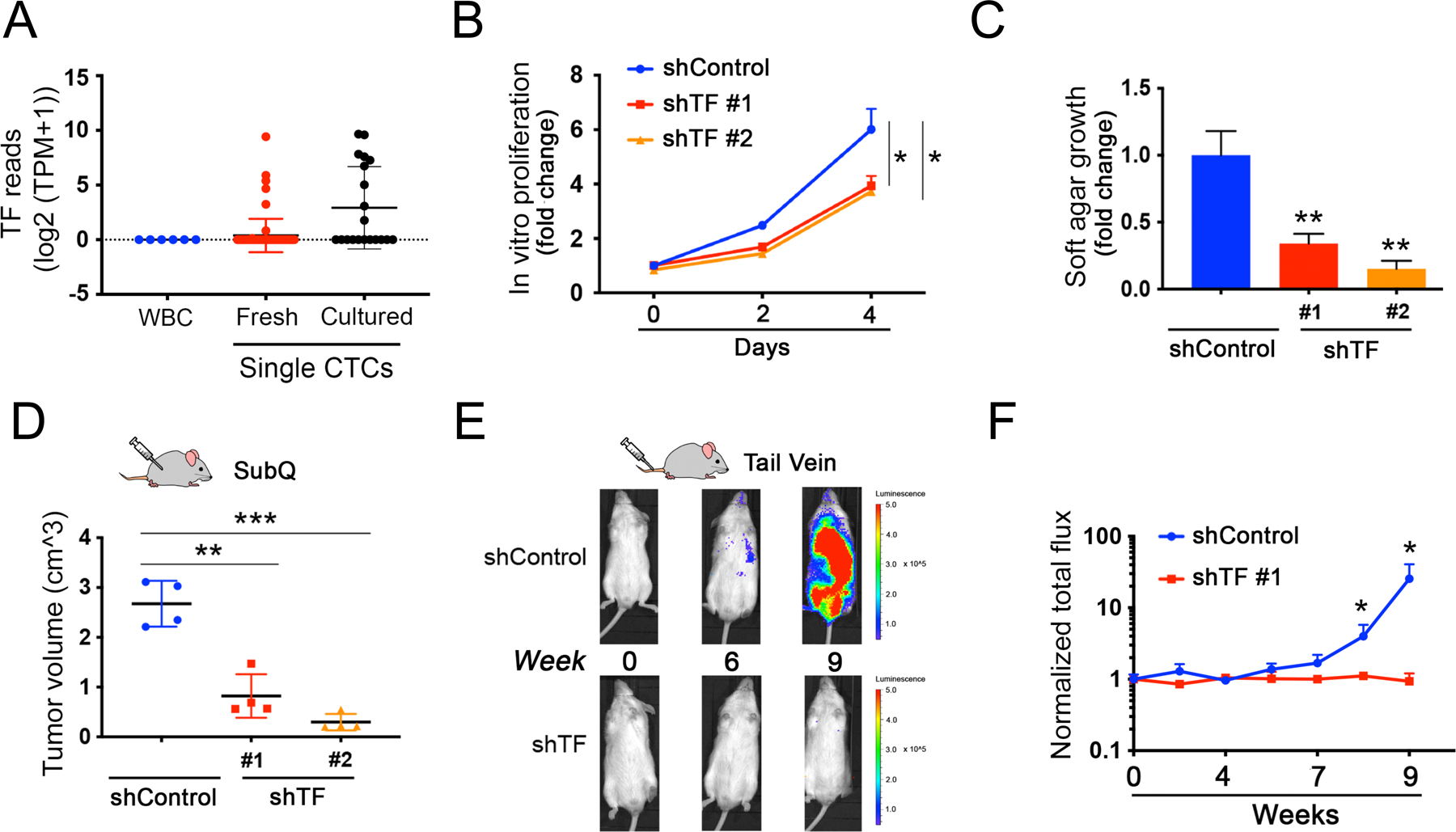
(A) Expression of TF transcripts within single melanoma CTCs freshly isolated from patients with metastatic melanoma by microfluidic negative depletion platform (n=76 single CTCs from 22 patients), or in CTCs that were incubated in culture medium for <8 weeks after microfluidic isolation. Short term culture ensures collection of viable CTCs with intact RNA, and precedes the initiation of in vitro proliferation (n=20 single CTCs). The fresh and cultured single CTCs are compared with contaminating leukocytes (WBC), from healthy donor blood identically processed through the microfluidic chip (WBC; n=6), Y axis, log2(TPM+1). TPM, Transcripts Per Million.
(B) Modest suppression of in vitro proliferation by Mel-167 CTCs, following TF-knockdown mediated by either of two independent shRNA constructs (knockdown efficacy shown in Fig S7A). compared with cells transfected with shControl. Two-sided Welch’s t-test was employed to assess the differences in proliferation rates between shTF and shControl at day 4. Y-axis: relative fold change normalized to day 0. *P < 0.05.
(C) Reduction in soft agar colony formation by Mel-167 melanoma CTCs, following TF knockdown using two independent shRNA constructs, versus shControl (see Figure S7A). Colonies were quantified by automated imaging at 4 weeks. Statistical significance from four independent experiments was assessed by two-sided Welch’s t-test. ** P < 0.01. Y-axis, relative fold change normalized to shControl.
(D) Suppression of subcutaneous melanoma formation by Mel-167 CTCs, following TF-Knockdown. Tumor volume following subcutaneous inoculation of tumor cells was quantified using 4 mice per experimental condition. Statistical significance was assessed by two-sided Welch’s t-test. **P < 0.01; ***P < 0.001.
(E-F) Abrogation of intravenous (tail vein) metastasis by Mel-167 melanoma CTCs following TF-Knockdown. (E) Metastatic burden was monitored in GFP-luciferase tagged CTCs using live imaging (IVIS), with representative images shown at time points 0, 6 and 9 weeks post injection. CTCs were infected prior to injection with either shTF or scrambled shRNA controls. (F) Time-course showing the quantification of metastatic tumors by in vivo luciferase imaging. Y-axis: averaged total flux of luciferase signal; shTF (n=4) and shControl (n=4) mice. Statistical significance was assessed by two-sided Welch’s t-test: P = 0.047 for week 8 comparisons and P = 0.046 for week 9 comparisons. * P < 0.05. Tissue-specific histological quantitation of metastases (lungs, liver, kidneys) is shown in Figures S7D–F.
Since TF is primarily a secreted protein, we tested whether a fraction might be retained intracellularly. We compared the liver cancer-derived HepG2 cells with the melanoma CTCs Mel-167, demonstrating comparable levels of TF mRNA (Figure S6A). Minimal baseline levels of TF protein are present in the culture media that is used to maintain either HepG2 or Mel-167, with or without addition of FBS or B27 supplements (Figure S6B). Following cell culture, TF is detectably secreted into the conditioned medium, and under comparable conditions, we calculate that Mel-167 CTCs secrete 45% of the TF secreted by HepG2 cells (Figure S6C). However, compared with HepG2 cells, Mel-167 CTCs retain 620% higher levels of TF intracellularly (Figure S6D) (calculated as 19.22 ng/ml from 1 million cells for Mel-167 versus 3.10 ng for HepG2 cells), with cell fractionation experiments showing most of the intracellular TF present in cytoplasmic and membrane fractions (Figures S6E). Intracellular retention of TF protein in Mel-167 CTCs may be partially explained by the presence of alternatively spliced transcripts, approximately 1.4% of which lack the secretion signal sequence which spans exons 1 and 2 (Figures S6F, S6G). Alternatively, it is also possible that an increase in secreted TF may result in more recycled TF-TFRC complex within the endosomal compartment.
To determine the functional consequences of aberrant TF expression in melanoma CTCs, we tested the effect of two independent shRNAs in Mel-167 CTCs. TF knockdown (TF-KD) using either of these shRNA constructs leads to a modest reduction in proliferation under baseline in vitro culture conditions (reduced by 34.5% for shTF#1, P = 0.0272 and 35.5% for shTF#2, P = 0.0384 at day 4 of growth when compared to shControl; Figures 4B, S7A, S7B), but a marked suppression of clonogenic activity by 65.9% for shTF#1 (P = 0.0026) and 84.8% for shTF#2 (P = 0.0013) in soft agar (Figures 4C, S7C). Subcutaneous tumor formation is suppressed by either shRNA construct in Mel-167 CTCs (a reduction in tumor size by 69.2% for shTF#1, P = 0.0011 and 88.9% for shTF#2 as compared to shControl, P = 0.0008; Figure 4D), and metastatic colonization following tail vein injection is abrogated, as measured by whole mouse imaging (IVIS) (Figures 4E, 4F), as well as histological analysis of lung, liver and kidney metastases (Figures S7D–F).
To confirm the specificity of this effect, we generated a synthetic TF cDNA, in which mutation of three 3rd-position nucleotides in the coding region of TF confers resistance to knockdown by shTF#1, without affecting expression levels (TF ALT, Figures S8A, S8B). In Mel-167, as well as in Mel-182–2, a second, independently isolated CTC line derived from another patient with metastatic melanoma, TF knockdown is effectively rescued by ectopic expression of TF ALT, resulting in restitution of TF mRNA and protein expression (Figures S8C, S8D, S8E, S8F). In both CTC-derived cell lines, TF depletion leads to the suppression in soft agar colony growth, which is largely rescued by overexpression of TF ALT (Mel-167: P = 0.0004 comparing shTF #1 to shTF#1 + Rescue; Mel-182–2: P = 0.0027 comparing shTF#1 to Control; P = 0.0159 comparing shTF#1 to shTF#1 + Rescue) (Figures S8G, S8H). Therefore, in melanoma CTCs that have acquired aberrant expression of TF, its suppression compromises tumorigenic phenotypes.
Transferrin modulates ferroptotic cell death
While the function of TF in regulating iron levels in the blood circulation is well established, it may also play a role in regulating intracellular iron through a sequestration mechanism, similar to iron-complexed ferritin, the primary source of intracellular iron storage(42). Depletion of intracellular iron may compromise key iron-dependent pathways, while its over-abundance may trigger iron-dependent cell death pathways, such as ferroptosis (43,44). Gene Set Enrichment Analysis (GSEA) analysis shows a predominant signaling pathway upregulated in TF-KD Mel-167 CTCs to be “REACTIVE_OXYGEN_SPECIES” (P = 0.0049, Figure 5A and Table S5). Indeed, TF-KD cells have a significant increase in reactive oxygen species (ROS) compared with shRNA controls, as measured by a fluorescent reporter (68% increase for shTF#1, P = 0.0028; 118% increase for shTF#2, P = 0.0104) (Figure 5B). Similarly, lipid peroxidation is strikingly increased in TF-KD melanoma CTCs, compared with controls (Figure 5C). This effect is iron-dependent because co-treatment of Mel-167 CTCs with the iron chelator deferoxamine (50 μM) results in restoration of lipid ROS to normal levels, comparable to those observed following TF rescue through expression in Mel-167 CTCs of the synthetic shRNA-resistant cDNA TFALT (Figure 5C). Similar effects on lipid peroxidation following TF-KD, and rescue by either desferoxamine or the TFALT construct are evident in the second CTC line Mel-182–2 (Figure S8I).
Figure 5. Transferrin modulates ferroptotic cell death.
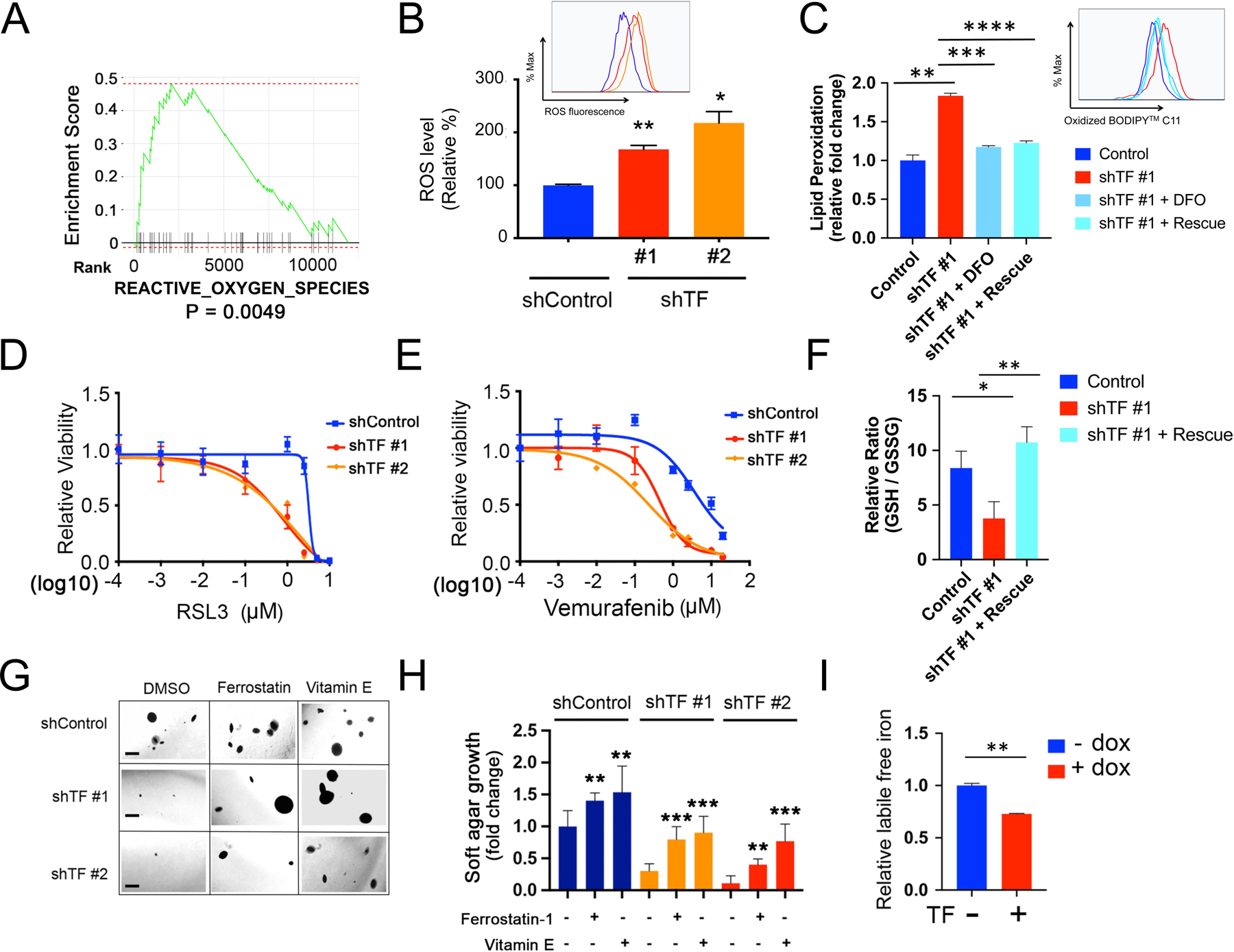
(A) GSEA pathway enrichment plot showing significant downregulation of “REACTIVE_OXYGEN_SPECIES” (ROS) pathway following TF-KD in Mel-167 melanoma CTCs, compared to shControl.
(B) Increased intracellular ROS in Mel-167 melanoma CTCs following TF-KD, using two independent shRNA constructs (#1, #2), compared with scrambled construct (shControl). ROS was quantified by flow cytometric analysis with a fluorescent ROS probe and the geometric mean of fluorescence signal was calculated for each group. The differences between the two groups were assessed by two-sided Welch’s t-test. statistical significance: P = 0.0028 for shTF#1 compared to shControl; P = 0.0104 for shTF#2 compared to shControl. ** P < 0.01; *P < 0.05.
(C) Quantification of lipid peroxidation levels by flow cytometry in Mel-167 CTCs following knockdown of TF (shTF#1), compared with shControl cells. The increase in lipid peroxidation induced by TF-KD is abolished either by the addition of the iron chelator deferoxamine (DFO; 50 μM with preincubation for 12 hours before flow cytometric assay), or by expression of TF ALT cDNA (rescue), a synthetic construct in which mutation of three 3rd position non-coding nucleotides renders TF resistant to targeting by shTF#1 without affecting expression levels (see Figure S8A, S8B). Lipid peroxidation is measured using BODIPY™ 581/591 C11 molecular sensor, and the fraction of cells positive for lipid peroxidation were calculated and data normalized to shControl. Y axis represents fold change. Statistical significance was assessed by two-sided Welch’s t-test. ** P < 0.01; *** P < 0.001; ****P < 0.0001.
(D) TF-KD enhances Mel-167 melanoma CTC cytotoxicity by the ferroptosis inducer RSL3. TF-KD was achieved using two different shRNAs, scrambled shControl. Y axis represents relative cell viability. X axis represents drug concentrations (μM) in log10 scale. Statistical significance was assessed by two-sided Welch’s t-test. When shTF#1 is compared to shControl: 1 μM, P = 0.0001; 2.5 μM, P = 0020. When shTF#2 is compared to shControl: 1 μM, P = 7.69 × 10−5; 2.5 μM, P = 0021; 5 μM, P = 0179.
(E) TF-KD increases sensitivity to vemurafenib in BRAFV600E-mutant melanoma Mel-167 CTCs. TF-KD was achieved using two different shRNAs and compared with scrambled shControl. Y axis represents relative cell viability. X axis represents drug concentrations (μM) in log10 scale. Statistical significance was assessed by two-sided Welch’s t-test. When shTF#1 is compared to shControl at different drug concentrations: 0.1 μM, P = 0.0246; 1 μM, P = 2.07 × 10−5; 2.5 μM, P = 2.14 × 10−6; 10 μM, P = 0.0001; 20 μM, P = 0.0007. When shTF#2 is compared to shControl: 0.01 μM, P = 0.0010; 0.1 μM, P = 0.0004; 1 μM, P = 9.38 × 10−6; 2.5 μM, P = 8.70 × 10−5; 10 μM, P = 0.0006; 20 μM, P = 0.0011.
(F) Reduced ratio of GSH/GSSG in TF-KD Mel-167, compared with vector Control. A synthetic TF ALT cDNA, resistant to shTF#1 knockdown, rescues the phenotype (see Figure S8A, S8B). Y axis, relative ratio between GSH/GSSG levels. Data are calculated based on three independent biological repeats and statistical significance was assessed by two-sided Welch’s t-test. P = 0.0216, comparing shTF#1 to Control and P = 0.0047 comparing shTF#1 + Rescue to shTF #1. * P < 0.05; ** P < 0.01.
(G) Suppression of soft agar colony formation by Mel-167 CTCs following TF-KD by two different shRNAs (Scrambled shRNA control). The clonogenic phenotype is rescued by two lipophilic anti-oxidants, Ferrostatin-1 (0.5 μM) and Vitamin E (20 μM). Representative images for each condition are shown. Scale bar: 500 μm.
(H) Quantitation of colony formation in soft agar shown in Figure 5F. Differences between the two groups were assessed by two-sided Welch’s t-test. Statistical significance: ** P < 0.01; *** P < 0.001.
(I) Quantitation of intracellular labile free iron in Mel-167 CTCs, using flow cytometry measurements of a fluorescent reporter (Goryo chemical). The normalized geometric mean of fluorescence intensity was calculated in CTCs expressing doxycline-inducible TF after 48 hours of dox treatment. Statistical significance was assessed by two-sided Welch’s t-test. ** P < 0.01. Similar results using Mel182–2 CTCs are shown in Figure S8N.
Consistent with these findings, TF-KD CTCs show increased sensitivity to the ferroptosis inducer RSL3 (45), compared with control, indicating that the increased ROS and lipid peroxidation are correlated with heightened susceptibility to ferroptosis (Figure 5D). Mel-167 CTCs, which carry the BRAFV600E mutation, also show increased sensitivity to vemurafenib following TF-KD (Figure 5E), suggesting that failure to suppress lipid peroxidation may also result in enhanced killing by BRAF inhibitors in melanoma CTCs. Previous studies have reported that BRAF inhibitor-resistant melanoma cells exhibit increased sensitivity to ferroptosis (46,47), suggesting cross-talk between these two druggable survival pathways. Ferroptosis itself is dependent on GPX4, which detoxifies lipid ROS at the expense of reduced glutathione (GSH), leading us to assess the GSH/GSSH ratio as a measure of redox balance. Indeed, the ratio of GSH/GSSG is significantly reduced in shTF-treated cells, an effect that is rescued by expression of the synthetic TF ALT construct (Figure 5F; total GSH and GSSG levels are shown in Figures S8J–K). Given the potent effect of TF-KD on soft agar clonogenic potential of melanoma CTCs, we tested a number of experimental rescue conditions. Both the anti-ferroptosis drug Ferrostatin-1 (0.5μM) and the lipophilic anti-oxidant Vitamin E (50μM) partially rescue the clonogenic suppression mediated by either of the two TF shRNA constructs (Figures 5G, 5H). Both of these anti-oxidants also have a moderate effect on baseline soft agar colony formation by melanoma CTCs. These observations are consistent with the role of TF expression in quenching intracellular free iron within melanoma CTCs.
To test the potential TF iron sequestration mechanism, we generated Mel-167 and Mel 182–2 CTCs with ectopic overexpression of TF. In both CTC lines, TF overexpression (Figures S8L, S8M) results in a significantly reduced pool of labile free iron, consistent with a sequestration mechanism (Figures 5I, S8N). To further test the functional effects of TF overexpression, we selected the CCLE melanoma cell line IGR-37, which does not express high levels of endogenous TF. Ectopic overexpression of TF in these cells leads to a reduction in lipid peroxidation (Figures S9A, S9B). Interestingly, ectopic expression of the mature and activated SREBF2 similarly reduces lipid peroxidation (Figures S9A, S9B). Ectopic overexpression of either TF or SREBF2 in IGR-37 melanoma cells also confers relative resistance to the ferroptosis inducer RSL3 (Figures S9C, S9D). All together, these observations support a model whereby TF expression regulates the intracellular labile free iron pool, impacting ROS and lipid peroxidation, with consequences for cellular susceptibility to ferroptosis.
The concordant expression of lipogenic and iron metabolic pathways observed in melanoma CTCs raises the possibility that these two pathways are mutually interegulated. We have shown that SREBF2 binds to the TF gene promoter and directly induces its expression, but modulation of TF levels themselves appears to affect SREBP targets, potentially through more indirect mechanisms. Indeed, TF-KD in melanoma CTCs leads to a reduction in expression of SREBP target genes: GSEA pathway analysis of RNA-seq data in TF-KD CTCs identifies lipogenesis as the most prominently downregulated pathway category, such as “SREBP_TARGET” (P=0.0024) (as well as “SREBF1/2_TARGET”, P=0.0022 for both pathways) and “CHOLESTEROL_HOMEOSTASIS” (P=0.0025) (Figures 6A, 6B; Table S5). Conversely, ectopic expression of TF leads to upregulation of the downstream SREBP target transcripts SCD and ACSL1 (Figures 6C, S8M). Furthermore, the suppression of ACSL1 protein expression resulting from shRNA knockdown of TF is fully rescued by expression of the non-degradable TF ALT construct (Figures S8E, S8F). Interestingly, addition of cholesterol (10μg/ml), an endpoint of lipogenic pathways, rescues TF-KD clonogenic suppression (Figures 6D, 6E). Thus, the iron homeostasis and lipogenic pathways appear to be mutually coregulated in melanoma CTCs, with SREBF2 directly activating the TF promoter, whereas TF expression may lead more indirectly to modulation of SREBP target gene expression and lipogenesis.
Figure 6. Transferrin expression modulates SREBP targets.
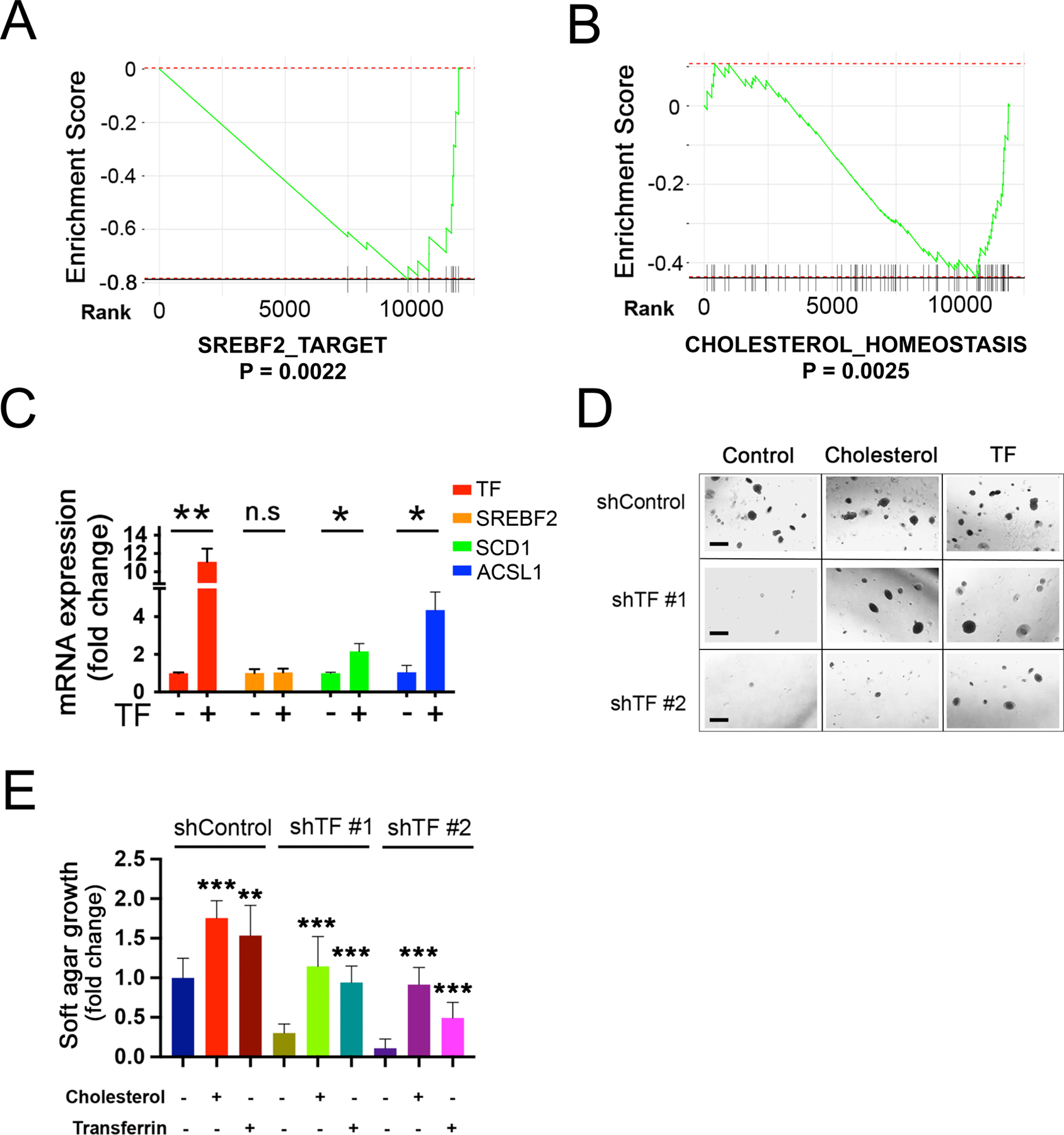
(A-B) Suppression of lipogenic pathways following TF-KD in Mel-167 melanoma CTCs. GSEA pathway enrichment plot showing significant downregulation of (A) “SREBP_2_TARGET” and (B) “CHOLESTEROL_HOMEOSTASIS”, in TF-KD CTCs, compared to shControl.
(C) Induction of downstream lipogenic effectors, following doxycycline-inducible expression of TF in Mel-167 CTCs (TF was induced within 96 hours post virus infection). Expression of SREBF2 mRNA and its downstream targets genes SCD and ACSL1 are shown (real time qPCR). Cells were grown under reduced nutrient conditions (RMPI with 2% B27) to sensitize SREBF2 activity. Y-axis: relative fold change in TF-overexpressing CTCs compared with control without dox treatment. Actin was used for internal normalization. Data were obtained from three independent biological repeats. Statistical significance was assessed by two-sided Welch’s t-test. P = 0.007 for TF; P = 0.868 for SREBF2; P = 0.039 for SCD; P = 0.017 for ACSL1. *P < 0.05; **P < 0.01.
(D-E) Rescue by exogenous cholesterol of soft agar colony formation defect in Me-167 melanoma CTCs following TF-KD. with two different shRNAs Scrambled shRNA was used as control (shControl). Cultures were incubated with either 10μg/ml cholesterol or 10 μg/ml recombinant TF protein (control). Representative images are shown in (D) (Scale bar, 500μm), quantification of colonies is shown in (E). The differences between two groups were assessed by two-sided Welch’s t-test. Statistical significance: ** P < 0.001; *** P < 0.0001.
Coregulation of SREBP and Iron Transport Pathways in Patient-derived Primary CTCs
To extend these findings from cultured melanoma CTCs to freshly isolated individual CTCs from patients with metastatic melanoma, we undertook comprehensive single cell RNA seq of 76 individual CTCs collected from 22 patients using microfluidic CTC-iChip isolation, followed by confirmation of cell identity by expression of melanoma lineage markers. (Figure 7A). Hierarchical clustering analysis reveals a well-demarcated subpopulation of CTCs, “cluster 2”, comprising 43 of the 76 single CTCs (57%), with concerted upregulation of lipogenic programs (“SREBF2_TARGET”, “ADIPOGENESIS” and “FATTY_ACID_METABOLISM”), iron homeostasis signatures (“IRON_ION_HOMEOSTASIS”) and proliferation (“MYC_TARGETS_V1 & V2”) (comparing Cluster 2 vs Cluster 1 CTCs, Figures 7A, S10A–D; Table S6). Cluster 2 CTCs also show signatures of increased energy production that is iron-dependent (“OXIDATIVE_PHOSPHORYLATION” (Figure 7A, Table S6). The enrichment of these pathways in Cluster 2 CTCs remains significant even if only CTCs from patients receiving immunotherapy were analysed (a total of 53 CTCs from 18 patients, Table S6). Since the majority of melanoma patients whose CTCs were analyzed here were enrolled on immune checkpoint therapies and some were treated with BRAF/MEK targeted therapies (Table S7), hence we were able to test whether these signatures were associated with responsive disease (RD) or progressive disease (PD), as measured by clinical imaging within a median of 74 days (−4 to 122) after CTC sampling (disease response was defined using RECIST1.1, see Table S7). Within a given patient blood draw containing two or more CTCs, a high (>60%) fraction of CTCs expressing signatures of “Lipogenesis” (SREBF2_TARGET), “Proliferation” (MYC_TARGETS_V2) and “Iron_Homeostasis” (IRON_ION_HOMEOSTASIS) (GSVA row Z-score >0) was highly associated with treatment failure, irrespective of treatment modality (Figure 7B). Breaking these down into individual gene expression signatures, for patients with a high “Lipogenesis” signature, 5/6 progressed on therapy, versus 0 of 10 patients with low signature CTCs (P=0.0014); for patients with high “Proliferation” CTCs, 4/5 progressed on treatment, compared to 1/11 patients with low signature CTCs (P = 0.0128); for patients with a high “Iron_Homeostasis” signature, 4/6 progressed, compared with 1/10 patients with low signature CTCs (P=0.0357); finally, for patients with a high “Oxi_Phos” signature, 3/4 progressed, compared with 2/12 patients with a low signature (P=0.0632) (Figure 7B). Thus, sampling CTC transcriptomic heterogeneity by single cell pathway-level analyses within a patient blood sample may identify subclonal populations of CTCs, with treatment-resistant signatures, correlated with adverse clinical outcome. Extending from these small pilot studies to large clinical datasets, we find that SREBF2 expression itself is highly correlated with reduced survival in patients with metastatic melanoma (TCGA high-purity metastases samples, P=0.0057), irrespective of treatment modality, an association that is not observed in localized primary tumors (TCGA high purity primary tumor, P=0.21) (Figure 7C, S10E, Table S8).
Figure 7. Coordinated upregulation of lipogenesis and iron homeostasis gene expression in patient-derived melanoma CTCs is correlated with poor clinical outcome.
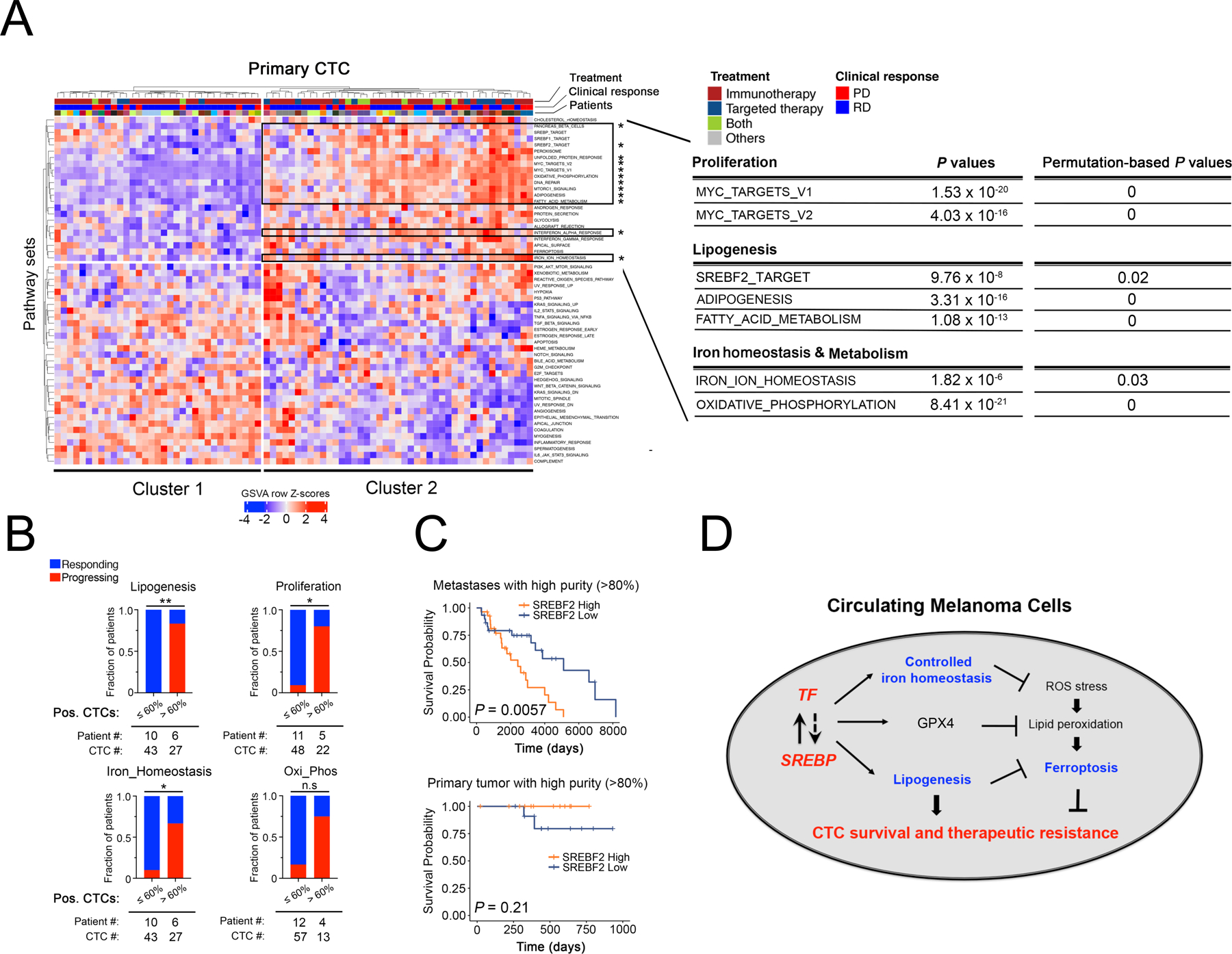
(A) Heatmap showing hierarchical clustering (Euclidean distance with complete linkage) of GSVA enrichment scores of pathways across 76 primary single CTCs obtained from 22 patients with metastatic melanoma (CTC lineage confirmed by expression of at least one melanoma-specific marker (24); see Figure S10 for CTC validation). The first row indicates treatment information of patients, from whom CTCs were isolated, that receive Immunotherapy (colored in dark red), Targeted therapy (dark blue), Both (Immunotherapy + Targeted therapy) or Others (grey). The second row indicates responding (blue) versus progressing (red) patients, and the third row indicates individual patients with color codes. The upper right block framed in black shows the subset of CTC samples (Cluster 2) with significantly enriched pathways marked by asterisks. Selected pathways upregulated in Cluster 2 CTC group vs Cluster 1 CTC group are listed on the right (Mean difference in GSVA enrichment scores between C1 and C2 > 0.20, FDR-adjusted P value < 0.05). Permutation-based P values were calculated as “the number of background P values lower than the observed P values from Figure 7A” divided by “the number of permutations which is 1,000”, as shown on the top right. GSVA scores of each pathway and expression levels of all genes within each selected pathway were listed in Supplementary Table S6.
(B) Stacked bar graphs showing the fraction of patients with either responsive disease (RD in blue) or progressing disease (PD in red), whose CTC populations were stratified by low (≤60%) or high (>60%) percentages of CTCs expressing markers of “Lipogenesis (SREBP_2_TARGET)” (upper left), “Proliferation (MYC_TARGETS_V2)” (upper right), “Iron_Homeostasis (Iron_Ion_Homeostasis)” (lower left), “Oxi_Phos (OXIDATIVE_PHOSPHORYLATION)”(lower right), using GSVA row Z score > 0 as a cutoff for each pathway signature. Only samples with 2 or more CTCs were included. Fisher’s exact test was performed to assess the association between CTC pathway signature expression and clinical outcome. * P < 0.05; ** P < 0.01. Clinical assessment of response or progression was ascertained after either immunotherapies or targeted therapies with a median time of 74 days following CTC collection.
(C) Kaplan-Meier plots of disease-specific survival in patients with either metastatic (upper panel) or primary melanoma (lower panel), classified according to SREBF2 mRNA expression. TCGA SKCM high purity melanoma samples (>80% tumor composition) were divided into “SREBF2 high” (SREBF2 mRNA expression higher than 75th percentile, orange) and “SREBF2 low” (SREBF2 mRNA expression lower than 25th percentile, blue) groups. Kaplan-Meier curves were plotted for disease specific survival (DSS) and P values were calculated using log-rank test. Y-axis, disease-specific survival probability; X-axis, time in days.
(D) Schematic model for coordinated expression of lipogenic and iron homeostatic pathways. The master lipogenic regulator SREBF2 directly induces expression of the iron carrier Transferrin, while TF mediated activation of SREBP signaling appears to be indirect. Increased intracellular TF reduces iron levels, ROS stress and lipid peroxidation, all of which serve to suppress ferroptosis and enhance CTC survival and drug resistance. Increased lipogenesis mediated by SREBP, including expression of the SREBF2 target GPX4 (Figure 3C) similarly reduces ferroptosis. This coordinated cross-talk mechanism between lipogenic and iron homeostatic pathways contributes to CTC-mediated tumorigenesis and therapeutic resistance.
DISCUSSION
Cancer heterogeneity is increasingly recognized as a major cause of therapeutic resistance, with outgrowth of either pre-existing or treatment-induced clonal subpopulations ultimately leading to disease progression (48). CTCs, comprising single metastatic precursor cells circulating in the bloodstream, thus provide an exceptional window into the diversity of drug-resistant phenotypes, while at the same time, offering insight into vulnerabilities of cancer cells as they briefly transit through the high oxygen blood environment (49). In this study using single cell RNA-Seq applied to melanoma CTCs, we have uncovered the aberrant coexpression of the lipogenic regulator SREBF2 and the iron-binding protein TF, modulating the cellular response to ferroptosis, with implications for both tumorigenesis and drug resistance (Figure 7D). In both melanoma CTC-derived long-term cultures, as well as in individual CTCs freshly isolated from patient-derived blood specimens, we find coregulation of lipogenic and iron homeostasis pathways. Together, they denote a subset of CTCs with treatment-resistant phenotypes, associated with an adverse clinical outcome. Before initiation of BRAF-targeted therapy, we established multiple clonally-derived CTC lines from a single melanoma patient, demonstrating a degree of cellular heterogeneity in intrinsic drug susceptibility that is correlated with endogenous SREBP activity. As these cultured CTCs are treated with vemurafenib, they display further overexpression of endogenous SREBP, and direct ectopic manipulation of SREBF2 profoundly modulates their drug sensitivity.
SREBPs are aberrantly activated in several cancer types, including subsets of glioblastoma, melanoma, kidney, prostate and breast cancers (32,50–53). While previous studies have linked PI3K and K-RAS oncogenic signals to SREBF1/2 activation (34,51,54), the actual consequences of SREBF1/2 signaling on tumorigenesis are uncertain. SREBF2 ChIP-Seq studies identify core target genes involved in cholesterol synthesis, glucose breakdown, and fatty acid synthesis, which may contribute to supporting the increased bioenergetic demands of proliferating tumor cells (33,55). In the context of melanoma CTCs, however, we unexpectedly observed that a substantial fraction of SREBF2 bound promoters regulate genes involved in iron homeostasis, in addition to classical lipogenic genes. Among these, TF is noteworthy both for its level of induction by SREBF2, as well as the dramatic consequences of its knockdown on tumorigenesis and metastasis phenotypes.
The intracellular functions of the iron binding transporter TF are poorly characterized. In CTCs with elevated endogenous TF expression, we find that its knockdown increases intracellular ROS and lipid peroxidation, lowering the GSH/GSSG ratio, and mediating a reduction in soft agar colony formation that is rescued by the lipophilic antioxidants Ferrostatin and Vitamin E. TF knockdown also enhances cellular sensitivity to the ferroptosis inducer RSL3, as well as to the BRAF inhibitor vemurafenib. Conversely, overexpression of TF in CTCs reduces labile free iron pools, consistent with a sequestration mechanism, and its ectopic expression in melanoma cells that lack endogenous TF expression leads to reduced lipid peroxidation and increased resistance to RSL3. While most TF protein is secreted from cells, a fraction is retained intracellularly, likely the result of alternative pre-mRNA splicing that excludes the 5’ secretion signal sequence. While we favor the concept that TF mediates iron sequestration intracellularly, we cannot exclude the possibility that it binds iron within the extracellular microenvironment, thereby reducing intracellular iron levels. As such, an iron transporting protein that is physiologically secreted by liver cells into the bloodstream appears to have been adapted by some cancer cells to help regulate intracellular iron levels.
The essential role of iron in heme biosynthesis and in multiple enzymatic reactions is well established, but the contribution of cellular iron levels to spontaneous and drug-induced ferroptosis is increasingly appreciated as a critical feature of cancer cell biology, which may be harnessed to modulate existing therapies (43,44,56). TF is physiologically produced by the liver and it regulates iron homeostasis through the Transferrin Receptor (TFRC), which is expressed in most tissues. While TFRC is overexpressed in many tumors (37,57), TF itself has not been implicated in malignancy. In TFRC-overexpressing cells, addition of iron-loaded TF to the culture medium increases iron import and appears to promote ferroptosis through iron overload (58). In contrast, in the setting of melanoma CTCs with endogenous expression of TF, it appears to function as a quencher of intracellular iron, leading to suppression of ferroptosis. Consistent with the importance of tightly regulated intracellular iron pools, we find that intracellular TF protein is abundant in melanoma CTCs and its suppression increases their levels of ROS, and oxidized lipids. Thus, melanomas CTCs appear to have hijacked this liver specific gene to modulate the availability of labile iron pools, oxidative stress and ferroptosis sensitivity.
The functional consequences of TF overexpression in melanoma CTCs appear to be considerable. Suppression of endogenous TF in these cells dramatically abrogates their blood-borne metastatic potential, and to a lesser extent their primary tumorigenesis. Soft agar clonogenic ability is also suppressed by TF knockdown, with a relatively modest effect on in vitro proliferation. Together, these observations highlight the important role played by ROS stress in controlling metastatic propensity, and they point to the unexpected role of a major iron transporting protein in mediating resistance to oxidative stress. The role of oxidative stress in the blood circulation as a critical suppressor of cancer metastasis has recently emerged, through the paradoxical role of anti-oxidants as pro-metastatic effectors (17,59–62). Indeed, while early studies had pointed to the cellular damage and potential tumorigenic effects of ROS, and hence the potential benefit of anti-oxidants (61,63–65), recent findings have demonstrated the positive impact of ROS in limiting blood-borne metastasis. In mouse models, systemic injections of the anti-oxidant N-acetyl-cysteine (NAC) enhances metastatic tumor formation following intravascular inoculation of primary human melanoma cells (17). In an endogenous mouse model of malignant melanoma, in vivo administration of NAC or the Vitamin E analog Trolox accelerates tumor progression and lymph node metastasis (66). Similarly, using single cell RNA-seq of prostate cancer CTCs, we previously noted overexpression of multiple gene signatures involved in anti-oxidative defense, including beta-hemoglobin, whose induction by KLF4 contributes to blood-borne metastasis (41,62). While this work was under revision, Ubellacker et al, described a model whereby metastasizing melanoma cells are initially shielded from ferroptosis as they spread through low oxygen tension lymphatic channels, enhancing their subsequent ability to resist ROS stress during subsequent intravascular dissemination(67). Among the mechanisms employed by circulating tumor cells to circumvent oxidative stress in the bloodstream, the SREBF2-mediated induction of TF constitutes a novel pathway, which may provide unique therapeutic opportunities.
In vitro, iron chelation using deferoxamine (DFO) suppresses ferroptosis, consistent with the iron requirement for lipid peroxidation(68). Altering serum iron levels to increase tumor cell ferroptosis is not feasible, however, inhibitors of lipogenesis may provide an intriguing opportunity to suppress the SREBP-TF pathway. Epidemiological studies have produced conflicting results as to the general effectiveness of statins on cancer outcomes(69–73). These inconsistent clinical findings may reflect the distinct lipogenesis dependencies of different cancer types, inter-patient heterogeneity, as well as different timing of drug administration as part of large observational clinical studies. Our results using melanoma CTC models raise the possibility that the short transit of tumor cells through the bloodstream may provide a unique window of opportunity to target these cells at a time of extraordinary ROS stress. As such, targeting the SREBP pathway in the context of melanoma with a high risk for metastatic recurrence may present a potential therapeutic strategy. Taken together, the cross-talk between SREBP and TF reveals a new and potentially therapeutically relevant vulnerability of highly refractory metastatic precursor cells.
MATERIALS AND METHODS
Experimental mouse model and human subjects
Written informed consent from patients with metastatic melanoma undergoing evaluation or treatment at the Massachusetts General Hospital Cancer Center was obtained, and blood collections (10–15ml) were performed as per IRB protocol (DF/HCC-0500), in accordance with the US ethical guidelines. CTCs were microfluidically enriched from peripheral blood samples, using high efficiency negative depletion of hematopoietic cells to isolate untagged and potentially viable cancer cells, as previously described (CTC-iChip)(22,23). All CTC lines were maintained under anchorage-independent culture condition as described previously (25). Standard melanoma cell lines IGR-37, SKML28, GAK and A375 were obtained from ATCC. CTC and ATCC cell lines used in this study have been authenticated using Short Tandem Repeat (STR) profilinng method (Genetica) and tested for mycoplasma contamination using Mycoalert™ kit (Lonza, Cat# LT07–218) before experiments. Mouse models of tumorigenesis and metastasis were generated using cultured Mel-167 and Mel-182–2 CTC lines in immunocompromised NOD-scid Il2rg−/− mice, according to guidelines approved by the MGH institutional Animal Care and Use committee (IACUC No:2010N000006).
Plasmid construction
FLAG-tagged SREBF2 and TF constructs were obtained from Addgene (SREBF2 #26807 and TF #67240) and subcloned into pInducer10 vector using restriction enzymatic digestion (5’ using Age1 and 3’ using Mlu1). Lentiviral shRNAs were purchased from TRC shRNA libraries of the Broad Institute. The targeting sequences for TF are: shTF #1 5-ACTACAATAAGAGCGATAATT-3 and shTF #2 5-TACACCAGAGGCAGGGGTATTT-3.
CTC ex vivo culture
For ex vivo culture of melanoma CTCs, microfluidically processed samples were incubated in hypoxic, anchorage independent conditions as described in Yu et al(25) with modifications. CTC cultures were grown in ultralow attachment plates (Corning) containing tumor sphere medium consisting of RPMI-1640 medium (with phenol red) supplemented with EGF (20ng/ml), basic FGF (20ng/ml), B27 (10ml, 50x in stock), 1x Antibiotic-antimycotic (Life Technologies), 5μg/ml Heparin (Stemcell Technologies) and 100nM Rock inhibitor Y-27632 (EMD Millipore). Cells were cultured in a humid 37oC incubator with 5% CO2 and 4% O2. Medium was replenished 2–3 times per week, until CTCs initiated in vitro proliferation (4–8 weeks) with the following components included in CTC culture media: Mel-182 and Pem-22 blood samples were processed through the CTC-iChip using a recently developed blood stabilization method (74) followed by 3-D fibrin matrigel culture initially and switched to anchorage independent condition after initial proliferation.
Mouse xenograft and metastases assays
For primary tumorigenesis assays, NOD-scid Il2rg−/− mice (6–8 weeks old, female) were injected subcutaneously in the left flank with cultured CTCs, and tumors were harvested when they reached 2 centimeters in diameter. For metastases assays, 100,000 to 500,000 cells were injected intravascularly (tail vein or intracardiac). Metastatic burden was monitored 2–3 times per week using in vivo luciferase imaging using the IVIS system (PerkinElmer). Mice were sacrificed at 8–10 weeks after tumor cell inoculation or at the first sign of discomfort as per IACUC guidelines.
Soft agar clonogenic assays
Cultured CTCs were resuspended in growth media containing 0.4% low-melting agarose (Sigma, type VII). Soft agarose (0.5 ml of 1% soft agarose solution) was layered and solidified at room temperature in 24-well plates before adding the cell-agar mixture. Freshly plated cells were incubated at 4°C for 6 minutes to ensure solidification of the soft agar, and 0.5 ml of culture medium was added to each well. Colony formation was assayed after 4 weeks by staining with colorimetric MTT assay. Plates were photographed under identical standardized settings and colonies were scored using an automated Matlab script.
siRNA and shRNA knockdown
Melanoma CTCs were seeded in 6-well plates the day before siRNA introduction by Lipofectamine RNAiMAX (Thermo Fisher Cat# 13778030) transfection. ON_TARGET plus smart pool siRNA was used to target SREBF1 (Dharmacon Cat# L-006891–00-0005) and SREBF2 (Cat# L-009549–00-0005). Growth media was replaced once after 12 hours of transfection and siRNA knockdown efficiency was assessed by Q-PCR 48 hours post transfection. shRNAs in lentiviral backbone against TF and scramble shRNA control vector were purchased from TRC shRNA library in Broad Institute. shRNA Lentivirus were produced and packaged in 293T cells as described previously(75). Virus transduction was carried out in CTCs incubated in sealed 6-well plates by centrifugation followed by puromycin (2 μg/ml) selection for 6–8 days.
In vitro drug sensitivity assays
Cultured CTCs were refreshed with growth media in suspension the day before drug exposure and then split into ultra-low attachment 96-well plates (Corning Costar-3474) at a density of 1000 cells per well. Serial dilutions of each drug were added to the media and cell viability was measured 5 days after drug exposure using CellTiter-Glo2.0 (Promega Ca#G9243).
ROS measurements
Control or TF-KD CTCs were incubated with fresh media the day before ROS detection. Cells with 1x ROS fluorescent probe (Abcam, cat #ab186029) were incubated at 37°C for 1 hour in CTC growth chamber. ROS quantification was performed using flow cytometry (calculated based on the geometric mean) and relative ROS level was calculated and normalized to control shRNA treated CTCs.
Lipid peroxidation assay
CTC cells were pre-incubated with fresh media 24 hours before the experiment. Cells were stained with 1x BODIPY™ 581/591 C11 lipid peroxidation sensor (Invitrogen, cat#D3861) for 30 minutes at 37°C. After the incubation, CTCs were washed with PBS and sent for flow cytometry quantification. The fraction of cells positive for oxidized BODIPY™ 581/591 C11 signal (shifts in emission peaks from 590nm to 510nm upon oxidation) was analyzed and normalized to control shRNA treated CTCs.
Real-time PCR and Western blot analyses
RNA was extracted by column purification kit (Qiagen RNeasy), followed by treatment with DNAse I, and oligo-dT based reverse transcription using Superscript III. Real-time quantitative PCR was performed using Sybr green reagents on an ABI 7500 fast real-time PCR platform. For Immunoblotting, cells were washed once with cold TBS (20mM Tris-HCl (pH 7.5), 150mM NaCl) and lysed using cell lysis buffer containing 1x TBS, 10mM β-glycerophosphate, 5 mM EGTA, 1mM Na4P2O7, 5mM NaF, 0.5% Triton X-100, 1mM Na3VO4, 1mM dithiothreitol and protease inhibitor cocktail (Roche). Following SDS-PAGE, antibody-tagged proteins were detected using Western Lightning Plus-ECL reagent (PerkinElmer) after SDS-PAGE. Primary antibodies used in the experiment are: rabbit anti-FLAG (1:1000, Invitrogen, Cat#PA1–984B); rabbit anti-TF (1:2000, Invitrogen, Cat#PA3–913); mouse anti-TFRC (1:2000, Life Technologies, Cat#136800) rabbit anti-GAPDH (1:2000, Cell Signaling Technology, Cat#2118S).
Single cell RNA library construction and sequencing
Freshly isolated individual CTCs were selected using a micromanipulator and introduced into a 0.5ml tube with 7ml of 1x lysis buffer (Takara, Cat#634891) containing recombinant RNase inhibitor (0.4U/ml; Takara, Cat#2313A), snap frozen and maintained at −80oC until processing. Single cell lysates were transferred into 96-well plates (Eppendorf, 951020401), and RNA extraction and clean up were carried out using Agencourt RNAClean XP SPRI beads (Beckman Coulter, A63987). Prior to library construction using the Nextera XT library Prep kit (Illumina, FC-131–1096), two steps of quality control were done to evaluate the concentration of each single cell, using Qubit dsDNA high sensitivity assay kit (Life Technologies, Q32854), according to the manufacturer’s instructions. Single cell CTC libraries were prepared using a modified Smart-Seq2 protocol (76). Combined libraries were sequenced on a NextSeq 500 sequencer (Illumina) using the 75 cycles kit, with paired-end 38-base-reads and dual barcoding.
Computational analyses
Detailed computational analytical methods can be found in in Supplementary Methods
Statistical analysis
Statistical analyses were performed using R 3.5.1. An empirical Bayes moderated t-test was used for two group comparison of gene expression values. Two-sided Welch’s t-test was used for two group comparison of GSVA enrichment scores.
Supplementary Material
Significance.
Through single cell analysis of primary and cultured melanoma CTCs, we have uncovered intrinsic cancer cell heterogeneity within lipogenic and iron homeostatic pathways that modulates resistance to BRAF inhibitors and to ferroptosis inducers. Activation of these pathways within CTCs is correlated with adverse clinical outcome, pointing to therapeutic opportunities.
Acknowledgements
We thank the patients who participated in this study and L. Libby for administrative support. This work was supported by grants from National Institute of Health (2RO1CA129933 to D.A.H, 2U01EB012493 to M.T, D.A.H, S.M, 5U01EB012493 to M.T, 5P41EB002503 to M.T), Howard Hughes Medical Institute (to D.A.H), ESSCO Breast Cancer Research Fund (to S.M), National Foundation for Cancer Research (to D.A.H), Wang Pediatric Brain Tumor Collaborative and MGH Claflin Distinguished Scholar Award (to SLT). MGH Research Scholar Award (to A.M.N). IBM funding (to G.G). National Science Foundation (PHY-1549535 to D.T.T), Stand Up to Cancer (SU2C, to D.T.T), Lustgarten Foundation (2015–002, to D.T.T) & ACD_Biotechne (to D.T.T)
Footnotes
Declaration of Competing Interests
Massachusetts General Hospital (MGH) has applied for patents regarding the CTC-iChip technology and CTC detection signatures. M.T., D.A.H., S.M. and D.T.T. are cofounders and have equity in Tell-Bio, which is not related to this work. D.T.T. has received consulting fees from EMD Millipore-Sigma, Ventana-Roche, Foundation Medicine Inc., and Merrimack pharmaceuticals. D.T.T. receives sponsored research support from ACD-Biotechne. D.T.T. is founder and has equity in PanTher Therapeutics and ROME Therapeutics, which are not related to this work. All authors interests were reviewed and are managed by MGH and Partners HealthCare in accordance with their conflict of interest policies.
Data and software availability
All RNA-seq and ChIP-seq data associated with this paper are deposited into GEO repository with the accession number GSE157745 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE157745). The whole exome sequence of melanoma CTC lines (as shown in Figure S3A) is in the process of uploading to SRA and the accession ID will be inserted in the proofs.
REFERENCES
- 1.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364(26):2507–16 doi 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012;367(2):107–14 doi 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- 3.Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012;380(9839):358–65 doi 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 4.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363(8):711–23 doi 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011;364(26):2517–26 doi 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 6.Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet 2014;384(9948):1109–17 doi 10.1016/S0140-6736(14)60958-2. [DOI] [PubMed] [Google Scholar]
- 7.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med 2015;372(26):2521–32 doi 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 8.Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372(4):320–30 doi 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 9.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med 2015;372(26):2509–20 doi 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015;348(6230):124–8 doi 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015;348(6230):69–74 doi 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 12.Johnson DB, Frampton GM, Rioth MJ, Yusko E, Xu Y, Guo X, et al. Targeted Next Generation Sequencing Identifies Markers of Response to PD-1 Blockade. Cancer Immunol Res 2016;4(11):959–67 doi 10.1158/2326-6066.CIR-16-0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolf Y, Bartok O, Patkar S, Eli GB, Cohen S, Litchfield K, et al. UVB-Induced Tumor Heterogeneity Diminishes Immune Response in Melanoma. Cell 2019;179(1):219–35 e21 doi 10.1016/j.cell.2019.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jimenez-Sanchez A, Memon D, Pourpe S, Veeraraghavan H, Li Y, Vargas HA, et al. Heterogeneous Tumor-Immune Microenvironments among Differentially Growing Metastases in an Ovarian Cancer Patient. Cell 2017;170(5):927–38 e20 doi 10.1016/j.cell.2017.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 2014;32(10):1020–30 doi 10.1200/JCO.2013.53.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, et al. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J Clin Oncol 2015;33(17):1889–94 doi 10.1200/JCO.2014.56.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015;527(7577):186–91 doi 10.1038/nature15726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu M, Stott S, Toner M, Maheswaran S, Haber DA. Circulating tumor cells: approaches to isolation and characterization. J Cell Biol 2011;192(3):373–82 doi 10.1083/jcb.201010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Allard WJ, Matera J, Miller MC, Repollet M, Connelly MC, Rao C, et al. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin Cancer Res 2004;10(20):6897–904 doi 10.1158/1078-0432.CCR-04-0378. [DOI] [PubMed] [Google Scholar]
- 20.Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med 2004;351(8):781–91 doi 10.1056/NEJMoa040766. [DOI] [PubMed] [Google Scholar]
- 21.Luo X, Mitra D, Sullivan RJ, Wittner BS, Kimura AM, Pan S, et al. Isolation and molecular characterization of circulating melanoma cells. Cell Rep 2014;7(3):645–53 doi 10.1016/j.celrep.2014.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ozkumur E, Shah AM, Ciciliano JC, Emmink BL, Miyamoto DT, Brachtel E, et al. Inertial focusing for tumor antigen-dependent and -independent sorting of rare circulating tumor cells. Sci Transl Med 2013;5(179):179ra47 doi 10.1126/scitranslmed.3005616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karabacak NM, Spuhler PS, Fachin F, Lim EJ, Pai V, Ozkumur E, et al. Microfluidic, marker-free isolation of circulating tumor cells from blood samples. Nat Protoc 2014;9(3):694–710 doi 10.1038/nprot.2014.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hong X, Sullivan RJ, Kalinich M, Kwan TT, Giobbie-Hurder A, Pan S, et al. Molecular signatures of circulating melanoma cells for monitoring early response to immune checkpoint therapy. Proc Natl Acad Sci U S A 2018;115(10):2467–72 doi 10.1073/pnas.1719264115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu M, Bardia A, Aceto N, Bersani F, Madden MW, Donaldson MC, et al. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science 2014;345(6193):216–20 doi 10.1126/science.1253533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Medford AJ, Dubash TD, Juric D, Spring L, Niemierko A, Vidula N, et al. Blood-based monitoring identifies acquired and targetable driver HER2 mutations in endocrine-resistant metastatic breast cancer. NPJ Precis Oncol 2019;3:18 doi 10.1038/s41698-019-0090-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jordan NV, Bardia A, Wittner BS, Benes C, Ligorio M, Zheng Y, et al. HER2 expression identifies dynamic functional states within circulating breast cancer cells. Nature 2016;537(7618):102–6 doi 10.1038/nature19328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cancer Genome Atlas N Genomic Classification of Cutaneous Melanoma. Cell 2015;161(7):1681–96 doi 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012;483(7391):603–7 doi 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 2013;14:7 doi 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Toth JI, Datta S, Athanikar JN, Freedman LP, Osborne TF. Selective coactivator interactions in gene activation by SREBP-1a and −1c. Mol Cell Biol 2004;24(18):8288–300 doi 10.1128/MCB.24.18.8288-8300.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen M, Zhang J, Sampieri K, Clohessy JG, Mendez L, Gonzalez-Billalabeitia E, et al. An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer. Nat Genet 2018;50(2):206–18 doi 10.1038/s41588-017-0027-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shimano H, Sato R. SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat Rev Endocrinol 2017;13(12):710–30 doi 10.1038/nrendo.2017.91. [DOI] [PubMed] [Google Scholar]
- 34.Ricoult SJ, Yecies JL, Ben-Sahra I, Manning BD. Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene 2016;35(10):1250–60 doi 10.1038/onc.2015.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell 2014;158(6):1431–43 doi 10.1016/j.cell.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Messori L, Kratz F. Transferrin: from inorganic biochemistry to medicine. Met Based Drugs 1994;1(2–3):161–7 doi 10.1155/MBD.1994.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shen Y, Li X, Dong D, Zhang B, Xue Y, Shang P. Transferrin receptor 1 in cancer: a new sight for cancer therapy. Am J Cancer Res 2018;8(6):916–31. [PMC free article] [PubMed] [Google Scholar]
- 38.Bloch B, Popovici T, Levin MJ, Tuil D, Kahn A. Transferrin gene expression visualized in oligodendrocytes of the rat brain by using in situ hybridization and immunohistochemistry. Proc Natl Acad Sci U S A 1985;82(19):6706–10 doi 10.1073/pnas.82.19.6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pascale RM, De Miglio MR, Muroni MR, Simile MM, Daino L, Seddaiu MA, et al. Transferrin and transferrin receptor gene expression and iron uptake in hepatocellular carcinoma in the rat. Hepatology 1998;27(2):452–61 doi 10.1002/hep.510270220. [DOI] [PubMed] [Google Scholar]
- 40.Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014;158(5):1110–22 doi 10.1016/j.cell.2014.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miyamoto DT, Zheng Y, Wittner BS, Lee RJ, Zhu H, Broderick KT, et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science 2015;349(6254):1351–6 doi 10.1126/science.aab0917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arosio P, Levi S. Ferritin, iron homeostasis, and oxidative damage. Free Radic Biol Med 2002;33(4):457–63 doi 10.1016/s0891-5849(02)00842-0. [DOI] [PubMed] [Google Scholar]
- 43.Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017;171(2):273–85 doi 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019;35(6):830–49 doi 10.1016/j.ccell.2019.04.002. [DOI] [PubMed] [Google Scholar]
- 45.Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol 2008;15(3):234–45 doi 10.1016/j.chembiol.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017;551(7679):247–50 doi 10.1038/nature24297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017;547(7664):453–7 doi 10.1038/nature23007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Holzel M, Bovier A, Tuting T. Plasticity of tumour and immune cells: a source of heterogeneity and a cause for therapy resistance? Nat Rev Cancer 2013;13(5):365–76 doi 10.1038/nrc3498. [DOI] [PubMed] [Google Scholar]
- 49.Miyamoto DT, Ting DT, Toner M, Maheswaran S, Haber DA. Single-Cell Analysis of Circulating Tumor Cells as a Window into Tumor Heterogeneity. Cold Spring Harb Symp Quant Biol 2016;81:269–74 doi 10.1101/sqb.2016.81.031120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bao J, Zhu L, Zhu Q, Su J, Liu M, Huang W. SREBP-1 is an independent prognostic marker and promotes invasion and migration in breast cancer. Oncol Lett 2016;12(4):2409–16 doi 10.3892/ol.2016.4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guo D, Prins RM, Dang J, Kuga D, Iwanami A, Soto H, et al. EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci Signal 2009;2(101):ra82 doi 10.1126/scisignal.2000446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee JH, Jeon YG, Lee KH, Lee HW, Park J, Jang H, et al. RNF20 Suppresses Tumorigenesis by Inhibiting the SREBP1c-PTTG1 Axis in Kidney Cancer. Mol Cell Biol 2017;37(22) doi 10.1128/MCB.00265-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Talebi A, Dehairs J, Rambow F, Rogiers A, Nittner D, Derua R, et al. Sustained SREBP-1-dependent lipogenesis as a key mediator of resistance to BRAF-targeted therapy. Nat Commun 2018;9(1):2500 doi 10.1038/s41467-018-04664-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guo D, Bell EH, Mischel P, Chakravarti A. Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr Pharm Des 2014;20(15):2619–26 doi 10.2174/13816128113199990486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS, et al. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci U S A 2003;100(21):12027–32 doi 10.1073/pnas.1534923100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019;569(7755):270–4 doi 10.1038/s41586-019-1170-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rychtarcikova Z, Lettlova S, Tomkova V, Korenkova V, Langerova L, Simonova E, et al. Tumor-initiating cells of breast and prostate origin show alterations in the expression of genes related to iron metabolism. Oncotarget 2017;8(4):6376–98 doi 10.18632/oncotarget.14093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol Cell 2015;59(2):298–308 doi 10.1016/j.molcel.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Takahashi N, Chen HY, Harris IS, Stover DG, Selfors LM, Bronson RT, et al. Cancer Cells Co-opt the Neuronal Redox-Sensing Channel TRPA1 to Promote Oxidative-Stress Tolerance. Cancer Cell 2018;33(6):985–1003 e7 doi 10.1016/j.ccell.2018.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, et al. Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 2018;33(5):890–904 e5 doi 10.1016/j.ccell.2018.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yun J, Mullarky E, Lu C, Bosch KN, Kavalier A, Rivera K, et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015;350(6266):1391–6 doi 10.1126/science.aaa5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zheng Y, Miyamoto DT, Wittner BS, Sullivan JP, Aceto N, Jordan NV, et al. Expression of beta-globin by cancer cells promotes cell survival during blood-borne dissemination. Nat Commun 2017;8:14344 doi 10.1038/ncomms14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Irani K, Xia Y, Zweier JL, Sollott SJ, Der CJ, Fearon ER, et al. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 1997;275(5306):1649–52 doi 10.1126/science.275.5306.1649. [DOI] [PubMed] [Google Scholar]
- 64.Chan DW, Liu VW, Tsao GS, Yao KM, Furukawa T, Chan KK, et al. Loss of MKP3 mediated by oxidative stress enhances tumorigenicity and chemoresistance of ovarian cancer cells. Carcinogenesis 2008;29(9):1742–50 doi 10.1093/carcin/bgn167. [DOI] [PubMed] [Google Scholar]
- 65.Kumar B, Koul S, Khandrika L, Meacham RB, Koul HK. Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res 2008;68(6):1777–85 doi 10.1158/0008-5472.CAN-07-5259. [DOI] [PubMed] [Google Scholar]
- 66.Le Gal K, Ibrahim MX, Wiel C, Sayin VI, Akula MK, Karlsson C, et al. Antioxidants can increase melanoma metastasis in mice. Sci Transl Med 2015;7(308):308re8 doi 10.1126/scitranslmed.aad3740. [DOI] [PubMed] [Google Scholar]
- 67.Ubellacker JM, Tasdogan A, Ramesh V, Shen B, Mitchell EC, Martin-Sandoval MS, et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature 2020;585(7823):113–8 doi 10.1038/s41586-020-2623-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012;149(5):1060–72 doi 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu B, Yi Z, Guan X, Zeng YX, Ma F. The relationship between statins and breast cancer prognosis varies by statin type and exposure time: a meta-analysis. Breast Cancer Res Treat 2017;164(1):1–11 doi 10.1007/s10549-017-4246-0. [DOI] [PubMed] [Google Scholar]
- 70.Abdel-Rahman O Statin treatment and outcomes of metastatic pancreatic cancer: a pooled analysis of two phase III studies. Clin Transl Oncol 2019;21(6):810–6 doi 10.1007/s12094-018-1992-3. [DOI] [PubMed] [Google Scholar]
- 71.Lytras T, Nikolopoulos G, Bonovas S. Statins and the risk of colorectal cancer: an updated systematic review and meta-analysis of 40 studies. World J Gastroenterol 2014;20(7):1858–70 doi 10.3748/wjg.v20.i7.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tsan YT, Lee CH, Ho WC, Lin MH, Wang JD, Chen PC. Statins and the risk of hepatocellular carcinoma in patients with hepatitis C virus infection. J Clin Oncol 2013;31(12):1514–21 doi 10.1200/JCO.2012.44.6831. [DOI] [PubMed] [Google Scholar]
- 73.Islam MM, Yang HC, Nguyen PA, Poly TN, Huang CW, Kekade S, et al. Exploring association between statin use and breast cancer risk: an updated meta-analysis. Arch Gynecol Obstet 2017;296(6):1043–53 doi 10.1007/s00404-017-4533-3. [DOI] [PubMed] [Google Scholar]
- 74.Wong KHK, Tessier SN, Miyamoto DT, Miller KL, Bookstaver LD, Carey TR, et al. Whole blood stabilization for the microfluidic isolation and molecular characterization of circulating tumor cells. Nat Commun 2017;8(1):1733 doi 10.1038/s41467-017-01705-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 2006;124(6):1283–98 doi 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- 76.Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 2019;176(1–2):404 doi 10.1016/j.cell.2018.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.