

Abstract
The use of targeted small molecule and immuno-therapeutics has been limited to date in pediatric oncology. Recently, the number of pediatric approvals has risen and regulatory initiatives in the United States and Europe have aimed to increase the study of novel anti-cancer therapies in children. Challenges of drug development in children include the rarity of individual cancer diagnoses and the high prevalence of difficult to drug targets, including transcription factors and epigenetic regulators. Ongoing pediatric adaptation of biomarker-driven trial designs and further exploration of agents targeting non-kinase drivers constitute high priority objectives for future pediatric oncology drug development.
Keywords: Pediatric oncology, targeted therapy, immunotherapy, drug development
Introduction
Today, over 80% of children with cancer survive for at least 5 years after diagnosis, the vast majority of whom are cured (https://seer.cancer.gov/archive/csr/1975_2015/). However, this success has not been uniform and many survivors of pediatric cancer face life-long sequelae of therapy.(1) While survival for children with the most common pediatric cancer, acute lymphoblastic leukemia, has improved from 10% to 90% through a series of clinical trials,(2) little improvement in survival has been achieved in certain other childhood malignancies. These include high grade gliomas and metastatic sarcomas, which remain incurable in the majority of patients. Further, as risk stratification has improved, defined subsets of patients with very high-risk molecular features of otherwise highly curable histologies have been identified.(2) Even among children with cancers for which outcomes have improved, few patients who relapse are cured.(3) Finally, while all pediatric cancers are rare by adult cancer definitions, some tumors including most carcinomas and melanoma occurring in pediatric populations have proven too rare to be successfully studied in clinical trials. Yet, in aggregate, these rare tumors comprise 11% of newly diagnosed malignancies in children, highlighting the need for novel trial designs to improve outcomes for these patients.(4)
As therapy has been intensified, the late effects of cytotoxic chemotherapy in children have become increasingly apparent. These include risks of cardiomyopathy from anthracyclines and chest radiotherapy and secondary malignancies from radiation, alkylating agents and topoisomerase inhibitors, as well as numerous others. Among childhood cancer survivors, the cumulative incidence of a chronic health condition is higher than 70% by 30 years after the cancer diagnosis, with 42% cumulative incidence of severe, disabling or life-threatening conditions.(1) As the use of targeted agents has increased, unique toxicities of these agents in the growing host have become apparent.(5–8) Today, drug development in pediatric oncology focuses on improving survival for those patients with largely incurable tumors as well as attempts to use novel agents to allow therapeutic substitution to improve the quality of survivorship.
Until the last decade, pediatric cancer drug development has focused on identification and intensification of cytotoxic therapy and improved supportive care. More recently, attention has turned to targeted small molecules and immunotherapy.(3) Still today, relatively few children with cancer are treated with targeted agents as standard of care in the frontline/curative-intent setting, with notable exceptions including those with Philadelphia chromosome positive acute lymphoblastic leukemia (Ph+ ALL) and chronic myelogenous leukemia (CML) treated with ABL-class inhibitors,(9) acute promyelocytic leukemia (APML) treated with ATRA,(10) B-cell lymphomas treated with anti-CD20 antibodies,(11) high-risk neuroblastoma treated with anti-GD2 antibodies,(12) and more recently, TRK fusion cancers treated with TRK inhibitors.(13) Particularly for children with ALL, however, this is changing with frontline trials evaluating the addition of either targeted small molecules or immunotherapeutics for the majority of patients (NCT02723994, NCT03959085, NCT03914625).
Traditionally, dose-finding phase 1 trials of targeted therapies in children followed standard 3+3 or rolling 6 designs originally developed for cytotoxic agents. These designs evaluate three to six patients per dose level and often included a larger number of dose levels starting well below the dose equivalent to the already defined dose in adults.(14,15) Based on data showing a tight correlation of the pediatric and adult maximum tolerated doses of kinase inhibitors, more efficient designs with fewer dose levels have been developed.(16,17) Pediatric dosing in early phase trials demonstrate that post-pubertal adolescents achieve similar drug levels as their adult counterparts.(18) There are increasing data to suggest that these patients could safely be enrolled on adult trials, possibly enrolling at a dose level behind the current adult cohort where no dose-limiting toxicities occurred or in a separate dosing cohort.(19) Additionally, dose finding studies have shown that pediatric recommended phase 2 doses (RP2Ds) in younger children are usually within 30% of the body surface area (BSA)-adjusted approved adult dose, limiting the need for extensive dose finding.(16,17) Despite this, most pediatric phase 1 studies still enroll patients with a broad range of histologic diagnoses and do not include biomarker selection, limiting the ability to evaluate efficacy in populations of greatest interest.(3)
Here we review the current FDA/EMA approved targeted drugs for the treatment of pediatric cancers and their development pathway. We also provide a perspective on the challenges of pediatric drug development moving forward, including the regulatory landscape, rarity of specific cancer subtypes in children, the relatively limited number of targetable mutations identified in pediatric cancers, difficulties in drugging non-kinase fusion oncoproteins, and the unique considerations of studying drugs in a developing host. We also highlight opportunities to overcome these challenges and continue to advance pediatric drug development in the era of targeted therapies including small molecule kinase inhibitors, immune-based, and more recently, epigenetic therapies.
Current FDA/EMA approved targeted drugs for pediatric cancer
The dual challenges of high costs of bringing a drug to market and the smaller number of pediatric patients with cancer compared to adults with cancer limit investment in this area. In this context, few drugs have achieved FDA or EMA approval specifically for the treatment of pediatric cancers: anti-GD2 monoclonal antibodies for neuroblastoma; and clofarabine and the chimeric antigen receptor T-cell [CAR-T] tisagenlecleucel for ALL. These drugs were developed for two of the most common pediatric cancers; development of agents specifically for the treatment of less common pediatric cancers has not yet been successful. Additional investment in preclinical work is necessary to expand the range of pediatric cancers for which novel therapies are ready to advance to the clinic. Further, additional incentives are necessary to encourage pharmaceutical companies to conduct clinical trials in rare pediatric cancer populations.
The vast majority of targeted therapies studied in children with cancer have been repurposed drugs initially developed for the treatment of adult cancers. Early phase clinical trials of repurposed adult cancer drugs in children with relapsed/refractory solid tumors without biomarker selection have demonstrated very little efficacy, with a recent meta-analysis demonstrating a 4% objective response rate and less than 4 month median progression-free survival, substantially worse than has been observed in recent pooled analysis of adult phase 1 trials showing a nearly 20% response rate.(3,20) There has not been a similar formal meta-analysis of phase 1 trials in pediatric hematologic malignancies. However, several recent pediatric phase 1 trials including several leukemia trials have demonstrated markedly greater response rates among children selected to have the target of interest, highlighting the importance of this approach.(13,21–26) By definition, development of repurposed drugs in children lags behind that in adults, with the median interval between the first studies in adults and those in children of 6.5 years.(27)
Prior to 2017, only 4 targeted therapies received a pediatric label indication, while 11 have achieved a US FDA label since 2017 (Table 1). Several features of these successful drugs are notable. First, in all cases except for pembrolizumab, the disease or biomarker of interest within childhood cancer had been identified prior to the initiation of pediatric clinical trials. This resulted in enrichment or exclusive enrollment of this population within the first studies. Second, exceptional response rates were observed early in drug development with eleven of fifteen of these eventually approved agents having an initial response rate greater than 50% in the targeted population in pediatric phase 1 or 2 trials (Table 1). Given the smaller number of children with cancer, successful development of targeted therapies with modest activity has not been possible, highlighting the need to prioritize early phase trials of potential transformational agents and design studies to provide early go/no-go decisions based on the presence of robust activity within the targeted patient population. A final challenge to pediatric drug development has been the cost and effort involved in seeking a pediatric label. Because of this, some drugs that have shown high response rates in the setting of academic or cooperative group trials (e.g., crizotinib for ALCL and IMT,)(25) remain off-label uses in pediatrics.
Table 1:
Targeted therapies approved by the FDA and/or EMA for pediatric cancer
| Year Approved | |||||
|---|---|---|---|---|---|
| Drug | Pediatric Indication | FDA | EMA | Pediatric Phase 1* Response Rate in Targeted Population | Number of Patients |
| Tretinoin | Acute promyelocytic leukemia (APML) | 1995 | NC | 83% (all CR)(101) | 6 |
| Everolimus | Subependymal giant cell astrocytoma (SEGA) | 2010 | 2011 | 75%(102) | 28 |
| Dinutuximab | Neuroblastoma | 2015 | 2015** | 66%(103) | 6 |
| Blinatumomab | Relapsed/refractory (R/R) B acute lymphoblastic leukemia (ALL) | 2016 | 2015 | 30% (all minimal residual disease negative)(21) | 23 |
| Pembrolizumab | R/R classic Hodgkin Lymphoma, primary mediastinal B-cell lymphoma, Merkel cell carcinoma, MSI-high tumors | 2017 | N/A | 60% (Hodgkin)(66) | 15 |
| Ipilimumab | Melanoma (≥12 years) | 2017 | 2017 | 0(67) | 12 |
| Gemtuzumab | R/R acute myeloid leukemia (AML) | 2017 | 2018*** | 28%(104) | 29 |
| Tisagenlecleucel | R/R B-ALL | 2017 | 2018 | 90%(105) | 30 |
| Dasatinib | CML / Ph+ ALL | 2017 | 2017 | 82% CCyR (CML chronic phase [CP])(22) | 17 |
| Imatinib | Ph+ ALL and chronic myeloid leukemia (CML) | 2017 | 2013 | 70% ALL, 83% CML CP(23) | 10 ALL, 14 CML |
| Nilotinib | CML | 2018 | 2017 | 90%(24) | 10 |
| Larotrectinib | TRK fusion solid tumors | 2018 | 2019 | 93%(13) | 15 |
| Entrectinib | TRK fusion solid tumors (≥12 years) | 2019 | 2020 | 100%(26) | 6 |
| Tazemetostat | Epithelioid sarcoma (≥16 years) | 2020 | N/A | 29%(106) | 7 |
| Selumetinib | Plexiform neurofibroma | 2020 | N/A | 71%(107) | 24 |
| Selpercatinib | RET-mutant/fusion thyroid cancer (≥12 years) | 2020 | N/A | Not yet reported | |
| Rituximab | Mature B-cell lymphomas | N/A | 2020 | 41%(108) | 87 |
if no pediatric phase 1 in targeted population, first published phase 2 response rate
Dinutuximab was approved in 2015, but authorization for commercialization was then withdrawn by the company. Dinutuximab beta was approved in 2017
Only children ≥ 15 years old
NC: no centralised procedure when approved (approved by each individual EU country)
N/A: not approved
Regulatory initiatives to augment pediatric oncology drug development
Until recently, the greatest challenge to pediatric drug development was access to investigational agents. Economics and the prevailing myth that an early bad outcome in a child might derail a drug may have discouraged the pharmaceutical industry from sponsoring pediatric trials or providing drug supply for investigator-initiated trials. Indeed, recent data demonstrate a disparity in rates of industry sponsorship of pediatric oncology trials, where only 16% of trials including minors were industry sponsored, compared to trials for adults with cancer (33%) or trials in other pediatric specialties (30%).(28) Over the past two decades, the FDA in the United States (US) and the EMA in Europe have passed legislation aimed at accelerating the development of drugs for children. Approaches have included both incentivizing and mandating sponsors to submit a pediatric development plan at the time of a new drug or biologic license application. Early incentives introduced by the 2002 US Best Pharmaceuticals for Children Act (BPCA; Public Law 107–109) granted a 6-month patent exclusivity extension to sponsors willing to develop appropriate drug formulations and complete pediatric clinical studies as outlined by the FDA. Soon after, the Pediatric Research Equity Act (PREA) [21U.S. Code 355B] codified requirements to perform such studies for agents undergoing FDA review for an adult indication. The EMA took a similar compulsory approach with the Pediatric Regulation [Regulation (EC) No 1901/2006 of the European Parliament and of the Council of 12 December 2006 on medicinal products for pediatric use] but required a more comprehensive Pediatric Investigation Plan (PIP) earlier in the drug development process, that had to be completed by the time the first marketing authorization was granted. The European regulation combined the obligation to submit the PIP for all drugs requesting marketing authorization with the incentive of a 6-month patent extension for drugs completing their PIP successfully, and included voluntary PIPs which allowed companies that might have been granted waivers to access the incentives if they committed to pediatric development.
However, the lack of harmonization across different regulatory agencies and, in particular, the differences in the timing and scope of the EMA PIP and FDA PREA requirements further complicated pediatric academic–industry partnerships and the drug development process.(29) Both mandatory programs were also undermined by the provision of automatic waivers for drugs addressing orphan diseases or histologies rarely occurring in children [section 505B(a)]. For targeted agents directed toward growth pathways relevant to adult malignancy, sponsors were released from pediatric development responsibilities based on the fact that these adult histologies (e.g. lung or prostate cancer) did not occur in children. According to one European review, from June, 2012 to June, 2015, 147 oncology class waivers were granted for 89 drugs, 48 (54%) of which had a mechanism of action of substantial interest to pediatrics.(30) As a corollary, while 126 drugs gained initial FDA approval for an oncologic diagnosis over the 20 year period from 1997 to 2017 in the US, only 6 of these agents included a pediatric indication.(27)
Newer legislation seeks to close these loopholes. In Europe, the EMA published a revised class waiver list in 2015 that came into effect in 2018. In the US, a recent amendment to PREA [Title V of the FDA Reauthorization Act of 2017 (FDARA; Public Law 115–52)] called the RACE for Children Act (Research to Accelerate Cures and Equity for Children Act), requires sponsors to submit an initial Pediatric Study Plan (iPSP) when the molecular target of an agent under evaluation is “substantially relevant to the growth and progression of a pediatric cancer.” To date, the Relevant Molecular Target List (RMTL) developed by members of the NCI, FDA and the pediatric oncology community contains more than 200 molecular targets involved in cell lineage, tumor growth and the microenvironment relevant to pediatric malignancy (https://www.fda.gov/about-fda/oncology-center-excellence/pediatric-oncology). To facilitate the appropriate preclinical development of RMTL drugs, the Foundation for the National Institutes of Health (FNIH) recently announced a Public-Private Partnership to foster industry and academic collaboration.
These initiatives aim to expedite pediatric access to drugs under development for the treatment of adult cancers. Beginning in 2012, the US Congress introduced the priority review voucher as a powerful new incentive through the Creating Hope Act, currently up for reauthorization. As opposed to the above legislation which seeks to encourage the study of drugs being developed for adult cancers, this act seeks to incentivize drug development specifically for pediatric cancer and other rare pediatric diseases. The voucher is awarded to a sponsor who successfully obtains FDA approval for a rare pediatric indication and can be used to either expedite the FDA review of a future drug or sold on the open market to raise capital. Successful development of both dinutuximab and tisagenlecleucel resulted in granting of such vouchers to their sponsors. In the case of dinutuximab, the sale of the priority review voucher was valued at $350 million.(31)
While these changes successfully address issues of early access to investigational products, with greater interest in pediatric trials to fulfill regulatory requirements, they also pose new challenges. In medical oncology, there may be a value proposition for both patient and industry around biosimilars,(32) and the market can support several agents in a class each with subtle differences in efficacy, toxicity and resistance mechanisms. Given the small numbers of patients available for early phase pediatric trials, however, there is little enthusiasm to repeat similarly designed trials using multiple agents with the same mechanism of action when the first demonstrated only modest activity. For example, single agent phase 1 trials have been conducted for multiple VEGFR blocking agents and antiangiogenic tyrosine kinase inhibitors including bevacizumab, aflibercept (NCT00622414), sorafenib, sunitinib (NCT00387920), pazopanib (NCT00929903), cediranib (NCT00354848), lenvatinib (NCT02432274) and cabozantinib (NCT01709435) each demonstrating sporadic responses and a signal of stable disease in sarcoma, such that there is little knowledge to be gained by additional single agent phase 2 trials of agents in class. Similarly, there can be contemporaneous trials of single agents all in the same therapeutic class such as the multiple single agent pediatric anti-programmed death-1 (antiPD-1) inhibitors trials currently or recently enrolling despite the lack of early results for immune checkpoint inhibitors in pediatrics. Numerous competing trials of agents in the same class may compromise accrual of children with rare diseases, delaying or preventing the ability to define efficacy within the pediatric population, as has occurred with pediatric studies of BRAF inhibitors for melanoma.(33,34)
Coordinating drug development in children across multiple agents in the same class raises a number of challenges, including a lack of clear standards to determine which agent is the “best-in-class” and an adequate mechanism to distribute the financial burdens and incentives across competing pharmaceutical companies. Several initiatives are ongoing to overcome these challenges by aligning key stakeholders (Figure 1). The Pediatric Cluster is a series of regular teleconferences between the FDA, EMA, Japan’s Pharmaceuticals and Medical Devices Agency (PMDA), Health Canada, and Australia’s Therapeutic Goods Administration (https://www.fda.gov/science-research/pediatrics/international-collaboration-pediatric-cluster). In 2018, these regulators began generating non-binding comments which are provided to drug sponsors after these meetings. Early inclusion of adolescents (12 years and older) on phase 1 trials has been encouraged by several working groups particularly for trials targeting known drivers of their disease or disease specific mutations.(19,35) In parallel, many pharmaceutical companies have hired pediatric oncologists and more actively engage in early interactions with academia. The Accelerate Platform has held a series of multi-stakeholder pediatric strategy forums which include involvement from the EMA and FDA, drug companies, patient advocacy groups and academic pediatric oncologists. To date, these meetings have focused on coordinating development across high priority pediatric targets including ALK inhibitors, mature B-cell malignancies, immune checkpoint inhibitors, acute myeloid leukemia, epigenetic modifiers and CAR-T cells.(36,37)
Figure 1:
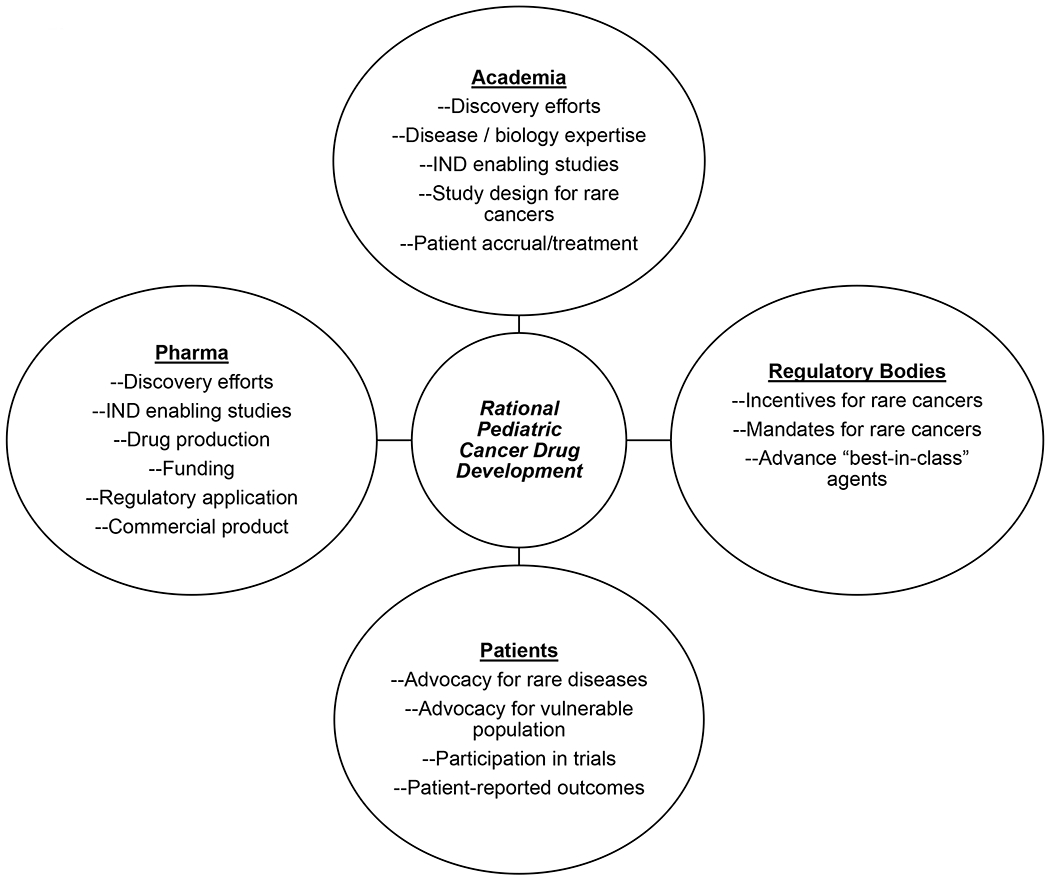
Key stakeholders contributing to rational pediatric cancer drug development.
Ideally, collaboration should begin even earlier in the drug development process. The rarity of pediatric cancers does not allow for routine clinical testing of all potential new agents thereby creating a greater reliance on prioritization based on scientific rationale and preclinical testing. For targeted therapies, it is important to assess the frequency and dependence on the target or pathway in the common malignancies of childhood. While the new US Childhood Cancer Data Initiative may provide new in silico tools that can inform pediatric cancer drug development, ultimately, pharmaceutical companies may have limited access to relevant tissue or appropriate model systems for testing and therefore collaboration with independent academic labs should be encouraged. In addition, the US Pediatric Preclinical Testing Consortium has a long track record of providing testing of novel agents in established models and similar work is being done in Europe by the ITCC.(38) New public-private partnerships such as that sponsored by the FNIH may continue to bridge this gap. While there is no predefined level of preclinical evidence needed to move an agent into the pediatric oncology clinic, preclinical testing can help to prioritize or deprioritize specific new agents or drug classes. The required level of evidence needed to move a drug forward depends upon the available histology and biomarker- specific data from adult patients, the agent’s toxicity profile and the level of unmet medical need in the potential pediatric target population.
With regard to unmet clinical need, potential for benefit and tolerance for risk, parent and patient advocacy organizations can provide the patient voice in early go/ no go discussions and as such, have become an integral part of the pediatric drug development process. Their contribution to multi-stakeholder initiatives is invaluable and they can provide insight about facilitating access to novel therapies, help ensure equipoise in randomized trials, facilitate research forums, fundraise to bridge specific needs in the field and advocate with lawmakers, regulators and health technology assessment bodies. As one recent example, passage of the US RACE for Children Act requiring sponsors to submit pediatric study plans based on relevance of the molecular target was achieved with the support of numerous advocacy groups.
Challenges of pediatric drug development and the opportunities they create
All pediatric cancers are rare diseases
Pediatric cancers occur in approximately 16 of 100,000 children between 1-18 years,(39) which as a whole meets the EU and US definitions of rare or orphan diseases.(40) Pediatric tumors include many histologically and biologically disparate tumors presenting unique challenges around patient numbers. As focus shifts increasingly to targeted therapy, conduct of studies of targeted agents can be challenging in the pediatric population. Identifying and selecting for subpopulations with specific mutations or biomarkers presents challenges around sample size and the power to detect an effect. Novel trial designs and approaches must be employed to obtain safety and preliminary efficacy data efficiently and from as few patients as possible. The majority of pediatric phase 1 dose finding studies utilize either the 3+3 or rolling 6 trial designs. These conservative trial designs, while easy to conduct, are less likely to determine the true maximally tolerated dose (MTD) and treat more patients at suboptimal dosing.(41–43) Utilization of adaptive model trial designs such as the continuous reassessment method (CRM) or Bayesian optimal interval (BOIN) can provide better dose finding accuracy using data from prior studies and thereby improving efficiency.(41,43,44) Additionally, these model-based designs are able to account for size-based dosing in non-pediatric friendly formulations such as pills.(43,45)
Historically, phase 1 studies in pediatrics have been agnostic of tumor type for the dose finding portion of the study. This approach allows for increased enrollment numbers and decreased time to determine the recommended phase 2 dose (RP2D). However, this method may limit opportunities for the desired patients with the targeted biologic changes in their tumor to enroll. Novel trial designs can enrich for the targeted population while maintaining efficiency. One such design permits enrollment of tumor regardless of mutational status, however patients with the desired tumor mutations targeted by the study drug can enroll either at the current dose escalation dose or, if there are no available slots, at one dose level below the current dose escalation level. This design is currently being employed in at least 2 pediatric trials (NCT03654716, NCT03936465). This enriches for the desired population while continuing to efficiently determine the RP2D and MTD. The Pediatric MATCH trial (NCT03155620) employs an alternative approach, incorporating dose finding and efficacy evaluation within a pediatric histology-agnostic basket trial. In subprotocols for which the adult MTD and RP2D are not well defined, a limited dose finding phase is implemented (NCT03213678, NCT 03698994). Such early phase trial designs allow for efficient determination of pediatric dosing while concurrently enriching for targeted tumor subtypes which are most likely to respond. In addition to increasing the chance to benefit enrolled patients, early efficacy signals in targeted tumor types on these studies may augment enthusiasm for patients and physicians to enroll, and if early evidence of activity is seen, for academia and pharma to continue to pursue pediatric development of the agent.
Umbrella and basket protocols allow for the application of precision medicine in a tumor-agnostic approach. This type of trial can increase accrual when compared to a histology-specific biomarker-positive design while increasing the chance of benefit to individual patients and providing early evidence of efficacy when compared to enrolling unselected cohorts of patients.(46) Notably, the pediatric phase 1/2 study of dabrafenib successfully utilized such a design, while the pediatric phase 1 study of vemurafenib failed to accrue while only enrolling a cohort of patients with BRAF-mutated melanoma.(33,34) Current pediatric trials such as the Pediatric MATCH (NCT03155620) and the European ESMART trial (NCT02813135) enroll patients with multiple relapsed/refractory pediatric cancer types and then assign patients to one of several treatment arms based on defined molecular alterations. Umbrella protocols such as these improve directed enrollment for tumor specific mutations to respective targeted agents and in some instances permit subsequent assignment to another therapy arm if initial treatment is not successful.
For agents of interest for diseases affecting young children, drug development plans should incorporate pediatric friendly formulations early in the development process to allow rapid initiation of pediatric drug development as soon as adult MTD is determined. When multiple drugs in the same class are in development, the availability of a pediatric friendly formulation can influence which agent(s) are selected for studies in children. Conversely, the selection of the agent(s) for studies in children should be early enough to allow for the development of pediatric formulations if none exist given the time and expense required. Oral formulations developed for fixed dosing in adults are often too large to allow for appropriate body surface area-based dosing for smaller children and limit the eligible population to those who can swallow pills. Also, multiple strength tablets or a liquid formulation are often needed to allow for appropriate dosing during the dose escalation phases, accounting for children with different body surface areas and allow suitable dose increases and reductions. Delays in developing a pediatric friendly formulation restrict enrollment on pediatric trials to older and larger children, inadequately representing younger patients and some pediatric histologies that occur almost exclusively in this age group. This point is highlighted by the pediatric development of larotrectinib, a selective tropomyosin receptor kinase (TRK) inhibitor. TRK-fusion cancers occur in very young children with the majority of pediatric cases in patients under 2 years of age, including glioma, fibrosarcoma and congenital mesoblastic nephroma. A pediatric friendly formulation of larotrectinib was developed early, permitting near simultaneous agent development in both pediatric and adult populations. For the pediatric study, almost 30% of the enrolled patients were less than 2 years of age and almost half of the responders had infantile fibrosarcoma.(13) As a result, an age- and histology-agnostic global approval was obtained in 2018. Without the pediatric formulation, these young patients would not have been included and the pediatric data not fulfilled the needs of children with TRK-fusion cancers. Notably, the TRK inhibitor entrectinib is only FDA approved for children 12 years and older despite an ongoing pediatric trial of this drug (NCT02650401), pediatric development has been slowed by delayed availability of a pediatric-friendly formulation.(26)
As pediatric cancer continues to be parsed into genomically-defined subtypes of already rare tumors, the importance of collaboration becomes even more critical. Pediatric cooperative groups have been integral in pediatric oncology for several decades. However, further international collaboration is crucial as targeted therapy is developed for molecular subtypes of pediatric tumors. Large multinational trials allow opportunity for enrollment of as many patients of a specific rare molecular subtype to garner enough power to be able to detect a difference in this population.(47) As discussed above, the rarity of specific subsets of pediatric cancer also requires coordination between regulators and sponsors when there are multiple agents in the same class seeking pediatric safety and efficacy data.
The quiet pediatric cancer genome
Compared to most adult malignancies, most pediatric cancer genomes are characterized by a low tumor mutational burden.(48,49) Instead, pediatric tumors often harbor structural variants, such as copy number alterations and translocations (see next section). When present, mutations recurrently observed in the same histology are thought to play an important role in the biology of the disease, particularly if few other genomic alterations are seen (e.g., SMARCB1 mutations in epithelioid sarcoma and malignant rhabdoid tumors, and BRAF mutations in melanoma, glioma, and Langerhans cell histiocytosis). These mutations may lead to specific vulnerabilities that may be exploited for therapeutic gain. For example, SMARCB1 inactivating mutations result in a dependency on EZH2,(50) leading to objective responses and recent regulatory approval for the EZH2 inhibitor tazemetostat for patients ≥16 years of age with epithelioid sarcoma.(51) Additionally, recent preclinical work suggests that SMARCB1 inactivating mutations result in derepression of endogenous retroviral elements leading to highly inflamed tumors that may be responsive to immunotherapy, despite their exceedingly low mutational burden.(52)
Nevertheless, in many pediatric cancers, there is not a clearly defined genomic target. In these cases, the field has moved towards targeting other vulnerabilities, including cell surface overexpression of specific antigens. As many pediatric cancers are embryonal cancers, developmentally regulated proteins with highly restricted expression may be present on the cell surfaceome and/or presented via the major histocompatability complex (MHC), allowing for immunotherapy approaches that minimize “on-target / off-tumor” toxicities. Targeting the cell surfaceome has led to several pediatric regulatory approvals of monoclonal antibodies, bispecific T cell engagers, and CAR-T cells. For example, the anti-GD2 chimeric antibody dinutuximab was shown to improve two year event-free survival by 20% in patients with newly diagnosed high-risk neuroblastoma when given during the post-consolidation phase of therapy.(12) The bispecific T-cell engager (BiTE) antibody targeting-CD19 and CD3 blinatumomab has improved disease-free survival and rates of minimal residual disease response when combined with standard relapse chemotherapy.(53) Another dual-affinity CD123 X CD3 antibody flotetuzumab is currently in phase 1 trials though the Pediatric Early Phase Clinical Trials Network for pediatric acute myelogenous leukemia (AML, NCT04158739).
Adoptive cell therapy with engineered autologous CAR-T cells against CD19 has demonstrated transformational activity in ALL, with response rates as high as 93% and 12-month event free survival of 50%, leading to FDA approval of tisagenlecleucel in 2017.(54–56) Epitope loss or downregulation are secondary resistance mechanisms(57,58) which are now being targeted clinically with dual targeted CAR-T products against CD19/CD22 or CD19/CD20 (NCT03241940, NCT03330691). Studies in adult patients are now evaluating the combination of checkpoint inhibition with CAR-T cell products to overcome resistance due to CAR-T cell exhaustion/senescence and failure of in vivo expansion, which can lead to relapses that retain CD19 expression (NCT03630159). Additional CAR constructs which target other cell surface antigens expressed on leukemia and lymphomas are actively in development and are of interest for AML, including in cases of lineage switch after treatment of ALL with CD19 targeted CAR-T cell products.(59)
Clinical trials of CAR-T cells targeting antigens on pediatric solid tumors have been relatively disappointing to date, highlighting the challenges of overcoming the immune suppressive tumor microenvironment.(60) While challenged by the need to subdivide already rare patient populations into HLA-specific cohorts, studies of cells engineered to express T-cell receptors recognizing developmentally restricted self-antigens in an MHC-dependent manner have shown promise in early studies for solid tumors and remain under investigation (NCT03697824).(61) Moreover, cell surface expression of specific antigens may be heterogeneous within the same histology (e.g., GD2 expression in pediatric sarcomas)(62) and clinical assays to select appropriate patients for therapies targeted to specific antigens are needed in these situations.
Another consequence of the quiet pediatric cancer genome may be fewer cancer neoantigens to generate an antitumor response in the context of immune checkpoint inhibition. Indeed, the clinical experience to date with this class of compounds in pediatrics has been disappointing. Expression of PD1 and its corresponding ligands PDL-1 and PDL-2 is low on most pediatric tumors.(63) Clinical responses to immune checkpoint inhibitors in children have largely been reported in pediatric tumors with higher PD1 expression such as lymphoma or with increased tumor mutational burden as seen in melanoma, tumors with high microsatellite instability or patients with congenital mismatch repair deficiency.(64–66) However, multiple early phase studies of immune checkpoint inhibitors in unselected pediatric patients have failed to demonstrate objective responses in the majority of patients enrolled.(65–67) While checkpoint monotherapy has been disappointing, there is interest in combination therapy of checkpoint inhibitors (NCT 03837899, NCT03130959, NCT02304458) or combinations with chemotherapy, radiation or other targeted radiation therapy (NCT02927769, NCT03445858, NCT02914405) which could improve the “cold” immune environment of pediatric cancers through increased antigen presentation and T-cell activation.(68,69)
The lower tumor mutational burden generally seen in pediatric cancers compared to adult cancers may ultimately be advantageous in the context of drug development. Owing to fewer secondary genomic events, pediatric tumors may tend to be more genomically homogeneous, potentially reducing the potential for confounding through activation of other pathways by passenger mutations. Likewise, primary resistance mutations may be less likely to be present de novo in tumors that are less genomically complex and that have not evolved over many years. For example, the response rate to the TRK inhibitor larotrectinib in patients with TRK fusion cancers is greater in children (94%) than in adults (71%) despite harboring the same biomarker.(70,71) Similarly, the response rates to crizotinib for ALK-fusion positive IMT (86%)(25) and ALCL (88%)(25) were numerically higher than for ALK-fusion lung cancer in adults (65-74%).(72,73) However, it is worth noting that ALK inhibitors have had very dissimilar results in pediatric tumors harboring different genomic aberrations, such as ALK-mutated or amplified neuroblastomas which show more resistance to ALK inhibitors. This further highlights the need for pediatric-specific drug development efforts which account for the common molecular alterations within common pediatric cancers.
Finally, it should be noted that most published sequencing data from pediatric cancers are from diagnostic samples. Enrichment of potentially targetable molecular alterations has been noted at relapse, such as RAS-MAPK activating alterations in neuroblastoma.(74) However, in the NCI-COG Pediatric MATCH study, still only 29% of patients had actionable mutations identified in relapse tissue.(75) Further evaluation of the evolution of the pediatric cancer genome over the course of therapy will be important to identify the frequency of the development of targetable alterations and whether the mutations that develop over the course of therapy are sufficiently clonal to serve as therapeutic targets. In addition, it will be especially important to collect and analyze relapse tissue and diagnostic tissue from patients with relapsed disease enrolling on targeted therapy trials to better characterize potential biomarkers. New approaches using cell-free DNA or circulating tumor DNA may provide a more feasible non-invasive means of detecting and monitoring mutational changes and burden during therapy.(76)
Targeting non-kinase fusion oncoproteins
A substantial proportion of pediatric, adolescent, and young adult cancers are characterized by recurrent chromosomal translocations that generate fusion oncoproteins. These fusion oncoproteins can be broadly classified as kinase fusions or non-kinase fusions, depending upon whether or not the fusion oncoprotein harbors a kinase domain. Kinase fusions typically result in an overexpressed and constitutively active kinase that drives the cancer. Key examples include NPM/ALK fusions in anaplastic large cell lymphoma or ETV6/NTRK3 fusions in infantile fibrosarcoma. In contrast, non-kinase fusions often result in an aberrant transcription factor that drives an abnormal gene expression program that characterizes a specific cancer. Key examples include EWSR1/FLI1 fusions in Ewing sarcoma and PAX3/FOXO1 fusions in alveolar rhabdomyosarcoma.
Kinase fusions represent the minority of fusion oncoproteins in the pediatric population but are illustrative of important principles of drug development that might be applied to development of drugs targeting non-kinase fusions. First, targeting kinase fusions with selective kinase inhibitors has resulted in prompt, dramatic, and often durable responses, that demonstrate the central role these fusions play as drivers of the disease.(13,25) Second, given the very high response rates associated with agents targeting these driver fusions, agents such as entrectinib and larotrectinib have received regulatory approval in adolescents and children based upon relatively small early phase clinical trials. Third, the therapeutic index has typically been wide, both due to increased kinase selectivity and due to the extreme sensitivity of these cancers to kinase inhibition, with responses observed across dose levels in first-in-human or first-in-child trials. Fourth, primary resistance is exceedingly rare, but secondary resistance to monotherapy kinase inhibition has been reported,(77–79) highlighting the potential role for combination strategies.
In this context, targeting non-kinase fusions may likewise provide important opportunities for developing drugs for the rare pediatric cancers driven by this type of fusion. To date, many of these cancers have been treated with non-specific cytotoxic chemotherapy, with some success seen in patients with localized disease and little success in patients with metastatic or recurrent disease. Due to difficulties targeting aberrant transcription factors (rather than kinases), attempts at targeted therapy have largely focused on effectors downstream of the oncoprotein. For example, activity of the IGF-1R pathway has been shown to be upregulated by the EWSR1/FLI1 fusion oncoprotein.(80) Early phase clinical trials of monoclonal antibodies directed against IGF-1R demonstrated that approximately 10% of patients with relapsed Ewing sarcoma responded.(81,82) This experience highlights the modest activity associated with targeting just one of many downstream effectors of the fusion oncoprotein, rather than targeting the fusion oncoprotein itself. Indeed, a recent randomized phase 3 trial failed to show an improvement in event-free survival when an IGF-1R monoclonal antibody was added to conventional chemotherapy for patients with newly diagnosed metastatic Ewing sarcoma.(83)
This type of disappointing outcome stands in stark contrast to the exciting results seen targeting kinase fusions and now motivates strategies to directly target non-kinase fusion oncoproteins. Key strategies under investigation in the laboratory and also now in the clinic include attempts to block the function of these oncoproteins, selectively reduce expression of the fusion oncogenes, or selectively degrade the fusion oncoprotein. As these non-kinase fusion oncoproteins are true drivers of their associated cancers, these strategies have shown promise in the laboratory,(84–87) though proof-of-concept is yet lacking in the clinic. Likewise, as these oncoproteins should not exist outside of cancer cells, the therapeutic index of selective approaches to targeting non-fusion kinases is expected to be wide. Many of the cancers driven by non-kinase fusions impact adolescents and young adults, providing an opportunity for age-agnostic drug development strategies that may facilitate accrual in these rare diseases. Some of the principles learned through the study of non-kinase fusions may extend to the evaluation of other oncoproteins that likewise serve as transcription factors driving abnormal gene expression progressions (e.g. high level MYCN amplification in neuroblastoma).
Assessing long-term toxicities of targeted therapies in growing and developing hosts without halting drug development
While most new anticancer drugs are more tolerable than conventional chemotherapeutics, they are not exempt from significant and severe toxicities in the short and long term. In the short-term, toxicities of targeted therapies in children are similar to those identified in adults.(17)
Only recently, efforts have started to improve our understanding of late toxicities of novel drugs in children with developing organs and systems.(88) Novel drugs can have an impact on transcriptional pathways, intracellular signaling pathways, epigenetic functions or the microenvironment, all of which are critical during growth and development of multiple organs. At the time of starting early phase pediatric trials, potential developmental toxicities are often considered but are difficult to assess and predict. Examples are risk of growth plate closure with antiangiogenics and sonic hedgehog inhibitors or risk of neurocognitive toxicities of ALK and TRK inhibitors. Experience gained over the last two decades shows that, while these concerns must be addressed during the development of novel agents, particularly once drugs move into phase 2 or phase 3 trials, they should not halt or delay the initiation of pediatric early phase trials.
The role of juvenile animal studies and its ability to predict toxicity in the pediatric oncology population has not been fully elucidated yet. For example, a review of 29 recently-developed anticancer medicines found that severe toxicities arising in the first-in-child trials could not be prevented by conducting standard juvenile animal studies.(89) Another review of drugs that received European approval and had Pediatric Investigational Plans showed that approximately 1 in 3 oncology medicines with PIPs had conducted juvenile animal studies, contributing to a better characterization of safety in some cases.(90) While these studies have proven useful in other pediatric disciplines, there has been general reluctance from pharma and academia to systematically incorporate these studies prior to initial clinical trials of new drugs for children with cancer. On the other hand, such studies have been supported by the EMA and on a case-by-case basis by the FDA.(91) More data are needed to fully ascertain the capacity of juvenile animal studies to identify, predict and hopefully prevent potential developmental toxicities in children.
At present, pediatric phase 1 and 2 trials provide limited opportunities to identify long-term toxicities since they are mostly conducted in patients with relapsed and refractory cancers with short life expectancy.(92,93) Even in highly successful trials with long-term responders, the relatively small and heterogeneous patient population, lack of randomized controls, and potential late effects of prior therapies limit the ability to evaluate long-term toxicities.(13,25,34) Hence, the main challenge (and opportunity) arises at the time of bringing these drugs upfront into first-line collaborative international trials by academic groups such as the Children’s Oncology Group (COG) and the European Society for Pediatric Oncology (SIOPE) where late effects can be systematically evaluated, particularly for tumor types occurring in younger children.
Potential late toxicities can affect virtually all organs. However, to date, very few have been reported. Table 2 shows potential toxicities in the growing/developing host associated with novel agents according to target organ. In many cases, the impact of these toxicities would also be influenced by the patient’s age and their developmental status. The following examples illustrate how these toxicities can be evaluated and risks mitigated.
Table 2:
Potential long-term toxicities of new targeted therapies in the developing host.
| Organ/system affected | Specific toxicity | Drugs |
|---|---|---|
| Growth and development | Short stature (due to IGF axis blockade) | TKI including BCR-ABL inhibitors |
| Short stature (growth plate closure) | Antiangiogenics Sonic hedgehog inhibitors |
|
| Reproductive | Testicular and ovarian toxicity | ALK inhibitors |
| Alterations of ovarian follicle development | Antiangiogenics | |
| Nervous system | Neurotoxicity | Blinatumomab ALK inhibitors CAR-T cells Anti-GD2 monoclonal antibodies |
| Visual disturbances | ALK inhibitors MEK inhibitors Anti-GD2 monoclonal antibodies |
|
| Neurocognitive disfunction and psychiatric conditions | ALK inhibitors (lorlatinib) | |
| Immune system | B-cell aplasia | Anti-CD19 CAR-T cells |
| Autoimmune conditions | Anti-PD-1/PD-L1 agents | |
| Endocrine system | Hypothyroidism | TKI |
| Hyperglycemia and metabolic syndrome | ALK, PI3K, MEK inhibitors | |
| Cardiovascular | Cardiac dysfunction | MEK inhibitors, TKI |
| Hypertension | Antiangiogenics |
Linear growth with imatinib and other tyrosine kinase inhibitors (TKI)
Imatinib and other ABL-kinase inhibitors were among the earliest targeted anticancer agents evaluated in pediatrics in the early 2000s. Some reports have highlighted growth impairment, particularly in pre-pubertal children. Shima et al. reported a decrease in height in more than 70% of children treated with imatinib, which was more pronounced in children starting imatinib before pubertal age.(5) Bone length and bone density were reduced after long term exposure in preclinical studies. The mechanism of this toxicity has not been fully elucidated, and could be related to off target effects on IGF-1 or PDGFR pathways.(6) Subsequently, early phase trials of new ABL TKIs, such as dasatinib and nilotinib have included growth measurements, circulating biomarkers and determinations of bone mineral density in all patients. The initial analysis from the dasatinib phase 2 trial reported bone growth and development toxicities in five of 113 patients treated (4%), all were grade 1–2.(47) In the nilotinib pediatric phase 2 trial, no impact on growth was reported,(94) however longer follow-up including analyses of growth biomarkers and bone mineral density studies will be required. A pediatric trial of bosutinib (NCT04258943), another more potent ABL TKI, has recently opened, with the potential advantage of reducing the risk of growth impairment.(7)
For other TKIs, particularly those with an antiangiogenic profile, abnormalities in the growth plates have been reported in a small but relevant proportion of patients. In the phase 1 trial of pazopanib, out of 25 patients included, one had physeal widening and three had growth plate widening.(95) As some of these agents move forward and are being evaluated in frontline studies, there is a greater need to continue growth plate monitoring.(96,97) While a pooled analysis of children receiving bevacizumab showed no negative effects on growth velocity, height or BMI,(98) longer follow up of patients treated with these drugs are needed to determine long-term effects on growth and development.
Neurocognitive and visual outcomes with crizotinib and other TKI
Crizotinib, an ALK inhibitor approved for ALK positive non-small cell lung cancer, showed promising preclinical and clinical activity in children with ALK mutated neuroblastomas and ALK-fused anaplastic large cell lymphomas and inflammatory myofibroblastic tumors. (25) Furthermore, the recently reported pediatric phase 1 trial of lorlatinib, a third generation ALK inhibitor by the New Advances in Neuroblastoma Therapy (NANT) consortium showed that a proportion of patients developed neurocognitive toxicities. Grade 1 events such as anxiety, cognitive disturbance, concentration or memory impairment were seen in pediatric patients and grade 1-4 events were noted in patients older than 18 years, particularly in a patient with an undiagnosed pre-existing psychiatric condition.(99) Given the known role of ALK in the developing embryonic and neonatal brain(100) and the occurrence of ocular and neurological toxicities in adult trials, enhanced monitoring of visual and neurocognitive outcomes was incorporated into the lorlatinib trial, which is now being moved forward to a frontline phase 3 trial by the COG. However, it is difficult to assess these outcomes in very young children with appropriate tools and scales and in the context of complex and aggressive multimodal therapy.
Risk of secondary malignancies and immune dysregulation with engineered cellular therapies
The development of CAR-T cells has revolutionized outcome for children with B-cell acute lymphoblastic leukemia,(55) leading to the approval of tisagenlecleucel. CAR-T cells directed against CD19 cause profound and potentially life-long B-cell aplasia which requires chronic immunoglobulin replacement. Late infections, second malignancies, immune-related events, neurologic and psychiatric events have been described.(8) Among 86 recipients of adult CAR-T cells surviving for a year or longer, 13 developed a second malignancy; but no data on pediatric patients have yet been reported. Following the regulatory requirements, a follow up study of patients treated with tisagenlecleucel was launched (NCT02445222) to address in depth long-term toxicities, paying particular attention to secondary malignancies, neurologic disorders, exacerbation of immune conditions or other emergent toxicities.
Duration of therapy is often as yet undefined for new targeted therapies, an aspect with crucial impact on potential for long-term toxicities. As novel drugs are being incorporated into frontline, the optimal duration of therapy should also be explored. Some targeted therapies will be given for a relatively short duration to facilitate further consolidation therapies, such as an ALK inhibitor facilitating remission in a patient with anaplastic large cell lymphoma who then goes to receive a hematopoietic stem cell transplant. For targeted agents developed against hematological conditions, such as inotuzumab (anti-CD22), blinatumomab (anti-CD19 BiTE) or anti-CD19 CAR-T, it remains essential to define the indications for hematopoietic stem cell transplantation after targeted therapies.
Likewise, a patient with an inoperable TRK-driven cancer could receive a TRK inhibitor until appropriate non-mutilating surgery can be conducted and then stop therapy. However, for most targeted drugs developed in early clinical trials, treatment is given until disease progression or unacceptable toxicity. In these cases, responding patients can often receive therapy for years, and the optimal timing to stop therapy remains unknown. When considering stopping therapy in responding patients, it will be important to define the likelihood of response if treatment re-challenge is needed for subsequent relapse for each targeted therapy and condition. Defining the shortest possible course of targeted drugs that minimizes long-term toxicities while has no impact on reducing survival outcomes remains a key challenge.
Towards a new model to address the need for long-term follow up
With numerous targeted agents being developed for pediatric cancers, some of them being taken to frontline therapy, it will be necessary to performed pooled analysis of long-term outcomes across all pediatric cancers, but also for drugs against a specific target (e.g. all patients treated with ALK inhibitors) or within a specific condition (e.g. all patients with neuroblastoma treated with novel agents). It is hence necessary to create an international data repository to collect information on long-term health in children that have received new anticancer therapies, across different targets, sponsors and tumor types, independently of specific regulatory requirements for each drug. The Accelerate platform has recently launched a working group on this matter (https://www.accelerate-platform.org/wp-content/uploads/sites/4/2019/07/Summary_7th-ACCELERATE-Paediatric-Oncology-Conference_14-15-Feb-2019.pdf). Pancare in Europe (www.pancare.eu) and the Childhood Cancer Survivor Study (CCSS) in North America are leading efforts involving all stakeholders, including empowering cancer survivors, to identify and mitigate long-term toxicities.
Conclusions
Only a limited number of targeted therapies have achieved regulatory approval or widespread use in pediatric oncology. Given recent regulatory changes, a broad range of drug classes and agents are of interest and under current investigation (Table 3). While the majority of currently approved targeted therapies for pediatric cancer are kinase inhibitors, agents are under investigation for a broader range of other targets, including epigenetic modifiers, anti-apoptotic proteins, and immunotherapies against cell surface proteins. New pharmaceutical strategies to directly target non-kinase oncoproteins, one of the most common oncogenic drivers in pediatric cancer, have shown promise in pre-clinical studies and have entered the clinic. Drug development for pediatric oncology presents a number of challenges including lower patient numbers compared with adult oncology, a generally quiet genome, and the need to develop strategies to target non-kinase fusion oncoproteins. However, these same challenges result in a number of opportunities including a mandate to coordinate drug development across national boundaries and sponsors, the potential for easier identification of true oncogenic drivers in pediatric cancer, and less acquired resistance. As novel therapies move to the frontline setting, it will be important to develop mechanisms to evaluate the long-term toxicities of these agents without halting new discoveries or use of promising agents in the clinic.
Table 3:
Mechanisms of action of selected targeted agents under investigation for the treatment of pediatric cancer
| Approval in Adults | ||||||
|---|---|---|---|---|---|---|
| Drug Class | Example Agent(s)* | Target | Pediatric Intended Use | FDA | EMA | Pediatric Trial(s) |
| Tyrosine kinase inhibitors | Midostaurin Gliteritinib |
FLT3 | FLT3-altered AML |
Y Y |
Y Y |
NCT03591510 NCT04293562 |
| Crizotinib | ALK, ROS1 | IMT, ALCL NB |
Y | Y |
NCT01979536 NCT03126916 |
|
| Pazopanib Regorafenib |
VEGFR, PDGFR | Sarcoma | Y Y |
Y Y |
NCT02180867 NCT02389244 |
|
| Cabozantinib | MET, VEGFR, PDGFR |
Y | Y |
NCT02867592 NCT02243605 |
||
| Downstream signaling pathway inhibitors | Temsirolimus | mTOR | RMS | Y | Y | NCT02567435 |
| Vemurafenib | BRAF V600 | Biomarker (+) | Y | Y | NCT03220035 | |
| Dabrafenib + Trametinib |
BRAF + MEK | Low grade glioma High grade glioma |
Y | Y |
NCT02684058 NCT03919071 |
|
| Cobimetinib | MEK | Langerhans cell Histiocytosis | Y | Y | NCT04079179 | |
| Ruxolitinib* | JAK | CRLF or JAK altered ALL | Y | Y | NCT02913261 | |
| Developmental pathway inhibitors | Vismodegib Sonidegib |
SMO | SHH-driven medulloblastoma | Y Y |
Y Y |
NCT01878617 NCT04402073 |
| Epigenetic therapy | Panobinostat Vorinostat |
Histone deacetylase | DIPG (H3K27M) NB AML ALL |
Y Y |
Y N |
NCT02717455 NCT02035137 NCT03263936 NCT02553460 |
| Pinometostat | DOT1L | KMT2A-rearranged ALL | N | N | NCT03724084 | |
| SNDX-5613 | Menin | KMT2A-rearranged ALL or AML | N | N | NCT04065399 | |
| Azacitidine Decitabine |
DNA Methyl-transferases | KMT2A-rearranged ALL or AML | Y Y |
Y Y |
NCT02828358 NCT03263936 |
|
| Molibresib | Bromodomain (BET) | NUT Carcinoma | N | N | NCT04116359 | |
| Seclidemstat INCB059872 |
LSD1 | Ewing Sarcoma | N N |
N N |
NCT03600649 NCT03514407 |
|
| Cell death pathway inhibitors | Venetoclax | BCL-2 | AML ALL, AML, NHL |
Y | Y |
NCT03194932 NCT03236857 |
| Non-kinase fusion inhibitors | TK216 | EWS-FLI1 | Ewing sarcoma | N | N | NCT02657005 |
| Cell lineage monoclonal antibodies and antibody drug conjugates |
Rituximab | CD20 | NHL | Y | Y | NCT03206671 |
| Gemtuzumab | CD33 | AML | Y | Y | NCT02724163 | |
| Inotuzumab ozogamicin | CD22 | ALL | Y | Y | NCT02981628 | |
| Brentuximab vedotin | CD30 | Hodgkin ALCL |
Y | Y |
NCT02166463 NCT01979536 |
|
| Targeted radionuclides | Iodine-131-meta-iodo-benzylguanidine (MIBG) | Norepinephrine transporter | NB | N | N | NCT03126916 |
Not intended to be a comprehensive list of all agents under investigation.
Statement of Significance.
Increasing attention to drug development for children with cancer by regulators and pharmaceutical companies holds the promise of accelerating the availability of new therapies for children with cancer, potentially improving survival and decreasing the acute and chronic toxicities of therapy. However, unique approaches are necessary to study novel therapies in children that take into account low patient numbers, the pediatric cancer genomic landscape and tumor microenvironment, and need for pediatric formulations. It is also critical to evaluate the potential for unique toxicities in growing hosts without impacting the pace of discovery for children with these life-threatening diseases.
Acknowledgments
Financial Support:TWL is supported by the Norma and Jim Smith Professorship of Clinical Excellence and Eugene P. Frenkel, M.D. Scholarship in Clinical Medicine. JGB is supported by the NIH/NCI grant CA 008748.
Conflicts of interest:
TWL has participated in advisory boards for Novartis, Bayer, Loxo Oncology/Eli Lilly, and Cellectis, and has research funding from Novartis, Bayer, and Pfizer. SGD has received fees for consulting and advisory board roles from Bayer and Loxo Oncology and has received travel expenses from Loxo Oncology, Roche/Genentech, and Salarius Pharmaceuticals. JGB has uncompensated advisory board roles with Abbvie, Springworks, Merck and BMS and has received travel expenses from Merck, Bayer, Amgen and Novartis. MEM has research funding from Bayer, and owns stock in Johnson and Johnson. LM has participated in advisory boards for Novartis, AstraZeneca, Roche/Genentech, Mundipharma, Bayer and Amgen; has received honoraria from Celgene and Novartis for an educational event and travel grants from Celgene, MundiPharma and Amgen. LM is also member of data monitoring committees for clinical trials sponsored by Novartis, Actuate Therapeutics, Shionogi, Incyte, the University of Southampton and the Royal Marsden NHS Foundation Trust and a member of the Executive Committee of SIOPEN (European neuroblastoma research cooperative group) which receives royalties for the sales of dinutuximab beta. His institution receives funding from sponsors for DMC participation, advisory roles and conducting industry-sponsored clinical trials.
References
- 1.Oeffinger KC, Mertens AC, Sklar CA, Kawashima T, Hudson MM, Meadows AT, et al. Chronic health conditions in adult survivors of childhood cancer. N Engl J Med 2006;355(15):1572–82 [DOI] [PubMed] [Google Scholar]
- 2.Hunger SP, Mullighan CG. Acute Lymphoblastic Leukemia in Children. N Engl J Med 2015;373(16):1541–52 [DOI] [PubMed] [Google Scholar]
- 3.Dorris K, Liu C, Li D, Hummel TR, Wang X, Perentesis J, et al. A comparison of safety and efficacy of cytotoxic versus molecularly targeted drugs in pediatric phase I solid tumor oncology trials. Pediatric blood & cancer 2017;64(3) [DOI] [PubMed] [Google Scholar]
- 4.Pappo AS, Furman WL, Schultz KA, Ferrari A, Helman L, Krailo MD. Rare Tumors in Children: Progress Through Collaboration. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2015;33(27):3047–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shima H, Tokuyama M, Tanizawa A, Tono C, Hamamoto K, Muramatsu H, et al. Distinct impact of imatinib on growth at prepubertal and pubertal ages of children with chronic myeloid leukemia. The Journal of pediatrics 2011;159(4):676–81 [DOI] [PubMed] [Google Scholar]
- 6.Tauer JT, Hofbauer LC, Jung R, Gerdes S, Glauche I, Erben RG, et al. Impact of long-term exposure to the tyrosine kinase inhibitor imatinib on the skeleton of growing rats. PloS one 2015;10(6):e0131192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tauer JT, Hofbauer LC, Jung R, Erben RG, Suttorp M. Micro-osmotic pumps for continuous release of the tyrosine kinase inhibitor bosutinib in juvenile rats and its impact on bone growth. Med Sci Monit Basic Res 2013;19:274–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cordeiro A, Bezerra ED, Hirayama AV, Hill JA, Wu QV, Voutsinas J, et al. Late Events after Treatment with CD19-Targeted Chimeric Antigen Receptor Modified T Cells. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 2020;26(1):26–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hijiya N, Schultz KR, Metzler M, Millot F, Suttorp M. Pediatric chronic myeloid leukemia is a unique disease that requires a different approach. Blood 2016;127(4):392–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abla O, Ribeiro RC. How I treat children and adolescents with acute promyelocytic leukaemia. British journal of haematology 2014;164(1):24–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Minard-Colin V, Auperin A, Pillon M, Burke A, Anderson JR, Barkauskas DA, et al. Results of the randomized Intergroup trial Inter-B-NHL Ritux 2010 for children and adolescents with high-risk B-cell non-Hodgkin lymphoma (B-NHL) and mature acute leukemia (B-AL): Evaluation of rituximab (R) efficacy in addition to standard LMB chemotherapy (CT) regimen. Journal of Clinical Oncology 2016;34(15_suppl):10507- [Google Scholar]
- 12.Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med 2010;363(14):1324–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laetsch TW, DuBois SG, Mascarenhas L, Turpin B, Federman N, Albert CM, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. The Lancet Oncology 2018;19(5):705–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skolnik JM, Barrett JS, Jayaraman B, Patel D, Adamson PC. Shortening the timeline of pediatric phase I trials: the rolling six design. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2008;26(2):190–5 [DOI] [PubMed] [Google Scholar]
- 15.Doussau A, Geoerger B, Jiménez I, Paoletti X. Innovations for phase I dose-finding designs in pediatric oncology clinical trials. Contemp Clin Trials 2016;47:217–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee DP, Skolnik JM, Adamson PC. Pediatric phase I trials in oncology: an analysis of study conduct efficiency. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2005;23(33):8431–41 [DOI] [PubMed] [Google Scholar]
- 17.Paoletti X, Geoerger B, Doz F, Baruchel A, Lokiec F, Le Tourneau C. A comparative analysis of paediatric dose-finding trials of molecularly targeted agent with adults' trials. Eur J Cancer 2013;49(10):2392–402 [DOI] [PubMed] [Google Scholar]
- 18.Momper JD, Mulugeta Y, Green DJ, Karesh A, Krudys KM, Sachs HC, et al. Adolescent dosing and labeling since the Food and Drug Administration Amendments Act of 2007. JAMA Pediatr 2013;167(10):926–32 [DOI] [PubMed] [Google Scholar]
- 19.Gaspar N, Marshall LV, Binner D, Herold R, Rousseau R, Blanc P, et al. Joint adolescent-adult early phase clinical trials to improve access to new drugs for adolescents with cancer: proposals from the multi-stakeholder platform-ACCELERATE. Ann Oncol 2018;29(3):766–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chakiba C, Grellety T, Bellera C, Italiano A. Encouraging Trends in Modern Phase 1 Oncology Trials. N Engl J Med 2018;378(23):2242–3 [DOI] [PubMed] [Google Scholar]
- 21.von Stackelberg A, Locatelli F, Zugmaier G, Handgretinger R, Trippett TM, Rizzari C, et al. Phase I/Phase II Study of Blinatumomab in Pediatric Patients With Relapsed/Refractory Acute Lymphoblastic Leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2016;34(36):4381–9 [DOI] [PubMed] [Google Scholar]
- 22.Zwaan CM, Rizzari C, Mechinaud F, Lancaster DL, Lehrnbecher T, Velden VHJvd, et al. Dasatinib in Children and Adolescents With Relapsed or Refractory Leukemia: Results of the CA180-018 Phase I Dose-Escalation Study of the Innovative Therapies for Children With Cancer Consortium. Journal of Clinical Oncology 2013;31(19):2460–8 [DOI] [PubMed] [Google Scholar]
- 23.Champagne MA, Capdeville R, Krailo M, Qu W, Peng B, Rosamilia M, et al. Imatinib mesylate (STI571) for treatment of children with Philadelphia chromosome-positive leukemia: results from a Children's Oncology Group phase 1 study. Blood 2004;104(9):2655–60 [DOI] [PubMed] [Google Scholar]
- 24.Hijiya N, Zwaan CM, Rizzari C, Foa R, Abbink F, Lancaster D, et al. Pharmacokinetics of Nilotinib in Pediatric Patients with Philadelphia Chromosome-Positive Chronic Myeloid Leukemia or Acute Lymphoblastic Leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research 2020;26(4):812–20 [DOI] [PubMed] [Google Scholar]
- 25.Mosse YP, Voss SD, Lim MS, Rolland D, Minard CG, Fox E, et al. Targeting ALK With Crizotinib in Pediatric Anaplastic Large Cell Lymphoma and Inflammatory Myofibroblastic Tumor: A Children's Oncology Group Study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2017;35(28):3215–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson GW, Gajjar AJ, Gauvain KM, Basu EM, Macy ME, Maese LD, et al. Phase 1/1B trial to assess the activity of entrectinib in children and adolescents with recurrent or refractory solid tumors including central nervous system (CNS) tumors. Journal of Clinical Oncology 2019;37(15_suppl):10009- [Google Scholar]
- 27.Neel DV, Shulman DS, DuBois SG. Timing of first-in-child trials of FDA-approved oncology drugs. Eur J Cancer 2019;112:49–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neel DV, Shulman DS, Ma C, Bourgeois F, DuBois SG. Sponsorship of oncology clinical trials in the United States according to age of eligibility. Cancer medicine 2020;9(13):4495–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomsen MDT. Global Pediatric Drug Development. Curr Ther Res Clin Exp 2019;90:135–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pearson ADJ, Pfister SM, Baruchel A, Bourquin JP, Casanova M, Chesler L, et al. From class waivers to precision medicine in paediatric oncology. Lancet Oncol 2017;18(7):e394–e404 [DOI] [PubMed] [Google Scholar]
- 31.Dulaney C United Therapeutics Sells Priority-Review Voucher to AbbVie for $350 Million. The Wall Street Journal 2015. Aug 15. [Google Scholar]
- 32.Nabhan C, Parsad S, Mato AR, Feinberg BA. Biosimilars in Oncology in the United States: A Review. JAMA oncology 2018;4(2):241–7 [DOI] [PubMed] [Google Scholar]
- 33.Chisholm JC, Suvada J, Dunkel IJ, Casanova M, Zhang W, Ritchie N, et al. BRIM-P: A phase I, open-label, multicenter, dose-escalation study of vemurafenib in pediatric patients with surgically incurable, BRAF mutation-positive melanoma. Pediatric blood & cancer 2018;65(5):e26947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kieran MW, Geoerger B, Dunkel IJ, Broniscer A, Hargrave DR, Hingorani P, et al. A Phase 1 and Pharmacokinetic Study of Oral Dabrafenib in Children and Adolescent Patients With Recurrent or Refractory BRAF V600 Mutation-Positive Solid Tumors. Clinical Cancer Research 2019;25(24):7294–302 [DOI] [PubMed] [Google Scholar]
- 35.Gore L, Ivy SP, Balis FM, Rubin E, Thornton K, Donoghue M, et al. Modernizing Clinical Trial Eligibility: Recommendations of the American Society of Clinical Oncology-Friends of Cancer Research Minimum Age Working Group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2017;35(33):3781–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pearson ADJ, Rossig C, Lesa G, Diede SJ, Weiner S, Anderson J, et al. ACCELERATE and European Medicines Agency Paediatric Strategy Forum for medicinal product development of checkpoint inhibitors for use in combination therapy in paediatric patients. Eur J Cancer 2020;127:52–66 [DOI] [PubMed] [Google Scholar]
- 37.Pearson ADJ, Scobie N, Norga K, Ligas F, Chiodin D, Burke A, et al. ACCELERATE and European Medicine Agency Paediatric Strategy Forum for medicinal product development for mature B-cell malignancies in children. Eur J Cancer 2019;110:74–85 [DOI] [PubMed] [Google Scholar]
- 38.Houghton PJ, Morton CL, Tucker C, Payne D, Favours E, Cole C, et al. The pediatric preclinical testing program: description of models and early testing results. Pediatric blood & cancer 2007;49(7):928–40 [DOI] [PubMed] [Google Scholar]
- 39.Steliarova-Foucher E, Colombet M, Ries LAG, Moreno F, Dolya A, Bray F, et al. International incidence of childhood cancer, 2001-10: a population-based registry study. Lancet Oncol 2017;18(6):719–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mariz S, Reese JH, Westermark K, Greene L, Goto T, Hoshino T, et al. Worldwide collaboration for orphan drug designation. Nature reviews Drug discovery 2016;15(6):440–1 [DOI] [PubMed] [Google Scholar]
- 41.Iasonos A, Wilton AS, Riedel ER, Seshan VE, Spriggs DR. A comprehensive comparison of the continual reassessment method to the standard 3 + 3 dose escalation scheme in Phase I dose-finding studies. Clin Trials 2008;5(5):465–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O'Quigley J, Pepe M, Fisher L. Continual reassessment method: a practical design for phase 1 clinical trials in cancer. Biometrics 1990;46(1):33–48 [PubMed] [Google Scholar]
- 43.Onar-Thomas A, Xiong Z. A simulation-based comparison of the traditional method, Rolling-6 design and a frequentist version of the continual reassessment method with special attention to trial duration in pediatric Phase I oncology trials. Contemp Clin Trials 2010;31(3):259–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Onar A, Kocak M, Boyett JM. Continual reassessment method vs. traditional empirically based design: modifications motivated by Phase I trials in pediatric oncology by the Pediatric Brain Tumor Consortium. J Biopharm Stat 2009;19(3):437–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Banerjee A, Jakacki RI, Onar-Thomas A, Wu S, Nicolaides T, Young Poussaint T, et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: a Pediatric Brain Tumor Consortium (PBTC) study. Neuro-oncology 2017;19(8):1135–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pearson AD, Herold R, Rousseau R, Copland C, Bradley-Garelik B, Binner D, et al. Implementation of mechanism of action biology-driven early drug development for children with cancer. Eur J Cancer 2016;62:124–31 [DOI] [PubMed] [Google Scholar]
- 47.Gore L, Kearns PR, de Martino ML, Lee, De Souza CA, Bertrand, et al. Dasatinib in Pediatric Patients With Chronic Myeloid Leukemia in Chronic Phase: Results From a Phase II Trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2018;36(13):1330–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma X, Liu Y, Liu Y, Alexandrov LB, Edmonson MN, Gawad C, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 2018;555(7696):371–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grobner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The landscape of genomic alterations across childhood cancers. Nature 2018;555(7696):321–7 [DOI] [PubMed] [Google Scholar]
- 50.Wilson BG, Wang X, Shen X, McKenna ES, Lemieux ME, Cho YJ, et al. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer cell 2010;18(4):316–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hoy SM. Tazemetostat: First Approval. Drugs 2020;80(5):513–21 [DOI] [PubMed] [Google Scholar]
- 52.Leruste A, Tosello J, Ramos RN, Tauziède-Espariat A, Brohard S, Han ZY, et al. Clonally Expanded T Cells Reveal Immunogenicity of Rhabdoid Tumors. Cancer cell 2019;36(6):597–612.e8 [DOI] [PubMed] [Google Scholar]
- 53.Brown PA, Ji L, Xu X, Devidas M, Hogan L, Borowitz MJ, et al. A Randomized Phase 3 Trial of Blinatumomab Vs. Chemotherapy As Post-Reinduction Therapy in High and Intermediate Risk (HR/IR) First Relapse of B-Acute Lymphoblastic Leukemia (B-ALL) in Children and Adolescents/Young Adults (AYAs) Demonstrates Superior Efficacy and Tolerability of Blinatumomab: A Report from Children's Oncology Group Study AALL1331. Blood 2019;134(Supplement_2):LBA-1-LBA- [Google Scholar]
- 54.Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 2017;129(25):3322–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med 2018;378(5):439–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. The Lancet 2015;385(9967):517–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nature medicine 2018;24(5):563–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer discovery 2015;5(12):1282–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bonifant CL, Tasian SK. The future of cellular immunotherapy for childhood leukemia. Curr Opin Pediatr 2020;32(1):13–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.DeRenzo C, Krenciute G, Gottschalk S. The Landscape of CAR T Cells Beyond Acute Lymphoblastic Leukemia for Pediatric Solid Tumors. Am Soc Clin Oncol Educ Book 2018;38:830–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ramachandran I, Lowther DE, Dryer-Minnerly R, Wang R, Fayngerts S, Nunez D, et al. Systemic and local immunity following adoptive transfer of NY-ESO-1 SPEAR T cells in synovial sarcoma. J Immunother Cancer 2019;7(1):276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dobrenkov K, Ostrovnaya I, Gu J, Cheung IY, Cheung NK. Oncotargets GD2 and GD3 are highly expressed in sarcomas of children, adolescents, and young adults. Pediatric blood & cancer 2016;63(10):1780–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pinto N, Park JR, Murphy E, Yearley J, McClanahan T, Annamalai L, et al. Patterns of PD-1, PD-L1, and PD-L2 expression in pediatric solid tumors. Pediatric blood & cancer 2017;64(11) [DOI] [PubMed] [Google Scholar]
- 64.Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson M, et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2016;34(19):2206–11 [DOI] [PubMed] [Google Scholar]
- 65.Davis KL, Fox E, Merchant MS, Reid JM, Kudgus RA, Liu X, et al. Nivolumab in children and young adults with relapsed or refractory solid tumours or lymphoma (ADVL1412): a multicentre, open-label, single-arm, phase 1-2 trial. Lancet Oncol 2020;21(4):541–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Geoerger B, Kang HJ, Yalon-Oren M, Marshall LV, Vezina C, Pappo A, et al. Pembrolizumab in paediatric patients with advanced melanoma or a PD-L1-positive, advanced, relapsed, or refractory solid tumour or lymphoma (KEYNOTE-051): interim analysis of an open-label, single-arm, phase 1-2 trial. Lancet Oncol 2020;21(1):121–33 [DOI] [PubMed] [Google Scholar]
- 67.Merchant MS, Wright M, Baird K, Wexler LH, Rodriguez-Galindo C, Bernstein D, et al. Phase I Clinical Trial of Ipilimumab in Pediatric Patients with Advanced Solid Tumors. Clinical cancer research : an official journal of the American Association for Cancer Research 2016;22(6):1364–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Heinhuis KM, Ros W, Kok M, Steeghs N, Beijnen JH, Schellens JHM. Enhancing antitumor response by combining immune checkpoint inhibitors with chemotherapy in solid tumors. Ann Oncol 2019;30(2):219–35 [DOI] [PubMed] [Google Scholar]
- 69.Seliger B Combinatorial Approaches With Checkpoint Inhibitors to Enhance Anti-tumor Immunity. Front Immunol 2019;10:999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tilburg CMv, DuBois SG, Albert CM, Federman N, Nagasubramanian R, Geoerger B, et al. Larotrectinib efficacy and safety in pediatric TRK fusion cancer patients. Journal of Clinical Oncology 2019;37(15_suppl):10010- [Google Scholar]
- 71.Hong DS, Kummar S, Farago AF, Lassen UN, Berlin J, Schilder RJ, et al. Larotrectinib efficacy and safety in adult TRK fusion cancer patients. Journal of Clinical Oncology 2019;37(15_suppl):3122- [Google Scholar]
- 72.Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med 2014;371(23):2167–77 [DOI] [PubMed] [Google Scholar]
- 73.Shaw AT, Kim DW, Nakagawa K, Seto T, Crinó L, Ahn MJ, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med 2013;368(25):2385–94 [DOI] [PubMed] [Google Scholar]
- 74.Eleveld TF, Oldridge DA, Bernard V, Koster J, Colmet Daage L, Diskin SJ, et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat Genet 2015;47(8):864–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Parsons DW, Janeway KA, Patton D, Coffey B, Williams PM, Hamilton SR, et al. Identification of targetable molecular alterations in the NCI-COG Pediatric MATCH trial. Journal of Clinical Oncology 2019;37(15_suppl):10011- [Google Scholar]
- 76.Andersson D, Fagman H, Dalin MG, Ståhlberg A. Circulating cell-free tumor DNA analysis in pediatric cancers. Mol Aspects Med 2020;72:100819. [DOI] [PubMed] [Google Scholar]
- 77.Hong DS, DuBois SG, Kummar S, Farago AF, Albert CM, Rohrberg KS, et al. Larotrectinib in patients with TRK fusion-positive solid tumours: a pooled analysis of three phase 1/2 clinical trials. Lancet Oncol 2020;21(4):531–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cocco E, Schram AM, Kulick A, Misale S, Won HH, Yaeger R, et al. Resistance to TRK inhibition mediated by convergent MAPK pathway activation. Nature medicine 2019;25(9):1422–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Drilon A, Nagasubramanian R, Blake JF, Ku N, Tuch BB, Ebata K, et al. A Next-Generation TRK Kinase Inhibitor Overcomes Acquired Resistance to Prior TRK Kinase Inhibition in Patients with TRK Fusion-Positive Solid Tumors. Cancer discovery 2017;7(9):963–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Prieur A, Tirode F, Cohen P, Delattre O. EWS/FLI-1 silencing and gene profiling of Ewing cells reveal downstream oncogenic pathways and a crucial role for repression of insulin-like growth factor binding protein 3. Molecular and cellular biology 2004;24(16):7275–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pappo AS, Patel SR, Crowley J, Reinke DK, Kuenkele KP, Chawla SP, et al. R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: results of a phase II Sarcoma Alliance for Research through Collaboration study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2011;29(34):4541–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Juergens H, Daw NC, Geoerger B, Ferrari S, Villarroel M, Aerts I, et al. Preliminary efficacy of the anti-insulin-like growth factor type 1 receptor antibody figitumumab in patients with refractory Ewing sarcoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2011;29(34):4534–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.DuBois S, Bender JG, Buxton A, Laack N, Randall L, Chen H, et al. Randomized phase 3 trial of ganitumab added to interval compressed chemotherapy for patients with newly diagnosed metastatic Ewing sarcoma: a report from the Children’s Oncology Group (COG). Connective Tissue Oncology Society Annual Meeting; 2019; Tokyo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Knott MML, Holting TLB, Ohmura S, Kirchner T, Cidre-Aranaz F, Grunewald TGP. Targeting the undruggable: exploiting neomorphic features of fusion oncoproteins in childhood sarcomas for innovative therapies. Cancer Metastasis Rev 2019;38(4):625–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sankar S, Theisen ER, Bearss J, Mulvihill T, Hoffman LM, Sorna V, et al. Reversible LSD1 inhibition interferes with global EWS/ETS transcriptional activity and impedes Ewing sarcoma tumor growth. Clinical cancer research : an official journal of the American Association for Cancer Research 2014;20(17):4584–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Erkizan HV, Kong Y, Merchant M, Schlottmann S, Barber-Rotenberg JS, Yuan L, et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing's sarcoma. Nature medicine 2009;15(7):750–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Claveau S, Nehlig E, Garcia-Argote S, Feuillastre S, Pieters G, Girard HA, et al. Delivery of siRNA to Ewing Sarcoma Tumor Xenografted on Mice, Using Hydrogenated Detonation Nanodiamonds: Treatment Efficacy and Tissue Distribution. Nanomaterials (Basel) 2020;10(3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chow EJ, Antal Z, Constine LS, Gardner R, Wallace WH, Weil BR, et al. New Agents, Emerging Late Effects, and the Development of Precision Survivorship. Journal of Clinical Oncology 2018;36(21):2231–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Visalli T, Bower N, Kokate T, Andrews PA. Lack of value of juvenile animal toxicity studies for supporting the safety of pediatric oncology phase I trials. Regul Toxicol Pharmacol 2018;96:167–77 [DOI] [PubMed] [Google Scholar]
- 90.Duarte DM. Use of juvenile animal studies to support oncology medicine development in children. Reprod Toxicol 2015;56:97–104 [DOI] [PubMed] [Google Scholar]
- 91.Silva-Lima B, Due Theilade-Thomsen M, Carleer J, Vidal JM, Tomasi P, Saint-Raymond A. Juvenile animal studies for the development of paediatric medicines: a description and conclusions from a European Medicines Agency workshop on juvenile animal testing for nonclinical assessors. Birth Defects Res B Dev Reprod Toxicol 2010;89(6):467–73 [DOI] [PubMed] [Google Scholar]
- 92.Carceller F, Bautista FJ, Jiménez I, Hladun-Álvaro R, Giraud C, Bergamaschi L, et al. Prognostic factors of overall survival in children and adolescents enrolled in dose-finding trials in Europe: An Innovative Therapies for Children with Cancer study. European Journal of Cancer 2016;67:130–40 [DOI] [PubMed] [Google Scholar]
- 93.Kim A, Fox E, Warren K, Blaney SM, Berg SL, Adamson PC, et al. Characteristics and outcome of pediatric patients enrolled in phase I oncology trials. Oncologist 2008;13(6):679–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hijiya N, Maschan A, Rizzari C, Shimada H, Dufour C, Goto H, et al. Phase 2 study of nilotinib in pediatric patients with Philadelphia chromosome-positive chronic myeloid leukemia. Blood 2019;134(23):2036–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Glade Bender JL, Lee A, Reid JM, Baruchel S, Roberts T, Voss SD, et al. Phase I pharmacokinetic and pharmacodynamic study of pazopanib in children with soft tissue sarcoma and other refractory solid tumors: a children's oncology group phase I consortium report. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2013;31(24):3034–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Voss SD, Glade-Bender J, Spunt SL, DuBois SG, Widemann BC, Park JR, et al. Growth plate abnormalities in pediatric cancer patients undergoing phase 1 anti-angiogenic therapy: a report from the Children's Oncology Group Phase I Consortium. Pediatric blood & cancer 2015;62(1):45–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chisholm JC, Merks JHM, Casanova M, Bisogno G, Orbach D, Gentet JC, et al. Open-label, multicentre, randomised, phase II study of the EpSSG and the ITCC evaluating the addition of bevacizumab to chemotherapy in childhood and adolescent patients with metastatic soft tissue sarcoma (the BERNIE study). Eur J Cancer 2017;83:177–84 [DOI] [PubMed] [Google Scholar]
- 98.Müller HL, Merks JHM, Geoerger B, Grill J, Hargrave D, Glade Bender J, et al. Integrated analysis of long-term growth and bone development in pediatric and adolescent patients receiving bevacizumab. Pediatric blood & cancer 2019;66(2):e27487. [DOI] [PubMed] [Google Scholar]
- 99.Goldsmith KC, Kayser K, Groshen SG, Chioda M, Thurm HC, Chen J, et al. Phase I trial of lorlatinib in patients with ALK-driven refractory or relapsed neuroblastoma: A New Approaches to Neuroblastoma Consortium study. Journal of Clinical Oncology 2020;38(15_suppl):10504- [Google Scholar]
- 100.Iwahara T, Fujimoto J, Wen D, Cupples R, Bucay N, Arakawa T, et al. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene 1997;14(4):439–49 [DOI] [PubMed] [Google Scholar]
- 101.Takitani K, Tamai H, Morinobu T, Kawamura N, Miyake M, Fujimoto T, et al. Pharmacokinetics of all-trans retinoic acid in pediatric patients with leukemia. Japanese journal of cancer research : Gann 1995;86(4):400–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, et al. Everolimus for Subependymal Giant-Cell Astrocytomas in Tuberous Sclerosis. New England Journal of Medicine 2010;363(19):1801–11 [DOI] [PubMed] [Google Scholar]
- 103.Handgretinger R, Baader P, Dopfer R, Klingebiel T, Reuland P, Treuner J, et al. A phase I study of neuroblastoma with the anti-ganglioside GD2 antibody 14.G2a. Cancer Immunol Immunother 1992;35(3):199–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Arceci RJ, Sande J, Lange B, Shannon K, Franklin J, Hutchinson R, et al. Safety and efficacy of gemtuzumab ozogamicin in pediatric patients with advanced CD33+ acute myeloid leukemia. Blood 2005;106(4):1183–8 [DOI] [PubMed] [Google Scholar]
- 105.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014;371(16):1507–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chi SN, Bourdeaut F, Laetsch TW, Fouladi M, Macy ME, Makin GW, et al. Phase I study of tazemetostat, an enhancer of zeste homolog-2 inhibitor, in pediatric pts with relapsed/refractory integrase interactor 1-negative tumors. Journal of Clinical Oncology 2020;38(15_suppl):10525- [Google Scholar]
- 107.Dombi E, Baldwin A, Marcus LJ, Fisher MJ, Weiss B, Kim A, et al. Activity of Selumetinib in Neurofibromatosis Type 1–Related Plexiform Neurofibromas. New England Journal of Medicine 2016;375(26):2550–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Meinhardt A, Burkhardt B, Zimmermann M, Borkhardt A, Kontny U, Klingebiel T, et al. Phase II Window Study on Rituximab in Newly Diagnosed Pediatric Mature B-Cell Non-Hodgkin's Lymphoma and Burkitt Leukemia. Journal of Clinical Oncology 2010;28(19):3115–21 [DOI] [PubMed] [Google Scholar]