

Abstract
Although immunotherapy has revolutionized cancer care, patients with pancreatic ductal adenocarcinoma (PDA) rarely respond to these treatments, a failure that is attributed to poor infiltration and activation of T cells in the tumor microenvironment (TME). We performed an in vivo CRISPR screen and identified lysine demethylase 3A (KDM3A) as a potent epigenetic regulator of immunotherapy response in PDA. Mechanistically, KDM3A acts through Krueppel-like factor 5 (KLF5) and SMAD family member 4 (SMAD4) to regulate the expression of the epidermal growth factor receptor (EGFR). Ablation of KDM3A, KLF5, SMAD4, or EGFR in tumor cells altered the immune TME and sensitized tumors to combination immunotherapy, while treatment of established tumors with an EGFR inhibitor erlotinib prompted a dose-dependent increase in intratumoral T cells. This study defines an epigenetic-transcriptional mechanism by which tumor cells modulate their immune microenvironment and highlights the potential of EGFR inhibitors as immunotherapy sensitizers in PDA.
Keywords: Pancreatic cancer, tumor microenvironment, immunotherapy, KDM3A, EGFR
INTRODUCTION
Immune checkpoint blockade (ICB) has impacted cancer care for multiple malignancies, including melanoma, non-small cell lung cancer, and renal cell carcinoma (1,2). Nevertheless, most patients with these malignancies fail to achieve potent and durable responses to ICB-based immunotherapy. Moreover, many cancer types, including pancreatic ductal adenocarcinoma (PDA), exhibit negligible responses to ICB in the absence of microsatellite instability (1–3). This poor response has been attributed to immunosuppressive conditions in the tumor microenvironment (TME), including the scarcity and dysfunction of infiltrating CD8 T cells, as increased abundance and activity of tumor infiltrating CD8 T cells enhances immunotherapy across different types of cancer (1,4–6). It has become increasingly clear that tumor cells play a central role in establishing a permissive immune TME by releasing secreted factors that can actively drive the exclusion of T cells and resistance to immunotherapy (7–9). Although several cancer cell intrinsic pathways have been reported to influence the composition of the immune TME (10–19), the key genetic and epigenetic drivers of this process remain largely unknown.
In the present study, we performed a targeted in vivo CRISPR screen and identified lysine demethylase 3A (KDM3A) as a potent epigenetic regulator of the immune TME. Cancer cells lacking KDM3A gave rise to tumors with marked alterations in lymphoid and myeloid infiltration and profound improvements in their response to combination immunotherapy. Unbiased molecular profiling suggested that KDM3A acts through Krueppel-like factor 5 (KLF5) and SMAD family member 4 (SMAD4) to regulate the expression of the epidermal growth factor receptor (EGFR), and functional studies revealed that ablation of tumor cell intrinsic KLF5, SMAD4, or EGFR resulted in increased T cell infiltration and improved immunotherapy responsiveness in previously-refractory tumors. Moreover, treatment of established tumors with the EGFR inhibitor erlotinib was sufficient to promote a T-cell-inflamed TME. Given the known requirement for EGFR signaling during pancreatic tumorigenesis (20–23) and erlotinib’s status as an FDA-approved therapy for PDA (24), our results highlight the potential use of this drug as an immunotherapy-sensitizing agent for this treatment-refractory disease.
RESULTS
An in vivo CRISPR screen identifies epigenetic regulators of tumor immunity in PDA
We previously established a group of clonal congenic primary tumor cell lines from the KPCY mouse model of spontaneous PDA (12,25). Upon implantation into immunocompetent C57BL/6 mice, these clones recapitulate the heterogeneity of the immune TME seen in human PDA, with some clones giving rise to T-cell-inflamed tumors and others giving rise to non-T-cell-inflamed tumors (12). Importantly, genomic and epigenetic differences, rather than tumor mutational burden, account for these observed differences in immune cell infiltration (12). To better understand the epigenetic differences distinguishing T-cell-inflamed and non-T-cell-inflamed samples, we used ATAC-seq (assay for transposase-accessible chromatin coupled with sequencing) to compare the two groups. This analysis revealed different chromatin accessibility patterns between T-cell-inflamed and non-T-cell-inflamed samples, and that differences in the epigenetic landscape of cancer cells correlate with differences in the corresponding TME (Supplementary Fig. 1A–B). This finding is consistent with recent studies describing functions of epigenetic regulators in modulating anti-tumor immunity in other types of cancer (26–30).
T-cell-inflamed and non-T-cell-inflamed tumors derived from our panel of PDA tumor cell clones exhibit heterogenous responses to a combination immunotherapy regimen consisting of gemcitabine (G), abraxane (A), CD40 agonistic antibody (F), CTLA4 blocking antibody (C) and PD1 blocking antibody (P), hereafter referred to as GAFCP (10–12). GAFCP therapy improves clinical outcomes in preclinical mouse models of PDA (31,32), and a similar regimen has shown promising results and is currently being tested in a Phase 2 clinical trial of patients with metastatic PDA (NCT03214250) (33). When challenged with GAFCP, PDA tumor cell clone 6419c5 – a non-T-cell-inflamed tumor with abundant myeloid cells and a paucity of tumor-infiltrating T cells (12) – responded modestly to GAFCP therapy, with no tumor regressions (Supplementary Fig. 1C). Nevertheless, RNA sequencing (RNA-seq) of tumor cells sorted from subcutaneously implanted tumors that were treated with either control or GAFCP therapy revealed significant transcriptional differences (Supplementary Fig. 1D–E). Specifically, tumor cells that persisted following GAFCP treatment were enriched for gene signatures similar to those enriched in untreated T cell low clones compared to T cell high clones (Supplementary Fig. 1F). These results indicate that the transcriptional networks underlying T cell infiltration status mirror those that define sensitivity to immunotherapy. Thus, we concluded that this model would be a suitable platform for investigating other epigenetic and transcriptional regulators that shape the immune TME and influence response to immunotherapy in PDA.
To systematically interrogate tumor cell intrinsic epigenetic factors modulating anti-tumor immunity, we devised an in vivo focused CRISPR screen strategy (Fig. 1A). First, we engineered the 6419c5 tumor cell clone to stably express Cas9 and then transduced it with a sgRNA library (34) consisting of 5100 sgRNAs targeting 850 epigenetic factors and RNA-binding factors, as well as 100 non-targeting control sgRNAs. We passaged the transduced cells in vitro for 12 days to allow for genome editing and then transplanted the cells into immunocompetent C57BL/6 mice. After 11 days, mice were treated with either vehicle or GAFCP therapy for an additional 12–14 days. Samples were collected at two timepoints prior to implantation (“T0” and “pre-injection) and from control- and GAFCP-treated tumors at the end of the experiment. Following sequencing, the representation of each sgRNA was measured for each sample and compared pairwise with all other samples (Supplementary Table1).
Figure 1. A genetic screen identifies KDM3A as a tumor cell intrinsic epigenetic regulator suppressing anti-tumor immunity in pancreatic cancer.
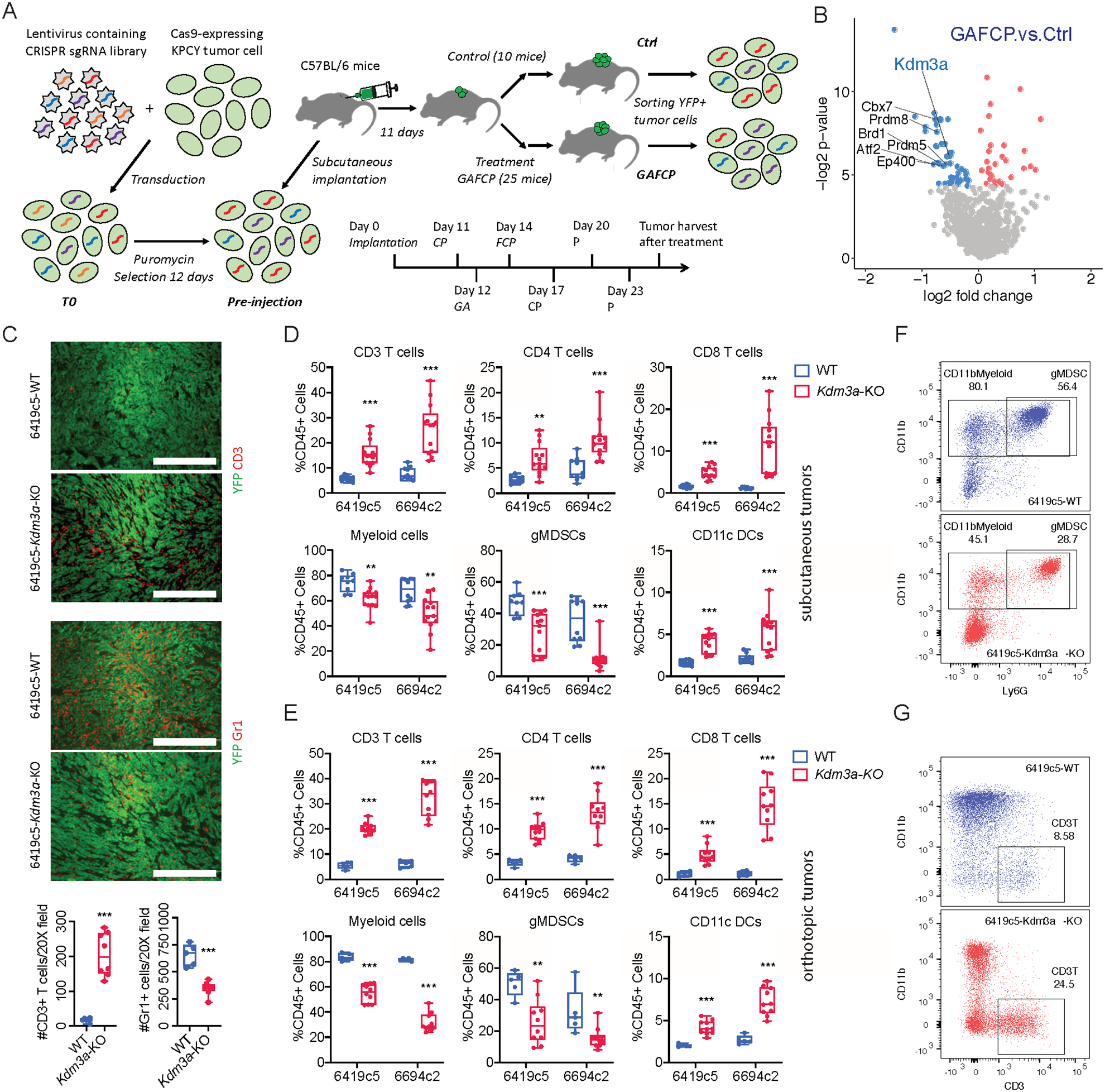
A, Diagram showing the strategy of an in vivo CRISPR-based genetic screen. B, Volcano plot showing the comparison between GAFCP and Ctrl groups. Each dot represents a gene whose sgRNAs were enriched in Ctrl (blue) or GAFCP (red) groups, with p-value < 0.05. Top candidate epigenetic regulators whose sgRNAs were enriched in the Ctrl group (p-value < 0.025 and >3 sgRNAs meeting a statistical cutoff) are highlighted. C, Top: representative immunofluorescent staining images of CD3+ T cells and Gr1+ myeloid cells in Kdm3a-WT or Kdm3a-KO tumors stained for CD3 or Gr1 (red) and YFP (green). Bottom: quantification of CD3+ T cells and Gr1+ myeloid cells comparing Kdm3a-WT and Kdm3a-KO tumors (counts taken from n=5–8 images/group; data combined from two independent 6419c5 knockout clones). D, Flow cytometric analysis of tumor infiltrating immune cells in subcutaneously implanted Kdm3a-WT and Kdm3a-KO 6419c5 and 6694c2 tumors (n=9–13/group). E, Flow cytometric analysis of tumor infiltrating immune cells in orthotopically implanted Kdm3a-WT and Kdm3a-KO 6419c5 and 6694c2 tumors (n=5–10/group). F-G, Representative flow plots showing the abundance of myeloid cells (F) and T cells (G) within total CD45+ leukocytes in Kdm3a-WT (blue) or Kdm3a-KO (red) tumors. In C, statistical differences were calculated using unpaired Student’s t-test. In D-E, statistical differences were calculated using Multiple T test. p<0.05 was considered statistically significant, * indicates p<0.05, ** p<0.01, and *** p< 0.001.
To confirm the robustness of the screening platform and strategy, we first identified genes whose sgRNAs were depleted from tumor cells in both the Ctrl and GAFCP groups compared to the pre-injection sample, as such genes would likely be important for tumor growth and progression in vivo. As sgRNAs targeting DNA methyltransferase 1 (Dnmt1) were the most highly depleted (Supplementary Fig. 2A–B and Supplementary Tables 2–3), we then knocked out Dnmt1 (Dnmt1-KO) in two PDA tumor cell clones, 6419c5 and 6694c2 (Supplementary Fig. 2C). This resulted in a decreased ability to form tumors (Supplementary Fig. 2D), confirming the importance of Dnmt1 in tumor growth. Moreover, flow cytometry revealed increased infiltration of T cells and decreased infiltration of myeloid cells in Dnmt1-KO tumors (Supplementary Fig. 2E–F). T cell depletion partially restored the Dnmt1-KO cells’ tumorigenic and growth capacities (Supplementary Fig. 2G–H), consistent with recent findings in other cancers showing that DNMT1 promotes tumor growth in part by suppressing anti-tumor immunity (35). Thus, our screening strategy can detect genes that control anti-tumor immunity to modulate tumor growth in vivo.
Tumor cell intrinsic lysine demethylase 3A (KDM3A) suppresses anti-tumor immunity in PDA
We next identified genes targeted by sgRNAs that are depleted from tumor cells in the GAFCP group compared to the Ctrl group. After filtering by p-value (< 0.025) and the presence of >3 sgRNAs meeting a statistical cutoff, we nominated 7 epigenetic regulators as potential suppressors of immunotherapy response (Fig. 1B, Supplementary Fig. 3A, and Supplementary Table 4). To determine whether the hits in our screen regulate immune cell composition in the TME, we individually knocked out all seven genes in 6419c5 (Supplementary Fig. 3B). Given that the makeup of the immune TME, particularly T cells and myeloid cells, determines the responsiveness of PDA tumors to GAFCP treatment, we screened the tumors derived from the mutant clones for changes in immune infiltrates by flow cytometry. In 5 of the 7 mutants (Prdm8, Atf2, Kdm3a, Ep400, and Prdm5), we observed a significant increase in intratumoral T cells and a significant decrease in intratumoral myeloid cells (Supplementary Fig. 3C), suggestive of a reprogramming of the immune landscape. We then deleted the top three hits from the screen (Prdm8, Atf2, Kdm3a) in a second PDA tumor cell clone (6694c2) and observed a similar change in immune infiltration in Kdm3a mutants but not in the other two genes (Supplementary Fig. 3D–F). KDM3A has recently been shown to promote PDA progression in animal models (36). Moreover, an analysis of TCGA data revealed KDM3A as the only gene among the 7 candidates whose elevated expression in PDA correlates with poor clinical outcome (Supplementary Fig. 3G–H). Thus, we focused our attention on the immune TME regulatory activities of KDM3A.
Next, we performed a more detailed characterization of KDM3A’s impact on the tumor immune microenvironment. In subcutaneous tumors, Kdm3a loss was associated with an increase in tumor-infiltrating T cells and dendritic cells (DCs) and a concomitant decrease in myeloid cells compared to wildtype (WT) tumors (Fig. 1C–D and Supplementary Fig. 3I). This trend was confirmed in an orthotopic transplantation model (Fig. 1E–G and Supplementary Fig. 3J). Recent studies identified subtypes of fibroblasts in PDA tumors which may have important implications for tumor immunity (37,38). Utilizing surface markers identified by one of these reports, we analyzed the abundance of different fibroblast populations in Kdm3a-WT and Kdm3a-KO tumors and observed a decrease of myCAFs following deletion of tumor cell intrinsic KDM3A (Supplementary Fig. 3K). Importantly, subcutaneously implanted Kdm3a-KO tumors acquired a marked sensitivity to GAFCP therapy compared to WT tumors, with most exhibiting a complete regression that led to a marked extension of survival (Fig. 2A–C). These results demonstrate that KDM3A is an epigenetic suppressor of anti-tumor immunity and that loss of KDM3A reprograms the immune TME to a T-cell-inflamed state, thereby conferring sensitivity to combination immunotherapy.
Figure 2. Genetic ablation of tumor cell intrinsic KDM3A promotes a T-cell-inflamed TME in PDA.
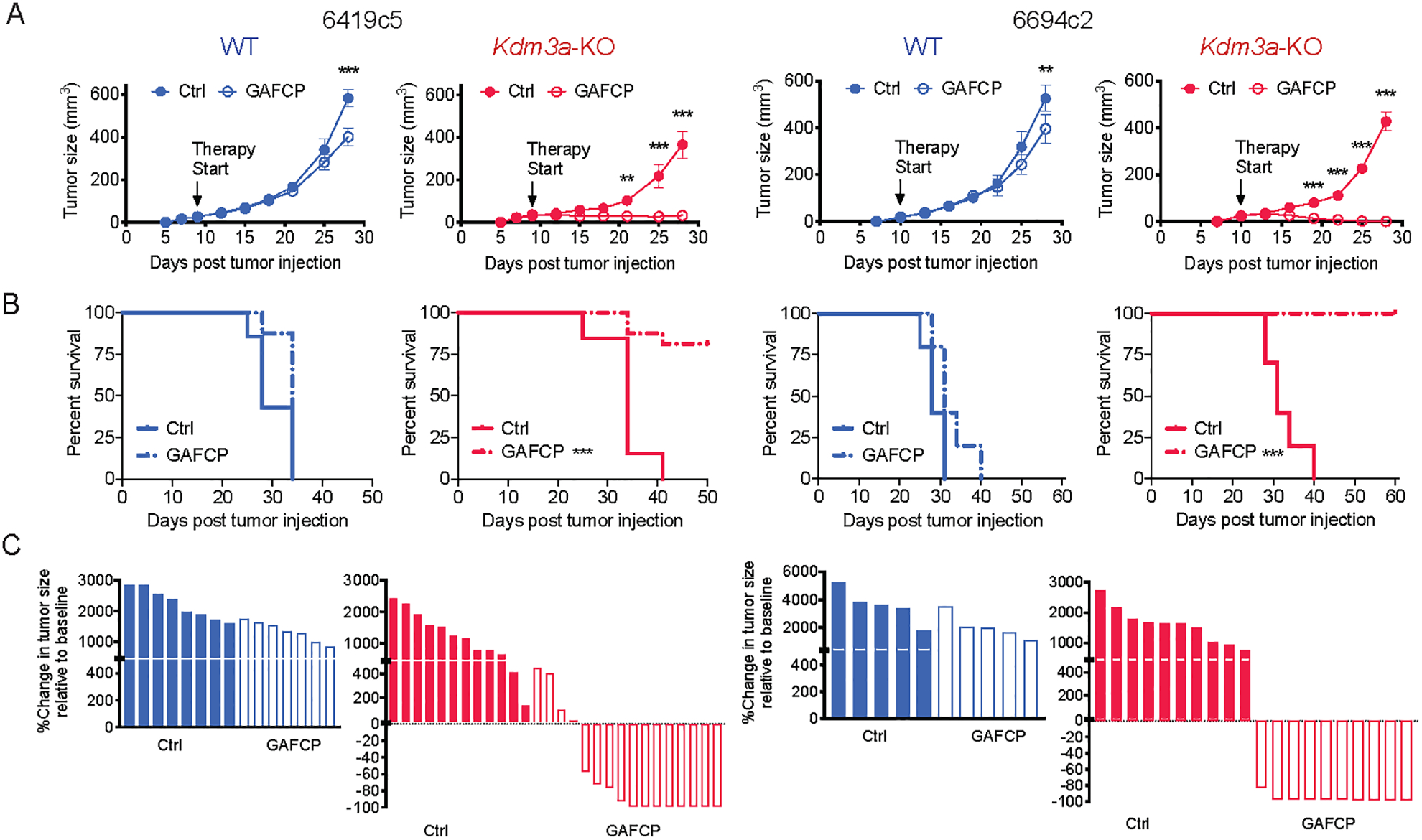
A, Growth curves of Kdm3a-WT or Kdm3a-KO tumors from two non-T-cell-inflamed lines (6419c5 and 6694c2) with or without GAFCP treatment. B, Overall survival curves of animals with Kdm3a-WT or Kdm3a-KO tumors with or without GAFCP treatment. C, Waterfall plots showing change in tumor size of Kdm3a-WT or Kdm3a-KO tumors relative to the start of treatment measured with or without GAFCP treatment. In A-C, tumor cells were subcutaneously implanted into C57BL/6 mice (2 knockout tumor cell clones for each parental PDA tumor cell clone were used, n=5–16/group). In A, statistical differences were calculated using two-way ANOVA with multiple comparisons. In B, statistical differences were calculated using Log-rank (Mantel-Cox) test. p<0.05 was considered statistically significant, * indicates p<0.05, ** p<0.01, and *** p< 0.001.
Transcriptional and epigenetic profiling identifies KLF5 and SMAD4 transcription factors as potential mediators of KDM3A activity
In previous work, transcriptional profiling of sorted tumor cells identified several gene signatures that distinguish T cell high clones from T cell low clones (12). For example, T cell low clones are enriched for TGFβ signaling and MYC signatures, while T cell high clones are enriched for interferon response signatures (12). When we performed RNA-seq and Gene Set Enrichment Analysis (GSEA) (39) on sorted tumor cells from WT versus Kdm3a-KO tumors (Supplementary Fig. 4A), we found that they enriched for molecular features similar to those that defined T cell low and T cell high tumor cells, including TGFβ signaling and MYC signatures in WT tumor cells and interferon response signatures in Kdm3a-KO tumor cells (Supplementary Fig. 4B–C). Kdm3a-KO tumor cells also expressed higher levels of PD-L1 and MHC class I in vivo (Supplementary Fig. 4D–E). When treated with interferon-gamma in culture, however, induction of MHC I and PD-L1 occurred to a similar degree in Kdm3a-KO cells and WT cells, suggesting that increased MHC class I expression by Kdm3a-KO cells is a consequence of the T-cell-inflamed TME (Supplementary Fig. 4F). Collectively, these results suggest that KDM3A regulates gene networks like those that arise spontaneously during the formation of T cell inflamed and non-T cell inflamed tumors.
To identify transcriptional regulators mediating KDM3A’s effects, we probed the epigenetic changes associated with KDM3A loss. Previous studies have identified KDM3A as a histone demethylase that co-localizes with and removes methyl groups from di-methylated histone H3 lysine residue 9 (H3K9me2) (40–45), a histone modification associated with transcriptional repression (46–50). To unbiasedly examine global changes in histone modifications, we performed histone mass spectrometry analysis. No significant differences were observed between WT and Kdm3a-KO cells (Supplementary Fig. 4G), suggesting that KDM3A loss impacts H3K9me2 levels at a local rather than a global level.
To characterize such locus-specific effects, we performed chromatin immunoprecipitation followed by next-generation sequencing (ChIP-seq) for both H3K9me2 and KDM3A. We observed that both signals were largely distributed in distal intergenic regions (Supplementary Fig. 4H), as previously reported (51–54). Motif analysis revealed a strong signal for the consensus DNA binding sequence recognized by members of the KLF family of transcriptional regulators. KDM3A peaks that were lost in Kdm3a-KO cells enriched for the KLF motif (Fig. 3A), which is highly conserved across multiple KLF family members (55). Accordingly, genomic regions enriching KDM3A ChIP-seq signals also included the KLF motif, consistent with a previous study in prostate cancer cells (51) (Fig. 3B). Furthermore, we found that peaks associated with H3K9ac, a classic marker of active promoters (56,57), that were decreased in Kdm3a-KO cells also enriched for the KLF motif (Fig. 3C).
Figure 3. Transcriptional and epigenetic profiling identifies KLF5 and SMAD4 as potential transcriptional regulators downstream KDM3A.
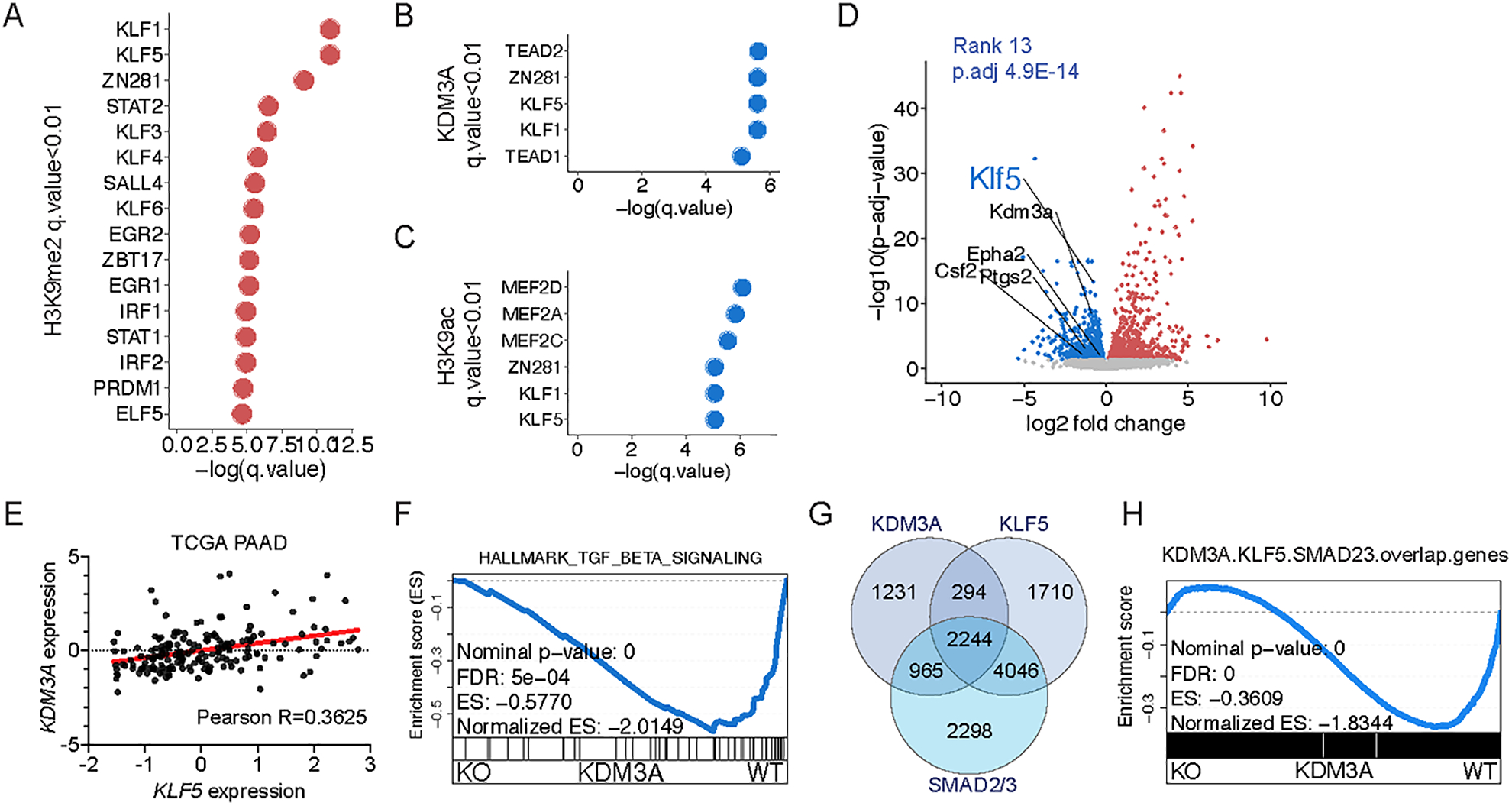
A-C, Graphs showing the top enriched motifs of transcriptional regulators in genomic regions with lost H3K9me2, gained H3K9ac, and gained KDM3A ChIP-seq signals in Kdm3a-WT tumor cells compared to Kdm3a-KO tumor cells (q-value < 0.01 following the DREME and Tomtom based motif analysis, using ChIP-seq analysis of both 6419c5 and 6694c2 tumor cell clones). Two knockout clones from each parental tumor cell clone were pooled together for ChIP-seq analysis (n=2/group, 8 samples in total). D, Volcano plot illustrating the differential gene expression between sorted YFP+ tumor cells from Kdm3a-WT or Kdm3a-KO tumors (6419c5 and 6694c2 tumor cell clones, 2 knockout clones of each parental tumor cell clone were used, n=3/group and 18 samples in total). Each dot represents a gene (Kdm3a-WT (blue) or Kdm3a-KO (red), with p.adj-value < 0.05). Several factors known to drive a non-T-cell-inflamed TME in PDA are highlighted. E, Dot plot showing the expression of KDM3A and KLF5 in TCGA PAAD dataset. F, Leading-edge plot of GSEA result highlighting Hallmark_TGF_beta_signaling. G. Venn diagram showing the overlapping gene targets of KDM3A, KLF5, and SMAD2/3. H, Leading-edge plot of GSEA result highlighting overlap genes of KDM3A, KLF5, and SMAD2/3 target genes.
We focused our attention on Klf5 as it was the mostly downregulated member of the KLF family in Kdm3a-KO cells (Fig. 3D and Supplementary Fig. 4I) and its expression positively correlated with the expression of KDM3A in human PDA samples (Fig. 3E). An analysis of TCGA data also showed that KLF5 expression in PDA correlates with poor clinical outcome (Supplementary Fig. 4J). Further examination of the RNA-seq data also identified SMAD4, which has recently been implicated in suppressing anti-tumor immunity in PDA and other types of cancers (11,58,59), as a possible mediator of KDM3A’s immune regulatory activity. Specifically, genes whose expression decreased upon KDM3A loss were enriched for both KLF and SMAD4 DNA binding motifs (Supplementary Fig. 4K), as well as the SMAD4-activating TGFβ Hallmark gene set (Fig. 3F). Interestingly, Kdm3a-KO cells expressed lower levels of factors that have previously been shown to promote a T cell low microenvironment in PDA (Fig. 3D), including Epha2, Ptgs2, and Csf2 (11,60,61).
To determine whether KDM3A functionally interacts with KLF5 or SMAD4 we performed several experiments. First, we performed an IP-mass spectrometry experiment to identify KDM3A-interacting proteins. However, we did not observe any direct interaction between KDM3A and KLF5 or SMAD4 (Supplementary Table 5 and Supplementary Fig. 4L). To investigate whether KLF5, SMAD4, and KDM3A modulates a shared group of genes, we analyzed recently published ChIP-seq datasets of KLF5 and SMAD2/3 (closely related to SMAD4) in mouse pancreatic tumor cells (62). We observed that KDM3A, KLF5, and SMAD2/3 share significant overlap of gene targets (Fig. 3G). Importantly, GSEA indicated that the shared targets by these proteins are downregulated following ablation of KDM3A (Figure 3H). Taken together, our transcriptomic and epigenomic analyses nominated KLF5 and SMAD4 as transcriptional regulators that may coordinate the output of KDM3A’s epigenetic program.
Loss of tumor cell intrinsic KLF5 or SMAD4 promotes a T-cell-inflamed TME
To functionally examine the role of KLF5 and SMAD4 on anti-tumor immunity, we used CRISPR to knock out Klf5 and Smad4 – along with several other transcription factors downregulated with Kdm3a loss – in T cell low clone 6694c2 (Supplementary Fig. 5A). These cells were implanted into immunocompetent mice, and immune infiltrates in the resultant tumors were examined by flow cytometry. Here, similar to our findings with KDM3A, we observed a significant increase of T cells and a concomitant decrease of myeloid cells with loss of KLF5 or SMAD4 (Fig. 4A and Supplementary Fig. 5B). Importantly, none of the other transcription factors representing potential KDM3A mediators yielded consistent changes in immune infiltration other than SMAD3, a binding factor of SMAD4 (Supplementary Fig. 5C). The effect of KLF5 on immune infiltration was also confirmed using an orthotopic model and in a second T cell low clone (Fig. 4B–D and Supplementary Fig. 5D–F). These results demonstrate that KLF5 and SMAD4 act within tumor cells to promote intratumoral infiltration of myeloid cells and exclusion of T cells.
Figure 4. Genetic ablation of tumor cell intrinsic KLF5 and SMAD proteins promotes a T-cell-inflamed TME in PDA.
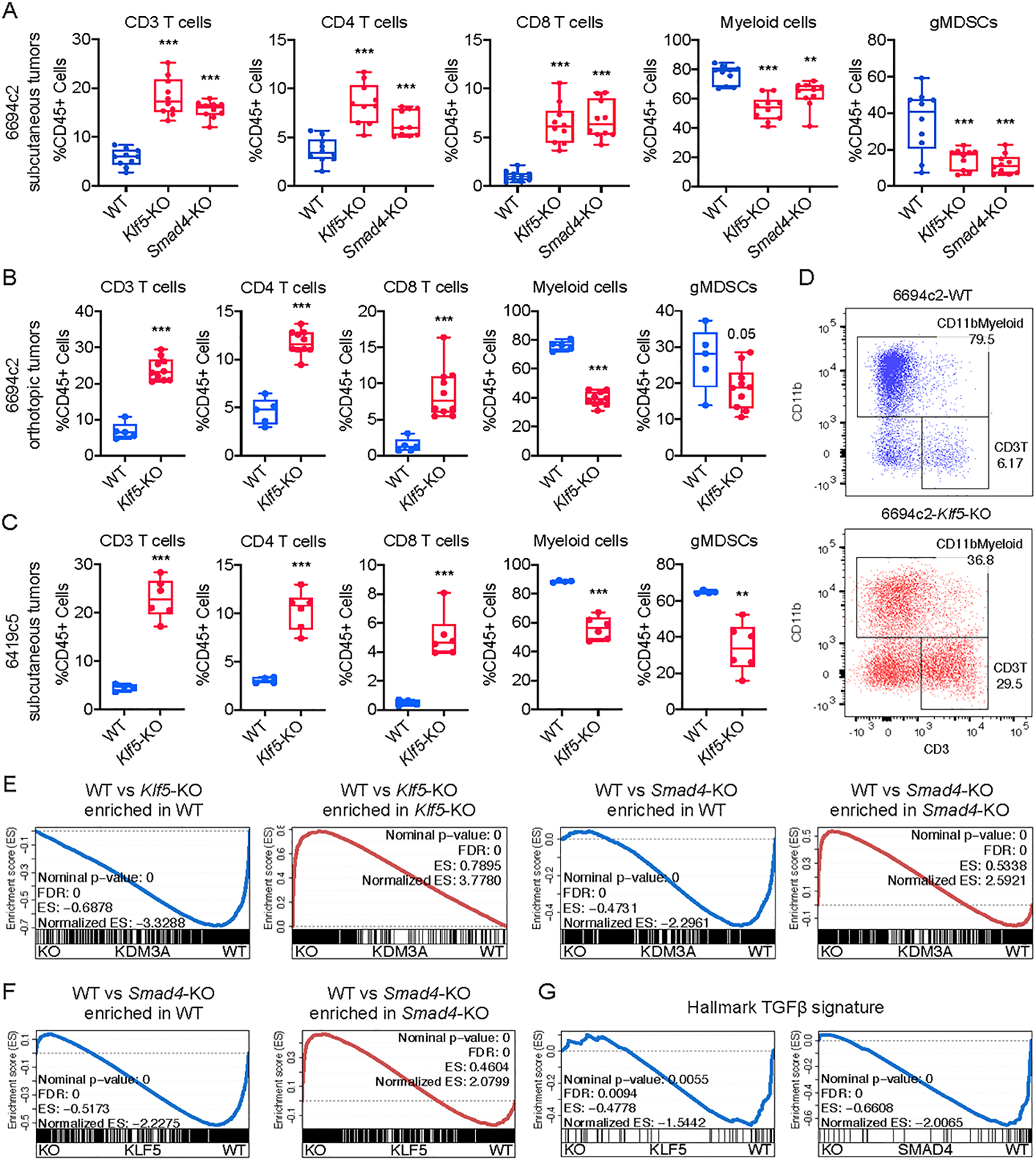
A, Flow cytometric analysis of immune cells in subcutaneously implanted WT, Klf5-KO or Smad4-KO 6694c2 tumors (n=10/group, two knockout clones/gene). B, Flow cytometric analysis of immune cells in orthotopically implanted WT and Klf5-KO 6694c2 tumors (n=5–10/group, two knockout clones/gene). C, Flow cytometric analysis of immune cells in subcutaneously implanted WT and Klf5-KO 6419c5 tumors (n=4–6/group, two knockout clones/gene). D, Representative flow plots showing the abundance of myeloid cells and T cells within total CD45+ leukocytes in Klf5-WT (blue) or Klf5-KO 6694c2 (red) tumors. E, Leading-edge plots from the GSEA analysis highlighting the enrichment of KLF5 and SMAD4 gene signatures based on the RNA-seq analysis of Kdm3a-WT or Kdm3a-KO tumor cells (gene signatures were generated based on p.adj-value < 0.01 and fold change > 2). F, Leading-edge plots from the GSEA analysis highlighting the enrichment of the SMAD4 gene signature based on the RNA-seq analysis of Klf5-WT or Klf5-KO tumor cells (gene signatures were generated based on p.adj-value < 0.01 and fold change > 2). G, Leading-edge plots from the GSEA analysis highlighting the enrichment of the Hallmark_TGF_beta_signaling geneset. In A, statistical differences were calculated using one-way ANOVA with multiple comparisons. In B-C, statistical differences were calculated using unpaired Student’s t-test. p<0.05 was considered statistically significant, * indicates p<0.05, ** p<0.01, and *** p< 0.001.
Next, we compared the transcriptional profiles of tumor cells sorted from subcutaneously implanted Kdm3a-KO, Smad4-KO, and Klf5-KO tumors (Supplementary Fig. 5G). GSEA analysis revealed that loss of Klf5 or Smad4 enriched for the gene signature defining Kdm3a-KO cells (Fig. 4E). Furthermore, ablation of Klf5 resulted in transcriptional alterations that recapitulated those seen with loss of Smad4 (Fig. 4F). Moreover, deletion of either KLF5 or SMAD4 resulted in a negative enrichment of the TGFβ signature, as was observed for KDM3A (Fig. 4G). Collectively, these results suggest that KLF5, SMAD4, and KDM3A coordinately regulate transcription in tumor cells to suppress anti-tumor immunity.
The KDM3A-KLF5-SMAD4 transcriptional axis converges on the epidermal growth factor receptor (EGFR)
Since KDM3A, KLF5, and SMAD4 broadly regulate a shared transcriptional network, we sought to identify downstream pathways and gene targets on which all three converge. The 75 genes that were consistently decreased following ablation of Kdm3a, Klf5, or Smad4 enriched most significantly for the KRAS signaling gene set (Fig. 5A–C and Supplementary Fig. 5H). Interestingly, genes encoding members of the epidermal growth factor (EGF) family of growth factors, as well as the EGF receptor (EGFR) itself, were among those showing the most consistent downregulation following ablation of these genes (Fig. 5B and Supplementary Fig. 6A). Several additional findings led us to focus on EGFR as a potential mediator of the KDM3A-KLF5-SMAD4 axis: (i) TGFβ treatment, which activates SMAD4 transcriptional activity, induced the expression of Egfr (Supplementary Fig. 6B); (ii) analysis of the Cancer Dependency Map Portal (DepMap) (63) revealed EGFR to exhibit the greatest co-dependency with KLF5, but not SMAD4 or KDM3A, for survival across tumor lines as compared to any other gene (Fig. 5D); and (iii) in PDA patient samples, EGFR expression correlated with KDM3A and KLF5 expression (Fig. 5E) and worse clinical outcome (Supplementary Fig. 6C).
Figure 5. Molecular profiling identifies EGFR as a tumor cell intrinsic factor regulating the immune TME and sensitivity to combination immunotherapy in PDA.
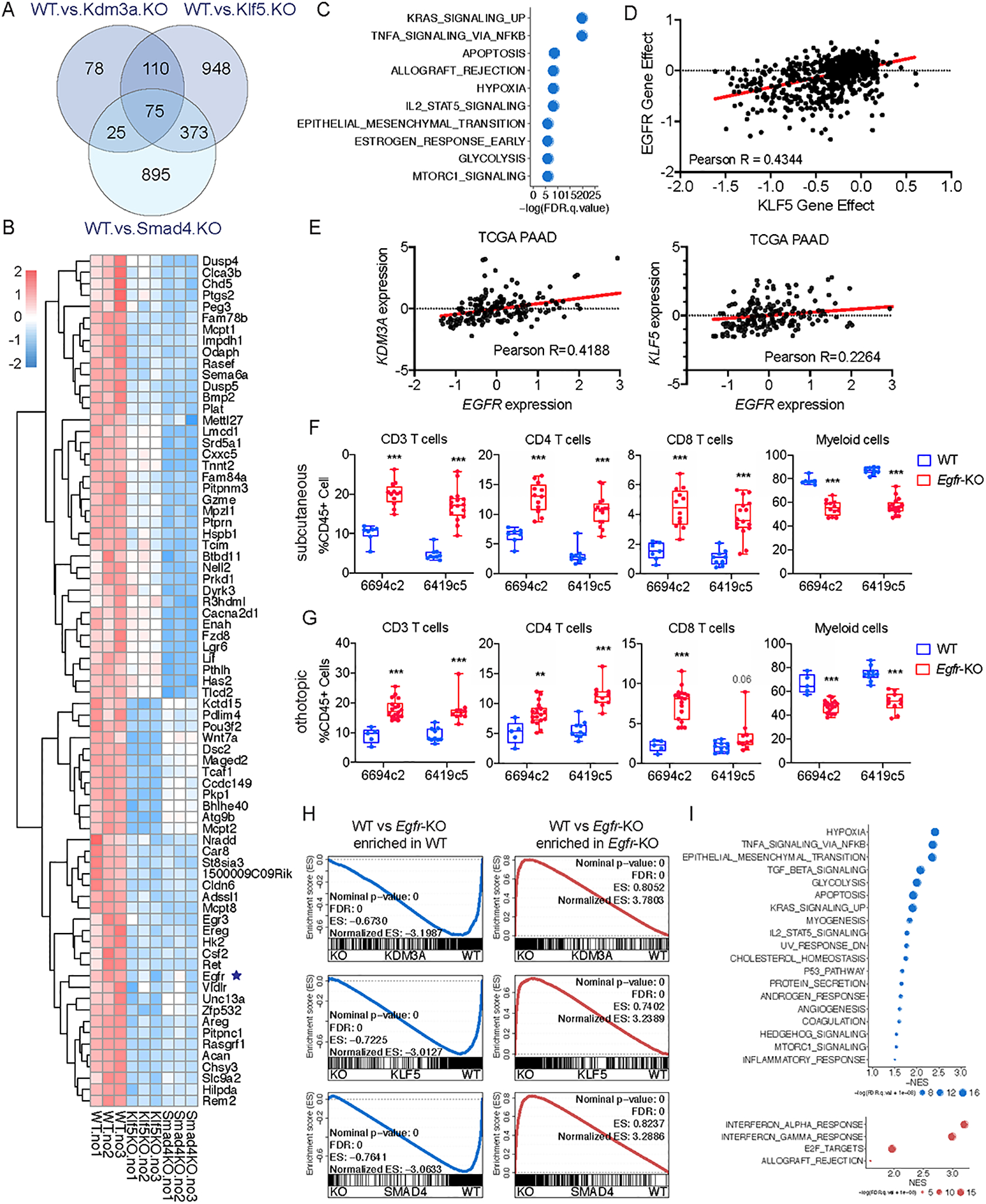
A, Venn diagram showing the overlap of differentially expressed genes in Klf5-KO, Kdm3a-KO, and Smad4-KO tumor cells compared to WT. B, Heatmap showing expression of the 75 overlapped genes in A. C, Graph showing the top enriched hallmark genesets based on the GSEA of overlapped genes identified in A. D, Dot plot showing the dependency scores for EGFR and KLF5 across all tumor cell lines in the Project Achilles/Cancer Dependency Map Portal (DepMap). E, Dot plot showing the expression of EGFR, KDM3A and KLF5 in the TCGA PAAD dataset. F, Flow cytometric analysis of immune cells in subcutaneously implanted WT and Egfr-KO 6419c5 and 6694c2 tumors (n=7–16/group, 2 knockout clones from each parental tumor cell clone). G, Flow cytometric analysis of immune cells in orthotopically implanted WT and Egfr-KO 6419c5 and 6694c2 tumors (n=5–18/group, 2 knockout clones from each parental tumor cell clone). H, Leading-edge plots from the GSEA analysis highlighting the enrichment of EGFR gene signature based on RNA-seq analysis of Kdm3a-KO, Klf5-KO, and Smad4-KO tumor cells (gene signatures were generated based on p.adj-value < 0.01 and fold change > 2). I, Graph showing top enriched functional pathways from GSEA using Hallmark genesets. In F-G, statistical differences were calculated using Multiple T test. p<0.05 was considered statistically significant, * indicates p<0.05, ** p<0.01, and *** p< 0.001.
To determine whether EGFR has a role in anti-tumor immunity, we knocked out EGFR in two PDA tumor cell clones and assessed the immune infiltration of the resulting tumors from both subcutaneous and orthotopic tumor models. As observed previously with KDM3A, SMAD4, and KLF5, loss of EGFR rewired the immune TME from a non-T-cell-inflamed state to a T-cell-inflamed state (Fig. 5F–G and Supplementary Fig. 6D–F). Remarkably, transcriptomic analysis of Egfr-KO cells revealed dysregulation of the same genes and pathways that were perturbed in Kdm3a, Klf5, and Smad4-KO cells (Fig. 5H–I), including downregulation of the TGFβ gene set and induction of interferon response gene sets. Importantly, among the 75 genes that were decreased by loss of Kdm3a, Klf5, and Smad4, 70 genes were also decreased following ablation of EGFR (Supplementary Fig. 6G). In addition, the tumor cell intrinsic expression of two immune modulatory secreted factors (CSF2 and PTGS2) that we and others have previously shown to promote a non-T-cell-inflamed TME (11,60,61) in PDA was also decreased across all four mutants compared to wild type tumor cells (Fig. 3A, Fig. 5B, and Supplementary Fig. 6G). Taken together, these results suggest that KDM3A, KLF5, SMAD4, and EGFR converge on a conserved transcriptional program in cancer cells, which in turn influences the composition of the tumor immune microenvironment.
EGFR inhibition sensitizes tumors to combination immunotherapy
Clones lacking Klf5, Smad4, or Egfr gave rise to tumors with a reduction in myeloid cells and an increase in T cells. Hence, we sought to determine whether these tumors responded to GAFCP immunotherapy, as we had earlier shown for Kdm3a mutants (Fig. 2). We found that tumors derived from Klf5-, Smad4-, or Egfr-KO tumor cells all became sensitized to GAFCP therapy, leading to tumor regression and improved animal survival, whereas parental cells showed minimal response (Fig. 6A). Therefore, the changes in immune infiltrates caused by loss of KLF5, SMAD4 or EGFR are sufficient to sensitize tumors to the effects of combination immunotherapy.
Figure 6. Ablation of tumor cell intrinsic KLF5, SMAD4 and EGFR increases the sensitivity of PDA tumors to a combination immunotherapy.
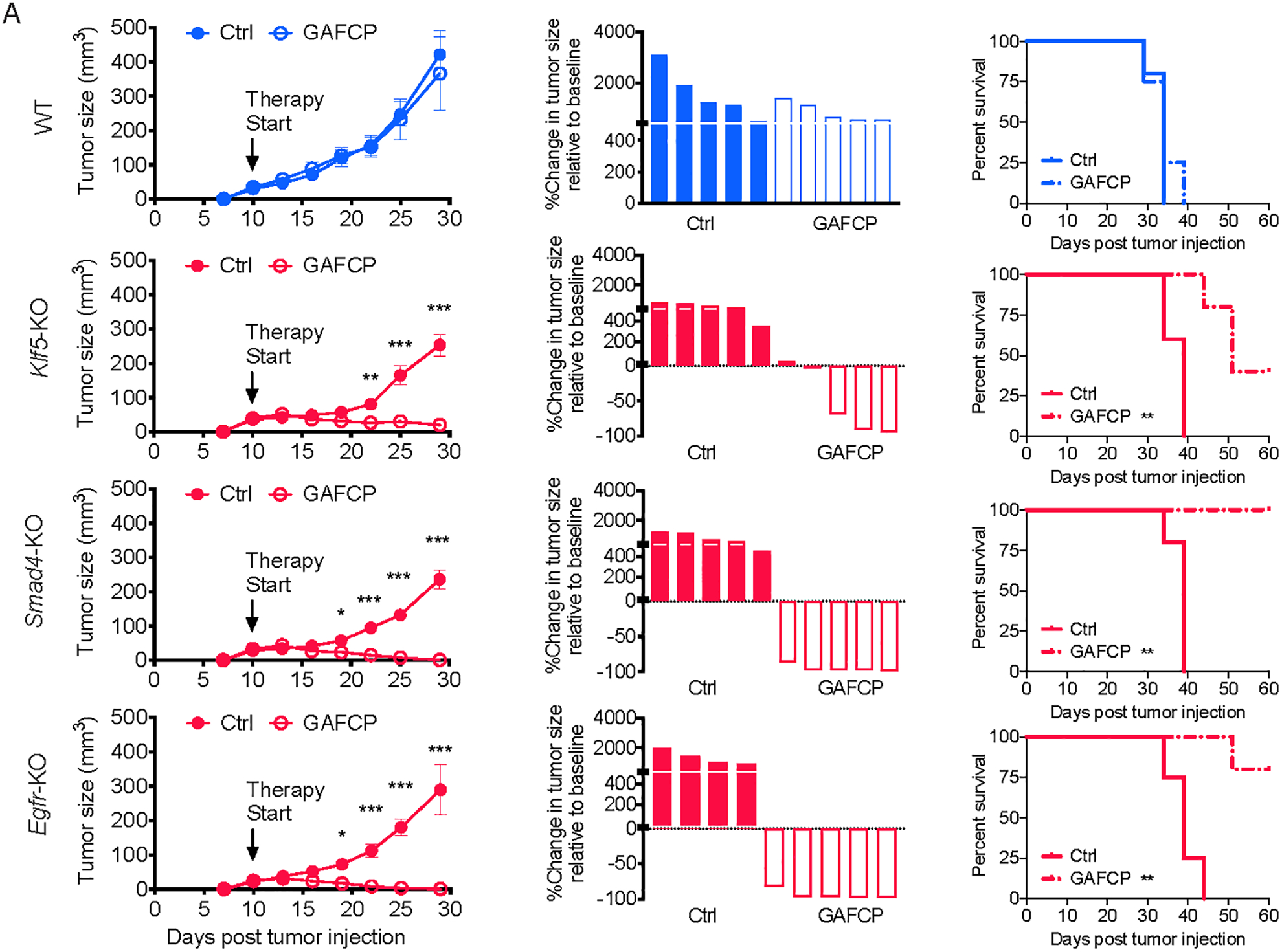
A, Growth curves, waterfall plots, and overall survival curves of WT, Klf5-KO, Smad4-KO, Egfr-KO tumors with or without GAFCP treatment. Tumor cells were subcutaneously implanted into C57BL/6 mice (2 knockout tumor cell clones were pooled together for this experiment, n=5/group). In A, statistical differences were calculated using two-way ANOVA with multiple analysis (tumor growth curves) and Log-rank (Mantel-Cox) test (survival curves).
EGFR signaling has been shown to play a central role in PDA biology (20–24,64,65). Erlotinib, a tyrosine kinase inhibitor (TKI) of EGFR, is approved for the treatment of PDA patients based on a small but significant clinical benefit (24). Importantly, these modest clinical effects were observed in the absence of any adjunctive immunotherapies. Given that deletion of the Egfr gene results in a T-cell-inflamed TME, we hypothesized that pharmacologic inhibition of EGFR might similarly shift the tumor immune landscape. Consistent with this hypothesis, we found that erlotinib treatment of subcutaneous or orthotopic 6694c2 tumors prompted a dramatic and dose-dependent increase in intratumoral T cells and decrease in intratumoral myeloid cells (Fig. 7A–B and Supplementary Fig. 7A–B). A more modest trend was observed in tumors derived from clone 6419c5 (Supplementary Fig. 7C–D), a difference that may be explained by the high abundance of gMDSCs in these tumors, a feature that has been associated with resistance to EGFR inhibitors (66). Furthermore, we examined the immune infiltration of 6694c2 tumors following a short-term treatment of erlotinib (5 days) and observed increased infiltration of T cells (Supplementary Fig. 7E). Therefore, pharmacologic inhibition of EGFR modulates the immune TME and may create a permissive environment for effective immunotherapy in non-T-cell-inflamed (“cold”) PDA tumors.
Figure 7. Erlotinib treatment increases T cell infiltration in PDA tumors and enhances the sensitivity of PDA tumors to a combination immunotherapy.
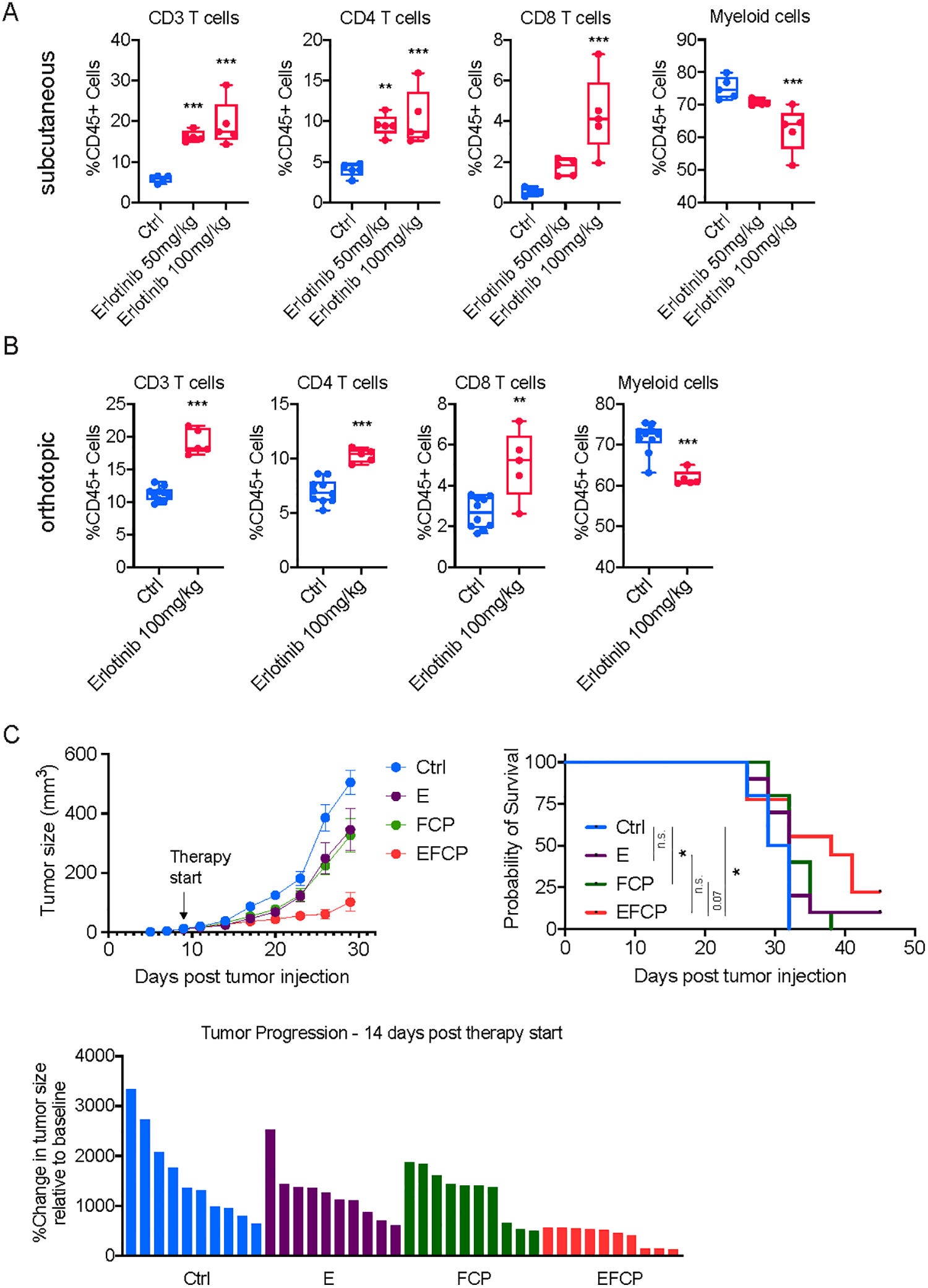
A, Flow cytometric analysis of immune cells in subcutaneously implanted 6694c2 tumors treated with either vehicle or erlotinib (n=5/group). B, Flow cytometric analysis of immune cells in orthotopically implanted 6694c2 tumors treated with either vehicle or erlotinib (n=5/group). For subcutaneous tumors, treatment started on day 10 post-implantation, and tumors were collected for flow analysis 10 days later. For orthotopic tumors, treatment started on day 7 post-implantation and tumors were harvested for flow analysis 10 days later. C, Growth curve, overall survival curve, and waterfall plot showing change in tumor size relative to the start of treatment of tumors from a non-T-cell-inflamed tumor cell clone, 6694c2, treated with vehicle, erlotinib (E), FCP, or erlotinib combined with FCP treatment (EFCP). In A, statistical differences were calculated using one-way ANOVA with multiple comparisons. In B, statistical differences were calculated using unpaired Student’s t-test. In C, statistical differences were calculated using two-way ANOVA with multiple comparisons and Log-rank (Mantel-Cox) test. p<0.05 was considered statistically significant, * indicates p<0.05, ** p<0.01, and *** p< 0.001.
To directly examine whether erlotinib treatment could enhance the efficacy of combination immunotherapy, we combined erlotinib (E) with CD40 agonist (F), PD-1 blockade (P), and CTLA-4 blockade (C). We observed a significant delay in tumor growth and improved clinical outcomes in mice treated with EFCP compared to the mice treated with only erlotinib (E) or FCP (Fig. 7C). This result directly demonstrates that erlotinib can enhance the responsiveness of a non-T-cell-inflamed PDA tumors to combination immunotherapy. This delay in tumor growth translated into a significant survival benefit for animals treated with EFCP compared to vehicle control and a trend towards improved survival from the addition of erlotinib to FCP immunotherapy (p=0.07, Fig. 7C). The effects on tumor growth persisted at later time points, as an analysis of animals surviving 20 days after initiation of therapy revealed persistently smaller tumors and one regression (Supplementary Fig. 7F). Together, these data suggest a potential new therapeutic opportunity for an otherwise treatment-refractory tumor type, providing a rationale for further preclinical, and even clinical, testing of this combination regimen.
DISCUSSION
Tumor heterogeneity describes distinct features that include both tumor cell intrinsic and extrinsic properties (67). Functionally, heterogeneous immune TMEs lead to distinct responses to immunotherapy (4). Recent studies have highlighted the role of tumor cell intrinsic factors in modulating both the abundance and activation of T cells and the consequences for productive anti-tumor immunity (7–9). A full understanding of these molecular regulators could inform novel therapeutic strategies to enhance the efficacy of existing immunotherapies. This principle is particularly relevant to PDA, which is refractory to most existing treatment options, including ICB-based immunotherapy (68). CRISPR loss-of-function genetic screening has been used to discover novel molecular regulators of anti-tumor immunity. Specifically, two recent studies utilized in vivo CRISPR screen strategies to unbiasedly identify potential therapeutic targets in melanoma and lung cancer (30,69). The current study is, to our knowledge, the first in vivo screen to identify tumor cell-intrinsic factors that can mediate anti-tumor immunity in pancreatic cancer.
We and others have shown that genomic mutations are unlikely to explain immune TME heterogeneity (10–12,19,60,61,70,71). Based on our observation that T-cell-inflamed and non-T-cell-inflamed tumor cells possess different epigenetic landscapes, we designed a screen to identify epigenetic factors important for immunotherapy responsiveness in vivo. Using this approach, together with transcriptional and epigenetic profiling and functional validation studies, we conclude that KDM3A, KLF5, and SMAD4 transcriptionally converge on EGFR to maintain a T cell low TME and drive resistance to immunotherapies. Loss of SMAD4 is frequently observed in human PDA samples, suggesting that it functions as a tumor suppressor (70), yet our study suggests that ablation of tumor cell-intrinsic SMAD4 promotes anti-tumor immunity. These seemingly discrepant observations may be due to the different functions of SMAD4 during tumor initiation versus progression. Nevertheless, the fact that SMAD4 loss is common in PDA, while responses to ICB are virtually non-existent in microsatellite-stable PDA patients, indicates that SMAD4 loss is insufficient to sensitize PDA tumors to either PD1 or CTLA4 inhibition. In the future, it will be important to evaluate whether immunotherapy-responsive PDA tumors are enriched for SMAD4 alterations. In addition, we observed that KDM3A loss resulted in a depletion of myCAFs from the TME, which could have an indirect effect on immune populations. Future work is needed to define the precise mechanism by which KDM3A, SMAD4, and KLF5 regulate anti-tumor immunity and whether this epigenetic-transcriptomic axis plays a role in other cancer types.
Our findings are in line with recent reports that small molecule inhibitors of EGFR enhance the efficacy of immunotherapy. For example, one recent study demonstrated that EGFR inhibition increased the sensitivity of EGFR-mutated non-small cell lung cancer to ICB-based immunotherapy (72). Mechanistically, we found that EGFR inhibition led to a downregulation of known immunosuppressive molecules, including PTGS2 and CSF2 (Supplementary Fig 6G) (11,30,60,61,73), as well as an induction of type I interferon signaling in tumor cells(Figure 5I), matching the observation from another recent study (74). Thus, there are at least two possible mechanisms by which EGFR may alter T cell infiltration:(i) inhibition of immunosuppressive molecules and (ii) activation of IFN signaling.
Importantly, the EGFR inhibitor erlotinib is FDA-approved for treating pancreatic cancer in combination with gemcitabine (24). Although EGFR signaling has been shown to play an important role in pancreatic cancer progression (20–24), erlotinib’s clinical benefit in PDA has been limited (22,24). The current study provides a rationale for further examining whether the combination of EGFR inhibition with immunotherapies will elicit a more potent response, not just in pancreatic cancer but also other KRAS-driven cancers such as lung cancer. In this respect, it is noteworthy that the combination therapy used in this study (GAFCP) is similar to one being evaluated in a phase II clinical trial for PDA (NCT03214250) and for which promising phase I data have been reported (33). As resistance to EGFR-targeting tyrosine kinase inhibitors is frequently observed (75–77), a deeper understanding of the molecular mechanisms underlying EGFR-mediated control of anti-tumor immunity is needed. In particular, the finding that different PDA tumor clones exhibit variable immune responses following EGFR inhibition suggests that inter-tumoral heterogeneity could lead to varied responses in patients. Consequently, elucidating the mechanisms underlying EGFR-dependent immune infiltration may help identify those patients most likely to respond to the immune sensitizing effects of EGFR inhibitors as well as facilitate the development of alternative therapies for those non-responders. Furthermore, it is also important to better understand the molecular changes in immune cells, especially CD8 T cells, following the treatment of EGFR inhibitors.
Accumulating evidence shows that different tumors exploit distinct cellular and molecular mechanisms to establish non-T-cell-inflamed TMEs (12,78). Consequently, further subtyping of non-T-cell-inflamed tumors and identification of subtype-specific immunosuppressive modules will be important for precision medicine and improved efficacy of immunotherapy. This might be particularly relevant to our observation that different T cell low PDA tumors vary in their sensitivity to erlotinib treatment. Since it has been previously shown that an increased abundance of gMDSCs is associated with therapy resistance, non-T-cell-inflamed tumors having an overabundance of this cell type might derive particular benefit from treatment with anti-gMDSCs agents (79).
MATERIALS AND METHODS
Animals
All mouse experiment procedures used in this study were performed following the National Institutes of Health guidelines. All mouse procedure protocols utilized in this study were in accordance with, and with the approval of, the Institutional Animal Care and Use committee (IACUC) of the University of Pennsylvania. All wild-type C57BL/6 were purchased from The Jackson Laboratory and/or bred and maintained at the University of Pennsylvania.
PDA tumor cell clones and cell lines
Mouse pancreatic tumor cell clones were generated in our lab recently(12), and were examined by the Research Animal Diagnostic Laboratory (RADIL) at the University of Missouri, using the Infectious Microbe PCR Amplification Test (IMPACT) II. PDA tumor cells were cultured in standard cell culture medium including DMEM (high glucose without sodium pyruvate, Corning, 10–017-CV) with 10% heat-inactivated FBS (Genesee Scientific 25–514) and Glutamax (Thermo 35050061). These tumor cell clones were used for less than 20 passages. YFP labeling of these tumor cell clones were derived from their genetic background (these tumor cell clones were derived from the KPCY mice as described in our previous report (25)). We used 293T cells (Clontech, 632180) for lentivirus packaging.
Implantation of tumor cells
Pancreatic tumor cells were dissociated into single cell suspension with 0.25% trypsin for 3 minutes at room temperature (Gibco), washed with serum-free Dulbecco’s Modified Eagle’s medium (DMEM) twice, and counted in preparation for subcutaneous or orthotopic implantations. 2.0×105 PDA tumor cells were implanted subcutaneously and 5.0×104 PDA tumor cells were implanted orthotopically into the pancreas of 6–8-week old female C57BL/6 mice as previously described. Tumors were harvested 17–24 days following implantation for flow analysis, fixation, and other described experiments. Endpoint criteria included tumor volume exceeding 500 mm3, severe cachexia, or weakness and inactivity.
Subcutaneous tumor growth and regression assessments
For tumor growth assessment, tumors were measured every 3–4 days. Tumor length and width were examined with calipers, and tumor volumes were then calculated as length*width2/2. Tumor volumes of 500 mm3 were used as an endpoint for overall survival analysis following our mouse protocols. Tumor progression and/or regressions and were calculated using the initial tumor size at the start of treatment to tumor size at the end point of analysis for calculation.
Treatments and T-cell depletions in mice
For therapy treatment, Gemcitabine (Hospira) and Nab-paclitaxel (Abraxane, Celgene) were both purchased from the Hospital of the University of Pennsylvania Pharmacy. Gemcitabine (G) was procured as pharmaceutical grade suspension at 38 mg/mL, and then diluted to 12 mg/mL in PBS and administered at 120 mg/kg via intraperitoneal (i.p.) injection. Abraxane (A) was purchased as a pharmaceutical grade powder resuspended at 12 mg/mL in PBS for i.p. injection at dose of 120 mg/kg. Vehicle control mice received the equivalent to nab-paclitaxel dose of human albumin (huAlb; Sigma). For anti-CD40 agonist treatment, mice were injected i.p. with 100 μg of either agonistic CD40 rat anti-mouse IgG2a (clone FGK45, endotoxin-free) or the isotype control IgG2a (clone 2A3, BioXcell) 48hrs after chemotherapy. For checkpoint blockade treatment, mice were injected i.p. with 200 μg of anti–PD-1 (clone RMP 1–14, BioXcell) and 200 μg anti–CTLA-4 (clone 9H10, BioXcell), beginning from described therapy start timepoint, with 6 and 3 doses, respectively. Control mice received the isotype control IgG2a (clone 2A3, BioXcell) on treatment days. Therapy was started when tumor was 3–5 mm size. CD4+ and CD8+ T cells were depleted using i.p. injections of 200 mg anti-CD4 (clone GK1.5, BioXcell) and anti-CD8 (clone 2.43, BioXcell), three days prior tumor implantation, and every three days for the duration of the experiment. Control groups received IgG2b isotype control (BioXcell). For erlotinib treatment, we prepared 500mg erlotinib (HY-50896) in DMSO and then diluted the drug in corn oil (5ml DMSO + 45mL corn oil) for a final concentration of 10 mg/mL. We delivered 100 mL and 200 mL of stock by gavage for 50 mg/kg and 100 mg/kg doses, respectively, daily.
CRISPR-Cas9 screen analysis
Cas9 protein was introduced to the 6419c5 PDA tumor cell clone by transduction of the lentiCas9-Blast plasmid (Addgene plasmid #52962) and selected with 10 μg/mL Blasticidin. Tumor cells were transduced with sgRNA lentivirus library at MOI of 0.2. 1000x coverage of the sgRNA library in tumor cells were maintained during in vitro cell culture. Enough tumor cells were implanted into mice to maintain 500x coverage of the sgRNA library. Tumor cells were collected for library preparation to maintain at least 500x coverage of the sgRNA library in each sample. Genomic DNA from collected tumor cells was harvested using the QIAamp DNA mini kit (Qiagen), and libraries were prepared following previously described protocols (80). Briefly, DNA containing sgRNAs were amplified over 25 cycles with Herculase II fusion DNA polymerase (Agilent) following manufacturer provided protocol with PCR.1 forward and reverse primer. 3 μg of genomic DNA were used for each PCR reaction, and several PCR reactions were run in parallel for each collected sample for maintaining library coverage. Product of PCR.1 reaction was pooled for each sample, and 5 μl of each pooled PCR.1 sample was used as a template for PCR.2. For PCR.2, template was amplified over 7 cycles with PCR.2 forward and reverse primers, and the resulting product were purified with the QIAquick PCR purification kit (Qiagen) followed by gel extracted with the QIAquick gel extraction kit (Qiagen). The barcoded libraries were pooled at an equal molar ratio and sequenced on a NextSeq500/550 (Illumina, 75 cycles High Output kit v2.0) to generate 75 bp single end reads. MAGeCK software was used for screen analysis (81). Briefly, the sequencing data were de-barcoded and merged, and the 20 bp sgRNA sequence was aligned to the reference sgRNA library without allowing for any mismatches. The read counts were calculated for each sgRNA using the method normalizing to the non-targeting sgRNAs. Differential analysis of sgRNA and targeted genes was also done following the MAGeCK instructions with standard parameters.
RNA-seq analysis
RNA samples were extracted from sorted YFP+ tumor cells from subcutaneous tumors, using the Qiagen RNeasy Micro Kit following manufacturer’s instructions. RNA was sent out to a commercial company, Novogene (California, USA), for library preparation and high-throughput sequencing using Illumina sequencers to generated paired-end results. Raw counts of gene transcripts were obtained using alignment-independent tool, Salmon (https://combine-lab.github.io/salmon/), using standard settings. The raw count matrix was subsequently imported into R-studio (R version 3.3.3) and used as input for DESeq2 following the vignette of the package for normalization and differential gene expression analysis. Salmon was used to normalize and quantitate gene expression in transcripts-per-million (tpm) through quasi-alignment. Differentially expressed genes were used as input for GSEA and other functional analysis. Differentially expressed genes were also used as input for online EnrichR analysis following provided instructions (82,83). Detailed Scripts and parameters used for each steps of analysis could be provided by reasonable request to the authors.
ATAC-seq analysis
For library construction, 50,000 tumor cells sorted from subcutaneously implanted tumors were used as input as previously described (12). The libraries were prepared as previously described with minor modifications (84). Briefly, nuclei were isolated from sorted cells using a solution of 10 mM Tris-HC pH 7.4, 10 mM NaCl, 3 mM MgCl2, and 0.1% IGEPAL CA-630. Immediately following nuclei isolation, the transposition reaction was conducted using Tn5 transposase and TD buffer (Illumina) for 45 min. at 37 degree. Transposed DNA fragments were purified using a Qiagen MinElute Kit, barcoded with primers based on Illumina TruSeq indices, and PCR amplified for 12 cycles using NEBNext High Fidelity 2x PCR master mix (New England Biolabs). Libraries were purified by extraction from a 6% TBE gel, followed by column purification with the Qiagen PCR Cleanup kit. Sequencing was performed using a HiSeq2500 (Illumina) on rapid run mode to generate 50 bp paired end reads. For analysis, after adapter trimming with cutadapt, reads were aligned to the mouse reference genome (mm10) by Bowtie2. MACS2 was applied to identify accessible chromatin (85). Peaks were subsequently merged using BEDTools and ATAC-seq read counts were calculated in the merged peaks for every sample (86). The resulting count table was used to identify differentially accessible loci with edgeR, which were then used for functional analysis by GREAT analysis (version 3.0.0) (87–89). Detailed Scripts and parameters used for each steps of analysis could be provided by reasonable request to the authors.
ChIP-seq analysis
For ChIP, about 1.5 × 107 cells were cultured on 15 cm plates with normal culture medium. Medium was changed to DMEM without serum first and then cells were crosslinked with 1.1% formaldehyde for 10 min at room temperature. Then samples were quenched with 125 mM glycine for 5 min at room temperature on plate. Cells were then washed with cold PBS three times and then lysed with 1 mL nuclei isolation buffer for 15 min on ice (20mM PIPES, 85mM KCl, and 0.5% NP-40). Nuclei were spun down at 300 g for 5 min at 4 degree and resuspended in 500 uL ChIP lysis buffer with proteinase inhibitor (1% SDS, 10 mM EDTA pH 8, 50 mM Tris-HCl pH 8.0). Samples were then sonicated with a Diagenode Bioruptor at high intensity setting for 15 X 5 min cycles of 30 sec on/off, to yield about 300bp fragments. After centrifugation at 4 degree with 13,500 rpm for 10 min, soluble chromatin was then diluted using ChIP dilution buffer (0.01% SDS, 1.2 mM EDTA pH 8, 16.7 mM Tris-HCl pH 8, 167 mM NaCl, and 1.1% Triton X 100), and pre-cleared with magnetic Protein G Dynabeads (Invitrogen, 10004D). Immunoprecipitation with anti-H3K9me2 (Abcam, ab32521), anti-H3K9ac (Abcam, ab4441), anti-KDM3A (Novus Biologicals, NB100–77282) was performed overnight at 4 degree with rotation, with 10% sample kept as input DNA. Products were recovered with magnetic Protein G Dynabeads and washed sequentially using ChIP Low Salt Buffer (0.1% SDS, 2.0 mM EDTA pH 8.0, 20 mM Tris-HCl pH 8.0, 150 mM NaCl, 1.0% Triton X 100), ChIP High Salt Buffer (0.1% SDS, 2.0 mM EDTA pH 8.0, 20 mM Tris-HCl pH 8.0, 500 mM NaCl, 1.0% Triton X 100), Lithium Chloride Buffer (1 mM EDTA pH 8.0, 10 mM Tris-HCl pH 8.0, 250 mM LiCl, 1% NP-40, 1% sodium deoxycholate), and TE buffer (1 mM EDTA pH 8.0, 10 mM Tris-HCl pH 8.0) 10 min each buffer at room temperature. Samples were eluted twice using ChIP Elution Buffer (2% SDS, 100 mM NaHCO3) at room temperature for 15 min each elution with rotation. Eluted sample DNA was incubated with RNase A (0.2 mg/mL) overnight at 65°C, then incubated with Proteinase K (0.2 mg/mL) for 2 hours at 45°C. DNA samples were purified using QIAquick PCR Purification Kit (Qiagen) and used for preparation of ChIP-seq libraries. Libraries were prepared following the NEBNext Ultra II DNA Library Prep Kit (New England Biolabs) protocol, using NEBNext Multiplex Oligos Index Primers Sets. Libraries were pooled and sequenced on the NextSeq500/550 (Illumina, 75 cycles High Output kit v2.0) to generate 75 bp single end reads. Sequencing reads were aligned to the mouse (mm10) using Bowtie2 with default settings (90). Resulted sam files were processed and marked with duplicates using SAMtools (91). Peaks calling was performed using MACS2 (85) with default settings. Peaks were subsequently merged using BEDTools and read counts were calculated in the merged peaks for every sample (86). The resulting count table was then used to find differentially genomic regions with edgeR (87,88). Differential genomic regions were then transformed to bed files and used for genomic region annotation using the ChIP-seeker analysis tool (92) as well as the motif analysis using the MEME suite (93). The DREME analysis and the Tomtom analysis were used in the MEME suite for motif discovery and motif comparison (94,95). The HOCOMOCO core collection of motifs (version 11) were used for the motif enrichment analysis. Detailed Scripts and parameters used for each steps of analysis could be provided by reasonable request to the authors.
Cancer Dependency Map Portal data analysis
The depmap portal (https://depmap.org/portal/) and the CRISPR (Avana) Public 19Q3 dataset were used for this analysis. No samples were excluded from the dataset in this analysis. Following the depmap instruction, the dependency scores of genes were downloaded and then plotted as dot plots. Pearson correlation was calculated for all the plots using Prism.
Lentiviral transduction of tumor cells for CRISPR-mediated ablation
The CRISPR vector, lentiCRISPR v2, was a gift from Feng Zhang (Addgene plasmid # 52961). The vector and pVSVg and psPAX2 lentiviral packaging plasmids (Addgene) were co-transfected into 293T cells (Clontech, 632180) using PEI reagent (Polysciences, 23966–2). Lentiviral particles were collected 48 hours after transfection and filtered for usage. Tumor cells transduced either with Cas9-Puro (control, ctrl) or Cas9-guide-Puro (knockout, KO) (ctrl is from Addgene, #52961, and KOs were cloned following the instruction from Addgene) were selected with 8 μg/mL puromycin (Invitrogen, A1113803). Single-cell clones were picked from bulk knockout cell line using single-cell sorting using BD Jazz FACS sorting machine in to 96-well plates. Knockout efficiencies were assessed by gene-specific qPCR analysis of target gene. CRISPR sgRNA sequences used were:
Dnmt1-sg1-TGA-AAC-TTC-ACC-TAG-TTC-CG; Dnmt1-sg2-CAA-ATA-GAT-CCC-CAA-GAT-CC;
Atf2-sg1-TGG-ACG-AAC-GAT-AGC-TGA-TG; Atf2-sg2-CGT-TCG-TCC-AGC-ATC-ATT-AC;
Brd1-sg1-TAA-TTG-AGC-TGC-TGC-GCA-AG; Brd1-sg2-GAG-CGT-AGC-AGC-ACA-GTT-AG;
Cbx7-sg1-GGA-AGA-GAG-GTC-CGA-AAC-CC; Cbx7-sg2-CCA-GAC-GTT-GTG-CAG-ACC-CC;
Ep400-sg1-TCG-GAA-CAT-GTA-GGG-CCG-GC; Ep400-sg2-CAG-AAG-CCG-ACC-CCT-TTA-AG;
Prdm5-sg1-GCT-CGA-TTC-ACA-CTG-TGG-AC; Prdm5-sg2-GAG-TTC-AAA-TGC-GAG-AAC-TG;
Prdm8-sg1-GCT-CTT-CCG-CTC-GTC-CGA-TG; Prdm8-sg2-TGT-TCT-ACC-GCT-CTC-TCC-GC;
Kdm3a-sg1-TAC-AGG-ATG-TTA-ACA-GTC-TT; Kdm3a-sg2-AAC-TCT-TCA-AGT-CAA-CTG-TG;
Tead2-sg1-GAA-GAC-GAG-AAC-GCG-AAA-GC; Tead2-sg2-TTC-GAG-CCA-AAA-CCT-GAA-TA;
Mycn-sg1-CGA-GTA-CGT-GCA-CGC-CCT-AC; Mycn-sg2-GCC-GAC-TCT-CGC-TTG-TTC-AC;
Prdm16-sg1-CTA-CGA-GAG-TCC-TCC-ATA-CC; Prdm16-sg2-CCC-GAT-TTC-CAT-CTT-CCG-CT;
Trim28-sg1-GGA-CCT-GCT-AAG-ACT-CGA-GA; Trim28-sg2-ATA-ATT-CTC-CAC-GAT-GTC-TT;
Klf5-sg1-CGC-GTG-TTT-CAG-ATC-GTC-TC; Klf5-sg2-TGC-GAA-CCC-GGC-CCG-CGA-CG;
Smad3-sg1-GCT-CCA-TGG-CCC-GTA-ATT-CA; Smad3-sg2-ACC-TAC-CTG-GAA-TAT-TGC-TC;
Smad4-sg1-GCC-AAG-TAA-TCG-CGC-ATC-AA; Smad4-sg2-TCC-GTT-GAT-GCG-CGA-TTA-CT;
Egfr-sg1-CGG-TCA-GAG-ATG-CGA-CCC-TC; Egfr-sg2-ACT-GCC-CAT-GCG-GAA-CTT-AC.
Quantitative PCR (qPCR) analysis for gene expression
RNA was prepared from cultured PDA tumor cells using the RNeasy Mini Kit or RNeasy Micro Kit (Qiagen). cDNA was generated using High-capacity cDNA Reverse Transcription Kit from 1 ug RNA in 20 ul reaction volume and diluted 1:10 for qPCR analysis (Life Technologies). qPCR analysis was performed using 2 ul diluted cDNA with biological and technical replicates using SsoAdvanced SYBR reagent (Bio-Rad) and Bio-Rad qPCR platform, and results were normalized to the expression of Tbp using the Bio-Rad software. Primer sequences utilized for qPCR were:
Dnmt1-F-AAG-AAT-GGT-GTT-GTC-TAC-CGA-C; Dnmt1-R-CAT-CCA-GGT-TGC-TCC-CCT-TG;
Atf2-F-CCG-TTG-CTA-TTC-CTG-CAT-CAA; Atf2-R-TTG-CTT-CTG-ACT-GGA-CTG-GTT;
Brd1-F-AAC-ACT-GAC-CTA-CGC-ACA-AGC; Brd1-R-GCC-TCT-CGC-TGT-TCT-CCT-TAT-T;
Cbx7-F-TGC-GGA-AGG-GCA-AAG-TTG-AAT; Cbx7-R-ACA-AGG-CGA-GGG-TCC-AAG-A;
Ep400-F-CGG-TTC-TCA-GGA-TAA-ACT-GGC; Ep400-R-CAC-CTC-CGC-TCT-TGA-GCA-A;
Prdm5-F-GAG-AAG-CGA-ATG-CCT-GAA-GAC; Prdm5-R-CTC-CCA-CGT-ACC-TCC-CAC-A;
Prdm8-F-ATG-GAG-GAT-TCA-GGC-ATC-CAG; Prdm8-R-GGA-CCG-AAT-ATG-GCG-TTC-TCT;
Kdm3a-F-GTG-ACA-CAA-CCA-TTT-TCA-ACC-TG; Kdm3a-R-CAC-CCT-GTT-GGC-AGT-TCT-TC;
Tead2-F-GAA-GAC-GAG-AAC-GCG-AAA-GC; Tead2-R-GAT-GAG-CTG-TGC-CGA-AGA-CA;
Mycn-F-ACC-ATG-CCG-GGG-ATG-ATC-T; Mycn-R-AGC-ATC-TCC-GTA-GCC-CAA-TTC;
Prdm16-F-CCC-CAC-ATT-CCG-CTG-TGA-T; Prdm16-R-CTC-GCA-ATC-CTT-GCA-CTC-A;
Trim28-F-CGG-CGC-TAT-GGT-GGA-TTG-T; Trim28-R-GGT-TAG-CAT-CCT-GGG-AAT-CAG-AA;
Klf5-F-CCG-GAG-ACG-ATC-TGA-AAC-ACG; Klf5-R-GTT-GAT-GCT-GTA-AGG-TAT-GCC-T;
Smad3-F-CAC-GCA-GAA-CGT-GAA-CAC-C; Smad3-R-GGC-AGT-AGA-TAA-CGT-GAG-GGA;
Smad4-F-ACA-CCA-ACA-AGT-AAC-GAT-GCC; Smad4-R-GCA-AAG-GTT-TCA-CTT-TCC-CCA;
Egfr-F-GCC-ATC-TGG-GCC-AAA-GAT-ACC; Egfr-R-GTC-TTC-GCA-TGA-ATA-GGC-CAA-T;
Tbp-F-AGA-ACA-ATC-CAG-ACT-AGC-AGC-A; Tbp-R-GGG-AAC-TTC-ACA-TCA-CAG-CTC.
Flow cytometry of implanted tumors
For flow cytometric analyses, subcutaneous or orthotopic tumors were chopped into small pieces and digested in collagenase (1 mg/mL in DMEM; Sigma-Aldrich) at 37 degree for 45 minutes and filtered through a 70-μM cell strainer to generate single cell suspensions. Single cell suspensions were then stained with fluorescence labelled antibodies on ice for 40 minutes and washed twice with cold PBS with 5% FBS for flow cytometric analysis. Cells were then analyzed by flow cytometry using BD FACS (BD Biosciences) and FlowJo software (Treestar). CD44 was used as a marker for T cell activation given our prior studies demonstrating that CD44 is consistently differentially expressed in CD8 T cells from naturally occurring T-cell-low and T-cell-high tumors (12)
Gating Strategies for immune cells:
myeloid cells - Live CD45+CD11b+; granulocytic (g)MDSCs/neutrophils - Live CD45+CD11b+Gr1+; macrophages - Live CD45+F4/80+CD11b+; CD11c+ dendritic cells - Live CD45+F4/80– CD11c+; CD103+ dendritic cells - Live CD45+CD11b–F4/80–CD11c+ CD103+; T cells - Live CD45+CD11b–F4/80–NKp46–CD3+; CD4+ T cells - Live CD45+CD11b–F4/80–NKp46–CD3+CD4+; CD8+ T cells - Live CD45+CD11b–F4/80–NKp46–CD3+CD8+.
Antibodies used for flow analysis:
CD279 (PD-1) FITC (29F.1A12) Biolegend 135214, CD335 (NKp46) PE (29A1.4) Biolegend 137604, CD103 PE/Dazzle 594 (2E7) Biolegend 121430, CD3e PE/Cy5 (145–2C11) Biolegend 100310, CD45 AF700 (30-F11) Biolegend 103128, CD8a PE/Cy7 (53–6.7) Biolegend 100722, Ly-6G V450 (1A8) BD 560603, H-2Kb/H-2Db (MHCI) AF647 (28-8-6) Biolegend 114612, F4/80 APC/Cy7 (BM8) Biolegend 123118, CD11b PerCP-Cy5.5 (M1/70) BD 550993, CD11c BV605 (N418) Biolegend 117334, Ly-6C BV570 (HK1.4) Biolegend 128030, CD4 BV650 (RM4–5) Biolegend 100546, CD44 APC Biolegend 103012.
TGFβ treatment
PDA tumor cells were plated into 6-well plates and 24 hours later were treated with either citric acid (control) or 10 ng/mL TGFβ (CST) over 3 days for RNA collection for RNA-seq analysis.
In vitro treatment with IFNγ for MHC I expression analysis
Kdm3a-WT and Kdm3a -KO PDA tumor cells were treated with 100ng/mL of IFNγ (Peprotech) in cell culture medium for 24 hours. Tumor cells were then trypsinized from culture plates and re-suspended in PBS with 5% FBS for staining of antibodies. Tumor cells were then stained with MHCI (Biolegend 114612) antibody on ice for 40 minutes and washed twice with cold PBS with 5% FBS for flow cytometric analysis using BD FACS LSR machine (BD Biosciences) and FlowJo software (Treestar).
Immunofluorescent and immunohistochemistry staining
For CD3 and Gr1 staining, collected implanted tumor tissues were fixed in Zn-formalin for 24 hours and embedded in paraffin. Sections were deparaffinized, rehydrated, and prepared by antigen retrieval for 6 minutes each, and then blocked in 5% donkey serum (Sigma, D9663) for 1 hour at room temperature, incubated with primary antibodies overnight at 4°C, washed with 0.1% PBST (PBS with Tween-20), incubated with secondary antibodies for 1 hour at room temperature, and then washed and mounted. Slides were visualized and imaged using an Olympus IX71 inverted multicolor fluorescent microscope and a DP71 camera. For CD3 and Gr1 staining quantification, stained cells were counted for CD3+ T cells manually in 5–8 fields per sample.
Primary antibodies: CD3 (Abcam ab5690, 1:100 dilution) and Gr-1 (eBioscience 14-5931-85, 1:50 dilution), YFP (Abcam, ab6673). Secondary antibodies were purchased from Invitrogen (A-11055, A-21207, A-21209) and were used as 1:250 dilution for all staining.
Immunoblotting
For whole cell lysates, cells were washed with PBS and lysed in RIPA lysis buffer. Extracted proteins were separated by SDS-PAGE, transferred to PVDF membrane, blocked in 5% non-fat milk in PBS with 0.1% Tween-20, probed with primary antibodies, and detected with horseradish peroxidase-conjugated secondary antibodies (Jackson Immunoresearch). Primary antibodies used include: KDM3A (Novus Biologicals, NB100–77282), alpha-TUBULIN (CST, 3873S).
Histone extraction, derivatization, mass spectrometry, and PTM mark quantification
For extraction, histones were acid extracted according to standard protocols. Briefly, nuclei were isolated from cells lysed with a hypotonic lysis buffer. Histones were extracted from the nuclei with 0.2N H2SO4, precipitated in 33% trichloroacetic acid (TCA), washed with acetone, and resuspended in diH2O. For derivatization, histones were derivatized according to standard protocol(96). Briefly, 5–10ug of acid-extracted histones were resuspended in 100mM ammonium bicarbonate (pH 8.0) and mixed with freshly prepared propionic anhydride with acetonitrile for 15 min. at 37 °C. Histones were then digested with trypsin (enzyme: sample ratio 1:20) overnight at 37 °C. The peptides were then desalted and stored dried. They were resuspended in 0.1% formic acid just before mass spectrometry. For direct injection–MS, samples were placed in a TriVersa NanoMate (Advion) and acquired either manually or by using a sequence coordinated with MS acquisition by a contact closure. The NanoMate was set up with a spray voltage of 1.7 kV and a gas pressure of 0.5 psi. Samples were acquired in the Orbitrap Fusion Tribrid (Thermo Scientific). All scans were acquired in the orbitrap, at 240,000 resolution for the full MS and at 120,000 resolution for MS/MS. The AGC target for the tSIM-MSX scans was set to 10E6. The full description of the DI-MS acquisition method has been previously described63. For histone peptide quantification, raw files were searched with a modified version of the software EpiProfile 2.064. Histone peptides are collected in MS scans, and isobaric peptides are collected in targeted pre-set MS/MS scans. The software reads the intensities from MS scans to calculate the percentage of all peptides with the same amino acid sequence. The unique fragment ions in the MS/MS scans are extracted to discriminate isobaric peptide intensities from the MS scans. The software EpiProfileLite is available at https://github.com/zfyuan/EpiProfileLite, with user guide.
Immunoprecipitation
For immunoprecipitation (IP) experiments, Dynabeads Protein G (Invitrogen) were first crosslinked with either rabbit IgG or KDM3A (Novus Biologicals, NB100–77282) antibodies at 6ug antibodies/50ul beads according to standard protocols. Briefly, antibodies and beads were mixed for 1 hr at room temperature, then incubated with 20mM DMP in 0.2M triethanolamine for 40min at room temperature. The reaction was quenched with 100mM Tris-HCl pH 8.0 for 1hr at room temperature. The crosslinked beads were then washed 3x with 0.1M glycine pH 2.5, then washed 2x with PBST and stored in PBST at 4°C for future use. For the IP, cultured cells were washed in cold PBS and nuclei were isolated as previously described. Nuclei were then lysed in lysis buffer (20 mM Tris pH 7.4, 0.15 M NaCl, 1.5mM MgCl2, 1 mM EDTA, 1% glycerol, 1%NP-40) with an EDTA-free protease inhibitor mixture (Roche Applied Science) and precleared by incubating with Dynabeads Protein G (Invitrogen) for 1 hr at room temperature. Precleared nuclear lysates were then incubated with crosslinked beads overnight at 4°C. After washing with lysis buffer, protein was eluted 2x from the beads with 100 mM glycine pH 2.5, which was then neutralized with 4 μl 1M Tris-HCl pH 8.5. Protein was then denatured by boiling with SDS protein loading buffer. Prepared samples were then either subjected to immunoblotting, as previously described, or LC/MS-MS analysis.
LC-MS/MS Analyses and Data Processing
Liquid chromatography tandem mass spectrometry (LC-MS/MS) analysis was performed as previously described (97) by the Proteomics and Metabolomics Facility at the Wistar Institute using a Q Exactive HF mass spectrometer (ThermoFisher Scientific) coupled with a Nano-ACQUITY UPLC system (Waters). Samples were reduced with TCEP, alkylated with iodoacetamide, digested in-gel with trypsin and injected onto a UPLC Symmetry trap column (180 μm i.d. × 2 cm packed with 5 μm C18 resin; Waters). Tryptic peptides were separated by reversed phase HPLC on a BEH C18 nanocapillary analytical column (75 μm i.d. × 25 cm, 1.7 μm particle size; Waters) using a 95 min gradient formed by solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in acetonitrile). A 30-min blank gradient was run between sample injections to minimize carryover. Eluted peptides were analyzed by the mass spectrometer set to repetitively scan m/z from 400 to 2000 in positive ion mode. The full MS scan was collected at 60,000 resolution followed by data-dependent MS/MS scans at 15,000 resolution on the 20 most abundant ions exceeding a minimum threshold of 10,000. Peptide match was set as preferred, exclude isotopes option and charge-state screening were enabled to reject singly and unassigned charged ions.
Peptide sequences were identified using MaxQuant v1.6.8.0 (98). MS/MS spectra were searched against a UniProt mouse protein database (10/01/2019) using full tryptic specificity with up to two missed cleavages, static carboxamidomethylation of Cys, and variable oxidation of Met and protein N-terminal acetylation. Consensus identification lists were generated with false discovery rates of 1% at protein, and peptide levels.
Data resources
The RNA-seq and ChIP-seq data, fastq files and processed count tables, have been deposited at the Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/) with following access numbers: GSE146832, GSE146833, GSE146834, GSE156889.
Software and Statistical analysis
PRISM software and R were used for data processing, statistical analysis, and result visualization (http://www.graphpad.com). The R language and environment for graphics (https://www.r-project.org) was used in this study for the bioinformatics analysis of CRISPR screen, RNA-seq, and ChIP-seq data. The R packages used for all analysis described in this manuscript were from the Bioconductor and CRAN. Statistical comparisons between two groups were performed using Student’s unpaired t test. For comparisons between multiple groups, one-way ANOVA with Tukey’s HSD post-test was used. For survival comparison between two groups, log-rank p-values of Kaplan-Meier curves were determined in GraphPad Prism 8 (GraphPad). On graphs, bars represent either range or standard error of mean (SEM), as indicated in legends. For all figures, p<0.05 was considered statistically significant, * indicates p<0.05, **p<0.01, and ***p<0.001.
Supplementary Material
STATEMENT OF SIGNIFICANCE.
Pancreatic ductal adenocarcinoma (PDA) remains refractory to immunotherapies. Here, we performed an in vivo CRISPR screen and identified an epigenetic-transcriptional network that regulates anti-tumor immunity by converging on EGFR. Pharmacologic inhibition of EGFR is sufficient to rewire the immune microenvironment. These results offer a readily accessible immunotherapy-sensitizing strategy for PDA.
ACKNOWLEDGMENTS
We thank members of the Stanger laboratories for technical help and scientific discussions. We also thank members of the flow cytometry and cell sorting resource laboratory for technical help with flow analysis and sorting.
Conflict of Interest: Dr. Stanger has received research funding from Boehringer-Ingelheim and serves as a consultant to iTeos Therapeutics. Dr. Vonderheide has received research funding from Apexigen, Fibrogen, Inovio, Janssen, and Lilly and consulting fees or honoraria from Apexigen, AstraZeneca, Celgene, Genentech, Janssen, Lilly, Medimmune, Merck and Verastem.
This work was supported by grants from NIH (CA229803, DK083355, CA224970), the Abramson Family Cancer Research Institute, the Abramson Cancer Center, and the NIH/Penn Center for Molecular Studies in Digestive and Liver Diseases. BA Garcia acknowledges National Institutes of Health (NIH) grants: CA196539 and U54CA232568. as well as St.Jude Collaborative A11576.
REFERENCES
- 1.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sanmamed MF, Chen L. A Paradigm Shift in Cancer Immunotherapy: From Enhancement to Normalization. Cell. 2018;175:313–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24:541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spranger S Mechanisms of tumor escape in the context of the T-cell-inflamed and the non-T-cell-inflamed tumor microenvironment. Int Immunol. 2016;28:383–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li J, Stanger BZ. The tumor as organizer model. Science. 2019;363:1038–9. [DOI] [PubMed] [Google Scholar]
- 8.Spranger S, Gajewski TF. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat Rev Cancer. 2018;18:139–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wellenstein MD, de Visser KE. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity. 2018;48:399–416. [DOI] [PubMed] [Google Scholar]
- 10.Li J, Yuan S, Norgard RJ, Yan F, Yamazoe T, Blanco A, et al. Tumor Cell–Intrinsic USP22 Suppresses Antitumor Immunity in Pancreatic Cancer. Cancer Immunol Res. 2019;8:282–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Markosyan N, Li J, Sun YH, Richman LP, Lin JH, Yan F, et al. Tumor cell-intrinsic EPHA2 suppresses anti-tumor immunity by regulating PTGS2 (COX-2). J Clin Invest. 2019;129:3594–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity. 2018;49:178–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN promotes resistance to T cell–mediated immunotherapy. Cancer Discov. 2016;6:202–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Welte T, Kim IS, Tian L, Gao X, Wang H, Li J, et al. Oncogenic mTOR signalling recruits myeloid-derived suppressor cells to promote tumour initiation. Nat Cell Biol. 2016;18:632–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–5. [DOI] [PubMed] [Google Scholar]
- 16.Liao W, Overman MJ, Boutin AT, Shang X, Zhao D, Dey P, et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell. 2019;35:559–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang G, Lu X, Dey P, Deng P, Wu CC, Jiang S, et al. Targeting YAP-dependent MDSC infiltration impairs tumor progression. Cancer Discov. 2016;6:80–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kortlever RM, Sodir NM, Wilson CH, Burkhart DL, Pellegrinet L, Brown Swigart L, et al. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell. 2017;6:1301–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sodir NM, Kortlever RM, Barthet VJA, Campos T, Pellegrinet L, Kupczak S, et al. Myc instructs and maintains pancreatic adenocarcinoma phenotype. Cancer Discov. 2020;10:588–607. [DOI] [PubMed] [Google Scholar]
- 20.Ardito CM, Grüner BM, Takeuchi KK, Lubeseder-Martellato C, Teichmann N, Mazur PK, et al. EGF Receptor Is Required for KRAS-Induced Pancreatic Tumorigenesis. Cancer Cell. 2012;22:304–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Navas C, Hernández-Porras I, Schuhmacher AJ, Sibilia M, Guerra C, Barbacid M. EGF Receptor Signaling Is Essential for K-Ras Oncogene-Driven Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2012;22:318–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nedaeinia R, Avan A, Manian M, Salehi R, Ghayour-Mobarhan M. EGFR as a Potential Target for the Treatment of Pancreatic Cancer: Dilemma and Controversies. Curr Drug Targets. 2014;15:1293–301. [DOI] [PubMed] [Google Scholar]
- 23.Ng SSW, Tsao MS, Nicklee T, Hedley DW. Effects of the epidermal growth factor receptor inhibitor OSI-774, Tarceva, on downstream signaling pathways and apoptosis in human pancreatic adenocarcinoma. Mol Cancer Ther. 2002;1:777–83. [PubMed] [Google Scholar]
- 24.Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960–6. [DOI] [PubMed] [Google Scholar]
- 25.Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burr ML, Sparbier CE, Chan KL, Chan YC, Kersbergen A, Lam EYN, et al. An Evolutionarily Conserved Function of Polycomb Silences the MHC Class I Antigen Presentation Pathway and Enables Immune Evasion in Cancer. Cancer Cell. 2019;36:385–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. 2015;527:249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell. 2018;174:549–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pan D, Kobayashi A, Jiang P, De Andrade LF, Tay RE, Luoma AM, et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science. 2018;359:770–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li F, Huang Q, Luster TA, Hu H, Zhang H, Ng WL, et al. In Vivo Epigenetic CRISPR Screen Identifies Asf1a as an Immunotherapeutic Target in Kras-Mutant Lung Adenocarcinoma. Cancer Discov. 2020;10:270–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Winograd R, Byrne KT, Evans RA, Odorizzi PM, Meyer ARL, Bajor DL, et al. Induction of T-cell immunity overcomes complete resistance to PD-1 and CTLA-4 blockade and improves survival in pancreatic carcinoma. Cancer Immunol Res. 2015;3:399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Byrne KT, Vonderheide RH. CD40 Stimulation Obviates Innate Sensors and Drives T Cell Immunity in Cancer. Cell Rep. 2016;15:2719–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O’Hara MH, O’Reilly EM, Rosemarie M, Varadhachary G, Wainberg ZA, Ko A, et al. Abstract CT004: A Phase Ib study of CD40 agonistic monoclonal antibody APX005M together with gemcitabine (Gem) and nab-paclitaxel (NP) with or without nivolumab (Nivo) in untreated metastatic ductal pancreatic adenocarcinoma (PDAC) patients. Cancer Res. 2019;79:CT004. [Google Scholar]
- 34.Shi Z, Xu S, Xing S, Yao K, Zhang L, Xue L, et al. Mettl17, a regulator of mitochondrial ribosomal RNA modifications, is required for the translation of mitochondrial coding genes. FASEB J. 2019;33:13040–50. [DOI] [PubMed] [Google Scholar]
- 35.Goel S, Decristo MJ, Watt AC, Brinjones H, Sceneay J, Li BB, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature. 2017;548:471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dandawate P, Ghosh C, Palaniyandi K, Paul S, Rawal S, Pradhan R, et al. The Histone Demethylase KDM3A, Increased in Human Pancreatic Tumors, Regulates Expression of DCLK1 and Promotes Tumorigenesis in Mice. Gastroenterology. 2019;157:1646–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov [Internet]. American Association for Cancer Research; 2019. [cited 2020 Aug 26];9:1102–23. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31197017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chakravarthy A, Khan L, Bensler NP, Bose P, De Carvalho DD. TGF-β-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat Commun [Internet]. Nature Publishing Group; 2018. [cited 2020 Aug 26];9:4692. Available from: http://www.nature.com/articles/s41467-018-06654-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamane K, Toumazou C, Tsukada Y ichi, Erdjument-Bromage H, Tempst P, Wong J, et al. JHDM2A, a JmjC-Containing H3K9 Demethylase, Facilitates Transcription Activation by Androgen Receptor. Cell. 2006;125:483–95. [DOI] [PubMed] [Google Scholar]
- 41.Goda S, Isagawa T, Chikaoka Y, Kawamura T, Aburatani H. Control of histone H3 lysine 9 (H3K9) methylation state via cooperative two-step demethylation by jumonji domain containing 1A (JMJD1A) homodimer. J Biol Chem. 2013;288:36948–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tateishi K, Okada Y, Kallin EM, Zhang Y. Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature. 2009;458:757–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuroki S, Matoba S, Akiyoshi M, Matsumura Y, Miyachi H, Mise N, et al. Epigenetic regulation of mouse sex determination by the histone demethylase Jmjd1a. Science. 2013;341:1106–9. [DOI] [PubMed] [Google Scholar]
- 44.Li J, Yu B, Deng P, Cheng Y, Yu Y, Kevork K, et al. KDM3 epigenetically controls tumorigenic potentials of human colorectal cancer stem cells through Wnt/β-catenin signalling. Nat Commun. 2017;8:15146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cloos PAC, Christensen J, Agger K, Helin K. Erasing the methyl mark: Histone demethylases at the center of cellular differentiation and disease. Genes Dev. 2008;22:1115–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hublitz P, Albert M, Peters AHFM. Mechanisms of transcriptional repression by histone lysine methylation. Int J Dev Biol. 2009;53:335–54. [DOI] [PubMed] [Google Scholar]
- 47.Peters AHFM, Kubicek S, Mechtler K, O’Sullivan RJ, Derijck AAHA, Perez-Burgos L, et al. Partitioning and Plasticity of Repressive Histone Methylation States in Mammalian Chromatin. Mol Cell. 2003;12:1577–89. [DOI] [PubMed] [Google Scholar]
- 48.Rice JC, Briggs SD, Ueberheide B, Barber CM, Shabanowitz J, Hunt DF, et al. Histone Methyltransferases Direct Different Degrees of Methylation to Define Distinct Chromatin Domains. Mol Cell. 2003;12:1591–8. [DOI] [PubMed] [Google Scholar]
- 49.Tachibana M, Sugimoto K, Fukushima T, Shinkai Y. SET Domain-containing Protein, G9a, is a Novel Lysine-preferring Mammalian Histone Methyltransferase with Hyperactivity and Specific Selectivity to Lysines 9 and 27 of Histone H3. J Biol Chem. 2001;276:25309–17. [DOI] [PubMed] [Google Scholar]
- 50.Tachibana M, Sugimoto K, Nozaki M, Ueda J, Ohta T, Ohki M, et al. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 2002;16:1779–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilson S, Fan L, Sahgal N, Qi J, Filipp FV. The histone demethylase KDM3A regulates the transcriptional program of the androgen receptor in prostate cancer cells. Oncotarget. 2017;8:30328–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell. 2007;129:823–37. [DOI] [PubMed] [Google Scholar]
- 53.Filion GJ, Van Steensel B. Reassessing the abundance of H3K9me2 chromatin domains in embryonic stem cells. Nat Genet. 2010;42:4. [DOI] [PubMed] [Google Scholar]
- 54.Wen B, Wu H, Shinkai Y, Irizarry RA, Feinberg AP. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat Genet. 2009;41:246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McConnell BB, Yang VW. Mammalian Krüppel-Like factors in health and diseases. Physiol Rev. 2010;90:1337–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liang G, Lin JCY, Wei V, Yoo C, Cheng JC, Nguyen CT, et al. Distinct localization of histone H3 acetylation and H3-K4 methylation to the transcription start sites in the human genome. Proc Natl Acad Sci U S A. 2004;101:7357–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–81. [DOI] [PubMed] [Google Scholar]
- 58.Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554:538–543. [DOI] [PubMed] [Google Scholar]
- 59.Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, et al. Tumor-Derived Granulocyte-Macrophage Colony-Stimulating Factor Regulates Myeloid Inflammation and T Cell Immunity in Pancreatic Cancer. Cancer Cell. 2012;21:822–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-Induced GM-CSF Production Promotes the Development of Pancreatic Neoplasia. Cancer Cell. 2012;21:836–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.David CJ, Huang Y-H, Chen M, Su J, Zou Y, Bardeesy N, et al. TGF-β Tumor Suppression through a Lethal EMT. Cell [Internet]. Elsevier; 2016. [cited 2020 Sep 5];164:1015–30. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26898331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet. 2017;49:1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang Y, Velez-Delgado A, Mathew E, Li D, Mendez FM, Flannagan K, et al. Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut. 2017;66:124–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bruns CJ, Solorzano CC, Harbison MT, Ozawa S, Tsan R, Fan D, et al. Blockade of the epidermal growth factor receptor signaling by a novel tyrosine kinase inhibitor leads to apoptosis of endothelial cells and therapy of human pancreatic carcinoma. Cancer Res. 2000;60:2926–35. [PubMed] [Google Scholar]
- 66.Feng PH, Yu CT, Chen KY, Luo CS, Wu SM, Liu CY, et al. S100A9+ MDSC and TAM-mediated EGFR-TKI resistance in lung adenocarcinoma: The role of RELB. Oncotarget. 2018;9:7631–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Runa F, Hamalian S, Meade K, Shisgal P, Gray PC, Kelber JA. Tumor Microenvironment Heterogeneity: Challenges and Opportunities. Curr Mol Biol Reports. 2017;3:218–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Morrison AH, Byrne KT, Vonderheide RH. Immunotherapy and Prevention of Pancreatic Cancer. Trends in Cancer. 2018;4:418–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature. 2017;547:413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Balli D, Rech AJ, Stanger BZ, Vonderheide RH. Immune cytolytic activity stratifies molecular subsets of human pancreatic cancer. Clin Cancer Res. 2017;23:3129–38. [DOI] [PubMed] [Google Scholar]
- 71.Spranger S, Luke JJ, Bao R, Zha Y, Hernandez KM, Li Y, et al. Density of immunogenic antigens does not explain the presence or absence of the T-cell-inflamed tumor microenvironment in melanoma. Proc Natl Acad Sci U S A. 2016;113:E7759–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sugiyama E, Togashi Y, Takeuchi Y, Shinya S, Tada Y, Kataoka K, et al. Blockade of EGFR improves responsiveness to PD-1 blockade in EGFR-mutated non-small cell lung cancer. Sci Immunol. 2020;5:eaav3937. [DOI] [PubMed] [Google Scholar]
- 73.Zelenay S, van der Veen AG, Böttcher JP, Snelgrove KJ, Rogers N, Acton SE, et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell [Internet]. Elsevier; 2015. [cited 2020 May 11];162:1257–70. Available from: https://www.cell.com/cell/fulltext/S0092-8674(15)01028-4?_returnURL=https%3A%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS0092867415010284%3Fshowall%3Dtrue [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gong K, Guo G, Panchani N, Bender ME, Gerber DE, Minna JD, et al. EGFR inhibition triggers an adaptive response by co-opting antiviral signaling pathways in lung cancer. Nat Cancer [Internet]. Nature Publishing Group; 2020. [cited 2020 Aug 26];1:394–409. Available from: http://www.nature.com/articles/s43018-020-0048-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jänne PA, Chih-Hsin Yang J, Kim DW, Planchard D, Ohe Y, Ramalingam SS, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med. 2015;372:1689–99. [DOI] [PubMed] [Google Scholar]
- 76.Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;12:735–42. [DOI] [PubMed] [Google Scholar]
- 77.Sequist LV, Soria JC, Goldman JW, Wakelee HA, Gadgeel SM, Varga A, et al. Rociletinib in EGFR-mutated non-small-cell lung cancer. N Engl J Med. 2015;372:1700–9. [DOI] [PubMed] [Google Scholar]
- 78.Kim IS, Gao Y, Welte T, Wang H, Liu J, Janghorban M, et al. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat Cell Biol. 2019;21:1113–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tavazoie MF, Pollack I, Tanqueco R, Ostendorf BN, Reis BS, Gonsalves FC, et al. LXR/ApoE Activation Restricts Innate Immune Suppression in Cancer. Cell. 2018;172:825–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014;15:554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV., et al. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10:1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Quinlan AR, Hall IM. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Robinson MD, McCarthy DJ, Smyth GK. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2009;26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yu G, Wang LG, He QY. ChIP seeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015;31:2382–2383. [DOI] [PubMed] [Google Scholar]
- 93.Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, et al. MEME Suite: Tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bailey TL. DREME: Motif discovery in transcription factor ChIP-seq data. Bioinformatics. 2011;27:1653–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS. Quantifying similarity between motifs. Genome Biol. 2007;8:R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sidoli S, Bhanu NV, Karch KR, Wang X, Garcia BA. Complete workflow for analysis of histone post-translational modifications using bottom-up mass spectrometry: From histone extraction to data analysis. J Vis Exp. 2016;17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Seo JH, Agarwal E, Bryant KG, Caino MC, Kim ET, Kossenkov AV, et al. Syntaphilin Ubiquitination Regulates Mitochondrial Dynamics and Tumor Cell Movements. Cancer Res [Internet]. Cancer Res; 2018. [cited 2020 Sep 4];78:4215–28. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29898993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol [Internet]. 2008. [cited 2020 Sep 4];26:1367–72. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19029910 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.