

Abstract
Autism spectrum disorder (ASD) is characterized by a complex polygenic background, but with the unique feature of a subset of cases (~15%‐30%) presenting a rare large‐effect variant. However, clinical interpretation in these cases is often complicated by incomplete penetrance, variable expressivity and different neurodevelopmental trajectories. NRXN1 intragenic deletions represent the prototype of such ASD‐associated susceptibility variants. From chromosomal microarrays analysis of 104 ASD individuals, we identified an inherited NRXN1 deletion in a trio family. We carried out whole‐exome sequencing and deep sequencing of mitochondrial DNA (mtDNA) in this family, to evaluate the burden of rare variants which may contribute to the phenotypic outcome in NRXN1 deletion carriers. We identified an increased burden of exonic rare variants in the ASD child compared to the unaffected NRXN1 deletion‐transmitting mother, which remains significant if we restrict the analysis to potentially deleterious rare variants only (P = 6.07 × 10−5). We also detected significant interaction enrichment among genes with damaging variants in the proband, suggesting that additional rare variants in interacting genes collectively contribute to cross the liability threshold for ASD. Finally, the proband's mtDNA presented five low‐level heteroplasmic mtDNA variants that were absent in the mother, and two maternally inherited variants with increased heteroplasmic load. This study underlines the importance of a comprehensive assessment of the genomic background in carriers of large‐effect variants, as penetrance modulation by additional interacting rare variants to might represent a widespread mechanism in neurodevelopmental disorders.
Keywords: ASD, mtDNA, NRXN1, penetrance, rare variants
1. INTRODUCTION
Autism Spectrum Disorder (ASD) is a heterogeneous neurodevelopmental disorder with a high prevalence (>1%) and a remarkable social burden, with no effective pharmacological treatments. 1 Despite the high heritability, the vast majority of genetic risk factors still remains unknown and only about 15%‐30% of ASD individuals have an identifiable genetic cause. 2
Several years of investigation has led to considerable progress in the identification of a large number of risk genes and the delineation of a heterogeneous and complex genetic architecture. It is now evident that ASD, like other neuropsychiatric disorders, has a polygenic basis, but with the peculiarity of a subset of cases where a large‐effect variant is present. Advance in microarrays and whole‐exome sequencing (WES) enabled the discovery of many rare variants of large effect, both structural and single‐nucleotide variants, which pinpointed more than 100 high‐confidence specific genes and genetic loci. 3 , 4 , 5 Even if the identification of rare genic likely pathogenic mutations can be extremely informative, their translation into clinical settings is not straightforward, as many of them are characterized by incomplete penetrance and variable expressivity. It is possible that these rare large‐effect variants may act affecting neurodevelopmental processes common to different disorders and the final clinical manifestation rely on the presence of additional risk variants that allow crossing the threshold for clear neuropsychiatric disorders, ultimately defining the specific phenotypic trajectory in the carrier. There are several examples where the penetrance of a ‘pathogenic’ copy number variants (CNV) is increased by the presence of additional risk CNVs. 6
Rare intragenic deletions affecting Neurexin‐1 (NRXN1) represent the prototype example of susceptibility variants for neurodevelopmental disorders with such complexity. 7 , 8 NRXN1 belongs to the neurexin family encoding for an evolutionarily conserved presynaptic cell adhesion molecules involved in formation and maintenance of synaptic connections and vesicular neurotransmitter release. NRXN1 is transcribed in neurons from two independent promoters which generate a longer alpha (α) and a shorter beta (β) isoform, composed of distinct extracellular domains but with an identical intracellular sequence. Moreover, through extensive alternative splicing, thousands of isoforms are produced and differentially expressed throughout the brain, with a likely role as surface recognition molecules that specify synapses. 9 The NRXN1 locus is extremely prone to non‐recurrent deletions with different size and breakpoint location, resulting from chromosomal rearrangements due to genomic instability. Rare intragenic deletions spanning NRXN1 have been described in individuals with ASD, Attention‐Deficit/Hyperactivity Disorder (ADHD), intellectual disability (ID), epilepsy, schizophrenia and bipolar disorder, but also in unaffected parents, siblings and healthy controls, suggesting reduced penetrance and the contribution of other interacting genetic and/or environmental factors that influence clinical features and severity. 7 , 8 The enhanced frequency of additional pathogenic CNVs in cohorts of patients carrying NRXN1 deletions has added support to the role of secondary and independently segregating genetic risk factors in the definition of the final phenotype in children with inherited deletions. Previous studies suggested that deletion extent and exon content may also play a role in this clinical heterogeneity; however, there is no consensus on the different penetrance of 5′ NRXN1 deletions (exons 1‐6, NM_001135659.2) versus 3′ NRXN1 deletions (exons 7‐24, NM_001135659.2). Specifically, one study proposed a lower penetrance of 3′ NRXN1 deletions as they are more frequently co‐occurring with another rare and often pathogenic CNV, 7 while a more recent study reported a higher penetrance of 3′ deletions, given the much higher frequencies in cases versus controls and the higher de novo occurrence. 8
In order to identify pathogenic CNVs in ASD probands, we performed array‐based comparative genomic hybridization (aCGH) on a cohort of 104 ASD individuals from 89 Italian families. A 3′ NRXN1 deletion was identified in a trio family in which a girl with ASD inherited the deletion from the unaffected mother. To further explore the impact of genetic background on the penetrance of NRXN1 deletions, a detailed clinical evaluation of the girl and her parents was combined with genetic analysis including (a) WES, in order to characterize the background of rare variants and (b) deep sequencing of the entire mitochondrial genome, with the aim of detecting rare pathogenic mutations and evaluating the burden of low‐heteroplasmy variants. Mitochondrial DNA (mtDNA) defects could, indeed, represent an overlooked contributing factor to ASD susceptibility, by reducing mitochondrial function sufficiently to fall below the brain's bioenergetics threshold. 10 Moreover, a recent study has provided evidence for a coordinated down‐regulation of synaptic and mitochondrial function genes in post‐mortem brain of ASD subjects, 11 suggesting that a mitochondrial dysfunction might enhance the clinical outcome of NRXN1 deletions.
2. EXPERIMENTAL SECTION
2.1. Participants
A total of 89 Italian families with one or more children with an ASD diagnosis were recruited at the UOSI Disturbi dello Spettro Autistico, IRCCS Istituto delle Scienze Neurologiche (Bologna, Italy).
Assessment and a deep phenotypic characterization of probands were done using a set of standardized clinical tests to evaluate the presence and severity of ASD (ADOS, CARS and M‐CHAT), to assess both developmental/cognitive levels (PEP‐3, Leiter‐R, Griffith Scales, or Wechsler Scales) and adaptive behaviour (Vineland Adaptive Behavior Scale, VABS) as well as discrete and clinical signs like mimicry, hyperactivity, sensory abnormalities and symptoms onset. Moreover, probands underwent EEG and MRI. Subclinical features in relatives were assessed using the Social and Communication Disorders Checklist and The Broad Autism Phenotype Questionnaire.
The ASD sample includes 78 males and 26 females, from 18 multiplex and 71 simplex families. DNA samples from both proband's parents were available for 87 families, and from a single parent for the remaining ones. All DNA samples were extracted from whole blood.
All participants provided a written informed consent to participate to this study. This study was approved by the local Ethical Committee (Comitato Etico di Area Vasta Emilia Centro (CE‐AVEC); code CE 14060). All research was performed in accordance with the relevant guidelines and regulations.
2.2. Copy number variant analysis
Array‐based comparative genomic hybridization (aCGH) was performed on 104 ASD individuals (89 probands, 13 siblings, 1 cousin and one uncle) and 5 siblings with another neurodevepmental disorders, using the SurePrint G3 Unrestricted CGH ISCA v2, 8x60K (Agilent Technologies) or the SurePrint G3 Human CGH Microarray 8x60K (Agilent Technologies), following manufacturer's instructions. Scan images were analysed through the Agilent CytoGenomics 5.0.1.6 software and aberrations were called by the ADM1 algorithm with a threshold of 6.0 and at least three consecutive oligonucleotides with similar log2 ratios.
All CNVs were compared to those collected in different public databases: Database of Genomic Variants (DGV, http://projects.tcag.ca/variation/), DECIPHER (https://decipher.sanger.ac.uk/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and database of human CNVs hosted by IRCCS OASI Maria SS. (Troina, Italy). The American College of Medical Genetics (ACMG) guidelines were used for CNV interpretation and reporting. 12 According to these criteria, the only identified pathogenic CNV is a deletion involving the NRXN1 gene in a girl with ASD from a simplex family. Parental inheritance and validation of NRXN1 deletion were carried out with quantitative PCR (qPCR) using SsoAdvanced™ Universal SYBR® Green Supermix (BIORAD). The assay was performed in triplicate, with four sets of primers corresponding to the region of interest and another mapping to a control region on FOXP2 gene at 7q31.1. The number of copies of each amplified fragment was calculated using the ddCt method.
2.3. Whole‐exome analysis
DNA from ASD proband and parents was subjected to exome capture using NimbleGen SeqCap EZ MedExome enrichment kit (Roche), followed by paired‐end reads sequencing on an Illumina NextSeq550 (Illumina Inc, San Diego, CA, USA). Exomes had a read depth (DP) of 10× or more for 90% of the total exome coverage and 20x or more for 80%. Coverage statistics and comparison of coverage between samples was performed using QualiMap, 13 showing a mean coverage depth of 120‐122X for the three samples and a mean quality mapping is 58. Data analysis was performed using CoVaCS, 14 a pipeline exploiting a consensus call‐set approach from three different algorithms (GATK, Varscan and Freebayes) to generate a final set of high‐confidence variants. All variants were annotated with ANNOVAR, using RefSeq for gene‐based annotation (position, nomenclature, gene name, gene function). In order to remove low‐quality variants, genotypes were required to have DP ≥ 10, and Genome Quality (GQ) ≥20. A minor allele frequency (MAF) threshold of ≤0.5% in gnomAD exome (https://gnomad.broadinstitute.org/) 15 (and <1% in gnomAD genome and the 1000 Genomes Project 16 ) was chosen. We selected exonic and splicing variants, excluding synonymous variants. Likely deleterious variants were prioritized to capture Likely Gene Disrupting (LGD) and damaging missense mutations. LGD mutations include stop‐gain, stop‐loss, frameshift and splicing mutations, while missense mutations were defined damaging if they satisfy at least two of the following criteria: SIFT ≤ 0.05, Polyphen2 (HDIV) ≥ 0.95, Mutation Assessor score ≥ 2, CADD ≥ 15, placental mammal PhyloP ≥ 2.4 and vertebrate PhyloP ≥ 4. 17 Mutation intolerant genes were defined by two metrics: residual variance to intolerance score (RVIS ≤ 20th percentile) 18 and probability of loss‐of‐function intolerance (pLI score ≥ 0.9). 19 To select genes previously associated with ASD, we used the SFARI gene database and its scoring system, including 4 categories: S (syndromic), 1 (high confidence), 2 (strong candidate) and 3 (suggestive evidence) (https://gene.sfari.org/, Release: 2019 Q4). Brain expressed, synaptic and postsynaptic density (PSD) genes were defined as previously described. 20 , 21 , 22 Likely damaging de novo variants have been validated by Sanger sequencing method.
2.4. Deep sequencing of mitochondrial genome
Direct sequence analysis of the entire mtDNA molecule was performed on total DNA extracted from blood, by next generation sequencing (NGS) approach. 23 Briefly, the mitochondrial genome was amplified in two long‐range PCR, the NGS library constructed by Nextera XT (Illumina) and paired‐end sequenced on NextSeq Instrument (Illumina), using an High Output Kit (300 cycle). Fastq files were analysed with an in‐house pipeline, integrating three different callers (MToolBox, Unified Genotyper of GATK and DetermineVariants) 24 , 25 , 26 to detect low‐level heteroplasmy.
2.5. Mitochondrial DNA quantification
MtDNA content was assessed on total DNA extracted from blood, using a multiplex probe‐based real‐time PCR method, 27 co‐amplifying a mitochondrial gene (MT‐ND2) and a nuclear gene (FASLG). All three individuals were compared with age‐matched control groups of healthy individuals.
3. RESULTS
3.1. Comparative genomic hybridization array CGH
Comparative genomic hybridization array CGH was performed in 104 individuals with ASD.
The only pathogenic CNV identified was a deletion of ~811 kb at 2p16.3 (NC_000002.11:g.50170766_50982172del) (Figure 1A) involving exons from 7 to 23 of the NRXN1 gene (NM_001135659.2) in a trio family. qPCR in all family members confirmed the presence of the NRXN1 intragenic deletion in the proband and showed that the CNV is inherited from the unaffected mother (Figure S1). We identified three other rare CNVs in this family (Table S1), but none of them is considered to be clinically relevant.
Figure 1.

NRXN1 deletion in the ASD proband. A, UCSC hg19 screenshot showing the NRXN1 maternally inherited deletion detected by array CGH in the female proband and validated by qPCR in the family. qPCR probes used for validation and inheritance testing are shown in green. B, Schematic representation outlining domains structure of neurexin alpha and neurexin beta protein variants. Canonical splice sites (SS) of neurexins are indicated by arrows. Protein region affected by deletion is highlighted in red
3.2. Clinical characterization of the family with the NRXN1 deletion
No familiarity for ASD, congenital malformations or intellectual disability was reported. The female proband was born without pre‐, peri‐ or post‐natal relevant findings. Birth weight was 3690 kg. Development of socio‐communicative and motor abilities was reported to be slightly delayed until 18 months of age with acquisition of some words. At 18‐19 months of age, parents reported a regression of the acquired socio‐communicative abilities: the girl stopped responding to simple commands and to her name. Eye‐contact was lacking, while social isolation started to be more evident. Imitation skills, communicative gestures and language stopped. She started to show hyperactivity, short attention span and motor stereotypies such as hand flapping when excited. She manifested sensory interests manipulating materials mostly to get visual, acoustic and tactile stimulation (ie passing a hair or thread upon her lips, thread waving and ripping paper in thin stripes) and a restricted interest for hair and threads. At 4.6 years old, language expression was limited to 4 single words, while language comprehension seemed to be relatively better. Hyperactivity appeared to be slightly reduced. No epileptic seizures were reported. Diagnosis of ASD was made at 35 months old through clinical observation, the Childhood Autism Rating Scale‐Second edition (CARS2‐ST) 28 and the Autism Diagnostic Observation Schedule (ADOS‐2: module 1) 29 (Table 1). Both diagnostic tests indicated the presence of severe clinical signs of autism. No cognitive or psychomotor development level could be assessed using standardized scales due to the lack of the child's compliance. At 4.8 years of age, the assessment of adaptive behaviour (Vineland Adaptive Behavior Scale) 30 showed a significant delay in Communication, Daily Living Skills, Socialization and Motor Skills domains (age equivalent = 1.6 years old). Neurological examination showed: macrocrania, speech delay, aloneness, stereotypies as turning around herself, and no neuromotor signs. The proband underwent an awake and sleep EEG at the age of 2.8 years old, showing only an unspecific predominance of a slow background activity in the right temporal regions. Parents did not provide consent to perform a brain MRI.
Table 1.
Summary of clinical data
| Mother | Father | Proband | |
|---|---|---|---|
| Age at first assessment | NA | NA | 23 mo |
| Sex | F | M | F |
| Microarray | NRXN1 del/+ | NRXN1 +/+ | NRXN1 del/+ |
| Morphology | |||
| Growth at birth | NA | NA | Weight = 3.69 kg (50%ile); length = 56 cm (85%‐97%ile) |
| Head circumference (%ile) | NA | NA | Birth = 38.2 cm (>98%ile); 31 mo = 53 cm (>98%ile); 60 mo = 54.5 cm (>98°perc.) |
| Neurodevelopment | |||
| Full‐scale IQ | NA | NA | NA‐NC |
| Speech delay | ‐ | ‐ | + |
| Adaptive level of functioning* | NA | NA | Vineland Adaptive Behavior Scales: CA: 4.8 y; Global AE: 1.6 y |
| Clinical diagnosis | NA | NA | Autism spectrum disorder |
| Autism scales | |||
| CARS2‐ST | NA | NA | Total score = 46 (severe autistic symptoms) Autism cut‐ off: 30 |
| ADOS‐2 (module 1) | NA | NA | Total score = over Autism cut‐off, Comparison score = 10 (severe autism) |
| Broad autism phenotype | |||
| Broad Autism Phenotype Questionnaire (BAPQ) | Within normal range, Scores under cut‐off for Aloof , Rigid, and Pragmatic language | Within normal range, Scores under cut‐off for Aloof , Rigid, and Pragmatic language | NA |
| Social and Communication Disorder Checklist (SCDC) | T = 4 (<cut‐off: 9) | T = 2 (<cut‐off:9) | NA |
| Neurological | |||
| EEG | NA | NA | Aspecific abnormalities: slow activity in right temporal regions |
| Congenital | |||
| Other medical | ‐ | stuttering | ‐ |
Abbreviations: ±, positive/negative for attribute; AE, age equivalent; CA, chronological age; NA, information not available; NC, non‐collaborative patient.
Both parents were evaluated for the presence of subclinical neurocognitive or neuropsychiatric features. Scores for the broad autism phenotype were in the normal range (Social and Communication Disorders Checklist and The Broad Autism Phenotype Questionnaire 31 , 32 ). To contextualize their phenotypic profiles beyond questionnaires, we also investigated their education status: they both reached a good education level (high school diploma and bachelor's degree, respectively) with no need for special education or services. No family history of psychiatric disorders was present, except for the paternal grandfather who was reported to take depression medication.
3.3. Whole‐exome sequencing
A trio‐based whole‐exome sequencing (WES) approach was undertaken for this family. We focused our analysis on rare variants (MAF ≤ 0.5%), and more specifically on those predicted to have a functional effect, including LGD variants and missense variants defined damaging, according to a combination of prediction algorithms. 17 We compared the load of rare variants between the proband and her mother: the proband has a higher number of rare variants compared with the NRXN1 deletion‐transmitting mother (1036 versus 573). This difference remains significant by considering the putative damaging variants only (303 in the proband vs 212 in the mother, χ2 = 16.08, P = 6.07 × 10−5). We then tested for transmission disequilibrium of damaging variants from the parents to the proband and we detected a preferential transmission of damaging variants from the father (203 transmitted vs 165 untransmitted variants, TDT P = 0.048) but not from the mother.
Genes with at least one LGD or putative damaging missense variant or CNV were analysed using the STRING database 33 to test the presence of an enrichment for functionally related networks of genes (Figure 2). This analysis identified a significant 1.2‐fold enrichment (223 edges vs 184 expected) in interactions among the 294 genes identified in the proband (P = 0.003, one‐tailed hypergeometric test), while no significant interaction enrichment was identified in NRXN1 deletion‐transmitting unaffected mother (84 edges vs 86 expected, P = 0.62).
Figure 2.

STRING network of predicted protein‐protein interactions for genes harbouring LGD or likely damaging missense variants identified by WES in the female proband. The network of predicted NRXN1 associations has been magnified. The edges represent the predicted functional associations and line colour indicates the type of interaction evidence: Red line—presence of fusion evidence; Green line—neighbourhood evidence; Blue line—cooccurrence evidence; Purple line—experimental evidence; Yellow line—text‐mining evidence; Light blue line—database evidence; Black line—co‐expression evidence. NRXN1 and CNTNAP5 show homology, co‐expression and text‐mining interaction evidence; NRXN1 and KIRREL2 show text‐mining interaction evidence; NRXN1 and NCAN show co‐expression and text‐mining interaction evidence; NRXN1 and TULP1 show co‐expression and experimental interaction evidence
Next, we looked for specific sequence variants identified in the proband that might contribute to the clinical manifestation of the NRXN1 deletion in the affected girl compared with the unaffected carrier mother. We first tested the possible presence of compound heterozygosity in NRXN1, but we found no rare functional nucleotide variants on the non‐deleted allele. Then, we focused on four categories of rare variants: (a) likely damaging de novo variants, (b) recessive‐acting variants (homozygous and compound heterozygous variants), (c) likely deleterious variants in genes previously implicated in ASD (SFARI genes) that are intolerant to functional variation (RVIS ≤ 20th percentile and/or pLI score ≥ 0.9) and (d) likely deleterious variants in genes with a predicted interaction with NRXN1 in STRING. We identified one de novo stop‐gain in CDC25C (NP_073720.1:p.(Ser143Xaa)) and de novo missense in WASHC5 (NP_001317538.1:p.(Asp254Gly)), 2 homozygous missense variants in ZFP37 and DCLRE1A and two compound heterozygous variants in IFT80, and 5 missense variants in SFARI ASD candidate genes intolerant to mutation (CNTNAP5, ERBIN, SYNE, HERC2 and TANC2); all of them, except for TANC2, were of paternal origin in brain expressed genes. Furthermore, we highlighted 4 genes with damaging missense variants that are predicted to interact with NRXN1 (CNTNAP5, TULP1, NCAN and KIRREL2) (Table 2). Among them, the missense variant in CNTNAP5 is likely to exert a significant role, given that CNTNAP5 is itself a previously known ASD candidate gene, likely to be intolerant to mutations (RVIS percentile = 8.4) (Figure 2). Finally, we investigated the mother's burden of rare variants in these risk categories and, although we are not able to identify de novo and compound heterozygous variants, there is still a higher number of variants in the proband compared with the mother (11 vs 6 variants) (Table S2).
Table 2.
Rare LGD and predicted damaging missense variants in four‐risk categories identified in the ASD proband
| Gene base change | Identified Variants | SFARI gene score | pLI score | RVIS percentile | PSD/Synaptic/ Brain expressed 20 , 21 , 22 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| AminoAcid Change | Effect | Inheritance | dbSNP | MAF in gnomAD (exome) | Gene | |||||
| a) De novo variants | ||||||||||
| NC_000005.9:g.137627774G>T | NP_073720.1:p.(Ser143Xaa) | Stopgain | De Novo | CDC25C | 3.01E‐08 | 46.96 | Brain expressed | |||
| NC_000008.10:g.126079907T>C | NP_001317538.1:p.(Asp254Gly) | Missense | De Novo | WASHC5 | 4.81E‐13 | 1.94 | PSD/ Brain expressed | |||
| b) Homozygous and compound heterozygous variants | ||||||||||
| NC_000003.11:g.160000321A>T | NP_001177171.1:p.(Asp350Glu) | Missense | Maternal | IFT80 | 1.44E‐10 | 50.94 | Brain expressed | |||
| NC_000003.11:g.160037568T>C | NP_001177171.1:p.(Thr176Ala) | Missense | Paternal | rs146065418 | 0.0004781 | IFT80 | 1.44E‐10 | 50.94 | Brain expressed | |
| NC_000009.11:g.115812140G>T | NP_001269444.1:p.(Leu64Met) | Missense | Paternal/ Maternal | rs151180938 | 0.0029 | ZFP37 | 4.11E‐09 | 45.02 | Brain expressed | |
| NC_000010.10:g.115602192T>A | NP_055696.3:p.(Ile859Phe) | Missense | Paternal/ Maternal | rs11196530 | 0.002732 | DCLRE1A | 4.68E‐08 | 70.92 | Brain expressed | |
| c) Variants in SFARI Gene intolerant to mutation a | ||||||||||
| NC_000002.11:g.125660581G>A | NP_570129.1:p.(Ala1186Thr) | Missense | Paternal | rs114400050 | 0.00231 | CNTNAP5 | 3 | 0.10 | 8.42 | Brain expressed |
| NC_000005.9:g.65321695C>G | NP_001006600.1:p.(Leu304Val) | Missense | Paternal | rs148121803 | 0.0001956 | ERBIN | 2 | 0.99 | 63.5 | PSD/ Synaptic/ Brain expressed |
| NC_000006.11:g.152642393C>G | NP_149062.1:p.(Asp5335His) | Missense | Paternal | SYNE1 | 3‐S | 3.75E‐27 | 7.64 | PSD/ Brain expressed | ||
| NC_000015.9:g.28479425G>A | NP_004658.3:p.(Leu1337Phe) | Missense | Paternal | rs145594989 | 0.002391 | HERC2 | S | 1 | 0.16 | Brain expressed |
| NC_000017.10:g.61498241A>G | NP_079461.2:p.(Tyr1633Cys) | Missense | Maternal | rs765761955 | 0.000004011 | TANC2 | 1 | 1 | 0.33 | Brain expressed b |
| d) Variants in genes interacting with NRXN1# | ||||||||||
| NC_000002.11:g.125660581G>A | NP_570129.1:p.(Ala1186Thr) | Missense | Paternal | rs114400050 | 0.00231 | CNTNAP5 | 3 | 0.10 | 8.42 | Brain expressed |
| NC_000006.11:g.35467787T>C | NP_001276324.1:p.(Lys436Arg) | Missense | Maternal | rs62636511 | 0.00001989 | TULP1 | 0.81 | 65.05 | ||
| NC_000019.9:g.19335134C>T | NP_004377.2:p.(Arg224Cys) | Missense | Maternal | NCAN | 0.15 | 6.15 | PSD/ Synaptic/ Brain expressed | |||
| NC_000019.9:g.36351545C>A | NP_001316459.1:p.(Arg252Ser) | Missense | Paternal | rs73928337 | 0.001664 | KIRREL2 | 8.95E‐06 | 93.31 | Brain expressed | |
pLI score ≥ 0.9 and/or RVIS percentile ≤ 20 (underlined); #Predicted protein‐protein interaction in STRING.
Brain expressed according to 51 ; Variants validated by Sanger sequencing are highlighted in bold.
3.4. Mitochondrial DNA analyses
We carried out deep sequencing of the entire mtDNA in the ASD proband and her parents (Table S3 and S4). Both the proband and her mother showed all defining variants of haplogroup H13a1a1 of European ancestry, whereas the father's variants identified the haplogroup L2c2b1b background of African ancestry. None of the rare variants were previously reported (private or unique to an individual) or predicted as pathogenic.
Taking advantage of the high mean coverage in sequencing (16721X in the proband, 12090X in the mother and 19027X in the father), we were able to detect variants with a low‐level heteroplasmy (between 0.2% and 15%), in all three individuals (Table S3 and S4). Two of these were present in the proband and inherited from the mother: the synonymous variant NC_012920.1(MT‐ND2):m.4847C>T and the non‐coding NC_012920.1(MT‐HV1):m.16092T>C , one of the hypervariable domains of the control region. The heteroplasmy of these variants was higher in the proband (13.6% and 12.7%, respectively) as compared with the mother (2.2% and 2.1%, respectively). Furthermore, other five variants with a very low‐level of heteroplasmy (0.2%‐0.7%) were found exclusively in the proband: the NC_012920.1(MT‐RNR2):m.1906G>A, NC_012920.1(MT‐RNR2):m.2009G>A and NC_012920.1(MT‐TL1):m.3242G>A, which were never reported in over 50.000 sequence (GenBank), whereas the missense NC_012920.1(MT‐CO3):p.(Val208Ile) and the non‐coding NC_012920.1(MT‐HV1):m.16348C>T variants were previously reported three and six times, respectively. The missense NC_012920.1(MT‐CO3):p.(Val208Ile) variant, even if never reported as pathogenic for any disease, was predicted to be deleterious. The mother presented only one private low‐level heteroplasmic variant, the missense NC_012920.1(MT‐ND5):p.(Phe326Ser,) (2.2%), never reported in GenBank and also predicted to be deleterious. The father presented four low‐level heteroplasmic variants: the missense NC_012920.1(MT‐CYB):p.(Gly251Asp) (0.6%), reported 6 time in GenBank and predicted to be deleterious, and the non‐coding variants NC_012920.1(MT‐HV1):m.16192C>T (0.4%), NC_012920.1(MT‐HV1):m.16256C>T (0.8%), NC_012920.1(MT‐HV1):m.16291C>T (0.9%), very frequent in GenBank (2029, 1645, 1341, respectively).
To verify the remote possibility of biparental inheritance, as recently reported, 34 we also compared the proband and her father mtDNA sequences. With the exception of the reference sequence private variants, 35 the proband and her father did no share any other variants, not even at low‐level heteroplasmy (Table S3 and S4).
Last, the mtDNA content of the proband and her parents was comparable to the range of age‐matched healthy individuals (Figure S2).
4. DISCUSSION
In this study, we identified an intragenic NRXN1 deletion in a female proband with ASD, who has inherited it from the unaffected mother. We have thus characterized rare variants in the nuclear and mitochondrial genome of this family, in order to investigate their contribution towards the manifestation of the ASD phenotype in NRXN1 deletion carriers.
The NRXN1 deletion identified in this family can be classified as a 3’deletion, 8 as it overlaps exons from 7 to 23 (NM_001135659.2). The deletion gives rise to a putative in‐frame transcript, lacking the majority of NRXN1 protein domains (from Gly311 to Leu1445), specifically all α‐neurexin LNS‐domains (laminin/neurexin/sex hormone‐binding globulin domains) except the first one, and the two intercalated epidermal growth factor (EGF)‐like domains, while maintaining the transmembrane and intracellular C‐terminal domain (Figure 1B). Moreover, the deletion impacts the canonical splice sites (SS2 to SS6), including SS4, thought to represent a key mechanism for the regulation of NRXN‐ligand interactions at synapses. 9 As the 3’deletion identified in our proband is in‐frame, it is possible that the phenotypic effect of this deletion may arise by two concurrent mechanisms: haploinsufficiency due to lack of wild‐type NRXN1α isoforms, and a dominant‐negative activity of the mutant splice isoform, as suggested by a recent study using induced pluripotent stem cell (hiPSC)‐derived neurons from subjects with heterozygous intragenic deletions. 36
The proband's phenotype is mainly compatible with clinical features of 3’ deletion carriers, as the girl with ASD has macrocephaly (10.3% of 3’deletion carriers had macrocephaly in comparison with only 1.2% of 5’ deletion carriers) and DD (more frequently presents in probands with 3’ deletions). 8 Given the importance of collecting phenotype information also on parents to look for possible endophenotypes, we have also evaluated both parents for the presence of broad autism phenotype signs, but scores in questionnaires (SCDC, BAPQ) were not consistent with any ASD traits in either of them.
Whole‐exome sequencing analysis in all family members identified a higher number of rare coding variants in the ASD child in comparison with the NRXN1 deletion‐transmitting mother and this difference remains statistically significant if we restrict the analysis to rare likely deleterious mutations only, suggesting that additional rare variants may contribute collectively to push the genetic liability beyond the threshold for ASD. Moreover, a significant interaction enrichment was detected among genes with damaging variants (CNVs and SNVs) in the proband, supporting a cumulative effect of interacting genes affected by mutations to the phenotype. Interestingly, among the NRXN1 interactions detected by STRING in the ASD proband, there is a predicted interaction with CNTNAP5, a functionally intolerant gene previously identified as an ASD candidate gene, which harbours a paternally inherited putative damaging missense variant. CNTNAP5 encodes for contactin‐associated protein‐like 5, a member of the neurexin family involved in cell adhesion and intercellular communication in the vertebrate nervous system. 37 CNTNAP5 is classified as a suggestive candidate gene for ASD (SFARI score 3), as a rare deletion and missense variants in CNTNAP5 have been identified in subjects with ASD. 38 Similarly to this report, a previous study identified a maternally inherited missense variant in CNTNAP5 segregating with a NRXN3 paternal deletion in two ASD siblings, 39 supporting a combined role of neurexins and contactin‐associated proteins in ASD risk.
In addition to CNTNAP5, damaging missense variants were identified in four other mutation intolerant genes, previously implicated in ASD: TANC2, ERBIN, SYNE1 and HERC2. 40 , 41 , 42 , 43 , 44 , 45 , 46 , 47 , 48 , 49 , 50 While TANC2 and ERBIN have been involved in idiopathic ASD susceptibility with high confidence (SFARI score 1 and 2, respectively), SYNE1 and HERC2 gene have been mostly involved in syndromic ASD. Moreover, TANC2 and ERBIN are interesting functional candidate genes. TANC2 is highly expressed in the human developing brain and encodes for a postsynaptic scaffold protein involved in dendritic spines and excitatory synapses regulation. 51 ERBIN encodes a postsynaptic protein which binds ERBB2 playing an important role during brain development and regulation of synaptic plasticity in the adult brain. 52 ERBIN is also implicated in dendritic morphogenesis by regulating localization and function of δ‐Catenin in hippocampal neurons. 53
Two de novo novel putative damaging variants were also identified in the proband: a stop‐gain variant located on CDC25C exon 5 (NP_073720.1:p.(Ser143Xaa)) and a predicted damaging missense variant in WASHC5 exon 9 (NP_001317538.1:p.(Asp254Gly)). Although neither of them have been previously implicated in ASD, both of them are expressed in the brain 22 and therefore they could contribute to the proband phenotype. It should be noted that heterozygous missense variants in WASHC5 are associated with autosomal dominant spastic paraplegia 8 (SPG8), a progressive upper‐motor neurodegenerative disease, 54 while biallelic pathogenic variants are also associated with Ritscher‐Schinzel Syndrome, a clinically recognizable condition characterized by distinctive craniofacial features, cerebellar defects and cardiovascular malformations. 55 The WASHC5 variant (NP_001317538.1:p.(Asp254Gly)) is predicted to be deleterious, is novel and it is located in the spectrin‐like repeat domain, thus, we cannot rule out a pathological role for this mutation for a spastic paraplegia phenotype, which usually has on onset in adult life. Moreover, although the proband did not show typical dysmorphic craniofacial features of individuals with Ritscher‐Schinzel, she presented macrocrania.
Recessive‐acting variants were identified in three genes (ZFP37, DCLRE1A and IFT80), all of which are brain‐expressed 22 but neither of them have been previously implicated in neurodevelopmental phenotypes.
Finally, the proband did not carry any pathogenic mutation in her mtDNA. However, we found five variants with low‐level heteroplasmy (ranging from 0.2% to 0.7%) that were absent in the mother, and two maternally inherited variants, which increased their heteroplasmic load in the proband as compared to the mother. The burden of low‐level heteroplasmic mtDNA variants, both inherited or de novo, also known as universal heteroplasmy, 56 might contribute to the risk of developing ASD, but further analyses on large cohorts are needed to validate this hypothesis. The mtDNA copy number was also uninformative.
In conclusion, we have characterized a trio family in which a large 3’ exonic NRXN1 deletion is transmitted from an unaffected mother to a child with ASD. Exonic NRXN1 deletions represent the prototype of incomplete penetrant ASD‐associated susceptibility variants as they are often inherited from unaffected or mildly affected parents, but they are still considered pathogenic. The key finding is the presence of an increased burden of exonic rare variants in the affected proband compared to the unaffected deletion‐transmitting mother, supporting the hypothesis that the NRXN1 deletion sensitizes the genome to a clinical manifestation, but other genetic contributors are necessary to cross the threshold for a phenotypic manifestation (Figure 3). 57 Moreover, the reduced penetrance of the NRXN1 deletion in the unaffected mother is consistent with a female protective effect: females would require an excess burden of deleterious CNVs and SNVs to reach the ASD diagnostic threshold. 58 Therefore, in this family, the paternal‐inherited rare variants in ASD‐related or functionally constrained genes and de novo rare variants identified in the proband may have additive effects acting on a sensitized background caused by haploinsufficiency and/or a dominant‐negative activity at the NRXN1 locus. This observation is in line with the hypothesis that the determinants of psychiatric traits are multifactorial even in the context of a large‐effect variant. These modifying determinants may include the genetic background of common polygenic variants as well as rare variants. It has been recently reported that the increased burden of rare likely deleterious variants enhances the expression of neurodevelopmental phenotypes in probands with 16p21.1 deletions and in probands with other gene disruptive variants compared with their carrier family members 59 ; hence, in this study, we have focused our attention on the background of rare variants. Our results are consistent with the hypothesis that the burden of rare variants contributes in defining the phenotypic trajectory in carriers of a large‐effect variant, thus, this may represent a widespread mechanism that modulates the penetrance and the expressivity of disease‐associated variants. However, it has been shown that also common polygenic variation contributes additively to ASD risk, even in cases that carry a strongly acting variant. 60 , 61 A limitation to our study is therefore that we were not able to test the potential contribution of common variation in modulating the penetrance of the NRXN1 deletion in this family.
Figure 3.
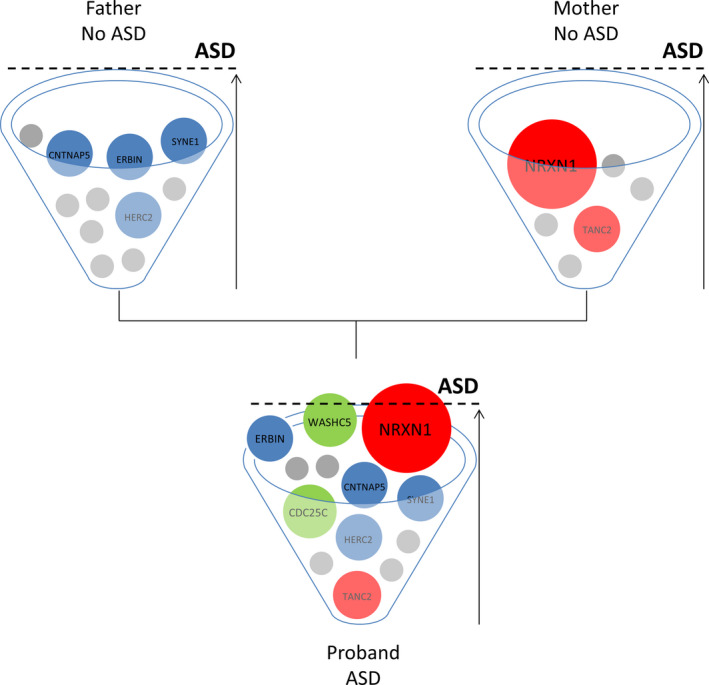
Multi‐factorial threshold model for ASD in this family. Each family member has an ASD risk cup with balls representing risk variants that contribute to ASD with variable degrees of impact. In both parents, the burden of risk variants is not enough to develop ASD, while in the child the ASD threshold is reached as a combination of strong and weak, inherited and de novo genetic variants. The NRXN1 deletion is depicted as a strong, primary contributing factor to reaching the ASD threshold in the ASD child, but not sufficient alone to develop ASD in the deletion carrier mother 57
Further investigation in a large dataset will be necessary to properly evaluate the cumulative effects of rare deleterious and common variants to ASD risk in NRXN1 deletion carriers; family‐based samples will be particularly informative, as they allow intrafamilial comparison of phenotypic features and inheritance pattern of specific variants.
This study underlines the importance of a comprehensive assessment of the genomic landscape of ASD individuals even when a ‘likely pathogenic’ variant has been already identified, as it is apparent that multiple rare variants contribute in conjunction to the overall genetic risk and the final clinical outcome. It is time to move from a genetic to a genomic perspective, shifting from a single variant analysis to an integrated view of many variants of different origin (nuclear and mitochondrial), types (CNVs and SNVs), inheritance pattern (de novo and inherited), frequency (rare and common, heteroplasmic and homoplasmic) and effect sizes, considering the role of protein‐protein interactions.
CONFLICT OF INTEREST
The authors confirm that there are no conflicts of interest.
AUTHOR CONTRIBUTIONS
Cinzia Cameli: Data curation (equal); Formal analysis (equal); Investigation (equal); Software (equal); Validation (lead); Visualization (equal); Writing‐original draft (supporting). Marta Viaggiano: Data curation (equal); Formal analysis (equal); Investigation (equal); Software (equal); Validation (supporting); Visualization (equal). Magali Jane Rochat: Formal analysis (equal); Funding acquisition (equal); Investigation (equal); Resources (supporting); Visualization (equal); Writing‐original draft (supporting). Alessandra Maresca: Formal analysis (equal); Funding acquisition (equal); Investigation (equal); Writing‐original draft (supporting). Leonardo Caporali: Formal analysis (equal); Investigation (equal); Writing‐original draft (supporting). Claudio Fiorini: Formal analysis (equal); Investigation (equal); Writing‐original draft (supporting). Flavia Palombo: Investigation (equal); Software (equal). Pamela Magini: Investigation (equal). Renée Concetta Duardo: Investigation (equal). Fabiola Ceroni: Validation (supporting). Maria Cristina Scaduto: Investigation (equal); Resources (supporting). Annio Posar: Investigation (equal); Resources (supporting). Marco Seri: Supervision (equal). Valerio Carelli: Methodology (equal); Project administration (equal); Supervision (equal); Writing‐review & editing (equal). Paola Visconti: Methodology (equal); Project administration (equal); Resources (lead); Supervision (equal); Writing‐review & editing (equal). ELENA BACCHELLI: Conceptualization (equal); Formal analysis (equal); Funding acquisition (equal); Methodology (equal); Project administration (equal); Software (equal); Supervision (equal); Visualization (equal); Writing‐original draft (lead); Writing‐review & editing (equal). Elena Maestrini: Conceptualization (equal); Methodology (equal); Project administration (equal); Supervision (equal); Writing‐review & editing (equal).
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge all the subjects who have participated in the study. We thank the Centro Interdipartimentale di Ricerche sul Cancro ‘Giorgio Prodi' (CIRC), University of Bologna for Illumina Sequencing Service and CINECA for computational resources.
Cameli C, Viggiano M, Rochat MJ, et al. An increased burden of rare exonic variants in NRXN1 microdeletion carriers is likely to enhance the penetrance for autism spectrum disorder. J Cell Mol Med. 2021;25:2459–2470. 10.1111/jcmm.16161
Cinzia Cameli and Marta Viggiano contributed equally to this work.
Funding informationThis research was funded by Italian Ministry of Health, grant number GR‐2013‐02357561, and by RFO (University of Bologna).
Contributor Information
Elena Bacchelli, Email: elena.bacchelli@unibo.it.
Elena Maestrini, Email: elena.maestrini@unibo.it.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available in the supplementary material of this article.
REFERENCES
- 1. Maenner MJ, Shaw KA, Baio J, et al. Prevalence of autism spectrum disorder among children aged 8 years ‐ autism and developmental disabilities monitoring network, 11 sites, United States, 2016. MMWR Surveill Summ. 2020;69(4):1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sullivan PF, Geschwind DH. Defining the genetic, genomic, cellular, and diagnostic architectures of psychiatric disorders. Cell. 2019;177(1):162‐183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pinto D, Delaby E, Merico D, et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet. 2014;94(5):677‐694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Coe BP, Stessman HAF, Sulovari A, et al. Neurodevelopmental disease genes implicated by de novo mutation and copy number variation morbidity. Nat Genet. 2019;51(1):106‐116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Satterstrom FK, Kosmicki JA, Wang J, et al. Large‐scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell. 2020;180(3):568‐584.e523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Girirajan S, Rosenfeld JA, Cooper GM, et al. A recurrent 16p12.1 microdeletion supports a two‐hit model for severe developmental delay. Nat Genet. 2010;42(3):203‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lowther C, Speevak M, Armour CM, et al. Molecular characterization of NRXN1 deletions from 19,263 clinical microarray cases identifies exons important for neurodevelopmental disease expression. Genet Med. 2017;19(1):53‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cosemans N, Vandenhove L, Vogels A, et al. The clinical relevance of intragenic NRXN1 deletions. J Med Genet. 2020. [DOI] [PubMed] [Google Scholar]
- 9. Sudhof TC. Synaptic neurexin complexes: a molecular code for the logic of neural circuits. Cell. 2017;171(4):745‐769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pei L, Wallace DC. Mitochondrial etiology of neuropsychiatric disorders. Biol Psychiatry. 2018;83(9):722‐730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schwede M, Nagpal S, Gandal MJ, et al. Strong correlation of downregulated genes related to synaptic transmission and mitochondria in post‐mortem autism cerebral cortex. J Neurodev Disord. 2018;10(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kearney HM, Thorland EC, Brown KK, Quintero‐Rivera F, South ST. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med. 2011;13(7):680‐685. [DOI] [PubMed] [Google Scholar]
- 13. Konechnikov K, Conesa A, García‐Alcalde F. Qualimap 2: advanced multi‐sample quality control for high‐throughput sequencing data. Bioinformatics (Oxford, England). 2016;32(2):292‐294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chiara M, Gioiosa S, Chillemi G, et al. CoVaCS: a consensus variant calling system. BMC Genom. 2018;19(1):120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Karczewski KJ, Francioli LC, Tiao G, et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss‐of‐function intolerance across human protein‐coding genes. BioRxiv. 2019;531210 [Google Scholar]
- 16. Auton A, Brooks LD, Durbin RM, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yuen RKC, Thiruvahindrapuram B, Merico D, et al. Whole‐genome sequencing of quartet families with autism spectrum disorder. Nat Med. 2015;21(2):185‐191. [DOI] [PubMed] [Google Scholar]
- 18. Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013;9(8):e1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285‐291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bayés À, van de Lagemaat LN, Collins MO, et al. Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat Neurosci. 2011;14(1):19‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jansen A, Dieleman GC, Smit AB, et al. Gene‐set analysis shows association between FMRP targets and autism spectrum disorder. Eur J Hum Genet. 2017;25(7):863‐868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kang HJ, Kawasawa YI, Cheng F, et al. Spatio‐temporal transcriptome of the human brain. Nature. 2011;478(7370):483‐489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Caporali L, Iommarini L, La Morgia C, et al. Peculiar combinations of individually non‐pathogenic missense mitochondrial DNA variants cause low penetrance Leber's hereditary optic neuropathy. PLoS Genet. 2018;14(2):e1007210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Calabrese C, Simone D, Diroma MA, et al. MToolBox: a highly automated pipeline for heteroplasmy annotation and prioritization analysis of human mitochondrial variants in high‐throughput sequencing. Bioinformatics. 2014;30(21):3115‐3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nat Genet. 2011;43(5):491‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Weissensteiner H, Forer L, Fuchsberger C, et al. mtDNA‐Server: next‐generation sequencing data analysis of human mitochondrial DNA in the cloud. Nucleic Acids Res. 2016;44(W1):W64‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Giordano C, Iommarini L, Giordano L, et al. Efficient mitochondrial biogenesis drives incomplete penetrance in Leber's hereditary optic neuropathy. Brain. 2014;137(Pt 2):335‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schopler E, Van Bourgondien ME, Wellman GJ, Love SR. CARS2 Childhood Autism Rating Scale, 2nd ed. Los Angeles, CA: Western Psychological Services; 2010. [Google Scholar]
- 29. Lord C, Rutter M, Dilavore PC, Risi S, Gotham K, Bishop SL. Autism Diagnostic Observation Schedule, 2nd ed. Torrance, CA: Western Psychological Services; 2012. [Google Scholar]
- 30. Sparrow SS, Balla DA, Cicchetti DV. Vineland Adaptive Behavior Scales. Circle Pines, MN: American Guidance Service; 1984. [Google Scholar]
- 31. Skuse DH, Mandy WP, Scourfield J. Measuring autistic traits: heritability, reliability and validity of the Social and Communication Disorders Checklist. Br J Psychiatry. 2005;187:568‐572. [DOI] [PubMed] [Google Scholar]
- 32. Hurley RS, Losh M, Parlier M, Reznick JS, Piven J. The broad autism phenotype questionnaire. J Autism Dev Disord. 2007;37(9):1679‐1690. [DOI] [PubMed] [Google Scholar]
- 33. Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein‐protein association networks with increased coverage, supporting functional discovery in genome‐wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607‐D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Luo S, Valencia CA, Zhang J, et al. Biparental inheritance of mitochondrial DNA in humans. Proc Natl Acad Sci U S A. 2018;115(51):13039‐13044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet. 1999;23(2):147. [DOI] [PubMed] [Google Scholar]
- 36. Flaherty E, Zhu S, Barretto N, et al. Neuronal impact of patient‐specific aberrant NRXN1alpha splicing. Nat Genet. 2019;51(12):1679‐1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Traut W, Weichenhan D, Himmelbauer H, Winking H. New members of the neurexin superfamily: multiple rodent homologues of the human CASPR5 gene. Mamm Genome. 2006;17(7):723‐731. [DOI] [PubMed] [Google Scholar]
- 38. Pagnamenta AT, Bacchelli E, de Jonge MV, et al. Characterization of a family with rare deletions in CNTNAP5 and DOCK4 suggests novel risk loci for autism and dyslexia. Biol Psychiatry. 2010;68(4):320‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vaags A, Lionel A, Sato D, et al. Rare deletions at the neurexin 3 locus in autism spectrum disorder. Am J Hum Genet. 2012;90(1):133‐141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. De Rubeis S, He X, Goldberg AP, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515(7526):209‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Krumm N, Turner TN, Baker C, et al. Excess of rare, inherited truncating mutations in autism. Nat Genet. 2015;47(6):582‐588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Iossifov I, O’Roak BJ, Sanders SJ, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515(7526):216‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu Y, Liang Y, Cicek AE, et al. A statistical framework for mapping risk genes from de novo mutations in whole‐genome‐sequencing studies. Am J Hum Genet. 2018;102(6):1031‐1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Guo H, Bettella E, Marcogliese PC, et al. Disruptive mutations in TANC2 define a neurodevelopmental syndrome associated with psychiatric disorders. Nat Commun. 2019;10(1):4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yu T, Chahrour M, Coulter M, et al. Using whole‐exome sequencing to identify inherited causes of autism. Neuron. 2013;77(2):259‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. O’Roak BJ, Vives L, Girirajan S, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485(7397):246‐250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wu H, Li H, Bai T, et al. Phenotype‐to‐genotype approach reveals head‐circumference‐associated genes in an autism spectrum disorder cohort. Clin Genet. 2020;97(2):338‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li J, Shi M, Ma Z, et al. Integrated systems analysis reveals a molecular network underlying autism spectrum disorders. Mol Syst Biol. 2014;10:774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ruzzo EK, Pérez‐Cano L, Jung J‐Y, et al. Inherited and de novo genetic risk for autism impacts shared networks. Cell. 2019;178(4):850‐866.e826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stessman HAF, Xiong BO, Coe BP, et al. Targeted sequencing identifies 91 neurodevelopmental‐disorder risk genes with autism and developmental‐disability biases. Nat Genet. 2017;49(4):515‐526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Han S, Nam J, Li Y, et al. Regulation of dendritic spines, spatial memory, and embryonic development by the TANC family of PSD‐95‐interacting proteins. J Neurosci. 2010;30(45):15102‐15112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Huang YZ, Won S, Ali DW, et al. Regulation of neuregulin signaling by PSD‐95 interacting with ErbB4 at CNS synapses. Neuron. 2000;26(2):443‐455. [DOI] [PubMed] [Google Scholar]
- 53. Arikkath J, Israely I, Tao Y, Mei L, Liu X, Reichardt LF. Erbin controls dendritic morphogenesis by regulating localization of delta‐catenin. J Neurosci. 2008;28(28):7047‐7056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ginanneschi F, D'Amore A, Barghigiani M, Tessa A, Rossi A, Santorelli FM. SPG8 mutations in Italian families: clinical data and literature review. Neurol Sci. 2020;41(3):699‐703. [DOI] [PubMed] [Google Scholar]
- 55. Elliott AM, Simard LR, Coghlan G, et al. A novel mutation in KIAA0196: identification of a gene involved in Ritscher‐Schinzel/3C syndrome in a First Nations cohort. J Med Genet. 2013;50(12):819‐822. [DOI] [PubMed] [Google Scholar]
- 56. Payne BAI, Wilson IJ, Yu‐Wai‐Man P, et al. Universal heteroplasmy of human mitochondrial DNA. Hum Mol Genet. 2013;22(2):384‐390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hoang N, Cytrynbaum C, Scherer SW. Communicating complex genomic information: a counselling approach derived from research experience with Autism Spectrum Disorder. Patient Educ Couns. 2018;101(2):352‐361. [DOI] [PubMed] [Google Scholar]
- 58. Jacquemont S, Coe B, Hersch M, et al. A higher mutational burden in females supports a "female protective model" in neurodevelopmental disorders. Am J Hum Genet. 2014;94(3):415‐425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pizzo L, Jensen M, Polyak A, et al. Rare variants in the genetic background modulate cognitive and developmental phenotypes in individuals carrying disease‐associated variants. Genet Med. 2019;21(4):816‐825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Robinson EB, Samocha KE, Kosmicki JA, et al. Autism spectrum disorder severity reflects the average contribution of de novo and familial influences. PNAS. 2014;111(42):15161‐15165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Weiner DJ, Wigdor EM, Ripke S, et al. Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat Gen. 2017;49(7):978‐985. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.