

Abstract
Recent evidence has elucidated how multipotent blood progenitors transform their identities in the thymus and undergo commitment to become T cells. Together with environmental signalling, a core group of transcription factors have essential roles in this process by directly activating and repressing specific genes. Many of these transcription factors also function again, but controlling different genes, in later T cell development. Here, we review how these transcription factors work to change the activities of specific genomic loci during early intrathymic development to establish T lineage identity. We introduce the key regulators and highlight newly emergent insights into the rules that govern their actions. Whole-genome deep sequencing-based analysis has revealed unexpectedly rich relationships between inherited epigenetic states, transcription factor-DNA binding affinity thresholds, and influences of given transcription factors on the activities of other factors in the same cells. Together, these mechanisms determine T cell identity and make the lineage choice irreversible.
Introduction
The T cell developmental programme is initiated by a prolonged iteration of environmental signals within the thymus, and by the transcription factor expression changes that are induced in a stepwise manner by those signals. Genetic and molecular biological evidence has identified many of these regulatory factors, and recent advances have shed light on the actual mechanisms that underlie their actions across the genome to result in the generation of T cell precursors. This Review introduces the main transcriptional regulators of T cell development and summarizes recent evidence for the ways in which they work. The results indicate general lessons that can be applied more broadly to immune cell development. As described below, developing T cells need to balance precursor expansion with quality-controlled differentiation. This need is met by a system in which chromatin-based epigenetic constraints and transcription factor interactions modulate the activities of the factors involved, which not only regulate the speed of differentiation but also give the factors highly stage-specific roles. Similar mechanisms might operate in other systems of immune cell development that are not as well understood yet.
This review introduces the main known transcriptional regulators that initiate T cell development. It describes work drawn mostly from the mouse, where extensive test perturbations have been carried out, which has shed light on the mechanisms of action of these factors. The review will then connect the actions of the ensemble of these factors into the two major processes that govern the pace and progression of T-lineage differentiation. Importantly, evidence shows that the factors work in highly context-dependent ways, as their activities are modulated, potentiated, and re-deployed by interactions with chromatin states and with each other. These interaction effects contribute strongly to the irreversibility of T-lineage commitment.
Overview of early T cell development
Multipotent blood progenitor cells launch the T cell programme when they migrate into the thymus and receive Notch pathway [G] signals from the thymic microenvironment. The thymus is specialized to make T cells, and its stroma provides extracellular signals to progenitor T cells, including stem cell factor (SCF, also known as Kit ligand), FLT3 ligand, IL-7 and Notch ligands1,2. T cell development begins in cells that lack the expression of both CD4 and CD8, known as double-negative (DN) cells, which subsequently acquire CD4 and CD8 expression to become double-positive (DP) cells and then differentiate into mature CD4 or CD8 single-positive (SP) cells (FIG. 1). Very few progenitor cells migrate into the thymus per day, but they proliferate extensively while initiating the T cell differentiation program; then they undergo T cell lineage commitment followed by T cell receptor (TCR) gene rearrangements at the DN and DP stages3. DP cells that successfully express a functional αβTCR are subjected to positive selection [G] and negative selection [G], then differentiate into CD4+ helper T cells or CD8+ cytotoxic T cells4. Early differentiation transitions are accompanied by extensive proliferation. This generates enough distinct TCR+ cells so that later steps in the thymus can select those with useful TCR recognition specificities and consign the rest to death. However, the expression of TCR itself depends on recombination processes that are confined to non-cycling cells5. Therefore, the T cell programme must not only equip cells with the expression of genes needed for conferring TCR activity, but also allow time for extensive proliferation to occur before the cells express TCR. Cells enter the thymus as multipotent precursors, but after several days of intrathymic proliferation they give up alternative lineage potentials, undergoing commitment, before TCR expression.
Figure 1 |.
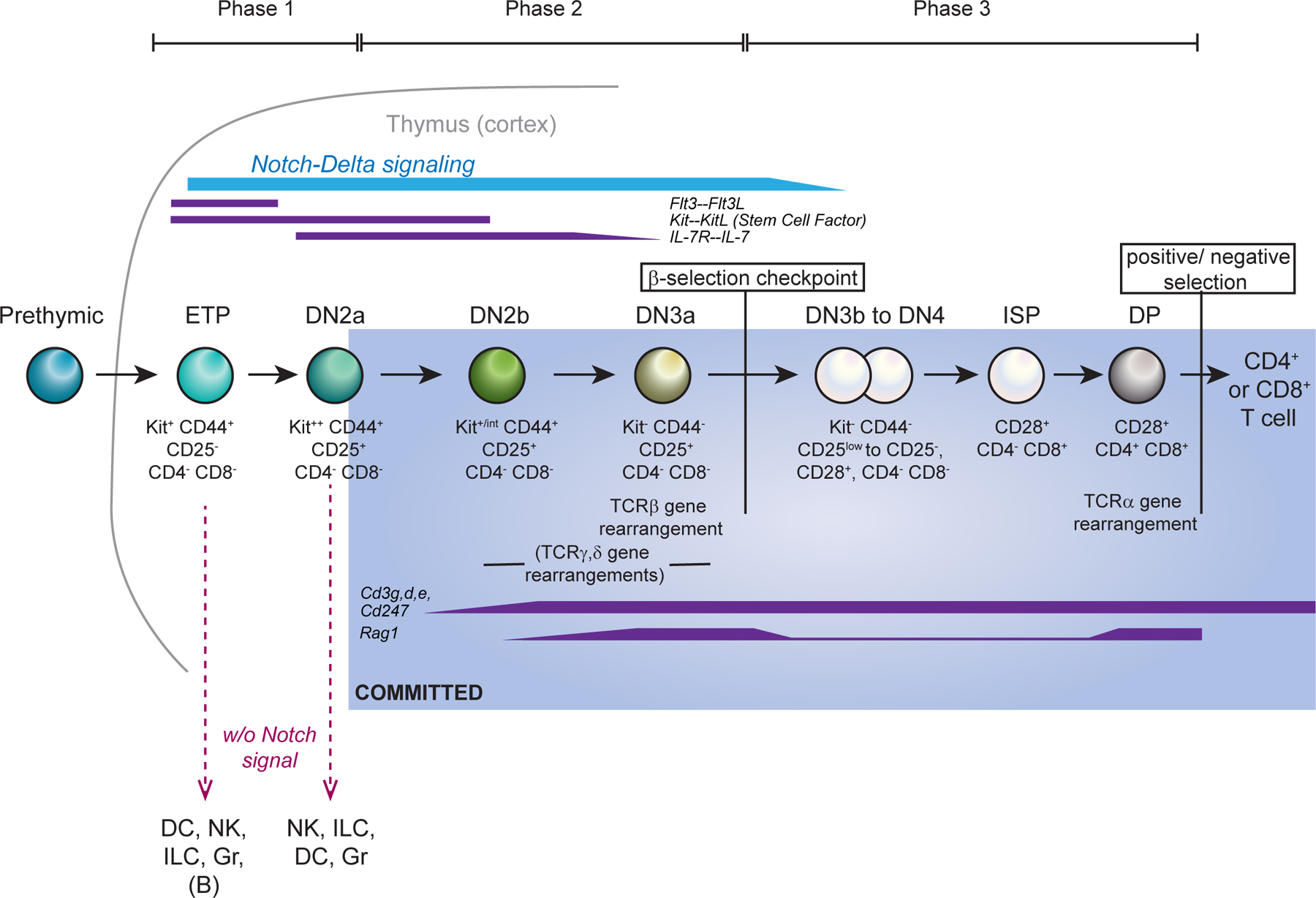
Schematic of early T cell developmental stages in mice.
Stages of development within the mouse thymus are shown up to TCRαβ-dependent positive selection, including the developmental checkpoints and major cell surface phenotype markers. Top: phases of responsiveness to indicated growth and survival signals from the environment. Middle: Approximate timing of TCR gene rearrangements is shown below the cells; “TCRβ gene rearrangement” indicates the stage when V-DJβ rearrangement can produce a complete TCRβ chain. Expression of genes encoding RAG1 and RAG2 recombinases and CD3 components is also depicted. Blue shading shows stages after commitment to the T cell lineage, as defined by loss of ability to generate non-T cells when placed in an alternative lineage-promoting environment. This coincides with expression of BCL11B at single-cell level159. Bottom: main alternative fates accessible to the indicated pre-commitment cell types if they are withdrawn from Notch signalling. The ability to generate B cells is apparently confined to the most immature ETPs; cells through DN2 stage are also reported to generate mast cells and macrophages under Notch withdrawal conditions (not depicted), and commitment timing is considerably earlier for fetal thymocytes than for postnatal thymocytes (reviewed in 2,164).
Murine DN thymocytes are divided into multiple phenotypically distinct stages defined by the expression of CD44, growth factor receptor Kit (CD117), and CD252,6,7 (FIG. 1; details given in BOX 1). All of the stages before the expression of TCR proteins are referred to as pro-T cells. To undergo conversion into definitive T lineage cells, the DN pro-T cells must activate growth factor receptor and signalling mediator genes associated with T cell function, encoding kinases such as LCK, ITK, and ZAP70, and adaptor molecules such as LAT and GADS (GRAP2), as well as the genes involved in TCR gene rearrangement, cell surface TCR complex assembly, and TCR-dependent selection. The IL-7 receptor (IL-7R) and signalling genes including those encoding the CD3 and CD247 invariant chains of the TCR complex are mostly upregulated starting in the DN2a or DN2b stages, whereas the genes associated with TCR rearrangement and expression are upregulated soon afterwards, in DN2b to DN3a stages (FIG. 1). DN stages can be separated into three phases based on known precursor–product relationships, the cells’ requirements for extracellular signals, and their commitment status2. Phase 1, including ETP (DN1) and DN2a stages, encompasses proliferation of uncommitted T cell precursors and is Notch dependent. Phase 2 includes T cell lineage-committed DN2b and DN3a stages, in which cells are more Notch dependent but proliferate less and undergo TCR gene rearrangement. Phase 3 includes DN3b and DN4 stages, through which the T lineage committed cells, proliferating in response to pre-TCR or γδ TCR signals, finally lose Notch dependency.
BOX 1 |. Early T-cell stages in the mouse.
The earliest T cell precursors in the thymus are known as early T cell progenitors (ETPs) or Kithi DN1 (Kit++CD44+CD25−) cells. At the following DN2a (Kit++CD44+CD25+) stage, expression of the T cell lineage marker CD25 is induced, but the cells can still gain access to alternative, non-T cell, fates if removed from Notch signalling. At the transition from DN2a to DN2b (Kit+CD44+CD25+) stages, marked by a decrease in Kit expression, pro-T cells become intrinsically committed to the T cell lineage. They then start preparing for Tcrb gene rearrangement, which mostly occurs at the DN3a (Kit−CD44−CD25+) stage. In parallel, some cells rearrange the TCRγ and TCRδ genes instead, which results in differentiation to the γδ T cell lineage. Successful rearrangement of the Tcrb locus encodes a TCRβ chain that can complex with an invariant surrogate α-chain and signalling partners, to form a pre-TCR. Pre-TCR assembly triggers activation to the DN3b stage (known as β-selection), strong proliferation, and progression through DN4 (Kit−CD44−CD25−) and immature SP stages to subsequent DP stages, during which the Tcra locus rearranges to generate the TCRα chain for the mature αβ TCR. CD4+CD8+ DP cells then undergo positive and negative selection based on their αβ TCR, giving rise to all later types of αβ T cell. In human pro-T cells, the cell surface markers that define these stages differ from those of mouse pro-T cells, but the commitment event to the T cell lineage is similarly marked by a specific phenotypic change: from CD34+CD7+CD1a− cells to CD34+CD7+CD1a+ cells165–167.
Transcription factors and chromatin
Two groups of transcription factors
Haematopoiesis has provided classic paradigms of lineage-determining transcription factors. Some can promote a cell identity ectopically, like C/EBPα and PU.1 for myeloid fates8–10. Some are distinctive positive regulators of cell-type specific genes that are uniquely expressed in the given lineage, like PAX5 and EBF1 (also known as COE1) for B cells11,12. But for early T cell development, no cell type-specific transcription factor set is known with ‘master-like’ activity in these senses. The factors that promote the T cell programme each individually bind the same motifs as related factors in other programmes. Nevertheless, in their combinatorial actions, overlapping with each other and with Notch signalling, these factors establish T-cell identity. These include basic-helix-loop-helix (bHLH) factors E2A and HEB, PU.1, GATA3, TCF1, BCL11b, Runx family factors, Ikaros family factors and others, all indispensable for appropriate T cell development2,6,7,13,14 (FIG. 2). Many of these factors are also expressed throughout later T cell stages and have roles in later developmental choices.
Figure 2 |.
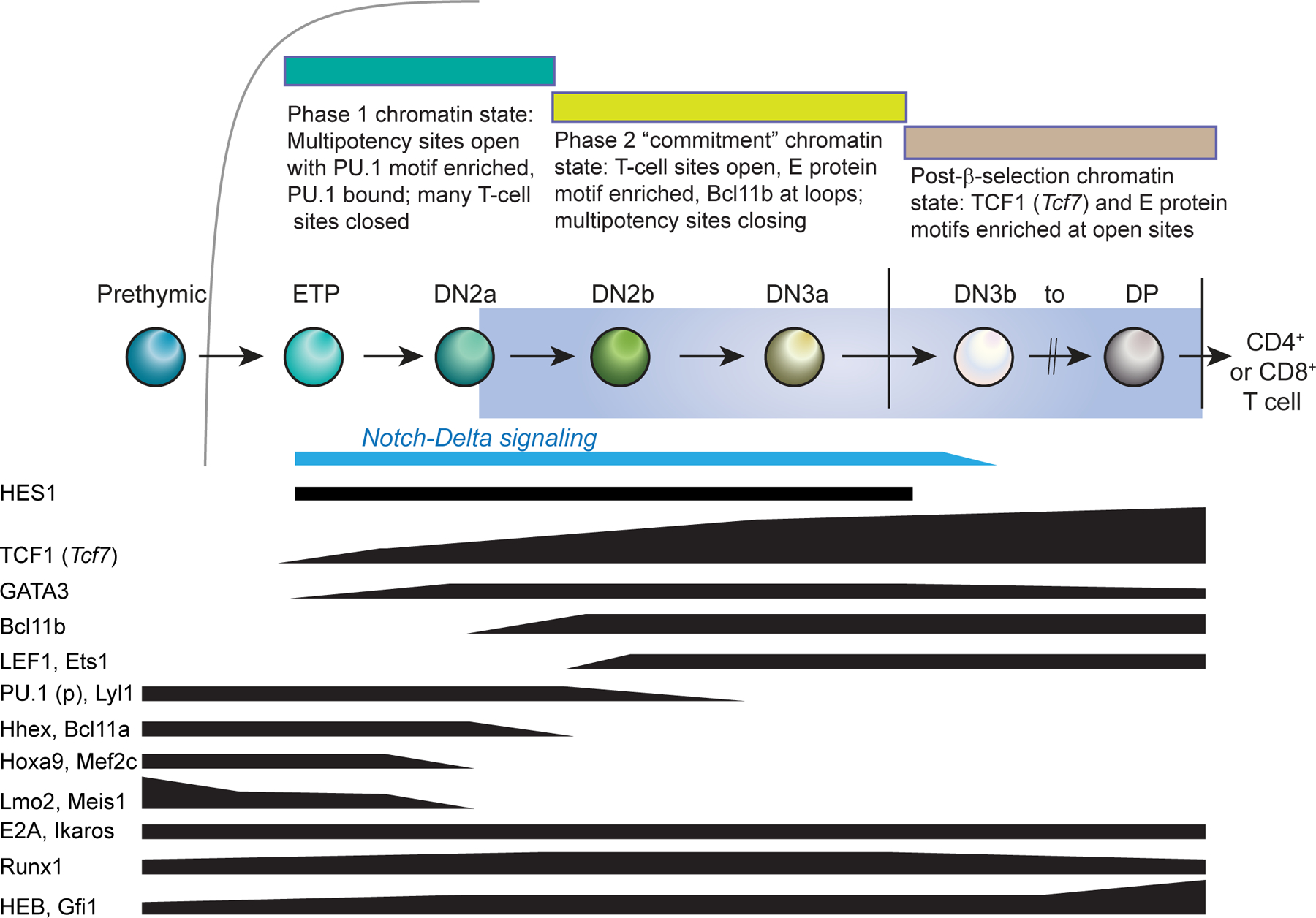
Major changes in epigenetic state and transcription factor expression in mouse pro-T cells.
The figure summarizes changes in genomic accessibility patterns and patterns of transcription factor expression based on data in REFS18,25 and REFS23,34, respectively. Indicated levels approximate a logarithmic scale. Labels indicate RNA expression except for PU.1 (p), which designates PU.1 protein. Whereas Spi1 RNA (encoding PU.1) is expressed like HHEX and BCL11A (not shown), the PU.1 protein persists longer due to its high stability16. Note the overlap in expression of progenitor transcription factors (such as LYL1 and PU.1 protein) and T cell lineage specification transcription factors (such as TCF1 and BCL11B) in late ETPs, DN2a cells and DN2b cells. This overlap extends through multiple cell cycles and has been validated at the single-cell level16.
These T-cell transcription factors are not activated on an empty regulatory background. Instead, the early pro-T cells continue to express a distinctive group of Phase 1 transcription factors that is normally associated with HSPCs in the bone marrow (FIG. 2): PU.1 (encoded by Spi1), LMO2 (also known as RBTN2), MEF2C, BCL11A, HHEX, LYL1, GFI1B, ERG, MYCN, and initially also HOXA9 and MEIS12,15,16. Forced experimental expression of LMO2 can upregulate these factors17. Furthermore, the preferred binding motif for PU.1 is the most highly enriched motif among all of the chromatin sites that are selectively open before T cell lineage commitment18–20. These results suggest that PU.1 and LMO2 actively help to maintain the precursor state. The genes encoding all of these factors are turned off around the time of commitment, albeit with individual kinetics (for example, LMO2 earlier, ERG later)2,15. However, as confirmed by single-cell transcriptome analyses in both mouse and human systems, the Phase 1 transcription factors provide the context within which the initial regulatory events specific for T cell development begin16,21,22.
Recent advances in genome-wide and multi-omics approaches and single-cell analyses have led to the discovery of key features in the molecular mechanisms by which both the T-cell associated and the progenitor-cell associated transcription factors control early T cell development. Unexpectedly stage-dependent biochemical roles of these transcription factors enable the developing cells to satisfy both the need for extensive precursor expansion and the need for coordinated differentiation once they reach the right stage.
A substantial shift in epigenome organization
The cell biological changes that are observed during T cell development, including changes in developmental potential and cell surface markers, result from sequential transformations of chromatin organization and genome activity in the cells as they pass through the pro-T cell stages. From the time that ETPs enter the thymus to the time that DP cells finish TCR gene rearrangement, the organization of the genome into topologically interacting DNA loops, the particular regions of chromatin that are accessible, and the patterns of DNA methylation and histone modification, all change markedly18,23–25. The epigenetic state in any given stage makes it easy for cells to maintain current patterns of gene expression as a default, whereas the changes in epigenetic state between stages are caused by the actions of transcription factors, probably requiring specific combinations of transcription factors acting together to trigger switches. The new chromatin states are important for biasing T cell development towards irreversible commitment because they in turn constrain future transcription factor actions, as discussed below.
Detailed analysis of genome-wide chromatin accessibility and 3D organization from haematopoietic stem cells (HSCs) to the DP thymocyte stage has shown that the greatest number of changes in chromatin site accessibility occur between DN2 and DN3 pro-T cell stages, which is the transition associated with T cell commitment18 (FIG. 2). This transition is also associated with widespread changes in histone modification and genome-wide transcriptional patterns23. In terms of chromatin accessibility (as measured by DNase-seq or ATAC-seq) and in terms of the transcription factor motifs enriched at genome-accessible sites, cells from the DN2b stage onwards cluster together with mature T cells, whereas ETPs (Kit+ DN1 cells) and DN2a cells consistently cluster with prethymic haematopoietic stem and progenitor cells (HSPCs)18,25. Thus, although early pro-T cells in Phase 1 stages proliferate in the thymus and receive intrathymic Notch signals, they do not convert to a definitive T cell lineage epigenetic state until the DN2b stage. This is because several factors in addition to Notch determine the timing of epigenetic change.
In the next section, the regulatory drivers that promote T-cell identity choice are profiled in detail. The first group are those activated early in thymus-settling progenitors, namely Notch signalling itself and the two T-cell specification transcription factors, TCF1 and GATA3. Then, stably-expressed factors that must collaborate with TCF1 and GATA3 to execute the T cell program are described: E proteins, Ikaros proteins, and Runx proteins. Finally, we introduce BCL11B, the regulatory factor most closely linked to lineage commitment in murine pro-T cells. The activities of these T-lineage programme supporting factors are then described in an ensemble as they are initially opposed by the progenitor-associated factors and then as they overcome that resistance to promote T-cell commitment.
Notch signals in T cell development
The most important intercellular signalling pathway required for T cell development is the Notch pathway, and one of the most important roles of the thymus is providing Notch ligands to the T progenitor cells. Conditional deletion of Notch1 in haematopoietic precursors, or of the Notch ligand Delta like 4 (Dll4) in thymic epithelium, leads to a complete block of T cell development accompanied by the appearance of B cells in the thymus26,27. By contrast, providing Notch signalling to fetal liver or bone marrow-derived non-T progenitor cells induces T cell development in vitro, or ectopically in vivo28,29. Notch family molecules themselves function both as transmembrane cell-surface receptors and as transcription factors30. Engagement of cell surface Notch by Notch ligands on neighboring cells triggers the proteolytic release of the intracellular domain of Notch (ICN), which undergoes nuclear translocation to become a coactivator for the DNA binding protein, RBPJ. Notch promotes pro-T cell developmental progression and survival through phases 1 and 2, and also supports viability and proliferation competence into β-selection31,32.
In early T cell development, Notch signalling helps to activate expression of genes encoding several functionally important transcription factors, including Gata3, Tcf7 (encoding TCF1) and later, Bcl11b. Well-studied mechanisms for Notch1 activity emphasize its function as a positive transcriptional regulator, through the recruitment of Mastermind-like factors to the RBPJ–ICN complex33. However, Notch pathway signalling can also trigger transcriptional repression, at least in part because Notch directly activates genes encoding transcriptional repressors from the HES family of class V helix-loop-helix (HLH) transcription factors. Hes1 is strongly activated throughout the ETP to DN3 stages of early T cell development23,34 and plays a role in T cell commitment as well as in pro-T cell survival35. Repression of Pten by Hes1 enhances the latent proliferation competence of DN3 cells, which is unleashed when they leave the pro-T cell stages via β-selection [G]36.
The dominant roles of Notch signalling in early T cell development have been extensively studied14,33; however, there are still several important questions to be answered. First, how does Notch signalling specifically induce the T cell programme in the thymus, as distinct from its instructive roles in many other cell fate decisions in a multiplicity of embryological contexts30,37? Second, recent evidence shows that future T cell precursors need to be primed by Notch signalling even before they reach the thymus38,39; what then makes intrathymic presentation of Notch ligands necessary for T cell specification? Also, as Notch1 is expressed in prethymic and intrathymic T cell precursors alike, why does Notch signalling have stage-specific activities in early T cell development that differ between Phase 1 and Phase 240,41? Notch signalling controls the expression of Il2ra (encoding CD25), Dtx1 (encoding Deltex 1), Notch3, Hes1 and Ptcra (encoding pre-TCRα) in DN cells, but many of these target genes are not activated immediately in ETPs, but only in DN2b and DN3 cells. Conversely, Nrarp, a negative feedback regulator of Notch signalling, is one of the earliest Notch-activated genes in ETPs, but its expression is shut off at the T cell lineage commitment checkpoint (between DN2 and DN3 stages)2,42. Specific mechanisms must exist to select distinct T cell lineage-specific and stage-specific Notch target genes even within the same cell lineage.
Some of the answers clearly involve the fact that Notch signals are received in the context of different combinations of other transcription factors, which are upregulated and downregulated in overlapping waves as T cell development proceeds2,16,23,25,34 (FIG. 2). At each stage of T cell development, some of the key factors that collaborate with Notch to control the target genes of that stage are encoded by genes that were themselves induced in response to the Notch signals in previous stages.
T cell specification transcription factors
In ETPs, Notch signalling activates Tcf7 (encoding TCF1) and Gata3, which are crucial regulatory genes for T cell specification2. Tcf7 and Gata3 seem to be upregulated within early ETPs, almost as early as the canonical Notch target gene Hes116. These factors are indispensable for initiating the T cell programme. Although their expression is not maximally activated until later in T cell development, knocking out either Tcf7 or Gata3 markedly decreases the survival as well as differentiation of ETPs43,44. Expression of GATA3 reaches a plateau at the end of the DN2a stage, whereas TCF1, which is already strongly expressed in DN2a cells, increases further in expression at later stages. Both GATA3 and TCF1 positively regulate T cell identity genes and contribute to the later activation of a third T cell lineage-associated regulatory gene, Bcl11b, concomitant with lineage commitment at the transition from Phase 1 to Phase 2.
TCF1
The high mobility group (HMG) box transcription factor, TCF1 (encoded by Tcf7) is a direct target gene of Notch signalling in ETPs. When thymic progenitors recognize Notch ligands from the thymic microenvironment, ICN is translocated to the nucleus and directly binds to the upstream region (–31.5kb) of the Tcf7 locus with RBPJ, as part of the mechanism that activates Tcf7 expression44,45. Interestingly, the involvement of Tcf7 in Notch signalling is stage-specific, probably due to additional regulatory inputs and TCF1 feedback itself, which may replace Notch in Phase 2 and later stages45,46.
Both loss-of-function and gain-of-function experiments identify TCF1 as a key T cell developmental regulator20,44,45. As TCF1 is also an important mediator of WNT signalling [G] in other systems, many years of debate have revolved around whether signalling through WNT pathway components is required to instruct initiation of T cell differentiation47; the balance of evidence now suggests that it does not45,48,49. Tcf7-mutant (TCF1-deficient) progenitor cells can migrate into the thymus44,45,48, but they are profoundly affected in terms of T cell development in adult mice, with survival and differentiation defects from the earliest stage of intrathymic T cell development44,45. Most molecular characterization of TCF1 to date has focused on post-commitment gene disruption phenotypes or biochemical analyses of TCF1 roles in DP cells (which constitute 85% of thymocytes); thus, the functions of TCF1 in earlier T cell development remain to be fully understood. Other complexities involve roles of TCF1 isotypes with distinct functions and the partial redundancy of TCF1 with its paralogue LEF1, which is expressed after T cell lineage commitment50,51. However, forced expression of full-length Tcf7 in prethymic progenitors is sufficient to trigger aspects of T cell development in the absence of Notch signalling, which confirms that it can activate a subset of T cell signature genes, including Gata3, Bcl11b, Il2ra, Lck, Cd3g and Rag245.
As the epigenetic state changes during early T cell development, recent evidence indicates that TCF1 functions as a pioneer-like factor52 to establish aspects of the T cell lineage-specific chromatin landscape, starting at the earliest stages of T cell development20. In a comparison of chromatin accessibility across T cell development from HSPCs to CD4+ SP T cells and CD8+ SP T cells, TCF1-binding motifs were found to be enriched at the genomic regions that are more ‘open’ in ETPs than in pre-thymic progenitors, and especially at sites that remain open in later T development. TCF1-binding motifs were also enriched at many groups of sites that are more accessible at later T cell stages. The few DP cells that emerge in Tcf7-knockout mice have defective T cell lineage chromatin landscapes and gene expression profiles20, and DP cells in which Tcf7 was acutely deleted have reduced genomic accessibility at TCF1-binding sites53. In addition, forced ectopic expression of Tcf7 in fibroblasts20 also showed a potential pioneering activity for TCF1. TCF1 seemed to be able to bind to closed chromatin, erase locally pre-existing repressive histone modifications, and generate de novo accessible chromatin at its target sites, to induce a part of the T cell lineage-specific epigenetic and transcriptome profiles in these fibroblasts20. Hence, Tcf7, which is an early direct Notch target gene in the T cell lineage programme, encodes a factor that is functionally important to initiate the T cell-specific chromatin landscape and to establish T cell lineage identity.
GATA3
GATA3 belongs the GATA family of zinc finger transcription factors and has two highly conserved type IV zinc fingers, each of which is followed by a conserved basic region. GATA3 expression is detected in a subset of HSCs and has important roles throughout T cell development in the thymus and periphery54,55. However, GATA3 is also important in multiple other tissues, ranging from the developing jaw to mammary glands, kidney and sympathetic neurons56–58. Loss-of-function mutations of Gata3 are associated with an autosomal dominant disease known as HDR syndrome that is characterized by hypoparathyroidism, deafness and renal disease59. Therefore, GATA3 has crucial dose-dependent and cell context-dependent roles in different developmental contexts.
Under physiological conditions, the very low levels of Gata3 in pre-thymic progenitor cells are upregulated by Notch signalling upon migration into the thymus. Notch signalling and GATA3 then seem to collaborate, most likely in a feed-forward relay, to exclude the B cell fate in ETP and DN2 stages60–62. The molecular mechanism through which Notch signalling induces up-regulation of Gata3 transcription is not fully understood, owing to the complexity of Gata3 regulatory elements63,64; in part, Gata3 transcription could be indirectly mediated by TCF145. Levels of GATA3 are tightly regulated during early T cell development, and both overexpression and knockout of GATA3 can be toxic in ETPs65,66. However, Gata3 expression gradually increases during Phase 1 and then regulates several checkpoints of early T cell development, including T cell lineage commitment and β-selection, at the transition from Phase 1 to Phase 2 and from Phase 2 to Phase 3, respectively2.
GATA3 has distinct roles in a succession of distinct T cell developmental choices long after its role in specifying the precursors of all T cells 55,67, and it also supports other lymphoid fates, especially that of group 2 innate lymphoid cells (ILC2s)68. In αβTCR+ thymocytes, GATA3 promotes the CD4+ fate over the CD8+ fate and subsequently promotes a T helper 2 (TH2) cell fate over TH1 cell or TH17 cell fates. GATA3 binds to substantially different genomic sites in Phase 1 pro-T cells than in later T cell developmental stages, which may help to explain its variety of roles23,69. This context dependence of GATA3 contrasts with the apparent ability of TCF1 to open many of the same ‘T cell lineage’ genomic sites in a fibroblast genome as those it binds in thymocytes. Interactions with other transcription factors could affect the genomic target preferences of GATA3 at different developmental stages (see below). Another mechanism that may contribute could be that GATA3 is functionally affected by post-translational modification, controlled by environmental signalling. In mature TH2 cells and ILC2s, GATA3 undergoes distinct post-translational modifications, including acetylation of lysine, phosphorylation of serine and threonine, and methylation of arginine, that not only enhance its expression but also control the nuclear translocation of GATA3, and organization of GATA3 complexes70–75. Thus, extracellular signalling from the different types of vascular endothelial cells and thymic epithelial cells in the thymus76 may be important to control post-translational modifications of GATA3 and thus affect its function in a stage-specific manner.
E proteins in T cell versus ILC fate
The Notch-induced factors, HES1, TCF1 and GATA3, operate in a rich context of other transcription factors already present in the prethymic precursors. Among the most important of these are E proteins. E proteins, especially E2A, are already highly expressed in lymphoid-competent precursors before the cells enter the thymus77–79. There is strong molecular evidence that they contribute to both Phase 1 and later events in T cell development80. However, their most prominent role is to activate definitive T cell lineage genes in Phase 2.
E proteins belong to the class I basic helix-loop-helix (bHLH) family of transcription factors81 and have essential roles in the generation of HSCs, lymphoid-primed multipotent progenitors (LMPPs), B cells and T cells81,82. Several E proteins arise through alternative transcriptional start site usage or differential splicing from E protein loci, including Tcf3 (E2A, encoding E12 and E47), Tcf4 (E2–2, encoding E2–2can and E2–2alt) and Tcf12 (HEB, encoding HEBcan and HEBalt). E proteins associate into homodimers or heterodimers with other (b)HLH factors and bind to a consensus E-box motif. E protein function is antagonized by class IV HLH family ‘inhibitor of DNA-binding’ (ID) factors, which lack the basic domain required for DNA binding but form stable E protein–ID heterodimers that cannot bind DNA. Among E protein dimers, the E2A homodimer and E2A–HEB heterodimer have crucial roles in early T cell development 83,84. E2A is essential not only for proper Notch1 expression in LMPP and pro-T cells, but also for promoting T cell lineage commitment cooperatively with Notch signalling85–87. In fact, the arrest of T cell development at the lineage commitment checkpoint in Tcf3-deficient thymocytes can be partially rescued by the introduction of ICN85.
E proteins and their ID family antagonists have a central role in defining the fate choice between T cells and ILCs. ID2 is a crucial regulator of ILC and NK cell development, and it is highly expressed in common helper ILC precursors88–91, as described later92. Id2 expression, either intrinsic or exogenously introduced, enables fetal thymic pro-T cells to generate NK cells93. The role of ID2 in ILCs and NK cells seems to be fully explained by its ability to suppress the DNA binding activity of E2A and HEB80,94, as either inappropriate expression of Id2 or deletion of both Tcf3 and Tcf12 genes similarly induces abnormally increased development of at least two classes of ILCs — ILC2s and lymphoid tissue inducer (LTi)-like cells — in the thymus80.
In developing T cell precursors themselves, however, the roles of E proteins remain highly dynamic through T cell development and in mature T cell function. E2A itself is expressed constantly throughout, but HEB, ID3 and other heterodimerization partners have fluctuating levels of expression, with ID3 being transiently upregulated in each response to TCR signalling95–97. E2A and HEB activities are crucial for TCR gene rearrangement98. In fetal thymocytes, which develop on an accelerated schedule, E-protein complexes assemble even in the ETP stage to establish T cell lineage chromatin landscapes around T cell signature loci, including Notch1, Rag1, Rag2, Ptcra, Cd3g, Cd3d, Cd3e and Tcrb80, probably through direct binding based on data from postnatal DN3 cells96. E2A is also important to control expression levels of the dose-dependent factor GATA3 around the T cell lineage commitment checkpoint, specifically to limit expression levels of Gata3 at the DN2 stage to enable optimal T cell lineage specification65. High E protein levels in DN3 and DP thymocytes arrest development in those stages until cells achieve successful pre-TCR or TCR signalling99. However, after successful TCRβ rearrangement, pre-TCR signalling-mediated induction of Id3 expression attenuates E protein activity, resulting in a decrease of Notch1 expression87. E protein activity increases again after β-selection and has crucial roles at the DP stage99, in close partnership with TCF153, but is again transiently neutralized during positive selection, and in maturation of CD8+ SP lineage cells100,101.
In summary, in pro-T cells, E proteins control stage-specific expression of Notch and of indispensable T cell lineage genes, modulate Notch-induced expression of GATA3, and ultimately repress intrathymic development of ILCs to establish T cell identity.
Ikaros and Runx factors in early T cells
Like Notch and E proteins, Ikaros family and Runx family transcription factors are already strongly expressed by precursors before they enter the thymus and from the ETP stage through T cell lineage commitment. Together with the zinc finger repressor GFI1102 and MYB103,104, they are extremely important for the early T cell programme105–107 despite their relatively small changes in expression level from Phase 1 to Phase 2. They are bifunctional, both repressing and activating target genes. Ikaros family factors function as tumour suppressors and to enforce developmental checkpoints108,109, specifically by making the activation and repression of different waves of regulatory genes more switch-like, not gradual, from one developmental stage to the next110. Among Runx family factors, RUNX1 is expressed very highly in DN pro-T cells and has been found to have a key role contributing to the activities of both Phase 1- and Phase 2-specific factors, as described below92,111. Individual members of the Ikaros and Runx families have different patterns of expression105,112. However, on the whole, both the Ikaros and Runx family factors have their effects reinforced and backed up by the highly overlapping expression of different family members in the same early T cells. Knockout of a single family member results in a limited phenotype, whereas the importance of these factor families emerges clearly from studies perturbing the whole family at once, such as multiple family member knockouts, knockout of the Runx-family shared cofactor CBFβ, or overexpression of dominant negatives107,113,114.
BCL11B in T cell lineage commitment
The highly T cell lineage-specific factor BCL11B is not activated immediately with TCF1 and GATA3, but seems to be expressed for the first time in late DN2a stage, marking the Phase 1 to Phase 2 transition. It then becomes essential for specific aspects of T cell lineage commitment, for all αβ T cell development, and for successful passage through β-selection, which are virtually eliminated in Bcl11b knockout mice (reviewed in 115,116), although certain fetal γδ T cell subsets in mice are less BCL11B-dependent117–119. BCL11B has roles in the brain and several other tissues as well as T cells, but in haematopoiesis it is restricted to T cell lineage cells and ILC2s 116,120. Once induced at the late DN2a stage, BCL11B expression is then sustained at some level in essentially all αβ T cell effector lineages, including NKT cells, regulatory T cells, cytotoxic T cells and peripheral effector TH cells121, where it often sets thresholds for effector responses122. In a human patient, a heterozygous missense mutation in the 2nd zinc finger of BCL11B protein (N441K, involved in DNA binding) caused severe T cell immunodeficiency as well as neurological defects. The resulting mutant BCL11B protein had a dominant negative activity and somehow blocked effective BCL11B DNA binding, despite expression of wild-type BCL11B protein from the other allele123. Targeted mutations have shown that the DNA binding and N-terminal repression domains of BCL11B are both crucial for T cell commitment, and the C-terminal zinc finger also becomes important at later stages of T cell development during the CD4+ versus CD8+ lineage choice124,125.
The functional importance of BCL11B at the T cell lineage commitment checkpoint has been demonstrated by analysis of Bcl11b-deficient mice and cells. Conditional deletion of Bcl11b in haematopoietic cells induces developmental arrest of T cells in the thymus at a distorted DN2–DN3 stage with some DN2a-like features92,118,126,127, and also induces or allows the abnormal activation of gene expression associated with ILC1s, NK cells and/or myeloid cells118,128. Bcl11b-deficient DN2a cells generated CD11c+ and/or NK1.1+ cells even in the presence of Notch signalling92,118,127,128, and Mac-1+ and/or Gr-1+ cells in the absence of Notch signals118,127. Thus, BCL11B is indispensable for appropriate lymphopoiesis in human and mouse.
Bifunctional like Ikaros and Runx factors, BCL11B also seems to have an active role in chromatin organization, as deletion of Bcl11b starting in DP thymocytes disrupted the promoter–enhancer interactions that control dichotomous Cd4 or Cd8a expression in positive selection125. A genome-wide survey of developmental changes in chromatin looping topology from HSPCs to DP cells showed that the newly interacting DNA sites after T cell lineage commitment were substantially enriched for BCL11B binding18. When Bcl11b expression was disrupted (albeit later, in naive mature CD4+ T cells), chromatin interactions and loop formations were globally reduced near BCL11B-deprived loop anchor sites18.
The mechanisms through which BCL11B regulates specific target genes in early pro-T cells include both direct, binding site-specific recruitment of chromatin-modifying proteins and indirect effects via gene network circuitry. Interestingly many binding sites occupied by BCL11B may themselves be determined by its collaboration with other factors, since they are most enriched for Ets-Runx motifs, and many of the genomic sites bound by BCL11B are also bound by RUNX192. During T cell lineage commitment, BCL11B is important both for activation and for repression of target genes92,126, as it forms complexes with both SWI/SNF [G] and nucleosome remodeling deacetylase [G] (NuRD) complexes129–131. In pro-T cells and a pro-T cell like line, BCL11B was shown by proteomic analysis to interact with the NuRD complex, Polycomb repressor complex 1 (PRC1), Rest transcriptional repressor complex and KDM1A histone demethylase complexes, as well as the transcription factor RUNX192. BCL11B seems to act directly to recruit these factors to many genomic sites, as shown by ChIP-seq analysis of control and Bcl11b-deficient cells to compare binding patterns of BCL11B partners in the presence and absence of BCL11B itself92. Overall, as is seen for many other transcription factors, BCL11B protein and its associated complexes occupied genomic sites near to both non-regulated genes and BCL11B -regulated genes alike. However, the regions near to functionally BCL11B-regulated target loci were distinguished by enrichment of specific sites where the recruitment of cofactors such as RUNX1 and NuRD complex components was dependent on BCL11B or redistributed by BCL11B92. Thus, BCL11B works at many of its functional target sites by correctly directing the nucleation of complexes with RUNX1 and chromatin-modulating complexes to establish the activation and repression of genes in a T cell lineage-specific pattern. In summary, BCL11B has essential roles to exclude T cell progenitors at the transition of Phase 1 to Phase 2 from potential access to alternative lineages, and it also functions to maintain T cell lineage-specific higher-order chromatin structure after Phase 2.
PU.1 and resistance to commitment
At least two general mechanisms slow the initial response of cells to the advent of the T cell specification regulatory factors, TCF1, GATA3, and Notch signalling. One mechanism involves the starting epigenetic state of the cells as they enter the thymus, in which substantial genomic regions around key T cell specific genes are inaccessible due to repressive marks on histones, DNA methylation, compaction of chromatin, and/or intranuclear localization18,23,24. One of the genes that is under strong cis-acting constraint by an initially closed epigenetic state is Bcl11b itself132,133, as discussed below. The other restrictive mechanism is the activity of a robust, alternative gene regulatory network operating in the T cell precursors initially, involving Phase 1 transcription factors such as PU.1. Recent single-cell transcriptome analysis using sensitive methodology has confirmed that the individual ETPs that enter the T cell programme do so initially while expressing an extensive set of Phase 1 transcription factors16. Regulatory linkages between these transcription factors show that they actively sustain a distinct subcircuit within the larger gene regulatory network of T cell specification134. The Phase 1 network opposes progression to T cell commitment and operates throughout the Phase 1 pro-T cell stages, sustaining multipotency while allowing multiple cell cycles prior to full TCR gene rearrangement. The cells need to de-activate this initial network in order to progress through commitment.
The best-studied member of the Phase 1 transcription factors in pro-T cells is PU.1. PU.1 (encoded by Spi1) is an ETS-family transcription factor with a broad range of roles in haematopoiesis. It regulates lineage specification of macrophages, granulocytes, dendritic cells and B cells. However, PU.1 is also modestly but consistently expressed in Phase 1 stages of T cell development, where it supports the proliferation of Phase 1 cells to maintain the size of the T cell progenitor pool before β-selection3,135–137. It is required to generate early T cell precursors, as Spi1-deficient HSCs fail to contribute to the T cell lineage138. Despite modest levels of Spi1 expression in Phase 1 in terms of RNA copies per cell16, the stability of PU.1 at the protein level139 enables it to have a substantial impact on the genomic activity of early pro-T cells within the thymus.
PU.1 directly regulates many target genes in the Phase 1 state, activating several that encode other Phase 1 transcription factors, such as Bcl11a, Lmo2 and Mef2c, which may themselves have roles in controlling T cell differentiation. The preferences of PU.1 binding to different sites in pro-T cells are largely based on site affinities, current PU.1 concentration, and degrees of initial site accessibility in chromatin19 (FIG. 3A,B). PU.1 can function as a pioneer-like factor, recognizing target sites in closed chromatin and recruiting other factors for lineage determination140–143. It induces chromatin accessibility at a subset of initially closed sites when it binds, which is associated with activating its target genes19,111,144. However, PU.1 binding site choices across the genome are also affected by cell type-specific binding partners145 with which PU.1 forms cell type-specific protein complexes. Such partners include C/EBPα and NF-κB in myeloid cells, and IRF4, IRF8 or E proteins in B lineage cells. In pro-T cells, ChIP-seq and proteomic analysis of PU.1-interacting molecules have shown that PU.1 forms functional protein complexes with RUNX1 and SATB192. Chromatin sites that are open specifically during the Phase 1 pro-T cell stages are highly enriched for the PU.1 recognition motif18–20, and ChIP-seq shows that PU.1 binds to tens of thousands of sites in the genomes of ETPs and DN2a cells, with the number of sites decreasing as PU.1 protein levels decrease in DN2b. Upon acute expression, PU.1 can open chromatin at its binding sites, and during T cell lineage commitment, its downregulation is associated with the closure of a large fraction of its open binding sites, contributing to the loss of expression of linked Phase 1-specific target genes19.
Figure 3 |.
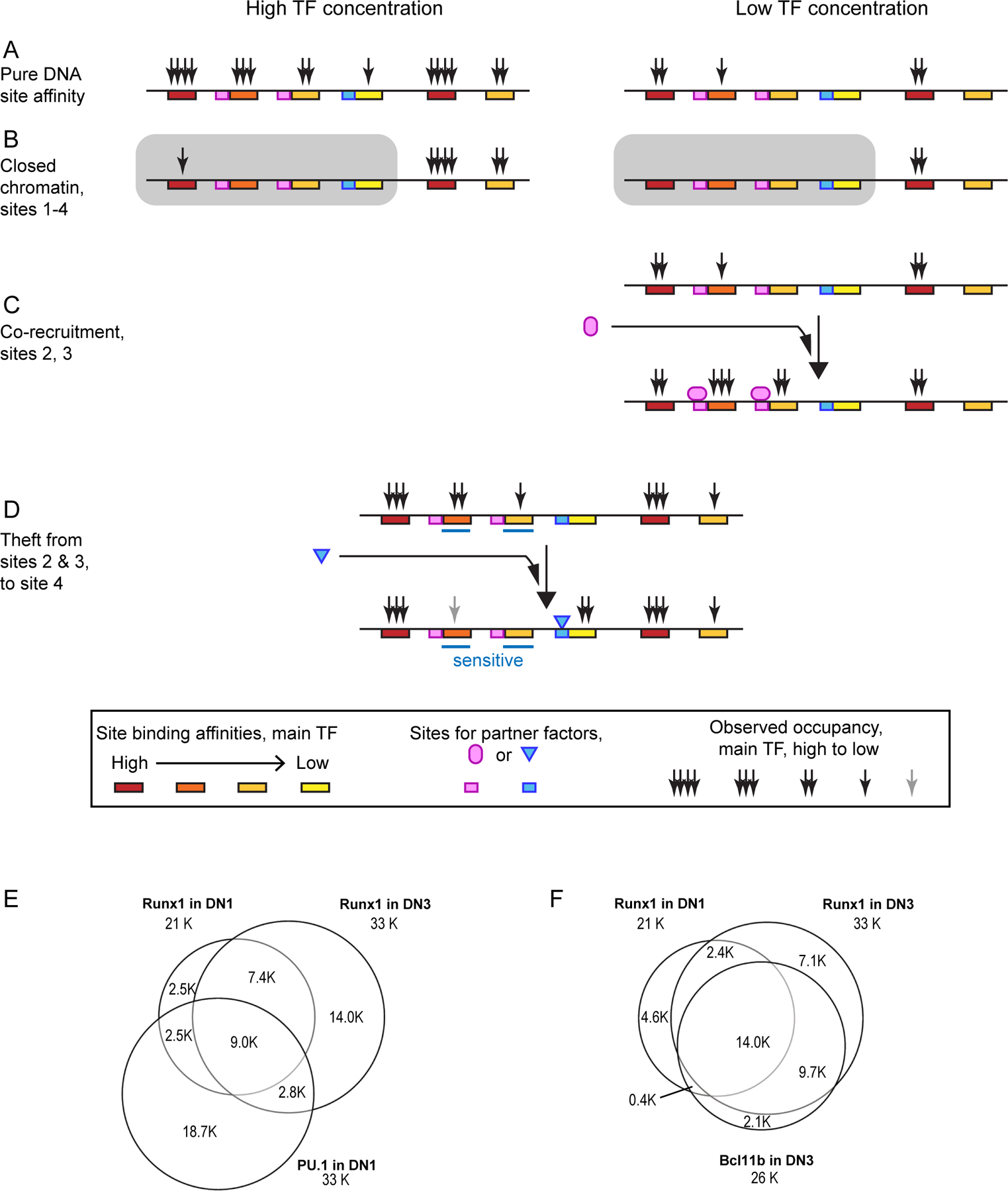
Conditionality of transcription factor binding at genomic sites.
Schematic illustrations of how the same transcription factor may differentially occupy genomic sites based on their intrinsic affinities for binding by the factor, their chromatin accessibility status, and their comparative advantage when a second transcription factor with its own binding specificities can interact with the first factor. Schematics in (A-D) are drawn from examples in REFS19,111,144. (A) Default occupancy patterns for an idealized transcription factor on six sites that it recognizes with different intrinsic affinities, at different expression levels of the transcription factor. (B) Alterations of the default occupancy pattern in a cell type where some sites are occluded by closed chromatin. This part of the figure schematizes results seen for PU.1 in pro-T cells19. Sites in closed chromatin may still be bound at high transcription factor concentrations if they have high-affinity motifs19. (C) Cooperative recruitment: the ability of a potential interaction partner (light magenta) to enhance occupancy of marginal sites by the main transcription factor by coordinated binding. (D) Cofactor “theft”: loss of binding by the main transcription factor from a subset of occupancy sites (“sensitive sites”), observed when certain partners (blue) recruit it to some alternative site(s). The same transcription factor can have either role in different contexts. (E, F) Impacts of the mechanisms described on the actual patterns of occupancy by RUNX1 before (E) and after (F) T cell lineage commitment. (E) Biased overlap of pre-commitment pattern of RUNX1 with sites occupied by PU.1 (data from REF111). (F) Extensive interaction of sites occupied by RUNX1 with BCL11B binding after commitment (data from REF92).
PU.1 is not only a strong “placeholder” for chromatin sites that are kept open during precommitment stages; it also potently influences the site choices of other transcription factors during the precommitment stage. As described in BOX 2, PU.1 can competitively recruit factors including RUNX1, SATB1 and, to some extent, GATA3, in the process depleting them from the genomic sites that they would otherwise occupy after PU.1 is downregulated111 (FIG. 3C–F). This expands the effects of PU.1 beyond the genomic sites that it binds directly.
BOX 2 |. Cofactor re-deployment.
Transcription factors work together at active enhancers to increase each other’s likelihood of occupancy, by opening chromatin, through direct protein–protein interactions, or both53,168–173. However, the recruitment of transcription factors to new sites may result in the loss of those transcription factors from other genomic sites (FIG. 3C,D), which means that stably expressed transcription factors can have stage-specific effects. For example, the bifunctional transcription factor RUNX1 associates with PU.1 and contributes to PU.1-mediated gene regulation in Phase 1; positively regulates Bcl11b expression at the late DN2a stage; and after commitment to the T cell lineage, collaborates functionally with BCL11B in both activation and repression of target genes92,111,159. Whereas Runx1 expression increases only moderately from Phase 1 to Phase 2, RUNX1 binding sites change markedly92,111 (FIG. 3E,F; FIG. 4). These binding site choices not only overlap with, but also are strongly affected by, PU.1 in Phase 1 and BCL11B and other factors in Phase 292,111.
When PU.1 is forcibly expressed in post-commitment DN3 cells, it primarily activates its own local binding targets, but also represses other genes even without obvious local DNA binding. This repression is often associated with PU.1-induced loss of RUNX1 from sites that RUNX1 was otherwise occupying111,135,174. In normal post-commitment pro-T cells, in which RUNX1 often binds with BCL11B at regulated and unregulated loci alike, the specific subset of RUNX1-binding sites that disappear or redistribute if BCL11B is removed are enriched at loci that change expression upon Bcl11b disruption92. Similar results have been reported for the deployment of heart specification factors during cardiogenesis174, for the action of Ikaros in tethering NuRD complexes to restrain leukemic transformation175, and for the effect of T-bet on GATA3 in developing human TH1 cells176. The system-level re-deployment of cofactors by lineage-determining transcription factors could contribute to many unknown phenomena ascribed to secondary or tertiary effects of transcription factors.
In Phase 1 pro-T cells within the thymus, PU.1 supports proliferation while inhibiting differentiation. Using a Cre-encoding retroviral vector or CRISPR/Cas9 systems for Phase 1-specific disruption of the Spi1 gene111,135, loss of PU.1 seemed to enable Phase 1 cells to progress to Phase 2 more rapidly than control cells, but also reduced their proliferation and survival, greatly decreasing the number of cells that were eventually recovered. Nevertheless, PU.1 expression needs to be downregulated during T cell lineage commitment. Inefficient silencing of Spi1, or abnormal expression of PU.1 or its positively regulated target genes, can cause T cell leukemia2,131,146,147. The ability of pro-T cells to avoid myeloid differentiation during PU.1-expressing stages depends on their sustained response to Notch signals46,148,149; thus it may be important for Spi1 to be silenced before the cells lose Notch responsiveness during β-selection.
In addition to PU.1, other members of the Phase 1 transcription factor network including BCL11A, LYL1 and LMO2 have important roles in T cell progenitor survival and/or expansion prior to TCR gene rearrangement17,150,151. Thus, the Phase 1 state may delay differentiation and sustain multipotency partly as a side effect of these population-sustaining activities, which are important for the ultimate yield of T cells.
How commitment is established
During commitment to the T cell lineage, pro-T cells relinquish access to at least three types of alternative regulatory states: myeloid and dendritic cells, ILCs and NK cells, or continuation as a multipotent progenitor. Evidence currently suggests that loss of access to the PU.1-dependent myeloid and dendritic cell fates is attributable to silencing of Spi1, whereas loss of access to ILC and NK cell fates is mediated by a stage-specific function of E proteins, reinforced by a gene network effect of newly expressed BCL11B.
There is good agreement between the naturally occurring downregulation of PU.1 expression and the loss of access to myeloid cell fates during T cell lineage commitment152–156. If Notch signalling is removed or attenuated from PU.1-expressing ETPs and DN2a cells, they can generate myeloid and dendritic cells; Phase 2 cells, which have naturally downregulated PU.1 expression, do not, but the reintroduction of PU.1 restores the myeloid potential of these cells46,148,149. Therefore, downregulation of PU.1 expression in Phase 1 cells is one of the crucial events that excludes myeloid and dendritic cell potential at the T cell lineage commitment checkpoint. Notch signalling itself does not repress Spi1, but GATA3, RUNX1, TCF1 and certain cis-regulatory elements around the Spi1 locus have been reported to participate in Spi1 silencing62,147,157,158. However, the molecular genetic mechanisms explaining how PU.1 expression is repressed in a stage-specific manner are still not fully clear, and downregulation of other Spi1 activators in the Phase 1 transcription factor set might also be involved.
A relatively small number of T cell lineage-associated regulatory genes are upregulated in the commitment transition. Gene expression evidence indicates that there is a change in E protein activity at this point96, despite only a small increase in the level of the E protein HEB and no increase in the level of E2A. Instead, the major regulatory genes that undergo a much sharper increase in expression from Phase 1 to Phase 2 are Bcl11b, Ets1 and Lef1. Commitment of murine pro-T cells coincides at the single-cell level with the abrupt onset of Bcl11b expression159.
Bcl11b activation is a result of the combined action of Notch signalling, TCF1, GATA3 and RUNX1159, but its activation is slowed by its trapping initially in a repressive chromatin configuration132,133 (reviewed in160). Current evidence suggests that TCF1 and GATA3 are needed specifically during the ETP stage159 to sensitize the Bcl11b locus for a slow-acting chromatin opening mechanism, which is accelerated by Notch signalling and enhanced by a far-distal enhancer132 that initiates a chromatin compartment flip18. A specifically activated long noncoding RNA from the enhancer complex region, ThymoD, is also required161. These changes open the chromatin at the Bcl11b locus, multiple loops are established from enhancer regions to the Bcl11b promoter18, and Bcl11b is finally activated. It may be down-modulated transiently after TCR signalling but remains expressed in T cells thereafter.
Once expressed, BCL11B directly represses certain genes that have constitutive functions in ILCs and NK cells but are needed only during antigen-activated effector function in T cells92. Among these BCL11B-repressed genes, Id2 and Zbtb16 (encoding PLZF) are functionally important direct targets. As discussed above, suppression of Id2 expression is crucial for T cell development, in particular to maintain E protein activity, and to avoid inappropriate development of ILCs in the thymus80. In fact, down-regulation of Id2 or Zbtb16 expression reverses part of the abnormal phenotype of Bcl11b-deficient pro-T cells92. Thus, BCL11B functions in T cell lineage commitment through three mechanisms: direct activation and repression of target genes involving several different protein complexes, large-scale chromatin organization, and repression of Id2 and Zbtb16 expression.
The global impact of commitment on gene expression is more than the advent or loss of a single key transcription factor like BCL11B or PU.1, respectively. Importantly, other transcription factors shift their deployment across the genome, even those like GATA3 and RUNX1 that have been present throughout the commitment transition. RUNX1 binds to markedly different genomic sites before and after commitment (FIG. 3E,F). Multiple changes of binding by these transcription factors occur around developmentally regulated genes, including coordinated gains of occupancy around some activated genes and losses of occupancy around some down-regulated genes (FIG. 4). This could reflect regional accessibility changes due to the global switch from a progenitor-like chromatin state to a definitive T cell lineage chromatin state. Thus, not only the direct transcriptional effects of factors such as PU.1 and BCL11B, but also indirect effects mediated by both cofactor redeployment and epigenetic accessibility changes (FIG. 3), likely contribute to regulation of specific loci (FIG. 4).
Figure 4 |.
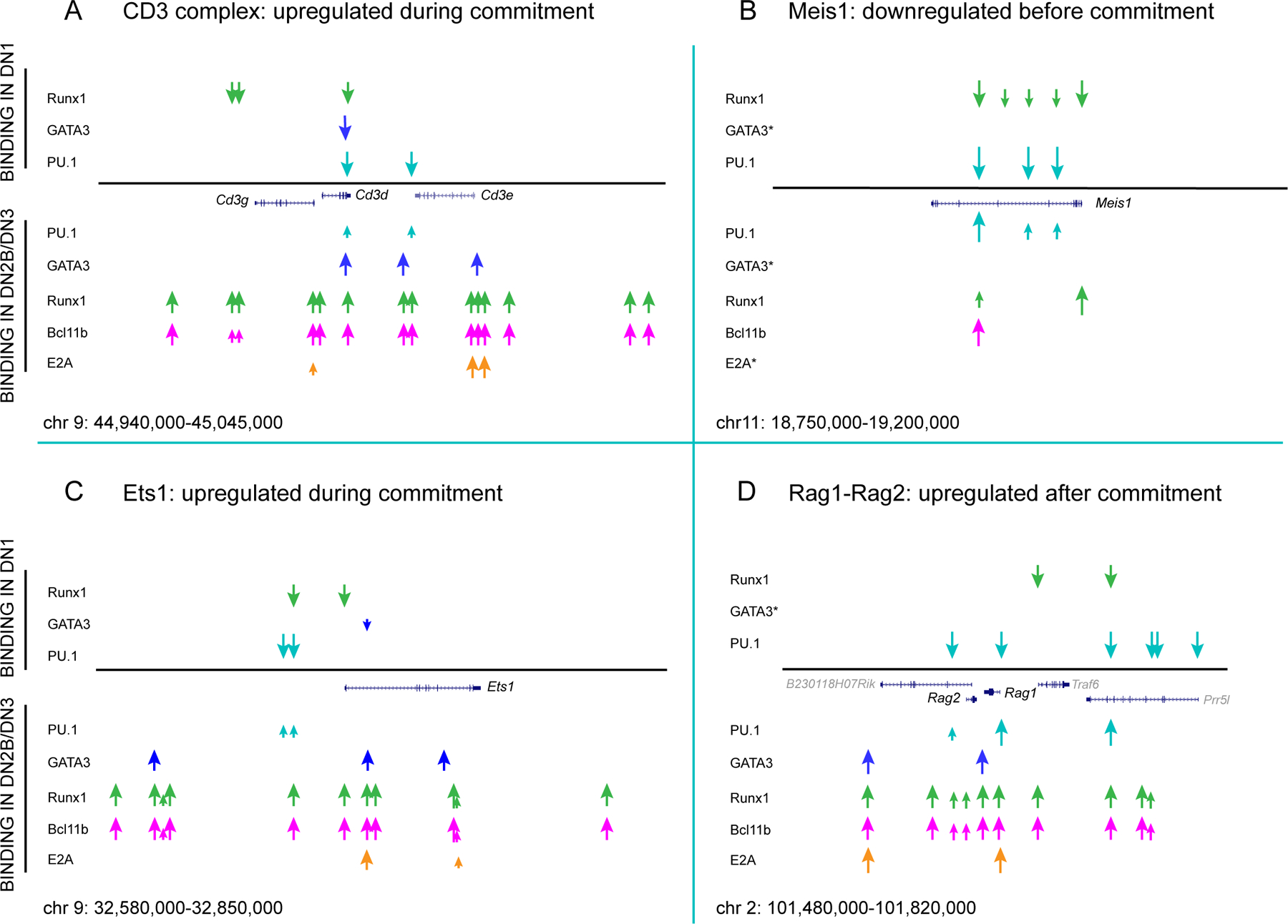
Transcription factor binding changes at key developmentally regulated loci.
Summary schematics are shown for transcription factor occupancies observed by ChIP-seq at indicated loci before T cell lineage commitment (in ETPs (DN1 cells)) and after T cell lineage commitment (in DN2b and DN3 cells), comparing PU.1 in ETP samples, BCL11B in DN2b samples, E2A in DN3 samples (Rag2-knockout thymocytes), and GATA3 and RUNX1 in both ETPs and DN2b–DN3 samples. Original data were from REFS23,92,96,111, aligned after re-mapping to the mm10 build of the mouse genome. Cd3gde cluster genes (panel A), Ets1 (panel C), and Rag1-Rag2 (panel D) are upregulated sharply from DN2a to DN2b stages, whereas the progenitor cell regulatory gene Meis1 (panel B) is downregulated before T cell lineage commitment. Genomic regions depicted are shown at the bottom of each panel in mm10 coordinates, and transcription factor binding positions are shown to scale. Smaller arrows indicate low detected occupancy of the indicated transcription factor. Note the changes in binding patterns of GATA3 and RUNX1 from pre- to post-commitment despite modest changes in expression. Bifunctional transcription factor RUNX1 undergoes single-site changes in occupancy at some loci but multi-site increases in occupancy at others, suggesting that its binding is regulated by broader genomic domain opening.
Conclusions
Notch signalling powerfully directs multipotent progenitors into the T cell pathway by activating genes encoding transcription factors that propagate a cascade of regulatory changes, both activating and repressive, to transform cell identity. The overall shifts in cellular developmental potential result from multi-step expression changes of groups of transcription factors, both activating T cell factors and repressing progenitor-specific factors. The coordinated timing of multiple changes in transcriptional regulators is responsible for T cell lineage commitment rather than the advent of a single “master regulator” during commitment. Coordinated action of these transcription factors is also likely to catalyze the epigenetic changes that transform default genomic accessibility profiles. Further, gene regulatory networks that mobilize intermediate transcription factors also contribute to the global state switches.
The sequence of regulatory changes, their relation to changes in target gene expression, and their relation to stepwise changes in developmental potential, as reviewed here, are characterized for early T cell development in a level of detail that is uncommon in mammalian systems. These changes can thus be instructive for other stem-cell based systems. One notable feature of T cell development is the long overlap, over multiple cell cycles, between expression of various transcription factors associated with multipotency or immaturity and various transcription factors “specifically” linked to T cell development (FIG. 2). The results reviewed here emphasize that the context of other transcription factors can substantially modify the way any given transcription factor is deployed genomically. Such an effect could underlie the way that E proteins, despite near-constant expression throughout pro-T cell development, become dominant regulators of signature genes that are turned on only after T cell lineage commitment. It also explains how the same factor, such as GATA3, can regulate different genes to control different developmental branch points, even within the same lineage. An intriguing possibility is that competitive transcription factor interactions could also contribute to the high dosage-dependency that is seen for many of the transcription factors in this system.
The evidence available now provides a strong vantage point for tackling the developmental decisions that need future elucidation. Key unanswered questions include the specific repressive mechanisms that determine the shutoff of progenitor genes. Also unclear is how distinct branches of T cell-related development diverge, specifically how the cells choose to adopt molecularly distinct fates. The αβ and γδ T cell lineages may begin to separate at the DN2 stage, whereas T cell and ILC precursors, which can be distinguished by levels of ID2 expression, presumably define an even earlier branchpoint. Another question is how developing T cells gain access to alternative, stereotypic, effector gene programmes, even though these programmes remain latent until different developmental milestones in αβ T cell, γδ T cell, invariant NKT cell and ILC lineages (see 160). Finally, for humans, the success of rejuvenation of T cell development in disease or aging may depend on understanding the control of progenitor expansion prior to TCR expression in the human thymus. Mouse evidence suggests that these stages could account for at least half of all the cell cycles that precursors undergo before becoming mature T cells3,162,163. Thus, transiently promoting the pre-commitment state and guiding cells through the commitment transition accurately, to stimulate precursor expansion while avoiding leukemogenesis and lineage infidelity, could become a clinical tool of the future.
Acknowledgements
We thank M. Romero-Wolf for helpful discussions and suggestions, and J. Ungerbäck, X. Wang, M. A. Yui, and present and former members of the Rothenberg group, whose helpful discussion and published and unpublished data were important for the ideas in this Review. We apologize to colleagues whose relevant work could not be cited owing to space constraints. The authors gratefully acknowledge support from the JSPS KAKENHI Grant Number JP19H03692, The Mochida Memorial Foundation for Medical and Pharmaceutical Research, The Naito Foundation, and The Takeda Science Foundation (to H.H.), from the USPHS to E.V.R. (R01AI135200, R01HL119102, R01HD100039, and R01HD076915), and from the Albert Billings Ruddock Professorship to E.V.R.
Glossary
- Notch pathway
Notch designates a cell surface receptor (Notch 1–4 family in mammals) that interacts with cell-bound ligands of the Delta (Delta-like in mammals) and Serrate (Jagged in mammals) families. Originally discovered through its potent role in fruitfly development, Notch signaling controls important embryological switchpoints for generation of various cell types in organisms of all kinds.
- Positive selection
Once immature thymocytes in the DP stage have expressed a complete TCRαβ complex, the cells are doomed to die unless that TCR can interact with cell-surface molecules on thymic epithelial cells. Positive selection is the TCR-dependent rescue of the cells from death, and the choice of helper or killer fate that results from that rescue.
- Negative selection
If the newly-expressed TCR on DP and immature single-positive thymocytes interacts too strongly with surface molecules on thymic antigen-presenting cells, the cells are induced to commit suicide rather than enabled to survive and mature. Negative selection designates this TCR stimulation-dependent suicide.
- WNT signalling
Signalling through the WNT pathway is a multi-step developmental pathway, often involved in tissue stem-cell self-renewal and in embryonic pattern formation in many organisms. In mammals, a soluble ligand from the large WNT family binds to a cell-surface receptor (FZD family), which enables a protein called β-catenin to avoid degradation in the cytoplasm and translocate to the nucleus where it becomes a coactivator for transcription factors of the TCF / LEF family.
- β-selection
β-selection is the first step of T-cell development that is dependent on a form of the TCR, in this case a special immature form of the TCR consisting of only a TCRβ-chain plus an invariant Pre-TCRα surrogate chain. This complex is generated when DN3a thymocytes successfully rearrange the genes encoding the TCRβ chain, and its assembly is required to enable the cells to proliferate and differentiate further to become DP thymocytes.
- SWI/SNF
The SWI/SNF complex is a nucleosome remodeling protein complex in eukaryotic cells that generally opens chromatin to allow greater transcription factor access. This is thought to be an important step involved in transcriptional activation of many genes.
- Nucleosome remodeling deacetylase (NuRD complex)
The NuRD complex is a nucleosome remodeling protein complex that is recruited by many transcription factors and is often involved in target gene repression. Although the histone deacetylase activity in the complex is often used for repression, the complex as a whole can be involved in a variety of transcriptional regulatory activities.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Hosokawa H & Rothenberg EV Cytokines, Transcription Factors, and the Initiation of T-Cell Development. Cold Spring Harb. Perspect. Biol 10(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yui MA & Rothenberg EV Developmental gene networks: a triathlon on the course to T cell identity. Nat. Rev. Immunol 14, 529–45 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu M et al. The earliest thymic progenitors in adults are restricted to T, NK, and dendritic cell lineage and have a potential to form more diverse TCRbeta chains than fetal progenitors. J. Immunol 175, 5848–56 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Singer A, Adoro S & Park JH Lineage fate and intense debate: myths, models and mechanisms of CD4- versus CD8-lineage choice. Nat. Rev. Immunol 8, 788–801 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Desiderio S Temporal and spatial regulatory functions of the V(D)J recombinase. Semin. Immunol 22, 362–9 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Rothenberg EV, Moore JE & Yui MA Launching the T-cell-lineage developmental programme. Nat. Rev. Immunol 8, 9–21 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang Q, Jeremiah Bell J & Bhandoola A T-cell lineage determination. Immunol. Rev 238, 12–22 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Romanoski CE, Link VM, Heinz S & Glass CK Exploiting genomics and natural genetic variation to decode macrophage enhancers. Trends Immunol 36, 507–18 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laiosa CV, Stadtfeld M & Graf T Determinants of lymphoid-myeloid lineage diversification. Annu. Rev. Immunol 24, 705–38 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Takahashi K & Yamanaka S A decade of transcription factor-mediated reprogramming to pluripotency. Nat. Rev. Mol. Cell. Biol 17, 183–93 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Schebesta A et al. Transcription factor Pax5 activates the chromatin of key genes involved in B cell signaling, adhesion, migration, and immune function. Immunity 27, 49–63 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Lin YC et al. A global network of transcription factors, involving E2A, EBF1 and Foxo1, that orchestrates B cell fate. Nat. Immunol 11, 635–43 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seo W & Taniuchi I Transcriptional regulation of early T-cell development in the thymus. Eur. J. Immunol 46, 531–8 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Thompson PK & Zúñiga-Pflücker JC On becoming a T cell, a convergence of factors kick it up a Notch along the way. Semin. Immunol 23, 350–9 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Rothenberg EV, Ungerback J & Champhekar A Forging T-Lymphocyte Identity: Intersecting Networks of Transcriptional Control. Adv. Immunol 129, 109–74 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou W et al. Single-cell analysis reveals regulatory gene expression dynamics leading to lineage commitment in early T cell development. Cell Syst 9, 321–337 e9 (2019).This study focuses on patterns of transcription factor expression in the earliest stages of T cell differentiation and their relationship to developmental potentials at the single-cell level. The authors combine single-molecule FISH and RNA-seq to gain sufficient sensitivity to overcome “drop-out problems” and measure expression of multiple transcription factor genes in the same single cells.
- 17.Cleveland SM et al. Lmo2 induces hematopoietic stem cell-like features in T-cell progenitor cells prior to leukemia. Stem Cells 31, 882–94 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu G et al. Transformation of Accessible Chromatin and 3D Nucleome Underlies Lineage Commitment of Early T Cells. Immunity 48, 227–242 e8 (2018).This study specifically notes the dominance of PU.1 motifs in the regions of chromatin that are open early, a genome-wide pattern of PU.1 occupancy of open sites giving way to Bcl11b occupancy of open sites, and evidence for a functional role for Bcl11b as a factor associated with CTCF-anchored genomic loops in T-lineage cells.
- 19.Ungerbäck J et al. Pioneering, chromatin remodeling, and epigenetic constraint in early T-cell gene regulation by SPI1 (PU.1). Genome Res 28, 1508–1519 (2018).This study uses a motif quality metric for a global dissection of how PU.1 works in open chromatin maintenance, pioneering activity, and gene regulation in early pro-T cells. It shows that PU.1 respects chromatin constraints as an “affinity penalty” in genomic site binding choice, but can bind to closed sites if they are sufficiently high affinity.
- 20.Johnson JL et al. Lineage-Determining Transcription Factor TCF-1 Initiates the Epigenetic Identity of T Cells. Immunity 48, 243–257 e10 (2018).This study establishes TCF1 as a controller of open chromatin states in T lineage cells. By global genomic analysis, the authors show high enrichment of TCF1 motifs in T-lineage identity-associated chromatin sites of two types: one set becoming accessible initially in ETPs, and a larger set becoming accessible around or after commitment.
- 21.Lavaert M et al. Integrated scRNA-Seq Identifies Human Postnatal Thymus Seeding Progenitors and Regulatory Dynamics of Differentiating Immature Thymocytes. Immunity 52, 1088–1104 e6 (2020). [DOI] [PubMed] [Google Scholar]
- 22.Le J et al. Single-Cell RNA-Seq Mapping of Human Thymopoiesis Reveals Lineage Specification Trajectories and a Commitment Spectrum in T Cell Development. Immunity 52, 1105–1118 e9 (2020).Refs. 21 and 22, Lavaert et al. and Le et al., provide complementary single-cell transcriptome analyses of pro-T cells in postnatal human thymus. Lavaert et al. focus on potential regulatory network linkages and Le et al. focus on lineage commitment. These two reports elegantly document broad similarities but some differences between early human and mouse stages.
- 23.Zhang JA, Mortazavi A, Williams BA, Wold BJ & Rothenberg EV Dynamic transformations of genome-wide epigenetic marking and transcriptional control establish T cell identity. Cell 149, 467–82 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ji H et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature 467, 338–342 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoshida H et al. The cis-Regulatory Atlas of the Mouse Immune System. Cell 176, 897–912 e20 (2019).In this comprehensive survey, the authors show that enhancer activity correlates better with gene expression than promoter accessibility, and that E protein and TCF family motifs predominate in enhancer sites becoming accessible after T-lineage commitment. In a global analysis to implicate specific TFs in chromatin accessibility change, evidence is shown that Bcl11b may act primarily as a repressor.
- 26.Radtke F et al. Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity 10, 547–58 (1999). [DOI] [PubMed] [Google Scholar]
- 27.Hozumi K et al. Delta-like 4 is indispensable in thymic environment specific for T cell development. J. Exp. Med 205, 2507–13 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmitt TM & Zúñiga-Pflücker JC Induction of T cell development from hematopoietic progenitor cells by Delta-like-1 in vitro. Immunity 17, 749–56 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Hozumi K, Abe N, Chiba S, Hirai H & Habu S Active form of Notch members can enforce T lymphopoiesis on lymphoid progenitors in the monolayer culture specific for B cell development. J. Immunol 170, 4973–9 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Bray SJ Notch signalling: a simple pathway becomes complex. Nat. Rev. Mol. Cell. Biol 7, 678–89 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Yashiro-Ohtani Y, Ohtani T & Pear WS Notch regulation of early thymocyte development. Semin Immunol 22, 261–9 (2010). [DOI] [PubMed] [Google Scholar]
- 32.Ciofani M & Zúñiga-Pflücker JC A survival guide to early T cell development. Immunol Res 34, 117–132 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Radtke F, Macdonald HR & Tacchini-Cottier F Regulation of innate and adaptive immunity by Notch. Nat. Rev. Immunol 13, 427–37 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Mingueneau M et al. The transcriptional landscape of αβ T cell differentiation. Nat. Immunol 14, 619–32 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Obaldia ME et al. T cell development requires constraint of the myeloid regulator C/EBP-α by the Notch target and transcriptional repressor Hes1. Nat. Immunol 14, 1277–84 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wong GW, Knowles GC, Mak TW, Ferrando AA & Zúñiga-Pflücker JC HES1 opposes a PTEN-dependent check on survival, differentiation, and proliferation of TCRβ-selected mouse thymocytes. Blood 120, 1439–48 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Muñoz-Descalzo S, de Navascues J & Martinez Arias A Wnt-Notch signalling: an integrated mechanism regulating transitions between cell states. Bioessays 34, 110–8 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Chen ELY, Thompson PK & Zúñiga-Pflücker JC RBPJ-dependent Notch signaling initiates the T cell program in a subset of thymus-seeding progenitors. Nat. Immunol 20, 1456–1468 (2019).The authors develop a mouse strain in which the Notch-interacting transcription factor RBPJ can be deleted and then inducibly restored at will. The results show not only the expected requirement for Notch signaling in all intrathymic stages, but also that a Notch signal via RBPJ needs to be delivered to bone marrow multipotent progenitors even before they reach the thymus.
- 39.Yu VW et al. Specific bone cells produce DLL4 to generate thymus-seeding progenitors from bone marrow. J. Exp. Med 212, 759–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hirano K et al. Delta-like 4-mediated Notch signaling is required for early T-cell development in a three-dimensional thymic structure. Eur. J. Immunol 45, 2252–62 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Wolfer A, Wilson A, Nemir M, MacDonald HR & Radtke F Inactivation of Notch1 impairs VDJβ rearrangement and allows pre-TCR-independent survival of early αβ lineage thymocytes. Immunity 16, 869–879 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Yun TJ & Bevan MJ Notch-regulated ankyrin-repeat protein inhibits Notch1 signaling: multiple Notch1 signaling pathways involved in T cell development. J. Immunol 170, 5834–41 (2003). [DOI] [PubMed] [Google Scholar]
- 43.Hosoya T et al. GATA-3 is required for early T lineage progenitor development. J. Exp. Med 206, 2987–3000 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Germar K et al. T-cell factor 1 is a gatekeeper for T-cell specification in response to Notch signaling. Proc. Natl. Acad. Sci. U S A 108, 20060–5 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weber BN et al. A critical role for TCF-1 in T-lineage specification and differentiation. Nature 476, 63–8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Del Real MM & Rothenberg EV Architecture of a lymphomyeloid developmental switch controlled by PU.1, Notch and Gata3. Development 140, 1207–19 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Staal FJT & Sen JM The canonical Wnt signaling pathway plays an important role in lymphopoiesis and hematopoiesis. Eur. J. Immunol 38, 1788–94 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu Z et al. Cutting Edge: β-Catenin-Interacting Tcf1 Isoforms Are Essential for Thymocyte Survival but Dispensable for Thymic Maturation Transitions. J. Immunol 198, 3404–3409 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jeannet G et al. Long-term, multilineage hematopoiesis occurs in the combined absence of β-catenin and γ-catenin. Blood 111, 142–149 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Tiemessen MM et al. The nuclear effector of Wnt-signaling, Tcf1, functions as a T-cell-specific tumor suppressor for development of lymphomas. PLoS Biol 10, e1001430 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu S et al. The TCF-1 and LEF-1 transcription factors have cooperative and opposing roles in T cell development and malignancy. Immunity 37, 813–26 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zaret KS & Carroll JS Pioneer transcription factors: establishing competence for gene expression. Genes Dev 25, 2227–41 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Emmanuel AO et al. TCF-1 and HEB cooperate to establish the epigenetic and transcription profiles of CD4+CD8+ thymocytes. Nat. Immunol 19, 1366–1378 (2018).The authors demonstrate the intimate cooperation between TCF1 and the bHLH factor HEB in establishing open chromatin and driving gene expression in DP stage thymocytes. TCF1 is shown to enhance the binding of HEB to a large fraction of its genomic sites and to promote HEB protein accumulation via antagonization of Notch signaling, which triggers HEB degradation.
- 54.Frelin C et al. GATA-3 regulates the self-renewal of long-term hematopoietic stem cells. Nat. Immunol 14, 1037–44 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tindemans I, Serafini N, Di Santo JP & Hendriks RW GATA-3 function in innate and adaptive immunity. Immunity 41, 191–206 (2014). [DOI] [PubMed] [Google Scholar]
- 56.Hasegawa SL et al. Dosage-dependent rescue of definitive nephrogenesis by a distant Gata3 enhancer. Dev.Biol 301, 568–577 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lim KC et al. Gata3 loss leads to embryonic lethality due to noradrenaline deficiency of the sympathetic nervous system. Nat.Genet 25, 209–212 (2000). [DOI] [PubMed] [Google Scholar]
- 58.Kouros-Mehr H, Slorach EM, Sternlicht MD & Werb Z GATA-3 maintains the differentiation of the luminal cell fate in the mammary gland. Cell 127, 1041–55 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Van Esch H et al. GATA3 haplo-insufficiency causes human HDR syndrome. Nature 406, 419–22 (2000). [DOI] [PubMed] [Google Scholar]
- 60.Hozumi K et al. Notch signaling is necessary for GATA3 function in the initiation of T cell development. Eur. J. Immunol 38, 977–85 (2008). [DOI] [PubMed] [Google Scholar]
- 61.Garcia-Ojeda ME et al. GATA-3 promotes T-cell specification by repressing B-cell potential in pro-T cells in mice. Blood 121, 1749–59 (2013). [DOI] [PubMed] [Google Scholar]
- 62.Scripture-Adams DD et al. GATA-3 dose-dependent checkpoints in early T cell commitment. J. Immunol 193, 3470–91 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hosoya T et al. Global dynamics of stage-specific transcription factor binding during thymocyte development. Sci. Rep 8, 5605 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ohmura S et al. Lineage-affiliated transcription factors bind the Gata3 Tce1 enhancer to mediate lineage-specific programs. J. Clin. Invest 126, 865–78 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xu W et al. E2A transcription factors limit expression of Gata3 to facilitate T lymphocyte lineage commitment. Blood 121, 1534–42 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Taghon T, Yui MA & Rothenberg EV Mast cell lineage diversion of T lineage precursors by the essential T cell transcription factor GATA-3. Nat. Immunol 8, 845–55 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hosoya T, Maillard I & Engel JD From the cradle to the grave: activities of GATA-3 throughout T-cell development and differentiation. Immunol. Rev 238, 110–25 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhu J GATA3 Regulates the Development and Functions of Innate Lymphoid Cell Subsets at Multiple Stages. Front. Immunol 8, 1571 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wei G et al. Genome-wide analyses of transcription factor GATA3-mediated gene regulation in distinct T cell types. Immunity 35, 299–311 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nakayama T et al. Th2 Cells in Health and Disease. Annu. Rev. Immunol 35, 53–84 (2017). [DOI] [PubMed] [Google Scholar]
- 71.Furusawa J et al. Critical role of p38 and GATA3 in natural helper cell function. J. Immunol 191, 1818–26 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hosokawa H et al. Methylation of Gata3 protein at Arg-261 regulates transactivation of the Il5 gene in T helper 2 cells. J. Biol. Chem 290, 13095–103 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hosokawa H et al. Akt1-mediated Gata3 phosphorylation controls the repression of IFNgamma in memory-type Th2 cells. Nat. Commun 7, 11289 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hosokawa H et al. Functionally distinct Gata3/Chd4 complexes coordinately establish T helper 2 (Th2) cell identity. Proc. Natl. Acad. Sci. U S A 110, 4691–6 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yamagata T et al. Acetylation of GATA-3 affects T-cell survival and homing to secondary lymphoid organs. EMBO J 19, 4676–87 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Buono M et al. A dynamic niche provides Kit ligand in a stage-specific manner to the earliest thymocyte progenitors. Nat. Cell Biol 18, 157–67 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Semerad CL, Mercer EM, Inlay MA, Weissman IL & Murre C E2A proteins maintain the hematopoietic stem cell pool and promote the maturation of myelolymphoid and myeloerythroid progenitors. Proc. Natl. Acad. Sci. U.S.A 106, 1930–1935 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang Q et al. E47 controls the developmental integrity and cell cycle quiescence of multipotential hematopoietic progenitors. J. Immunol 181, 5885–5894 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dias S, Mansson R, Gurbuxani S, Sigvardsson M & Kee BL E2A proteins promote development of lymphoid-primed multipotent progenitors. Immunity 29, 217–227 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Miyazaki M et al. The E-Id Protein Axis Specifies Adaptive Lymphoid Cell Identity and Suppresses Thymic Innate Lymphoid Cell Development. Immunity 46, 818–834 e4 (2017).ILCs lack E protein dependent recombination of antigen receptor genes, because E protein activity in ILCs is tightly inhibited by Id2. The authors demonstrate that inappropriate expression of Id2 or deletion of both Tcf3 and Tcf12 genes induces abnormal development of ILCs within the thymus. Thus, Id2 acts primarily via E protein neutralization to segregate T cells and ILCs.
- 81.Murre C Helix-loop-helix proteins and the advent of cellular diversity: 30 years of discovery. Genes Dev 33, 6–25 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Braunstein M & Anderson MK HEB in the spotlight: Transcriptional regulation of T-cell specification, commitment, and developmental plasticity. Clin. Dev. Immunol 2012, 678705 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bain G et al. E2A deficiency leads to abnormalities in alphabeta T-cell development and to rapid development of T-cell lymphomas. Mol. Cell Biol 17, 4782–91 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wojciechowski J, Lai A, Kondo M & Zhuang Y E2A and HEB are required to block thymocyte proliferation prior to pre-TCR expression. J. Immunol 178, 5717–26 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ikawa T, Kawamoto H, Goldrath AW & Murre C E proteins and Notch signaling cooperate to promote T cell lineage specification and commitment. J. Exp. Med 203, 1329–42 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pereira de Sousa A et al. Inhibitors of DNA binding proteins restrict T cell potential by repressing Notch1 expression in Flt3-negative common lymphoid progenitors. J. Immunol 189, 3822–30 (2012). [DOI] [PubMed] [Google Scholar]
- 87.Yashiro-Ohtani Y et al. Pre-TCR signaling inactivates Notch1 transcription by antagonizing E2A. Genes Dev 23, 1665–76 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zook EC & Kee BL Development of innate lymphoid cells. Nat. Immunol 17, 775–82 (2016). [DOI] [PubMed] [Google Scholar]
- 89.Moro K et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 463, 540–4 (2010). [DOI] [PubMed] [Google Scholar]
- 90.Koues OI et al. Distinct gene regulatory pathways for human innate versus adaptive lymphoid cells. Cell 165, 1134–46 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Boos MD, Yokota Y, Eberl G & Kee BL Mature natural killer cell and lymphoid tissue-inducing cell development requires Id2-mediated suppression of E protein activity. J. Exp. Med 204, 1119–1130 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hosokawa H et al. Bcl11b sets pro-T cell fate by site-specific cofactor recruitment and by repressing Id2 and Zbtb16. Nat. Immunol 19, 1427–1440 (2018).This study reveals the molecular basis of direct and indirect Bcl11b actions to promote T cell identity and to avoid acquisition of alternative lineage phenotypes in the thymus. Bcl1b forms multiple repressor complexes to regulate its direct targets. Among Bcl11b-repressed targets, Id2 and Zbtb16 (PLZF) are shown to be functionally important to control distinct alternative programs.
- 93.Ikawa T, Fujimoto S, Kawamoto H, Katsura Y & Yokota Y Commitment to natural killer cells requires the helix-loop-helix inhibitor Id2. Proc. Natl. Acad. Sci. U.S.A 98, 5164–5169 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zook EC et al. Transcription factor ID2 prevents E proteins from enforcing a naive T lymphocyte gene program during NK cell development. Sci. Immunol 3(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Braunstein M & Anderson MK HEB in the spotlight: Transcriptional regulation of T-cell specification, commitment, and developmental plasticity. Clin Dev Immunol 2012, 678–705 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Miyazaki M et al. The opposing roles of the transcription factor E2A and its antagonist Id3 that orchestrate and enforce the naive fate of T cells. Nat. Immunol 12, 992–1001 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jones ME & Zhuang Y Stage-specific functions of E-proteins at the β-selection and T-cell receptor checkpoints during thymocyte development. Immunol. Res 49, 202–15 (2011). [DOI] [PubMed] [Google Scholar]
- 98.Jones ME & Zhuang Y Regulation of V(D)J recombination by E-protein transcription factors. Adv. Exp. Med. Biol 650, 148–56 (2009). [DOI] [PubMed] [Google Scholar]
- 99.Jones ME & Zhuang Y Acquisition of a functional T cell receptor during T lymphocyte development is enforced by HEB and E2A transcription factors. Immunity 27, 860–70 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bain G et al. Regulation of the helix-loop-helix proteins, E2A and Id3, by the Ras-ERK MAPK cascade. Nat. Immunol 2, 165–171 (2001). [DOI] [PubMed] [Google Scholar]
- 101.Jones-Mason ME et al. E protein transcription factors are required for the development of CD4+ lineage T cells. Immunity 36, 348–61 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Phelan JD et al. Growth factor independent-1 maintains Notch1-dependent transcriptional programming of lymphoid precursors. PLoS Genet 9, e1003713 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Emambokus N et al. Progression through key stages of haemopoiesis is dependent on distinct threshold levels of c-Myb. EMBO J 22, 4478–4488 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Allen RD III, Bender TP & Siu G c-Myb is essential for early T cell development. Genes Dev 13, 1073–1078 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Georgopoulos K The making of a lymphocyte: the choice among disparate cell fates and the IKAROS enigma. Genes Dev 31, 439–450 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ebihara T, Seo W & Taniuchi I Roles of RUNX Complexes in Immune Cell Development. Adv. Exp. Med. Biol 962, 395–413 (2017). [DOI] [PubMed] [Google Scholar]
- 107.Guo Y, Maillard I, Chakraborti S, Rothenberg EV & Speck NA Core binding factors are necessary for natural killer cell development, and cooperate with Notch signaling during T cell specification. Blood 112, 480–492 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Geimer Le Lay AS et al. The tumor suppressor Ikaros shapes the repertoire of Notch target genes in T cells. Sci. Signal. STKE 7, ra28 (2014). [DOI] [PubMed] [Google Scholar]
- 109.Winandy S, Wu L, Wang JH & Georgopoulos K Pre-T cell receptor (TCR) and TCR-controlled checkpoints in T cell differentiation are set by Ikaros. J. Exp. Med 190, 1039–48 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Arenzana TL, Schjerven H & Smale ST Regulation of gene expression dynamics during developmental transitions by the Ikaros transcription factor. Genes Dev 29, 1801–16 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hosokawa H et al. Transcription Factor PU.1 Represses and Activates Gene Expression in Early T Cells by Redirecting Partner Transcription Factor Binding. Immunity 48, 1119–1134 e7 (2018).This study highlights an additional mechanism of transcription factor network-mediated gene regulation, without direct DNA binding. PU.1 directly activates its target genes in part by recruiting Runx1 and Satb1. However, the result is to “steal” Runx1 and Satb1 from sites linked to T-lineage related genes, thus repressing some of these genes, indirectly, without direct DNA binding.
- 112.David-Fung ES et al. Transcription factor expression dynamics of early T-lymphocyte specification and commitment. Dev. Biol 325, 444–67 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Talebian L et al. T-lymphoid, megakaryocyte, and granulocyte development are sensitive to decreases in CBFβ dosage. Blood 109, 11–21 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cortes M, Wong E, Koipally J & Georgopoulos K Control of lymphocyte development by the Ikaros gene family. Curr. Opin. Biol 11, 167–171 (1999). [DOI] [PubMed] [Google Scholar]
- 115.Liu P, Li P & Burke S Critical roles of Bcl11b in T-cell development and maintenance of T-cell identity. Immunol. Rev 238, 138–149 (2010). [DOI] [PubMed] [Google Scholar]
- 116.Kominami R Role of the transcription factor Bcl11b in development and lymphomagenesis. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci 88, 72–87 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kastner P et al. Bcl11b represses a mature T-cell gene expression program in immature CD4+CD8+ thymocytes. Eur. J. Immunol 40, 2143–54 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Li L, Leid M & Rothenberg EV An early T cell lineage commitment checkpoint dependent on the transcription factor Bcl11b. Science 329, 89–93 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shibata K et al. IFN-γ-producing and IL-17-producing γδ T cells differentiate at distinct developmental stages in murine fetal thymus. J. Immunol 192, 2210–8 (2014). [DOI] [PubMed] [Google Scholar]
- 120.De Obaldia ME & Bhandoola A Transcriptional regulation of innate and adaptive lymphocyte lineages. Annu. Rev. Immunol 33, 607–42 (2015). [DOI] [PubMed] [Google Scholar]
- 121.Avram D & Califano D The multifaceted roles of Bcl11b in thymic and peripheral T cells: impact on immune diseases. J. Immunol 193, 2059–65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fang D et al. Bcl11b, a novel GATA3-interacting protein, suppresses Th1 while limiting Th2 cell differentiation. J. Exp. Med 215, 1449–1462 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Punwani D et al. Multisystem Anomalies in Severe Combined Immunodeficiency with Mutant BCL11B. N. Engl. J. Med 375, 2165–2176 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hirose S et al. Bcl11b prevents the intrathymic development of innate CD8 T cells in a cell intrinsic manner. Int. Immunol 27, 205–15 (2015). [DOI] [PubMed] [Google Scholar]
- 125.Kojo S et al. Priming of lineage-specifying genes by Bcl11b is required for lineage choice in post-selection thymocytes. Nat. Commun 8, 702 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Longabaugh WJR et al. Bcl11b and combinatorial resolution of cell fate in the T-cell gene regulatory network. Proc. Natl. Acad. Sci. U S A 114, 5800–5807 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ikawa T et al. An essential developmental checkpoint for production of the T cell lineage. Science 329, 93–6 (2010). [DOI] [PubMed] [Google Scholar]
- 128.Li P et al. Reprogramming of T cells to natural killer-like cells upon Bcl11b deletion. Science 329, 85–89 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kadoch C & Crabtree GR Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci. Adv 1, e1500447 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Cismasiu VB et al. BCL11B functionally associates with the NuRD complex in T lymphocytes to repress targeted promoter. Oncogene 24, 6753–6764 (2005). [DOI] [PubMed] [Google Scholar]
- 131.Hosokawa H et al. Cell type-specific actions of Bcl11b in early T-lineage and group 2 innate lymphoid cells. J. Exp. Med (2019).Bcl11b is expressed in pro-T cells and ILC2 cells among hematopoietic cells. Id2 is an important repressed target gene of Bcl11b in pro-T cells, however, Id2 must be co-expressed with Bcl11b in ILC2s. This study show that, in the two lymphocyte lineages, Bcl11b regulates distinct sets of genes by organizing distinctive protein complexes and binding to different genomic regions.
- 132.Ng KK et al. A stochastic epigenetic switch controls the dynamics of T-cell lineage commitment. Elife 7(2018).This paper shows that cis-acting epigenetic constraints cause delays on the order of days in the activation of the Bcl11b gene for T-lineage commitment. The authors establish two-color Bcl11b reporter mice, where the two Bcl11b allelles are tagged with two different fluorescent proteins, to monitor activation status and enhancer dependence of Bcl11b alleles separately.
- 133.Li L et al. A far downstream enhancer for murine Bcl11b controls its T-cell specific expression. Blood 122, 902–11 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rothenberg EV Dynamic control of the T-cell specification gene regulatory network. Curr. Opin. Syst. Biol 18, 62–76 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Champhekar A et al. Regulation of early T-lineage gene expression and developmental progression by the progenitor cell transcription factor PU.1. Genes Dev 29, 832–48 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rothenberg EV, Hosokawa H & Ungerback J Mechanisms of Action of Hematopoietic Transcription Factor PU.1 in Initiation of T-Cell Development. Front. Immunol 10, 228 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kawamoto H et al. Extensive proliferation of T cell lineage-restricted progenitors in the thymus: an essential process for clonal expression of diverse T cell receptor beta chains. Eur. J. Immunol 33, 606–15 (2003). [DOI] [PubMed] [Google Scholar]
- 138.Scott EW, Simon MC, Anastasi J & Singh H Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science 265, 1573–7 (1994). [DOI] [PubMed] [Google Scholar]
- 139.Kueh HY, Champhekar A, Nutt SL, Elowitz MB & Rothenberg EV Positive feedback between PU.1 and the cell cycle controls myeloid differentiation. Science 341, 670–3 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Heinz S et al. Effect of natural genetic variation on enhancer selection and function. Nature 503, 487–92 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Heinz S et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–89 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ostuni R et al. Latent enhancers activated by stimulation in differentiated cells. Cell 152, 157–71 (2013). [DOI] [PubMed] [Google Scholar]
- 143.Escalante CR et al. Crystal structure of PU.1/IRF-4/DNA ternary complex. Mol. Cell 10, 1097–105 (2002). [DOI] [PubMed] [Google Scholar]
- 144.Minderjahn J et al. Mechanisms governing the pioneering and redistribution capabilities of the non-classical pioneer PU.1. Nat. Commun 11, 402 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Carotta S et al. The transcription factor PU.1 controls dendritic cell development and Flt3 cytokine receptor expression in a dose-dependent manner. Immunity 32, 628–641 (2010). [DOI] [PubMed] [Google Scholar]
- 146.Seki M et al. Recurrent SPI1 (PU.1) fusions in high-risk pediatric T cell acute lymphoblastic leukemia. Nat. Genet 49, 1274–1281 (2017). [DOI] [PubMed] [Google Scholar]
- 147.Rosenbauer F et al. Lymphoid cell growth and transformation are suppressed by a key regulatory element of the gene encoding PU.1. Nat. Genet 38, 27–37 (2006). [DOI] [PubMed] [Google Scholar]
- 148.Franco CB et al. Notch/Delta signaling constrains reengineering of pro-T cells by PU.1. Proc. Natl. Acad. Sci. U S A 103, 11993–8 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Laiosa CV, Stadtfeld M, Xie H, de Andres-Aguayo L & Graf T Reprogramming of committed T cell progenitors to macrophages and dendritic cells by C/EBPα and PU.1 transcription factors. Immunity 25, 731–44 (2006). [DOI] [PubMed] [Google Scholar]
- 150.Yu Y et al. Bcl11a is essential for lymphoid development and negatively regulates p53. J. Exp. Med 209, 2467–83 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zohren F et al. The transcription factor Lyl-1 regulates lymphoid specification and the maintenance of early T lineage progenitors. Nat. Immunol 13, 761–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yui MA, Feng N & Rothenberg EV Fine-scale staging of T cell lineage commitment in adult mouse thymus. J. Immunol 185, 284–93 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Masuda K et al. T cell lineage determination precedes the initiation of TCR beta gene rearrangement. J. Immunol 179, 3699–706 (2007). [DOI] [PubMed] [Google Scholar]
- 154.Bell JJ & Bhandoola A The earliest thymic progenitors for T cells possess myeloid lineage potential. Nature 452, 764–7 (2008). [DOI] [PubMed] [Google Scholar]
- 155.Wada H et al. Adult T-cell progenitors retain myeloid potential. Nature 452, 768–772 (2008). [DOI] [PubMed] [Google Scholar]
- 156.De Obaldia ME, Bell JJ & Bhandoola A Early T-cell progenitors are the major granulocyte precursors in the adult mouse thymus. Blood 121, 64–71 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Zarnegar MA, Chen J & Rothenberg EV Cell-type-specific activation and repression of PU.1 by a complex of discrete, functionally specialized cis-regulatory elements. Mol. Cell. Biol 30, 4922–39 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Huang G et al. PU.1 is a major downstream target of AML1 (RUNX1) in adult mouse hematopoiesis. Nat. Genet 40, 51–60 (2008). [DOI] [PubMed] [Google Scholar]
- 159.Kueh HY et al. Asynchronous combinatorial action of four regulatory factors activates Bcl11b for T cell commitment. Nat. Immunol 17, 956–65 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Rothenberg EV Programming for T-lymphocyte fates: modularity and mechanisms. Genes Dev 33, 1117–1135 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Isoda T et al. Non-coding Transcription Instructs Chromatin Folding and Compartmentalization to Dictate Enhancer-Promoter Communication and T Cell Fate. Cell 171, 103–119 e18 (2017).The authors focus on the distal superenhancer region of the Bcl11b gene and show that it harbors a functionally important lncRNA that acts positively on gene regulation. The authors find a severe, allele-specific loss of Bcl11b expression when lncRNA transcription is truncated, associated with disrupted looping of the superenhancer to the gene body.
- 162.Manesso E, Chickarmane V, Kueh HY, Rothenberg EV & Peterson C Computational modelling of T-cell formation kinetics: output regulated by initial proliferation-linked deferral of developmental competence. J. R. Soc. Interface 10, 20120774 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Krueger A, Ziętara N & Łyszkiewicz M T Cell Development by the Numbers. Trends Immunol 38, 128–139 (2017). [DOI] [PubMed] [Google Scholar]
- 164.Cumano A et al. New Molecular Insights into Immune Cell Development. Annu. Rev. Immunol 37, 497–519 (2019). [DOI] [PubMed] [Google Scholar]
- 165.Casero D et al. Long non-coding RNA profiling of human lymphoid progenitor cells reveals transcriptional divergence of B cell and T cell lineages. Nat. Immunol 16, 1282–91 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ha VL et al. The T-ALL related gene BCL11B regulates the initial stages of human T-cell differentiation. Leukemia 31, 2503–2514 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Canté-Barrett K et al. Loss of CD44dim Expression from Early Progenitor Cells Marks T-Cell Lineage Commitment in the Human Thymus. Front. Immunol 8, 32 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Garvie CW, Pufall MA, Graves BJ & Wolberger C Structural analysis of the autoinhibition of Ets-1 and its role in protein partnerships. J. Biol. Chem 277, 45529–36 (2002). [DOI] [PubMed] [Google Scholar]
- 169.Pufall MA & Graves BJ Ets-1 flips for new partner Pax-5. Structure 10, 11–4 (2002). [DOI] [PubMed] [Google Scholar]
- 170.Soufi A, Donahue G & Zaret KS Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell 151, 994–1004 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Iwafuchi-Doi M & Zaret KS Cell fate control by pioneer transcription factors. Development 143, 1833–7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Chronis C et al. Cooperative binding of transcription factors orchestrates reprogramming. Cell 168, 442–459 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Weikum ER, Knuesel MT, Ortlund EA & Yamamoto KR Glucocorticoid receptor control of transcription: precision and plasticity via allostery. Nat. Rev. Mol. Cell. Biol 18, 159–174 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Luna-Zurita L et al. Complex interdependence regulates heterotypic transcription factor distribution and coordinates cardiogenesis. Cell 164, 999–1014 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zhang J et al. Harnessing of the nucleosome-remodeling-deacetylase complex controls lymphocyte development and prevents leukemogenesis. Nat. Immunol 13, 86–94 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kanhere A et al. T-bet and GATA3 orchestrate Th1 and Th2 differentiation through lineage-specific targeting of distal regulatory elements. Nat. Commun 3, 1268 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]