

Abstract
The infiltration of chronic lymphocytic leukemia (CLL) cells into lymphoid organs correlates with disease severity. CXCL12 is a key chemotactic factor for the trafficking of CLL. Tissue factor pathway inhibitor (TFPI) is a serine protease inhibitor and plays a role in CXCL12-mediated hematopoietic stem cell homing. We aim to explore the role of TFPI in CXCL12-mediated migration of CLL cells. In this study, plasma TFPI concentrations were measured by ELISA. CLL cells were isolated from patients and used for trans-endothelial migration (TEM) assays. Quantitative RT-PCR and Western blotting were used to detect the expression of CXCR7, CXCR4 and β-catenin. Immunofluorescence and co-immunoprecipitation was used to detect the binding of TFPI and glypican-3 (GPC3). We found that plasma TFPI levels in CLL patients were higher than in healthy controls, particularly in the patients with advanced disease. TFPI enhanced CXCL12-mediated TEM of CLL cells by increasing the expression of the CXCL12 receptor CXCR7, but not of the CXCL12 receptor CXCR4. The effect of TFPI on TEM was abolished by the CXCR7 inhibitor, CCX771, while the CXCR4 inhibitor AMD3100 strongly increased TEM. TFPI co-localized with GPC3 on the cell surface. An antibody to GPC3, HS20, decreased CXCR7 expression and abolished the effect of TFPI on TEM. TFPI activated β-catenin and the Wnt/β-catenin inhibitor IWP4 repressed the effect of TFPI on CXCR7 expression and TEM. We conclude that TFPI may contribute to organ infiltration in CLL patients.
Subject terms: Biomarkers, Risk factors
Introduction
With an age-adjusted incidence of 4–5 per 100,000 per year chronic lymphocytic leukemia (CLL) is the most common type of leukemia in the western world1. Infiltration of CLL cells in lymphoid tissues is a key element of the disease pathogenesis2. CLL cells infiltrate primary and secondary lymphoid organs where they are protected from apoptosis through crosstalk with stromal cells3. Critically, CLL cells in lymphoid niches are protected against cytotoxic effects of many chemotherapeutics and likely cause minimal residual disease and future relapse4. Growing evidence indicates that lymphocyte trafficking plays a critical role in the pathophysiology of CLL5. Interfering with CLL cell migration or retention in lymphoid tissues could thus improve the efficacy of conventional therapy of CLL patients.
The C-X-C motif chemokine ligand 12 (CXCL12), also called chemokine stromal-derived factor 1 (SDF1α), is constitutively secreted by fibroblasts and stromal cells and attracts leukemic cells to the specific microenvironment necessary for their survival6. A recent study showed increased levels of CXCL12 in serum of symptomatic late stage CLL patients in comparison to patients with early stage CLL7. The chemokine receptor CXCR4 was the first identified receptor for CXCL128, and CXCR7, also known as RDC1 or atypical chemokine receptor 3 (ACKR3), was recently reported as a receptor for CXCL129. CXCR7 has a stronger affinity for CXCL12 than CXCR410, and ligand binding to CXCR7 activates alternative signaling pathways regulating cellular adhesion, proliferation and dissemination of tumor cells11 as well as differentiation of mature B cells12. Furthermore, CXCR7 has been shown to potentiate and regulate trans-endothelial migration (TEM) of circulating tumor cells leading to enhanced extravasation13. However, the role of CXCR7 in CLL cell trafficking has not been elucidated, and further understanding of this receptor may facilitate the development of novel therapies.
CLL cell homing involves cell adhesion molecules and chemotactic factors which are highly influenced by endothelial cells14. It is well known that endothelial cells are the main source of tissue factor (TF) pathway inhibitor (TFPI), which is the primary inhibitor of the initiation of blood coagulation and inhibits both TF-factor (F) VIIa-dependent FXa generation and free FXa15. The interaction between TF-FVIIa and TFPI promote tumor cell adhesion and migration16. TFPI inhibits primary and metastatic tumor growth and represses endothelial proliferation in vitro17. We have previously reported that TFPI is involved in cell migration in breast cancer18.
A recent study showed that TFPI increases hematopoietic stem cell homing by binding to glypican-3 (GPC3), a heparan sulfate proteoglycan19, and regulates the activity of CXCL1220. GPC3 regulates canonical Wnt/β-catenin signaling21, which is activated in hematopoietic malignancies and contributes to tumor recurrence22. The activation of Wnt/β-catenin leads to the accumulation of β-catenin and favors its translocation to the nucleus as a cofactor activating the transcription of Wnt/β-catenin target genes23.
We investigated the role of TFPI in the trans-endothelial migration of CLL cells. The effect of TFPI was analyzed in detail with respect to regulation of the key chemotactic factor CXCL12 and its receptors. The role of the potential TFPI receptor, GPC3, and the involvement of the Wnt/β-catenin signaling pathway were examined. We have unraveled a novel mechanism of action of TFPI that correlates with its higher levels in patients with advanced stage CLL.
Materials and methods
Human samples, cell lines and reagents
All CLL patients and healthy controls provided written informed consent using protocols approved by the Regional Committee for Medical and Health Research Ethics (Approval No. 2016/947/REC South-East A) and in accordance with the principles of the Declaration of Helsinki. Patients were diagnosed according to the International Workshop on Chronic Lymphocytic Leukemia 2008 criteria24.
To obtain peripheral blood mononuclear cells (PBMCs), heparinized blood samples from CLL patients and buffy coats obtained from healthy donors at the Blood Transfusion Centre of the Oslo University Hospital were centrifuged using Lymphoprep (Alere Technologies AS, Oslo, Norway) in SepMate 50 mL tubes (Stem Cell Technologies, Cambridge, UK). CLL and normal B cells were purified from the PBMCs by negative selection of MACS B cell Isolation Kit II (Miltenyi Biotec, Auburn, CA, USA) by using AutoMACS cell separator (Miltenyi Biotec, Bergisch Gladbach, Germany). The amount of antibodies and beads was reduced to 1/10 for CLL B cell isolation. The purities of CLL or normal B cell enrichments were 95% or greater, determined by flow cytometry analysis staining for CD19+ cells. The purified CLL cells and normal B cells were incubated in RPMI1640 medium (Lonza, Verviers, Belgium) supplemented with 10% fetal bovine serum (FBS) (Lonza), 100 U/mL penicillin and 100 µg/mL streptomycin (Lonza).
The CLL cell line HG3 (DSMZ-German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany) and the chronic myeloid leukemia blast crisis cell line K562 provided by the Institute of Hematology of the Chinese Academy of Medical Sciences (Tianjin, China) were grown in flasks containing RPMI1640 medium (Lonza) supplemented with 10% FBS (Lonza), 100 U/mL penicillin and 100 µg/mL streptomycin (Lonza).
Human umbilical vein endothelial cells (HUVECs, CC-2517, Lonza) were grown in complete endothelial cell growth medium MV2 (Promocell, Heidelberg, Germany).
Human recombinant TFPI (rTFPI) and human recombinant CXCL12 (rCXCL12) were obtained from R&D Systems (Minneapolis, MN, USA). CCX771, a novel specific CXCR7 inhibitor was provided by ChemoCentryx (Mountain View, CA, USA). AMD3100, a specific inhibitor for CXCR4 was obtained from Merck KgaA (Darmstadt, Germany). HS20, a human monoclonal antibody against the heparan sulphate chains of GPC325 was kindly provided by Dr. Mitchell Ho, U.S.National Cancer Institute (NCI). IWP4, a potent inhibitor of Wnt/β-catenin signaling was obtained from Tocris Bioscience (Bristol, UK).
Free TFPI ELISA antigen detection
Citrated venous blood samples were collected from 36 CLL patients (25 males and 11 females, mean age 65 years, range 38–80 years). Patients were also clinically staged according to the Binet staging system26,27 with the following distribution: stage A, 21 (58.3%), stage B, 7 (19.4%) and stage C, 8 (22.2%). Citrated venous blood samples from 34 healthy subjects (17 males and 17 females, mean age 42 years, range 24–68 years) were used as controls. Plasma samples were collected by centrifugation at 2500×g for 15 min at 20 °C. Plasma aliquots were frozen and stored at − 80 °C until being assayed. The commercial enzyme-linked immunosorbent assay (ELISA) Asserachrom Free TFPI (Diagnostica Stago, Asnières, France) was used to measure the concentration of full-length TFPI in the plasma according to the manufacturer’s protocol28.
Transendothelial migration (TEM) assays
TEM assays were performed in transwell insert plates (96-well, 3 µm pore size; Costar, Corning). Approximately 30 000 HUVECs were seeded into the transwell the day before the chemotaxis assay and incubated overnight to generate a HUVEC monolayer. Fresh CLL or normal B cells were incubated in RPMI1640 medium with 10% FBS and treated with rTFPI at the indicated concentrations for 24 h before the migration assays. In some experiments, the CLL cells were pre-treated with HS20 or IWP4 for 1 h prior to rTFPI treatment. Then the cells were washed with RPMI1640 without serum and 3.75 × 105 CLL cells or normal B cells were resuspended in 75 µL RPMI1640 supplemented with 1% BSA (Sigma Aldrich, MO, USA) and placed in the upper chamber of transwell inserts. Inserts were placed in the wells containing 235 µL medium alone (basal) or medium with 400 ng/mL rCXCL12. Stimuli were applied at optimal concentrations determined by previous titration. Plates were centrifuged shortly (for 1 s at 150 g) to spin down the cells onto the filter and migration proceeded for 3 h in the incubator (37 °C, 5% CO2). Migrated cells were harvested from the lower chamber and counted by flow cytometer MACS Quant (Miltenyi Biotec GMbH, Bergisch Gladbach, Germany). Cell migration capacity was expressed either as the percentage of migrated cells, or as a fold change, which is defined as the number of migrated cells in the presence of rCXCL12 divided by the number of migrated cells in the absence of rCXCL12.
Western blot analysis
Protein extracts were prepared as previously described29. 20–50 µg proteins were resolved by SDS-PAGE, transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad Laboratories, Hercules, CA, USA). Then the PVDF membranes were cut along the 75 kDa level or between 75 and 50 kDa according to the protein standards (Bio-Rad Laboratories). The upper parts of the membranes were incubated with the antibodies against active β-catenin (Cell Signaling Technology, Boston, MA, USA) or β-catenin (Novus Biologicals); the lower parts of the membranes were incubated with the antibodies against CXCR7 (Novus Biologicals, Centennial, CO, USA), CXCR4 (Abcam, Cambridge, UK) or β-actin (Cell Signaling Technology). Detections were performed as previously described29.
Flow cytometry
CLL cells were isolated from CLL patients and treated with 200 ng/mL rTFPI for 24 h. Then the cells were washed and blocked by human FcR blocking reagent (Miltenyi Biotec, Bergisch Gladbach, Germany) for 10 min at room temperature. Afterwards, the cells were stained with PE-conjugated anti-human CXCR7 antibody (Biolegend, San Diego, CA) for 1 h. PE-conjugated mouse IgG2b was used as isotype control (Biolegend). The expression of CXCR7 was measured by a FACS Calibur flow cytometry (Becton Dickinson, Franklin Lakes, NJ, USA) and analyses were carried out using FlowJo software (Becton Dickinson).
RNA isolation, cDNA synthesis and relative quantification of mRNA
Total RNA was extracted, quantified and reversely transcribed to cDNA as described before30. Quantitative RT-PCR was used to measure the relative mRNA expression of CXCR7 (ACKR3, Hs00604567_m1, Applied Biosystems) and CXCR4 (Hs00237052_m1, Applied Biosystems). Ct values were normalized against the endogenous control TBP (Applied Biosystems). Negative controls without cDNA were always included.
Immunofluorescence staining and confocal microscopy studies
Expression of TFPI and GPC3 in HG3 cells before or after rTFPI treatment was examined by immunostaining. 20 µL cell suspension were spread on a slide and air-dried for 10 min. Cells were fixed by ethanol for 10 min at 4 °C and incubated with anti-TFPI (ADG72, Sekisui Diagnostics, Pfungstadt, Germany) and anti-GPC3 (1G12, Santa Cruz, CA, USA) primary antibodies overnight at 4 °C. Normal rabbit IgG (Cell Signaling Technology) and mouse IgG1 (R&D Systems Inc., MN, USA) were used as controls. Following washing with Dulbecco’s phosphate buffered saline (Sigma Aldrich, MO, USA) in 2% fetal calf serum, cells were stained with Alexa Fluor 488-labeled donkey anti-rabbit and Alexa Fluor 568-labeled donkey anti-mouse secondary antibodies (Thermo Fisher Scientific, Eugene, OR, USA) for 90 min at room temperature. Nuclei were stained with diamidino-2-phenylindole (ProLong Gold antifade reagent, Thermo Fisher Scientific). Fluorescence images were acquired by using a Leica TCS SP8 confocal microscope equipped with a 40 × 1.30 NA oil immersion lens, a UV (405 nm) laser and a CW (Continuous Wavelength) white light laser. Sequential acquisition scanning was carried out at 1024 × 1024 pixel resolution. For each of the cells, 10–20 Z-scans spanning the entire cell were generated. Z-stack images were acquired by using ImageJ software.
Co-immunoprecipitation
The total protein content of HG3 and K562 cell lysate was measured with the Pierce BCA protein assay Kit (Thermo Scientific Pierce, Rockford, IL, USA). 3 mg of total proteins from HG3 and K562 cells were used for co-immunoprecipitation (Co-IP) experiments. Co-IP was performed using the Co-IP Kit (Thermo Scientific Pierce) following the manufacturer’s protocol. Briefly, 20 mg of TFPI antibody (MAB2974, R&D Systems Inc., MN, USA) and isotype control mouse IgG2α (R&D systems) were incubated with AminoLink Plus coupling resin for 2 h at room temperature. The antibody-coupled resins were incubated with 500 µL of the whole protein lysates overnight at 4 °C. The resins were washed and the protein complexes bound to the antibody were eluted. Subsequent Western blot analysis was done as described before29. The pulling down of GPC3 was detected by using sheep anti-GPC3 (AF2119, R&D systems). The detection of TFPI with goat anti-TFPI antibody (AF2974, R&D systems) was used as a control for IP.
Statistics
We performed statistical analysis using Prism (version 8.0.1, Graphpad software, San Diego, CA, USA) and SPSS version 25.0.0.1. For in vitro data, mean ± SEM of n = 3 or more determinations are shown. We used two-tailed Student’s t test or one-way ANOVA test. For comparison of plasma TFPI levels, TFPI values were slightly skewed as evaluated by the Kolmogorov Smirnov test and were log transformed prior to UNIANOVA, which was age and Bonferroni-adjusted. Differences were considered statistically significant at a two-tailed value of P < 0.05.
Results
Higher plasma TFPI concentration was detected in CLL patients with advanced disease
The main characteristics of all the untreated CLL patients are described in Table 1. CLL patients exhibited elevated TFPI levels (median: 16.9 ng/mL, range: 8.5–27.2 ng/mL) as compared to healthy controls (median: 10.8 ng/mL, range: 5.7–21.4 ng/mL; Fig. 1). No differences in sex distribution between patients and controls were detected, but patients were older and age was used in adjustment of p-values. To address the severity of CLL, we applied the Binet staging system which relates to organ infiltration. The results showed that patients with Binet stage C (median: 21.5 ng/mL, range: 13.8–27.2 ng/mL) had higher plasma TFPI level than patients with Binet stage A (median: 15.8 ng/mL, range: 9.8–27.0 ng/mL) and Binet stage B (median: 16.1 ng/mL, range: 8.5–19.0 ng/mL) (Fig. 1), suggesting that higher plasma TFPI relates to the severity of organ infiltration.
Table 1.
Characteristics of all the untreated CLL patients stratified according to Binet stages.
| Variables | All patients (n = 36) | Binet stage A (n = 21) | Binet stage B (n = 7) | Binet stage C (n = 8) |
|---|---|---|---|---|
| Age (> 65 years) | 18 | 11 | 2 | 5 |
| Gender (M/F) | 25/11 | 14/7 | 6/1 | 5/3 |
| Homology with germ line (≥ 96%) | 10 | 3 | 2 | 5 |
| Lymphocytes (> 50 × 109/L) | 15 | 3 | 6 | 6 |
| Hemoglobin (< 10 g/dL) | 4 | 0 | 0 | 4 |
| Platelet (< 100 × 109/L) | 2 | 0 | 0 | 2 |
Figure 1.
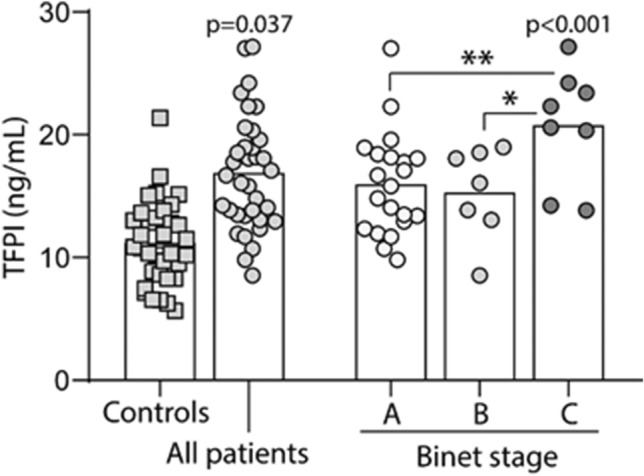
Plasma TFPI concentration increased in patients with CLL and related to CLL progression. Increased plasma TFPI level was observed in CLL patients compared to healthy controls. Compared to the patients with Binet stage A and B, the patients with Binet stage C showed the highest TFPI concentration in the plasma. Binet stages represent the severity of organ infiltration. Data are presented as dot plots and columns represent median levels. Concentrations are expressed as ng/mL. p values in the graph represent differences vs. controls. *p < 0.05, **p < 0.01. All statistical comparisons were adjusted for age using UNIANOVA with Bonferroni adjustment on log transformed TFPI values.
TFPI enhanced CXCL12-mediated TEM of CLL cells
To explore the possible role of TFPI in organ infiltration of CLL patients, we first investigated whether rTFPI treatment affects the migration of CLL cells in response to CXCL12 compared to normal B cells. After exposure to exogenous rTFPI, CLL or normal B cells were applied to the TEM assay. Results showed that CXCL12 significantly induced TEM of CLL (Fig. 2A) and normal B cells (Fig. 2B). These findings indicated that CXCL12 was an efficient chemoattractant for both CLL and normal B cells. However, rTFPI significantly enhanced the CXCL12-mediated TEM of CLL cells (Fig. 2A), while no effect of rTFPI was found on normal B cells (Fig. 2B). Furthermore, in CLL cells, the CXCL12-mediated migration was increased by rTFPI in a dose-dependent manner (Fig. 2C). We concluded that TFPI enhanced the CXCL12-mediated TEM of CLL cells but had no effect on normal B cells despite of their higher migration capacity in response to CXCL12.
Figure 2.

TFPI enhances CXCL12-mediated TEM of CLL cells, but not normal B cells. (A) Fresh CLL cells were pre-treated with or without 200 ng/mL rTFPI for 24 h and then washed and applied to the TEM assay for 3 h with or without 400 ng/mL recombinant CXCL12 added to the lower chamber. The percentage was determined by counting the migrated fraction of the input cells. Data are presented as mean ± SEM; n = 8 individuals, **P < 0.01, ****P < 0.0001. (B) Normal B cells were also treated with or without rTFPI and used in the TEM assay as described above. Data are presented as mean ± SEM; n = 4 individuals, **P < 0.01. (C) Fresh CLL cells were treated with 0, 50, 100 and 200 ng/mL rTFPI for 24 h and then washed and applied to the TEM assay as described above. The number of migrated cells in the lower chamber was counted and cell migration to CXCL12 was expressed as fold change relative to control. Data are presented as mean ± SEM; n = 5 individuals, **P < 0.01, ***P < 0.001.
TFPI upregulated the expression of receptor CXCR7, but not CXCR4 in CLL cells
To understand how TFPI affects CXCL12-mediated TEM of CLL cells, we investigated the expression of CXCR4 and CXCR7 in the CLL cells. Western blots showed that after 24 h of rTFPI exposure, CXCR7 expression was dose-dependently upregulated in the CLL cells, while CXCR4 expression showed no increase (Fig. 3A–C). Flow cytometry analysis showed that surface expression of CXCR7 was increase by 24 h treatment of rTFPI (Supplementary Fig. 8). Moreover, Western blots showed that the expression of CXCR7 was increased over time after rTFPI exposure, but no obvious change in CXCR4 expression was observed (Fig. 3D–F). Quantitative RT-PCR showed a 16% increase of CXCR7 mRNA level 15 min after rTFPI exposure, but only a 4% increase of CXCR4 mRNA (Fig. 3G,H). To further address the involvement of CXCR7 in the TFPI-mediated TEM, CCX77131, a small molecule with high affinity and selectivity for CXCR7 was used. In the presence of CCX771, the enhanced TEM induced by rTFPI was strongly repressed, while CCX771 alone did not repress the migration potential of CLL cells (Fig. 3I). Surprisingly, the CXCR4 inhibitor AMD310032 strongly increased CXCL12-mediated TEM both with and without TFPI (Fig. 3J). We concluded that CXCR7, but not CXCR4 is involved in the effect of TFPI on the TEM of CLL cells.
Figure 3.

CXCR7 plays a key role in the effect of TFPI on the CXCL12-mediated TEM of CLL cells. (A) Western blotting and (B, C) quantification of CXCR7 and CXCR4 expression, relative to β-actin, after treating CLL cells with different doses of rTFPI for 24 h. n = 3 individuals, *P < 0.05. (D) Western blotting and (E, F) quantification of CXCR7 and CXCR4 expression, relative to β-actin, after treating the CLL cells with 200 ng/mL rTFPI for 0, 15, 60, 180, 360 and 960 min. n = 4 individuals, **P < 0.01. (G, H) mRNA expression of CXCR7 and CXCR4 was detected by quantitative PCR, relative to TBP, after treating the CLL cells with 200 ng/mL rTFPI for 0, 15, 60, 180, 360 and 960 min. n = 4 individuals; *P < 0.05. (I) After 24 h exposure of 200 ng/mL rTFPI, CLL cells were treated with 2 µM CCX771 for 1 h before they were used in the TEM assay. DMSO was used as control. n = 6 individuals; *P < 0.05, **P < 0.01. (J) After 24 h exposure of 200 ng/mL rTFPI, CLL cells were treated with 5 µg/mL AMD3100 for 1 h before they were applied to the transwells for TEM assay. For panels (I, J), results were presented as described in Fig. 1A. n = 6 individuals; *P < 0.05.
The binding of TFPI to GPC3 on the surface of CLL cells was involved in the TFPI-mediated TEM
Western blot analyses showed a dose-dependent accumulation of TFPI protein in the cell lysates after rTFPI treatment for 24 h (Fig. 4A,B), which indicates that exogenous TFPI binds to or enters the CLL cells. Therefore, we hypothesized that a potential receptor for TFPI exists on the surface of CLL cells. Since TFPI is known to bind to cell membrane heparan sulphate proteoglycans, such as GPC320,33, we examined the expression of GPC3 in CLL cells by immunofluorescence. Immunostaining showed that TFPI and GPC3 were co-expressed on the surface of the CLL cell line HG3 and stronger TFPI staining on the cell surface was detected after adding exogenous rTFPI (Fig. 4C). Co-IP experiments were performed to investigate the binding between TFPI and GPC3 in the CLL cell line HG3 and the myeloid leukemic cell line K562. The antibody against TFPI was used to precipitate the protein complex, and then immunoblots were performed to detect GPC3 protein. Results showed the co-precipitation of GPC3 along with TFPI in both HG3 and K562 cells (Fig. 4D). To assess whether GPC3 is involved in the effect of TFPI on regulating CXCR7 expression and increasing CXCL12-mediated TEM, HS20, a specific antibody against the heparan sulphate chains of GPC3, was applied to block the binding of TFPI to GPC3. Western blotting showed that rTFPI treatment increased the expression of CXCR7 and that pre-treatment with HS20 abolished the effect of TFPI, while HS20 itself did not affect CXCR7 expression (Fig. 4E,F). Furthermore, TEM results showed that HS20 pre-treatment abolished the effect of TFPI on TEM (p = 0.0074), although HS20 itself has no significant effect on the migration of CLL cells (p = 0.5) (Fig. 4G). We also analyzed the effect of addition of TFPI to HS20 compared to the effect of HS20 alone, however, no significant difference was found (p = 0.65).
Figure 4.

TFPI binds to GPC3 on the membrane of CLL cells. (A) Western blotting and (B) quantification of TFPI protein in the cell lysates of CLL patients, relative to β-actin, after 0, 50, 100 and 200 ng/mL rTFPI treatment. n = 3 individuals; *P < 0.05. (C) After 24 h treatment of 200 ng/mL rTFPI, co-localization of TFPI (red) and GPC3 (green) on the surface of human CLL cell line HG3 was assessed by immunofluorescence staining. Nuclei were stained with DAPI (blue). (D) Co-IP was performed in the HG3 and K562 cells. Immunoblots showed the bait protein TFPI and the pull-down of the prey protein GPC3. (E) Western blotting and (F) quantification of CXCR7 expression, relative to β-actin, after CLL cells were pre-treated with GPC3 antibody HS20 (100 µg/mL) for 1 h prior to 24 h treatment of 200 ng/mL rTFPI. Human IgG was used as control since HS20 is isolated from human serum. n = 4 individuals; *P < 0.05, **P < 0.01. (G) After CLL cells were pre-treated with HS20 before rTFPI treatment, the cells were washed and applied to TEM assay. Human IgG was used as control. Results were presented as we described in Fig. 2A. n = 10 individuals; **P < 0.01.
Activation of β-catenin was involved in the TFPI-mediated TEM
Since Wnt/β-catenin signaling is known to be regulated by GPC321,25,34, we examined if the binding of TFPI to GPC3 activates the Wnt/β-catenin signaling pathway in CLL cells. Western blotting showed that active β-catenin was detected 15 min after rTFPI exposure and was further increased over time (Fig. 5A,B). To confirm the involvement of Wnt/β-catenin signaling in the effect of TFPI, we used IWP4, a potent inhibitor for the palmitoylation of Wnt proteins. Quantitative PCR showed that CXCR7 mRNA expression was increased by rTFPI and this effect was abolished by IWP4 (Fig. 5C). Consistently, Western blotting showed that the effect of TFPI on CXCR7 expression was repressed by IWP4 (Fig. 5D,E). TEM results showed that IWP4 alone did not affect the migration of CLL cells, but pre-treating the CLL cells with IWP4 prior to rTFPI exposure fully prevented the effect of rTFPI on CLL cell migration (Fig. 5F).
Figure 5.

Activation of β-catenin is involved in the effect of TFPI on the expression of CXCR7 and the CXCL12-mediated TEM of CLL cells. (A) Western blotting and (B) quantification of the active β-catenin expression, relative to β-actin, after treating the CLL cells from patients with 200 ng/mL rTFPI for 0, 15, 60, 180 and 360 min. n = 3 individuals; *P < 0.05 (C) Quantitative PCR was used to detect the mRNA expression of CXCR7, relative to TBP, after CLL cells were pre-treated with IWP4 (5 µM) for 30 min prior to 24 h treatment of 200 ng/mL rTFPI, with DMSO as a control. n = 4 individuals; *P < 0.05, **P < 0.01. (D) Western blotting and (E) quantification of CXCR7 expression, relative to β-actin, after CLL cells were pre-treated with 5 µM IWP4 for 30 min prior to 24 h treatment of 200 ng/mL rTFPI, with DMSO as a control. n = 3 individuals; *P < 0.05. (F) CLL cells were pre-treated with 5 µM IWP4 for 30 min prior to 24 h treatment of 200 ng/mL rTFPI, with DMSO as a control. Then CLL cells were washed and applied to TEM assay. TEM results were presented as we described in Fig. 2A. n = 10 individuals; **P < 0.01.
Discussion
TFPI was previously thought to be limited to its key role in regulating coagulation, but our data suggest that TFPI is involved in the migration of CLL cells. We found that plasma TFPI concentrations in CLL patients were higher than in the healthy controls, with particularly high levels in Binet stage C. It cannot be explained by the increase of platelets which are the main source of TFPI in the blood cells because most of CLL patients with Binet stage C have thrombocytopenia. Then it suggests that patients with higher levels of TFPI are more prone to tissue infiltration of CLL. In this study, the low numbers of patients in different stages has been a limitation. However, in our in vitro TEM study, we found consistently that TFPI enhanced CXCL12-mediated cell migration through binding to GPC3, activating β-catenin, and finally upregulating CXCR7 expression in the CLL cells.
A previous study showed that TFPI improves the migration and homing of hematopoietic stem cells through TFPI-mediated inhibition of the cleavage of CXCL12 by CD2620. However, we observed that TFPI increases CLL cell migration by upregulating the expression of CXCR7, a receptor for CXCL12. Either one or both pathways may operate in a given cell depending on the cell type.
CXCR7 together with CXCR4 are the two CXCL12 receptors, which play critical roles in CXCL12-mediated cell migration. It is known that CXCR4 is highly expressed by CLL cells and is involved in their migration35. Previous studies showed that CXCR7 plays a role in the migration of U937 leukemic cells and CD34+ stem cells36. Our study showed that CXCR7 is also expressed by CLL cells along with CXCR4. Interestingly, despite the higher levels of CXCR4 in CLL cells35, we observed that CLL cells migrate with lower efficiency and potency to CXCL12 than normal B cells, which is consistent with the previous studies37–39. Even though CXCR7 may be physically associated with CXCR440, CXCR7 can independently induce cell signaling, provide survival advantage and participate in CXCL12-mediated migration41,42. In our study, the expression of CXCR7, not CXCR4, was upregulated by TFPI in CLL cells and thus enhanced the CXCL12-mediated cell migration. Our results are consistent with the study of hematopoietic stem cells which showed that the expression of CXCR4 is not affected by TFPI20. Moreover, the inhibition of CXCR7 abolished the effect of TFPI on the cell migration, supporting that CXCL12/CXCR7 interaction plays an important role in the TFPI-induced migration of CLL cells. In contrast, the CXCR4 inhibitor AMD3100 increased the migration potential of CLL cells. This is probably because AMD3100 also improves the binding of CXCL12 to CXCR743, which improves the cell migration. However, CXCR7 inhibition itself does not repress cell migration suggesting that in addition to CXCL12/CXCR7 chemotaxis, other mechanisms might be involved in the regulation of CLL cell migration.
In our study, pre-treatment of CLL cells with recombinant TFPI increased TFPI protein in the cell lysates, suggesting that exogenous TFPI probably binds to CLL cells. Heparan sulphate proteoglycans are implicated in the binding and internalization of recombinant forms of TFPI, depending on their C-terminal polybasic portions44. GPC3 is a family member of heparan sulphate proteoglycans45. We found the co-expression of TFPI and GPC3 on the surface of CLL cells and co-IP confirmed the binding between the two proteins, which is consistent with previous studies on the hepatocellular carcinoma cell line HepG233 and hematopoietic stem cells20. Moreover, after adding exogenous TFPI to CLL cells, more TFPI protein was observed on the cell membrane, partially co-localized with GPC3, which indicates that exogenous TFPI from stromal cells may affect the cell function by binding to the membrane protein GPC3. The heparan sulphate chains on GPC3 are located close to the C terminus and the cell surface, which mediate the interaction of GPC3 with other cell membrane proteins46. Therefore it is suggested that TFPI probably binds to GPC3 through its heparan sulphate chains. This hypothesis was supported by the results showing that the GPC3 antibody HS20, which recognizes the heparan sulphate chains and inhibits the activity of GPC347, abolished the effect of TFPI on the expression of CXCR7 and the migration of CLL cells.
Accumulating evidence indicates that GPC3 activates the canonical Wnt signaling pathway in hepatocellular carcinoma cells21. Our data showed that the binding of TFPI to GPC3 on the CLL cells activated β-catenin, which is a key mediator of the canonical Wnt signaling pathway48. Researchers has found that Wnt3, Wnt5b, Wnt6, Wnt10a, Wnt14, and Wnt16, as well as the Wnt receptor Fzd3, are highly expressed in CLL, compared with normal B cells, which suggests that the Wnt signaling pathway is active in CLL49 and contributes to the survival of CLL cells50. In our study, a potent inhibitor for the palmitoylation of Wnt proteins were used to block the activation of Wnt/β-catenin pathway and we found that the effect of TFPI on both CXCR7 expression and TEM were impaired. It suggests that the activation of canonical Wnt/β-catenin pathway is involved in the CLL cell migration.
In conclusion, our results indicate that TFPI may enhance the capacity of CLL cells to transmigrate through multiple vascular endothelial beds and potentially contribute to organ infiltration.
Supplementary Information
Acknowledgements
The work was supported by the South-Eastern Norway Regional Health Authority in Norway (Grant No. 2016080). The development of the HS20 human monoclonal antibody was surported by the Intramural Research Program of the National Institutes of Health (NIH), National Cancer Institute (NCI) (Z01 BC010891 and ZIA BC010891 to MH). We thank Marie-Christine Mowinckel, Vigdis Bjerkeli, Kari Otterdal, Anna Lång and Yang Jin for their technical assistance. We thank Eva Marie Jacobsen for clinical support.
Author contributions
X.Y.C. designed and performed the experiments, analysed the data, wrote the manuscript; G.E.T. diagnosed CLL, studied patient characteristics, provided patient samples, and edited the manuscript; S.M.K., A.E.A.D. and N.I. contributed to the experiment design and paper revision; C.F.M. contributed to experiments and paper revision; L.S. and Z.X.J. were involved in the experiment design and paper revision; T.U. analysed the patient data and edited the manuscript. J.C. provided CXCR7 inhibitor CCX771 and was involved in the relative experiment design; M.H. supplied GPC3 antibodies and discussed the analysed data; P.M.S. supervised all the research and edited the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Xue Yan Cui, Email: x.y.cui@medisin.uio.no.
Per Morten Sandset, Email: p.m.sandset@medisin.uio.no.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-84695-8.
References
- 1.Hallek M, Shanafelt TD, Eichhorst B. Chronic Lymphocytic Leukaemia. New York: Springer; 2018. [DOI] [PubMed] [Google Scholar]
- 2.Fecteau JF, Kipps TJ. Structure and function of the hematopoietic cancer niche: Focus on chronic lymphocytic leukemia. Front. Biosci. 2012;4:61–73. doi: 10.2741/s251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Panayiotidis P, Jones D, Ganeshaguru K, Foroni L, Hoffbrand AV. Human bone marrow stromal cells prevent apoptosis and support the survival of chronic lymphocytic leukaemia cells in vitro. Br. J. Haematol. 1996;92(1):97–103. doi: 10.1046/j.1365-2141.1996.00305.x. [DOI] [PubMed] [Google Scholar]
- 4.Hayden RE, Pratt G, Roberts C, Drayson MT, Bunce CM. Treatment of chronic lymphocytic leukemia requires targeting of the protective lymph node environment with novel therapeutic approaches. Leuk. Lymphoma. 2012;53(4):537–549. doi: 10.3109/10428194.2011.610014. [DOI] [PubMed] [Google Scholar]
- 5.Davids MS, Burger JA. Cell trafficking in chronic lymphocytic leukemia. Open J. Hematol. 2012;3:S1. doi: 10.13055/ojhmt_3_S1_03.120221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoellenriegel J, Zboralski D, Maasch C, et al. The Spiegelmer NOX-A12, a novel CXCL12 inhibitor, interferes with chronic lymphocytic leukemia cell motility and causes chemosensitization. Blood. 2014;123(7):1032–1039. doi: 10.1182/blood-2013-03-493924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Agarwal A, Cooke L, Riley C, Qi W, Mount D, Mahadevan D. Genetic and cytokine changes associated with symptomatic stages of CLL. Leuk. Res. 2014;38(9):1097–1101. doi: 10.1016/j.leukres.2014.05.017. [DOI] [PubMed] [Google Scholar]
- 8.Bleul CC, Fuhlbrigge RC, Casasnovas JM, Aiuti A, Springer TA. A highly efficacious lymphocyte chemoattractant, stromal cell-derived factor 1 (SDF-1) J. Exp. Med. 1996;184(3):1101–1109. doi: 10.1084/jem.184.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Torossian F, Anginot A, Chabanon A, et al. CXCR7 participates in CXCL12-induced CD34+ cell cycling through beta-arrestin-dependent Akt activation. Blood. 2014;123(2):191–202. doi: 10.1182/blood-2013-05-500496. [DOI] [PubMed] [Google Scholar]
- 10.Burns JM, Summers BC, Wang Y, et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med. 2006;203(9):2201–2213. doi: 10.1084/jem.20052144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hattermann K, Held-Feindt J, Lucius R, et al. The chemokine receptor CXCR7 is highly expressed in human glioma cells and mediates antiapoptotic effects. Cancer Res. 2010;70(8):3299–3308. doi: 10.1158/0008-5472.CAN-09-3642. [DOI] [PubMed] [Google Scholar]
- 12.Infantino S, Moepps B, Thelen M. Expression and regulation of the orphan receptor RDC1 and its putative ligand in human dendritic and B cells. J. Immunol. 2006;176(4):2197–2207. doi: 10.4049/jimmunol.176.4.2197. [DOI] [PubMed] [Google Scholar]
- 13.Zabel BA, Lewen S, Berahovich RD, Jaen JC, Schall TJ. The novel chemokine receptor CXCR7 regulates trans-endothelial migration of cancer cells. Mol. Cancer. 2011;10:73. doi: 10.1186/1476-4598-10-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartmann TN. CLL cells under flow. Blood. 2014;123(23):3533–3534. doi: 10.1182/blood-2014-04-565358. [DOI] [PubMed] [Google Scholar]
- 15.Wood JP, Ellery PE, Maroney SA, Mast AE. Biology of tissue factor pathway inhibitor. Blood. 2014;123(19):2934–2943. doi: 10.1182/blood-2013-11-512764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischer EG, Riewald M, Huang HY, et al. Tumor cell adhesion and migration supported by interaction of a receptor-protease complex with its inhibitor. J. Clin. Invest. 1999;104(9):1213–1221. doi: 10.1172/JCI7750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hembrough TA, Ruiz JF, Swerdlow BM, et al. Identification and characterization of a very low density lipoprotein receptor-binding peptide from tissue factor pathway inhibitor that has antitumor and antiangiogenic activity. Blood. 2004;103(9):3374–3380. doi: 10.1182/blood-2003-07-2234. [DOI] [PubMed] [Google Scholar]
- 18.Stavik B, Skretting G, Aasheim HC, et al. Downregulation of TFPI in breast cancer cells induces tyrosine phosphorylation signaling and increases metastatic growth by stimulating cell motility. BMC Cancer. 2011;11:357. doi: 10.1186/1471-2407-11-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shimizu Y, Suzuki T, Yoshikawa T, et al. Cancer immunotherapy-targeted glypican-3 or neoantigens. Cancer Sci. 2018;109(3):531–541. doi: 10.1111/cas.13485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khurana S, Margamuljana L, Joseph C, Schouteden S, Buckley SM, Verfaillie CM. Glypican-3-mediated inhibition of CD26 by TFPI: A novel mechanism in hematopoietic stem cell homing and maintenance. Blood. 2013;121(14):2587–2595. doi: 10.1182/blood-2012-09-456715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Capurro M, Martin T, Shi W, Filmus J. Glypican-3 binds to Frizzled and plays a direct role in the stimulation of canonical Wnt signaling. J. Cell Sci. 2014;127(Pt 7):1565–1575. doi: 10.1242/jcs.140871. [DOI] [PubMed] [Google Scholar]
- 22.Krishnamurthy N, Kurzrock R. Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat. Rev. 2018;62:50–60. doi: 10.1016/j.ctrv.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ring A, Kim YM, Kahn M. Wnt/catenin signaling in adult stem cell physiology and disease. Stem Cell Rev. 2014;10(4):512–525. doi: 10.1007/s12015-014-9515-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: A report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111(12):5446–5456. doi: 10.1182/blood-2007-06-093906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao W, Kim H, Feng M, et al. Inactivation of Wnt signaling by a human antibody that recognizes the heparan sulfate chains of glypican-3 for liver cancer therapy. Hepatology. 2014;60(2):576–587. doi: 10.1002/hep.26996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rai KR, Sawitsky A, Cronkite EP, Chanana AD, Levy RN, Pasternack BS. Clinical staging of chronic lymphocytic leukemia. Blood. 1975;46(2):219–234. doi: 10.1182/blood.V46.2.219.219. [DOI] [PubMed] [Google Scholar]
- 27.Binet JL, Auquier A, Dighiero G, et al. A new prognostic classification of chronic lymphocytic-leukemia derived from a multivariate survival analysis. Cancer. 1981;48(1):198–206. doi: 10.1002/1097-0142(19810701)48:1<198::AID-CNCR2820480131>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 28.Dahm A, Van Hylckama VA, Bendz B, Rosendaal F, Bertina RM, Sandset PM. Low levels of tissue factor pathway inhibitor (TFPI) increase the risk of venous thrombosis. Blood. 2003;101(11):4387–4392. doi: 10.1182/blood-2002-10-3188. [DOI] [PubMed] [Google Scholar]
- 29.Cui XY, Skretting G, Jing Y, Sun H, Sandset PM, Sun L. Hypoxia influences stem cell-like properties in multidrug resistant K562 leukemic cells. Blood Cells Mol. Dis. 2013;51(3):177–184. doi: 10.1016/j.bcmd.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 30.Cui XY, Tinholt M, Stavik B, et al. Effect of hypoxia on tissue factor pathway inhibitor expression in breast cancer. J. Thromb. Haemost. 2016;14(2):387–396. doi: 10.1111/jth.13206. [DOI] [PubMed] [Google Scholar]
- 31.Zabel BA, Wang Y, Lewen S, et al. Elucidation of CXCR7-mediated signaling events and inhibition of CXCR4-mediated tumor cell transendothelial migration by CXCR7 ligands. J. Immunol. 2009;183(5):3204–3211. doi: 10.4049/jimmunol.0900269. [DOI] [PubMed] [Google Scholar]
- 32.Stamatopoulos B, Meuleman N, De Bruyn C, et al. AMD3100 disrupts the cross-talk between chronic lymphocytic leukemia cells and a mesenchymal stromal or nurse-like cell-based microenvironment: Pre-clinical evidence for its association with chronic lymphocytic leukemia treatments. Haematologica. 2012;97(4):608–615. doi: 10.3324/haematol.2011.052779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mast AE, Higuchi DA, Huang ZF, Warshawsky I, Schwartz AL, Broze GJ., Jr Glypican-3 is a binding protein on the HepG2 cell surface for tissue factor pathway inhibitor. Biochem J. 1997;327(Pt 2):577–583. doi: 10.1042/bj3270577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song HH, Shi W, Xiang YY, Filmus J. The loss of glypican-3 induces alterations in Wnt signaling. J. Biol. Chem. 2005;280(3):2116–2125. doi: 10.1074/jbc.M410090200. [DOI] [PubMed] [Google Scholar]
- 35.Mohle R, Failenschmid C, Bautz F, Kanz L. Overexpression of the chemokine receptor CXCR4 in B cell chronic lymphocytic leukemia is associated with increased functional response to stromal cell-derived factor-1 (SDF-1) Leukemia. 1999;13(12):1954–1959. doi: 10.1038/sj.leu.2401602. [DOI] [PubMed] [Google Scholar]
- 36.Melo RCC, Ferro KPV, Duarte A, Olalla Saad ST. CXCR7 participates in CXCL12-mediated migration and homing of leukemic and normal hematopoietic cells. Stem Cell Res. Ther. 2018;9(1):34. doi: 10.1186/s13287-017-0765-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gu B, Dao LP, Wiley J. Impaired transendothelial migration of B-CLL lymphocytes: A defect linked to low L-selectin expression. Leuk. Lymphoma. 2001;42(1–2):5–12. doi: 10.3109/10428190109097671. [DOI] [PubMed] [Google Scholar]
- 38.Trinidad EM, Ballesteros M, Zuloaga J, Zapata A, Alonso-Colmenar LM. An impaired transendothelial migration potential of chronic lymphocytic leukemia (CLL) cells can be linked to ephrin-A4 expression. Blood. 2009;114(24):5081–5090. doi: 10.1182/blood-2009-03-210617. [DOI] [PubMed] [Google Scholar]
- 39.O'Hayre M, Salanga CL, Kipps TJ, Messmer D, Dorrestein PC, Handel TM. Elucidating the CXCL12/CXCR4 signaling network in chronic lymphocytic leukemia through phosphoproteomics analysis. PLoS ONE. 2010;5(7):e11716. doi: 10.1371/journal.pone.0011716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levoye A, Balabanian K, Baleux F, Bachelerie F, Lagane B. CXCR7 heterodimerizes with CXCR4 and regulates CXCL12-mediated G protein signaling. Blood. 2009;113(24):6085–6093. doi: 10.1182/blood-2008-12-196618. [DOI] [PubMed] [Google Scholar]
- 41.Wang Y, Li G, Stanco A, et al. CXCR4 and CXCR7 have distinct functions in regulating interneuron migration. Neuron. 2011;69(1):61–76. doi: 10.1016/j.neuron.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen Q, Zhang M, Li Y, et al. CXCR7 mediates neural progenitor cells migration to CXCL12 independent of CXCR4. Stem Cells. 2015;33(8):2574–2585. doi: 10.1002/stem.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kalatskaya I, Berchiche YA, Gravel S, Limberg BJ, Rosenbaum JS, Heveker N. AMD3100 is a CXCR7 ligand with allosteric agonist properties. Mol. Pharmacol. 2009;75(5):1240–1247. doi: 10.1124/mol.108.053389. [DOI] [PubMed] [Google Scholar]
- 44.Ahamed J, Belting M, Ruf W. Regulation of tissue factor-induced signaling by endogenous and recombinant tissue factor pathway inhibitor 1. Blood. 2005;105(6):2384–2391. doi: 10.1182/blood-2004-09-3422. [DOI] [PubMed] [Google Scholar]
- 45.Filmus J, Capurro M. The role of glypicans in Hedgehog signaling. Matrix Biol. 2014;35:248–252. doi: 10.1016/j.matbio.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 46.Capurro M, Shi W, Izumikawa T, Kitagawa H, Filmus J. Processing by convertases is required for glypican-3-induced inhibition of Hedgehog signaling. J. Biol. Chem. 2015;290(12):7576–7585. doi: 10.1074/jbc.M114.612705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gao W, Xu Y, Liu J, Ho M. Epitope mapping by a Wnt-blocking antibody: evidence of the Wnt binding domain in heparan sulfate. Sci. Rep. 2016;6:26245. doi: 10.1038/srep26245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang K, Wang X, Zhang H, et al. The evolving roles of canonical WNT signaling in stem cells and tumorigenesis: Implications in targeted cancer therapies. Lab. Invest. 2016;96(2):116–136. doi: 10.1038/labinvest.2015.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu D, Zhao Y, Tawatao R, et al. Activation of the Wnt signaling pathway in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA. 2004;101(9):3118–3123. doi: 10.1073/pnas.0308648100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang L, Shalek AK, Lawrence M, et al. Somatic mutation as a mechanism of Wnt/beta-catenin pathway activation in CLL. Blood. 2014;124(7):1089–1098. doi: 10.1182/blood-2014-01-552067. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.