

There is a Blood Commentary on this article in this issue.
Key Points
SCD mice exhibit a myocardium vulnerable to VT.
IL-18 is a novel therapeutic target for sickle cell cardiomyopathy.
Abstract
Previous reports indicate that IL18 is a novel candidate gene for diastolic dysfunction in sickle cell disease (SCD)–related cardiomyopathy. We hypothesize that interleukin-18 (IL-18) mediates the development of cardiomyopathy and ventricular tachycardia (VT) in SCD. Compared with control mice, a humanized mouse model of SCD exhibited increased cardiac fibrosis, prolonged duration of action potential, higher VT inducibility in vivo, higher cardiac NF-κB phosphorylation, and higher circulating IL-18 levels, as well as reduced voltage-gated potassium channel expression, which translates to reduced transient outward potassium current (Ito) in isolated cardiomyocytes. Administering IL-18 to isolated mouse hearts resulted in VT originating from the right ventricle and further reduced Ito in SCD mouse cardiomyocytes. Sustained IL-18 inhibition via IL-18–binding protein resulted in decreased cardiac fibrosis and NF-κB phosphorylation, improved diastolic function, normalized electrical remodeling, and attenuated IL-18–mediated VT in SCD mice. Patients with SCD and either myocardial fibrosis or increased QTc displayed greater IL18 gene expression in peripheral blood mononuclear cells (PBMCs), and QTc was strongly correlated with plasma IL-18 levels. PBMC-derived IL18 gene expression was increased in patients who did not survive compared with those who did. IL-18 is a mediator of sickle cell cardiomyopathy and VT in mice and a novel therapeutic target in patients at risk for sudden death.
Visual Abstract

Introduction
Despite significant progress in extending the lifespan of patients with sickle cell disease (SCD),1 prognosis remains poor and the average lifespan remains in the 40s.1 On the basis of autopsy studies, the main causes of premature death in patients are acute chest syndrome (ACS), pulmonary hypertension (PH), and sudden death.1-5 Although fatalities from ACS and PH are well characterized, the etiology of sudden death is unknown but includes potential contributions from arrhythmias that would lead to sudden cardiac death.
Previous case series reported increased QT dispersion and prolongation, which imply the risk of developing fatal arrhythmias in SCD.6-10 Other case series reported the presence of ventricular arrhythmias in patients.11,12 Associations between an increased QTc interval with hemolysis, increased free heme levels, and higher mortality were reported in a large patient cohort over 5 years.13 However, mechanisms that contribute to prolonged QTc, changes to action potential (AP), and arrhythmogenic propensity are unknown.
A significant subset of patients exhibit myocardial fibrosis and diastolic dysfunction, the latter representing an independent risk factor for mortality in SCD.14,15 Fibrosis independently increases the risk of ventricular arrhythmias in patients with cardiomyopathy. A previous report observed myocardial hypertrophy, increased mass in the left ventricle (LV), and diastolic dysfunction with preserved systolic function with fibrosis in SCD mice.16 Although underlying mechanisms for these cardiovascular findings are unclear, sickled red cells are considered partly causal for systemic inflammation and interleukin-18 (IL-18)–mediated inflammasome signaling.17-20 We recently reported IL18 as a novel candidate gene associated with diastolic dysfunction in patients with SCD and the observation that IL18 single nucleotide polymorphisms were associated with expression quantitative trait loci.21 We reported increased IL18 gene expression levels in the hearts of SCD vs control (CTR) mice. Additional association of promoter single nucleotide polymorphisms in the IL18 gene have been previously reported with sudden cardiac death in a population with heart failure not related to SCD.22 We hypothesized that IL-18 contributes to the development of murine sickle cell cardiomyopathy, which increases susceptibility to ventricular tachycardia (VT). We characterized the cardiac electrophysiological phenotype of the humanized SCD mouse model and evaluated the molecular underpinnings that bridge relationships between IL-18–mediated inflammation, action potential duration (APD) and QTc prolongation, and ventricular arrhythmias.
Methods
Patient cohorts
UCH cohort.
Adult patients (age 18 years or older) from the University of Chicago Hospital (UCH) were recruited from an outpatient program. They provided written consent to participate in the study, which had approval from the institutional review board (http://clinicaltrials.gov/ct2/show/NCT01044901). A total of 23 clinically stable African American patients (including those with homozygous hemoglobin S [HbSS], sickle cell hemoglobin C [HbSC], and beta thalassemia demonstrated by high-performance liquid chromatographic separation or gel electrophoresis) were included. Cardiovascular magnetic resonance imaging with late gadolinium enhancement (LGE)23 sequence and blood draw were prospectively performed on the same day. Patients were excluded only if they were clinically unstable, which was defined as having vaso-occlusive crisis, ACS, or unscheduled blood transfusions within 3 weeks of the study. Details of imaging and microarray processing have been published previously.14
UIC cohort.
All adult patients with SCD (age 18 years or older) from the University of Illinois at Chicago (UIC; n = 153) since 2010 were prospectively recruited and provided written consent to participate with institutional review board approval. Patients were recruited from outpatient clinics in steady-state condition (no vaso-occlusive crises for 3 weeks). Details of clinical data collection are provided in the supplemental Data (available at the Blood Web site). RNA from peripheral blood mononuclear cells (PBMCs) was evaluated for gene expression profiles using microarrays. In contrast to the UCH cohort, circulating IL-18 levels in patients from the UIC cohort were measured from plasma samples, available for a subset of patients and drawn on the same day as PBMCs (as described previously24). Complete details regarding arrays, hybridization, labeling, quality control, and analyses for both cohorts are provided in the supplemental Data.
Circulating plasma IL-18 levels in mice and patients.
Within the UIC cohort, correlation between circulating plasma IL-18 levels (Cosmo Bio) in patients and QTc intervals was assessed. Total IL-18 (Abcam) as well as IL-18-binding protein (IL-18BP) levels (R&D) were measured in the serum of CTR and SCD mice after administration of IL-18BP or vehicle for 4 weeks. Free serum IL-18 levels were calculated as previously described.25
Animal studies.
Homozygous Townes SCD model mice and non-sickling CTR mice were obtained from The Jackson Laboratory (stock #013071). Four sets of experiments (Parts I-IV) were performed using 8- to 12-week-old male mice, unless otherwise indicated. Animals were handled in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All protocols were approved by animal welfare committees at Indiana University, University of Wisconsin, Rhode Island Hospital, and the University of Arizona.
Part I. Surface ECG recording and programmed ventricular stimulation in vivo.
Surface electrocardiograms (ECGs) were recorded, and arrhythmia inducibility was determined in CTR and SCD mice at baseline under general anesthesia with isofurane by using described methods.26
Part II. Measurement of ventricular CV.
LV conduction velocity (CV) was measured (supplemental Data) in anesthetized CTR (n = 5) and SCD (n = 5) mice at baseline by using a flexible multielectrode array system (Flex-MEA, 72 electrodes; Multichannel Systems, Reutlingen, Germany) according to the manufacturer’s instructions.
Part III. Optical mapping and ventricular stimulation ex vivo.
Acute effects of IL-18, dose-dependent effects of IL-18 on APD prolongation, and VT induction were initially studied in isolated and perfused SCD hearts (n = 4). Because there were frequent early afterdepolarizations (EADs) at higher doses, 2 ng/mL was used to study effects on APD prolongation and 5 ng/mL was used to study VT induction. VTs were defined from ECGs with more than 3 consecutive arrhythmic beats.
Hearts from CTR and SCD male mice also underwent optical mapping using a voltage sensitive dye (di-4 ANEPPS; Invitrogen), as described.26 For simultaneous voltage and Ca2+ mapping, RH237 and Rhod-2 were used, as described.27 Sample activation potential traces and activation maps with 150 ms pacing were made for both CTR and SCD hearts. IL-18 was further perfused for 60 minutes, and fluorescence signals were recorded every 2 minutes in the latter subset of mice (supplemental Data). APD and Ca2+ duration (CaD) were measured at 90% repolarization (APD90) and 90% recovery (CaD90).
Part IV. Optical mapping, echocardiogram, and fibrosis: chronic effects of IL-18BP.
In separate experiments, vehicle (phosphate-buffered saline) or IL-18BP (0.5 mg/kg; R&D)28 were injected intraperitoneally into 10- to 14-week-old CTR and SCD mice every 2 days for 28 days. Another group of 6-month-old SCD mice were injected with identical doses and duration of vehicle or IL-18BP to evaluate the effect of age. Echocardiograms, fibrosis levels, and optical mapping were assessed as terminal phenotypes. Recombinant IL-18 was further perfused for 60 minutes to study APD VT induction (supplemental Data).
Patch clamping in vitro and statistics.
Cardiac myocytes were isolated from hearts of CTR and SCD mice at baseline by standard enzymatic techniques, and patch clamp recordings of transient outward potassium current (Ito) channels (supplemental Figure 7) were made with an Axopath-200B amplifier (Molecular Devices, San Jose, CA), as described previously29 (supplemental Data). Details of statistical testing and analyses are provided in the supplemental Data. P values ≤ .05 were considered significant.
Results
Murine model of SCD
Altered conduction and APDs in SCD mice.
Hearts were stimulated at the center of the LV with a concentric bipolar electrode to measure APD90 and longitudinal and transverse conduction velocity (Figure 1). Compared with CTR mice, SCD mice showed significantly longer APDs and greater APD dispersion (Figure 1A-B,E-G). CV was slower in SCD mice in the longitudinal direction (Figure 1C) but not in the transverse direction (Figure 1D). The rise time of AP upstroke was unchanged (supplemental Figure 1A).
Figure 1.
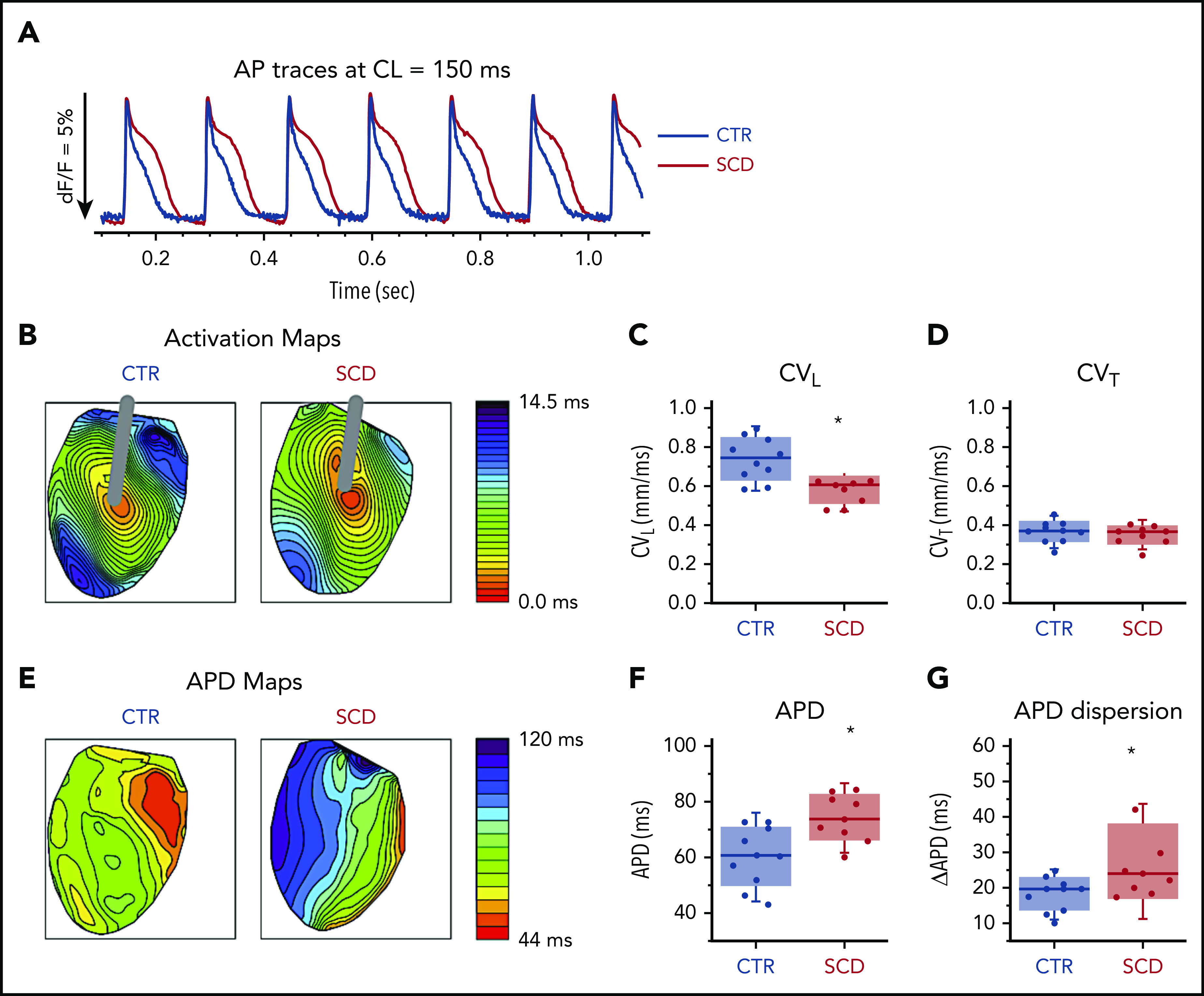
Prolongation of APD and slow conduction in hearts of SCD mice. (A) Representative AP traces from CTR (black) and SCD (red) hearts. (B) Activation maps from CTR (left) and SCD hearts (right). The center of the LV was stimulated with a concentric bipolar electrode to measure longitudinal conduction velocity (CVL) and transverse conduction velocity (CVT). (C-D) CVL of SCD hearts was slower than that for CTR hearts (0.75 ± 0.11 ms in CTR vs 0.58 ± 0.07 ms in SCD mice; P = .002), and CVT was not significantly different between CTR and SCD hearts (0.37 ± 0.056 ms in CTR vs 0.35 ± 0.05 ms in SCD mice; P = .51). *P < .05. Error bars represent standard error. (E) APD maps from the LV in CTR and SCD hearts. (F) APDs of SCD hearts were prolonged (60.4 ± 10.7 ms in CTR vs 74.5 ± 8.4 ms in SCD; P = .006). (G) Spatial dispersion of APDs was also increased in SCD hearts (18.3 ± 4.8 ms in CTR vs 27.6 ± 10.9 ms in SCD; P = .025) (n = 10 for CTR group; n = 9 for SCD group). Median values displayed with whiskers using the Tukey method. CL, cycle length.
Increased VT induction in SCD mice with pacing in vivo.
Inducibility testing for arrhythmias (supplemental Figure 1B) revealed a significantly greater propensity for polymorphic VT in SCD mice (77.8%) compared with CTR mice (11.1%) with pacing (P = .0018). SCD mice exhibited frequent VTs that lasted longer during both burst and premature programmed electrical stimulation protocols (sample tracings are illustrated in supplemental Figure 1C).
Acute IL-18 perfusion prolongs APD and triggers VT ex vivo.
Patients with SCD17 exhibit significantly increased circulating IL-18 levels compared with healthy controls. Because our data were consistent with ranges found in both acute25 and chronic inflammatory states, we tested dose responses to higher levels of recombinant IL-18 in CTR and SCD mice. Exogenous perfusion of IL-18 during optical mapping resulted in dose-dependent APD prolongation associated with frequent EADs and VTs (supplemental Figure 2A) in SCD mice. At higher concentrations (>5 ng/mL), frequent EADs and VTs were seen shortly after IL-18 perfusion (<10 minutes); APD measurements were not possible because of frequent EADs and VTs. We compared the effect of 2 ng/mL of IL-18 on APD in the hearts of SCD vs CTR mice, a dose previously reported as low normal in patients with SCD.17 Exogenous perfusion of IL-18 (2 ng/mL) resulted in acute APD prolongation with frequent atrioventricular block (Figure 2A). Within 60 minutes, IL-18 perfusion led to the development of frequent premature ventricular contractions (PVCs) and short-lived VTs in SCD mice alone (n = 6 of 6 SCD vs 0 of 6 in the hearts of CTR mice). Because of the presence of frequent triggered activities and VT, APDs were compared after 20-minute perfusion of IL-18 at 2 ng/mL. Figure 2B displays paired dot plots of APD before and after 20 minutes of IL-18 perfusion. IL-18 prolonged APD in both CTR and SCD mice with a greater change in SCD mice (Figure 2B). Analysis of APs revealed that a new PVC was initiated before the previous one fully repolarized, suggesting that IL-18 resulted in APD prolongation and EAD (Figure 2C). The activation maps (Figure 2D) revealed that VTs were initiated by PVCs and maintained by multiple focal activity. The long-lasting VTs were maintained by reentry anchored in the right ventricle (RV) (supplemental Movie 1). Although acute IL-18 exposure did not alter CV at the basic cycle length, AP upstroke rise time was increased significantly by IL-18 (supplemental Figure 2B-D), suggesting that the Na+ channel altered by IL-18 may potentiate reentry formation in SCD hearts.
Figure 2.
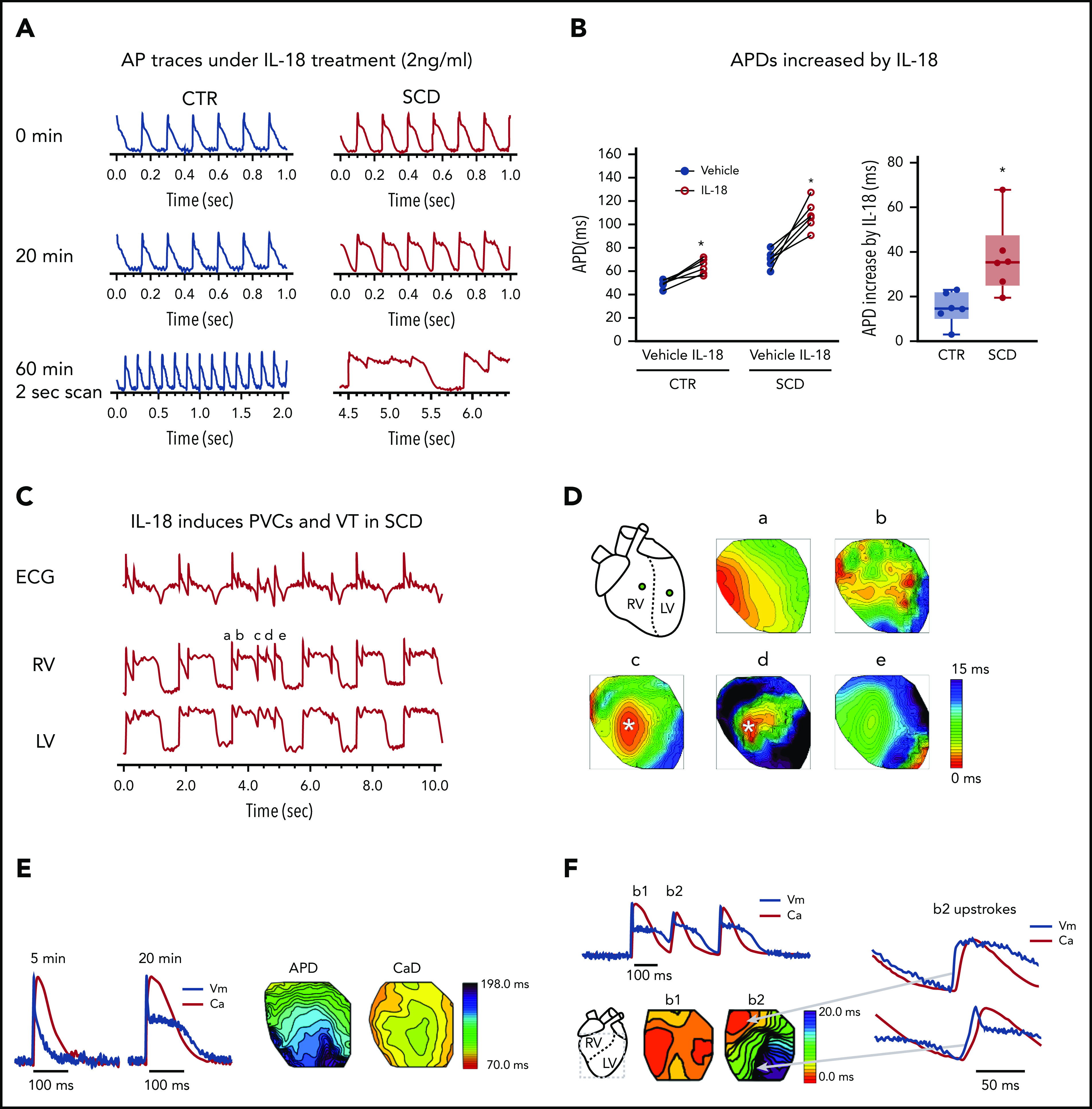
Excessive APD prolongation and frequent arrhythmias (PVCs and VT) in the presence of IL-18 in murine SCD hearts. (A) Representative AP traces under IL-18 (2 ng/mL) in both CTR and SCD hearts after IL-18 perfusion over 1 hour. Frequent PVCs and VT were developed from APD prolongation in SCD (n = 6 of 6) hearts compared with CTR (n = 0 of 6) hearts. (B) APDs were prolonged by IL-18 in both CTR (P = .004) and SCD hearts (P = .003) but were greater in SCD hearts (right panel, ΔAPD = 14.9 ± 7.2 ms in CTR vs 37.6 ± 16.6 ms in SCD; P = .009) (n = 5 mice per group). *P < .05, Error bars represent standard error. Median values displayed with whiskers using the Tukey method. (C) Representative PVC and VT traces induced by IL-18 in SCD hearts. (D) Activation maps of PVCs (marked in panel C) showed focal activations originating from the RV (concentric pattern of c, d, and e beats) that propagated and formed reentry. (E) IL-18 (5 ng/mL) prolongs APD (black) for a substantially longer time than CaD (red; from 75.0 ± 10.1 ms vs 87.1 ± 3.77 ms to 143.9 ± 16.2 ms vs 95.8 ± 3.4 ms; P < .01; n = 4 hearts). (F) Voltage precedes Ca2+ during PVC upstroke. Top: Sample traces of Vm (black) and Ca2+ (red) of PVC; bottom: corresponding activation maps of sinus rhythm (beat 1 [b1]) and PVC (b2). Right: Vm (black) precedes Ca2+ (red) during the upstroke of PVC. Spontaneous Ca2+ release preceding Vm upstrokes was not seen from n = 13 of 13 episodes of VTs recorded (see additional examples in supplemental Figure 3).
We further examined whether altered Ca2+ handling in SCD mice promotes APD prolongation, triggered activity, and reentry formation by simultaneously mapping voltage (Vm) and Ca2+ transients (see “Methods”). Initially, CaD was longer than APD (Figure 2E, left) but, with exposure to IL-18, APD was more prolonged than CaD (Figure 2E, right; APD − CaD = −12.0 ± 8.3 ms before and 48.1 ± 13.7 ms after IL-18; P < .01). In addition, triggered activities observed in SCD mice did not show spontaneous Ca2+ release leading to PVCs (Figure 2F) with Vm upstrokes preceding Ca2+ rise (supplemental Figure 3 shows Vm and Ca2+ examples during VT). These data strongly support the notion that altering membrane ion channels may be responsible for IL-18–mediated APD prolongation and frequent VTs in SCD.
Chronic IL-18 inhibition attenuates triggered VT.
To test the hypothesis that increased IL-18 levels in SCD contribute to increased risk of arrhythmias and represent a novel therapeutic target, we injected vehicle or recombinant IL-18BP into CTR and SCD mice for 4 weeks. We measured mouse serum levels of IL-18 and IL-18BP, a native protein which binds to free IL-18 and prevents it from activating its receptor. Total circulating serum IL-18 levels were not different among all groups of mice (supplemental Figure 4A). Even though serum IL-18BP levels are nonsignificant, they tended to be higher in groups receiving injections of IL-18BP (supplemental Figure 4B). Free circulating IL-18 levels trended higher in SCD (vs CTR) mice, which supports published reports of increased IL-18 expression in the hearts of SCD (vs CTR) mice.21 Compared with vehicle, IL-18BP significantly reduced free IL-18 in SCD mice (analysis of variance [ANOVA] P = .026; Figure 3A), confirming the known function of IL-18BP as a decoy receptor for free IL-18.
Figure 3.
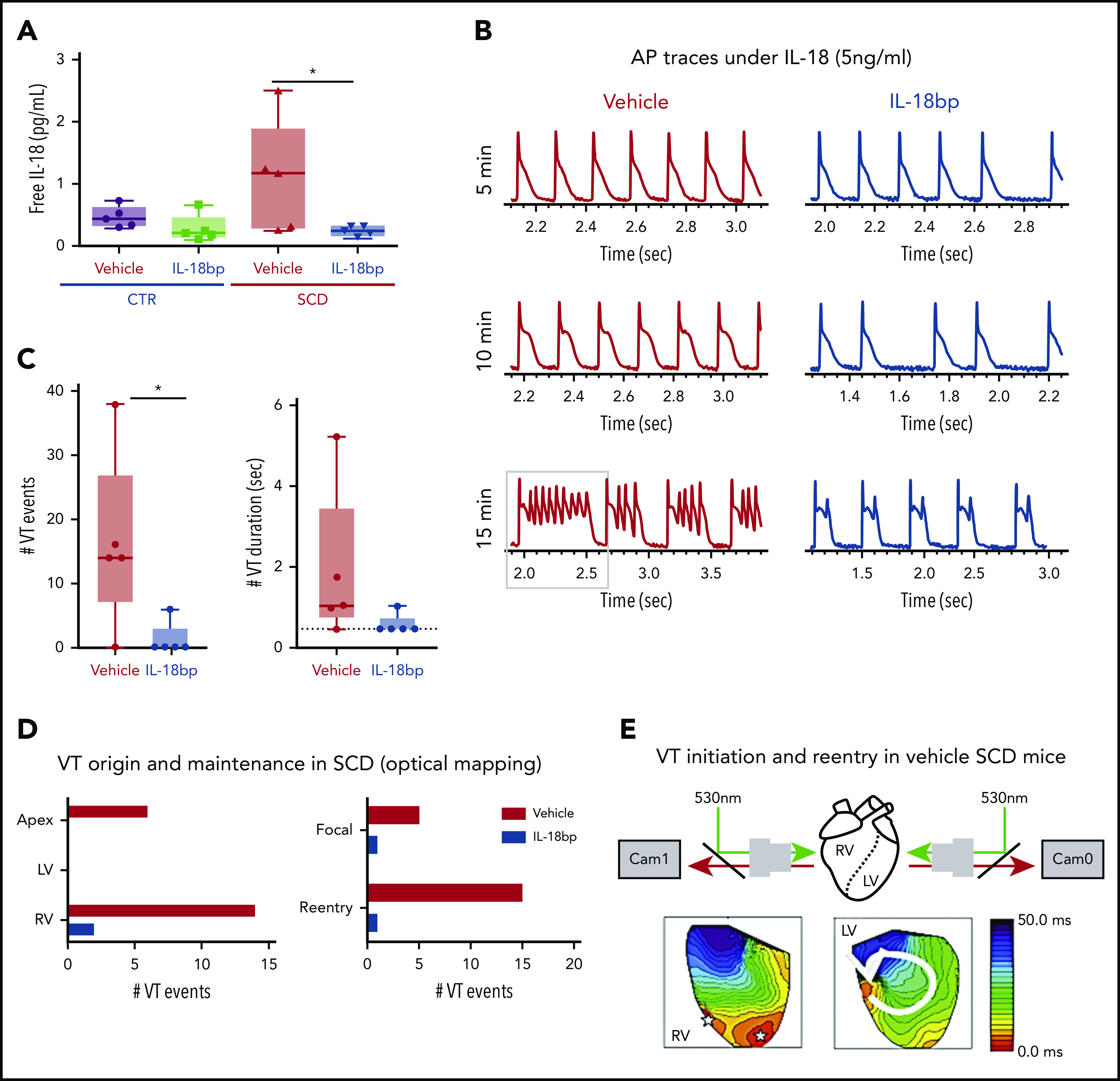
IL-18BP treatment prevents VT initiations under IL-18 exposure in murine SCD hearts. (A) Free IL-18 levels were higher in SCD mice compared with CTR mice, both with exposure to vehicle for 4 weeks, and reduced with IL-18BP exposure for 4 weeks in SCD mice compared with vehicle administration (CTR + vehicle, 0.46 ± 0.07 pg/mL; CTR + IL-18BP, 0.28 ± 0.09 pg/mL; SCD + vehicle, 1.13 ± 0.35 pg/mL; SCD + IL-18BP, 0.24 ± 0.03 pg/mL; ANOVA P = .026; n = 5 for each group). (B) Representative AP and VT traces under IL-18 in vehicle and IL-18BP treated hearts in the presence of 5 ng/mL IL-18. PVCs and VT were frequently induced in vehicle-treated hearts (n = 4 of 5) compared with IL-18BP-treated hearts (n = 1 of 5). (C) IL-18BP treatment suppressed incidence of VT events and VT duration. (D) Focal activity from RV and reentry formations were markedly reduced in the IL-18BP group. (E) Activation maps from the RV and LV showed that focal activity from the RV formed reentry to initiate a transient VT shown in panel B (gray box represents the VT episode that was used for activation maps shown in panel E). Median values displayed with whiskers using the Tukey method. *P < .05. Horizontal bar in panels A and C signify mean of values. Error bars represent standard error. Cam0, camera 0; Cam1, camera 1.
Chronic administration of IL-18BP also prevented IL-18–mediated VT events in the hearts of SCD mice during optical mapping. Specifically, IL-18 perfusion in isolated mouse hearts at higher concentrations (5 ng/mL) resulted in a rapid onset of APD prolongation (Figure 3B; supplemental Figure 2A) associated with frequent PVCs and VT in vehicle-treated SCD mice (n = 4 of 5 mice) within 10 minutes, whereas only 1 SCD mouse heart treated with IL-18BP exhibited VT (Figure 3B). Both VT events and durations were suppressed in the IL-18BP treatment group (Figure 3C).
RV and apex as sources for arrhythmias.
Activation maps from the RV and LV showed focal activity from the RV, and the apex formed reentry and initiated VT in SCD mice with vehicle (Figure 3E), whereas chronic exposure to IL-18BP significantly reduced this activity (Figure 3C-D). Because of activity triggered by IL-18 perfusion in the vehicle group, it was difficult to reliably measure APD after the onset of frequent PVCs and VT, and the early phase of IL-18 perfusion showed a trend toward less prolongation of APDs in mice treated with IL-18BP vs vehicle (supplemental Figure 5A). There was significant improvement in AP rise time and increased CV with IL-18BP (supplemental Figure 5B-C).
IL-18 reduces Ito in SCD cardiomyocytes.
Reverse transcriptase-polymerase chain reaction validated recently published cardiac ion channel expression profiling from RNA sequencing for hearts of SCD and CTR mice16 and revealed that SCD mice had reduced expression levels of myocardial KCND2 and KCND3 genes (encoding Kv4.2 and Kv4.3 that underlie Ito channels) (Figure 4A), consistent with our observation of APD prolongation. Expression of other surveyed sodium and potassium channels previously associated with VT30 were not significantly altered (supplemental Figure 6).
Figure 4.
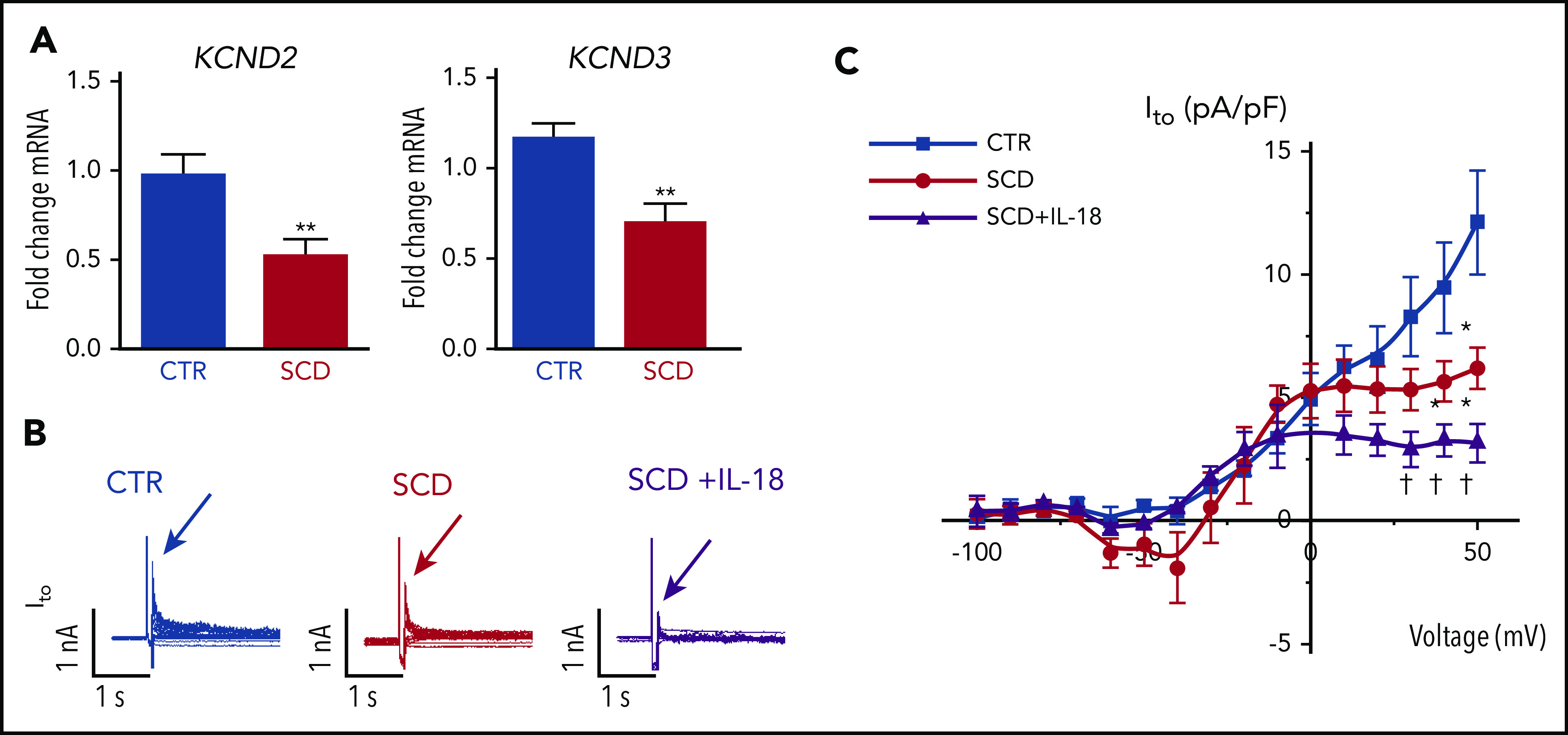
IL-18 effects on Ito in murine SCD hearts. (A) KCND2 and KCND3 gene expression levels were lower in SCD mice compared with CTR mice (n = 5 mice for both groups). (B) Representative Ito traces from CTR and SCD myocytes. (C) Current-voltage curve of Ito shows that the SCD heart has lower Ito amplitude above 0 mV, which is further reduced by IL-18 (25 ng/mL) (n = 5 mice for CTR group; n = 6 mice for SCD group). *P < .05; **P < .01 compared with CTR group. †P < .05, compared with SCD group. Error bars in panels A and C represent standard error. nA, nanoampere; pA, picoampere; pF, picofarad.
Patch clamping was performed in isolated cardiomyocytes to assess for Ito from hearts of SCD and CTR mice (supplemental Figure 7). We observed reduced peak Ito in SCD vs CTR mice, which was further decreased by IL-18 exposure (Figure 4B-C). Steady-state current (Iss) was increased at baseline in SCD vs CTR mouse myocytes, but administration of IL-18 did not have significant effects (supplemental Figure 8A). Similar effects were also seen in CTR mice in which IL-18 exposure resulted in reduced Ito (supplemental Figure 8B-C).
Chronic IL-18 inhibition attenuates murine sickle cell cardiomyopathy and improves diastolic function.
Consistent with results we observed in a report of Berkeley SCD mice,16 we also observed increased patchy myocardial collagen deposition and fibrosis in both ventricles in the Townes model of SCD compared with CTR mice (Figure 5). Along with decreased burden of arrhythmia, exposure to chronic IL-18BP resulted in reduced cardiac fibrosis and improved diastolic function in SCD mice. Specifically, IL-18 inhibition led to a reduced percentage of the area of fibrosis measured by Masson Trichrome staining in the total myocardium vs vehicle (Figure 5A-E); reduction in fibrosis was observed across both the RV and the LV and in perivascular as well as interstitial tissues. These patterns were also visualized in older mice (age 6 months; supplemental Figure 9A). Echocardiography-detected diastolic measures of the ratio (E/e′) of mitral valve peak inflow velocity of early filling (E) over early mitral annular valve velocity (e′) were also reduced by IL-18 inhibition vs vehicle (Figure 5F), which reflects reduced LV filling pressures. Both LV end-systolic (supplemental Figures 9B and 10) and end-diastolic volumes (Figure 5G) were significantly smaller after IL-18BP treatment, suggesting attenuated LV dilation and remodeling. No changes in systolic function were observed in SCD mice after IL-18BP therapy including fractional shortening and cardiac output (supplemental Figures 9C-D and 10). Moreover, the ratio of heart weight to body weight was significantly different among the 4 groups (ANOVA P < .01; supplemental Figure 11): IL-18BP administration tended to attenuate heart hypertrophy in SCD mice (heart weight/body weight: SCD + vehicle, 6.56 ± 0.15 mg/g, SCD + IL-18BP, 5.70 ± 0.25 mg/g).
Figure 5.
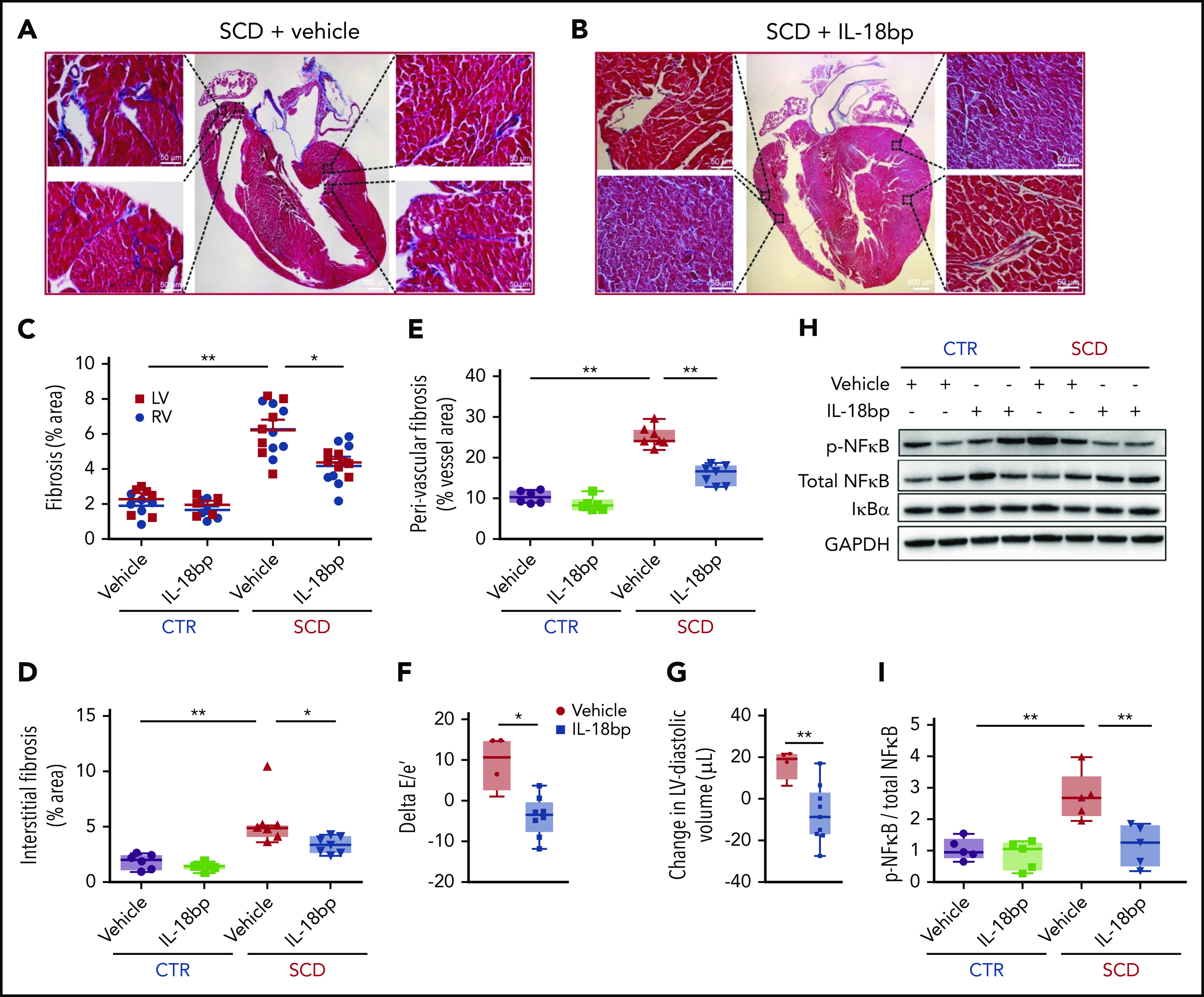
IL-18BP reduces fibrosis and improves function in SCD hearts. (A-B) Representative pictures of Masson trichrome staining with vehicle vs IL-18BP-exposed hearts in SCD mice. (C-E) Fibrosis area percentage was significantly higher in SCD mice LV and RV tissues compared with CTR heart tissues (C) and was reduced in SCD mice with exposure to IL-18BP (total myocardium: CTR + vehicle, 2.08% ± 0.25% [n = 6]; CTR + IL-18BP, 1.80% ± 0.18% [n = 6]; SCD + vehicle, 6.25% ± 0.29% [n = 7]; SCD + IL-18BP, 4.27% ± 0.29% [n = 7]; ANOVA P < .01). (C) IL-18BP significantly reduced fibrosis levels in both the LV and the RV (ANOVA P < .01) as well as (D) in both perivascular and interstitial myocardium (ANOVA P < .01) and (E) perivascular myocardium (ANOVA P < .01) in SCD mice. ANOVA and post hoc *P < .05, **P < .01. (F) Diastolic function was improved (reduced delta E/e′ or smaller E/e′ after IL-18BP treatment) with exposure to IL-18BP in SCD mice (ANOVA P = .04) (n = 4 for vehicle and n = 9 for IL-18BP group). (G) SCD mice exposed to IL-18BP for 4 weeks manifested a reduced change in LV end diastolic volume (eg, smaller volumes) vs vehicle (n = 4 for vehicle; n = 9 for IL-18BP group). (H-I) Chronic IL-18 inhibition is associated with reduced activated NF-κB protein levels in SCD mouse hearts. Phosphorylated NF-κB (p-NF-κB) is higher in SCD hearts (*P < .05) compared to CTR group and inhibited (**P < .01) in SCD with IL-18BP injection (n = 5 mice per group). Median values displayed with whiskers using the Tukey method.
Inflammation in SCD hearts.
Given the established role of IL-18 in the inflammasome, NF-κB and I-κBα levels were next assessed in young and older mice. Although total NF-κB and I-κBα remained unchanged, phosphorylated NF-κB levels and the ratio of phosphorylated NF-κB to total NF-κB (Figure 5H-I) were significantly higher in the hearts of young and older (supplemental Figure 12) SCD mice and were effectively reduced by IL-18BP (ANOVA P < .01). These data suggest that IL-18 may be mediating cardiac inflammation and fibrosis in SCD mice, in part, via NF-κB activation.
Human sickle cell cardiomyopathy
IL18 levels and fibrosis in the UCH cohort.
On the basis of the acute and chronic cardiac effects of IL-18 in SCD mice, we correlated IL-18 levels in patients with SCD with clinical traits. In a historical UCH cohort,23 patients prospectively underwent cardiac magnetic resonance imaging with LGE to detect myocardial fibrosis revealing 25% of patients (n = 8 of 32) with LGE. Evaluation of PBMC-derived gene expression profiling in these patients with and without evidence of myocardial fibrosis revealed 84 differentially expressed genes (Figure 6A; supplemental Table 1). We observed higher IL18 gene expression as 1 of the top 2 differentially regulated genes in patients with LGE, which is consistent with reports of IL-18 as a profibrotic inflammatory mediator.31 These data support circulating IL18 gene expression levels as a marker for cardiac fibrosis.
Figure 6.
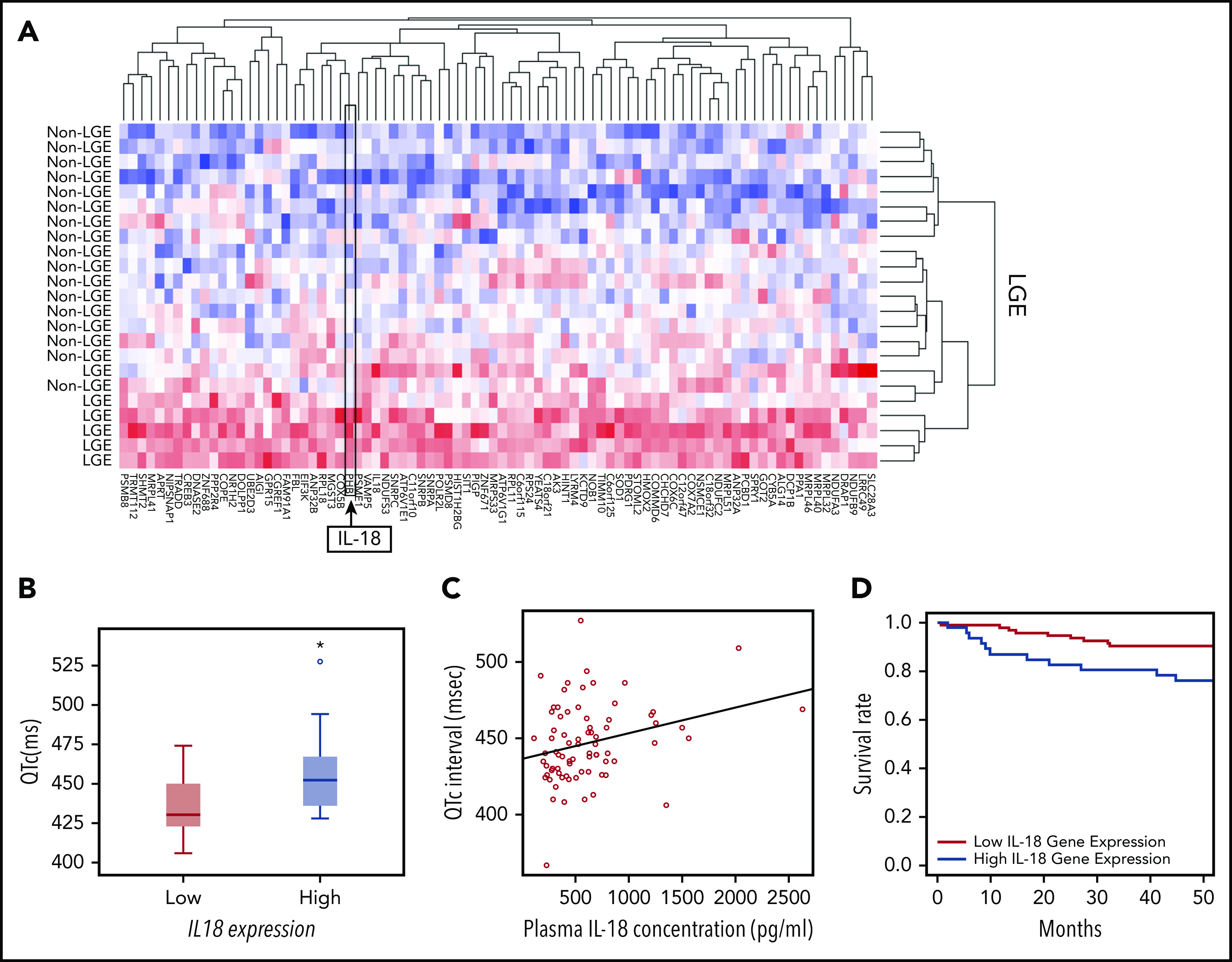
IL-18 levels are associated with fibrosis, prolonged QTc, and mortality in patients with SCD. (A) Microarrays performed on PBMC-derived RNA in patients with SCD revealed a unique set of genes that were differentially regulated between patients who exhibited evidence of myocardial fibrosis on cardiac magnetic resonance imaging scans and those who did not. IL18 was 1 of the genes within this signature. (B) Gene expression profiling from PBMC-derived RNA of patients with SCD revealed longer QTc intervals associated with the patients with higher expression of IL18 (n = 8) compared with those with lower levels (n = 18) (*P = .047). Horizontal bar signifies mean of values. Error bars represent standard error. (C) Circulating plasma IL-18 levels are significantly correlated with QTc intervals in patients with SCD (n = 74; r = 0.53; P = .015). (D) Patients with SCD with increased circulating PBMC-derived IL18 expression had a significantly increased risk of death compared with those who had lower levels (P = .017).
IL18 expression and levels with QTc in the UIC cohort.
Using available PBMC-derived genome-wide microarray data from a separate published UIC cohort,32 we evaluated for relationships between IL18 gene expression and QTc. Among all patients with the HbSS genotype who underwent an ECG prospectively within 1 year after blood collection, significantly higher QTc intervals (median QTc, 452.0 ± 32.0 ms vs 430.5 ± 29.0 ms; P = .047) were observed in patients classified as having higher IL18 gene expression (Figure 6B).
Plasma IL-18 levels also exhibited a significant positive correlation with QTc intervals (ρ = 0.26; P = .015; n = 74; Figure 6C) and left atrial volumes (ρ = 0.51; P = .05; n = 74; supplemental Figure 13). The former association was strengthened (P = .008) and the latter was no longer significant (P = .45; supplemental Table 2) after adjustment for covariates. PBMC-derived IL18 gene expression was not correlated with plasma IL-18 protein levels (r = −0.36; P = .304; n = 63) in a subset of these patients (for which both gene expression and plasma were available).
IL18 levels and mortality in the UIC cohort.
In the portion of the UIC cohort for which vital status was available, PBMC-derived IL18 gene expression was evaluated among patients who were prospectively followed for survival. By using a Cox proportional hazard model adjusted for age, sex, hemoglobin genotype, and hydroxyurea use, we observed that increased IL18 expression was significantly associated with an increased mortality risk (P = .012; supplemental Table 3). In addition, patients in the highest tertile of IL18 expression had a threefold risk of death compared with patients in the lowest tertile (hazard ratio, 3.06; 95% confidence interval, 1.22-7.68; P = .017; Figure 6D). These data cumulatively indicate that circulating IL-18 levels may represent a novel biomarker for cardiovascular and mortality risk in SCD.
Discussion
Sickle cell cardiomyopathy is characterized by increased LV mass, fibrosis, diastolic dysfunction, and prolonged QTc.14,15,33-35 In this study, the humanized SCD mouse model mimics prolonged APD and fibrosis with a predisposition toward inducible VT. Mechanistically, prolonged APD in SCD mice reflects the contribution of reduced cardiac KCND2 and KCND3 expression and Ito function. Moreover, exogenous acute administration of IL-18 results in further APD prolongation and frequent VTs that originate predominantly in the RV in SCD mice. Cumulatively, these acute IL-18 effects are driven, in part, by inflammatory-mediated reductions in Ito function and are enabled by baseline pathological cardiac structural (fibrosis) and electrophysiological remodeling (baseline Ito reduction) in SCD hearts. Sustained IL-18 inhibition in SCD mice leads to attenuated IL-18–induced VT activity, decreased fibrosis, improved diastolic function, and reduced cardiac phosphorylated NF-κB levels. Patients with SCD with higher IL18 gene expression also exhibited an increased prevalence of myocardial fibrosis, longer QTc intervals, and increased mortality. Based on known associations between features of sickle cell cardiomyopathy and mortality in patients,9,36 the findings of inducible polymorphic VT in SCD mice suggest the mechanistic and pathologic link between IL-18–mediated inflammation and arrhythmic propensity to poor outcomes and sudden cardiac death in patients.
A previous study reported sudden death with ECG-documented ventricular fibrillation in 1 SCD mouse.16 Our study demonstrates prolonged APD and inducible VT (via pacing or IL-18) as novel additional phenotypes of nearly all murine SCD hearts and implicates a vulnerable myocardium. APD prolongation and increased dispersion in SCD mice can precipitate repolarization heterogeneity and reentry formation leading to VT. The molecular and electrophysiological remodeling within SCD hearts resembles the concept of “reduced repolarization reserve”37, an environment that promotes long QT-related arrhythmias such as Torsades de Pointes with additional stress. Specifically, APD prolongation is reflective of significant reductions in baseline Ito current and cardiac KCND2 and KCND3 expression in SCD mice. These genes encode the subunits of the voltage-gated potassium channels Kv4.2 and Kv4.3 that conduct fast transient outward current (Ito,f). Functional knockout of Kv4.2 in mice previously demonstrated prolonged QT, atrioventricular block, and frequent VT38,39 as seen in SCD mice. In contrast to Ito downregulation, we found that Iss (steady-state K+ current) is slightly increased in SCD mice. Because of its slow activation (30 times slower than Ito), the contribution of Iss to the very short APD in mice is thought to be limited.40 In addition, Iss was not previously detected in humans, suggesting that Iss is not the major contributor to QT prolongation and arrhythmogenesis in SCD.41 Although our polymerase chain reaction data do not suggest that several other channels are involved, it is possible that trafficking, membrane retention, and posttranslational modification of ion channels and Ca2+ handling proteins may contribute to arrhythmias in SCD. Although our simultaneous voltage and Ca2+ mapping did not show either persistent Ca2+ elevation during long APD or spontaneous Ca2+ release during triggered activity, it is still possible that intracellular Ca2+ can contribute to arrhythmogenesis through activation of membrane ion channels such as phosphorylation of sodium, L-type calcium, transient outward potassium and inward rectifier potassium channels by CaMKII.42 Further studies are needed to assess the proarrhythmic potential of other ion channels and Ca2+ handling modification in SCD mice such as those that compare RV and LV cardiomyocytes in SCD.
This study demonstrates the pathological roles (acute and chronic) and biomarker potential of IL-18 in sickle cell cardiomyopathy, linking it to Ito function and NF-κB activation. Associations of IL18 gene expression with fibrosis, QTc intervals, and patient mortality further support the pathological role of IL-18 in SCD. The latter findings support our previous observations of the association between IL18 gene expression levels with diastolic dysfunction in patients with SCD.21 Plasma IL-18 levels, however, do not correlate with PBMC-derived IL18 gene expression, likely reflecting a combination of small sample size, heterogeneity in the source of plasma IL-18 protein, and differences between transcriptional and translational regulation of IL-18 in SCD. Whereas plasma IL-18 levels originate from several cells (including PBMCs and endothelial and immune cells43-45), which dilutes the potential associations of IL-18, observed IL18 gene expression associations may reflect the role of PBMCs in the development of inflammation and fibrosis in the sickle myocardium. IL-18 inhibition studies in mice also bolster the idea of targeting IL-18 as a novel therapeutic for improving cardiac function and preventing VT. Studies of IL-18BP emphasize the possible role of IL-18 receptor signaling, but the mechanisms of the acute effects of IL-18 on Ito via IL-18 receptor signaling or via direct interactions with the channel remain uncharacterized.
VT predominantly originates from the RV in SCD mice. Differences in the electrophysiological remodeling between the RV and LV that may help explain this susceptibility have not been characterized. Although fibrosis patterns were not different between the RV and LV (which could explain the arrhythmogenic propensity), we speculate whether PH and increased RV afterload may be contributing factors. Previous reports show the RV as the source of pacing-induced VT and spontaneous ventricular fibrillation in animal models of PH and RV failure,46,47 with attenuation of VT after treating PH. PH is a well-established risk factor for mortality in patients with SCD,48,49 although SCD mice develop spontaneous PH with aging.50 Cumulatively, these data raise the idea of PH as a contributing factor for IL-18–mediated VT susceptibility in SCD. IL-18 has also been implicated in the development of PH in animal models,51 which mechanistically links these clinical manifestations. Previous associations between prolonged QTc, diastolic dysfunction, fibrosis, and IL18 gene expression levels with markers of hemolytic burden in patients with SCD7,9,17,36,52,53 suggest a contributing role for hemolysis in VT vulnerability.
In summary, SCD mice develop a cardiomyopathy with an acquired long-QT syndrome phenotype, which results in a predisposition to inducible VT. This electrical phenotype is associated with increased IL-18 and myocardial fibrosis, augmenting the propensity for ventricular arrhythmias. With “sudden death” characterized as one of the top causes of death, we stress the need for further studies to link IL-18 to prolonged QTc and fatal cardiac arrhythmias in patients with SCD.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Natalia Arana, Hector Quijada, Michelle Shi, Kyle Weigand, Josh Strom, Jon Groth, Vishal Mali, and Oluwaseun Kafisanwo for assistance with the mouse study.
This study was funded by grants from the National Center for Research Resources/National Center for Advancing Translational Sciences, National Institutes of Health (NIH) (UL1RR029879), the National Heart, Lung, and Blood Institute (R01 HL111656, R01 HL127342 [R.F.M.], and R01 136603 [A.A.D.]), the National Institute of General Medical Sciences (K23 GM112014) (J.D.D.), by the American Heart Association (14CRP18910051) (A.A.D.), and by the American Thoracic Society Foundation/Pulmonary Hypertension Association (A.A.D.).
Footnotes
Four articles have been cited in this article for data sets we used in our study.13,14,24,32 There is no overlap between what has been published and what we analyzed in this study; the details are below in terms of what we leveraged from each published article.13,14,24,32 We used the data sets from Indik et al13 to evaluate QTc associations with IL18 gene expression data from Desai et al.32 We used the data from Desai et al14 to evaluate cardiac fibrosis data with gene expression data from Desai et al.24 We used the data from Desai et al32 to filter and evaluate IL18 gene expression associations with survival. We used the gene expression data from Desai et al24 for association with cardiac fibrosis data from Desai et al.14
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: A.G., Y.-D.F., and A.A.D. conceived and designed the study, analyzed the data, and drafted the manuscript; T.Y.K. and A.X. collected and analyzed the data; K.B., J.D.D., and J.G.N.G. collected data and drafted the manuscript; I.G., H.T., E.J., S.C.-Y., J.H.I., G.S., J.C., G.G., C.H., M.M.K., D.S.P., T.Z., J.X.-J.Y., P.B., G.K., R.K., and R.F.M. collected data; Y.K. helped draft the manuscript; S.C.D. helped design the study and draft the manuscript; and B.-R.C. helped collect and analyze the data and draft the manuscript.
Conflict-of-interest disclosure: A.A.D. served on the advisory board for Novartis. The remaining authors declare no competing financial interests.
Correspondence: Ankit A. Desai, Indiana University, Krannert Institute of Cardiology, Department of Medicine, 950 W. Walnut St, R3 Building, Room 408, Indianapolis, IN 46202; e-mail: ankdesai@iu.edu.
REFERENCES
- 1.Hamideh D, Alvarez O. Sickle cell disease related mortality in the United States (1999-2009). Pediatr Blood Cancer. 2013;60(9):1482-1486. [DOI] [PubMed] [Google Scholar]
- 2.Fitzhugh CD, Lauder N, Jonassaint JC, et al. Cardiopulmonary complications leading to premature deaths in adult patients with sickle cell disease. Am J Hematol. 2010;85(1):36-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Graham JK, Mosunjac M, Hanzlick RL, Mosunjac M. Sickle cell lung disease and sudden death: a retrospective/prospective study of 21 autopsy cases and literature review. Am J Forensic Med Pathol. 2007;28(2):168-172. [DOI] [PubMed] [Google Scholar]
- 4.Darbari DS, Kple-Faget P, Kwagyan J, Rana S, Gordeuk VR, Castro O. Circumstances of death in adult sickle cell disease patients. Am J Hematol. 2006;81(11):858-863. [DOI] [PubMed] [Google Scholar]
- 5.Manci EA, Culberson DE, Yang YM, et al. ; Investigators of the Cooperative Study of Sickle Cell Disease . Causes of death in sickle cell disease: an autopsy study. Br J Haematol. 2003;123(2):359-365. [DOI] [PubMed] [Google Scholar]
- 6.Holloman KL, Johnson CS, Haywood LJ. Electrocardiogram analysis in adult patients with sickle cell disease. J Natl Med Assoc. 1987;79(8):809-814. [PMC free article] [PubMed] [Google Scholar]
- 7.Liem RI, Young LT, Thompson AA. Prolonged QTc interval in children and young adults with sickle cell disease at steady state. Pediatr Blood Cancer. 2009;52(7):842-846. [DOI] [PubMed] [Google Scholar]
- 8.Mueller BU, Martin KJ, Dreyer W, Bezold LI, Mahoney DH. Prolonged QT interval in pediatric sickle cell disease. Pediatr Blood Cancer. 2006;47(6):831-833. [DOI] [PubMed] [Google Scholar]
- 9.Upadhya B, Ntim W, Brandon Stacey R, et al. Prolongation of QTc intervals and risk of death among patients with sickle cell disease. Eur J Haematol. 2013;91(2):170-178. [DOI] [PubMed] [Google Scholar]
- 10.Fontaine JM, Ofili EO, Adenaike MB, VanDecker W, Haywood LJ. Clinical assessment of the risk for sudden cardiac death in patients with sickle cell anemia. J Natl Med Assoc. 2008;100(4):360-368. [DOI] [PubMed] [Google Scholar]
- 11.Maisel A, Friedman H, Flint L, Koshy M, Prabhu R. Continuous electrocardiographic monitoring in patients with sickle-cell anemia during pain crisis. Clin Cardiol. 1983;6(7):339-344. [DOI] [PubMed] [Google Scholar]
- 12.Chacko P, Kraut EH, Zweier J, Hitchcock C, Raman SV. Myocardial infarction in sickle cell disease: use of translational imaging to diagnose an under-recognized problem. J Cardiovasc Transl Res. 2013;6(5):752-761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Indik JH, Nair V, Rafikov R, et al. Associations of prolonged QTc in sickle cell disease. PLoS One. 2016;11(10):e0164526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Desai AA, Patel AR, Ahmad H, et al. Mechanistic insights and characterization of sickle cell disease-associated cardiomyopathy. Circ Cardiovasc Imaging. 2014;7(3):430-437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Niss O, Quinn CT, Lane A, et al. Cardiomyopathy with restrictive physiology in sickle cell disease. JACC Cardiovasc Imaging. 2016;9(3):243-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bakeer N, James J, Roy S, et al. Sickle cell anemia mice develop a unique cardiomyopathy with restrictive physiology. Proc Natl Acad Sci U S A. 2016;113(35):E5182-E5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cerqueira BA, Boas WV, Zanette AD, Reis MG, Goncalves MS. Increased concentrations of IL-18 and uric acid in sickle cell anemia: contribution of hemolysis, endothelial activation and the inflammasome. Cytokine. 2011;56(2):471-476. [DOI] [PubMed] [Google Scholar]
- 18.Dutra FF, Alves LS, Rodrigues D, et al. Hemolysis-induced lethality involves inflammasome activation by heme. Proc Natl Acad Sci U S A. 2014;111(39):E4110-E4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sundd P, Gladwin MT, Novelli EM. Pathophysiology of sickle cell disease. Annu Rev Pathol. 2019;14(1):263-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pitanga TN, Oliveira RR, Zanette DL, et al. Sickle red cells as danger signals on proinflammatory gene expression, leukotriene B4 and interleukin-1 beta production in peripheral blood mononuclear cell. Cytokine. 2016;83:75-84. [DOI] [PubMed] [Google Scholar]
- 21.Duarte JD, Desai AA, Sysol JR, et al. Genome-wide analysis identifies IL-18 and FUCA2 as novel genes associated with diastolic function in African Americans with sickle cell disease. PLoS One. 2016;11(9):e0163013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hernesniemi JA, Karhunen PJ, Oksala N, et al. Interleukin 18 gene promoter polymorphism: a link between hypertension and pre-hospital sudden cardiac death: the Helsinki Sudden Death Study. Eur Heart J. 2009;30(23):2939-2946. [DOI] [PubMed] [Google Scholar]
- 23.Mehra MR, Uber PA, Walther D, et al. Gene expression profiles and B-type natriuretic peptide elevation in heart transplantation: more than a hemodynamic marker. Circulation. 2006;114(1 suppl):I21-I26. [DOI] [PubMed] [Google Scholar]
- 24.Desai AA, Zhou T, Ahmad H, et al. A novel molecular signature for elevated tricuspid regurgitation velocity in sickle cell disease. Am J Respir Crit Care Med. 2012;186(4):359-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Novick D, Schwartsburd B, Pinkus R, et al. A novel IL-18BP ELISA shows elevated serum IL-18BP in sepsis and extensive decrease of free IL-18. Cytokine. 2001;14(6):334-342. [DOI] [PubMed] [Google Scholar]
- 26.Rutledge CA, Ng FS, Sulkin MS, et al. c-Src kinase inhibition reduces arrhythmia inducibility and connexin43 dysregulation after myocardial infarction. J Am Coll Cardiol. 2014;63(9):928-934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi BR, Salama G. Simultaneous maps of optical action potentials and calcium transients in guinea-pig hearts: mechanisms underlying concordant alternans. J Physiol. 2000;529(pt 1):171-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang X, Hagberg H, Mallard C, et al. Disruption of interleukin-18, but not interleukin-1, increases vulnerability to preterm delivery and fetal mortality after intrauterine inflammation. Am J Pathol. 2006;169(3):967-976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.El Gebeily G, Fiset C. 4-Hydroxytamoxifen inhibits K(+) currents in mouse ventricular myocytes. Eur J Pharmacol. 2010;629(1-3):96-103. [DOI] [PubMed] [Google Scholar]
- 30.Yang KC, Kyle JW, Makielski JC, Dudley SC Jr.. Mechanisms of sudden cardiac death: oxidants and metabolism. Circ Res. 2015;116(12):1937-1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xing SS, Bi XP, Tan HW, Zhang Y, Xing QC, Zhang W. Overexpression of interleukin-18 aggravates cardiac fibrosis and diastolic dysfunction in fructose-fed rats. Mol Med. 2010;16(11-12):465-470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Desai AA, Lei Z, Bahroos N, et al. Association of circulating transcriptomic profiles with mortality in sickle cell disease. Blood. 2017;129(22):3009-3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gladwin MT, Sachdev V. Cardiovascular abnormalities in sickle cell disease. J Am Coll Cardiol. 2012;59(13):1123-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Manci EA, Hillery CA, Bodian CA, Zhang ZG, Lutty GA, Coller BS. Pathology of Berkeley sickle cell mice: similarities and differences with human sickle cell disease. Blood. 2006;107(4):1651-1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vinchi F, De Franceschi L, Ghigo A, et al. Hemopexin therapy improves cardiovascular function by preventing heme-induced endothelial toxicity in mouse models of hemolytic diseases. Circulation. 2013;127(12):1317-1329. [DOI] [PubMed] [Google Scholar]
- 36.Indik JH, Nair V, Rafikov R, et al. Associations of prolonged QTc in sickle cell disease. PLoS One. 2016;11(10):e0164526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roden DM. Taking the “idio” out of “idiosyncratic”: predicting torsades de pointes. Pacing Clin Electrophysiol. 1998;21(5):1029-1034. [DOI] [PubMed] [Google Scholar]
- 38.Barry DM, Xu H, Schuessler RB, Nerbonne JM. Functional knockout of the transient outward current, long-QT syndrome, and cardiac remodeling in mice expressing a dominant-negative Kv4 alpha subunit. Circ Res. 1998;83(5):560-567. [DOI] [PubMed] [Google Scholar]
- 39.Guo W, Li H, London B, Nerbonne JM. Functional consequences of elimination of i(to,f) and i(to,s): early afterdepolarizations, atrioventricular block, and ventricular arrhythmias in mice lacking Kv1.4 and expressing a dominant-negative Kv4 alpha subunit. Circ Res. 2000;87(1):73-79. [DOI] [PubMed] [Google Scholar]
- 40.Brouillette J, Clark RB, Giles WR, Fiset C. Functional properties of K+ currents in adult mouse ventricular myocytes. J Physiol. 2004;559(pt 3):777-798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sabir IN, Killeen MJ, Grace AA, Huang CL. Ventricular arrhythmogenesis: insights from murine models. Prog Biophys Mol Biol. 2008;98(2-3):208-218. [DOI] [PubMed] [Google Scholar]
- 42.Bers DM, Grandi E. Calcium/calmodulin-dependent kinase II regulation of cardiac ion channels. J Cardiovasc Pharmacol. 2009;54(3):180-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu C-L, Ren J, Wang Y, et al. Adipocytes promote interleukin-18 binding to its receptors during abdominal aortic aneurysm formation in mice. Eur Heart J. 2020;41(26):2456-2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu L, Fang C, Fu W, et al. Endothelial cell-derived IL-18 released during ischemia reperfusion injury selectively expands T peripheral helper cells to promote alloantibody production. Circulation. 2020;141(6):464-478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meda Spaccamela V, Valencia RG, Pastukhov O, et al. High levels of IL-18 and IFN-γ in chronically inflamed tissue in chronic granulomatous disease. Front Immunol. 2019;10:2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tanaka Y, Takase B, Yao T, Ishihara M. Right ventricular electrical remodeling and arrhythmogenic substrate in rat pulmonary hypertension. Am J Respir Cell Mol Biol. 2013;49(3):426-436. [DOI] [PubMed] [Google Scholar]
- 47.Umar S, Lee JH, de Lange E, et al. Spontaneous ventricular fibrillation in right ventricular failure secondary to chronic pulmonary hypertension. Circ Arrhythm Electrophysiol. 2012;5(1):181-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gladwin MT, Sachdev V, Jison ML, et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med. 2004;350(9):886-895. [DOI] [PubMed] [Google Scholar]
- 49.Klings ES, Machado RF, Barst RJ, et al. ; American Thoracic Society Ad Hoc Committee on Pulmonary Hypertension of Sickle Cell Disease . An official American Thoracic Society clinical practice guideline: diagnosis, risk stratification, and management of pulmonary hypertension of sickle cell disease. Am J Respir Crit Care Med. 2014;189(6):727-740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hsu LL, Champion HC, Campbell-Lee SA, et al. Hemolysis in sickle cell mice causes pulmonary hypertension due to global impairment in nitric oxide bioavailability. Blood. 2007;109(7):3088-3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morisawa D, Hirotani S, Oboshi M, et al. Interleukin-18 disruption suppresses hypoxia-induced pulmonary artery hypertension in mice. Int J Cardiol. 2016;202:522-524. [DOI] [PubMed] [Google Scholar]
- 52.Sachdev V, Machado RF, Shizukuda Y, et al. Diastolic dysfunction is an independent risk factor for death in patients with sickle cell disease. J Am Coll Cardiol. 2007;49(4):472-479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Niss O, Fleck R, Makue F, et al. Association between diffuse myocardial fibrosis and diastolic dysfunction in sickle cell anemia. Blood. 2017;130(2):205-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.