

Abstract
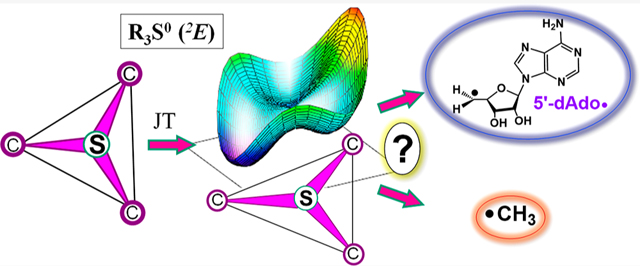
Catalysis by canonical radical S-adenosyl-l-methionine (SAM) enzymes involves electron transfer (ET) from [4Fe–4S]+ to SAM, generating an R3S0 radical that undergoes regioselective homolytic reductive cleavage of the S–C5′ bond to generate the 5′-dAdo· radical. However, cryogenic photo-induced S–C bond cleavage has regioselectively yielded either 5′-dAdo· or ·CH3, and indeed, each of the three SAM S–C bonds can be regioselectively cleaved in an RS enzyme. This diversity highlights a longstanding central question: what controls regioselective homolytic S–C bond cleavage upon SAM reduction? We here provide an unexpected answer, founded on our observation that photoinduced S–C bond cleavage in multiple canonical RS enzymes reveals two enzyme classes: in one, photolysis forms 5′-dAdo·, and in another it forms ·CH3. The identity of the cleaved S–C bond correlates with SAM ribose conformation but not with positioning and orientation of the sulfonium center relative to the [4Fe–4S] cluster. We have recognized the reduced-SAM R3S0 radical is a (2E) state with its antibonding unpaired electron in an orbital doublet, which renders R3S0 Jahn–Teller (JT)-active and therefore subject to vibronically induced distortion. Active-site forces induce a JT distortion that localizes the odd electron in a single priority S–C antibond, which undergoes regioselective cleavage. In photolytic cleavage those forces act through control of the ribose conformation and are transmitted to the sulfur via the S–C5′ bond, but during catalysis thermally induced conformational changes that enable ET from a cluster iron generate dominant additional forces that specifically select S–C5′ for cleavage. This motion also can explain how 5′-dAdo· subsequently forms the organometallic intermediate Ω.
Graphical Abstract
INTRODUCTION
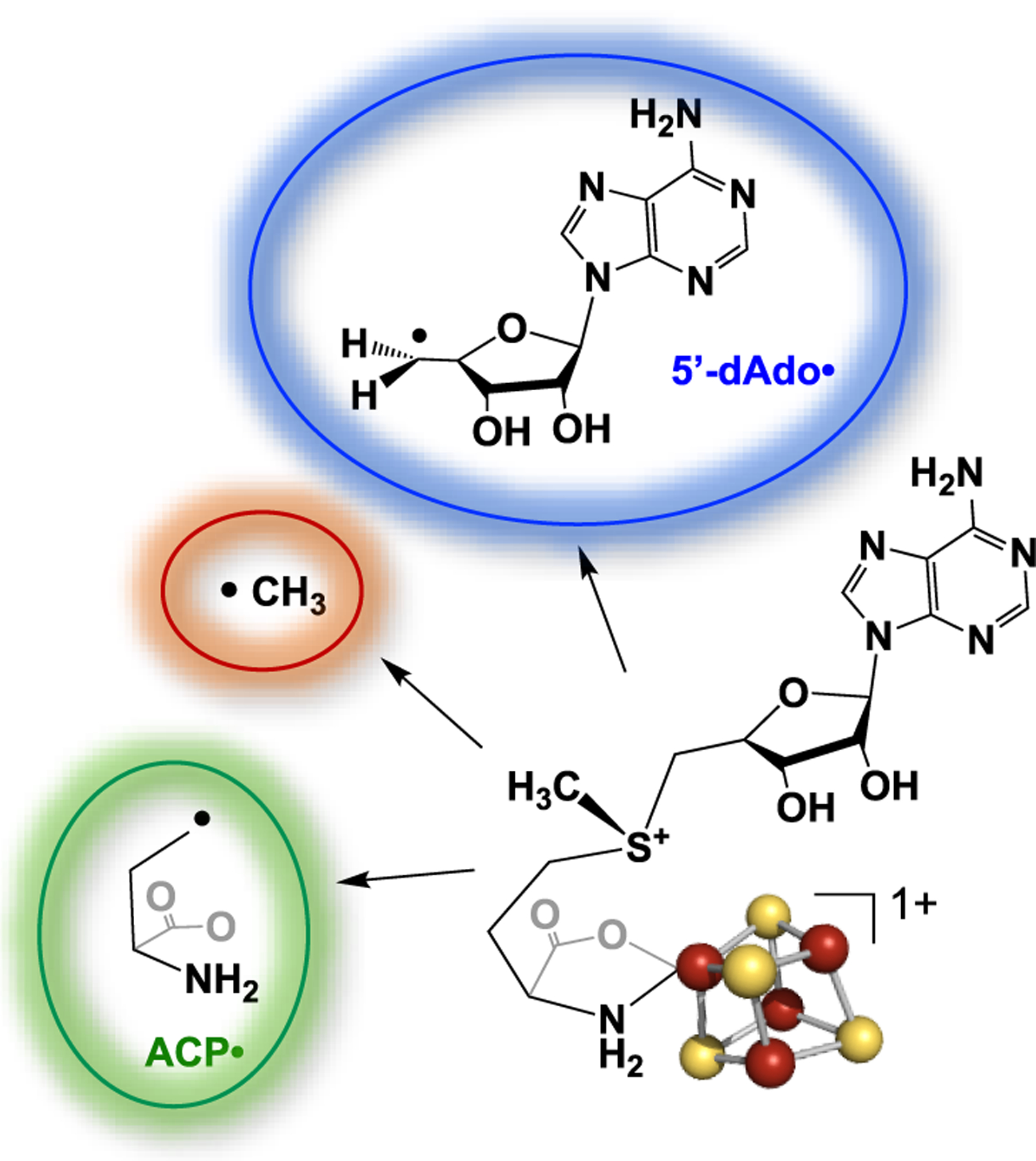
Radical S-adenosyl-l-methionine (radical SAM, RS) enzymes use SAM as either a cofactor or cosubstrate to initiate the wide range of reactions catalyzed by this enzyme superfamily.1–5 SAM binds in the active site of RS enzymes in close contact with the catalytically essential [4Fe–4S]+ cluster,6,7 with the SAM amino and carboxylate moieties coordinated to the unique iron of the cluster.8–11 It had been widely accepted that catalysis in the RS superfamily involves electron transfer (ET) from a reduced [4Fe–4S]+ to the sulfonium ion of SAM, inducing homolytic cleavage of the 5′C–S bond and thus liberating the 5′-dAdo· radical to abstract an H atom from the substrate.1,2,6,7 This mechanism was recently superseded with the discovery that 5′-dAdo· created by SAM reductive cleavage first generates an organometallic intermediate Ω in which the 5′C of this radical forms a bond to the unique iron of the [4Fe–4S] cluster, with subsequent homolysis of the Fe–C bond of Ω liberating 5′-dAdo·.12 The Ω intermediate was first trapped and characterized by electron nuclear double resonance (ENDOR) and electron paramagnetic resonance (EPR) spectroscopies when pyruvate formate-lyase activating enzyme (PFL-AE) underwent rapid freeze quench (RFQ) 500 ms after initiation of catalysis.12 We subsequently established that this intermediate is formed in a diverse suite of RS enzymes catalyzing the widest range of reactions, demonstrating that Ω plays a central role in the radical initiation mechanism by this enzyme superfamily.13 Suess and co-workers have recently taken a major step toward a synthetic model for Ω through synthesis of alkyl-[4Fe–4S]3+ clusters.14
The realization that Ω generates a 5′-dAdo· by homolytic Fe–C bond cleavage deepened the parallelism, first recognized by Frey,15–17 between the two classes of enzymes that utilize 5′-dAdo· for H atom abstraction: the RS superfamily and the adenosylcobalamin (or B12) dependent radical enzymes, with the latter utilizing 5′-dAdo· liberated by the homolysis of the Co(III)-5′-dAdo bond.18 The similarity in structure between adenosylcobalamin and the RS Ω intermediate, with both species having a direct metal–carbon bond to the 5′-C of a 5′-deoxyadenosyl moiety, raised questions regarding similarities in reactivity.19 Specifically, we asked whether Ω, like B12, might undergo photolysis to cleave the metal–carbon bond to generate a 5′-dAdo· radical. To date, attempts to photolyze the Fe–C5′ of the Ω have been inconclusive. However, as part of this experimental approach, we carried out the photolysis of the [4Fe–4S]+/SAM complex of PFL-AE at 12 K in the EPR cavity and were surprised to find that 450 nm light quantitatively induced ET from [4Fe–4S]1+ to the sulfonium sulfur of SAM with resulting homolysis of the S–C5′ bond, which liberated the 5′-dAdo· while forming the EPR-silent [4Fe–4S]2+ state.20
As a further surprise, when similar photolysis experiments were carried out with HydG, an RS enzyme involved in the maturation of [FeFe]-hydrogenase, ET from the [4Fe–4S]+ cluster to SAM cleaved its S–CH3 bond to regioselectively generate a trapped methyl radical rather than cleaving the S–C5′ bond to generate 5′-dAdo·.21 Thus, photolysis of RS [4Fe–4S]+/SAM complexes may regioselectively yield either 5′-dAdo· or ·CH3, depending on the enzyme.20,21 Indeed, all three bonds to the sulfonium sulfur of SAM can be regioselectively cleaved in an enzyme under the appropriate circumstances (Figure 1), as the noncanonical RS enzyme Dph2 cleaves the S–Cγ bond to generate the amino-carboxypropyl radical (ACP·).22 This diversity in S–C bond cleavage highlights a longstanding central question in the mechanism of RS enzymes: what controls the regioselectivity of S–C bond cleavage upon reduction of SAM?
Figure 1.
Structure of the complex of SAM with a [4Fe–4S]+ RS cluster illustrating the three alternative radical products resulting from the reductive cleavage of SAM.
To address this question, we have carried out a broad study of the photolysis of canonical RS enzymes containing the [4Fe–4S]+/SAM complex. An advantage of this approach is that, unlike enzymatic catalysis, which results solely in cleavage of the S–C5′ bond of SAM, photolysis offers instructive comparisons between RS enzymes that cleave two different S–C bonds, S–C5′ and S-CH3. In addition, there is a second advantage. Frey and co-workers recognized that conformational changes involving coordination of the sulfonium to the unique Fe of the [4Fe–4S]+ cluster are necessary to energetically drive the enzyme-catalyzed SAM reductive cleavage process,23–25 whereas the photochemical pathway simplifies the process by using light to drive the ET from the [4Fe–4S]+ cluster to the sulfonium center, without these changes.
We here report the regioselectivity of photoinduced reductive cleavage of SAM in eight different RS enzymes. We find a direct correlation between the S–C bond cleaved and the SAM ribose ring conformation, which known X-ray crystal structures of SAM-bound RS enzymes show to be controlled by interactions in the active site. Our analysis, supported by density functional theory (DFT) calculations, led us to recognize that the lowest unoccupied molecular orbital (LUMO) of the SAM sulfonium R3S+ moiety is orbitally degenerate, and thus the R3S0 radical center produced by reduction of SAM is Jahn–Teller (JT) active.26,27 As a consequence, the R3S0 radical is exquisitely poised for active-site control of regioselective S–C bond cleavage. These interactions direct the JT distortion of the R3S0 radical and localize an antibonding electron in a single S–C bond for cleavage, but with key differences between photoinduced and catalytic bond cleavage. We further recognized that this JT mechanism provides a plausible explanation of how the 5′-dAdo· generated catalytically is able to form the Fe–C5′ bond of the organometallic intermediate Ω subsequent to SAM cleavage. The studies described here thus provide fundamental new insights into the overall mechanism of RS enzymes, and in particular the origin of regioselectivity during reductive cleavage of SAM.
MATERIALS AND METHODS
Materials.
[Methyl-13C]-l-methionine and 2,8-D2-1′,2′,3′,4′,5′,5″-D6-adenosine 5′-triphosphate salt solution were purchased from Cambridge Isotope Laboratories, Inc., and [methyl-d3]-methionine was obtained from CDN Isotopes.
Enzymatic SAM Synthesis.
Natural abundant and isotopically labeled SAMs were synthesized following a previously published protocol.13,28 The preparation of S-adenosyl-l-ethionine (SAE) and its behavior in RS enzymes as a catalytically competent SAM analogue are described elsewhere.29
Preparation of Proteins.
Growth, purification, and reconstitution of RNR-AE, PFL-AE, lysine 2,3-aminomutase (LAM), OspD, PoyD, and CaSPL were carried out following published protocols with no modification.13 Preparation of HydG was achieved using procedures detailed in our recent publication.21
Expression and Preparation of HydE.
HydE was expressed, purified, and reconstituted following previously published protocols30 with minor modifications. In short, the codon-optimized hydE gene from Clostridium acetobutylicum (CahydE) was cloned into a Novagen pET23b vector and subsequently transformed into BL21-(DE3)-ΔiscR competent cells to express this protein with a C-terminal His6-tag. The transformed cells were streaked on fresh LB agar plates containing 50 μg/mL of ampicillin and 30 μg/mL of kanamycin. Single colonies from these plates were used for pilot expression trials, where pre- and postinduction samples were compared on an sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel. A glycerol stock made from the colonies that showed the best overexpression was used to streak fresh LB agar plates, and single colonies picked from these were used to start 50 mL overnight cultures. An aliquot (8 mL) of the overnight culture was used to inoculate each of the six Fernbach flasks containing 1.5 L of low-salt phosphate-buffered LB media supplemented with the same antibiotics as mentioned earlier. The cultures were grown at 37 °C while shaking (180 rpm), to an OD600 = 0.5, at which point protein expression was induced by addition of isopropyl β-d-1-thiogalactopyranoside (IPTG) (1 mM), followed by addition of ferric ammonium citrate (0.3 mM final concentration) and then incubation for an additional 4 h at 30 °C at the same shaker speed. After this aerobic incubation period, the cultures were cooled down to room temperature, prior to being transferred to a 4 °C refrigerator to be sparged with N2 overnight. This phase was followed by centrifugation (6000 rpm, 10 min, 4 °C). The supernatant was decanted, and the wet cell pellet was immediately flash-frozen in liquid nitrogen and stored at −80 °C until lysis and purification.
Expression of Thermotoga maritima HydE (TmHydE) was achieved as follows. The TmhydE gene was cloned into a pET21b vector followed by transformation into BL21-(DE3)-RIL competent cells. The transformed cells were spread on LB agar plates containing ampicillin (50 μg/mL). Initiation of culture growth and protein expression were performed following the procedure described earlier.
Purification and Reconstitution.
Both protein isolation and reconstitution processes were carried out in an anaerobic Coy chamber (Glass Lakes, MI) maintained at ≤20 ppm oxygen. Cell pellets (20 g) containing HydE from either organism were resuspended in 40 mL of buffer A (50 mM Tris pH 8.0, 250 mM KCl, and 5% glycerol) supplemented with 10 mM imidazole, phenylmethylsulfonyl fluoride (PMSF) (1 mM), 1% (v/v) Triton-X100, 9 mg of lysozyme, and trace amounts of DNaseI and RNase A. This mixture was stirred at room temperature for 1 h, followed by centrifugation (60 min, 18 000 rpm, and 4 °C); the clarified lysate containing the soluble protein was then decanted. The supernatant was loaded onto a HisTrap Ni-NTA affinity column pre-equilibrated with Buffer A supplemented with 10 mM imidazole. Using a GE Healthcare AKTA Basic FPLC, a step wash method was employed to elute the protein with buffer A containing different concentrations of imidazole (0, 12.5, 33, 106, and 250 mM). Fractions (10 mL) were collected during elution and analyzed by SDS-PAGE, revealing that pure HydE eluted at 106 mM imidazole. Colored fractions that showed the best purity on the gel were combined and then buffer-exchanged using a Sephadex-G25 desalting column into imidazole-free buffer (Buffer A). The protein was then concentrated to 3 mL using 30 kDa MWCO Amicon spin filters (Millipore). Aliquots of the purified concentrated protein were then flash-frozen in liquid nitrogen prior to storage in the −80 °C freezer. Iron quantitation and protein concentration analyses were accomplished via flame atomic absorption spectroscopy and Bradford assay, respectively.
To increase the content of catalytically essential clusters, in vitro reconstitutions were carried out following a previously published protocol with no modifications.30 Typically, the iron content in the protein was 7.9 ± 0.2 after reconstitution.
Exchanging Protein into D2O Buffer.
All described manipulations were performed in an anaerobic Coy chamber maintained at ≤10 ppm oxygen and 2.1% hydrogen. An aliquot (500 μL) of CaHydE that had been purified and reconstituted in H2O buffer (50 mM Tris pH 8.0, 250 mM KCl, 5% glycerol), was diluted into 4.5 mL of D2O buffer (50 mM Tris pD 8.0, 250 mM KCl, 5% glycerol, D atom >99%) in a Millipore spin concentrator (30 kDa MWCO). The diluted mixture was concentrated to 500 μL, at which point 4.5 mL more D2O buffer was added. This process was repeated 2 more times to bring the estimated %D to >95%, and finally the protein was concentrated to the starting volume. EPR samples were prepared soon after, following the procedure described below.
Photolysis of HydE To Remove Copurified SAM.
An aliquot (100 μL) of HydE (550 μM) was reduced with 3 mM sodium dithionite (NaDT), centrifuged (5 min, 13 000 rpm), and loaded into an EPR tube. This tube was then placed in an ice water bath maintained at ~0 °C. The sample was irradiated using the 450 nm laser, which was placed ~1 cm away from the tube. The photolysis was carried out for a total of 20 min, prior to determining whether the SAM had successfully been reacted through reductive cleavage; the presence of an axial EPR spectrum for the N-terminal cluster was an indication that the clusters were SAM-free.30
Photolysis Sample Preparation.
All samples were prepared in an anaerobic Coy chamber. PFL-AE, LAM, SPL, RNR-AE, OspD, PoyD, HydE, and HydG samples were prepared by reducing each enzyme with sodium dithionite (NaDT): NaDT was added to 0.5–1 mM protein to a final DT concentration of 3–5 mM. Reduction was followed by addition of SAM and appropriate buffers: 50 mM Tris pH 8.0, 250 mM KCl, 10% glycerol for HydE and HydG; 50 mM HEPES pH 8.0, 100 mM KCl, 5% glycerol for OspD and PoyD; 50 mM EPPS pH 8.0, 1 mM dithiothreitol (DTT), 10 μM PLP for LAM; 50 mM Tris pH 8.5, 200 mM NaCl, 1 mM DTT for RNR-AE; 50 mM Tris pH 7.5, 100 mM NaCl, 5% glycerol, and 1.0 mM DTT for PFL-AE; and 20 mM sodium phosphate pH 7.5, 350 mM NaCl, 5% glycerol for SPL, to give final protein concentrations of 500 μM (for PFL-AE, RNR-AE, HydE, HydG, LAM, and SPL) or ~300 μM for OspD and PoyD. The final concentrations of the other components (SAM and NaDT) were 5.5 and 3.0 mM, respectively. Each sample was centrifuged (5 min, 13 000 rpm), loaded into a Q-band EPR tube, and flash-frozen in liquid nitrogen.
EPR and ENDOR Measurements.
X-band and Q-band continuous wave (CW) spectroscopies were conducted using a Bruker ESP 300 spectrometer and a Bruker EMX spectrometer, respectively, both equipped with continuous helium flow Oxford cryostats. Photolysis was carried out for varying times using a 450 nm Thorlabs diode laser situated ≲1 cm from samples in the EPR cavity maintained at 12 K. Generally, the integrity of a SAM-bound cluster prior to photolysis was checked at 12 K, photolysis was performed, and then the temperature was raised to 40 K to characterize radical species. The modulation amplitude was normally set to 10 G, while the power was at 1 or 2 mW.
JT Effect and DFT Calculations.
The JT effect is associated with nonlinear molecules of idealized symmetry that exhibit an electronically degenerate state that is vibronically coupled to molecular vibrations.27 In the present case we consider the R3S0 radical with idealized trigonal symmetry that exhibits a 2-fold degenerate 2E state coupled to e vibration(s). We recognize that, in such radicals, incorporation of spin–orbit coupling on S abolishes this degeneracy even in idealized trigonal symmetry, and the radical actually is technically described as being subject to the so-called pseudo-JT effect.26,27,31 Such a treatment would be required to describe the EPR spectrum of a freeze-trapped R3S0 state.32,33 However, for the present purpose of understanding regioselective bond cleavage, the spin–orbit coupling energies are so small that we may ignore this elaboration.
To illuminate the behavior of the R3S0 radical, we have taken an indirect approach to density functional theory (DFT) computations. Such computations employ a single-determinant wave function, whereas description of the JT-active R3S0 radical state requires a multireference wave function approach, the entry level of which is the complete active space self-consistent field (CASSCF) method.32–34 It would be a distraction in this first discussion of RS regioselectivity to incorporate such higher-level computations; they will be a subject of later treatment. Instead, for illustrative purposes we examine here the DFT-derived LUMOs of the non JT-active R3S+ parent cation, which are representative of the corresponding highest occupied molecular orbitals (HOMOs) of the JT-active neutral R3S0 radical itself (see the Supporting Information).
All DFT computations were carried out with the ORCA 4.0.1 software package developed by Neese.34 For the small sulfur-centered molecule models, the starting geometry was created in Avogadro35 completely in silico. Geometry and single-point calculations utilized the B3LYP/G hybrid functional36–38 and the Ahlrichs’ valence triple-ξ with a polarization function basis set.39 For calculations of the SAM molecule, the crystal structure of the cofactor served as the starting geometry and hydrogens were added, assuming neutral pH for simplicity. These models were subjected to the calculations described earlier as well as with Becke’s functional for exchange along with Perdew’s functional for correlation (BP86) for comparison. The molecular orbitals were visualized as Gaussian cubes in Pymol40 with an isosurface density of 0.08 au.
RESULTS
Photolytic SAM Cleavage.
We previously demonstrated that SAM-bound [4Fe–4S]+ clusters in RS enzymes are photochemically active, with low-temperature blue-light photolysis of PFL-AE:SAM giving S–C5′ bond cleavage to yield the long-sought but elusive 5′-dAdo·, while the same process applied to HydG:SAM resulted in S–CH3 bond cleavage to generate a ·CH3 trapped within the enzyme active site.20,21 Here we carry out photolysis of six additional RS enzymes, finding that all fall into one of these two categories.
Intracavity 12K photolysis of [4Fe–4S]+:SAM in RNR-AE, OspD, and CaSPL generates the multiline 1H hyperfine pattern of 5′dAdo· as originally reported with PFL-AE (Figure 2). As confirmation of the photolytic liberation of 5′-dAdo·, photolysis of reduced enzymes complexed with adenosyl-[2,8-D2-1′,2′,3′,4′,5′,5″-D6]-SAM collapses this pattern into a broad single peak (Figure 2). However, although the spectra of unlabeled and D8 5′-dAdo· formed with PFL-AE are “clean”, the corresponding spectra for RNR-AE, OspD, and CaSPL are overlaid with a weak hyperfine-split signal from a small amount of minor radicals presumably generated through H atom abstraction by 5′-dAdo· from nearby protein residues in these substrate-free samples.
Figure 2.
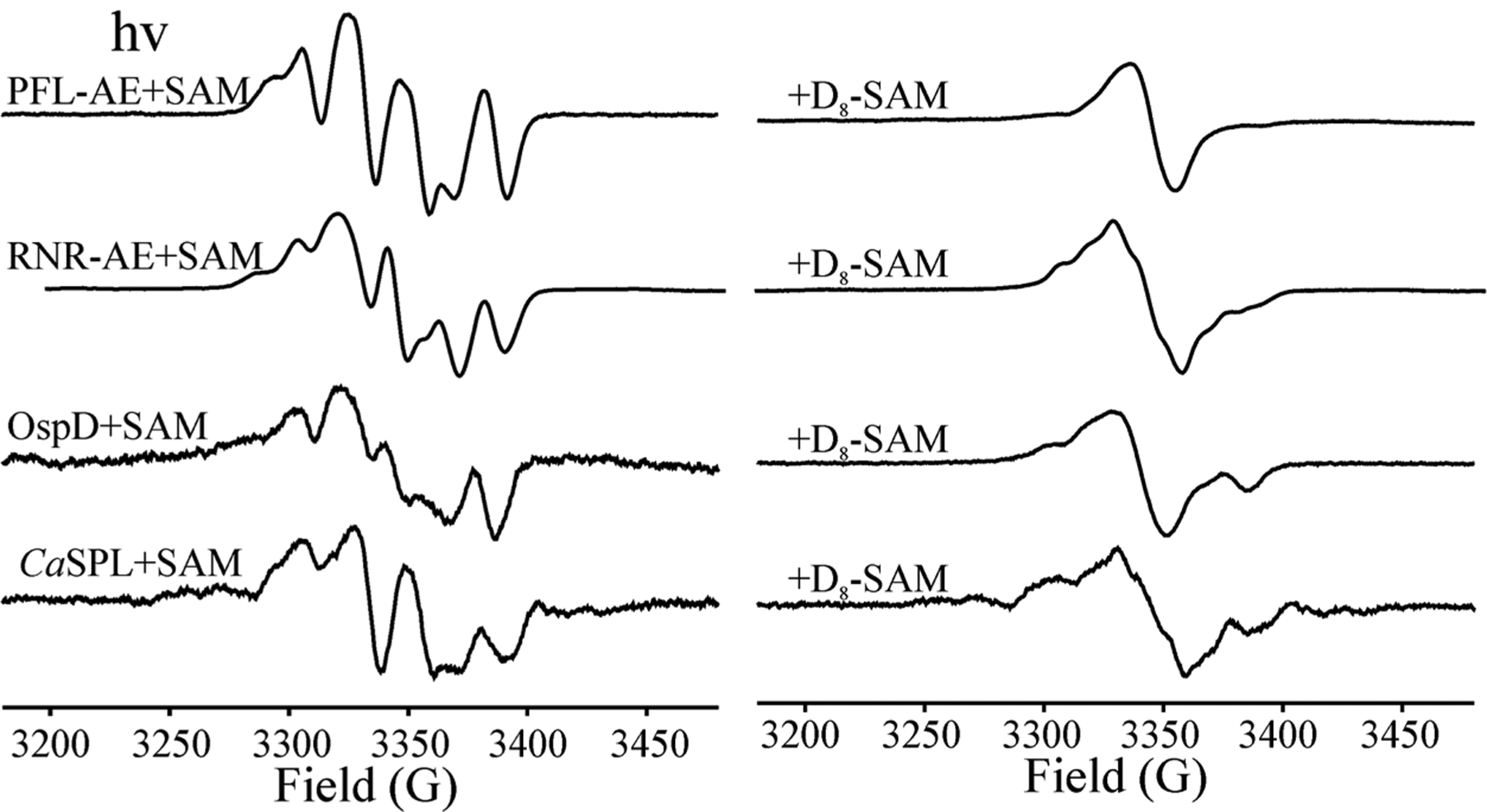
X-band EPR spectra of 5′dAdo· obtained from the photolysis of reduced RS enzymes PFL-AE, RNR-AE, OspD, and SPL with SAM (left) that collapse to singlets with adenosyl-2,8-D2-[1′,2′,3′,4′,5′,5″-D6]-SAM (D8-SAM) (right). Conditions: microwave frequency, 9.37 GHz; modulation amplitude, 5 G; T = 40 K.
Interestingly, the EPR signal of 5′-dAdo· trapped in PFL-AE differs in small ways from those of CaSPL, OspD, and RNR-AE (Figure S1 and Table S1). The simulations show that the difference of the EPR spectra for 5′-dAdo· trapped in the different protein environments is attributable to variations in the hyperfine coupling to H–C4′ (Figure S1 and Table S1) that result from variation in the tilt of the C5′–H2 fragment relative to the ribose ring. This is presumably induced by van der Waals interactions between 5′-dAdo· and its differing protein environments. Changes in the tilt correspond to changes in the dihedral angle between the odd-electron 2pz(C5′) orbital and the H–C4′ bond, which change the hyperfine coupling to that hydrogen.
In contrast to the enzymes just discussed, the cryogenic photolysis of [4Fe–4S]1+:SAM in HydG liberates ·CH3, which exhibits a 1:3:3:1 quartet hyperfine pattern in the 40 K spectrum, indicative of rapid tumbling in the frozen matrix at this temperature.20,21 Again, confirmation of this assignment is given by use of CD3–SAM, which collapses the quartet into a singlet as previously published (Figure 3). The three additional RS enzymes studied herein, the epimerase PoyD, lysine 2,3-aminomutase (LAM), and TmHydE hydrogenase maturase, also undergo photolysis to cleave the SAM S–CH3 bond to generate the 1:3:3:1 intensity pattern of ·CH3 (Figure 3).
Figure 3.

X-band EPR spectra of ·CH3 from photolysis of reduced RS enzymes HydG, PoyD, LAM, TmHydE, and CaHydE with SAM and CD3-SAM. All samples are prepared with H2O, except that the sample for the bottom spectra is prepared in D2O solvent. EPR conditions: microwave frequency, 9.37 GHz; modulation amplitude, 5 G; T = 40 K.
The photolysis of reduced CaHydE with SAM liberates an organic radical with a slightly more complex EPR spectrum that clearly can be decomposed into the ·CH3 quartet and a contribution of a second radical. The photolytic liberation of · CH3 in CaHydE is confirmed by the collapse of the features associated with the ·CH3 quartet when reduced CaHydE:CD3–SAM is photolyzed, as well as by the appearance of an additional 13C coupling when CaHydE:13CH3–SAM is photolyzed (Figure S2).
Insight into the nature of the additional radical species formed during photolysis of CaHydE:SAM is provided by carrying out the photolysis in D2O solvent, which eliminates the EPR signal of the second radical ·X species and reveals the clean quartet structure of ·CH3 (Figure 3). We interpret this observation as indicating that ·X is generated as ·CH3 abstracts a solvent-exchangeable proton from a nearby protein residue and that this reaction is significantly slowed in D2O due to the solvent kinetic isotope effect.
The photolytic reductive cleavage of the SAM analogue S-adenosyl-l-ethionine (SAE) bound to the [4Fe–4S]1+ cluster was studied in both PFL-AE, where photolysis of the SAM complex forms 5′-dAdo·, and in HydG, where SAM is photolytically cleaved to ·CH3. In neither enzyme does the Me → Et substitution change the regioselectivity of reductive S–C bond cleavage. In PFL-AE, photolysis with both SAM and SAE liberates 5′dAdo· (Figure 4). Correspondingly, photolysis of [4Fe–4S]1+:SAE in HydG liberates a novel organic radical with a multiline pattern that differs from the 5′-dAdo· EPR spectrum observed after photolyzing [4Fe–4S]+:SAE in PFL-AE. This multiline signal is associated with the ethyl radical, as shown by the simulation of the EPR spectrum on the basis of a ·CH2CH3 structure that has the carbon spin center coupled to two α-protons with the expected anisotropic hyperfine coupling and three methyl β-protons with the expected isotropic couplings, with the magnitude for each proton depending on the angle of its C–H bond with respect to carbon radical 2pz orbital (see legend in Figure 4). Thus, the radical photolytically liberated from photolyzing SAE in HydG is indeed ·CH2CH3. The largest methyl proton hyperfine coupling, aiso = 110 MHz, implies a conformation in which the C–H bond of that methyl proton lies nearly parallel to the α-carbon 2pz orbital.
Figure 4.

X-band EPR spectra of radical species from the photolysis of reduced PFL-AE and HydG with SAM and SAE, with an EPR spectrum simulation for ·CH2CH3 (red): LW = 27 MHz, g = [2.003, 2.003, 2.001], two α-protons from ·CH2− with A(1Ha) = [80, 40, 60] MHz, (α, β, γ) = (−60, 0, 0), A(1Hb) = [80, 40, 60], (α, β, γ) = (60, 0, 0), and three β-protons from the methyl group with aiso(1Ha, 1Hb, 1Hc) = 110, 20, and 20 MHz. EPR conditions: microwave frequency, 9.37 GHz; modulation amplitude, 5 G; T = 40 K.
Protein Control of SAM Conformation.
The striking bifurcation in regioselectivity described earlier for cryogenic photoinduced reductive SAM cleavage, where some RS enzymes cleave S–C5′ while others cleave S–CH3, is surprising given that all these RS enzymes cleave the S–C5′ bond during enzyme catalysis.1,2 What is the origin of the two classes of regioselectivity? The most obvious possible source for the regioselectivity of photoinduced homolytic SAM S–C bond cleavage would be differences in distance(s)/angles that define the geometric relationship between the unique cluster Fe and the sulfonium of SAM. However, there is no such correlation (Table S2). Indeed, as shown in Figures 5 and S3, when one compares the SAM-bound structures of SPL,41 a photochemical 5′-dAdo·-former, and HydE,42 a ·CH3-former, we see that there is little alteration in the positioning and orientation of the sulfonium centers of SAM relative to the unique Fe of the [4Fe–4S] cluster. This positioning is anchored by the methionyl amino acid chelation of the unique cluster Fe on one end of SAM and by nonbonding interactions of the adenine with neighboring residues on the other end of SAM, as previously recognized and detailed by Vey and Drennan.43
Figure 5.

(Upper) Crystallographic structure of SAM bound to the [4Fe–4S] cluster in SPL (left: 4fhf.pdb), which undergoes photochemical liberation of 5′-dAdo·, and in HydE (right: 3iiz.pdb), which undergoes photochemical liberation of ·CH3. (Lower) Overlay of the two structures. Together, the figures show nearly identical positioning and orientation of the sulfonium center with respect to the unique Fe of the [4Fe–4S] cluster.
In contrast, as also noted by Vey and Drennan, the SAM ribose conformation does vary among RS enzymes; as seen in Figure 5, there are clear differences in the ribose conformations in the SPL and HydE SAM-bound structures.41,42 To explore whether such structural details of the enzyme-bound SAM correlate to photolytic regioselectivity, we began by comparing the SAM structure in PFL-AE,44 a 5′-dAdo·-forming host, and HydG,45 a ·CH3-former, with Figure S4 showing their ribose configurations from two perspectives. The ribose ring of SAM in PFL-AE has the 2′-endo conformation with an axial C4′–C5′ bond and with an OH–C3′–C4′–C5′ dihedral angle of ~140°. In contrast, the ·CH3-forming host HydG has the ribose ring in a 3′-endo conformation with an equatorial C4′–C5′ bond and, correspondingly, with a sharply smaller OH–C3′–C4′–C5′ dihedral angle of ~ 80°. This latter HydG ribose conformation is the most common in RS enzymes examined by Vey and Drennan43 and is that of the classical B-DNA conformation; the former PFL-AE ribose conformation is that in A-DNA.46
To determine whether such a difference in SAM ribose configuration is found for other members of the two classes of RS enzymes that differ in regioselective photoinduced S–C bond cleavage, we examined all five known SAM-bound structures of the enzymes studied here. In Figure 6 we orient all of the structures so that the ribose C2′, C1′, and ring O atoms overlay. On the left, the figure shows that, when SAM of PFL-AE44 (5′-dAdo·-forming) and HydG45 (·CH3-forming) are so oriented and overlaid, they exhibit a dramatic axial versus equatorial orientation of C5′. The right side of the figure likewise aligns and overlays the five published SAM-bound structures of the enzymes studied here (PFL-AE (3cb8.pdb),44 HydG (4wcx.pdb),45 SPL (4fhf.pdb),41 HydE (3iiz.pdb),42 and LAM (2a5h.pdb)47), revealing that the two regioselectivity classes exhibit the same sharp axial/equatorial differences seen for PFL-AE and HydG (see also the OH–C3′–C4′–C5′ dihedral angles in Table S2). Moreover, members of each class exhibit the same SAM structure with minimal deviations: 5′-dAdo·-forming hosts have the 2′-endo ribose-ring conformation with an axial C4′–C5′ bond; ·CH3-forming hosts have a 3′-endo ribose ring with an equatorial C4′–C5′ bond.
Figure 6.
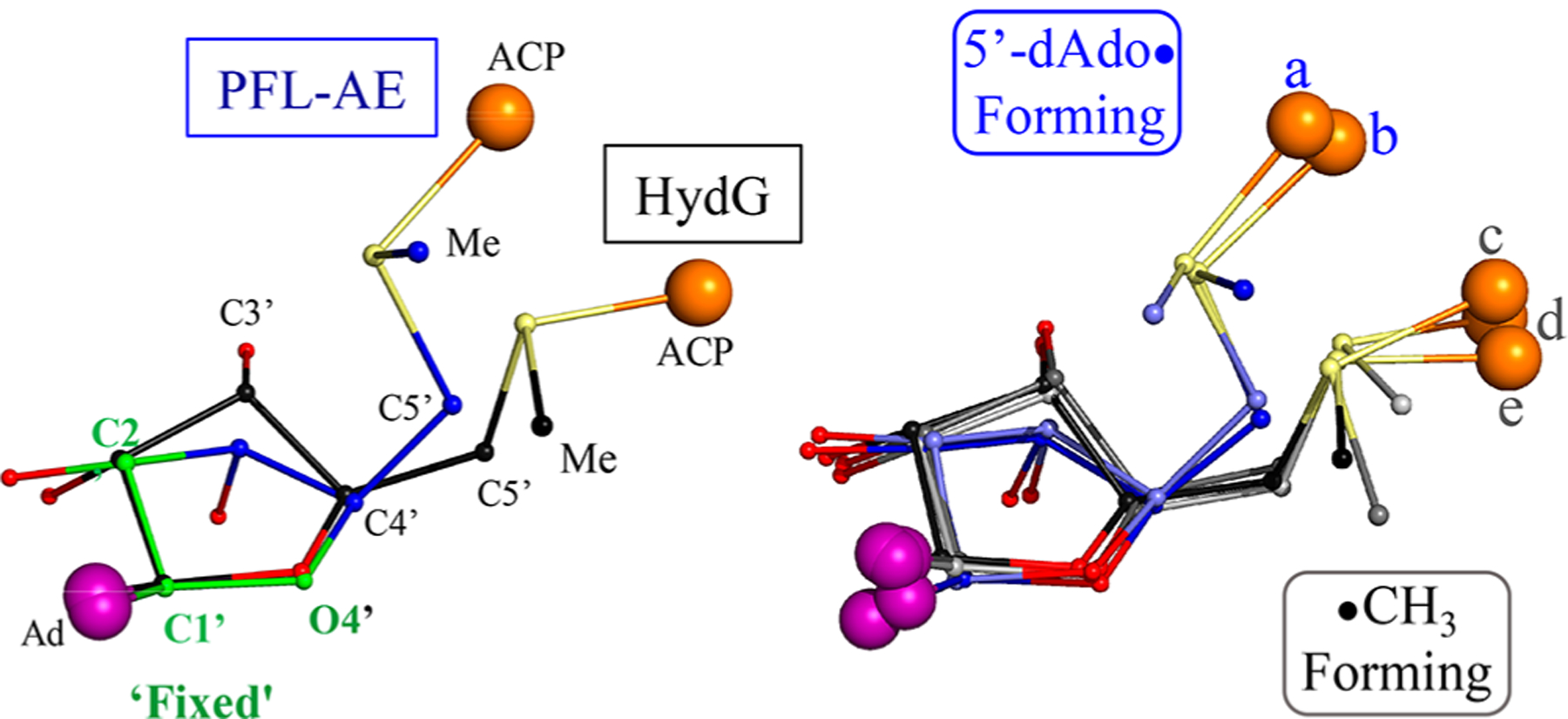
Comparisons of SAM configurations that yield photo-induced cleavage to form 5′-dAdo· or ·CH3, with orientation adjusted so that in all cases the ribose C2′, C1′, and ring O atoms overlay. (Left) Overlay of PFL-AE (3cb8.pdb) and HydG (4wcx.pdb). (Right) Overlay of (a) SPL (4fhf.pdb), (b) PFL-AE, (c) HydE (3iiz.pdb), (d) LAM (2a5h.pdb), (e) HydG, demonstrating the axial orientation of the C4′–C5′ bond in 5′-dAdo· formers and the equatorial orientation in ·CH3 formers.
To gain insight into the active-site influences that produce the SAM conformational differences exhibited by the members of the two classes of RS enzymes, we took the PDB coordinates of a SAM structure from each class (SPL41 from 5′-dAdo·-forming and HydE42 from ·CH3-forming) and carried out a DFT energy minimization to obtain the favored geometry in vacuo, namely, without environmental influences.48 The SAM structure from HydE was optimized with small changes in geometry, but that from SPL underwent a 2′-endo → 3′-endo flip of the ribose ring with concomitant axial → equatorial shift of the C4′–C5′ bond, and it converged to the same structure observed for SAM bound to HydE (Figure S5) and in most canonical RS enzymes considered by Vey and Drennan.43 In short, in HydE and the other two RS enzymes that form ·CH3 photochemically, SAM binds with little distortion from its intrinsically most stable conformation, whereas PFL-AE and SPL, which photochemically form 5′-dAdo· active-site interactions, force SAM to undergo a major change in ribose conformation upon binding.
ANALYSIS AND DISCUSSION
Here we have shown that cryogenic blue-light photolysis of eight distinct and functionally diverse RS enzymes leads to photoinduced reductive cleavage of SAM to generate carbon-centered radicals bound in the enzyme active sites. These results reveal that the surprising photochemistry previously observed for PFL-AE and HydG,20,21 in which a [4Fe–4S]+ cluster excited state transfers an electron to SAM, causing S–C bond cleavage, is observed broadly throughout the RS superfamily. However, the suite of enzymes divides into two classes, distinguished by which S–C bond is cleaved. In one class photoinduced S–C5′ bond cleavage forms 5′dAdo·, while the second class undergoes S–CH3 homolytic bond cleavage to form the ·CH3 radical, even though all the enzymes cleave the S–C5′ bond catalytically. The photochemical radical initiation described here is thus functionally richer and therefore turns out to be more mechanistically revealing than the enzyme-catalyzed cleavage of the SAM S–C5′ bond. The photolytic reaction also is conceptually simpler. Frey and co-workers previously recognized that enzymatic ET to the sulfonium moiety must be preceded by conformational changes that introduce direct bonding interactions between the sulfonium and the electron-donor [4Fe–4S]+ cluster that energetically drive the catalytic ET event.23–25 By disentangling the ET step from this process through use of light to drive ET, we have discerned previously unrecognized active-site influences on both the overall geometry of SAM binding and on the regioselectivity of the reductive S–C bond-cleavage step.
Alignment of the enzyme-bound SAM molecules from the available crystal structures of enzymes in these two RS classes shows that the conformation of SAM bound in the enzyme active site directly correlates with the identity of the S–C bond that is photolytically cleaved. The ribose of SAM in SPL and PFL-AE (5′-dAdo· formers) has the 2′-endo conformation with an axial C4′–C5′ bond, whereas in LAM, HydG, and HydE (·CH3 formers), the ribose ring is 3′-endo, with an equatorial C4′–C5′ bond (Figure 6). Thus, the overall configuration of the enzyme-bound SAM molecule is determined by interactions with surrounding protein residues in the active site, which in turn control the regioselectivity of photoinduced cleavage. This in turn raises the following questions: how do the interactions of SAM with the active-site environment determine the regioselectivity of photoinduced S–C reductive bond cleavage, and what can we learn from photoinduced reductive cleavage that sheds new insights into the regioselectivity of RS catalysis?
Origin of S–C Bond-Cleavage Regioselectivity.
Canonical RS enzymes reductively cleave only the S–C5′ bond of SAM during catalysis, whereas the cryogenic photoinitiated reductive cleavage of SAM bound to RS enzymes exhibits bifurcation in regioselectivity, with cleavage at either S–C5′ or S–CH3, depending on the enzyme, and correlated with the SAM ribose conformations. To determine what controls regioselectivity of the reductive cleavage of SAM, we first consider and reject an earlier proposed explanation for regioselectivity and then advance a previously unanticipated mechanism.
Colinear Fe–S–C Reactive Arrangement?
It has been hypothesized that the regioselectivity of RS catalytic S–C5′ bond cleavage to form 5′dAdo· results from a complex and stereospecific interaction between the [4Fe–4S]1+ cluster and SAM.49 The cleavage process was visualized as a radical displacement reaction, with regioselectivity arising from a colinear Fe–S–C5′ arrangement as shown in Scheme 1. It was further suggested that crystal structures of SAM-bound RS enzymes support this hypothesis.49 It was even speculated that there might be RS enzymes that position the sulfur–methyl bond of SAM colinear with the reactive iron of the cluster and thus produce methyl radicals by the displacement mechanism.49
Scheme 1.
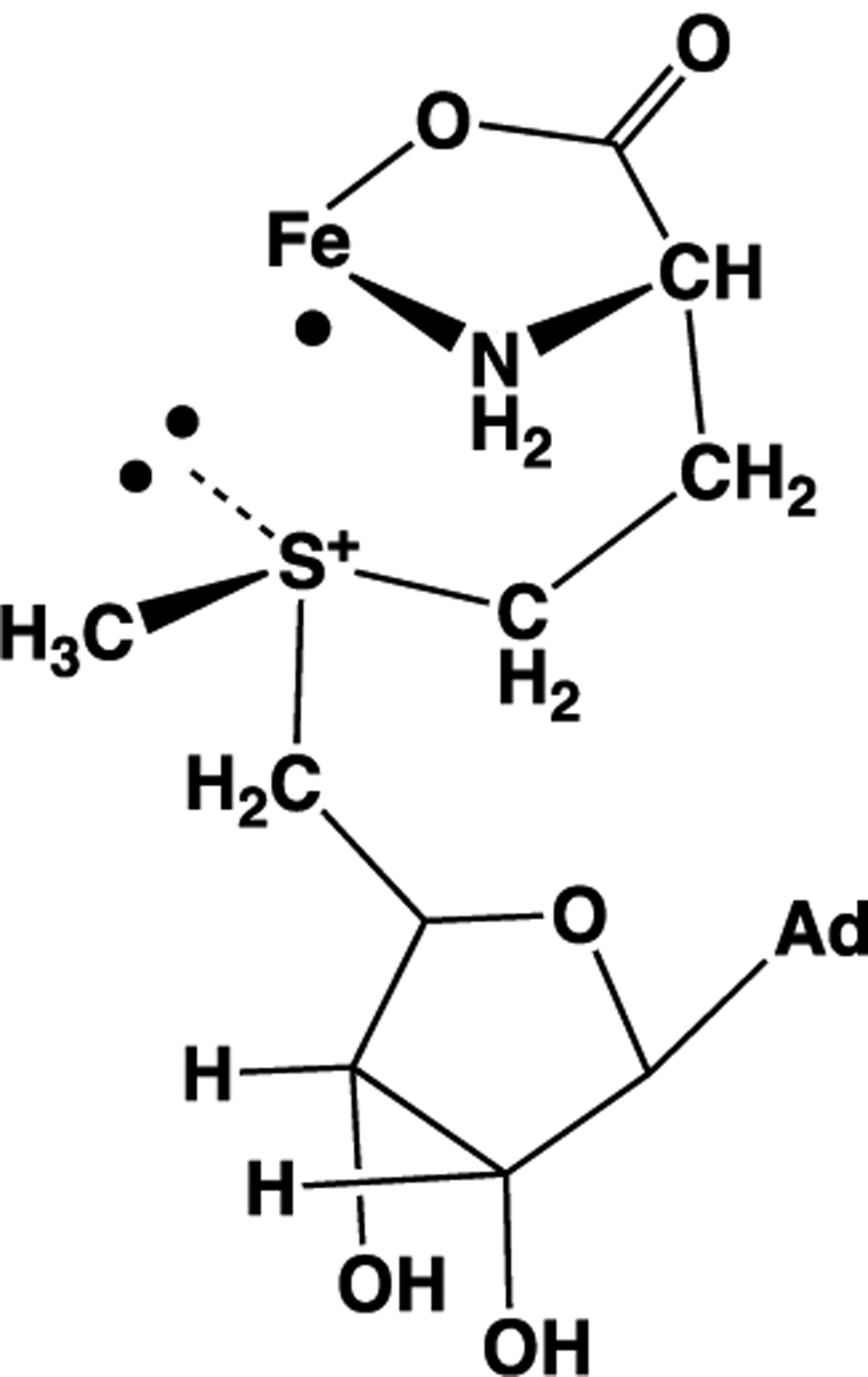
Radical Displacement Model; See Ref 47
Can this proposal explain the experimental results described here? In considering this question, we note that none of the five published RS enzyme:SAM structures for the enzymes in our study in fact has the truly colinear Fe–S–C5′ arrangement (Figure 5 and Table S2): the average Fe–S–C5′ angle is 148°, or 32° away from colinear. Focusing on the cryogenic photoinduced S–CH3 bond cleavage observed here in frozen RS enzyme solutions, the contradictions are more dramatic: those S–CH3 bonds are at nearly right angles to the Fe–S vector in all structures examined (Table S2), rather than colinear as required by the radical displacement mechanism (Scheme 1). Indeed, as noted earlier, the SAM active-site structures of both the 5′-dAdo· and ·CH3 formers exhibit similar orientations of the sulfonium adenosyl and methyl groups relative to the unique Fe (Figure 5). Thus, the radical displacement, colinear model cannot explain the clear bifurcation in S–C bond cleavage observed for the cryogenic photolysis of the diverse set of RS enzymes studied here, nor, for that matter, the overall results for photoinduced and catalytic radical formation. This conclusion is further supported in the next subsection.
Frey Model.
As first recognized by Frey and co-workers,23 with thorough investigations recently by Miller and Bandarian,50 the [4Fe–4S]+ cluster is not a sufficiently strong reductant to reduce the sulfonium of SAM in solution. In the Frey model, catalytic RS SAM cleavage overcomes this through Fe–S coordination, which stabilizes the S ↔ Fe cation–cation interaction by S → Fe σ-donation of the sulfur lone pair to an empty Fe orbital (Scheme 2). This enables Fe → S ET from a singly occupied Fe orbital donor to the LUMO of SAM. Frey and co-workers summarize their view as follows:23 “This suggests a mechanism for cleavage in which substrate binding induces a conformational change that brings the nonbonding electron pair of the sulfonium in proximity to one of the irons of the FeS cluster. This might raise the redox potential of AdoMet to that which would allow inner-sphere ET from the FeS cluster, with concerted cleavage of the carbon–sulfur bond.” In contrast, the putative active configuration of the colinear proposal represented by Scheme 149 precludes this S → Fe σ donation: the sulfonium lone-pair orbital is orthogonal to the Fe–S bonding vector.
Scheme 2.
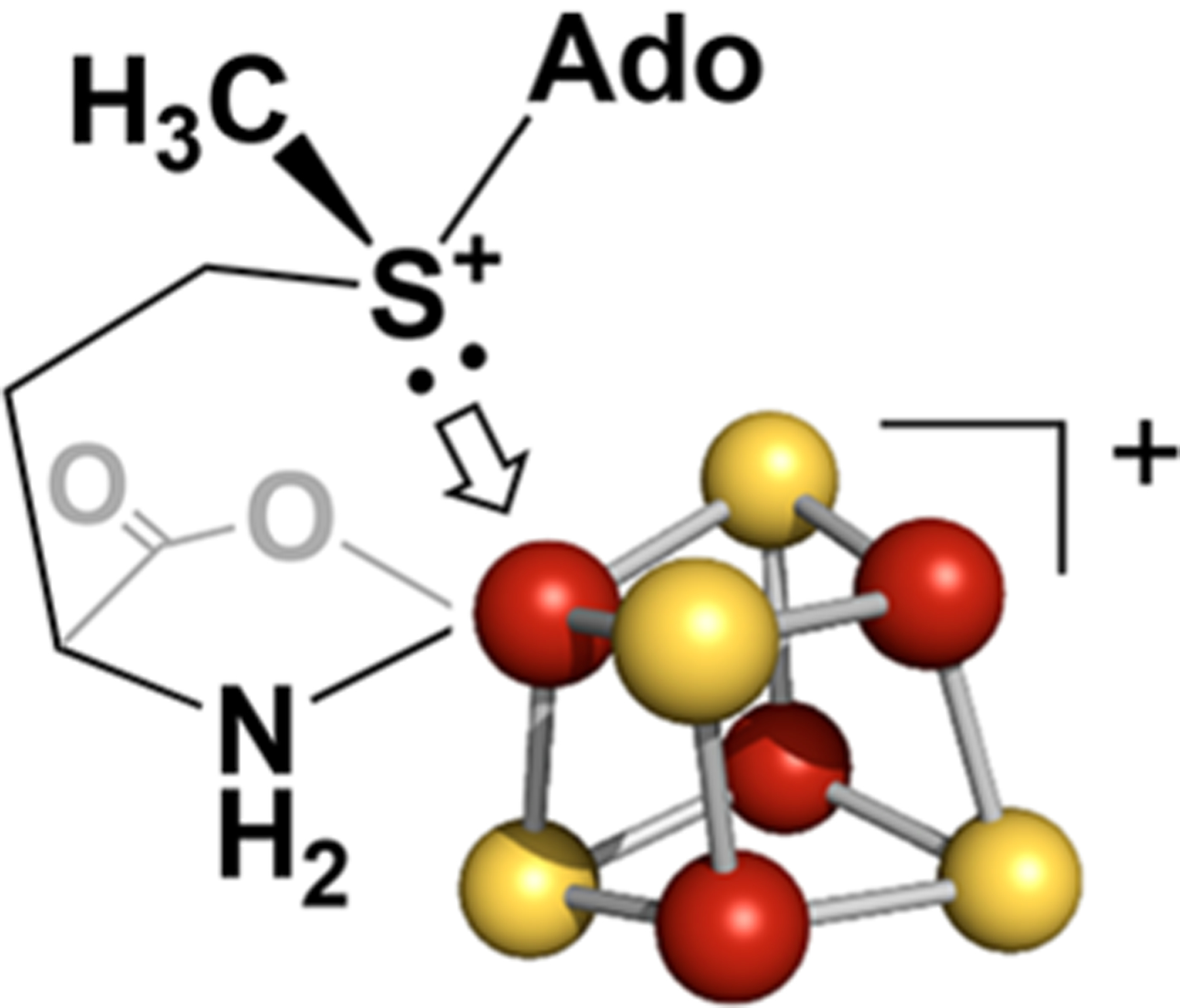
Frey Model for SAM σ-Donation; See Ref 23
The view that the energetic contributions from sulfur lone-pair donation to a cluster Fe are integral to RS enzyme function is supported by DFT computations of the properties of the simplest trialkyl sulfonium ion model for the sulfonium center in SAM: the trigonally symmetric (CH3)3S+ cation (Figure 7). The resulting MO diagram reveals that the HOMO is the sulfonium lone pair, the proposed donor to the unique Fe of the cluster, with S–C bonding orbitals over ~4 eV lower in energy, and it is readily shown that this precludes the type of electronic interactions that underlie the colinear model.
Figure 7.
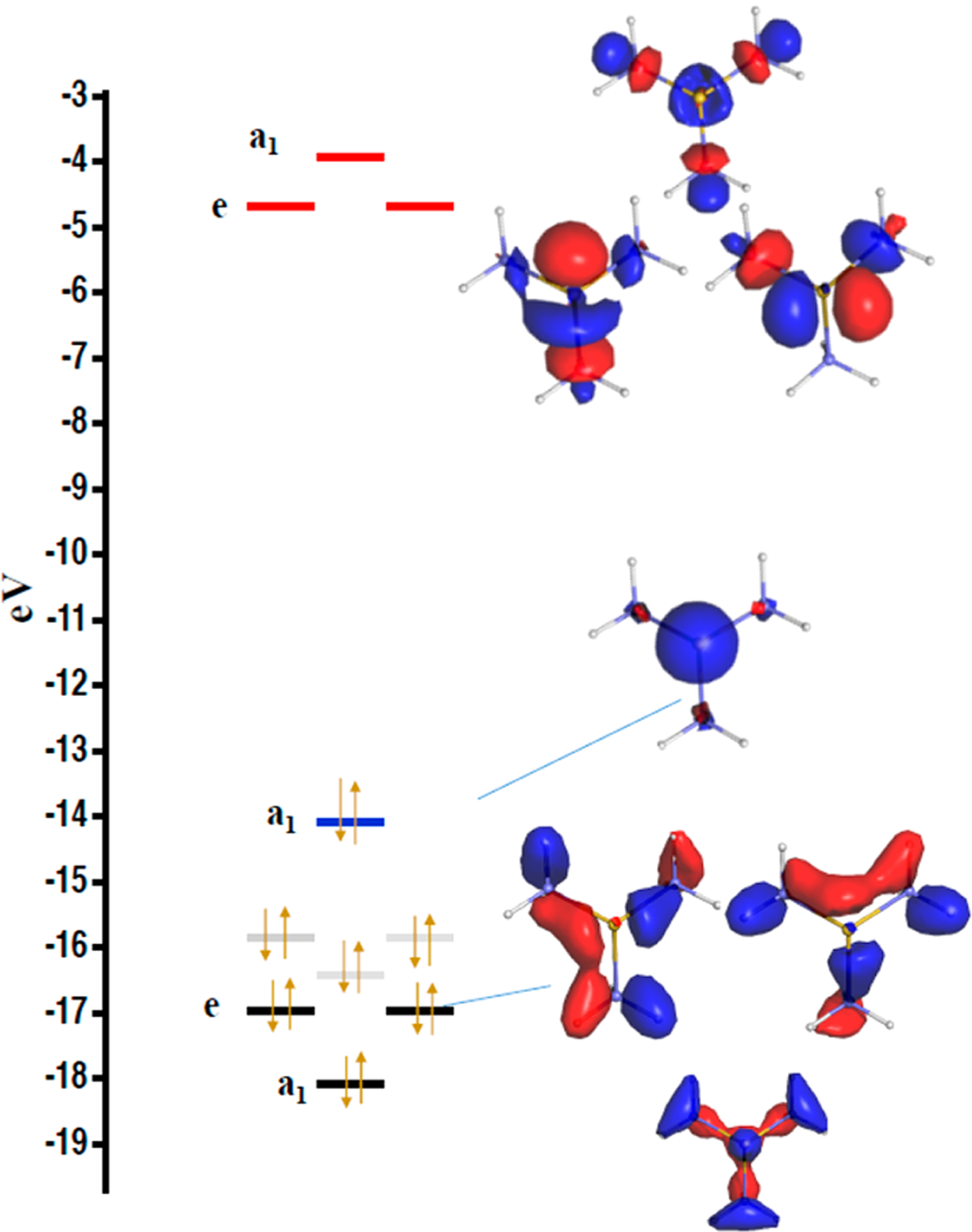
DFT computed energy levels (left) and MO diagrams (right) for S(CH3)3+. (Left) Black, carbon–sulfur based orbitals; gray, the highest-lying C–H based bonding orbitals; blue, sulfur lone pair HOMO; red, C–S based antibonding orbitals. (Right) Blue, positive orbital coefficients; red, negative orbital coefficients. The orbitals are visualized with an isodensity of 0.08 au in PyMol. For completeness we note that the components of a degenerate (e) level are not uniquely defined.
The Frey model further resolves a serious puzzle regarding how the C5′ of the 5′-dAdo· radical could proceed to form the Fe–C5′ bond of Ω after bond cleavage. When SAM adopts the catalytically active conformation, with the sulfonium lone pair pointing at the unique Fe, the DFT-optimized sulfonium structure indicates that the Fe–S–C5′ angle would be only ~108°. With this geometry, only modest conformational fluctuations would enable the formation of the Ω Fe–C5′ bond. In contrast, the colinear Fe–S–C5′ model has the sulfur placed directly in the way between C5′ and Fe, and thus it would block this reaction, precluding the formation of Ω. However, with its energetic focus, the Frey model did not address the question of why the S–C5′ bond is selectively cleaved.
Bonding Within (CH3)3S+/0 and the Origin of Regioselectivity.
The electronic structure of this sulfur center provides the foundation for an understanding of regioselective C–S bond cleavage. To begin, we note that the basic idea of independent SAM S–C bonds, with the S–C5′ antibonding orbital simply acting as the electron-acceptor orbital on SAM, is incorrect: a hypothesis of ET into an individual S–C antibond, either catalytically or via direct ET as occurs in these studies, oversimplifies the process and precludes our understanding of the S–C bond-cleavage step. It is not in general appropriate to consider the LUMOs of an R3S+ center as comprising three independent S–C antibonds, each potentially functioning as an independent electron-acceptor orbital, just as it is correspondingly inappropriate to consider the R3S+ center as comprising three independent S–C bonds (Figure 7).
To illustrate this remark, we begin by again considering the electronic structure of the (CH3)3S+ cation (Figures 7 and S6). First, as can be seen, S–C bonding is delocalized. There is a low-lying, doubly occupied, a1 S–C orbital uniformly delocalized among the three S–C bonds. Above this is a filled, doubly degenerate S–C e level whose nonuniquely defined components involve pairs of bonds and whose total density again is uniformly delocalized. The LUMO of this trigonally symmetric ion, which would accept the electron during reduction, is an orbitally degenerate doublet (e) whose overall density again is uniform (Figure 7). Correspondingly, one-electron reduction of an R3S+ with this idealized trigonal geometry at S would create an R3S0 radical whose unpaired electron occupies a doubly degenerate e level delocalized over the three S–C antibonds. To achieve regioselective bond cleavage, this added electron must somehow become localized in the priority S–C antibond.
Turning to regioselectivity in SAM reductive cleavage, neither the bonding to the sulfur nor its environment is symmetric. Thus, the question is which of these effects, or what combination of them, controls regioselective cleavage?
Enzyme Control of Regioselectivity Through the Jahn–Teller Effect.
The sulfur of SAM has three different substituents, but during catalysis the canonical RS enzymes cleave only the S–C5′ bond. This might have focused attention on the differing S–C bond energies of the three substituents as the source of S–C bond cleavage regioselectivity. However, although differing bond energies may play a role in the reductive cleavage of sulfonium salts with greatly differing substituents,51–53 our photolysis studies show this clearly is not determining for SAM cleavage in RS enzymes, as different circumstances lead to selective cleavage of a different S–C bond (Figure 1). Instead, having recognized that the basic idea of three independent SAM S–C bonding/antibonding orbitals is incorrect, further consideration of the electronic structure of trigonally symmetric R3S+/0 (e.g., Figure 7) leads to a previously unrecognized characteristic of this system that is the actual basis of active-site conformational control of regioselective S–C bond cleavage.
JT Effect and R3S0 S–C Bond Homolysis.
One-electron reduction of the R3S+ sulfonium cation adds an electron to its doubly degenerate S–C antibonding LUMO to give the R3S0 radical in a 2E state (Figures 7 and S6), which is subject to JT distortion through vibronic coupling to (e) vibrations.26,27 In this vibronic distortion the R3S0 triangle of carbons undergoes an equilateral → acute-isosceles transformation through changes in S–C bond lengths, in particular elongation of the apex bond, along with changes in C–S–C angles, as visualized in the cartoon of Scheme 3 (obtuse triangles also are possible).27 This splits the 2E orbital doublet, and the resultant lower-energy component of the doublet is the actual R3S0 HOMO, as also shown in Scheme 3. However, as we next explain, the JT-distorted state by itself could not exhibit regioselective homolysis. Regioselectivity is only introduced when environmental influences acting on R3S0 through the JT effect create and stabilize a component of the 2E doublet with a distortion that localizes the R3S0 antibonding electron in one particular SAM S–C priority bond, which proceeds to undergo homolysis. The difference in photoinduced regioselectivity in the two enzyme classes must then arise because a difference in forces operative at the sulfur within the two different SAM structures of Figure 6, as controlled by active-site interactions, leads to selection of different bonds for cleavage.
Scheme 3.

Splitting of 2E Orbital Degeneracy with JT Distortion
Before elaborating on this phenomenon, we note that the trigonal-pyramidal structure of molecules isoelectronic with the sulfonium cation, the simplest being NH3, has long been discussed in terms of vibronic coupling and the pseudo-JT effect,31,54 and likewise for the metastability of a radical somewhat analogous to R3S0, H3O.55 However, in previous studies of the reductive cleavage reactions of sulfonium salts,51–53,56 it has not been recognized that the neutral R3S0 radical exhibits a degenerate 2E state and its behavior is vibronically driven.
The simplest way to explain the combined operation of the JT effect and environmental forces in creating regioselective S–C bond homolysis of an R3S0 radical is through application of a formal JT model for a trigonal molecule in which a singly occupied orbital (e) doublet is vibronically coupled to a composite (e) vibration.27,57,58 Figure 8 shows the adiabatic potential energy surface (APES) for the JT-stabilized lower component of such a doublet (Scheme 3) at different levels of elaboration of the model. The radial coordinate in Figure 8 corresponds to the extent of distortion; the azimuthal angle corresponds to the direction of the distortion, which dictates the relative contributions to the JT-SOMO (SOMO = singly occupied molecular orbital) (Scheme 3) of the two orbital components of the (e) doublet. This figure visualizes the behavior of R3S0 as a high-energy but locally stable intermediate, with well-defined minima on the APES; for completeness, in Figure S8 we show the equivalent figure treating R3S0 as a dissociative state that would undergo prompt homolysis upon formation.
Figure 8.
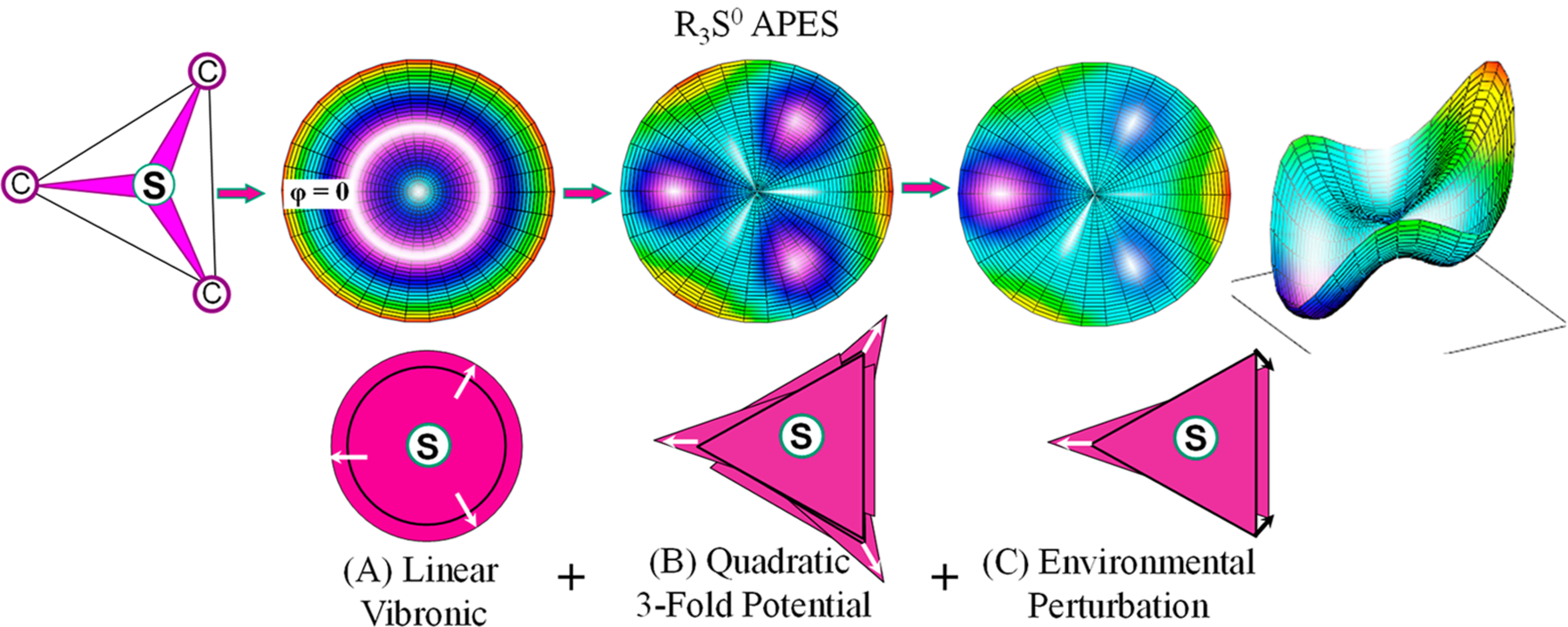
Upper row: (Left) Cartoon of trigonal R3S0 SAM sulfur center; (Others) contour plots of the APES (orange highest energy) for the JT distortion of R3S0 with a singly occupied E-doublet vibronically coupled to an e vibration, within a formal JT model that treats R3S0 as a stable intermediate for progressively more complex treatments (see Figure S8 for corresponding figure for R3S0 as a dissociative state); radial distance ⇔ JT distortion parameter, azimuthal angle, pseudorotation angle of triangle distortion. Lower Row: Cartoons representing the distribution of triangle orientations. (A) Linear JT vibronic coupling term yields rotationally symmetric APES (upper), and pseudorotating distortion with uniform distribution in orientation (lower). (B) Addition of quadratic vibronic coupling terms plus 3-fold potential of S–C bonds yields three energy minima on APES contour plot (upper) corresponding to three equivalent acute isosceles triangles (e.g., Scheme 3) oriented (lower) with the acute vertex (direction of elongation) pointed along the angle φ associated with that minimum: φ = 0, 2π/3, and 4π/3. (C) Inclusion of appropriate active-site distortion forces lowers the energy of a single minimum (upper, arbitrarily chosen here at φ = 0) with elongated priority S–C bond (lower) that ultimately undergoes regioselective homolysis, as illustrated by both contour and perspective plots.
The simplest formulation of this JT model, illustrated in Figure 8A, incorporates only linear electron–nuclear vibronic coupling, proportional to the extent of the distortion, which involves both elongation of the S–R bonds and changes in C–S–C angles. However, this treatment level is inadequate for the present purposes, as it yields a cylindrically symmetric APES (Figure 8A) corresponding to a freely pseudorotating JT distortion of the triangle without preference for one of the three S–C bonds (see Figure S9 for visualization of the pseudorotation). This treatment merely converts the electronic degeneracy of the orbitals into a vibronic degeneracy.
To properly apply the model to bond cleavage in the R3S0 radical requires incorporation of terms quadratic in the vibronic couplings (quadratic JT effect).26,27 In combination with the 3-fold potential associated with the S–C bonds, the quadratic JT effect introduces terms in the APES that are 3-fold symmetric in the azimuthal angle and generate three localized energy minima in the APES (Figure 8B). Each of these is associated with an R3S0 distortion of the equilateral triangle of carbons into an acute isosceles triangle through changes in C–S–C bond angles and S–C bond lengths, most importantly with elongation of the apex S–C bond (Scheme 3) specific to that minimum. As indicated in the figure, the acute vertex of the distorted triangle associated with each of the minima is directed along the radius of the APES at the angle φ associated with that minimum: φ = 0, 2π/3, and 4π/3. The distortion at a minimum localizes the vibronically coupled antibonding odd-electron density into the destabilized apex S–C bond and would lead to cleavage of that bond. However, this still does not pick out one bond of the three as having priority for ultimate dissociation. As evident in the center panel, this localization occurs with equal probability for the priority bond associated with each of the stabilized distortions. The result would be an equal probability of cleaving each of the three SAM S–C bonds.
Regioselectivity is only introduced through the action of symmetry-lowering forces that act on the R3S0 radical to generate an APES (Figure 8C) having a single global minimum in which the acute-isosceles distortion (arbitrarily chosen to be that at φ – 0) stabilizes a component of the orbital doublet with the R3S0 antibonding electron localized in the elongated (apex) S–C priority bond (Scheme 3), which would proceed to undergo regioselective homolytic cleavage. Such localization is visualized in our DFT examination of the evolution of the LUMO of the (CH3)3S+ center upon desymmetrizing it by stretching one of the three S–C bonds (Figure S7A–E). This LUMO models the SOMO of R3S0 along the reaction pathway to reductive cleavage and shows a localized priority S–C antibonding orbital (Figure 9), which would lead to cleavage of that bond. Thus, the key to understanding regioselective bond cleavage in RS enzymes is identification of the symmetry-lowering perturbations that act on the R3S0 SAM sulfur center through the JT effect, especially identification of the differences between photoinduced and catalytic cleavage.
Figure 9.
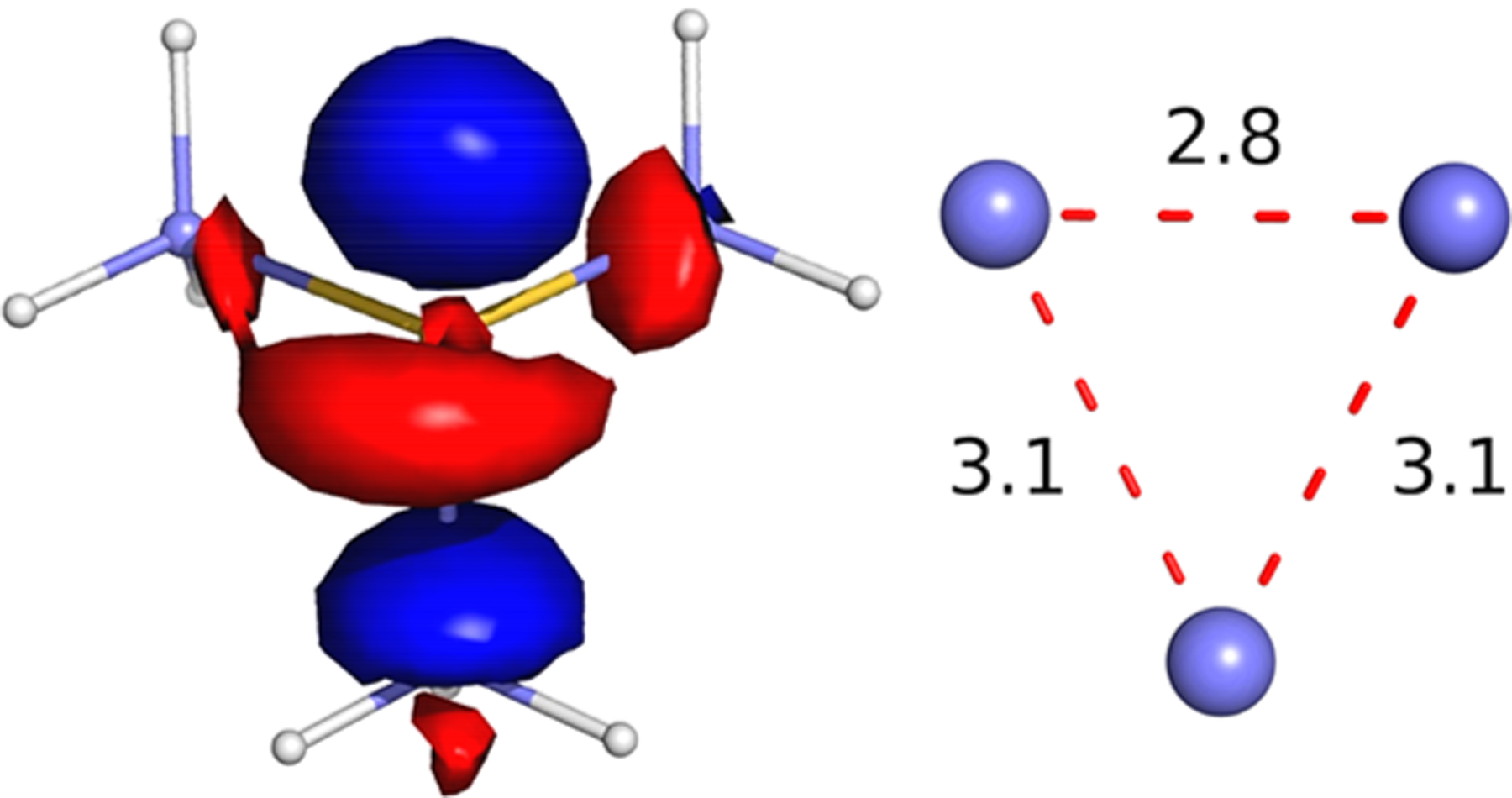
(Left) LUMO of (CH3)3S+ (see the Supporting Information) corresponding to the SOMO of the JT-distorted acute-isosceles R3S0 radical (Scheme 3) with a stretched apex priority S–C bond of 2.2 Å, showing strongly antibonding character of the priority (S[3p-like]–C[σ]) orbital at the apex of the triangle of S–C carbons (see the Supporting Information). (Right) Dimensions of the triangle of carbons in the distorted R3S0 radical.
JT Effect and Regioselectivity in Photoinduced S–C Bond Homolysis.
The present study of cryogenic photoinduced reductive cleavage of SAM bound to RS enzymes shows that the 5′-dAdo· and ·CH3 forming enzymes are distinguished by active sites that select alternative ribose conformations: 2′-endo/axial C4′–C5′ bond for the former, 3′-endo/equatorial C4′–C5′ for the latter (Figure 6). As noted earlier, these structural differences in the two classes do not alter the positioning of the two ends of SAM, and in particular only modestly alter the positioning and orientation of the sulfonium centers of SAM relative to the unique Fe of the [4Fe–4S] cluster (Figure 5). This suggests that the regioselectivity of cryogenic photoinduced reductive S–C bond cleavage is associated with a difference in forces operative at the sulfonium sulfur due to the distinct SAM ribose conformations shown in Figure 6, as transmitted to the sulfur through the S–C5′ bond. In short, we propose that the distinct SAM ribose conformations in the two photolysis product classes (5′-dAdo· and ·CH3 formers) generate symmetry-lowering perturbations that result in the localized JT distortion that selects a single S–C bond for cleavage, giving rise to the observed regioselectivity.
Further Observations on Active-Site Symmetry Lowering and Regioselectivity.
The finding that the added steric bulk of ethyl versus methyl as a SAM sulfur substituent (SAE vs SAM cosubstrates) has no effect on the regioselectivity of photo-induced bond cleavage supports the view that active-site forces function by controlling the ribose configuration of SAM and are not exerted at the methyl substituent. This of course is not a surprise, as a methyl group is too small to provide a handle for the protein environment to influence. The invariance of the photoinduced C–S bond-cleavage regioselectivity to replacement of the S substituent also is further support for the conclusion that bond energy differences in the S–C bonds of SAM are not determining for regioselectivity.
We further note that, as the parent SAM R3S+ cation is not JT-active, the environmental forces that control the JT distortion of the R3S0 radical need not cause distortions visible in an X-ray structure of SAM bound to an RS enzyme. While keeping this in mind, we have visualized how symmetry breaking might influence the R3S0 through examination of the LUMOs from DFT computations on a suite of sulfonium R3S+ cations (Figure S7A). These computations show that quite modest reductions from trigonal symmetry around the closed-shell sulfonium-ion sulfur can stabilize one component of the LUMO e-doublet level such that reduction would favor—or disfavor—cleavage of a particular S–C bond. Of course, the JT-active R3S0 center is much more influenced than the parent cation.
A particularly intriguing insight offered by this approach comes from looking at the LUMOs of SAM itself. Recognizing the inherent crystallographic uncertainties in the reported structures, we examined the sulfonium ion of the DFT-optimized structure of SAM in vacuo, namely, without the influence of environmental forces (Figure S7A). Despite the fact that sulfur has three distinct substituents, the geometry around sulfur is little distorted from trigonal pyramidal, and the LUMO (e) component orbitals remain nearly degenerate (splitting, 0.05 eV); nothing indicates which bond would cleave upon reduction. In short, the result of this exercise is in keeping with the idea that the unsymmetrically substituted sulfonium ion of free SAM nonetheless has little intrinsic propensity to reductively cleave a particular S–C bond and that regioselectivity is generated by forces on the sulfur center ultimately imposed by the protein environment.
JT Effect and Regioselectivity in Catalysis.
All canonical RS enzymes, as represented by those examined here, undergo S–C5′ cleavage during enzymatic catalysis independent of the conformation of SAM observed in the enzyme crystal structures (Figure 5), in sharp contrast to the photoinduced reaction with its two types of regioselectivity. How can one account for this fundamental difference in the two processes? Cryogenic photoinduced reductive cleavage of SAM and RS enzymatic catalysis both proceed via electron transfer from the [4Fe–4S]+ cluster to the SAM sulfonium center, yielding a common mechanistically central JT-active R3S° radical species that undergoes a distortion, manifested in S–C bond lengthening and subsequent homolytic cleavage. Thus, an understanding of the regioselectivity of S–C5′ bond cleavage in canonical RS enzymes must center on the environmental forces that act on the JT-active R3S0 active species to localize the antibonding electron in the S–C5′ bond for cleavage. We propose that different forces act on the JT-active R3S0 during catalytic S–C5′ bond cleavage than those outlined earlier that act during cryogenic photoinduced reductive SAM cleavage.
Forces acting on R3S0 in cryogenic photoinduced reductive cleavage necessarily are present prior to photolysis, being generated within the cryogenically fixed enzyme structures, as represented by the static X-ray crystal structures of the enzymes. The ribose structures of SAM differ in the two classes (5′-dAdo· and ·CH3 formers) of RS enzymes (Figures 5, S3, and S4), and this leads to the photoinduced cleavage of different S–C bonds. However, as revealed in a number of radical SAM enzymes whose SAM-bound structures have been determined in both the presence and absence of substrate, substrate binding alone does not induce a change in the SAM binding conformation.42,44,59,60 Rather, as proposed by Frey, catalytic bond cleavage within all canonical members of the RS superfamily requires a thermally induced conformational change that brings the SAM sulfur into contact with the unique cluster Fe, thereby enabling Fe → S ET. This motion, visualized in Figure 10, requires a swing of the sulfonium sulfur of SAM toward the unique Fe while the amino acid end remains anchored through coordination to that Fe and the adenine end remains fixed by interactions with its neighboring residues. This movement plausibly occurs with a flip of the ribose-ring conformation in one of the two classes, to give both classes equivalent ribose conformations. Such SAM conformational changes involved in bringing the sulfur into contact with Fe would clearly generate a set of forces at the sulfur of the R3S0 produced by ET, forces present only during catalysis, and only in the catalytically active configuration. We propose that it is this thermally induced conformational activation that determines the regioselective enzymatic S–C5′ bond cleavage in canonical RS enzymes.
Figure 10.
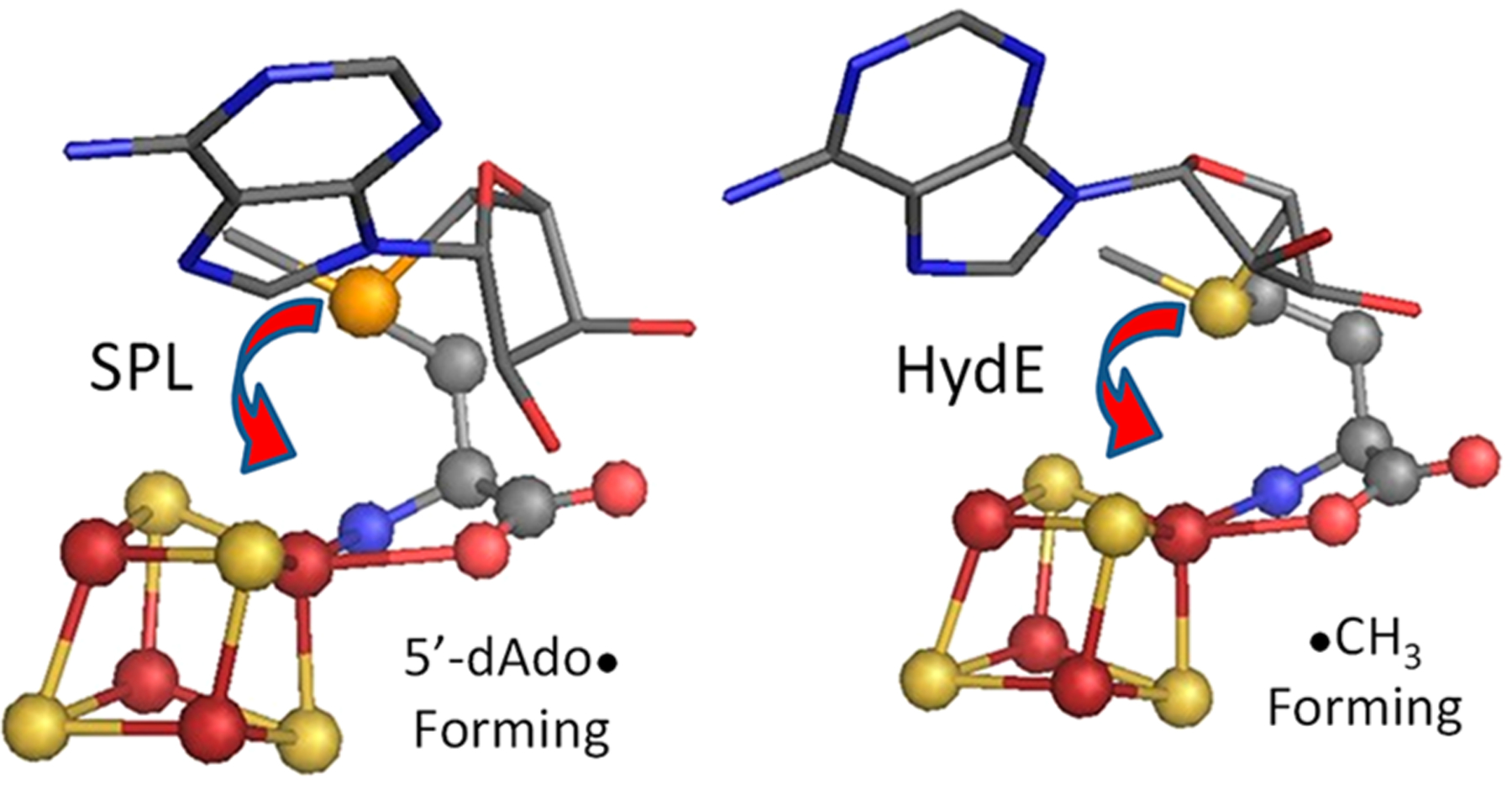
Crystallographic structures of SAM bound to the [4Fe–4S] clusters in SPL (4fhf.pdb; left) and in HydE (3iiz.pdb; right), as reoriented from Figure 5 to visualize the basis of conformational changes involved in creating an S–Fe contact in the two regioselectivity classes.
In short, the JT-active R3S0 radical center produced by reduction of the SAM sulfonium R3S+ moiety is exquisitely poised for effecting active-site control of regioselective S–C bond cleavage in RS enzymes. This active-site control manifests itself differently under the disparate conditions of cryogenic photolysis versus enzymatic catalysis.
CONCLUSIONS
Examination of cryogenic photoinduced SAM S–C bond cleavage in RS enzymes has led us to recognize the presence of two enzyme classes differing in SAM ribose conformation, with photolysis forming 5′dAdo· in one class and ·CH3 in the other. The different SAM ribose conformations (2′-endo/axial C4′–C5′ bond versus 3′-endo/equatorial C4′–C5′) directly correlate with the identity of the photocleaved S–C bond, yet the positioning and orientation of the sulfonium centers relative to the [4Fe–4S] cluster are largely the same in the two classes. Analysis of the S–C bonding in R3S+/0 centers led to recognition of the JT activity of mechanistically central R3S0 radicals and yielded insights into the sensitivity of reductive SAM cleavage to SAM ribose conformation, thereby leading to an understanding of S–C bond cleavage regioselectivity in both photoinduced and enzymatic radical formation.
Most importantly, these two aspects of the present report have together revealed that the coordinated SAM provides a uniquely sensitive platform for vibronically enabled protein control of enzymatic regioselective S–C bond cleavage. ET from the [4Fe–4S]1+ cluster to the SAM R3S+ sulfonium ion creates the JT-active 2E R3S0 radical state with the odd electron occupying a degenerate or near-degenerate (e)-symmetry antibonding orbital doublet. Such a center is subject to JT symmetry-lowering distortions that concomitantly split the (e)-symmetry antibonding orbital doublet, lowering the energy of the R3S0 radical (Scheme 3). By inducing such distortions, active-site forces select a priority S–C bond (Figure 8C) for antibonding electron localization (Figure 9), with attendant regioselective bond cleavage. As the SAM R3S+ parent cation center is not JT-active, such forces need not even cause substantial distortions visible in an X-ray structure of this state.
We suggest that the forces that generate S–C bond cleavage regioselectivity have two types of origin. The dichotomy in cryogenic photoinduced regioselectivities reported here arises from forces operative at the sulfur caused by active-site induced differences in SAM ribose conformation as visualized in X-ray structures and as transmitted to sulfur through the S–C5′ bond. Intriguingly, the intrinsically stable SAM conformation present in most RS enzymes, with a 3′-endo ribose ring and concomitant equatorial C4′–C5′ bond, gives rise to regioselective photocleavage of the S–CH3 bond, although all canonical RS enzymes regioselectively cleave the S–C5′ of SAM during catalysis. This suggests that the S–CH3 bond may be intrinsically favored for reductive cleavage. However, in enzyme-catalyzed S–C bond cleavage, thermally induced conformational changes are necessary to bring the sulfur into contact with the unique Fe, thereby enabling Fe → S ET in the presence of the substrate, as first recognized by Frey (Figure 10).23 These changes would generate forces at the JT-active R3S0 sulfur that are operative only in the catalytically active configuration in canonical RS enzymes and that cause regioselective enzymatic bond cleavage of the S–C5′ bond in all the RS enzymes. Both types of force are ultimately enabled by the anchoring of the two ends of SAM, with the amino acid end anchored by chelation of the unique cluster Fe and the adenine end anchored by nonbonding interactions with neighboring residues.
We further note that the enzymatically active configuration, involving σ-donation from the sulfonium lone pair to the unique iron of the [4Fe–4S]1+ cluster, requires an Fe–S–C5′ angle of ~108°, not 180°. This plausibly provides access of the product 5′-dAdo· C5′ to the unique Fe, thus offering an explanation for the stoichiometric formation of the Fe–C5′ bond of the organometallic intermediate Ω subsequent to SAM cleavage.
Whether the resulting mechanistically central R3S0 state is a high-energy, reactive intermediate (Figure 8) or a dissociative state that undergoes activationless bond cleavage (Figure S8) could be tested by time-resolved photo-EPR experiments and will be explored by high-level (CASSCF, see the Supporting Information) computations. The suggestions for the source(s) of the symmetry-lowering perturbations that control regioselectivity during photolysis can be tested by variations in the ribose ring, site-directed mutations in the active site, and high-level computations that include quantum mechanics/molecular mechanics (QM/MM) approaches. As a final observation, this work has deeply embedded the JT effect into enzymology, by revealing its controlling influence on the regioselectivity of S–C bond cleavage to generate the reactive 5′-dAdo· radical in radical SAM enzymes, one of the largest enzyme superfamilies across the tree of life.
Supplementary Material
ACKNOWLEDGMENTS
All work involving the preparation and handling of HydE and HydG was funded by the U.S. Department of Energy, Office of Basic Energy Sciences, Grant DE-SC0005404 (to J.B.B. and E.M.S.). All other work was funded by the NIH (GM 111097 to B.M.H. and GM 131889 to J.B.B.). R.J.J. is supported by the NIH (T32GM008382). We thank Prof. George Schatz (Northwestern) for the use of his computational cluster in performing DFT calculations and for helpful discussions.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c10925.
X-band EPR spectra, overlaid structure of SAM-bound [4Fe–4S] clusters, comparisons of SAM configurations that yield photo-induced cleavage, relaxed DFT-computed SAM cofactors, DFT computations, perspective views of selected orbitals, DFT-optimized sulfonium ions, EPR simulation parameters, spatial distance and angle parameters between the unique Fe and SAM/SeAM, APES for the JT distortion of a trigonal R3S0, pseudorotation of JT-distorted X3 molecule, and results of DFT optimizations (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.0c10925
The authors declare no competing financial interest.
REFERENCES
- (1).Frey PA; Hegeman AD; Ruzicka FJ The Radical SAM Superfamily. Crit. Rev. Biochem. Mol. Biol 2008, 43 (1), 63–88. [DOI] [PubMed] [Google Scholar]
- (2).Broderick JB; Duffus BR; Duschene KS; Shepard EM Radical S-adenosylmethionine enzymes. Chem. Rev 2014, 114 (8), 4229–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Nicolet Y Structure–function relationships of radical SAM enzymes. Nature Catalysis 2020, 3 (4), 337–350. [Google Scholar]
- (4).Landgraf BJ; McCarthy EL; Booker SJ Radical S-Adenosylmethionine Enzymes in Human Health and Disease. Annu. Rev. Biochem 2016, 85 (1), 485–514. [DOI] [PubMed] [Google Scholar]
- (5).Holliday GL; Akiva E; Meng EC; Brown SD; Calhoun S; Pieper U; Sali A; Booker SJ; Babbitt PC Chapter One—Atlas of the Radical SAM Superfamily: Divergent Evolution of Function Using a “Plug and Play” Domain. In Methods in Enzymology; Bandarian V, Ed.; Academic Press: 2018; Vol. 606, pp 1–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Henshaw TF; Cheek J; Broderick JB The [4Fe-4S]1+ Cluster of Pyruvate Formate-Lyase Activating Enzyme Generates the Glycyl Radical on Pyruvate Formate-Lyase: EPR-Detected Single Turnover. J. Am. Chem. Soc 2000, 122 (34), 8331–8332. [Google Scholar]
- (7).Dey A; Peng Y; Broderick WE; Hedman B; Hodgson KO; Broderick JB; Solomon EI S K-edge XAS and DFT Calculations on SAM Dependent Pyruvate Formate-Lyase Activating Enzyme: Nature of Interaction between the Fe4S4 Cluster and SAM and its Role in Reactivity. J. Am. Chem. Soc 2011, 133 (46), 18656–18662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Krebs C; Broderick WE; Henshaw TF; Broderick JB; Huynh BH Coordination of Adenosylmethionine to a Unique Iron Site of the [4Fe-4S] of Pyruvate Formate-Lyase Activating Enzyme: A Moessbauer Spectroscopic Study. J. Am. Chem. Soc 2002, 124 (6), 912–913. [DOI] [PubMed] [Google Scholar]
- (9).Walsby CJ; Hong W; Broderick WE; Cheek J; Ortillo D; Broderick JB; Hoffman BM Electron-Nuclear Double Resonance Spectroscopic Evidence That S-Adenosylmethionine Binds in Contact with the Catalytically Active [4Fe–4S]+ Cluster of Pyruvate Formate-Lyase Activating Enzyme. J. Am. Chem. Soc 2002, 124 (12), 3143–3151. [DOI] [PubMed] [Google Scholar]
- (10).Walsby CJ; Ortillo D; Broderick WE; Broderick JB; Hoffman BM An Anchoring Role for FeS Clusters: Chelation of the Amino Acid Moiety of S-Adenosylmethionine to the Unique Iron Site of the [4Fe-4S] Cluster of Pyruvate Formate-Lyase Activating Enzyme. J. Am. Chem. Soc 2002, 124 (38), 11270–11271. [DOI] [PubMed] [Google Scholar]
- (11).Silakov A; Lanz ND; Booker SJ Characterization of Radical S-Adenosylmethionine Enzymes and Intermediates in Their Reactions by Continuous Wave and Pulse Electron Paramagnetic Resonance Spectroscopies. In Future Directions in Metalloprotein and Metalloenzyme Research; Hanson G, Berliner L, Eds.; Springer International Publishing: Cham, 2017; pp 143–186. [Google Scholar]
- (12).Horitani M; Shisler K; Broderick WE; Hutcheson RU; Duschene KS; Marts AR; Hoffman BM; Broderick JB Radical SAM catalysis via an organometallic intermediate with an Fe-[5′-C]-deoxyadenosyl bond. Science 2016, 352 (6287), 822–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Byer AS; Yang H; McDaniel EC; Kathiresan V; Impano S; Pagnier A; Watts H; Denler C; Vagstad AL; Piel J; Duschene KS; Shepard EM; Shields TP; Scott LG; Lilla EA; Yokoyama K; Broderick WE; Hoffman BM; Broderick JB Paradigm Shift for Radical S-Adenosyl-l-methionine Reactions: The Organometallic Intermediate Ω Is Central to Catalysis. J. Am. Chem. Soc 2018, 140 (28), 8634–8638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).McSkimming A; Sridharan A; Thompson NB; Müller P; Suess DLM An [Fe4S4]3+−Alkyl Cluster Stabilized by an Expanded Scorpionate Ligand. J. Am. Chem. Soc 2020, 142 (33), 14314–14323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Moss M; Frey PA The role of S-adenosylmethionine in the lysine 2,3-aminomutase reaction. J. Biol. Chem 1987, 262 (31), 14859–14862. [PubMed] [Google Scholar]
- (16).Frey PA; Reed GH Lysine 2,3-aminomutase and the mechanism of the interconversion of lysine and ß-lysine. Adv. Enzymol. Relat. Areas Mol. Biol 2006, 66, 1–39. [DOI] [PubMed] [Google Scholar]
- (17).Frey PA Travels with Carbon-Centered Radicals. 5′-Deoxyadenosine and 5′-Deoxyadenosine-5′-yl in Radical Enzymology. Acc. Chem. Res 2014, 47 (2), 540–549. [DOI] [PubMed] [Google Scholar]
- (18).Bridwell-Rabb J; Grell TAJ; Drennan CL A Rich Man, Poor Man Story of S-Adenosylmethionine and Cobalamin Revisited. Annu. Rev. Biochem 2018, 87, 555–584. [DOI] [PubMed] [Google Scholar]
- (19).Broderick WE; Hoffman BM; Broderick JB Mechanism of Radical Initiation in the Radical S-Adenosyl-l-methionine Superfamily. Acc. Chem. Res 2018, 51 (11), 2611–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Yang H; McDaniel EC; Impano S; Byer AS; Jodts RJ; Yokoyama K; Broderick WE; Broderick JB; Hoffman BM The Elusive 5′-Deoxyadenosyl Radical: Captured and Characterized by Electron Paramagnetic Resonance and Electron Nuclear Double Resonance Spectroscopies. J. Am. Chem. Soc 2019, 141 (30), 12139–12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Yang H; Impano S; Shepard EM; James CD; Broderick WE; Broderick JB; Hoffman BM Photoinduced Electron Transfer in a Radical SAM Enzyme Generates an S-Adenosylmethionine Derived Methyl Radical. J. Am. Chem. Soc 2019, 141 (40), 16117–16124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Zhang Y; Zhu X; Torelli AT; Lee M; Dzikovski B; Koralewski RM; Wang E; Freed J; Krebs C; Ealick SE; Lin H Diphthamide biosynthesis requires an organic radical generated by an iron-sulphur enzyme. Nature 2010, 465 (7300), 891–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Cosper NJ; Booker SJ; Ruzicka F; Frey PA; Scott RA Direct FeS Cluster Involvement in Generation of a Radical in Lysine 2,3-Aminomutase. Biochemistry 2000, 39 (51), 15668–15673. [DOI] [PubMed] [Google Scholar]
- (24).Chen D; Walsby C; Hoffman BM; Frey PA Coordination and Mechanism of Reversible Cleavage of S-Adenosylmethionine by the [4Fe-4S] Center in Lysine 2,3-Aminomutase. J. Am. Chem. Soc 2003, 125 (39), 11788–11789. [DOI] [PubMed] [Google Scholar]
- (25).Wang SC; Frey PA Binding Energy in the One-Electron Reductive Cleavage of S-Adenosylmethionine in Lysine 2,3-Aminomutase, a Radical SAM Enzyme. Biochemistry 2007, 46 (45), 12889–12895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Bersuker IB; Polinger VZ Springer Series in Chemical Physics, Vol. 49: Vibronic Interactions in Molecules and Crystals; Springer: 1989. [Google Scholar]
- (27).Bersuker IB The Jahn–Teller Effect, first ed.; Cambridge University Press: Cambridge, 2006; p 616. [Google Scholar]
- (28).Byer AS; McDaniel EC; Impano S; Broderick WE; Broderick JB Chapter Ten—Mechanistic Studies of Radical SAM Enzymes: Pyruvate Formate-Lyase Activating Enzyme and Lysine 2,3-Aminomutase Case Studies. In Methods in Enzymology; Bandarian V, Ed.; Academic Press: 2018; Vol. 606, pp 269–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Impano S; Yang H; Shepard EM; Swimley R; Pagnier A; Broderick WE; Hoffman BM; Broderick JB S-Adenosyl-lethionine is a Catalytically Competent Analog of S-Adenosyl-lmethione (SAM) in the Radical SAM Enzyme HydG. Angew. Chem., Int. Ed 2020, DOI: 10.1002/anie.202014337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Betz JN; Boswell NW; Fugate CJ; Holliday GL; Akiva E; Scott AG; Babbitt PC; Peters JW; Shepard EM; Broderick JB [FeFe]-Hydrogenase Maturation: Insights into the Role HydE Plays in Dithiomethylamine Biosynthesis. Biochemistry 2015, 54 (9), 1807–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Bersuker IB Pseudo-Jahn–Teller Effect—A Two-State Paradigm in Formation, Deformation, and Transformation of Molecular Systems and Solids. Chem. Rev 2013, 113, 1351–1390. [DOI] [PubMed] [Google Scholar]
- (32).Sharma A; Roemelt M; Reithofer M; Schrock RR; Hoffman BM; Neese F EPR/ENDOR and Theoretical Study of the Jahn–Teller-Active [HIPTN3N]MoVL Complexes (L = N−, NH). Inorg. Chem 2017, 56 (12), 6906–6919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).McNaughton RL; Roemelt M; Chin JM; Schrock RR; Neese F; Hoffman BM Experimental and Theoretical EPR Study of Jahn-Teller Active [HIPTN3N]MoL Complexes (L = N2, CO, NH3). J. Am. Chem. Soc 2010, 132 (25), 8645–8656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Neese F The ORCA program system. Wiley Interdiscip. Rev.: Comput. Mol. Sci 2012, 2 (1), 73–78. [Google Scholar]
- (35).Hanwell MD; Curtis DE; Lonie DC; Vandermeersch T; Zurek E; Hutchison GR Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminf 2012, 4, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Becke AD Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A: At., Mol., Opt. Phys 1988, 38 (6), 3098–3100. [DOI] [PubMed] [Google Scholar]
- (37).Perdew JP Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B: Condens. Matter Mater. Phys 1986, 33 (12), 8822–8824. [DOI] [PubMed] [Google Scholar]
- (38).Lee C; Yang W; Parr RG Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B: Condens. Matter Mater. Phys 1988, 37 (2), 785–789. [DOI] [PubMed] [Google Scholar]
- (39).Schäfer A; Horn H; Ahlrichs R Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys 1992, 97 (4), 2571–2577. [Google Scholar]
- (40).The PyMOL Molecular Graphics System, Version 2.0; Schrödinger, LLC. [Google Scholar]
- (41).Benjdia A; Heil K; Barends TRM; Carell T; Schlichting I Structural insights into recognition and repair of UV-DNA damage by Spore Photoproduct Lyase, a radical SAM enzyme. Nucleic Acids Res. 2012, 40 (18), 9308–9318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Nicolet Y; Amara P; Mouesca JM; Fontecilla-Camps JC Unexpected electron transfer mechanism upon AdoMet cleavage in radical SAM proteins. Proc. Natl. Acad. Sci. U. S. A 2009, 106 (35), 14867–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Vey JL; Drennan CL Structural Insights into Radical Generation by the Radical SAM Superfamily. Chem. Rev 2011, 111 (4), 2487–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Vey JL; Yang J; Li M; Broderick WE; Broderick JB; Drennan CL Structural basis for glycyl radical formation by pyruvate formate-lyase activating enzyme. Proc. Natl. Acad. Sci. U. S. A 2008, 105 (42), 16137–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Dinis P; Suess DLM; Fox SJ; Harmer JE; Driesener RC; De La Paz L; Swartz JR; Essex JW; Britt RD; Roach PL X-ray crystallographic and EPR spectroscopic analysis of HydG, a maturase in [FeFe]-hydrogenase H-cluster assembly. Proc. Natl. Acad. Sci. U. S. A 2015, 112 (5), 1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Broyde SB; Stellman SD; Wartell RM The A and B conformations of DNA and RNA subunits. Potential energy calculations for dGpdC. Biopolymers 1975, 14 (12), 2625–2637. [DOI] [PubMed] [Google Scholar]
- (47).Lepore BW; Ruzicka FJ; Frey PA; Ringe D The x-ray crystal structure of lysine-2,3-aminomutase from Clostridium subterminale. Proc. Natl. Acad. Sci. U. S. A 2005, 102 (39), 13819–13824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).We were unable to find a structure of SAM crystallized by itself.
- (49).Kampmeier JA Regioselectivity in the Homolytic Cleavage of S-Adenosylmethionine. Biochemistry 2010, 49 (51), 10770–10772. [DOI] [PubMed] [Google Scholar]
- (50).Miller SA; Bandarian V Analysis of Electrochemical Properties of S-Adenosyl-l-methionine and Implications for Its Role in Radical SAM Enzymes. J. Am. Chem. Soc 2019, 141 (28), 11019–11026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Saeva FD; Morgan BP Mechanism of one-electron electrochemical reductive cleavage reactions of sulfonium salts. J. Am. Chem. Soc 1984, 106 (15), 4121–4125. [Google Scholar]
- (52).Wang X; Saeva FD; Kampmeier JA Photosensitized Reduction of Sulfonium Salts: Evidence for Nondissociative Electron Transfer. J. Am. Chem. Soc 1999, 121 (18), 4364–4368. [Google Scholar]
- (53).Donck S; Baroudi A; Fensterbank L; Goddard J-P; Ollivier C Visible-Light Photocatalytic Reduction of Sulfonium Salts as a Source of Aryl Radicals. Adv. Synth. Catal 2013, 355 (8), 1477–1482. [Google Scholar]
- (54).Wedler HB; Wendelboe P; Tantillo DJ; Power PP Second order Jahn–Teller interactions at unusually high molecular orbital energy separations. Dalton T 2020, 49 (16), 5175–5182. [DOI] [PubMed] [Google Scholar]
- (55).Ogurtsov I; Gorinchoy N; Balan I Vibronic origin of the H3O metastability. J. Mol. Struct 2007, 838 (1), 107–111. [Google Scholar]
- (56).Kampmeier JA; Hoque AKMM; Saeva FD; Wedegaertner DK; Thomsen P; Ullah S; Krake J; Lund T Regioselectivity in the Reductive Bond Cleavage of Diarylalkylsulfonium Salts: Variation with Driving Force and Structure of Sulfuranyl Radical Intermediates. J. Am. Chem. Soc 2009, 131 (29), 10015–10022. [DOI] [PubMed] [Google Scholar]
- (57).McNaughton RL; Chin JM; Weare WW; Schrock RR; Hoffman BM EPR Study of the Low-Spin [d3; S = 1/2], Jahn-Teller-Active, Dinitrogen Complex of a Molybdenum Trisamidoamine. J. Am. Chem. Soc 2007, 129 (12), 3480–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Gunderson WA; Suess DL; Fong H; Wang X; Hoffmann CM; Cutsail GE III; Peters JC; Hoffman BM Free H2 Rotation vs Jahn-Teller Constraints in the Nonclassical Trigonal (TPB)Co-H2 Complex. J. Am. Chem. Soc 2014, 136 (42), 14998–5009. [DOI] [PubMed] [Google Scholar]
- (59).Nicolet Y; Rubach JK; Posewitz MC; Amara P; Mathevon C; Atta M; Fontecave M; Fontecilla-Camps JC ”XRay Structure of the [FeFe]-Hydrogenase Maturase HydE from Thermotoga maritima. J. Biol. Chem 2008, 283, 18861–18872. [DOI] [PubMed] [Google Scholar]
- (60).Rohac R; Amara P; Benjdia A; Martin L; Ruffié P; Favier A; Berteau O; Mouesca J-M; Fontecilla-Camps JC; Nicolet Y ”Carbon-sulfur bond-forming reaction catalysed by the radical SAM enzyme HydE,. Nat. Chem 2016, 8, 491–500. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.