

Abstract
The immunosuppressive effects of transforming growth factor-beta (TGF-β) promotes tumor progression and diminishes response to therapy. In this study we used ID8-p53−/− tumors as a murine model of high grade serous ovarian cancer. A monoclonal antibody targeting all three TGF-β ligands was used to neutralize TGF-β. Ascites and omentum were collected and changes in T cell response were measured using flow. Treatment with anti-TGF-β therapy every other day following injection of tumor cells resulted in decreased ascites volume (4.1 vs 0.7mL; p < 0.001) and improved CD8:Treg ratio (0.37 vs 2.5; p = 0.02) compared to untreated mice. A single dose of therapy prior to tumor challenge, resulted in a similar reduction of ascites volume (2.7 vs. 0.67mL p = 0.002) and increased CD8:Tregs ratio (0.36 vs 1.49; p = 0.007), while also significantly reducing omental weight (114.9 vs 93.4mg; p = 0.017). Beginning treatment before inoculation with tumor cells and continuing for 6 weeks, we observe similar changes and prolonged overall survival (median 70 vs 57.5 days). TGF-β neutralization results in favorable changes to the T-cell response within the tumor microenvironment leading to decreased tumor progression in ovarian cancer. The utilization of anti-TGF-β therapy may be an option for management in ovarian cancer patients to improve clinical and warrants further investigation.
Keywords: TGF-β, ovarian cancer, immune suppression, ID8 cells, tumor immunity
Background
In the United States, ovarian cancer is the 5th most common gynecologic malignancy. It is estimated that in 2019, over 22,000 women will be diagnosed with ovarian cancer, resulting in nearly 14,000 deaths [1]. Upfront therapy in ovarian cancer results in remission rates of approximately 75%; however, the vast majority of these patients will recur within 1-2 years following treatment [2,3]. Recurrence is characterized by chemoresistant disease, with limited options for curative second line therapy [4]. Current research is aimed at identifying biomarkers that are both prognostic as well as potential targets for novel therapies [5].
The transforming growth factor-beta (TGF-β) family of proteins (TGF-β 1-3) regulate a wide variety of physiologic functions including cell growth, cell differentiation, and immune responsiveness [6]. However, abnormal signaling via the TGF-β pathway has also been implicated in tumor progression. Elevated levels of TGF-β have been associated with increased angiogenesis, immunosuppression, and changes in the extracellular matrix that promote tumor implantation and growth [7,8]. In ovarian cancer specifically, increased activation of the TGF-β pathway is linked to epithelial-mesenchymal transition, activation of tumor associated fibroblasts, and increased tumor dissemination, leading to advanced disease and poorer outcomes. Published literature has identified a role for TGF-β in the seeding and dissemination of peritoneal tumors, including ovarian cancer [9-12]. Furthermore, increased expression of TGF-β receptor II (TβRII) is seen in patients with suboptimal debulking in late stage ovarian cancer [12].
A key mechanism in TGF-β promoted tumor growth is its ability to generate an immunosuppressive tumor microenvironment. TGF-β affects both innate and adaptive immunity to decrease host immune responses to tumor antigens [9]. Unfavorable changes in natural killer cells, dendritic cell activity, T-cell function, and cytokine activity occur as a response to increased TGF-β signaling within ovarian cancer [9,13-16]. Furthermore, elevated expression of intratumoral TGF-β suppresses cytotoxic T cell function through increased recruitment of regulatory T cells (Tregs) into the tumor microenvironment [14,15]. In ovarian cancer, the recruitment of Tregs occurs independently of any changes that might be present in the peripheral blood. Not surprisingly, an increased number of tumor-infiltrating Tregs is associated with increased tumor growth and decreased overall survival in ovarian cancer and other malignancies [17]. FoxP3 expression is a classic marker of Tregs and has been shown to be an independent prognostic factor in ovarian cancer, as high FoxP3 levels corelate with decreased progression free and overall survival [18]. In addition, a decrease in the ratio of CD8 T cells to Tregs has also been shown to be associated with poorer outcome in ovarian cancer [19].
The administration of anti-TGF-β biologics has been studied as a therapeutic option in a variety of malignancies [20-22]. In particular, therapy aimed at improving tumor immunity by silencing the TGF-β signaling pathway is of interest and has been combined with immune checkpoint inhibition to improve responses in colon, bladder, and head and neck cancers [23-27]. Given the documented roles of TGF-β and Tregs in promoting ovarian cancer [17], these studies suggest a potential role for anti-TGF-β therapy in ovarian cancer, as this approach may impeded tumor progression by blocking the immunosuppressive properties of TGF-β within the tumor microenvironment and restoring the ability of cytolytic T effector cells and NK cells to mediate tumor clearance.
In this study, we use a murine ovarian cancer cell line, ID8 with the p53 gene knocked out (ID8-p53−/−) which is a better model of high grade serous ovarian cancer (HGSOC) than the commonly used ID8 parental cell line [28], to study the impact of TGF-β neutralization via an anti-mouse monoclonal antibody targeting all three TGF-β ligands. We hypothesize that neutralization of TGF-β via a monoclonal antibody targeting TGF-β ligands, will result in decreased tumor burden as assessed via ascites volume and omental weights. Given the documented immunosuppressive effects of TGF-β in ovarian cancer, we also predict that TGF-β neutralization will lead to increased tumor immunogenicity, specifically increasing the number of CD8 T cells and reducing the number of Tregs.
Materials and Methods
Cell culture and reagents
ID8-p53−/− cell lines, provided by Dr. Iain McNeish (Wolfson Wohl Cancer Research Centre, Institute of Cancer Sciences, University of Glasgow, United Kingdom) were grown in Dulbecco’s Modified Eagle Medium (Thermo Fisher) with 4% FBS. Due to the fact that there are no agreed STR profiles for murine cells, we currently use best practice techniques – single cell clones, multiple cryovials, and no cryovial used for more than 6 passages. Cultures were maintained with PenStrep antibiotic for control of bacterial growth. L-glutamine and Insulin-Transferrin-Selenium were added for additional growth support. Cells used had undergone between 3 to 5 passage prior to utilization in studies. The monoclonal antibody which neutralizes all three isoforms of TGF-β, 1D11, was purchased from BioxCell [29]. Liposomal clodronate for in vivo macrophage depletion was purchased from Fisher Scientific.
CAGA12 luciferase reporter assay
Cultured ID8-p53−/− cells were plated at 2 × 104 cells/well in a 24-well plate in serum-free Opti-MEM media (Thermo Fisher). Cells were co-transfected for 24hrs with 100ng of the CAGA12-luciferase transcriptional reporter construct and 10ng of a Renilla luciferase plasmid construct (Promega). The TGF-β inducible CAGA12-luciferase reporter construct consists of a series of 12 repeats of a specific DNA sequence that has been identified as a Smad3/4 binding element in the plasminogen activator inhibitor-1 promoter region [30]. Renilla luciferase was used in each experiment to control for transfection efficiency. Cells were stimulated with 20ng of TGF-β (R&D Systems). An equivalent amount of monoclonal antibody (Anti-TGF-β 1,2,3) was added to measure its inhibitory effects in both the presence and absence of TGF-β stimulation. Twenty-four hours later, luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega).
Mice
Wild-type (WT) C57BL/6 female mice were purchased from Charles River. MISIIR mice and LysM-Cre TGFβ were kindly provided Dr. Denise Connolly (Fox Chase Cancer Center, Philadelphia, PA) and Dr. Brent Carter (University of Alabama at Birmingham, Birmingham, AL) respectively. All mice were maintained under pathogen-free specifications within animal facilities. All mice were 7-8 weeks in age when they arrived at the animal facility.
ID8-p53−/− syngeneic tumor model
Both wild type C57BL/6 and LysM-Cre TGFβ mice were injected intraperitoneally with 7 × 106 ID8-p53−/− cells suspended in 100μL of phosphate buffered sulfate (PBS) when animals were between 8-9 weeks of age. Mice were euthanized at day 7 or day 42 post tumor injection. Peritoneal cavities were inspected for tumor metastasis and the presence of ascites at referenced time points. Whole omentum were collected and weighed from each mouse. Ascites was collected with paracentesis performed at the time of sacrifice.
Bioluminescent Imaging
Wild type C57BL/6 mice were injected with ID8-p53−/− cells expressing luciferase. Seven days following tumor challenge, these mice were anesthetized; injected with D-luciferin (150mg/kg body weight); and imaged via the BioRad Lumina III at 15 minutes thereafter for luciferase activity.
Anti-TGF-β treatment
Treatment with the TGF-β monoclonal antibody was administered intraperitoneally. Each dose was 500μg of the monoclonal antibody in 100μL of buffer. Three different dosing regimens were used. For schedule #1: C57BL/6 mice were given one dose every other day starting 8 days after injection of tumor cells and completed treatment at day 42. For schedule #2: C57BL/6 mice were given a single dose 18-24 hours prior to tumor cell injection. For schedule #3: mice were treated with one dose prior to tumor challenge, then received one additional dose every other day for 42 days. MISIIR-TAg mice began receiving anti-TGF-β doses every other day for 35 days when they were 13 weeks old.
Survival study
Wild type C57BL/6 mice were injected with 7 × 106 ID8-p53−/− cells suspended in 100μL of phosphate buffered sulfate (PBS) at 8-9 weeks of age. Mice received 500μg of anti-TGF-β therapy via peritoneal injection starting the day prior to tumor cell injection and continuing every other day for 6 weeks. Mice were examined daily and individual animals were euthanized when their tumor burden limited mobility and/or prevented them from eating or consuming water.
Macrophage depletion study
Macrophage depletion was accomplished using liposomal clodronate (Encapsula NanoSciences). Briefly, liposomes encapsulating clodronate are taken up by macrophages via phagocytosis, which subsequently releases clodronate intracellularly, inducing macrophage apoptosis. To eliminate peritoneal macrophages, 100 μL of liposomal clodronate was injected intraperitoneally (i.p.) one day prior to tumor cell challenge and then again 4 days later to ensure depletion of any macrophages that were recruited following tumor challenge.
Flow cytometry
Omental tumors were harvested and mechanically homogenize in omental digestion buffer (DMEM 25Mm HEPES, 3.5 % fatty acid free BSA plus 0.5mg/ml collagenase + 70ug/ml DNAse-I; Corning Costar). Digestion was completed over 30 minutes at room temperature and fragments were filtered over nylon mesh prior to washing with staining buffer (1X PBS, 2% BCS, and 2mM EDTA), followed by erythrocyte lysis. Cells were stained with panel of antibodies targeting a variety of cell surface and intracellular markers. Data were collected using BD FACS Canto II (BD Biosciences), DIVA software 6.1.3 and analyzed with FlowJo version 9.9.6.
Statistical Analysis
Unpaired parametric Student t-test or non-parametric Mann-Whitney U tests were performed on GraphPad Prism version 7.0 to analyze the data, unless otherwise indicated in the text.
Results
ID8-p53−/− cells are responsive to TGF-β stimulation
The ID8-p53−/− cell line was evaluated for response to TGF-β stimulation using a TGF-β responsive reporter gene [30]. Cells were cultured with exogenous TGF-β in the presence or absence of anti-TGF-β monoclonal antibody. The ratio of CAGA12 to renilla luciferase activity was increased in the presence of exogenous TGF-β indicating an increase in transcription of TGF-β responsive genes (0.005 vs 0.222; P < 0.001). This effect was abrogated when cells were co-cultured with anti-TGF-β monoclonal antibody, reducing luciferase activity back to baseline (Fig. 1A).
Figure 1.

Establishment of intraperitoneal tumor model using ID8-p53−/− cells occurs within 7 days. (A) In vitro, ID8-p53−/− cell line is responsive to TGF-β stimulation, which is reduced in the presence of a TGF-β monoclonal antibody. (B) Bioluminescent imaging of mice 7 days after tumor cell injection with luciferase tagged ID8-p53−/− cells. (C) Histological comparison of normal mouse omentum (left) and mouse omentum following i.p. injection of tumor cells (right). (D) Comparison of normal mouse omentum weights to omental weights in the tumor model after 7 days. (E) Significant increase in mean expression for all three TGF-β ligand isoforms and both receptors, with trending increase in TGF-β score demonstrated via nanostring. (** p < 0.01, **** p < 0.0001, bars represent SEM)
Establishment of ID8-p53−/− tumor results in increased TGF-β protein expression within omentum
To understand the early changes that occur as ID8-p53−/− tumors grow within the omentum, ID8-p53−/− cells tagged with luciferase were injected i.p. into wildtype C57Bl/6 mice and analyzed via bioluminescent imaging (BLI) as previously described. Tumor growth within the peritoneal cavity was localized predominantly to the omentum area (Fig. 1B). To corroborate bioluminescent evaluations of tumor growth patterns, omental tissues were harvested and prepared for histology. Tumor-free omentum sections showed diffuse, preserved adipocytes, whereas in ID8-p53−/− injected mice, lymphoid and tumor cells were readily detectable in the omentum (Fig. 1C). This increase in cellularity was reflected in omental weights, which significantly increased in the 7 days following injection of tumor cells, as compared to those from tumor-free mice (Fig 1D). Omental samples from both groups were analyzed via Nanostring using both the PanCancer and Immune panels. The cumulative expression of TGF-β 1,2, and 3 ligands and TβR I and II is significantly increased 7 days post-tumor challenge compared to tumor-free mice (P < 0.0001) (Fig. 1E). These results show that 7 days following tumor challenge, ID8-p53−/− cells establish tumors within the omentum and this is accompanied by an increase in gene expression of TGF-β ligands and receptors.
Treatment with anti-TGF-β monoclonal antibody reduces ascites and improves tumor immunity
Given the role of TGF-β in this model of ovarian cancer, we asked if neutralizing TGF-β after tumor establishment would slow tumor progression in this model. To accomplish this, we treated mice with anti-TGF-β monoclonal antibody via i.p. injection. Based on previous studies [31-33], 500μg of anti-TGF-β was injected every other day beginning on day 7 after tumor injection and continued for 5 weeks (Fig. 2A). From a total of 28 mice (n = 14 each; treated and untreated), 24 produced measurable ascites that was collected; 2 animals in each arm did not have any appreciable ascites. There was a significant reduction in mean ascites volume in mice receiving anti-TGF-β therapy (Fig. 2B). The omentum represented the only evidence of visible tumor growth; no other organs were involved and the peritoneum was also negative for gross disease. Omental weights were statistically similar between untreated and treated mice (Fig. 2C).
Figure 2.

Treatment with anti-TGF-β monoclonal antibody in the ID8-p53−/− tumor model. (A) Schema of treatment dosing. (B) Ascites volume is significantly reduced following treatment with anti-TGF-β antibody. (C) Omental weights are similar between treated and untreated mice. (D-F) Favorable changes in T-cell response – (D) trending increase in proportion of CD8 T cells, (E) decrease in Tregs and (F) increased in CD8:Treg ratio, occur following treatment. (* p < 0.05, *** p < 0.001, ns - non-significant, bars represent SEM; dotted line – mean weight of naïve mouse omentum).
TGF-β is known to create an immunosuppressive environment contributing to tumor progression in a variety of malignancies, including ovarian cancer [33,34]. Specifically, it has been shown that increased TGF-β secretion within the tumor microenvironment recruits regulatory T-cells (Tregs), which ultimately results in diminished cytotoxic T-lymphocyte function, leading to increased tumor progression, poorer response to therapy, and overall worse outcomes [15,34]. We investigated the effect of anti-TGF-β therapy on tumor immunity by evaluating the proportion of CD8 T cells and Tregs within the omentum (Fig. 2D-F). Within the omentum, there was an increase in CD8 T cells and a decrease in Tregs, resulting in a significantly increased CD8 to Treg ratio within mouse omentum following treatment with anti-TGF-β therapy. Thus, although therapeutic administration of anti-TGF-β therapy did not result in inhibition of established tumor growth, significant reductions in ascites volume and improvements in anti-tumor immunity were seen, suggesting that there is potential for TGF-β targeting to induce beneficial immunological changes, even in the absence of significantly delayed tumor outgrowth.
Prophylactic treatment with anti-TGF-β antibody improves tumor immunity and decreases ascites volume and tumor burden
TGF-β production plays a crucial role in the generation of metastatic disease, particularly within the omentum [12,22]. In our transplanted tumor model, we are able to time TGF-β neutralization in relation to i.p. inoculation with tumor cells, to determine if changes in the dosing schedule differentially impacted tumor outgrowth. Therefore, we next sought to evaluate the ability of anti-TGF-β therapy prior to tumor injection to slow growth within the peritoneal cavity. A single 500μg dose of anti-mouse TGF-β was injected i.p. and 18-24 hours later, ID8-p53−/− cells were injected into the peritoneal cavity. Six weeks later, omentum were harvested and weighed to evaluate for tumor growth (Fig. 3A). Despite only receiving a single dose of neutralizing anti-TGF-β prior to tumor challenge, we witnessed a significant reduction in ascites volume (Fig. 3B) in the treatment group similar to what was demonstrated with 5 weeks of therapy administration that started on day 7 post-tumor challenge. The average ascites volume was 2.7mL in untreated mice compared to 0.67mL (P = 0.0024) in mice receiving prophylactic therapy. All of the untreated mice generated a collectable volume of ascites (minimum 0.5mL) whereas 50% (7/14) of treated mice did not produce a measurable amount of ascites. While the reduction in ascites volume was comparable to the effect seen with therapeutic administration of anti-TGF-β therapy, prophylactic administration resulted in significant omental weights differences. Omental weights were significantly lower in treated mice compared to untreated mice (84.3mg vs 124.9mg; P = 0.004) (Fig. 3C). Once again, we quantified the proportion of CD8 T cells and Tregs within the omentum via flow cytometry (Fig. 3D-F). Six weeks after tumor cell injection, there were improvements in tumor immunity in mice receiving the single prophylactic anti-TGF-β dose the day prior to tumor challenge. Treated mice omentum had an increased proportion of CD8 T cells and decreased Tregs. The mean ratio of CD8 T cells to Tregs was also significantly increased in treated mice. . These beneficial effects are sustained six weeks out from tumor exposure without any additional therapy.
Figure 3.

Inhibition prior to tumor cell injection results in reduced tumor burden as well as ascites. (A) Schema of prophylactic dosing (B) Similar decease in ascites volume is seen with anti-TGF-β antibody administered prior to tumor challenge compared to treatment over five weeks. (C) Reduction in omental weights with single dose of anti-TGF-β antibody prior to tumor cell injection. (D-F) Again, favorable changes in T-cell response (D) increased proportion of CD8 T cells, (E) decrease in Tregs and (F) increased in CD8:Treg ratio, occur with this treatment strategy. (* p < 0.05, ** p < 0.01, bars represent SEM; dotted line – mean weight of naïve mouse omentum).
Continuous treatment with anti-TGF-β antibody reduces tumor progression and increases tumor immunity and overall survival.
To explore the potential effects of TGF-β neutralization as part of a maintenance therapy regimen, we treated mice every other day with anti-TGF-β starting 18-24 hours prior to injection of ID8-p53−/− cells into the peritoneal cavity. Treatment was continued for 6 weeks, at which time, mice were evaluated for ascites production and tumor burden (Fig. 4A). As in our previous studies, we found a reduction in ascites volume with treatment (Fig. 4B). Hemorrhagic ascites fluid was produced in all untreated mice, but in only 50% (5/10) of mice receiving therapy. TGF-β neutralization throughout the duration of tumor challenge led to a decrease in the average omental weight (Fig. 4C). Analysis of T cell populations within omentum once again demonstrated a similar improvement in tumor immunity (Fig. 4D-F).
Figure 4.

Continuous therapy with anti-TGF-β monoclonal antibody starting prior to tumor cell injection and continuing for 6 weeks. (A) Maintenance therapy schema. Decrease in both (B) ascites volume and (C) omental weights. (D-F) Consistent pattern of T cell profile with treatment including (D) CD8 T cells, (E) Tregs and (F) CD8:Treg ratio. (G) Overall survival is improved (median survival of 57.5 days vs 70 days) with continuous anti-TGF-β therapy (* p < 0.05, ** p < 0.01, bars represent SEM; dotted line – mean weight of naïve mouse omentum).
Given the reduction in tumor progression that resulted from this approach, we investigated if this treatment strategy could also improve overall survival in our mouse model. Mice were treated for six weeks with every other day dosing of i.p. anti-TGF-β monoclonal antibody beginning the day prior to introducing ID8-p53−/− tumor cells to the peritoneal cavity. Following completion of therapy, mice were monitored for deteriorating condition. At the time when mice were sacrificed, they each had developed large volume ascites and were moribund due to immobility and poor intake. Median overall survival for treated mice was 70 days compared to 57.5 days in untreated mice (Fig. 4G). Neutralizing TGF-β activity prior to tumor challenge followed by continued treatment results in significant reduction of both tumor size (P = 0.004) and ascites volume (P = 0.0025), while improving anti-tumor immunity. These results indicate that anti-TGF-β as maintenance therapy resulted in decreased tumor burden which leads to prolonged overall survival in our mouse model.
TGF-β neutralization reduces metastatic disease in MISIIR mice
To evaluate the effect of TGF-β neutralization on the development of ovarian cancer, we used MISIIR-TAg mice which spontaneously develop palpable abdomino-pelvic ovarian tumors by 13 weeks of age and show full tumor growth and metastasis at 20 weeks of age [35]. We therefore initiated anti-TGF-β therapy at 13 weeks, coincident with early signs of tumor growth, administering 500μg of anti-TGF-β monoclonal antibody via intraperitoneal injection every other day for five weeks. At 18 weeks of age, animals were euthanized and ovarian tumors and omentum were collected and weighed (Fig. 5A). All mice were alive at 18 weeks with large bilateral ovarian tumors. No ascites was noted at the time of sacrifice, consistent with previously published literature for this mouse model [35]. In both groups, ovarian masses were noted in bilateral ovaries (Fig. 5B). Large hemorrhagic and necrotic components were also observed in these tumors. No other peritoneal disease was seen. Enlarged bilateral ovaries were readily identified and removed for weighing. The total tumor weight for each mouse was recorded as the sum of both ovaries. Mean primary tumor weights were similar between treated and untreated mice (Fig. 5C). The omentum is the most frequent location of metastatic disease in this model [35]. When comparing omental weights (Fig. 5D), we noted a significant reduction in mice treated with anti-TGF-β. These data show that a therapy based on TGF-β neutralization results in decreased metastatic tumor burden in a mouse model with spontaneously generated ovarian cancer, but does not impact primary tumor growth.
Figure 5.
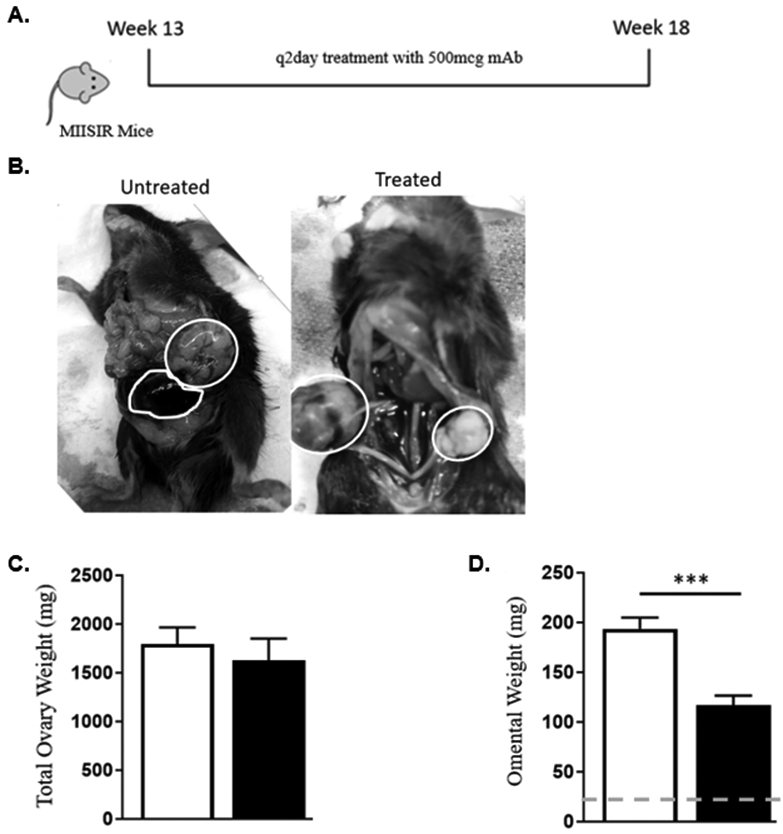
Treatment with anti-TGF-β monoclonal antibody in a spontaneously generating ovarian cancer mouse model. (A) Treatment schema for MISIIR mice model (B) Representative photos of bilateral ovarian tumors (white circles) in both treated and untreated mice. (C) Comparison of primary tumor site weights, sum of weights of bilateral ovaries. (D) Omental weights (*** p < 0.01, bars represent SEM; dotted line – mean weight of naïve mouse omentum).
Loss of macrophage-derived TGF-β results in reduction of tumor burden
Various cell populations are potential sources of TGF-β within the tumor microenvironment [36]. Macrophages are a crucial source of TGF-β and play an integral role in the establishment of tumors within the omentum [37]. Our nanostring data indicated a trending increase in the macrophage score in mice following 7 days of tumor challenge compared to tumor-free mice (4.82 vs 2.91; P = 0.23) (Fig. 6A). This is in agreement with other studies which have noted the contribution of macrophages in promoting peritoneal metastasis, specifically in ovarian cancer [38,39]. To evaluate the impact of macrophages on tumor growth during this 7-day period, we performed macrophage depletion via liposomal clodronate. To achieve this, we injected 500 μg of clodronate liposomes one day prior to tumor cell challenge then again 4 days later to reduce possible macrophage recruitment. Omental weights with reduced macrophages were significantly smaller compared to omentum following one week of tumor challenge, but increased in size compared to tumor naïve mice (Fig. 6B). We identified, via flow cytometry, that the total omental macrophages, (defined as CD11b+Ly6C-CD11c- cells), and omental tumor associated macrophages (TAMs) (defined as CD11b+Ly6C-CD11c-CD68+F4/80+ cells), were reduced after clodronate treatment. Monocytic myeloid derived suppressor cells (MDSCs), defined as CD11c-Ly6G-CD11b+Ly6C+, have been shown to play an integral role as drivers of immunosuppression in ovarian cancer [40]. This was consistent in our model after 7 days of tumor challenge, with a significantly higher percentage of MDSCs compared to tumor-free omentum. This cell population was unaffected by liposomal macrophage depletion and was similarly elevated (Fig. 6C).
Figure 6.
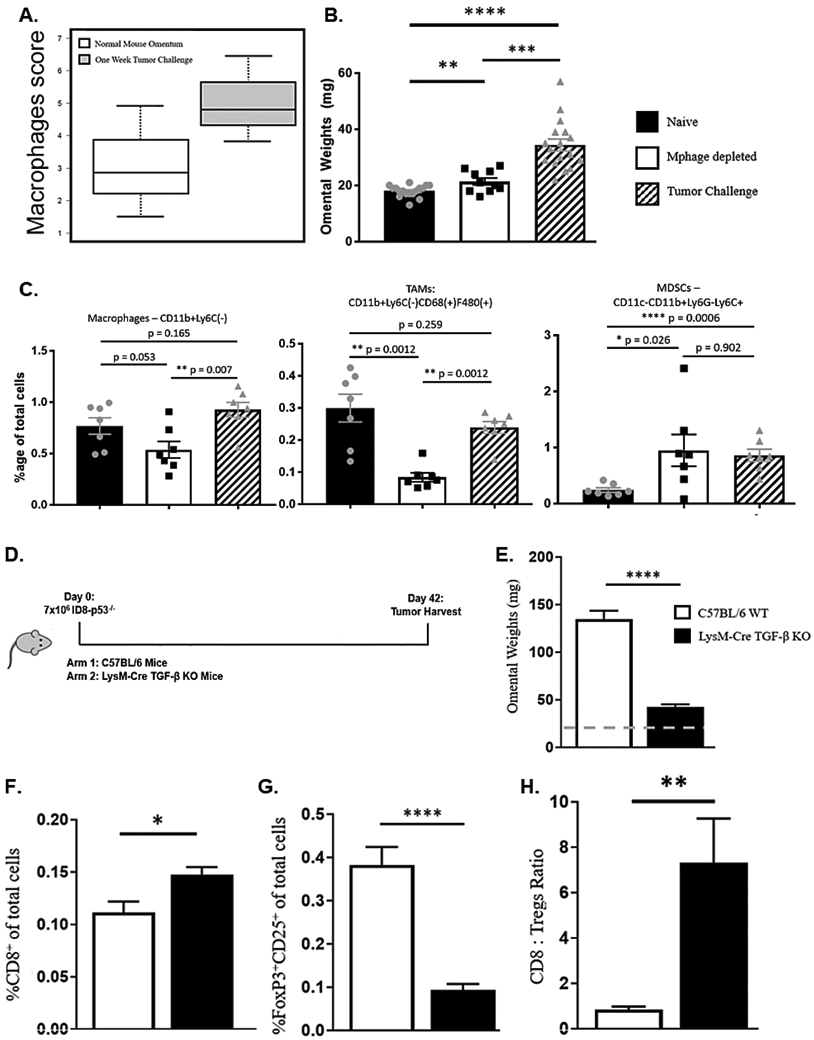
Constitutive loss of TGF-β production from macrophages results in significant decrease of tumor burden. (A) Macrophage score following one-week tumor challenge compared to tumor naïve omentum (2.91 vs 4.82; P = 0.23) (B) Omental weights of tumor-free mice compared to mice following 7 days of tumor challenge with and without depletion of macrophage cell population. (C) Macrophage subset populations within omentum following 7 days of tumor challenge with and without macrophage depletion. (D) Experimental schema for LysM-Cre TGF-β experiment. (E) Omental weight comparison. (F) CD8 T cells (G) Tregs (H) CD8:Treg ratio. (* p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, bars represent SEM; dotted line – mean weight of naïve mouse omentum).
To evaluate long term effects on tumor progression with the loss of TGF-β from the macrophages specifically, we injected ID8-p53−/− cells into wild type or in LysM-Cre TGF-β knockout C57BL/6 mice. The latter have a constitutive loss of TGF-β production specifically in macrophages. Mice were observed for 6 weeks at which time, omentum were collected and evaluated for weight and T cell composition (Fig. 6D). There was a significant reduction in omental weights in the LysM-Cre TGF-β knockout mice compare to the WT (Fig. 6E). This was accompanied by a significant increase in CD8 T cells and a decrease in the number of Tregs (Fig. 6F-G), which resulted in an increased CD8 to Tregs ratio (Fig. 6H). The lack of TGF-β production from macrophages, resulted in the largest reduction in mean omental weights (162.1mg vs. 42.1mg; P < 0.0001) and this was accompanied by favorable changes in the T cell population profile within this tumor model. These findings point to the role of macrophage-specific TGF-β production as a key contributor to promoting ovarian tumor growth and decreasing anti-tumor immunity in the ID8-p53−/− model.
Discussion
Elevated production of TGF-β has been identified as a marker of advanced stage malignancy and overall poorer prognosis in a variety of malignancies including ovarian cancer [6,41-43]. Increased signaling via the TGF-β pathway promotes angiogenesis, immunosuppression, and epithelial to mesenchymal transition, all of which contribute to tumor dissemination [7,8]. Given the documented role for TGF-β in tumor progression, it represents a potential target for therapeutic intervention.
In this study, we sought to investigate the effect of TGF-β neutralization in multiple, complementary ovarian cancer mouse models. TGF-β neutralization was accomplished via a monoclonal antibody targeting all three ligands of TGF-β. In the ID8-p53−/− model, anti-TGF-β therapy consistently reduced ascites volume, whether it was administered therapeutically (every other day starting on tumor challenge day 7) or prophylactically (18-24 hours prior to tumor cell injection). In ovarian cancer, ascites fluid allows for efficient exchange of inflammatory and mesothelial cells leading to tumor growth and invasion. In a study by Liao et al, TGF-β blockade was shown to reduce ascites volume via inhibition of vascular endothelial growth factor (VEGF) production and changes to the lymphatics that block ascites outflow and absorption [11]. Similarly, we found that ascites volume was reduced with continuous anti-TGF-β therapy following the establishment of omental tumors. Also, a single dose given just prior to i.p. injection of tumor cells results in reduces ascites volume sustained for 6 weeks following tumor challenge.
Intraperitoneal dissemination of ovarian cancer utilizes the mechanical flow of peritoneal fluid and has a preference for seeding and growth within the omentum [44-46]. Our data suggests that the timing of anti-TGF-β therapy is important with regards to cancer seeding and metastasizing. TGF-β activity plays an important role in the initial phases of tumor formation secondary to its immunosuppressive effects within the tumor microenvironment [22].
The presence of tumor infiltrating lymphocytes has been correlated with improved outcomes in ovarian cancer [47]. Whereas increased numbers of Tregs is associated with poorer outcomes and advanced stage disease through their suppressive effects on cytotoxic T cells. It has also been shown that TGF-β is critical in the induction of CD4+ T cells into T regulatory cells [14]. In our study we showed that anti-TGF-β therapy significantly improved the ratio of CD8 T cells to Tregs. This potentially points to utilizing anti-TGF-β therapy to improve responses to immunotherapy, which has had limited response in ovarian cancer patients [48]. This concept was demonstrated in preclinical models using M7824, a bifunctional fusion protein targeting both PD-L1 and TβRII. This therapy improved anti-tumor response in mouse models studying breast cancer and potentiated the effects of both chemotherapy and radiation [49].
Alterations to the innate immune system also contribute to immunosuppression in ovarian cancer. A synergistic interaction between immunosuppressive M2 macrophages and Tregs has been demonstrated and this is dependent on the macrophage release of TGF-β [50]. The loss of TGF-β from macrophages resulted in the most profound reduction in tumor burden in our ID8-p53−/− model, which further supports the claim that macrophages are crucial in the establishment ovarian tumors and more specifically, contribute to tumor seeding via TGF-β production.
Our study points to an important window of time as the tumor initially begins to grow where TGF-β production is key. Our data indicates that anti-TGF-β therapy is effective in preventing the establishment of tumors whether via tumor metastasis as seen in treatment of MISIIR mice or treatment prior to direct peritoneal injection of tumor cells. We show that neutralization of TGF-β as tumor cells are implanting within the peritoneum results in reduced tumor burden. Clinically, this is the goal of maintenance therapy in ovarian cancer.
In ovarian cancer, TGF-β plays an important role in creating an immunosuppressive tumor microenvironment leading to tumor progression. Our results indicate that neutralization of TGF-β is a potential targeted therapy that can improve outcomes in the management of ovarian cancer patients. Specifically, targeting the initial effects of TGF-β in establishing the tumor microenvironment results in decreased tumor dissemination and metastasis of disease. This directly correlates clinically with the strategy employed in maintenance therapy in the treatment of ovarian cancer. Clinical trials evaluating the efficacy of anti-TGF-β therapy are currently underway in a variety of malignancies including ovarian cancer. These results highlight areas for additional preclinical and translational research utilizing anti-TGF-β therapy in ovarian cancer patients.
Acknowledgments
Financial Support: This project was funded under the Norma Livingston Ovarian Cancer Foundation and the National Comprehensive Cancer Network (NCCN) Young Investigator Award.
Footnotes
Conflict of Interest: Rebecca Arend: Advisory board for Tesaro/GSK, Clovis, AZ, Merck
Research Support: VBL Therapeutics, LEAP
References
- 1.Society, A.C. Cancer Facts & Figures 2019. Availabe online: (accessed on May 27). [Google Scholar]
- 2.Barakat R; Berchuck A; Markman M; Randall ME Principles and Practice of Gynecologic Oncology, 6th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, 2013. [Google Scholar]
- 3.Heintz A; Odicino F; Maisonneuve P; Quinn M; Benedet J; Creasman W; Ngan H; Pecorelli S; Beller U Carcinoma of the Ovary. 2006, 95, S161–S192, doi:doi: 10.1016/S0020-7292(06)60033-7. [DOI] [PubMed] [Google Scholar]
- 4.Bast RC Jr.; Hennessy B; Mills GB The biology of ovarian cancer: new opportunities for translation. Nature reviews. Cancer 2009, 9, 415–428, doi: 10.1038/nrc2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanahan D; Weinberg RA The Hallmarks of Cancer. Cell 2000, 100, 57–70, doi: 10.1016/S0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 6.Bristow RE; Baldwin RL; Yamada SD; Korc M; Karlan BY Altered expression of transforming growth factor-beta ligands and receptors in primary and recurrent ovarian carcinoma. Cancer 1999, 85, 658–668. [DOI] [PubMed] [Google Scholar]
- 7.Massagué J; Cheifetz S; Laiho M; Ralph DA; Weis FM; Zentella A Transforming growth factor-beta. Cancer Surv 1992, 12, 81–103. [PubMed] [Google Scholar]
- 8.Sporn MB; Roberts AB Transforming growth factor-beta: recent progress and new challenges. J Cell Biol 1992, 119, 1017–1021, doi: 10.1083/jcb.119.5.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yigit R; Massuger LF; Figdor CG; Torensma R Ovarian cancer creates a suppressive microenvironment to escape immune elimination. Gynecologic oncology 2010, 117, 366–372, doi: 10.1016/j.ygyno.2010.01.019. [DOI] [PubMed] [Google Scholar]
- 10.Nakanishi Y; Kodama J; Yoshinouchi M; Tokumo K; Kamimura S; Okuda H; Kudo T The expression of vascular endothelial growth factor and transforming growth factor-beta associates with angiogenesis in epithelial ovarian cancer. International journal of gynecological pathology : official journal of the International Society of Gynecological Pathologists 1997, 16, 256–262. [DOI] [PubMed] [Google Scholar]
- 11.Liao S; Liu J; Lin P; Shi T; Jain RK; Xu L TGF-beta blockade controls ascites by preventing abnormalization of lymphatic vessels in orthotopic human ovarian carcinoma models. Clinical cancer research : an official journal of the American Association for Cancer Research 2011, 17, 1415–1424, doi: 10.1158/1078-0432.Ccr-10-2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Riester M; Wei W; Waldron L; Culhane AC; Trippa L; Oliva E; Kim S.-h.; Michor F; Huttenhower C; Parmigiani G, et al. Risk Prediction for Late-Stage Ovarian Cancer by Meta-analysis of 1525 Patient Samples. Journal of the National Cancer Institute 2014, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kao JY; Gong Y; Chen CM; Zheng QD; Chen JJ Tumor-derived TGF-beta reduces the efficacy of dendritic cell/tumor fusion vaccine. Journal of immunology (Baltimore, Md. : 1950) 2003, 170, 3806–3811. [DOI] [PubMed] [Google Scholar]
- 14.Chen W; Wahl SM TGF-beta: the missing link in CD4+CD25+ regulatory T cell-mediated immunosuppression. Cytokine & growth factor reviews 2003, 14, 85–89. [DOI] [PubMed] [Google Scholar]
- 15.Ghiringhelli F; Menard C; Terme M; Flament C; Taieb J; Chaput N; Puig PE; Novault S; Escudier B; Vivier E, et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. The Journal of experimental medicine 2005, 202, 1075–1085, doi: 10.1084/jem.20051511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kobie JJ; Wu RS; Kurt RA; Lou S; Adelman MK; Whitesell LJ; Ramanathapuram LV; Arteaga CL; Akporiaye ET Transforming growth factor beta inhibits the antigen-presenting functions and antitumor activity of dendritic cell vaccines. Cancer Res 2003, 63, 1860–1864. [PubMed] [Google Scholar]
- 17.Winkler I; Wilczynska B; Bojarska-Junak A; Gogacz M; Adamiak A; Postawski K; Darmochwal-Kolarz D; Rechberger T; Tabarkiewicz J Regulatory T lymphocytes and transforming growth factor beta in epithelial ovarian tumors-prognostic significance. Journal of ovarian research 2015, 8, 39, doi: 10.1186/s13048-015-0164-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolf D; Wolf AM; Rumpold H; Fiegl H; Zeimet AG; Muller-Holzner E; Deibl M; Gastl G; Gunsilius E; Marth C The expression of the regulatory T cell-specific forkhead box transcription factor FoxP3 is associated with poor prognosis in ovarian cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2005, 11, 8326–8331, doi: 10.1158/1078-0432.Ccr-05-1244. [DOI] [PubMed] [Google Scholar]
- 19.Leffers N; Gooden MJ; de Jong RA; Hoogeboom BN; ten Hoor KA; Hollema H; Boezen HM; van der Zee AG; Daemen T; Nijman HW Prognostic significance of tumor-infiltrating T-lymphocytes in primary and metastatic lesions of advanced stage ovarian cancer. Cancer immunology, immunotherapy: CII 2009, 58, 449–459, doi: 10.1007/s00262-008-0583-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haque S; Morris JC Transforming growth factor-beta: A therapeutic target for cancer. Human vaccines & immunotherapeutics 2017, 13, 1741–1750, doi: 10.1080/21645515.2017.1327107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fabregat I; Fernando J; Mainez J; Sancho P TGF-beta signaling in cancer treatment. Current pharmaceutical design 2014, 20, 2934–2947. [DOI] [PubMed] [Google Scholar]
- 22.Roane BM; Arend RC; Birrer MJ Review: Targeting the Transforming Growth Factor-Beta Pathway in Ovarian Cancer. 2019, 11, 668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tauriello DVF; Palomo-Ponce S; Stork D; Berenguer-Llergo A; Badia-Ramentol J; Iglesias M; Sevillano M; Ibiza S; Canellas A; Hernando-Momblona X, et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543, doi: 10.1038/nature25492. [DOI] [PubMed] [Google Scholar]
- 24.Ravi R; Noonan KA; Pham V; Bedi R; Zhavoronkov A; Ozerov IV; Makarev E; A, V.A.; Wysocki, P.T.; Mehra, R., et al. Bifunctional immune checkpoint-targeted antibody-ligand traps that simultaneously disable TGFbeta enhance the efficacy of cancer immunotherapy. Nature communications 2018, 9, 741, doi: 10.1038/s41467-017-02696-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mariathasan S; Turley SJ; Nickles D; Castiglioni A; Yuen K; Wang Y; Kadel EE III; Koeppen H; Astarita JL; Cubas R, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548, doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lan Y; Zhang D; Xu C; Hance KW; Marelli B; Qi J; Yu H; Qin G; Sircar A; Hernandez VM, et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-beta. Science translational medicine 2018, 10, doi: 10.1126/scitranslmed.aan5488. [DOI] [PubMed] [Google Scholar]
- 27.Holmgaard RB; Schaer DA; Li Y; Castaneda SP; Murphy MY; Xu X; Inigo I; Dobkin J; Manro JR; Iversen PW, et al. Targeting the TGFbeta pathway with galunisertib, a TGFbetaRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. Journal for immunotherapy of cancer 2018, 6, 47, doi: 10.1186/s40425-018-0356-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walton J; Blagih J; Ennis D; Leung E; Dowson S; Farquharson M; Tookman LA; Orange C; Athineos D; Mason S, et al. CRISPR/Cas9-Mediated Trp53 and Brca2 Knockout to Generate Improved Murine Models of Ovarian High-Grade Serous Carcinoma. Cancer research 2016, 76, 6118–6129, doi: 10.1158/0008-5472.CAN-16-1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dasch JR; Pace DR; Waegell W; Inenaga D; Ellingsworth L Monoclonal antibodies recognizing transforming growth factor-beta. Bioactivity neutralization and transforming growth factor beta 2 affinity purification. Journal of immunology (Baltimore, Md. : 1950) 1989, 142, 1536–1541. [PubMed] [Google Scholar]
- 30.Dennler S; Itoh S; Vivien D; ten Dijke P; Huet S; Gauthier JM Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. The EMBO journal 1998, 17, 3091–3100, doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie L; Tintani F; Wang X; Li F; Zhen G; Qiu T; Wan M; Crane J; Chen Q; Cao X Systemic neutralization of TGF-β attenuates osteoarthritis. Ann N Y Acad Sci 2016, 1376, 53–64, doi: 10.1111/nyas.13000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bedi A; Chang X; Noonan K; Pham V; Bedi R; Fertig EJ; Considine M; Califano JA; Borrello I; Chung CH, et al. Inhibition of TGF-β enhances the in vivo antitumor efficacy of EGF receptor-targeted therapy. Mol Cancer Ther 2012, 11, 2429–2439, doi: 10.1158/1535-7163.MCT-12-0101-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Terra M; Oberkampf M; Fayolle C; Rosenbaum P; Guillerey C; Dadaglio G; Leclerc C Tumor-Derived TGFbeta Alters the Ability of Plasmacytoid Dendritic Cells to Respond to Innate Immune Signaling. Cancer Res 2018, 78, 3014–3026, doi: 10.1158/0008-5472.Can-17-2719. [DOI] [PubMed] [Google Scholar]
- 34.Mule JJ; Schwarz SL; Roberts AB; Sporn MB; Rosenberg SA Transforming growth factor-beta inhibits the in vitro generation of lymphokine-activated killer cells and cytotoxic T cells. Cancer immunology, immunotherapy : CII 1988, 26, 95–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Connolly DC; Bao R; Nikitin AY; Stephens KC; Poole TW; Hua X; Harris SS; Vanderhyden BC; Hamilton TC Female mice chimeric for expression of the simian virus 40 TAg under control of the MISIIR promoter develop epithelial ovarian cancer. Cancer Res 2003, 63, 1389–1397. [PubMed] [Google Scholar]
- 36.Dahmani A; Delisle J-S TGF-β in T Cell Biology: Implications for Cancer Immunotherapy. Cancers 2018, 10, 194, doi: 10.3390/cancers10060194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sorensen EW; Gerber SA; Sedlacek AL; Rybalko VY; Chan WM; Lord EM Omental immune aggregates and tumor metastasis within the peritoneal cavity. Immunologic research 2009, 45, 185–194, doi: 10.1007/s12026-009-8100-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robinson-Smith TM; Isaacsohn I; Mercer CA; Zhou M; Van Rooijen N; Husseinzadeh N; McFarland-Mancini MM; Drew AF Macrophages mediate inflammation-enhanced metastasis of ovarian tumors in mice. Cancer Res 2007, 67, 5708–5716, doi: 10.1158/0008-5472.Can-06-4375. [DOI] [PubMed] [Google Scholar]
- 39.Wang E; Ngalame Y; Panelli MC; Nguyen-Jackson H; Deavers M; Mueller P; Hu W; Savary CA; Kobayashi R; Freedman RS, et al. Peritoneal and subperitoneal stroma may facilitate regional spread of ovarian cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2005, 11, 113–122. [PubMed] [Google Scholar]
- 40.Baert T; Vankerckhoven A; Riva M; Van Hoylandt A; Thirion G; Holger G; Mathivet T; Vergote I; Coosemans A Myeloid Derived Suppressor Cells: Key Drivers of Immunosuppression in Ovarian Cancer. Front Immunol 2019, 10, 1273–1273, doi: 10.3389/fimmu.2019.01273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hawinkels LJ; Verspaget HW; van der Reijden JJ; van der Zon JM; Verheijen JH; Hommes DW; Lamers CB; Sier CF Active TGF-beta1 correlates with myofibroblasts and malignancy in the colorectal adenoma-carcinoma sequence. Cancer science 2009, 100, 663–670, doi: 10.1111/j.1349-7006.2009.01100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Higashi T; Sasagawa T; Inoue M; Oka R; Shuangying L; Saijoh K Overexpression of latent transforming growth factor-beta 1 (TGF-beta 1) binding protein 1 (LTBP-1) in association with TGF-beta 1 in ovarian carcinoma. Japanese journal of cancer research : Gann 2001, 92, 506–515, doi: 10.1111/j.1349-7006.2001.tb01123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kjellman C; Olofsson SP; Hansson O; Von Schantz T; Lindvall M; Nilsson I; Salford LG; Sjogren HO; Widegren B Expression of TGF-beta isoforms, TGF-beta receptors, and SMAD molecules at different stages of human glioma. International journal of cancer 2000, 89, 251–258, doi:. [DOI] [PubMed] [Google Scholar]
- 44.Lee W; Ko SY; Mohamed MS; Kenny HA; Lengyel E; Naora H Neutrophils facilitate ovarian cancer premetastatic niche formation in the omentum. The Journal of experimental medicine 2019, 216, 176–194, doi: 10.1084/jem.20181170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tan DS; Agarwal R; Kaye SB Mechanisms of transcoelomic metastasis in ovarian cancer. The Lancet. Oncology 2006, 7, 925–934, doi: 10.1016/s1470-2045(06)70939-1. [DOI] [PubMed] [Google Scholar]
- 46.Sodek KL; Murphy KJ; Brown TJ; Ringuette MJJC; Reviews M Cell–cell and cell–matrix dynamics in intraperitoneal cancer metastasis. 2012, 31, 397–414, doi: 10.1007/s10555-012-9351-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang L; Conejo-Garcia JR; Katsaros D; Gimotty PA; Massobrio M; Regnani G; Makrigiannakis A; Gray H; Schlienger K; Liebman MN, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. The New England journal of medicine 2003, 348, 203–213, doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 48.Turner TB; Buchsbaum DJ; Straughn JM Jr.; Randall TD; Arend RC Ovarian cancer and the immune system - The role of targeted therapies. Gynecologic oncology 2016, 142, 349–356, doi: 10.1016/j.ygyno.2016.05.007. [DOI] [PubMed] [Google Scholar]
- 49.Lan Y; Zhang D; Xu C; Hance KW; Marelli B; Qi J; Yu H; Qin G; Sircar A; Hernández VM, et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-β. Science Translational Medicine 2018, 10, eaan5488, doi: 10.1126/scitranslmed.aan5488. [DOI] [PubMed] [Google Scholar]
- 50.Schmidt A; Zhang XM; Joshi RN; Iqbal S; Wahlund C; Gabrielsson S; Harris RA; Tegner J Human macrophages induce CD4(+)Foxp3(+) regulatory T cells via binding and re-release of TGF-beta. Immunology and cell biology 2016, 94, 747–762, doi: 10.1038/icb.2016.34. [DOI] [PubMed] [Google Scholar]