

Abstract
Sulfur dioxide (SO2) has emerged as a physiological relevant signaling molecule that plays a prominent role in regulating vascular functions. However, molecular mechanisms whereby SO2 influences its upper-stream targets have been elusive. Here we show that SO2 may mediate conversion of hydrogen peroxide (H2O2) to a more potent oxidant, peroxymonosulfite, providing a pathway for activation of H2O2 to convert the thiol group of protein cysteine residues to a sulfenic acid group, aka cysteine sulfenylation. By using site-centric chemoproteomics, we quantified >1000 sulfenylation events in vascular smooth muscle cells in response to exogenous SO2. Notably, ~42% of these sulfenylated cysteines are dynamically regulated by SO2, among which is cysteine-64 of Smad3 (Mothers against decapentaplegic homolog 3), a key transcriptional modulator of transforming growth factor β signaling. Sulfenylation of Smad3 at cysteine-64 inhibits its DNA binding activity, while mutation of this site attenuates the protective effects of SO2 on angiotensin II-induced vascular remodeling and hypertension. Taken together, our findings highlight the important role of SO2 in vascular pathophysiology through a redox-dependent mechanism.
Keywords: SO2, Sulfenylation, Smad3, Cysteine, Vascular remodeling
Graphical abstract
Highlights
-
•
Vascular smooth muscle cell-derived endogenous SO2 regulates vascular remodeling and hypertension in vivo.
-
•
SO2 may facilitate H2O2-mediated protein cysteine oxidation.
-
•
Chemoproteomics reveals Smad3C64 as a target of SO2-dependent sulfenylation.
-
•
Smad3C64 is required for SO2-dependent regulation of vascular functions in vivo.
Abbreviations
- AAT1
Aspartate aminotransferase 1
- AAT1f/f
AAT1 flox/flox
- APEX1
DNA-apurinic or apyrimidinic site endonuclease
- Bisulfite
HSO3‾
- C64
Cysteine-64
- C64S
Mutation of cysteine-64 to serine
- CFL1
Cofilin
- CO2
Carbon dioxide
- CuAAC
Copper catalyzed alkyne azide cycloaddition
- CV
Coefficient of variation
- DFI
Diagnostic fragment ion
- EMSA
Electrophoretic mobility shift assay
- GO
Gene ontology
- HCO3‾
Carbonates
- HE
Haematoxylin and eosin
- H2O2
Hydrogen peroxide
- (HOO)SO2‾
Peroxymonosulfurous acid anion
- HPLC-FD
High-performance liquid chromatography with fluorescence detector
- H2S
Hydrogen sulfide
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- LC-MS/MS
Liquid chromatography-tandem mass spectrometry
- MeCN
Acetonitrile
- NO
Nitric oxide
- OOH‾
Hydroperoxide ion
- PRDX
Peroxiredoxin
- PTPN1
Tyrosine-protein phosphatase non-receptor type 1
- ROS
Reactive oxygen species
- RT
Room temperature
- SBP
Systolic blood pressure
- SCX
Strong cation exchange
- shMOCK
A mock lentiviral vector containing a scramble shRNA sequence
- shRNA
Small hairpin RNA
- SM22α
Smooth muscle 22 alpha
- Smad3
Mothers against decapentaplegic homolog 3
- SO2
Sulfur dioxide
- SOH
Sulfenic acid
- SO2H
Sulfinic acid
- Sulfite
SO32‾
- TgAAT1
AAT1 transgene
- WT
Wild-type
- VSMC
Vascular smooth muscle cell
1. Introduction
Sulfur dioxide (SO2), a major environmental pollutant produced from industrial processes, has been historically considered to be a hazard to health. Epidemiological surveys suggest that an excess or long-term exposure to airborne SO2 results in detrimental effects and is associated with numerous human diseases [1]. Interestingly, SO2 can also be physiologically generated from sulfur-containing small molecules in biological systems. For instance, aspartate aminotransferase (AAT1) mediates transamination of l-cysteinesulfinate to yield β-sulfinylpyruvate, which then spontaneously decomposes to pyruvate and SO2 [2]. In addition, oxidation-dependent conversion of hydrogen sulfide (H2S) represents another route of endogenous SO2 production [3]. In physiological conditions, SO2 is hydrated rapidly to give sulfite (SO32‾) and bisulfite (HSO3‾), and the molar ratio of the latter two ionic forms is expected to be mostly 2.5:1 [4]. In this regard, we and others have confirmed the endogenous production of SO2 and/or its sulfite derivatives (SO32‾/HSO3‾) in body fluids and tissues [[5], [6], [7], [8], [9]].
In the past decade, emerging evidence has stablished that gaseous SO2 is likely to be a physiological relevant signaling molecule in mammals [5,[10], [11], [12], [13]]. Like other gasotransmitters such as H2S and NO (nitric oxide) [[14], [15], [16]], SO2 has a prominent role in regulating vascular functions and is involved in inflammatory responses. Mounting evidence has shown that exogenous infusion of SO2 donors (SO32‾/HSO3‾) can alleviate atherosclerosis [17], myocardial injury [18], ischemia reperfusion [19], hypoxic pulmonary hypertension [20] in vivo or ex vivo. However, whether and how vascular smooth muscle cell (VSMC)-derived endogenous SO2 affects vascular homeostasis under physiological and pathophysiological conditions in vivo remain largely unexplored. Alternatively, in attempts to explore a causal relationship between endogenous SO2 and its vascular functions, we previously developed several VSMC models by genetically manipulating the levels of SO2-generating enzymes, like AAT1, and showed that endogenous SO2 suppresses abnormal proliferation of VSMCs by inducing apoptosis and/or collagen degradation in cellulo [11,21,22]. Nonetheless, molecular mechanisms whereby endogenous SO2 influences its upper-stream targets have been elusive.
Here, by using newly developed transgenic mouse models, we first demonstrate that VSMC-derived endogenous SO2 indeed regulates vascular remodeling and hypertension in vivo. We further show that SO2 can augment hydrogen peroxide (H2O2) signal through peroxymonosulfite formation, leading to global and site-specific redox changes in cysteine residues across the VSMC proteome. Among these changes is SO2-dependent dynamic sulfenylation of C64 in Smad3 (Mothers against decapentaplegic homolog 3), a key transcriptional modulator of transforming growth factor β signaling. Finally, we demonstrate that this residue is required for SO2-mediated vascular remodeling and hypertension. Taken together, our findings highlight the important role of SO2 in vascular pathophysiology through a redox-dependent mechanism. More generally, the molecular link between SO2 and H2O2 may provide new insights into the mechanism underlying redox signaling and transduction.
2. Materials and methods
2.1. Chemicals
BTD, a benzothiazine-based probe specific for cysteine sulfenic acid, was prepared as previously described [23]; Iodoacetamide (IAM, V900335), Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA) (678937), and Sodium Ascorbate (A7631) were purchased from Sigma-Aldrich; DAz-2 (13382) and phosphine-biotin (13581) were purchased from Cayman Chemical; Light Azido-UV-Biotin (EVU102) and Heavy Azido-UV-Biotin (EVU151) were purchased from KeraFast; CuSO4 (C493-500) was purchased from Thermo Fisher Scientific; SS-1 (Fig. S4B), a chemoselective fluorescent probe for visualizing SO2 in situ was developed and kindly gifted by Professor Kun Li, Sichuan University (Sichuan, China) [24].
2.2. Antibodies
Anti-AAT1 (SAB2500473), Anti-AAT2 (AV43518), Anti-type I collagen (SAB4500362), Anti-type III collagen (SAB4500367) and secondary antibodies conjugated horseradish peroxidase (Anti-goat IgG-peroxidase antibody: A5420, Anti-mouse IgG-peroxidase antibody: A4416, and Anti-rabbit IgG-peroxidase antibody: A0545) were purchased from Sigma-Aldrich (St Louis, MO, USA); Anti-p-Smad3 (9520S) and Anti-Smad3 (9523S) were purchased from Cell Signaling Technology; Anti-type I collagen (ab34710), Anti-type III collagen (ab7778), Anti-p-Smad3 (ab52903), Anti-MCP-1 (ab9669), Anti-MMP-2 (ab92536) and Anti-CD31 (ab28364) were purchased from Abcam (Cambridge, MA, USA); Anti-GAPDH (KC-5G4) was purchased from Kangcheng (Shanghai, China); Anti-ICAM-1 (sc8439) was purchased from Santa Cruz Biotechnology (Dallas, TX, USA); Anti-CD31 (ZS15060) and Anti-Alpha-smooth muscle actin (α-SMA) were purchased from ZSGB-Bio (Beijing, China); All Alexa Fluor secondary antibodies were purchased from Invitrogen (Carlsbad, CA, USA).
Anti-AAT1 or anti-AAT2 was diluted at 1:1000 for western blotting and 1:100 for immunofluorescence analysis. Anti-p-Smad3 (9520S) was diluted at 1:500 for western blotting. Anti-p-Smad3 (ab52903) was diluted at 1:50 for immunofluorescence analysis. Anti-Smad3 was diluted at 1:1000 for western blotting and 1:50 for immunofluorescence analysis. Anti-type I collagen (ab34710) or anti-type III collagen (ab7778) was diluted at 1:1000 for western blotting. Anti-type I collagen (SAB4500362) or anti-type III collagen (SAB4500367) was diluted at 1:100 for immunofluorescence analysis. Anti-GAPDH was diluted at 1:5000 for western blotting analysis. Anti-ICAM-1, anti-MCP-1 or anti-MMP-2 was diluted at 1:100 for immunofluorescence analysis. Anti-CD31 or anti-α-SMA was diluted at 1:200 for immunofluorescence analysis. All three secondary antibodies conjugated peroxidase antibodies were diluted at 1:6000 for western blotting analysis. The Alexa Fluor secondary antibodies were diluted at 1:500 for immunofluorescence analysis.
2.3. Ethics approval
All animal experiments were approved by Peking University First Hospital Animal Ethics Committee (J201417) and complied with the US National Institutes of Health Guide for the Care and Use of Laboratory.
2.4. Animals
The VSMC-specific AAT1 transgenic mouse line was generated through microinjection of full-length mouse AAT1 cDNA. AAT1 gene expression was governed by the smooth muscle 22 alpha (SM22α) promoter. PCR for the genotyping of transgenic mice was conducted with the following primers: 5′-ACGGCAGAGGGGTGACATCA-3′ and 5′-CGCACAGGCATGGAGGACAA-3'.
The VSMC-specific AAT1 knockout mouse line was generated with Cre/LoxP technology. Tamoxifen-inducible SM22α-Cre transgenic (SM22α-CreERT2) mice and AAT1flox/flox (AAT1f/f) mice were produced by Cyagen Biosciences Inc. (Guangzhou, China). Crossing SM22α-CreERT2+ mice with AAT1f/f mice generate SM22α-CreERT2+ AAT1f/f mice with AAT1 gene specifically deleted in vascular smooth muscle cells (VSMC-AAT1−/−). Their littermates with SM22α-CreERT2- AAT1f/f genotype were chosen as control mice. PCR for the genotyping of this knockout mice was conducted with SM22α-CreERT2 primers (5′-TGTAGAGAAGGCACTTAGC-3′ and 5′-CCAGGTTACGGATATAGTTCA-3′) and AAT1f/f primers (5′-TAATTATGGCTGTATCCGGAGCTCAG-3′ and 5′-CCAAAGCAGTGGCAGAGATGAGTC-3′). Eight-week-old mice were intraperitoneally injected with tamoxifen (2.5 mg/day) dissolved in ethanol/peanut oil (1:19, v/v) for five consecutive days, three days apart and another five consecutive days [25,26].
Smad3C64S/WT mice were generated by Viewsolid Biotech (Beijing, China) using CRISPR/Cas9 technology [27]. One sgRNA was used to target exon 1 of Smad3 (5′-TGGTGTTCACGTTCTGCGTGG-3′). The genotypes of Smad3C64S/WT mice were verified by PCR amplification with primers (5′-GGCAAGTTCTCCAGAGTT-3′ and 5′-AGGCAAATCCGAAGGTTT-3′) and direct sequencing after the extraction of genomic DNA from mouse tail tissues. All these mouse lines were generated in a C57BL/6J background.
2.5. Angiotensin II-induced hypertension
In order to induce vascular remodeling [28] and hypertension, male mice at 10–12 weeks of age were subcutaneously implanted with osmotic minipumps (Alzet Model 2002; Alza Corp. Palo Alto, CA) to deliver Ang II (Kang Tai Co., Beijing, China) at a rate of 500 ng/kg/min for two weeks [29]. Starting on the first day of implantation of osmotic minipumps, the mice were intraperitoneally injected with vehicle or SO2 donors (Na2SO3: 68.04 mg/kg; NaHSO3: 18.72 mg/kg body weight, freshly dissolved in saline) daily in these two weeks [30].
2.6. Blood pressure assessment
Blood pressure was measured with two methods, including the tail-cuff technique (Blood Pressure Meter, BP-2010 Series, Softron, Tokyo, Japan) [31] and telemetric blood pressure assessment [29] (Data Science International, DSI, Tilburg, The Netherlands). For the latter, mice were anesthetized with sodium pentobarbital (50 mg/kg). A catheter was inserted into the left carotid artery and advanced to the thoracic aorta. The telemetric transducer (model HD-X11, DSI) was embedded in the abdominal subcutaneous. One week after telemetry implantation, the conscious mice were put on the DSI receiver platforms and their blood pressures were recorded continuously. Data were collected through Ponemah Physiology Platform software (version 6.20.00513.2, DSI).
2.7. Histopathology
Mice aortas and brown adipose tissues were fixed in 4% paraformaldehyde. Haematoxylin and eosin (HE) staining was applied to paraffin sections (5 μm) of brown adipose tissue. Weigert's elastic fiber reagent staining (Beijing Leagene Biological Technology Company, China) was applied to paraffin aorta sections. Four points at 12, 3, 6 and 9 o'clock clockwise were selected for each aorta section to measure the aortic medial thickness, and the average values were calculated. The aortic medial areas were obtained using classic Barth's methods.
2.8. Culture of VSMCs
The rat VSMC line (A7r5, CRL-1444) was purchased from National Infrastructure of Cell Line Resource (China) and cultured in DMEM containing 10% FBS at 37 °C in 5% CO2.
2.9. Genetic manipulation of AAT1 in VSMCs
An adenovirus vector carrying AAT1 cDNA (Vigene Bioscience, Jinan, China) was constructed to overexpress AAT1 [13]. Vehicle recombinant adenovirus was used as a control. In brief, VSMCs were seeded in 6-well plates to 60–80% confluence, and then infected with virus (multiplicity of infection: 20) for 48 h.
Lentivirus-delivered small hairpin RNA (shRNA) targeting AAT1 (Cyagen Bioscience, Guangzhou, China) was prepared to silence AAT1. A mock lentiviral vector containing a scramble shRNA sequence (shMOCK) was used as a negative control. In brief, VSMCs were seeded in 25 cm2 culture flasks to 50% confluence, and then infected with the lentiviral at the concentration of 1 × 106 TU/mL. After 72 h, VSMCs were treated with puromycin (2 μg/mL) for 2 weeks to screen cells stably transfected with lentivirus.
2.10. Construction of Smad3-deficient VSMCs
VSMCs were seeded in 25 cm2 culture flasks to 50% confluence, and then infected with the lentivirus-delivered Smad3 shRNA (Cyagen Bioscience, Guangzhou, China) at the concentration of 1 × 106 TU/mL. After 72 h, the transfected cells were incubated in DMEM containing 2 μg/mL of puromycin for 2 weeks to screen stably transformed cells.
2.11. Plasmid transfection
The pCMV-Entry vectors for wild-type (WT) Smad3 and C64S (mutation of cysteine-64 to serine) were purchased from OriGene Technologies (Rockville, MD, USA). Smad3-deficient VSMCs grown to 50% confluence were transfected with WT or C64S Smad3 plasmid using jetPEI™ transfection kit (Polyplutransfection, Illkirch, France).
2.12. HPLC-FD for measuring SO2
SO2 levels in plasma, aortic tissues and VSMC supernatants were measured by high-performance liquid chromatography with fluorescence detector (HPLC-FD, Agilent Technologies, Palo Alto, CA, USA) [22]. In brief, aortic tissues were homogenized in PBS using a tissue lyser, centrifuged, and the clear supernatants were collected. The sulfite in plasma, tissue homogenates or VSMC supernatants were reduced to sulfhydryl compounds by sodium borohydride, and then labeled with monobromobimane to generate fluorescent derivative. Perchloric acid were added to deproteinize. After centrifugation, the clear supernatants were neutralized by Tris-HCl (pH 3.0) and prepared for HPLC-FD. The fluorescent derivatives were separated by chromatographic column and detected at excitation/emission wavelength of 392/479 nm.
2.13. In situ fluorescent imaging of SO2
Endogenous SO2 in VSMCs was measured with the SS-1 probe as previously described [22]. VSMCs were treated with 10 μM probe for 1 h at 37 °C. Cells were then washed with PBS to remove excessive probe and fixed by paraformaldehyde. Blue fluorescence detected with an Olympus confocal laser scanning microscope was considered positive signals.
2.14. Immunofluorescence staining
Frozen sections of mouse aorta were blocked and incubated with primary antibodies overnight at 4 °C, respectively. The washed sections were then treated with secondary antibodies (Invitrogen, Carlsbad, CA, USA) for 1.5 h away from light. After washing, sections were stained with DAPI (ZSGB-Bio, ZLI-9557). The fluorescence signals were captured under an Olympus confocal laser scanning microscope.
VSMCs were fixed with 4% formaldehyde, washed with PBS, blocked and permeabilized, and then treated with Smad3 antibody (Cell Signaling Technology, CST, Danvers, MA, USA, 9523S) overnight at 4 °C [32]. Then, the cells were washed three times and treated with secondary antibody for 90 min. The fluorescence signals were also captured under microscope.
2.15. Western blotting
Western blotting was conducted following a standard protocol [22]. Nitrocellulose membranes were treated with primary antibodies as indicated for 16 h at 4 °C. Then, they were treated with their specific secondary antibodies conjugated horseradish peroxidase. Protein bands were observed with Enhanced Chemiluminescent Western Blotting Substrate Kit (GE Healthcare, Pittsburgh, PA, USA) on a FluorChem M MultiFluor System (Protein Simple, San Francisco, CA, USA). Uncropped images are provided in Fig. S9.
2.16. Sample preparation for quantitative analysis of the CRL-1444 sulfenylome (the entire complement of protein sulfenic acids)
CRL-1444 cells pre-stimulated with Ang II were treated with or without 100 μM SO2 donors (SO32‾: HSO3‾ = 2.5:1), harvested, lysed in pre-chilled and degassed HEPES (50 mM, pH 7.6) buffer containing 150 mM NaCl, 1% IGEPAL, 1 x protease and phosphatase inhibitors (Thermo Fisher Scientific, A32961), and 200 unit/mL catalase (Sigma-Aldrich), and processed according to a previously reported protocol [33]. In brief, cell lysates were first incubated with 5 mM of the SOH-specific probe BTD at 37 °C for 2 h with rotation and light protection, followed by iodoacetamide-based alkylation. After precipitation with a methanol-chloroform system, proteins were resuspended with 50 mM ammonium bicarbonate containing 0.2 M urea and digested with sequencing grade trypsin (Promega) at a 1:50 (enzyme/substrate) ratio overnight at 37 °C. The tryptic digests were desalted, evaporated to dryness, and resuspended in a water solution containing 30% acetonitrile (MeCN). CuAAC reaction was then performed by subsequently adding 1 mM either light or heavy Azido-UV-biotin (1 μL of a 40 mM stock), 10 mM sodium ascorbate (4 μL of a 100 mM stock), 1 mM TBTA (1 μL of a 50 mM stock), and 10 mM CuSO4 (4 μL of a 100 mM stock). After 2 h incubation at room temperature (RT), the light and heavy reaction mixtures were then combined, cleaned with strong cation exchange (SCX), and then subjected to the enrichment with streptavidin beads for 2 h at RT. Streptavidin beads were washed with 50 mM NaAc (pH4.5), 50 mM NaAc containing 2 M NaCl (pH 4.5), and deionized water twice each with vortexing and/or rotation to remove non-specific binding substances, then resuspended in 25 mM ammonium bicarbonate, photoreleased under 365 nm UV light for 2 h at RT with magnetic stirring. The supernatant was collected, dried under vacuum, and stored at −20 °C until LC-MS/MS analysis.
2.17. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis
The dried peptide samples were resuspended in 8 μL 0.1% formic acid (FA) and analyzed by LC-MS/MS on a Q ExactiveTM HF-X (Thermo Fisher Scientific) equipped with a UltiMateTM 3000 RSLCnano system (Thermo Fisher Scientific) for online sample handling and peptide separations. Then 6 μL of sample was loaded onto a fused-silica nano-ESI column (360 μm OD × 150 μm ID) with a needle tip (3–5 μM) packed to a length of 15 cm with a C18 reverse phase resin (AQ 1.9 μm, 120 Å, ReproSil-Pur).
The peptides were separated using an 88-min linear gradient from 6% to 95% buffer B (80% MeCN) equilibrated with buffer A (0.1% formic acid) at a flow rate of 600 nL/min across the column. The scan sequence for the Orbitrap began with an MS1 spectrum (resolution: 120,000, scan range: 350–1550 m/z, AGC target: 3 × 106, maximum injection time: 20 ms, dynamic exclusion: 15 s). The “Top25” precursors was selected for MS2 analysis, in which precursors were fragmented by higher-energy collisional dissociation (HCD) ((N)CE: 27, AGC target: 2 × 104, maximum injection time: 30 ms, resolution: 15,000, and isolation window: 1.6 Da). The thermo.raw files can be obtained from iProX database [34] (www.iprox.org, accession number IPX0002494000).
2.18. Peptide identification and quantification
Raw data files were searched against Rattus norvegicus Uniprot canonical database using PEAKS studio (A customized version based on v10.5). Data refinement was enabled to associate feature with chimera/mixed scan and to keep features with charge between 2 and 5. Precursor ion mass and fragmentation tolerance were set as 10 ppm and 0.2 Da, respectively. The maximum number of modifications and missed cleavages allowed per peptide were both set as three. Mass shifts of +15.9949 Da (methionine oxidation), +57.0214 Da (iodoacetamide alkylation) and +418.131 (C19H22N4O5S) for BTD-triazohexanoic acid were searched as variable modifications. A differential modification of 6.0201 Da on BTD-derived modification was used for stable-isotopic quantification. The FDRs were estimated by the program from the number and quality of spectral matches to the decoy database. The FDR threshold was set as 1%. Quantification results were obtained from three biological replicates.
2.19. Preparation of recombinant Smad3 proteins
Wild-type Smad3 recombinant protein (Smad3WT) and Smad3 in which C64 (Smad3C64S) was mutated to serine were purchased from Origene Technologies (Rockville, MD, USA). Briefly, the Smad3WT (or Smad3C64S) recombinant protein was produced with a TrueORFGold clone, RC208749 (or RC208749 C64S), encoding the full-length human SMAD3 (or with mutant C64S), with C-terminal Myc/DDK tag, from human HEK293 cells. These recombinant proteins were dissolved in solution containing 25 mM Tris-HCl (pH 7.3), 100 mM glycine, and 10% glycerol [35].
2.20. Validation of Smad3 sulfenylation
DAz-2 was used to label and detect Smad3 sulfenylation (Fig. S3) [36,37]. For cell lysates, VSMCs were lysed in non-denaturing lysis buffer (C1050, Applygen Technologies Inc., Beijing, China) supplemented with 5 mM DAz-2. After centrifugation, the supernatants were incubated at 37 °C for 2.5 h with gentle shaking. For aortic tissue homogenates, aortic tissues were homogenized in non-denaturing lysis solutions supplemented with 1 mM DAz-2 using a tissue lyser. After centrifugation at 16000 g for 4 min, the homogenates were further incubated at 37 °C for 2.5 h. The DAz-2-labeled protein samples were reacted with 250 μM phosphine-biotin for 2 h at 37 °C via the Staudinger-Bertozzi ligation. Acetone-based protein precipitation was used to remove excess biotin reagents from the reaction mixture. Afterward, the pelleted proteins were resuspended in non-denaturing lysis buffer and incubated with UltraLink™ Immobiolized NeutrAvidin™ (Thermo Fisher Scientific) at 4 °C for 4 h to capture biotinylated proteins. The NeutrAvidin-captured proteins were boiled in the non-denaturing sample buffer for 10 min, and then subjected to western blotting for detecting Smad3.
2.21. Colorimetric detection of Smad3-DNA binding activity
EpiQuik™ General Protein-DNA Binding Assay Kit (Epigentek, Farmingdale, NY, USA) was used to measure Smad3-DNA binding activity. After treatment, 23 μL of assay binding buffer, 20 ng of biotinylated oligonucleotides and 1 μg of purified Smad3 protein were added into strip well in turn, followed by the incubation for 60 min. The blank control only contained biotinylated oligonucleotides and assay binding buffer without purified protein. After incubation, each well was washed with 150 μL of washing buffer three times. Then, Smad3 antibody (1 μg/mL) was added and incubated for 1 h. After that, each well was washed with 150 μL of washing buffer four times. HRP-conjugated secondary antibody at 0.5 μg/mL was added, followed by the incubation for 0.5 h. Each well was then washed four times. Developing solution was added and incubated away from light until the blue color was developed in wells. Stop solution (50 μL) was added to stop the reaction. The absorbance was measured at 450 nm using a Thermo microplate reader and Skanlt RE 4.1 software.
2.22. Electrophoretic mobility shift assay (EMSA) detection of DNA binding activity of Smad3
Nuclear proteins were extracted by using the thermo nucleoprotein extraction kit according to the guide for users [38]. The probes (F: 5′-CCGCCAAGGGTCTAGACGACCGACC-3'; R: 5′-GGTCGGTCGTCTAGACCCTTGGCGG-3′) were annealed and end-labeled with DIG-ddUTP. The labeled probe was mixed with nuclear protein, and the mixture was incubated for 15 min. The protein-DNA complexes were loaded on a 5% polyacrylamide gel, electrically transferred onto a nylon membrane (Boehringer Mannheim Biochemica, Mannheim, Germany), and the chemiluminescent band was detected at the end of the experiment [39].
2.23. Statistical analysis
Both IBM SPSS 22 and GraphPad Prism 8 were used for statistical analysis. Data are shown as mean ± SD. The two-tailed Student's t-test was performed for the comparison between two groups, and ANOVA followed by Bonferroni post-doc analysis was performed for the comparison of multiple groups. Statistical significance is indicated by P < 0.05.
3. Results
3.1. VSMC-derived endogenous SO2 regulates vascular remodeling and hypertension in vivo
To test whether VSMC-derived endogenous SO2 affects vascular homeostasis under pathophysiological conditions in vivo, we first developed transgenic mice with VSMC-specific conditional knockout of AAT1, a key SO2-generating enzyme, through crossbreeding with wild type mice (aka AAT1f/f) where loxP sites were located on both sides of the second exon of AAT1 gene and SM22α-Cre-ERT2 transgenic mice (Fig. S1A). As expected, we observed a significant decrease in AAT1 protein expression in the aortic smooth muscle layers of VSMC-AAT1−/− mice (Fig. S1B). SO2 contents in both plasma and aorta of VSMC-AAT1−/− mice were also found to be lower than those of AAT1f/f mice (Fig. S1C and Fig. 1A). However, there was no significant difference in aortic AAT2 protein expression between VSMC-AAT1−/− mice and AAT1f/f mice (Fig. S1D). As compared to AAT1f/f, VSMC-AAT1−/− mice expressed in the aorta higher levels of collagen I/III, a hallmark of vascular remodeling (collagen I is associated with the tenacity and tensile strength of vascular wall, and collagen III is associated with the elasticity of vascular wall) (Fig. 1B). We next examined the effect of SO2 on blood pressure in AAT1 knockout mice using two orthogonal approaches, including the noninvasive tail-cuff [40] and invasive telemetry methods [41]. Notably, both experiments indicated that VSMC-AAT1−/− mice developed significantly higher systolic blood pressure (SBP) than the AAT1f/f animals (Fig. 1C and D).
Fig. 1.

VSMC-derived endogenous SO2 inhibits vascular remodeling and hypertension in vivo. (A) Comparison of endogenous SO2 levels in the aortic tissues AAT1f/f and VSMC-AAT1−/− mice (n = 6). SO2 was measured by HPLC-FD as described in the Materials and Methods. (B) Representative immunofluorescence staining of collagen I/III in aortas of AAT1f/f and VSMC-AAT1−/− mice. E, endothelium; M, media; and A, adventitia. Scale bar, 20 μm. (C) Comparison of the systolic blood pressure (SBP) of AAT1f/f and VSMC-AAT1−/− mice monitored by 24-h telemetry analysis (n = 4). Data represent mean values ± SEM. *P < 0.05. (D) Comparison of the SBP of AAT1f/f and VSMC-AAT1−/− mice measured by noninvasive tail-cuff method (n = 22). (E) Comparison of endogenous SO2 levels in the aortic tissues of wild type (WT) and TgAAT1 mice pretreated with or without Ang II (n = 10). (F) Representative immunofluorescence staining of aortic collagen I/III in WT or TgAAT1 mice. Mice were infused with Ang II or saline for two weeks. Scale bar, 20 μm. (G) Hart's staining of thoracic aorta (upper panel) and quantification (lower panels) the medial thickness and medial surface in WT and TgAAT1 mice infused with Ang II or saline (n = 12). Scale bar, 20 μm. (H) H&E staining of arterioles in brown fat of WT and TgAAT1 mice infused with Ang II or saline. Scale bar, 100 μm. (I) Comparison of changes in the SBP of WT and TgAAT1 mice upon Ang II stimulation (n = 12). SBP values were measured by noninvasive tail-cuff method. (J) Comparison of the SBP of WT and TgAAT1 mice after saline or angiotensin II infusion (n = 1). SBP values were measured by invasive 24-h telemetry analysis. The mice at 10 weeks of age were subcutaneously implanted with osmotic minipumps to deliver saline or angiotensin II. On the 7th day of saline or angiotensin II infusion, telemetry transmitters were implanted into the aortic arch of mice. On the 14th day of saline or angiotensin II infusion, the transmitters were turned on and data began to be collected. Blood pressure readings were acquired continuously for 24 h. For A, D, E, G, I and J, n value represents the number of animals used for each experiment, and data represent mean values ± SD. *P < 0.05. ns, not significant. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
On the other hand, we developed transgenic mice overexpressing AAT1, aka VSMC-TgAAT1 (Fig. S2A), as confirmed by the increased level of AAT1 in their aortic media (Fig. S2B). After 2-week Ang II infusion, a well-established approach to mimic pathophysiological hypertension, the aortic SO2 level also decreased substantially in wild-type (WT) mice, while such a pathophysiological change could be rescued in VSMC-TgAAT1 mice (Fig. 1E). Moreover, overexpression of AAT1 attenuated Ang II-induced upregulation of aortic collagen I/III, aortic medial thickness and medial surface (Fig. 1F and G). Notably, unlike WT mice, VSMC-TgAAT1 mice were resistant to Ang II-induced arterial remodeling, as measured by H&E staining (Fig. 1H). Noninvasive tail-cuff measurement and invasive telemetry analysis further indicated that VSMC-TgAAT1 mice were less prone to develop hypertension after Ang II infusion than wild type mice (Fig. 1I and J). Taken together, our results demonstrate that VSMC-derived SO2 regulates vascular remodeling and hypertension in vivo.
3.2. SO2 promotes dynamic changes in the sulfenylome of VSMCs
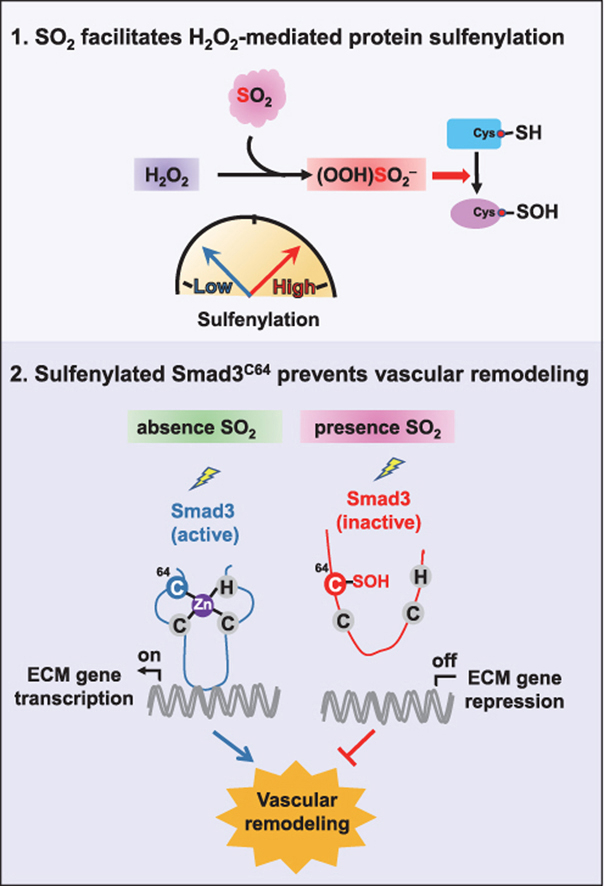
Inspired by the role of SO2 in regulating vascular functions, we next set up to get a better understanding of its underlying mechanism. It is known that SO2 reacts with sulfide to yield zero-valent sulfur [42], while it exhibits relatively low oxidative capacity as compared to other physiological oxidants, like hydrogen peroxide (H2O2). In other words, it is unlikely for SO2 to directly oxidize biomacromolecules like proteins. In this regard, we reason that SO2 may indirectly cause protein oxidation like carbon dioxide (CO2)/carbonates (HCO3‾) that rapidly reacts with H2O2 or hydroperoxide ion (OOH‾) to yield a more potent oxidant anion, peroxymonocabonate (HCO4‾) [43]. Notably, it is previously shown that CO2/HCO3‾ can potently facilitate H2O2-mediated oxidation and inactivation of many redox sensors (e.g., peroxiredoxins and PTP1B) via peroxymonocabonate formation [44,45]. Likewise, the SO2-mediated conversion of peroxide to peroxymonosulfite intermediate (HOO)SO2‾ (also known as peroxymonosulfurous acid anion) with a hydroperoxy group [[46], [47], [48]] may also provide a pathway for activation of peroxide for two-electron oxidation, though it can also form sulfate (Fig. 2A).
Fig. 2.

SO2 promotes dynamic changes in the sulfenylome of VSMCs. (A) Plausible mechanism of SO2-mediated formation of peroxymonosulfite. Sulfur atom is highlighted in red color. (B) Scheme of chemoselective labeling of protein sulfenic acid by BTD. (C) Schematic workflow for globally and site-specifically profiling protein sulfenylation using the BTD-based SulfenQ method. After 1 h pre-incubation with Ang II, VSMCs were treated with or without 100 μM SO2 donors (SO32‾: HSO3‾ = 2.5:1) for 2 h at 37 °C. Cell lysates were harvested, labeled with 5 mM BTD for 2 h at 37 °C, and then subjected to tryptic digestion. Equal amounts of peptides were reacted with light (for SO2) and heavy (for control) tagged azido-biotin with a photocleavable linker (Az-UV-biotin), respectively. The UV-cleavable biotinylated peptides were captured by streptavidin, and photoleased for LC-MS/MS analysis. (D) Rank order of the determined RSO2/control (RL/H) values of BTD-adducted peptides from VSMCs treated with or without exogenous SO2. GO (E) and KEGG (F) annotations enriched for protein targets of SO2-dependent dynamic sulfenylation. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Due to its intrinsic high reactivity and nucleophilicity, protein cysteine is the primary target for peroxide-dependent two-electron oxidation to give a variety of oxoforms [49]. Sulfenylation, the oxidation of a free cysteinyl thiol group to sulfenic acid (SOH), represents the first step in such oxidative processes and functions as a key mediator of redox signaling in cells [50,51]. Recent advances in the development of chemoselective probes and state-of-the-art chemoproteomics enable us to comprehensively identify and quantify distinct types of redox forms in complex proteomes [52]. For instance, building upon a highly efficient SOH-specific probe called BTD (Fig. 2B) [23], we previously developed a quantitative chemoproteomic method termed SulfenQ to globally profile cysteine sulfenylation in native or living biological systems [33]. Here we adopted this approach to investigate whether and how SO2 perturbs the Ang II-stimulated VSMC sulfenylome (Fig. 2C). Specifically, lysates obtained from the Ang II-stimulated CRL-1444 cells (VSMCs from rat thoracic aorta) treated with or without SO2 were labeled with BTD, processed into tryptic peptides, reacted with light or heavy azido biotin reagents with a photocleavable linker via copper catalyzed alkyne azide cycloaddition (CuAAC), and combined equally. Then, the probe modified peptides were cleaned with strong cation exchange (SCX), captured with streptavidin beads, photoreleased, and analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) for identification and quantification. The light/heavy (L/H SO2/Control) ratio calculated for each BTD-labeled cysteine provided a measure of its relative reactivity in SO2-treated samples versus controls.
Overall, we quantified 1137 site-specific sulfenylation events in 658 VSMC proteins across three biological replicates (Table S1). 81.4% of these cysteine-mediated redox events could be repeatedly quantified in at least two biological replicates with the medium coefficient of variation (CV) value of 15.2%, which demonstrated the high reproducibility of our analysis. Using a common criterion (1.5-fold change) in redox proteomics [53,54], 42.0% (478/1137) of BTD-labeled sulfenylated sites in a total of 372 proteins changed upon SO2 treatment (Fig. 2D), which included many well-known redox-sensitive cysteines. For example, significant SO2-dependent increases in sulfenylation occurred on functional cysteines in two key redox regulators, including C47 of PRDX6 (peroxiredoxin 6) and C92 of APEX1 (DNA-apurinic or apyrimidinic site endonuclease) [55], with RSO2/control values of 2.36 and 1.99, respectively (Fig. 2D, left inset), In addition, the down-regulated sulfenylation events were observed in the active sites of PRDX5 (peroxiredoxin 5, C99) and PTPN1 (tyrosine-protein phosphatase non-receptor type 1, also known as PTP1B, C215), an indication of overoxidation to sulfinic acid (SO2H, Fig. 2D, right inset). To get more biological insights into SO2-dependent sulfenylation, we used DAVID [56] to perform gene ontology (Fig. 2E) and KEGG (Kyoto Encyclopedia of Genes and Genomes, Fig. 2F) analyses. Of interest, sulfenylated proteins dynamically regulated by SO2 were bioinformatically enriched in diverse biological processes and pathways involving central metabolism, redox homeostasis, cytoskeleton organization and cellular responses to stresses (ischemia or hyperoxia). Together, our findings suggest that SO2 may facilitate H2O2-mediated protein cysteine oxidation through peroxymonosulfite formation, thereby promoting dynamic changes in the sulfenylome of VSMCs.
3.3. Smad3C64 is a functional target of SO2-dependent sulfenylation
Among all the dynamically regulated sulfenylated sites is C64 of Smad3 (Mothers against decapentaplegic homolog 3, RSO2/control = 1.86, Fig. 3A), a key transcription factor involved in the developmental and disease process in VSMCs [57]. Interestingly, we noted that the evolutionarily conserved Smad3C64 is adjacent to a conserved lysine residue that can stabilize a thiolate form of its nearby cysteine (Fig. 3B), further supporting the oxidative susceptibility of this site [58]. Further, we sought to use DAz-2, another SOH-specific probe [59], to orthogonally verify this finding made by a chemoproteomic analysis (Fig. S3). To this end, Smad3-deficient VSMCs (Smad3−/−) were expressed with wild-type Smad3 (Smad3WT) and its C64S mutant (Smad3C64S), respectively, preincubated with Ang II and then treated with or without exogenous SO2. As expected, DAz-2-labeled sulfenylation of wild-type Smad3 significantly increased upon SO2 treatment, whereas the formation of such an oxidative modification was largely prohibited by the C64S mutation (Fig. 3C). These results therefore demonstrate that C64 is the prominent sulfenylated cysteine in Smad3.
Fig. 3.

Smad3C64 is a functional target of SO2-mediated sulfenylation. (A) Left panel: Representative MS/MS spectrum of the peptide containing Smad3C64 labeled by BTD-derived modification. Diagnostic fragment ion (DFI) generated from the modified peptide via C–S cleavage is shown in pink color. Right panel: Representative extracted ion chromatogram showing for the change in BTD-labeled cysteine of Smad3 protein from VSMCs treated with or without SO2, with the profiles for light and heavy-labeled peptides in red and blue, respectively. (B) C64 of Smad3 is evolutionarily conserved in vertebrates and is adjacent to a lysine residue that is conserved as well. (C) DAz-2-labeled sulfenylation of Smad3 in Smad3-deficient VSMCs transfected with the wild-type (Smad3WT) or C64S mutated Smad3 (Smad3C64S) plasmid. Cells were treated with or without SO2 donors, harvested and lysed for DAz-2-based labeling and detection. For C, D, G, J, cell/tissue lysates were labeled with DAz-2, ligated with phosphine-biotin, captured with NeutrAvidin, and subjected to western blotting against Smad3. (D) DAz-2-labeled sulfenylation of Smad3 in vehicle- or AAT1-overexpressing VSMCs. Cells pre-stimulated with or without Ang II were harvested and lysed for DAz-2-based labeling and detection. (E) Immunofluorescence localization of Smad3 (red) in vehicle- and AAT1-overexpressing VSMCs with or without Ang II stimulation. Scale bar, 20 μm. (F) Western blots (left panel) and quantification (right panel) of collagen I/III in vehicle- and AAT1-overexpressing VSMCs with or without Ang II stimulation. (G) DAz-2-labeled sulfenylation of Smad3 in VSMCs infected with lentivirus containing MOCK or AAT1 shRNA. Cells pre-stimulated with Ang II were harvested and lysed for DAz-2-based labeling and detection. (H) Immunofluorescence localization of Smad3 (red) in VSMCs infected with lentivirus containing MOCK or AAT1 shRNA. Nuclei stained with DAPI are shown in blue color. Scale bar, 20 μm. (I) Protein expression of collagen I/III in VSMCs infected with lentivirus containing MOCK or AAT1 shRNA. (J) DAz-2-labeled sulfenylation of Smad3 in the aortic tissues of VSMC-TgAAT1 mice and WT mice infused with angiotensin II (500 ng/kg/min) for two weeks (n = 6). n value represents the number of animals used for the experiment. (K) Crystal structure of human Smad3 with its zinc finger motif being shown in a ball-stick style (PDB ID: 1OZJ). (L) EDTA abolishes SO2-dependent deactivation of Smad3WT DNA binding ability. Recombinant Smad3 proteins were treated with SO2 donors or vehicle in the presence or absence of EDTA (10 mM). Smad3-DNA binding activity was measured by ELISA. (M) SO2 inhibits DNA binding activity of wild-type Smad3 (Smad3WT) but not that of C64S mutant (Smad3C64S). Recombinant Smad3 proteins (Smad3WT and Smad3C64S) were treated with or without SO2 donors and subjected to an ELISA analysis. For C, D, F, G, I, J, L and M, data represent mean values ± SD. *P < 0.05. ns, not significant. For C, D, F, G, I, L and M, data are from three independent experiments. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
To further confirm the physiological relevance of Smad3 sulfenylation, we also set up to investigate whether it could also be perturbed by VSMC-derived endogenous SO2 both in cellulo and in vivo. First, VSMCs were infected with an AAT1-expressing recombinant adenovirus (Ad-AAT1), resulting in the overexpression of AAT1 and the increase of endogenous SO2 levels (Fig. S4). AAT1 overexpression significantly increased Smad3 sulfenylation, inhibited nuclear translocation of Smad3, and reduced the expression of collagen I/III in VSMCs (Fig. 3D–F). Second, VSMCs were transfected with lentivirus based AAT1 shRNA, resulting in the reduction of AAT1 protein expression and endogenous SO2 level with no change in AAT2 protein expression (Fig. S5). AAT1 knockdown reduced Smad3 sulfenylation, promoted Smad3 nuclear translocation and the downstream collagen I/III expressions in VSMCs (Fig. 3G–I). Third, we confirmed this redox event in the animal model of Ang II-induced arterial remodeling, as aortic Smad3 sulfenylation was significantly upregulated in VSMC-TgAAT1 mice compared with that in WT mice (Fig. 3J).
Notably, Smad3C64 is a conserved zinc-coordinating cysteine within the N-terminal Mad homology domain-1 (Fig. 3K), where zinc ion is bound to three cysteine residues (C64, C109 and C121) and one histidine (H126). This C3H1 type zinc finger is required for DNA binding [60], while oxidation of zinc-coordinating cysteine can lead to the loss of zinc ion and structural/functional changes in proteins [61]. We therefore sought to investigate the role of site-specific oxidation on Smad3-DNA binding (60). To this end, we purified wild-type Smad3 recombinant protein (Smad3WT) and Smad3 in which C64 was mutated to serine (Smad3C64S). Notably, EDTA, a chelator of metal ions to disrupt the formation of zinc finger motif, abolished SO2-dependent inhibition of Smad3WT DNA binding ability in vitro (Fig. 3L). Moreover, SO2 significantly inhibited DNA binding by Smad3WT protein, whereas its C64S mutant was resistant to SO2-mediated inhibition on the DNA binding activity (Fig. 3M). Collectively, these results indicate that SO2 inhibits the DNA binding ability of Smad3 through sulfenylation of C64.
3.4. Smad3C64 is required for SO2-mediated vascular remodeling and hypertension
The activation of VSMC Smad3 has been recognized as a pivotal mechanism underlying many vascular diseases including hypertension, arteriosclerosis and coronary artery disease [62,63]. Although it has been shown that SO2 can inhibit Smad3 phosphorylation [22], a hallmark of its activation, its underlying molecular mechanism remains largely unknown. Here we found that VSMCs overexpressing AAT1 (TgAAT1) became more resistant to Ang II-induced Smad3 phosphorylation than wild type VSMCs (Fig. 4A), providing a further support to the previous finding. Next, we sought to test whether C64 was involved in the SO2-dependent regulation of Smad3 in VSMCs. By using electrophoretic mobility shift assay, we found that the C64S mutation significantly attenuated the SO2-mediated downregulation of Smad3-DNA binding activity in Smad3WT VSMCs (Fig. 4B). Meanwhile, we noted that, upon SO2 treatment, collagen expression decreased in Smad3WT rather than Smad3C64S VSMCs (Fig. 4C).
Fig. 4.

Smad3C64 is required for SO2-mediated vascular remodeling and hypertension. (A) Western blots (upper panel) and quantification (lower panel) of Smad3 phosphorylation in the aortic tissues of wild type and TgAAT1 mice pre-stimulated with Ang II (n = 6). (B) SO2 inhibits DNA binding activity of wild-type Smad3 but not its C64S mutant. Nuclear proteins were extracted, and the protein-DNA complexes were measured by electrophoretic mobility shift assay. (C) Western blots (upper panel) and quantification (lower panel) of collagen I/III in Smad3-deficient VSMCs transfected with Smad3 WT or C64S plasmid. Cells were pre-incubated with or without Ang II for 1 h, followed by adding SO2 donors or vehicle. Data are from three independent experiments. (D) Representative immunofluorescence staining of phosphorylated Smad3 (p-Smad3), collagen I/III in the aortas of Smad3C64S/WT and Smad3WT/WT mice. During the two weeks of Ang II or saline infusion, the mice were treated daily with SO2 donors or saline. Scale bar, 20 μm. E, endothelium; M, media; and A, adventitia. (E) Hart's staining of thoracic aorta (upper panel) and quantification of the medial thickness (lower panel), n = 5–6. Scale bar, 500 μm. (F) Comparison of SBP measured for Smad3C64S/WT and Smad3WT/WT mice in response to Ang II infusion and/or SO2 donors (n = 5–6). Noninvasive tail-cuff method was used in the SBP measurements. For A, E and F, n value represents the number of animals used for each experiment; data represent mean values ± SD. *P < 0.05. ns, not significant.
To further test a role of Smad3C64 in vivo, we sought to generate a Smad3C64S knock-in mouse line using CRISPR-Cas9 technology (Fig. S6). Due to the low birth rates of homogeneous mice (Smad3C64S/C64S), heterogeneous mice (Smad3C64S/WT) were produced for this study (Fig. S7). After Ang II infusion, wild type and Smad3C64S/WT mice were treated with saline or SO2. Of note, although Ang II infusion significantly increased Smad3 phosphorylation and promoted collagen disposition and vascular remodeling in both Smad3WT/WT and Smad3C64S/WT mice, SO2 reversed these Ang II-dependent effects only in Smad3WT/WT mice but not in Smad3C64S/WT mice (Fig. 4D and E). Furthermore, SO2 significantly attenuated the Ang II-dependent increases in systolic blood pressure in Smad3WT/WT mice, whereas such protective effects were nearly abolished in Smad3C64S/WT mice (Fig. 4F). Taken together, we provide evidence that Smad3C64, as a dynamic sulfenylation target, can modulate sensitivity to SO2-mediated amelioration of vascular remodeling and hypertension.
3.5. Supplementation of SO2 ameliorates angiotensin II-induced vascular endothelial injury
Endothelial cells (ECs) have been known to regulate vascular tone and remodeling. To explore whether the reduction in SO2 caused by Ang II is limited to VSMCs and any influence of EC-mediated SO2 on vascular function, C57 mice were infused with Ang II and administrated with SO2 donor. We found that Ang II infusion not only downregulated AAT1 protein expression in VSMCs (Fig. S8A), but also reduced AAT1 protein level in ECs of mice aortic sections (Fig. S8B). Compared with control mice, Ang II infusion upregulated the expression of ICAM-1, MCP-1 and MMP-2, known markers of EC inflammation and injury, in endothelial layer of mice aortic sections (Figs. S8C–E). However, supplementation of SO2 donor could prevent the increases in the expressions of endothelial ICAM-1, MCP-1 and MMP-2 induced by angiotensin II in mice aortic sections (Figs. S8C–E). These results imply that angiotensin II-caused reduction in EC-derived SO2 promoted vascular endothelial injury, which might contribute to vascular remodeling.
4. Discussion
Studies of NO and H2S over the past decades have highlighted the fundamental importance of gaseous signaling molecules (also defined as gasotransmitters) in health and diseases [[64], [65], [66], [67], [68], [69]]. Akin to NO and H2S, SO2 has recently emerged as a mammalian gasotransmitter that plays a prominent role in vasorelaxation and is involved in inflammatory responses. Here, by genetically manipulating the levels of AAT1 in vivo, we further demonstrate that endogenous SO2 indeed regulates Ang II-vascular remodeling and induced hypertension, inspiring us to further pursue its underlying molecular mechanism as well as upper-stream targets of SO2.
Unlike NO and H2S that can directly modify sulfhydryl groups of cysteine residues in target proteins (the processes known as nitrosylation and persulfidation/sulfhydration, respectively), SO2 may rapidly react with H2O2/HOO‾ to yield (HOO)SO2‾, a much more potent oxidant, that can oxidize protein cysteine thiols to sulfenic acids. It is also likely for (HOO)SO2‾ to bind or react with endogenous metal ions (in either a free ionic format or a coordinative format within zinc finger motifs [70,71]) and generate even stronger oxidants including radical species. In other words, SO2 may function as an ‘enhancer’ of H2O2 signal in biological systems. Moreover, according to a previously reported kinetics study of the reaction between SO2 and H2O2 [48], decreasing pH from a physiological value (pH 7.4) to mild acidic conditions may increase the rate of (HOO)SO2‾ production by 2-3 orders of magnitude, thereby representing another layer for regulating this redox signaling in vivo. For instance, the pH-dependent formation of (HOO)SO2‾ may be attributed to the SO2-induced aggravative effects on myocardial ischemia-reperfusion injury [19] that typically exhibits reduced local pH value [72].
In addition, it is known that a large number of persulfidated/sulfhydrated (-SSH) cysteine residues can be co-modified by sulfenylation [73], due to the intrinsic relationship between these two post-translational modifications (PTMs) [74]. However, sulfenylation typically inhibits protein function, whereas persulfidation/sulfhydration often activates protein activity [75,76]. Thus, H2S and SO2 may reciprocally modulate diverse cellular processes by acting on crucial cysteine residues. Additionally, it is worth noting that SO2 likely mediates other types of redox reactions across complex proteomes, given that the valence state of sulfur atom in SO2 can fluctuate up and down [4]. For instance, SO2 likely react with, in addition to H2O2, other reactive oxygen species (ROS, e.g., hydroperoxide, HO•) to function as a transducer or enhancer of radical ROS signaling [77]. Alternatively, SO2 can be auto-oxidized or enzymatically oxidized to produce sulfur trioxide radical anion (SO3•‾) [78]. The latter can cause lipid peroxidation, likely rendering secondary oxidative damage [79].
Regardless, our previous attempts reveal that SO2-dependent sulfenylation indeed takes place in a few of functionally important protein targets, including AAT1 [80] and NF-κB p65 [81]. Here we used state-of-the-art chemoproteomic method [33] to globally and site-specifically quantify the Ang II-stimulated VSMC sulfenylome in response to exogenous SO2 donor. The analysis allowed us to identify Smad3 as a target of SO2-mediated sulfenylation. Notably, Smad3 is an important transcription factor that regulates diverse biological processes, such as proliferation, growth, apoptosis, differentiation, migration and inflammation in vascular systems [82]. Moreover, over-activation of Smad3 participates in the initiating mechanism of a variety of diseases, such as hypertension, heart failure, diabetic nephropathy, cancer, and so on [62,63].
Indirect evidence has shown that phosphorylation and transcriptional activity of Smad3 can be regulated in a redox-dependent manner [83,84]. Herein, the identification of a redox-sensitive cysteine C64 in Smad3 and the follow-up functional studies further highlights the redox-regulatory role of Smad3. Specifically, VSMC-derived SO2 promotes sulfenylation of Smad3C64 that leads to the disruption of zinc finger motif and the inhibition of Smad3-DNA binding activity. Of note, mutation of this site in Smad3 attenuates the protective effects of SO2 on angiotensin II-induced vascular remodeling and hypertension. Nonetheless, given that the SmadC64/WT knock-in mouse line used here is not VSMC-specific, we cannot rule out the possibility that the effects may involve actions in other cell/tissue types.
In addition to redox mechanisms, many other types of PTMs are also involved in controlling the activity of Smad3. For example, lysine acetylation of Smad3 promotes its activity [85], while ubiquitination in lysine residues at the C-terminal Mad homology domain-2 of Smad3 destabilizes its complex with Smad4 and reduces its translocation into the nucleus [86]. Hence, the crosstalk between sulfenylation and diverse other PTMs in regulating Smad3-mediated cellular signaling, particularly in vascular physiology, merits future investigations.
It is also important to note that we cannot rule out the possibility of Smad3-independent mechanisms being involved in the SO2-dependent regulation of vascular remodeling and hypertensions. In fact, our sulfenylome analysis reveals, in addition to Smad3, many other interesting candidates for future studies. For example, C80 of CFL1 (Cofilin, Table S1), a primary actin-regulatory protein, is likely to be a functional target of SO2-dependent sulfenylation, given that oxidation of this protein can inhibit its affinity for actin [87]. Moreover, CFL1 has been shown to mediate VSMC reorientation in a redox-dependent manner [88]. Although further functional studies are required to verify the roles of this and many other candidates in regulating vascular remodeling and hypertension, our work reveals the potential of quantitative sulfenylome analysis to uncover the upper-stream targets of SO2 and to further elucidate the mechanism underlying its physiological and pathophysiological effects.
Many complex factors such as nerves, body fluids, vascular function and vascular structure participate in blood pressure regulation. This study showed that AAT1 knockout reduced endogenous SO2 content, leading to excessive deposition of vascular collagen protein, damaging blood vessel structure, and increasing blood pressure. However, in TgAAT1 mice, although the level of endogenous SO2 increased, the vascular collagen content and vascular structure did not change significantly compared with the WT mice, and accordingly the blood pressure did not alter. Previous study (30) also showed that intraperitoneal injection of SO2 donor (85 mg/kg) in normal C57 mice for 1 week or 2 weeks did not affect the blood pressure, which was consistent with our results. Another study [89] showed that after 30 s of intravenous injection of SO2 donor to Wistar rats, the mean arterial pressure decreased significantly, and returned to normal level in about 10 min; while the serum SO2 level rose to the highest after about 1 min of administration with SO2 donor. After 2 h, the serum SO2 level dropped to normal level. The above studies indicate that the normal cardiovascular system is highly adaptable to changes in the internal and external environment. In the present study, overexpression of AAT1 in vascular smooth muscle cells produced a long-term increase in endogenous SO2 levels, which might induce other mechanisms to maintain physiological homeostasis. Therefore, there was no significant difference in blood pressure between TgAAT1 mice and WT mice in the absence of angiotensin II stimulation.
Vascular dysfunction is also an important pathogenesis of hypertension. Whether AAT1 knockout or overexpression causes changes in vascular function merits further studies. Data of vascular blood flow and dilation changes via Doppler or myography will be informative as a direct functional outcome of AAT1 manipulation.
5. Conclusions
Collectively, our results highlight a potentially important role for SO2-dependent site-specific sulfenylation in protecting against pathophysiological vascular remodeling and hypertension, and suggest that modulating Smad3 sulfenylation by upregulation of AAT1/SO2 might be a potential clinical prevention strategy for vascular diseases.
Funding
This work was supported by National Natural Science Foundation of China (81770422, 81921001, 81970424, 81770278, 21922702 and 82070445), Beijing Natural Science Foundation (7182168, 7191012, 7171010), Changjiang Scholar Program of Ministry of Education of China (Q2017004), and the State Key Laboratory of Proteomics (SKLP-K201703 and SKLP-K201804).
Author contributions
Y.H. performed the experiments, analyzed the data and wrote the manuscript. L.Z., Z.L., H.T., H.Z., D.B., Z.Z., Z.Z., P.Y. and X.Y. performed the experiments and analyzed the data. S.C. revised the manuscript. C.W., K.L., W.K. and C.T. designed the study. Y.J., R.B.F, and K.S.C. provided reagents; J.Y., J.D. and H.J. designed the study, supervised the research and wrote the manuscript.
Data and materials availability
The authors declare that all data and materials supporting the findings of this study are available within this article or from the corresponding authors on request.
Declaration of competing interest
The authors declare that they have no conflict of interest.
Acknowledgements
We thank Tuo Zhang and Longqin Sun from Beijing Qinglian Biotech Co., Ltd for their help and technical supports. We also thank Boyang Lv from Peking University First Hospital for her kind help in vessel experiment.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2021.101898.
Contributor Information
Jing Yang, Email: yangjing@ncpsb.org.cn.
Hongfang Jin, Email: jinhongfang51@126.com.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Sunyer J., Ballester F., Tertre A.L., Atkinson R., Ayres J.G., Forastiere F., Forsberg B., Vonk J.M., Bisanti L., Tenías J.M., Medina S., Schwartz J., Katsouyanni K. The association of daily sulfur dioxide air pollution levels with hospital admissions for cardiovascular diseases in Europe (the Aphea-ii study) Eur. Heart J. 2003;24:752–760. doi: 10.1016/s0195-668x(02)00808-4. [DOI] [PubMed] [Google Scholar]
- 2.Stipanuk M.H. Metabolism of sulfur-containing amino acids. Annu. Rev. Nutr. 1986;6:179–209. doi: 10.1146/annurev.nu.06.070186.001143. [DOI] [PubMed] [Google Scholar]
- 3.Mitsuhashi H., Yamashita S., Ikeuchi H., Kuroiwa T., Kaneko Y., Hiromura K., Ueki K., Nojima Y. Oxidative stress-dependent conversion of hydrogen sulfide to sulfite by activated neutrophils. Shock. 2005;24:529–534. doi: 10.1097/01.shk.0000183393.83272.de. [DOI] [PubMed] [Google Scholar]
- 4.Wang W., Wang B. SO2 donors and prodrugs, and their possible applications: a review. Front. Chem. 2018;6:559. doi: 10.3389/fchem.2018.00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Du S.X., Jin H.F., Bu D.F., Zhao X., Geng B., Tang C.S., Du J.B. Endogenously generated sulfur dioxide and its vasorelaxant effect in rats. Acta Pharmacol. Sin. 2008;29:923–930. doi: 10.1111/j.1745-7254.2008.00845.x. [DOI] [PubMed] [Google Scholar]
- 6.Luo L., Chen S., Jin H., Tang C., Du J. Endogenous generation of sulfur dioxide in rat tissues. Biochem. Biophys. Res. Commun. 2011;415:61–67. doi: 10.1016/j.bbrc.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 7.Ji A.J., Savon S.R., Jacobsen D.W. Determination of total serum sulfite by HPLC with fluorescence detection. Clin. Chem. 1995;41:897–903. [PubMed] [Google Scholar]
- 8.Balazy M., Abu-Yousef I.A., Harpp D.N., Park J. Identification of carbonyl sulfide and sulfur dioxide in porcine coronary artery by gas chromatography/mass spectrometry, possible relevance to EDHF. Biochem. Biophys. Res. Commun. 2003;311:728–734. doi: 10.1016/j.bbrc.2003.10.055. [DOI] [PubMed] [Google Scholar]
- 9.Meng Z., Li J., Zhang Q., Bai W., Yang Z., Zhao Y., Wang F. Vasodilator effect of gaseous sulfur dioxide and regulation of its level by Ach in rat vascular tissues. Inhal. Toxicol. 2009;21:1223–1228. doi: 10.3109/08958370902798463. [DOI] [PubMed] [Google Scholar]
- 10.Zhang R.Y., Du J.B., Sun Y., Chen S., Tsai H.J., Yuan L., Li L., Tang C.S., Jin H.F. Sulfur dioxide derivatives depress L-type calcium channel in rat cardiomyocytes. Clin. Exp. Pharmacol. Physiol. 2011;38:416–422. doi: 10.1111/j.1440-1681.2011.05528.x. [DOI] [PubMed] [Google Scholar]
- 11.Liu D., Huang Y., Bu D., Liu A.D., Holmberg L., Jia Y., Tang C., Du J., Jin H. Sulfur dioxide inhibits vascular smooth muscle cell proliferation via suppressing the Erk/MAP kinase pathway mediated by cAMP/PKA signaling. Cell Death Dis. 2014;5:e1251. doi: 10.1038/cddis.2014.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yao Q., Huang Y., Liu A.D., Zhu M., Liu J., Yan H., Zhang Q., Geng B., Gao Y., Du S., Huang P., Tang C., Du J., Jin H. The vasodilatory effect of sulfur dioxide via SGC/cGMP/PKG pathway in association with sulfhydryl-dependent dimerization. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016;310:R1073–R1080. doi: 10.1152/ajpregu.00101.2015. [DOI] [PubMed] [Google Scholar]
- 13.Zhang H., Huang Y., Bu D., Chen S., Tang C., Wang G., Du J., Jin H. Endogenous sulfur dioxide is a novel adipocyte-derived inflammatory inhibitor. Sci. Rep. 2016;6:27026. doi: 10.1038/srep27026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cao X., Ding L., Xie Z.Z., Yang Y., Whiteman M., Moore P.K., Bian J.S. A review of hydrogen sulfide synthesis, metabolism, and measurement: is modulation of hydrogen sulfide a novel therapeutic for cancer? Antioxidants Redox Signal. 2019;31:1–38. doi: 10.1089/ars.2017.7058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shackelford R., Ozluk E., Islam M.Z., Hopper B., Meram A., Ghali G., Kevil C.G. Hydrogen sulfide and DNA repair. Redox Biol. 2021;38:101675. doi: 10.1016/j.redox.2020.101675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuan S., Patel R.P., Kevil C.G. Working with nitric oxide and hydrogen sulfide in biological systems. Am. J. Physiol. Lung Cell Mol. Physiol. 2015;308:L403–L415. doi: 10.1152/ajplung.00327.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li W., Tang C., Jin H., Du J. Regulatory effects of sulfur dioxide on the development of atherosclerotic lesions and vascular hydrogen sulfide in atherosclerotic rats. Atherosclerosis. 2011;215:323–330. doi: 10.1016/j.atherosclerosis.2010.12.037. [DOI] [PubMed] [Google Scholar]
- 18.Chen S., Du J., Liang Y., Zhang R., Tang C., Jin H. Sulfur dioxide restores calcium homeostasis disturbance in rat with isoproterenol-induced myocardial injury. Histol. Histopathol. 2012;27:1219–1226. doi: 10.14670/HH-27.1219. [DOI] [PubMed] [Google Scholar]
- 19.Zhang S., Du J., Jin H., Li W., Liang Y., Geng B., Li S., Zhang C., Tang C. Endogenous sulfur dioxide aggravates myocardial injury in isolated rat heart with ischemia and reperfusion. Transplantation. 2009;87:517–524. doi: 10.1097/TP.0b013e318195fe82. [DOI] [PubMed] [Google Scholar]
- 20.Sun Y., Tian Y., Prabha M., Liu D., Chen S., Zhang R., Liu X., Tang C., Tang X., Jin H., Du J. Effects of sulfur dioxide on hypoxic pulmonary vascular structural remodeling. Lab. Invest. 2010;90:68–82. doi: 10.1038/labinvest.2009.102. [DOI] [PubMed] [Google Scholar]
- 21.Huang Y., Tang C., Du J., Jin H. Endogenous sulfur dioxide: a new member of gasotransmitter family in the cardiovascular system. Oxid. Med. Cell. Longev. 2016;2016:8961951. doi: 10.1155/2016/8961951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang Y., Shen Z., Chen Q., Huang P., Zhang H., Du S., Geng B., Zhang C., Li K., Tang C., Du J., Jin H. Endogenous sulfur dioxide alleviates collagen remodeling via inhibiting TGF-β/Smad pathway in vascular smooth muscle cells. Sci. Rep. 2016;6:19503. doi: 10.1038/srep19503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gupta V., Yang J., Liebler D.C., Carroll K.S. Diverse redoxome reactivity profiles of carbon nucleophiles. J. Am. Chem. Soc. 2017;139:5588–5595. doi: 10.1021/jacs.7b01791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang J., Li K., Hou J.-T., Lu C.-Y., Li L.-L., Yu K.-K., Yu X.-Q. A novel coumarin-based water-soluble fluorescent probe for endogenously generated SO2 in living cells. Sci. China (Chem.) 2017;60:793–798. [Google Scholar]
- 25.Hao H., Hu S., Wan Q., Xu C., Chen H., Zhu L., Xu Z., Meng J., Breyer R.M., Li N., Liu D.P., FitzGerald G.A., Wang M. Protective role of mPGES-1 (microsomal prostaglandin E synthase-1)-derived PGE2 (prostaglandin E2) and the endothelial EP4 (prostaglandin E receptor) in vascular responses to injury. Arterioscler. Thromb. Vasc. Biol. 2018;38:1115–1124. doi: 10.1161/ATVBAHA.118.310713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kühbandner S., Brummer S., Metzger D., Chambon P., Hofmann F., Feil R. Temporally controlled somatic mutagenesis in smooth muscle. Genesis. 2000;28:15–22. doi: 10.1002/1526-968x(200009)28:1<15::aid-gene20>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 27.Yang N., Smyllie N.J., Morris H., Gonçalves C.F., Dudek M., Pathiranage D.R.J., Chesham J.E., Adamson A., Spiller D.G., Zindy E., Bagnall J., Humphreys N., Hoyland J., Loudon A.S.I., Hastings M.H., Meng Q.J. Quantitative live imaging of Venus::BMAL1 in a mouse model reveals complex dynamics of the master circadian clock regulator. PLoS Genet. 2020;16 doi: 10.1371/journal.pgen.1008729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Earley S., Brayden J.E. Transient receptor potential channels in the vasculature. Physiol. Rev. 2015;95:645–690. doi: 10.1152/physrev.00026.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang L., Zhao X.C., Cui W., Ma Y.Q., Ren H.L., Zhou X., Fassett J., Yang Y.Z., Chen Y., Xia Y.L., Du J., Li H.H. Genetic and pharmacologic inhibition of the chemokine receptor CXCR2 prevents experimental hypertension and vascular dysfunction. Circulation. 2016;134:1353–1368. doi: 10.1161/CIRCULATIONAHA.115.020754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Q., Zhang L., Chen S., Huang Y., Li K., Yu X., Wu H., Tian X., Zhang C., Tang C., Du J., Jin H. Downregulated endogenous sulfur dioxide/aspartate aminotransferase pathway is involved in angiotensin II-stimulated cardiomyocyte autophagy and myocardial hypertrophy in mice. Int. J. Cardiol. 2016;225:392–401. doi: 10.1016/j.ijcard.2016.09.111. [DOI] [PubMed] [Google Scholar]
- 31.Earley S., Pauyo T., Drapp R., Tavares M.J., Liedtke W., Brayden J.E. TRPV4-dependent dilation of peripheral resistance arteries influences arterial pressure. Am. J. Physiol. Heart Circ. Physiol. 2009;297:H1096–H1102. doi: 10.1152/ajpheart.00241.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang N., Williams J., Pekovic-Vaughan V., Wang P., Olabi S., McConnell J., Gossan N., Hughes A., Cheung J., Streuli C.H., Meng Q.J. Cellular mechano-environment regulates the mammary circadian clock. Nat. Commun. 2017;8:14287. doi: 10.1038/ncomms14287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fu L., Liu K., Ferreira R.B., Carroll K.S., Yang J. Proteome-wide analysis of cysteine S-sulfenylation using a benzothiazine-based probe. Curr. Protein Pept. Sci. 2019;95:e76. doi: 10.1002/cpps.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma J., Chen T., Wu S., Yang C., Bai M., Shu K., Li K., Zhang G., Jin Z., He F., Hermjakob H., Zhu Y. iProX: an integrated proteome resource. Nucleic Acids Res. 2019;47:D1211–d1217. doi: 10.1093/nar/gky869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Göbl C., Morris V.K., van Dam L., Visscher M., Polderman P.E., Hartlmüller C., de Ruiter H., Hora M., Liesinger L., Birner-Gruenberger R., Vos H.R., Reif B., Madl T., Dansen T.B. Cysteine oxidation triggers amyloid fibril formation of the tumor suppressor p16INK4A. Redox Biol. 2020;28:101316. doi: 10.1016/j.redox.2019.101316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu J., Cheng Z., Reddie K., Carroll K., Hammad L.A., Karty J.A., Bauer C.E. RegB kinase activity is repressed by oxidative formation of cysteine sulfenic acid. J. Biol. Chem. 2013;288:4755–4762. doi: 10.1074/jbc.M112.413492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Svoboda L.K., Reddie K.G., Zhang L., Vesely E.D., Williams E.S., Schumacher S.M., O’Connell R.P., Shaw R., Day S.M., Anumonwo J.M., Carroll K.S., Martens J.R. Redox-sensitive sulfenic acid modification regulates surface expression of the cardiovascular voltage-gated potassium channel Kv1.5. Circ. Res. 2012;111:842–853. doi: 10.1161/CIRCRESAHA.111.263525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cao X., Nie X., Xiong S., Cao L., Wu Z., Moore P.K., Bian J.S. Renal protective effect of polysulfide in cisplatin-induced nephrotoxicity. Redox Biol. 2018;15:513–521. doi: 10.1016/j.redox.2018.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Du J., Huang Y., Yan H., Zhang Q., Zhao M., Zhu M., Liu J., Chen S.X., Bu D., Tang C., Jin H. Hydrogen sulfide suppresses oxidized low-density lipoprotein (ox-LDL)-stimulated monocyte chemoattractant protein 1 generation from macrophages via the nuclear factor κb (NF-κB) pathway. J. Biol. Chem. 2014;289:9741–9753. doi: 10.1074/jbc.M113.517995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krege J.H., Hodgin J.B., Hagaman J.R., Smithies O. A noninvasive computerized tail-cuff system for measuring blood pressure in mice. Hypertension. 1995;25:1111–1115. doi: 10.1161/01.hyp.25.5.1111. [DOI] [PubMed] [Google Scholar]
- 41.Van Vliet B.N., Chafe L.L., Antic V., Schnyder-Candrian S., Montani J.P. Direct and indirect methods used to study arterial blood pressure. J. Pharmacol. Toxicol. Methods. 2000;44:361–373. doi: 10.1016/s1056-8719(00)00126-x. [DOI] [PubMed] [Google Scholar]
- 42.Wang H., Dalla Lana I.G., Chuang K.T. Kinetics of reaction between hydrogen sulfide and sulfur dioxide in sulfuric acid solutions. Ind. Eng. Chem. Res. 2002;41:4707–4713. [Google Scholar]
- 43.Bakhmutova-Albert E.V., Yao H., Denevan D.E., Richardson D.E. Kinetics and mechanism of peroxymonocarbonate formation. Inorg. Chem. 2010;49:11287–11296. doi: 10.1021/ic1007389. [DOI] [PubMed] [Google Scholar]
- 44.Peskin A.V., Pace P.E., Winterbourn C.C. Enhanced hyperoxidation of peroxiredoxin 2 and peroxiredoxin 3 in the presence of bicarbonate/CO2. Free Radic. Biol. Med. 2019;145:1–7. doi: 10.1016/j.freeradbiomed.2019.09.010. [DOI] [PubMed] [Google Scholar]
- 45.Dagnell M., Cheng Q., Rizvi S.H.M., Pace P.E., Boivin B., Winterbourn C.C., Arnér E.S.J. Bicarbonate is essential for protein-tyrosine phosphatase 1b (PTP1B) oxidation and cellular signaling through EGF-triggered phosphorylation cascades. J. Biol. Chem. 2019;294:12330–12338. doi: 10.1074/jbc.RA119.009001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shostak S., Kim K., Horbatenko Y., Choi C.H. Sulfuric acid formation via H2SO3 oxidation by H2O2 in the atmosphere. J. Phys. Chem. 2019;123:8385–8390. doi: 10.1021/acs.jpca.9b05444. [DOI] [PubMed] [Google Scholar]
- 47.Hung H.M., Hoffmann M.R. Oxidation of gas-phase SO2 on the surfaces of acidic microdroplets: implications for sulfate and sulfate radical anion formation in the atmospheric liquid phase. Environ. Sci. Technol. 2015;49:13768–13776. doi: 10.1021/acs.est.5b01658. [DOI] [PubMed] [Google Scholar]
- 48.Hoffmann M.R., Edwards J.O. Kinetics of the oxidation of sulfite by hydrogen peroxide in acidic solution. J. Phys. Chem. 1975;79:2096–2098. [Google Scholar]
- 49.Held J.M., Gibson B.W. Regulatory control or oxidative damage? Proteomic approaches to interrogate the role of cysteine oxidation status in biological processes. Mol. Cell. Proteomics. 2012;11 doi: 10.1074/mcp.R111.013037. R111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paulsen C.E., Carroll K.S. Cysteine-mediated redox signaling: chemistry, biology, and tools for discovery. Chem. Rev. 2013;113:4633–4679. doi: 10.1021/cr300163e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shi T., Dansen T.B. Reactive oxygen species induced p53 activation: DNA damage, redox signaling, or both? Antioxidants Redox Signal. 2020;33:839–859. doi: 10.1089/ars.2020.8074. [DOI] [PubMed] [Google Scholar]
- 52.Shi Y., Carroll K.S. Activity-based sensing for site-specific proteomic analysis of cysteine oxidation. Acc. Chem. Res. 2020;53:20–31. doi: 10.1021/acs.accounts.9b00562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang J., Willems P., Wei B., Tian C., Ferreira R.B., Bodra N., Martínez Gache S.A., Wahni K., Liu K., Vertommen D., Gevaert K., Carroll K.S., Van Montagu M., Yang J., Van Breusegem F., Messens J. Mining for protein S-sulfenylation in Arabidopsis uncovers redox-sensitive sites. Proc. Natl. Acad. Sci. U. S. A. 2019;116:21256–21261. doi: 10.1073/pnas.1906768116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deng X., Weerapana E., Ulanovskaya O., Sun F., Liang H., Ji Q., Ye Y., Fu Y., Zhou L., Li J., Zhang H., Wang C., Alvarez S., Hicks L.M., Lan L., Wu M., Cravatt B.F., He C. Proteome-wide quantification and characterization of oxidation-sensitive cysteines in pathogenic bacteria. Cell Host Microbe. 2013;13:358–370. doi: 10.1016/j.chom.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Georgiadis M.M., Luo M., Gaur R.K., Delaplane S., Li X., Kelley M.R. Evolution of the redox function in mammalian apurinic/apyrimidinic endonuclease. Mutat. Res. 2008;643:54–63. doi: 10.1016/j.mrfmmm.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang da W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using david bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 57.Liu B., Pjanic M., Wang T., Nguyen T., Gloudemans M., Rao A., Castano V.G., Nurnberg S., Rader D.J., Elwyn S., Ingelsson E., Montgomery S.B., Miller C.L., Quertermous T. Genetic regulatory mechanisms of smooth muscle cells map to coronary artery disease risk loci. Am. J. Hum. Genet. 2018;103:377–388. doi: 10.1016/j.ajhg.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang J., Gupta V., Carroll K.S., Liebler D.C. Site-specific mapping and quantification of protein S-sulphenylation in cells. Nat. Commun. 2014;5:4776. doi: 10.1038/ncomms5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Leonard S.E., Reddie K.G., Carroll K.S. Mining the thiol proteome for sulfenic acid modifications reveals new targets for oxidation in cells. ACS Chem. Biol. 2009;4:783–799. doi: 10.1021/cb900105q. [DOI] [PubMed] [Google Scholar]
- 60.Chai J., Wu J.W., Yan N., Massagué J., Pavletich N.P., Shi Y. Features of a Smad3 MH1-DNA complex. Roles of water and zinc in DNA binding. J. Biol. Chem. 2003;278:20327–20331. doi: 10.1074/jbc.C300134200. [DOI] [PubMed] [Google Scholar]
- 61.Bae J.B., Park J.H., Hahn M.Y., Kim M.S., Roe J.H. Redox-dependent changes in RsrA, an anti-sigma factor in Streptomyces coelicolor: zinc release and disulfide bond formation. J. Mol. Biol. 2004;335:425–435. doi: 10.1016/j.jmb.2003.10.065. [DOI] [PubMed] [Google Scholar]
- 62.Wang W., Huang X.R., Canlas E., Oka K., Truong L.D., Deng C., Bhowmick N.A., Ju W., Bottinger E.P., Lan H.Y. Essential role of Smad3 in angiotensin II-induced vascular fibrosis. Circ. Res. 2006;98:1032–1039. doi: 10.1161/01.RES.0000218782.52610.dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Iyer D., Zhao Q., Wirka R., Naravane A., Nguyen T., Liu B., Nagao M., Cheng P., Miller C.L., Kim J.B., Pjanic M., Quertermous T. Coronary artery disease genes SMAD3 and TCF21 promote opposing interactive genetic programs that regulate smooth muscle cell differentiation and disease risk. PLoS Genet. 2018;14 doi: 10.1371/journal.pgen.1007681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pacher P., Beckman J.S., Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Farah C., Michel L.Y.M., Balligand J.L. Nitric oxide signalling in cardiovascular health and disease. Nat. Rev. Cardiol. 2018;15:292–316. doi: 10.1038/nrcardio.2017.224. [DOI] [PubMed] [Google Scholar]
- 66.Szabó C. Hydrogen sulphide and its therapeutic potential. Nat. Rev. Drug Discov. 2007;6:917–935. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- 67.Li Z., Polhemus D.J., Lefer D.J. Evolution of hydrogen sulfide therapeutics to treat cardiovascular disease. Circ. Res. 2018;123:590–600. doi: 10.1161/CIRCRESAHA.118.311134. [DOI] [PubMed] [Google Scholar]
- 68.Paul B.D., Snyder S.H. H2S: a novel gasotransmitter that signals by sulfhydration. Trends Biochem. Sci. 2015;40:687–700. doi: 10.1016/j.tibs.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yuan S., Pardue S., Shen X., Alexander J.S., Orr A.W., Kevil C.G. Hydrogen sulfide metabolism regulates endothelial solute barrier function. Redox Biol. 2016;9:157–166. doi: 10.1016/j.redox.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Conte D., Narindrasorasak S., Sarkar B. In vivo and in vitro iron-replaced zinc finger generates free radicals and causes DNA damage. J. Biol. Chem. 1996;271:5125–5130. doi: 10.1074/jbc.271.9.5125. [DOI] [PubMed] [Google Scholar]
- 71.Sarkar B. Metal replacement in DNA-binding zinc finger proteins and its relevance to mutagenicity and carcinogenicity through free radical generation. Nutrition. 1995;11:646–649. [PubMed] [Google Scholar]
- 72.Lemasters J.J., Bond J.M., Chacon E., Harper I.S., Kaplan S.H., Ohata H., Trollinger D.R., Herman B., Cascio W.E. The pH paradox in ischemia-reperfusion injury to cardiac myocytes. EXS. 1996;76:99–114. doi: 10.1007/978-3-0348-8988-9_7. [DOI] [PubMed] [Google Scholar]
- 73.Fu L., Liu K., He J., Tian C., Yu X., Yang J. Direct proteomic mapping of cysteine persulfidation. Antioxidants Redox Signal. 2020;33:1061–1076. doi: 10.1089/ars.2019.7777. [DOI] [PubMed] [Google Scholar]
- 74.Filipovic M.R., Zivanovic J., Alvarez B., Banerjee R. Chemical biology of H2S signaling through persulfidation. Chem. Rev. 2018;118:1253–1337. doi: 10.1021/acs.chemrev.7b00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Paul B.D., Snyder S.H. H₂S signalling through protein sulfhydration and beyond. Nat. Rev. Mol. Cell. Biol. 2012;13:499–507. doi: 10.1038/nrm3391. [DOI] [PubMed] [Google Scholar]
- 76.Zivanovic J., Kouroussis E., Kohl J.B., Adhikari B., Bursac B., Schott-Roux S., Petrovic D., Miljkovic J.L., Thomas-Lopez D., Jung Y., Miler M., Mitchell S., Milosevic V., Gomes J.E., Benhar M., Gonzalez-Zorn B., Ivanovic-Burmazovic I., Torregrossa R., Mitchell J.R., Whiteman M., Schwarz G., Snyder S.H., Paul B.D., Carroll K.S., Filipovic M.R. Selective persulfide detection reveals evolutionarily conserved antiaging effects of S-sulfhydration. Cell Metabol. 2019;30:1152–1170. doi: 10.1016/j.cmet.2019.10.007. e1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Flockhart B.D., Ivin K.J., Pink R.C., Sharma B.D. The nature of the radical intermediates in the reactions between hydroperoxides and sulphur dioxide and their reaction with alkene derivatives: electron spin resonance study. J. Chem. Soc. D Chem. Commun. 1971:339–340. [Google Scholar]
- 78.Mottley C., Mason R.P., Chignell C.F., Sivarajah K., Eling T.E. The formation of sulfur trioxide radical anion during the prostaglandin hydroperoxidase-catalyzed oxidation of bisulfite (hydrated sulfur dioxide) J. Biol. Chem. 1982;257:5050–5055. [PubMed] [Google Scholar]
- 79.Lizada M.C.C., Yang S.F. Sulfite-induced lipid peroxidation. Vol. 16. 1981. pp. 189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Song Y., Peng H., Bu D., Ding X., Yang F., Zhu Z., Tian X., Zhang L., Wang X., Tang C., Huang Y., Du J., Jin H. Negative auto-regulation of sulfur dioxide generation in vascular endothelial cells: AAT1 S-sulfenylation. Biochem. Biophys. Res. Commun. 2020;525:231–237. doi: 10.1016/j.bbrc.2020.02.040. [DOI] [PubMed] [Google Scholar]
- 81.Chen S., Huang Y., Liu Z., Yu W., Zhang H., Li K., Yu X., Tang C., Zhao B., Du J., Jin H. Sulphur dioxide suppresses inflammatory response by sulphenylating NF-κB p65 at Cys38 in a rat model of acute lung injury. Clin. Sci. (Lond.) 2017;131:2655–2670. doi: 10.1042/CS20170274. [DOI] [PubMed] [Google Scholar]
- 82.Yokote K., Kobayashi K., Saito Y. The role of Smad3-dependent TGF-beta signal in vascular response to injury. Trends Cardiovasc. Med. 2006;16:240–245. doi: 10.1016/j.tcm.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 83.Hseu Y.C., Yang T.Y., Li M.L., Rajendran P., Mathew D.C., Tsai C.H., Lin R.W., Lee C.C., Yang H.L. Chalcone flavokawain A attenuates TGF-β1-induced fibrotic pathology via inhibition of ROS/Smad3 signaling pathways and induction of Nrf2/ARE-mediated antioxidant genes in vascular smooth muscle cells. J. Cell Mol. Med. 2019;23:775–788. doi: 10.1111/jcmm.13973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Keshk W.A., Katary M.A. Transforming growth factor-β1/Smad3 signaling and redox status in experimentally induced nephrotoxicity: impact of carnosine. Indian J. Clin. Biochem. 2017;32:19–25. doi: 10.1007/s12291-016-0564-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Inoue Y., Itoh Y., Abe K., Okamoto T., Daitoku H., Fukamizu A., Onozaki K., Hayashi H. Smad3 is acetylated by p300/CBP to regulate its transactivation activity. Oncogene. 2007;26:500–508. doi: 10.1038/sj.onc.1209826. [DOI] [PubMed] [Google Scholar]
- 86.Tang L.Y., Yamashita M., Coussens N.P., Tang Y., Wang X., Li C., Deng C.X., Cheng S.Y., Zhang Y.E. Ablation of Smurf2 reveals an inhibition in TGF-β signalling through multiple mono-ubiquitination of Smad3. EMBO J. 2011;30:4777–4789. doi: 10.1038/emboj.2011.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Klamt F., Zdanov S., Levine R.L., Pariser A., Zhang Y., Zhang B., Yu L.R., Veenstra T.D., Shacter E. Oxidant-induced apoptosis is mediated by oxidation of the actin-regulatory protein cofilin. Nat. Cell Biol. 2009;11:1241–1246. doi: 10.1038/ncb1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Montenegro M.F., Valdivia A., Smolensky A., Verma K., Taylor W.R., San Martín A. Nox4-dependent activation of cofilin mediates VSMC reorientation in response to cyclic stretching. Free Radic. Biol. Med. 2015;85:288–294. doi: 10.1016/j.freeradbiomed.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Du S., Jin H., Liang Y., Zhao X., Wei H., Wang L., Du J., Tang C. Influence of sulfur dioxide and its derivatives on rats' blood pressure. J. Appl. Clin. Pediatr. 2008;23:22–24. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.