

Summary
DNA replication complexes (replisomes) routinely encounter proteins and unusual nucleic acid structures that can impede their progress. Barriers can include transcription complexes and R-loops that form when RNA hybridizes with complementary DNA templates behind RNA polymerases. Cells encode several RNA polymerase and R-loop clearance mechanisms to limit replisome exposure to these potential obstructions. One such mechanism is hydrolysis of R-loops by ribonuclease HI (RNase HI). Here, we examine the cellular role of the interaction between Escherichia coli RNase HI and the single-stranded DNA-binding protein (SSB) in this process. Interaction with SSB localizes RNase HI foci to DNA replication sites. Mutation of rnhA to encode an RNase HI variant that cannot interact with SSB but that maintains enzymatic activity (rnhAK60E) eliminates RNase HI foci. The mutation also produces a media-dependent slow-growth phenotype and an activated DNA damage response in cells lacking Rep helicase, which is an enzyme that disrupts stalled transcription complexes. RNA polymerase variants that are thought to increase or decrease R-loop accumulation enhance or suppress, respectively, the growth phenotype of rnhAK60E rep::kan strains. These results identify a cellular role for the RNase HI/SSB interaction in helping to clear R-loops that block DNA replication.
Abbreviated summary
DNA replication complexes routinely encounter impediments such as transcription complexes and R-loops that form when RNA hybridizes with complementary DNA templates behind RNA polymerases. We show that interaction between RNase HI and single-stranded DNA-binding protein localizes RNase HI to help clear R-loops that block DNA replication.
Introduction
Replisomes are protein complexes that catalyze high fidelity DNA replication at speeds approaching 1,000 bp/sec in bacteria (Chandler et al.,1975; O’Donnell et al., 2013). During the replication process replisomes encounter numerous impediments to their progress including protein/DNA complexes, non-duplex nucleic acid structures, and chromosomal damage (Mirkin & Mirkin, 2007). To overcome these obstacles, cells have evolved several systems that support replication on imperfect genomic templates. These include enzymes that dissociate protein/DNA complexes and resolve unusual nucleic acid structures, repair pathways that mitigate damaged DNA, and proteins that restructure collapsed replication forks.
RNA polymerase (RNAP) and transcription-dependent nucleic acid structures called R-loops are common barriers to replisome progress (Aguilera & Garcia-Muse, 2012; Helmrich et al., 2013). R-loops are structures that form when a nascent RNA hybridizes with the DNA template behind RNAP (Westover et al., 2004). Bacterial replisomes moves at rates that are ~10-20-times faster than RNAP and can encounter R-loops and/or RNAP from both head-on and co-directional collisions in bacteria (Merrikh et al., 2012). Replication-transcription collisions occur even in eukaryotes, where replication forks move at similar rates to RNAP and most replication and transcription reactions are temporally and spatially separated (Azvolinsky et al., 2009; Helmrich et al., 2011). Head-on collisions can result in replication fork arrest and activation of DNA-damage-dependent recombination (French, 1992; Deshpande & Newlon, 1996; Vilette et al., 1996; Prado & Aguilera, 2005; Mirkin & Mirkin, 2007; Wang et al., 2007). Replisome collisions with RNAP or R-loops can also lead to DNA breaks, genome rearrangements, increased mutagenesis, and activation of DNA damage responses (Huertas & Aguilera, 2003; Li & Manley, 2005; Tuduri et al., 2009; Wahba et al., 2011). These events can create double-strand DNA breaks (DSBs) when a switch in the DNA replication template forms a ssDNA gap that escapes repair prior to the next round of replication (Kuzminov, 2001; Pomerantz & O’Donnell, 2010).
Cells encode redundant pathways to minimize replisome encounters with RNAPs and R-loops and to repair the damage when such collisions occur. These pathways rely on DNA helicases, transcription-associated factors, nucleases, and DNA repair enzymes. One such contributor is ribonuclease HI (RNase HI), an enzyme that hydrolyzes RNA within RNA:DNA hybrids. Classical roles for RNase HI in bacteria include degradation of RNA primers used during DNA replication and suppression of origin-independent chromosomal DNA replication (Itoh & Tomizawa, 1980a; Ogawa & Okazaki, 1980; Ogawa et al., 1984; Alberts, 1987). A more recent role for RNase HI in removing R-loops that block replication forks has been identified (Itoh & Tomizawa, 1980b;Dutta et al., 2011; Merrikh et al., 2012). Genetic relationships link RNase HI with proteins that promote replication fork progression, that repair or restructure replication forks following R-loop induced DNA damage, or that prevent R-loop accumulation (Drolet et al., 1995; Hong et al., 1995; Itaya & Crouch, 1991; Hraiky et al., 2000; Harinarayanan & Gowrishankar, 2003; Sandler, 2005).
Through biochemical and structural studies, a stimulatory interaction formed between E. coli RNase HI and the single-stranded DNA binding protein (SSB) has been identified (Petzold et al., 2015). Stimulation requires docking of the intrinsically-disordered C-terminus of SSB (SSB-Ct) into a binding pocket on RNase HI. This binding mechanism is shared with several other proteins that also form complexes with SSB via the SSB-Ct (Shereda et al., 2008). In some cases, interactions with SSB have been shown to localize its interaction partners to DNA replication forks (Sun & Godson, 1996; Shereda et al., 2008; Marceau et al., 2011).
To probe possible cellular roles of the RNase HI/SSB interaction, we have compared the localization and activity of RNase HI to that of an RNase HI variant that has lost the ability to interact with SSB but that retains normal nuclease activity levels (RNase HI K60E (Petzold et al., 2015)). To do this we created an RNase HI fluorescent fusion protein that forms foci in the cell and colocalizes with a subunit of the DNA replication machinery in E. coli. In contrast, the RNase HI K60E fusion variant failed to form foci in E. coli. Thus, RNase HI localizes to sites of DNA replication in vivo via the interaction with SSB. Strains that substitute the rnhA gene (encodes RNase HI) with rnhAK60E have normal activities in RNA primer processing and in suppressing origin-independent DNA replication. However, a strain combining rnhAK60E with a mutation that inactivates the Rep DNA helicase, an enzyme that helps to promote replisome movement through transcription complexes (Boubakri et al., 2010; Guy et al., 2009), displays a plating deficiency on rich medium and an activated DNA damage response. rpoB mutations that produce RNAP variants that are thought to increase or decrease R-loop levels (Kogoma, 1994) enhance or suppress, respectively, the plating deficiency of rnhAK60E rep::kan cells. These data lead to a model in which interaction with SSB mediates RNase HI removal of transcription-dependent R-loop obstacles by localizing the enzyme to DNA replication sites.
Results
Interaction with SSB localizes RNase HI to sites of DNA replication in E. coli
SSB is concentrated at replication sites in bacterial cells through its binding to the extended tracts of ssDNA present at DNA replication forks (Meyer & Laine, 1990; Reyes-Lamothe et al., 2010; Reyes-Lamothe, 2012; Marceau, 2012). In some, but not all, instances, direct interaction with SSB also localizes SSB’s interaction partner proteins to DNA replication forks (Lecointe et al., 2007; Costes et al., 2010; Bentchikou et al., 2015). To determine whether RNase HI is localized to DNA replication sites in E. coli, we created a strain in which the chromosomal rnhA locus was replaced with an RNase HI-YPet fluorescent fusion protein and examined RNase HI-YPet focus formation using fluorescence microscopy. RNase HI-YPet foci were found in ~80% of the E. coli cells examined, with the majority of cells having either one or two foci (Figure 1A & B). This pattern is strikingly similar to that observed for several fluorescently-tagged DNA replication proteins (Reyes-Lamothe et al., 2008).
Figure 1. SSB-mediated localization of RNase HI to the replication fork.

Fluorescence microscopy studies of E. coli strains expressing the fluorescent fusion proteins RNase HI-YPet and β-clamp-mCherry. (A) Representative images showing RNase HI-YPet (left), β-clamp-mCherry (middle) and a merged image showing the overlap of RNase HI-YPet and β-clamp-mCherry (right) in strain VV11. Digital contrast enhancement was used for presentation purposes. Scale bar in the right image is 2 μm. (B) Detectable RNase HI-YPet and β-clamp-mCherry foci per cell are plotted as the frequency for the cell population. (C) Radial distribution function for RNase HI-YPet and β-clamp-mCherry representing colocalization (blue line). As comparison, g(r) is plotted for a set of randomly distributed spots in cells (grey line). Inset shows the distribution of nearest-neighbor distances between spots of RNase HI-YPet and β-clamp-mCherry. (D) Representative fluorescent image of showing RNase HI K60E-YPet fusion protein distributed throughout strain VV08. Imaging conditions and digital contrast enhancement used were as in 1B. Scale bar is 2 μm.
To determine whether RNase HI-YPet foci localize to DNA replication forks, the localization of RNase HI-YPet foci and a commonly used replication site marker, mCherry-fused DNA Pol III β-clamp (Reyes-Lamothe et al., 2008; Liu et al., 2010; Reyes-Lamothe et al., 2012; Moolman et al., 2014), were simultaneously measured (Figure 1A). Consistent with RNase HI-YPet foci forming at DNA replication forks, the median distance between a spot of RNase HI-YPet and the nearest mCherry-β-clamp spot is 202 nm (Figure 1C, inset). These values are slightly higher than those previously reported for replisomal components with the ε subunit of DNA Pol III and β-clamp, which have a median distance of 128 nm (Soubry et al., 2019). Consequently, we further tested for co-localization between RNase HI-YPet and β-clamp-mCherry by comparing the distribution of distances to a random distribution of spots in cells, represented by the radial distribution function g(r). Values of g(r) over 1 at short radial distances indicate that the two foci of the proteins overlap far more frequently than randomly distributed spots, supporting co-localization between RNase HI-YPet and β-clamp-mCherry (Figure 1C). In addition, we observed that 16.2% carried spots for β-clamp-mCherry but no RNase HI-YPet, compared with 2.4% of cases for cells with RNase HI-YPet spots but no β-clamp-mCherry, suggesting that RNaseHI is not present at the replication fork at all times or may be present at low levels that are not detected by fluorescence microscopy in some instances.
To better understand the activity of RNase HI, we used single-molecule microscopy to measure the RNase HI copy number per cell and determine the fraction of RNase HI molecules found in foci (Figure S1). To do this, we modified E. coli strain AB1157 to encode RNase HI-mNeonGreen at the rnhA locus. mNeonGreen was used in place of Ypet as it is currently the brightest monomeric fluorescent protein in use, which maximized detection of single molecules (Shaner et al., 2013). The intensity of a single molecule of RNase HI-mNeonGreen was estimated by measuring the last bleaching step in time traces of foci (Figure S1D & E). Dividing the integrated intensity of a cell by the value of a single molecule, we estimated an average of 82 ± 40 RNase HI molecules per E. coli cell (Figure S1F), of which 20 ± 13% were localized in foci (Figure S1). These data suggest that in many instances there are multiple copies of RNase HI localized in foci. In addition, a significant number of RNase HI molecules are available in the diffusive pool.
Prior structural and biochemical experiments defined the SSB-binding site on RNase HI and identified a point mutation in RNase HI (K60E) that eliminated interaction without altering nuclease activity in vitro (Petzold et al., 2015). The cellular localization of RNase HI K60E-YPet was examined next to determine whether RNase HI-YPet focus formation requires interaction with SSB. Unlike RNaseHI-YPet, foci were not observed for RNase HI K60E-YPet and the fluorescence signal of the variant was distributed throughout the cell (Figure 1D). This result indicates that RNase HI/SSB complex formation is necessary for recruitment and/or retention of RNase HI at DNA replication sites in E. coli.
SSB-mediated localization of RNase HI is not required to suppress origin-independent replication or to degrade lagging-strand Okazaki fragment primers
RNase HI has noted activities in three major cellular processes in E. coli: (1) suppression of origin-independent “constitutive stable DNA replication” (cSDR), (2) degradation of lagging-strand RNA primers used during DNA replication, and (3) removal of R-loop replication barriers. To test for possible roles of SSB-mediated localization of RNase HI in each of these activities, we created an E. coli strain with a mutated rnhA locus that encodes for the RNase HI K60E variant (rnhAK60E) and compared its phenotype to otherwise isogenic rnhA+ and rnhA::cat E. coli strains (Table S1). E. coli with the rnhAK60E mutation were phenotypically indistinguishable from rnhA+ and rnhA::cat cells in terms of growth rate, cell morphology and rich medium plating efficiency (Figures 2, 3 & S2). Given the lack of a phenotypic difference for the rnhA point mutation, we next combined the rnhAK60E mutation with mutations in other genes that have established genetic relationships with rnhA to test whether loss of RNase HI/SSB complex formation affects the pathways in which RNase HI is involved.
Figure 2. The rnhAK60E rep::kan strain is sensitive to growth on rich medium plates.
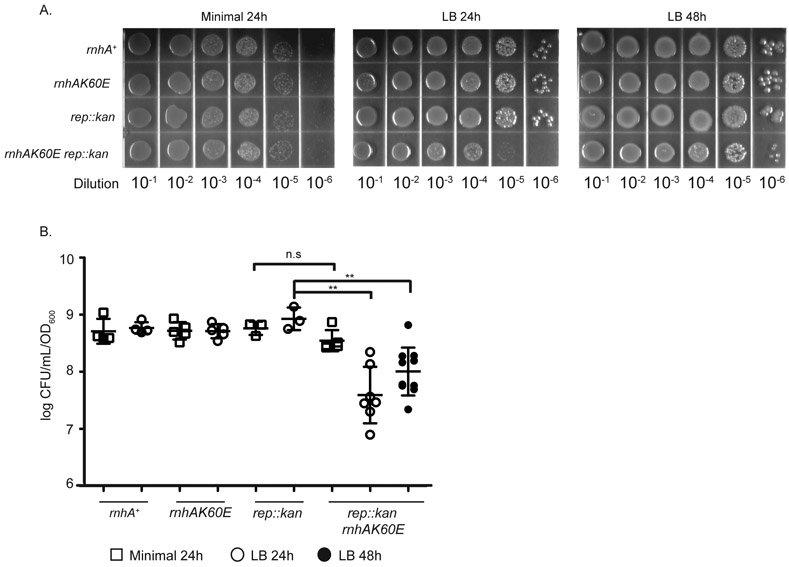
(A) Dilutions of overnight cultures grown in minimal medium (56/2) and plated on minimal (left) or LB media (middle and right). Plates were incubated at 37°C for 24 or 48 hours. The images are representative of plating experiments performed in triplicate. Strains are CP65, CP58, CP84, and CP86 from top to bottom. (B & C) The CFU/mL of each strain (normalized to OD600) is plotted from overnight cultures diluted and plated on minimal (squares) or LB (circles) media. Colonies were quantitated after growth at 37°C for 24 or 48 hours. Each symbol is a single culture and the mean CFU/mL for each strain is represented by a black line. Strains are CP65, CP58, CP84, and CP86 from left to right. Error bars indicate the standard deviation. ** = p-value < 0.005 and n.s = p-value > 0.05 using two-tailed t-test.
Figure 3. The rnhAK60E rep::kan strain has elevated SOS levels in rich medium.

(A) The relative fluorescent units (RFU) from plasmid-borne GFP driven by a recN promoter is plotted for each strain at mid-log phase and normalized to CFU/mL at the time of data collection. Bars represent the mean RFU/CFU/mL from biological and technical replicates of strains were grown in minimal medium (56/2) and LB. Error bars display the standard deviation of the mean. Mean values are written above each bar. Strains in this figure are CP127, CP128, CP129, and CP126 (from left to right). (B) Representative phase contrast images from strains grown in minimal medium (left) or LB (right). The strains were grown to early log phase and subsequently spotted on 2% agarose pads for imaging using a Nikon Eclipse Ti microscope equipped with a Photometrics CoolSNAP HQ2 charge-coupled-device (CCD) camera. (C) The cell lengths captured by MATLAB of microscope images. Cells were grown in minimal medium or LB and treated as in (B).
We first examined whether the rnhAK60E mutation impacted RNase HI suppression of cSDR in E. coli. In cSDR, R-loops are processed by RecA, DNA polymerase I and the primosome to allow for replication initiation away from oriC. RNase HI nuclease activity regulates this process by removing R-loops before the replisome can be loaded (Tokio Kogoma, 1978; von Meyenburg et al., 1987). Therefore the inactivation of rnhA preserves R-loops and can rescue cell growth in strains carrying otherwise lethal mutations in the oriC-replication initiation pathway (e.g. dnaA loss of function mutants) (Frey et al., 1981; Kogoma & von Meyenburg, 1983; de Massy et al., 1984; Lindahl & Lindahl, 1984; von Meyenburg et al., 1987; Carr & Kaguni, 1996). To test whether abrogation of SSB-mediated RNase HI localization facilitates cSDR similarly to rnhA::cat, we introduced the rnhAK60E allele into dnaA46(ts) cells. Similarly to rnhA+ dnaA46(ts), the rnhAK60E dnaA46(ts) strain was unable to grow under non-permissive conditions (42 °C), whereas the rnhA::cat dnaA46(ts) strain was able to grow at 42 °C (Figure S3). This result indicates that the RNase HI K60E variant maintains sufficient activity to remove R-loops required for cSDR. Thus, SSB-mediated localization of RNase HI is not required for inhibiting R-loop dependent replication initiation.
We next tested whether SSB-mediated RNase HI localization was important for degrading RNA primers used during canonical DNA replication. The nuclease activities of RNase HI and DNA polymerase I work together to remove RNA primers that initiate lagging-strand Okazaki fragments (Ogawa & Okazaki, 1984; Kitani et al., 1985; Crouch, 1990). Inactivation of rnhA leads to media- and temperature-dependent impaired growth of an E. coli strain carrying the polA12(ts) allele, which encodes a DNA polymerase I variant that has strongly reduced 5’-3’ exonuclease and polymerase activities at 42 °C (Uyemura & Lehman, 1976; Joyce et al., 1985;). To determine whether binding to SSB is required for primer-degradation by RNase HI, growth of a strain combining the rnhAK60E mutation with polA12(ts) was compared to rnhA+ polA12(ts) and rnhA::cat polA12(ts) strains. Growth of rnhAK60E polA12(ts) cells was indistinguishable from rnhA+ polA12(ts) cells at 30 or 42 °C in rich or minimal media (Figure S4). In contrast, the rnhA::cat polA12(ts) strain displayed media- and temperature-dependent growth defects as previously reported (Uyemura & Lehman, 1976; Joyce et al., 1985;). These data indicate that SSB-mediated RNase HI localization is not required for Okazaki fragment RNA primer processing.
Genetic interactions of rnhAK60E with DNA helicases
To examine the role of the RNase HI/SSB interaction in additional replication and repair processes, we tested the effects of combining rnhAK60E with deletions in several DNA replication/repair genes: rep, recBCD, recG, uvrD, or dinG (Table 1). These were chosen based on the reported synthetic lethal effects of rnhA deletion with rep (Sandler, 2005), recBCD (Itaya & Crouch, 1991; Kogoma et al., 1993; Hong et al., 1995; Kogoma, 1997b;), and recG (Hong et al., 1995) deletions and the proposed roles of UvrD and DinG helicases in DNA replication fork maintenance or repair (Kornberg & Baker, 1992; Sandler et al., 1996; Michel, 2005; Srivatsan et al., 2010; Boubakri et al., 2010; Dimude et al., 2015). Inactivation of several of these genes along with rnhA increases the possibility of collisions between replisomes and transcription-dependent R-loops and/or diminishes the capacity of cells to repair the resulting DNA damage from replication fork collapse (Kogoma et al., 1994; Hraiky et al., 2000; Wahba et al., 2011; Merrikh et al., 2012; Dimude et al., 2015).
Table 1.
Co-transduction analysis to determine the synthetic lethality of helicase mutations with rnhAK60E
| Donor Strain (relevant genotype) |
Recipient Strain (relevant genotype) |
Results (Screen/Selection) |
|
|---|---|---|---|
| (a) Rep | SS9364 (rep::kan ilv+) | CP70 (rnhA+, cat ilv-500::Tn10 ) | 86/101 (Kanr /ilv+) |
| SS9364 (rep::kan ilv+) | CP60 (rnhAK60E, cat ilv-500::Tn10 ) | 54/77 (Kanr /ilv+) | |
| SS9364 (rep::kan ilv+) | SS9180 (rnhA::cat ilv-500::Tn10) | 0/57 (Kanr /ilv+) | |
| CP165 (rnhA::cat zae502::Tn10) | CP65 (rnhA+) | 17/56 (Cmr/Tcr) | |
| CP165 (rnhA::cat zae502::Tn10) | CP84 (rep::kan) | 21/54 (Cmr/Tcr) | |
| (b) RecBCD | SS6046 (recBCD::cat proA+) | JC13509 (rnhA+ proA−) | 30/30 (Cmr/proA+) |
| SS6046 (recBCD::cat proA+) | CP62 (rnhAK60E proA::kan) | 28/30 (Cmr/proA+) | |
| SS6046 (recBCD::cat proA+) | SS10032 (rnhA::cat del(proA)kan) | 0/41 (UVS /proA+)* | |
| SS10032 (rnhA::cat del(proA)kan) | JC13509 (rnhA+) | 6/46 (Cmr/Kanr) | |
| SS10032 (rnhA::cat del(proA)kan) | SS7329 (recB270(ts) recC271(ts)) | 0/42 (Cmr /Kanr)** | |
| (c) RecG | CP79 (recG(kan ins) zic-4901::Tn10) | CP63 (rnhA+, cat ) | 52/54 (Kanr/Tcr) |
| CP79 (recG(kan ins) zic-4901::Tn10) | CP54 (rnhAK60E, cat) | 44/54(Kanr/Tcr) | |
| CP79 (recG(kan ins) zic-4901::Tn10) | CP154 (rnhA::cat) | 0/53 (Kanr/Tcr) | |
| CP165 (rnhA::cat zae502::Tn10) | CP64 (recG(kan ins)) | 0/12 (Cmr/Tetr) | |
| (d) UvrD | CP95 (uvrD::kan fadAB101::Tn10) | CP63 (rnhA+, cat ) | 23/103 (Kanr/Tcr) |
| CP95 (uvrD::kan fadAB101::Tn10) | CP54 (rnhAK60E, cat) | 27/103 (Kanr/Tcr) | |
| CP95 (uvrD::kan fadAB101::Tn10) | SS1651 (rnhA339::cat) | 0/104 (Kanr/Tcr) | |
| CP165 (rnhA::cat zae502::Tn10) | CP88 (uvrD::kan) | 0/58 (Cmr/Tcr) | |
| (e) DinG | CP96 (dinG::kan zbi-29::Tn10) | CP63 (rnhA+, cat ) | 23/31 (Kanr/Tcr) |
| CP96 (dinG::kan zbi-29::Tn10) | CP54 (rnhAK60E, cat) | 27/44 (Kanr/Tcr) | |
| CP96 (dinG::kan zbi-29::Tn10) | CP154 (rnhA::cat) | 30/48 (Kanr/Tcr) |
PCR to confirm rnhA locus and test recBCD genotype with UV sensitivity
Selection at 30°C, screen at 42°C
We first measured the co-transduction frequencies of rep::kan, recBCD::cat, or recG::kan into three strains with different rnhA backgrounds: rnhA+, rnhAK60E, or rnhA::cat. rep::kan, recBCD::kan, and recG::cat each transduced with similar frequencies into rnhA+ and rnhAK60E strains whereas they failed to transduce into rnhA::cat cells (Table 1). Another observation to note was that the rnhA::cat rep::kan synthetic lethality was dependent on transduction direction, a trait not observed with other synthetic lethal pairs (Table 1). This could be due to background differences between the rep::kan strain and the rnhA strains used in our study.
We expanded our screen to examine deletions of two other helicase genes: uvrD::kan and dinG::kan. The uvrD::kan rnhA::cat combination was synthetically lethal whereas the dinG::kan rnhA::cat, uvrD::kan rnhAK60E, and dinG::kan rnhAK60E strains were viable and had similar co-transduction frequencies to those observed with the rnhA+ recipient (Table 1). Synthetic lethality of the uvrD::kan rnhA::cat is interesting considering recent data demonstrating that UvrD has a direct role in promoting replication fork movement past transcription collisions whereas DinG operates more indirectly and could reduce the chance of collisions (Hawkins et al., 2019).
These results indicate that the rnhAK60E mutation is not synthetically lethal with rep, recBCD, recG, uvrD, or dinG deletions (Table 1). Thus, loss of RNase HI localization conferred by the rnhAK60E mutation is not synonymous with a loss of RNase HI activity when combined with mutations in DNA replication and repair helicases. Nonetheless, the viability of the rnhAK60E variant in these helicase mutant strains allowed us to probe the role of the RNase HI/SSB interaction under other cellular conditions.
The rnhAK60E rep::kan strain is sensitive to rich medium and is induced for SOS
Although the co-transduction experiments did not reveal synthetic lethal combinations between rnhAK60E and several different DNA repair gene deletions, a media-dependent growth defect was detected specifically with the rnhAK60E rep::kan strain (Figure 2). The rnhAK60E rep::kan strain plated with the same efficiency as wild-type, rnhAK60E, or rep::kan strains on minimal medium (normalized to OD600 (CFU/mL/OD)) but the double mutant has an approximately 10-fold reduced plating efficiency on LB after 24 hours relative to the other strains (Figure 2). The lower plating efficiency on LB improves somewhat after 48 hours, indicating that the phenotype is due to slow growth rather than cell death (Figure 2). Interestingly, the slow growth defect of rnhAK60E rep::kan cells is not obvious in liquid growth curves (Figure S2). Slow growth was unique to the rnhAK60E rep::kan strain since the other strains that were synthetic lethal with rnhA::cat (uvrD::kan, recG::kan, recBCD::cat) plated with indistinguishable efficiencies on minimal and LB plates with the rnhAK60E mutation (Figure S5).
With the increased frequency of DNA replication and other stresses that arise in rich nutrient conditions, rich-medium dependent growth defects can be indicative of dysfunctional genome maintenance pathways (Boubakri et al., 2010; Srivatsan et al., 2010). Such problems can also lead to an increase in DNA damage that induces the SOS response, a survival mechanism used by E. coli to regulate the expression of DNA repair genes (Janion, 2008; Walker et al., 2000; Michel, 2005). To test whether loss of RNase HI replication fork localization and/or Rep helicase activity induces the SOS response in E. coli, the strains were transformed with a reporter plasmid that has a recN promoter-GFP fusion (Chen et al., 2015) and GFP levels were measured during exponential growth. recN is among the first group of genes to be induced during SOS, making its expression an indicator of SOS status (Finch et al., 1985; Rostas et al., 1987).
The SOS level of the rnhAK60E strain was indistinguishable from wild-type cells in minimal and rich media whereas the rep::kan strain had ~3.5-fold higher GFP levels in both minimal and rich media (Figure 3). Interestingly, combining the rnhAK60E and rep::kan mutations resulted in the same GFP levels as the rep::kan strain in minimal medium but the GFP levels were 3.3-fold higher than rep::kan in rich medium. The rich medium-specific increase in SOS response for rnhAK60E rep::kan cells correlates with reduced plating efficiency for the strain on LB.
Cell morphologies of the strains were examined to determine whether they demonstrated signs of cell filamentation, a phenotypic consequence for the SOS response. rnhAK60E and rep::kan cells had lengths that were similar to wild-type E. coli, however, the rnhAK60E rep::kan cells were more frequently filamented (Figure 3B & C). In minimal medium, rnhAK60E rep::kan cells were slightly elongated in comparison to wild-type cells whereas growth in rich medium led to much more extreme cell filamentation (p-value < 0.001) (Figure 3B & C). These data show that abrogation of SSB-mediated RNase HI localization to the replication fork along with a loss of Rep helicase activity leads to an increase in DNA damage stress.
The rnhAK60E rep::kan phenotype is related to transcription-dependent R-loop removal.
RNase HI can remove transcription-dependent R-loops and the Rep helicase helps the replication fork progress through protein obstacles including stalled transcription complexes (Kogoma, 1978; Itoh & Tomizawa, 1980b; von Meyenburg et al., 1987; Guy et al., 2009; Boubakri et al., 2010). With the result that RNase HI is localized to the replication fork through interaction with SSB, it is possible that RNase HI and Rep collaborate to promote replisome progression when faced with R-loops and transcription complexes (Drolet et al., 1995; Hraiky et al., 2000; Boubakri et al., 2010; Dutta et al., 2011; McGlynn et al., 2012). To further test this hypothesis, we examined whether changes in RNAP activity impact the growth phenotype of rnhAK60E rep::kan cells. Two RNAP β subunit mutations were chosen for the analysis: rpoB2 is defective in termination whereas rpoB8 has a slower elongation rate than wild-type and is prone to termination (Jin et al., 1988; Jin et al., 1992). A previous study showed that combining an rnhA deletion with rpoB2 leads to a plating defect on rich medium and elevated SOS whereas combining an rnhA deletion with rpoB8 reduces SOS levels (Kogoma, 1994). These effects are thought to arise from rpoB2 and rpoB8 mutant RNAPs generating more or fewer R-loops, respectively, than wild-type RNAP, and with the abundant R-loops in the rpoB2 rnhA- cells acting as barriers that slow replication (Kogoma, 1994).
Consistent with the model for synergy between RNAP and RNase HI/Rep, a rnhAK60E rep::kan rpoB2 strain had a worsened plating efficiency relative to rnhAK60E rep::kan cells on both LB and minimal media plates (Figure 4). Even after a 48 hour incubation period the rnhAK60E rep::kan rpoB2 CFUs on LB did not increase (Figure 4). In contrast, the rnhAK60E rep::kan rpoB8 strain had the same plating efficiency as wild-type cells on minimal medium and LB, indicating that the rpoB8 mutation suppressed LB growth sensitivity in rnhAK60E rep::kan cells. When either rnhAK60E or rep::kan were combined individually with the rpoB point mutations there were no significant changes in the plating efficiency relative to rpoB2 or rpoB8 cells (Figure S6). These data demonstrate that an RNAP variant that is thought to produce high levels of R-loops exacerbates the impact of losing RNase HI localization and Rep helicase activity whereas an RNAP variant that reduces cellular R-loop levels counteracts the mutations.
Figure 4. The plating efficiency of rnhAK60E rep::kan rpoB mutants on minimal and rich media.
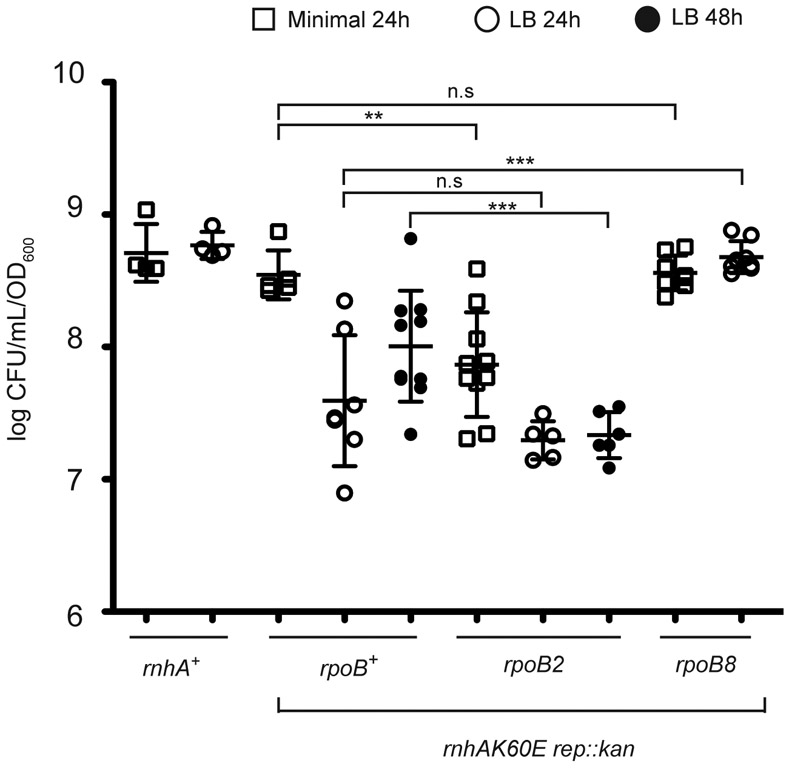
The CFU/mL of each strain (normalized to OD600) is plotted from overnight cultures diluted and plated on minimal (squares) or LB (circles) media. Colonies were quantitated after growth at 37°C for 24 or 48 hours. Each symbol is a single culture and the mean CFU/mL for each strain is represented by a black line. The strains are CP65, CP86, CP119, and CP124 from left to right. The error bars indicate the standard deviation. Significant difference levels (p-values) were determined between rnhAK60E rep::kan strains +/− rpoB mutants grown in the same medium using two-tailed t-test. * = p-value < 0.05; ** = p-value < 0.005; *** = p-value < 0.0005; n.s = p-value > 0.05
Discussion
E. coli RNase HI forms a direct interaction with SSB that stimulates RNase HI enzymatic activity in vitro (Petzold et al., 2015). In this report, the cellular roles of the RNase HI/SSB interaction have been examined. An E. coli RNase HI fluorescent fusion protein forms foci in cells whereas an enzymatically-active variant that cannot interact with SSB (RNase HI K60E) does not form foci. RNase HI foci colocalize with a component of the replisome, consistent with SSB-dependent accumulation of the enzyme at DNA replication sites in E. coli. The RNase HI K60E variant supported RNase HI-mediated suppression of cSDR and lagging-strand RNA primer degradation, indicating that localization was not required for these cellular functions. In contrast, cells encoding RNase HI K60E displayed a media-dependent plating defect and an activated SOS response when the gene encoding the Rep DNA helicase was deleted. This phenotype was linked to a deficiency in resolving transcription-derived R-loops by examining the effects of RNAP mutations that alter R-loop levels (Jin et al., 1988; Jin et al., 1992) on rnhAK60E rep::kan cells. Adding rpoB2, which is thought to increase the abundance of R-loops (Kogoma, 1994), further increased the plating defect of rnhAK60E rep::kan cells whereas adding rpoB8, which is thought to decrease R-loop abundance, suppressed the growth phenotype. These results support a model in which SSB-dependent replication fork localization of RNase HI assists in replication progression through transcription-derived R-loops.
Roles for RNase HI in resolving replication/transcription conflicts have been proposed, but the precise mechanisms that allow RNase HI to target specific R-loop challenges to replication fork progression have not been defined (Drolet et al., 1995; Hraiky et al., 2000; Li & Manley, 2005; Tuduri et al., 2009; El Hage et al., 2010; Gan et al., 2011; Dutta et al., 2011; Houlard et al., 2011; Wahba et al., 2011). Our results support a model in which binding to SSB recruits RNase HI to sites of replication in E. coli, which positions the enzyme to degrade R-loop impediment. Several other cellular factors that resolve R-loop/RNAP blockages similarly rely on protein interactions for recruitment to their site of action to mediate replication progress. These include Rep, DinG, and UvrD helicases in E. coli, that directly associate with RNAP or with replication fork-bound proteins (Trautinger et al., 2005; Baharoglu et al., 2010; Boubakri et al., 2010; Proshkin et al., 2010; Tehranchi et al., 2010; Dutta et al., 2011; Washburn & Gottesman, 2011). Rep is localized to sites of replication by interaction with the replicative DnaB helicase and DinG binds to SSB (Guy et al., 2009; Boubakri et al., 2010; Atkinson et al., 2011; Cheng et al., 2012; Syeda et al., 2019). UvrD associates with RNAP, which may aid in its removal of stalled transcription complexes or resolution of replication/transcription conflicts through other mechanisms (Epshtein et al., 2014; Hawkins et al., 2019; Kamarthapu et al., 2016; Sanders et al., 2017).
Our results show that simultaneous mislocalization of RNase HI and loss of Rep helicase result in defects in E. coli growth that are dependent on R-loop production by RNAP. Interestingly, full deletion of rnhA and uvrD is synthetically lethal as well (Table 1), but, unlike the situation with Rep, RNase HI mislocalization in the rnhAK60E mutant did not result in a measurable phenotype when coupled with a uvrD deletion (Table 1 and Figure 2). Furthermore, dinG was able to be deleted from an rnhA-deleted strain, consistent with a recent study suggesting that DinG may act less directly to resolve replication/transcription conflicts (Hawkins et al., 2019). Our results suggest that RNase HI functions synergistically with Rep, and likely UvrD to a lesser extent, to promote genome duplication through R-loop-dependent fork obstacles.
How might SSB-medicated RNase HI localization help to remove R-loop obstacles to the replication fork? In E. coli, the lagging strand template is exposed as ~1-2 kb long ssDNA segments at replication forks. SSB binding to this ssDNA positions numerous SSB molecules at each site of DNA replication (Figure 5). Since each SSB tetramer has four SSB-Ct protein interaction sites, the SSB/ssDNA structures at replication forks offers abundant bindings sites for partner proteins at each fork. As has been shown here, RNase HI forms foci at replication sites in a manner that requires interaction with SSB (Figure 1); several other SSB protein partners localize to the replication fork in the same manner (Sun & Godson, 1996; Glover & McHenry, 1998; Marceau et al., 2011; Wessel et al., 2013; Bhattacharyya et al., 2014). Thus, the interaction poises RNase HI at replication forks to concentrate its nuclease activity to loci adjacent to the advancing fork. Our data collectively support a model in which this localization allows RNase HI to hydrolyze R-loops encountered ahead of the replication fork to aid in clearing RNAP complexes that would otherwise impede replication progression. With its noted roles in clearing RNAP (Guy et al., 2009; Boubakri et al., 2010), Rep helicase appears to cooperate with RNase HI in this activity.
Figure 5. Schematic model for RNase HI and Rep helicase localization and action at sites of replication/transcription collision.

RNase HI (purple) is localized to the DNA replication fork by interaction with SSB (yellow). Rep helicase (grey) is localized by interaction with DnaB (orange). SSB-Ct tails are shown explicitly for only one SSB tetramer for clarity. DNA strands are shown in blue, RNA strands are shown in red, and DNA polymerases are shown in green. Several replisome components have been omitted or separated for clarity.
We note that SSB might also coat the exposed DNA strands of R-loops and that such an arrangement could also assist with recruiting RNase HI to R-loops. Since this would also impact cSDR and no deficiency in RNase HI inhibition of cSDR was detected with the RNase HI K60E variant, this recruitment strategy does not appear to be as important as the localization to replication forks for resolving replication/transcription conflicts.
The observations described here highlight parallels between bacterial RNase HI and human RNase H1 noted in recent studies. Human RNase H1 directly interacts with Replication Protein A, the functional equivalent of bacterial SSB, forming a complex that stimulates the activity of RNase H1 in vitro and that is critical for suppression of R-loop forming in vivo (Nguyen et al., 2017). Additionally, depletion of human RNase H1 leads to an accumulation of R-loops, slowed replication fork progression, and increased DNA damage (Parajuli et al., 2017). It therefore appears that SSB-mediated RNase H localization is a general phenomenon that cells have adapted to facilitate progression through R-loop challenges to replication processes in both bacterial and eukaryotic cells.
Experimental Procedures
Bacterial strains and growth
All bacterial strains are derivatives of E. coli K12 and are described in Table S1. Mutations were introduced into the strain of interest by P1 transduction (Willetts et al., 1969). P1 transductions were selected on 2% agar plates made with Luria broth (LB) or 56/2 minimal medium (Willetts et al., 1969). M9 was supplemented with glycerol (final concentration 0.2%); 100 μg/ml of amino acids threonine, leucine, proline, histidine and arginine; and thiamine (0.5 μg/ml). Ampicillin (10 μg/mL), tetracycline (10 μg/mL), kanamycin (50 μg/mL), chloramphenicol (25 μg/mL; 12.5 μg/mL for electroporation selection), rifampicin (50 μg/mL) were added when required. Mutations were confirmed after new strain generations by colony PCR and sequencing.
Strain preparation
General procedure for constructing the rnhAK60E chromosomal point mutation followed the λ-Red recombination method (Datsenko & Wanner, 2000). The rnhA gene was placed onto a pET15b with a 10 nt gap and Cmr cassette on the 3′ to generate the following product: pET15b-rnhA-10nt – FRT-cat- FRT. The pET15b-rnhA plasmid was used as a template for site directed mutagenesis as previously described to make the K60E point mutation (Petzold et al., 2015). In addition to the point mutation a restriction site polymorphism (XhoI) was inserted adjacent to K60E to allow for screening of the allele. Electrocompetent BW25113 cells with pKD46 (Datsenko & Wanner, 2000) were used to electroporate the rnhA-cat and rnhAK60E-cat DNA fragments. Cells that had incorporated the DNA were selected on LB with 12.5ug/mL chloramphenicol (LB + Cm). Colonies were purified on LB + Cm and colony PCR was performed on Cmr transformants to confirm the incorporation of Cmr cassette. For cells transformed with rnhAK60E-cat a restriction digest with XhoI was also used to check for the point mutation. Cells with the correct product size were sequenced at the rnhA locus by colony PCR. P1 lysates were prepared from rnhA-cat and rnhAK60E-cat cells and introduced into JC13509 selecting for Cmr. The protocol for P1 transduction has been described previously (Willetts et al., 1969). The Cmr transductants were again sequenced to confirm the rnhA genotype. The protocol for removing Cmr was performed as described in (Datsenko & Wanner, 2000).
All strains used for microscopy are AB1157 derivatives. Strain carrying a wildtype copy of RNase HI fused to YPet (RRL327) or mNeonGreen (VV14) were generated through lambda red recombination as previously described (Reyes-Lamothe et al., 2008). Briefly, primers L_rnhA_F (ccgcggcgatgaatcccacactggaagatacaggctaccaagttgaagttTCGGCTG GCTCCGCTGCTGG) and L_rnhA_R (cgaattcgccaggcggttggagccacccggcaatgtcgtaaacc acaggcCTTATGAATATCCTCCTTAG) and were used to amplify ypet and kanR from the plasmid pROD61 or mneongreen and kanR from the plasmid plasmid pVV04. Integration into the chromosome was done using plasmid pKD46, which expresses the lambda red genes under the control of arabinose (Datsenko & Wanner, 2000). The strain carrying the rnhAK60E allele fused to ypet (VV8) was generated by cloning rnhA-ypet kan with ~500 bp of flanking regions at either side, amplified from RRL327, into a plasmid carrying an R6K gamma origin using primers rnhA_seqF5 (TTTAGACGTCGGCTATCGGTGACCTTACAG) and rnhA_seqR3 (ATCAAGTGAATGTTTCTGCGC) (resulting in pVV10). Cloning was done using an AatII site at one end and TA cloning at the other end, and ligated into a fragment containing the R6K gamma origin amplified from pKD4 (Datsenko & Wanner, 2000). The point mutation was introduced by Quick Change mutagenesis using primers rnhA_K3EF (GCGCTGGAGGCGTTAGAAGAACATTGCGAAGTC) and rnhA_K3ER (GACTTCGCAATGTTCTTCTAACGCCTCCAGCGC). The resulting rnhAK60E-ypet kan was introduced in the chromosome, replacing the wildtype copy, using primers rnhA_seqF3 (GGCTATCGGTGACCTTACAG) and rnhA_seqF3.
The mcherry-dnaN fusion (RRL355) was constructed by lambda red using primers dnaN-NF (TATCAAAGAAGATTTTTCAAATTTAATCAGAACATTGTCATCGTAAACCTGTAGGCTGGAGCTGCTTCG) and dnaN-NR (ACCTGTTGTAGCGGTTTTAATAAATGCTCACGTTCTAC GGTAAATTTCATCGCGCTGCCAGAACCAGC) and pROD84 as a template, carrying a kanR mcherry followed by an 11 amino acid linker. The kanamycin cassette was removed by FLP recombinase expression from pCP20 (Datsenko & Wanner, 2000) resulting in strain RRL388. Fluorescent rnhA-ypet kanR fusion was moved into the strain carrying RRL388 by P1 transduction (resulting in strain VV11).
Growth curves
Strains were inoculated from single colonies in overnight liquid cultures of 56/2 minimal media with antibiotic where relevant (Willetts et al., 1969). After growth at 37°C for 16h, overnight cultures were used to inoculate 25mL of LB or 56/2 minimal media (100-fold dilution). OD600 was taken at t=0 and at subsequent 30 minute time points until several time points at stationary phase was reached. Time points from three independent experiments were plotted with standard deviation using GraphPad Prism.
CFU/mL/OD600 determination
Strains were inoculated from single colonies in overnight liquid cultures of 56/2 minimal medium with antibiotics when relevant (Willetts et al., 1969). After growth at 37°C for 16h the OD600 of overnight cultures was taken. Cultures were serially diluted in sterile saline and 10μL of the appropriate dilution was plated on LB or 56/2 minimal media. Colony counts were performed after the plates were incubated at 37°C for 24h and 48h. CFU/ml/OD600 values were determined by the following equation: [(dilution factor/volume plated) * colony count]/ OD600. CFU/ml/OD600 values were accumulated for at least three independent cultures and plotted with standard deviation using GraphPad Prism.
Phase Contrast Microscopy
Strains were inoculated from single colonies in overnight liquid cultures of 56/2 minimal medium with 100μg/mL ampicillin (Willetts et al., 1969). After growth at 37°C for 16h the overnight cultures were diluted 100-fold in 56/2 minimal medium or LB with ampicillin. 4μL of cells were spotted on fresh 2% agarose pads (1XPBS) and covered with a coverslip. Cells were imaged at 25°C using an inverted Nikon Eclipse Ti microscope equipped with a Photometrics CoolSNAP HQ2 charge-coupled-device (CCD) camera (Photometrics, Tucson, AZ). Images were acquired using a ×100 oil objective (Nikon Plan Apo 100/1.40 oil Ph3 DM) and the Nikon Instruments Software (NIS)-Elements Advanced Research (AR) microscope imaging software program (Version 4.000.07) (Nikon, Melville, NY). Data were collected on the EMCCD using an exposure time of 50 ms. Data from at least 200 cells from three independent cultures was collected and analyzed using MicrobeTracker to determine average cell length for each strain in minimal and rich media (Sliusarenko et al., 2011).
Fluorescence Microscopy
Imaging was performed at room temperature on an inverted Olympus IX83 microscope using a 100x oil objective lens (Olympus Plan Apo 100X NA 1.40 oil). Images were captured using an Andor Zyla 4.2 sCMOS camera. Z-stacks were done using a NanoScanZ piezo by Prior Scientific. Excitation was done from an iChrome Multi-Laser Engine (405nm 100mW, 488nm 100mW, 561nm 100mW, 640nm 70mW) from Toptica Photonics and a 405/488/561/640nm filter set (Chroma). Laser triggering was done through a real-time controller U-RTCE (Olympus). Experiments were done from a single-line cellTIRF illuminator (Olympus). Olympus CellSens 2.1 imaging software was used to control the microscope and lasers.
Before imaging cells were grown in LB for at least 5 hours then transferred to M9-Glycerol medium via a 1:1000 dilution. After being grown overnight cells were diluted again in M9-Glycerol and grown to an OD600 between 0.1- 0.2. Cells were spotted on a 1% agarose pad in M9-Glycerol. Imaging of YPet strains was done capturing for 500ms with 15% laser power (488nm laser), while imaging of mCherry was done using 500ms with 13% laser power (561nm laser). At each field of view a z-stack of 32 pictures with a 100nm step size was taken using brightfield illumination, which was later used for segmentation.
Spot counting and colocalization analysis
All analysis was done using custom scripts written in MATLAB (Mathworks). A 32-frame bright field Z-stacks was compressed to create a black and white phase contrast image for cell segmentation (Julou et al., 2013). Cells were segmented using SuperSegger software (Stylianidou et al., 2016). Spots were counted using a modified version of the previously developed tracking software (Uphoff et al., 2013). Spots were determined using an intensity threshold then further processed using a 2D-elliptical Gaussian fit. The extracted fitted parameters were; x-position, y-position, x-standard deviation, y-standard deviation, intensity and background. Co-localization analysis was done by measuring the distance between the positions of ribonuclease HI-YPet to the β-clamp-mCherry in two-colour experiments. If cells had multiple foci of the same protein, then the smallest distance was recorded, and the 2 spots measured removed so their positions would not be used again in further calculations.
Colocalization was characterized using radial distribution analysis (Zawadzki et al., 2015; Thrall et al., 2017). The radial distribution function g(r) displays the increased likelihood of ribonuclease HI-YPet at a distance r from a mCherry-β-clamp focus relative to random cellular localization. This measurement incorporates the cell shape and size of the cells analysed in the colocalization analysis. An equivalent number of random localizations were generated within the cell outline and the distance between them and the position of a β-clamp-mCherry focus (obtained from the data for a particular cell in the analysis) measured. We also measured the distance between two randomly generated localizations in the cell. This procedure was repeated for each cell. To account for variability, we simulated the single protein random localization and two protein random localization distributions 100 times. We then normalized the average number of simulated localizations per bin by the number of cells to determine the probability to be found in a certain bin, obtaining the single random spot distance distributions and the two random spot distance distributions. We followed the same normalization for the measured data to obtain the measured distance distributions. Histograms were generated for the measured and random distance distributions. Numbers for individual bins in the distribution of the experimental data were divided by the number in the corresponding bind for the one random spot distribution to give g(r). A g(r) value of 1 indicates no enrichment relative to a random distribution. We also divided the double random spot distribution by the single random spot distribution as a control.
Single molecule intensity and copy number analysis
Imaging was performed at room temperature on an inverted Nikon Ti2 microscope using a 100x oil objective lens (CFI APO 100x oil TIRF NA 1.49). Images were captured using an Andor iXon EM+ DU-897 EMCCD camera. Excitation was done from an OBIS 514nm LX 50mW laser delivered from an OBIS Galaxy laser beam combiner (Coherent) and a zet405/514/561x custom triple excitation set (Chroma). Laser triggering was done through a real-time controller NI DAQ (National Instruments). Experiments were done using HiLo illumination setup (Tokunaga et al., 2008) from a custom made TIRF setup. Molecular Devices MetaMorph imaging software was used to control the microscope and lasers. Videos for each strain included 500 frames. Image capture was done using continuous acquisition at 10 milliseconds frame rate (100% 515nm laser).
Initial spot positions were manually selected using the coordinates for localized bleaching in the image recorded by the acquisition software. Tracking was then done automatically using a previously developed custom program in MATLAB (Mathworks), ADEMS code (Miller et al., 2015) (freely available at https://sourceforge.net/projects/york-biophysics/). This program generated individual intensity traces. The change point detection algorithm, ischange, a built-in function in MATLAB was then used to find abrupt changes in the spot intensity and to determine the intensity change in the last step during bleaching (representing a single molecule). The maximum numbers of steps was restricted to 10. The intensity of the last step was used as the single molecule intensity.
An average frame projection of the videos was used to manually create single cell outlines. Individual cell were identified along with the cell lengths in pixels, the area in pixels and the total cell intensities for only the first frame of each video. To remove the endogenous cell background from our copy number calculations, the average pixel intensity was determined in AB1157 untagged cells. The single pixel background intensity was multiplied by the area of each individual cell and this total background intensity was removed from the total cell intensities. This provided the total intensity of our fluorescent protein per cell. This total intensity was divided by the single molecule intensity to give the copy number.
To determine the bound proportion, the cell background from our AB1157 measurements and the mean cell intensity for one pixel of the protein in question were removed from the first frame intensity from the single molecule intensity analysis intensity traces to get the intensity of each spot. The intensity was divided by our single molecule intensity data to get the number of molecules bound. Finally, this number was divided by the protein copy number to get the bound proportion.
SOS response assay
Strains were transformed with pEAW903, a pET21a plasmid with SuperGlo GFP (Qbiogene) under the control of the E. coli recN promoter (Ronayne et al., 2016). Strains were grown overnight in M63 minimal medium + 0.4% glucose supplemented with 0.2% casamino acids and ampicillin (Elbing & Brent, 2001). Overnight cultures were diluted to OD600 0.05 in minimal medium or LB with ampicillin and 200uL was added to the wells (in triplicate) of a 96-well black-side clear-bottom plate (Corning). A BioTek Synergy 2 plate reader was used for taking OD600 and fluorescence reads (excitation 485nm /emission 528nm) during the growth curve. The plate was maintained at 37°C with shaking set to ‘slow-continuously’ and an optical adhesive cover (Applied Biosystems). Once log phase was reached, the protocol was stopped and 10μL of cells was used for serial dilution and determining CFU/mL values. The plate was immediately returned to the plate reader and a new protocol was started to capture the growth curve through stationary phase.
P1 co-transduction frequency
All transductions were performed as described (Willetts et al., 1969). Transductions were plated on 56/2 minimal medium with the selection markers noted in Table 1. Transductants were grown at 37°C (unless otherwise noted) and purified on the same type of media on which they were selected. Purified transductants were then patch plated on 56/2 or LB (corresponding to initial media used for selection) and grown at 37°C. Patch plates were then used for replica plating to screen for co-transduction of experimental mutation with the selectable marker. Screening for recBCD genotype was done using UV sensitivity. Replicate plates were exposed to UV using a Spectrolinker XL-1000 UV crosslinker (Spectronics Corp.) and plates were kept in the dark until analyzed. Co-transduction frequencies are scored out of total transductants confirmed to have the initial selection marker and genotype of recipient strain after replica plating.
Supplementary Material
Acknowledgements
We thank Dr. Jeremy Schroeder and members of the Keck, Reyes-Lamothe, and Sandler laboratories for critical evaluation of the manuscript. This work was supported by a National Institutes of Health grant to JLK and SJS (R01 GM098885) and by a National Science Foundation Graduate Research Fellowship Program to CW (DGE-1256259).
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- Aguilera A, & Garcia-Muse T (2012). R loops: from transcription byproducts to threats to genome stability. Mol Cell, 46(2), 115–124. [DOI] [PubMed] [Google Scholar]
- Alberts BM (1987). Prokaryotic DNA replication mechanisms. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, Vol. 317, pp. 395–420. [DOI] [PubMed] [Google Scholar]
- Atkinson J, Gupta MK, & McGlynn P (2011). Interaction of rep and DnaB on DNA. Nucleic Acids Research, 39(4), 1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson J, Gupta MK, Rudolph CJ, Bell H, Lloyd RG, & McGlynn P (2011). Localization of an accessory helicase at the replisome is critical in sustaining efficient genome duplication. Nucleic Acids Research, 39(3), 949–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azvolinsky A, Giresi PG, Lieb JD, & Zakian VA (2009). Highly transcribed RNA polymerase II genes are impediments to replication fork progression in Saccharomyces cerevisiae. Molecular Cell, 34(6), 722–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baharoglu Z, Lestini R, Duigou S, & Michel B (2010). RNA polymerase mutations that facilitate replication progression in the rep uvrD recF mutant lacking two accessory replicative helicases. Mol Microbiol, 77(2), 324–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentchikou E, Chagneau C, Long E, Matelot M, Allemand JF, & Michel B (2015). Are the SSB-interacting proteins reco, RecG, PriA and the DnaB-interacting protein rep bound to progressing replication forks in Escherichia coli PLoS ONE, 10(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya B, George NP, Thurmes TM, Zhou R, Jani N, Wessel SR, … Keck JL (2014). Structural mechanisms of PriA-mediated DNA replication restart. Proc Natl Acad Sci U S A, 111(4), 1373–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boubakri H, de Septenville AL, Viguera E, & Michel B (2010). The helicases DinG, Rep and UvrD cooperate to promote replication across transcription units in vivo. The EMBO Journal, 29(1), 145–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce RP, & Howard-Flanders P (2003). Release of ultraviolet light-induced thymine dimers from DNA in E. coli K-12. 1964. DNA Repair, 2(1927), 1280–1287. [PubMed] [Google Scholar]
- Carr KM, & Kaguni JM (1996). The A184V missense mutation of the dnaA5 and dnaA46 alleles confers a defect in ATP binding and thermolability in initiation of Escherichia coli DNA replication. Molecular Microbiology, 20, 1307–1318. [DOI] [PubMed] [Google Scholar]
- Chandler M, Bird RE, & Caro L (1975). The replication time of the Escherichia coli K12 chromosome as a function of cell doubling time. Journal of Molecular Biology, 94(1), 127–132. [DOI] [PubMed] [Google Scholar]
- Chen SH, Byrne RT, Wood E. a., & Cox MM (2015). Escherichia coli radD (yejH) gene: a novel function involved in radiation resistance and double-strand break repair. Molecular Microbiology, 95(January), 754–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z, Caillet A, Ren B, & Ding H (2012). Stimulation of Escherichia coli DNA damage inducible DNA helicase DinG by the single-stranded DNA binding protein SSB. FEBS Letters, 586(21), 3825–3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costes A, Lecointe F, McGovern S, Quevillon-Cheruel S, & Polard P (2010). The C-terminal domain of the bacterial SSB protein acts as a DNA maintenance hub at active chromosome replication forks. PLoS Genet, 6(12), e1001238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouch RJ (1990). Ribonuclease H: from discovery to 3D structure. New Biol, 2(9), 771–777. [PubMed] [Google Scholar]
- Datsenko KA, & Wanner BL (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A, 97(12), 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Massy B, Fayet O, & Kogoma T (1984). Multiple origin usage for DNA replication in sdrA(rnh) mutants of Escherichia coli K-12. Initiation in the absence of oriC. Journal of Molecular Biology, 178(2), 227–236. [DOI] [PubMed] [Google Scholar]
- Deshpande a M., & Newlon CS (1996). DNA replication fork pause sites dependent on transcription. Science (New York, N.Y.), 272(5264), 1030–1033. [DOI] [PubMed] [Google Scholar]
- Dimude JU, Stockum A, Midgley-Smith SL, Upton AL, Foster HA, Khan A, … Rudolph CJ (2015). The Consequences of Replicating in the Wrong Orientation: Bacterial Chromosome Duplication without an Active Replication Origin. MBio, 6(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drolet M, Phoenix P, Menzel R, Masse E, Liu LF, & Crouch RJ (1995). Overexpression of RNase H partially complements the growth defect of an Escherichia coli delta topA mutant: R-loop formation is a major problem in the absence of DNA topoisomerase I. Proc Natl Acad Sci U S A, 92(8), 3526–3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta D, Shatalin K, Epshtein V, Gottesman ME, & Nudler E (2011). Linking RNA polymerase backtracking to genome instability in E. coli. Cell, 146(4), 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Hage A, French SL, Beyer AL, & Tollervey D (2010). Loss of Topoisomerase I leads to R-loop-mediated transcriptional blocks during ribosomal RNA synthesis. Genes & Development, 24(14), 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbing K, & Brent R (2001). Media Preparation and Bacteriological Tools. In Current Protocols in Molecular Biology. [DOI] [PubMed] [Google Scholar]
- Epshtein V, Kamarthapu V, McGary K, Svetlov V, Ueberheide B, Proshkin S, … Nudler E (2014). UvrD facilitates DNA repair by pulling RNA polymerase backwards. Nature, 505(7483), 372–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch PW, Chambers P, & Emmerson PT (1985). Identification of the Escherichia coli recN gene product as a major SOS protein. Journal of Bacteriology, 164(2), 653–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French S (1992). Consequences of replication fork movement through transcription units in vivo. Science, 258(5086), 1362–1365. [DOI] [PubMed] [Google Scholar]
- Frey J, Chandler M, & Caro L (1981). The initiation of chromosome replication in a dnaAts46 and a dnaA+ strain at various temperatures. Mol Gen Genet, 182(2), 364–366. [DOI] [PubMed] [Google Scholar]
- Gan W, Guan Z, Liu J, Gui T, Shen K, Manley JL, & Li X (2011). R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes and Development, 25(19), 2041–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover BP, & McHenry CS (1998). The chi psi subunits of DNA polymerase III holoenzyme bind to single-stranded DNA-binding protein (SSB) and facilitate replication of an SSB-coated template. J Biol Chem, 273(36), 23476–23484. [DOI] [PubMed] [Google Scholar]
- Guy CP, Atkinson J, Gupta MK, Mahdi A. a., Gwynn EJ, Rudolph CJ, … McGlynn P (2009). Rep Provides a Second Motor at the Replisome to Promote Duplication of Protein-Bound DNA. Molecular Cell, 36(4), 654–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harinarayanan R, & Gowrishankar J (2003). Host factor titration by chromosomal R-loops as a mechanism for runaway plasmid replication in transcription termination-defective mutants of Escherichia coli. Journal of Molecular Biology, 332(1), 31–46. [DOI] [PubMed] [Google Scholar]
- Hawkins M, Dimude JU, Howard JAL, Smith AJ, Dillingham MS, Savery NJ, … McGlynn P (2019). Direct removal of RNA polymerase barriers to replication by accessory replicative helicases. Nucleic Acids Research, 47(10), 5100–5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmrich A, Ballarino M, Nudler E, & Tora L (2013). Transcription-replication encounters, consequences and genomic instability. Nat Struct Mol Biol, 20(4), 412–418. [DOI] [PubMed] [Google Scholar]
- Helmrich A, Ballarino M, & Tora L (2011). Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol Cell, 44(6), 966–977. [DOI] [PubMed] [Google Scholar]
- Hinds T, & Sandler SJ (2004). Allele specific synthetic lethality between priC and dnaAts alleles at the permissive temperature of 30°C in E. coli K-12. BMC Microbiology, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong X, Cadwell GW, & Kogoma T (1995). Escherichia coli RecG and RecA proteins in R-loop formation. Embo J, 14(10), 2385–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houlard M, Artus J, Léguillier T, Vandormael-Pournin S, & Cohen-Tannoudji M (2011). DNA-RNA hybrids contribute to the replication dependent genomic instability induced by Omcg1 deficiency. Cell Cycle, 10(1), 108–117. [DOI] [PubMed] [Google Scholar]
- Hraiky C, Raymond MA, & Drolet M (2000). RNase H overproduction corrects a defect at the level of transcription elongation during rRNA synthesis in the absence of DNA topoisomerase I in Escherichia coli. J Biol Chem, 275(15), 11257–11263. [DOI] [PubMed] [Google Scholar]
- Huertas P, & Aguilera A (2003). Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol Cell, 12(3), 711–721. [DOI] [PubMed] [Google Scholar]
- Itaya M, & Crouch RJ (1991). Correlation of activity with phenotypes of Escherichia coli partial function mutants of rnh, the gene encoding RNase H. Molecular & General Genetics : MGG, 227(3), 433–437. [DOI] [PubMed] [Google Scholar]
- Itaya Mitsuhiro, & Crouch RJ (1991). A combination of RNase H (rnh) and recBCD or sbcB mutations in Escherichia coli K 12 adversely affects growth. MGG Molecular & General Genetics, 227, 424–432. [DOI] [PubMed] [Google Scholar]
- Itoh T, & Tomizawa J (1980a). Formation of an RNA primer for initiation of replication of ColE1 DNA by ribonuclease H. Proceedings of the National Academy of Sciences of the United States of America, 77(5), 2450–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T, & Tomizawa J (1980b). Formation of an RNA primer for initiation of replication of ColE1 DNA by ribonuclease H. Proc Natl Acad Sci U S A, 77(5), 2450–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janion C (2008). Inducible SOS response system of DNA repair and mutagenesis in Escherichia coli. International Journal of Biological Sciences, 4(6), 338–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin DJ, Burgess RR, Richardson JP, & Gross C. a. (1992). Termination efficiency at rho-dependent terminators depends on kinetic coupling between RNA polymerase and rho. Proceedings of the National Academy of Sciences of the United States of America, 89(4), 1453–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Ding Jun, Cashel M, Friedman DI, Nakamura Y, Walter WA, & Gross CA (1988). Effects of Rifampicin resistant rpoB mutations on antitermination and interaction with nusA in Escherichia coli. Journal of Molecular Biology, 204(2), 247–261. [DOI] [PubMed] [Google Scholar]
- Joyce CM, Fujii DM, Laks HS, Hughes CM, & Grindley ND (1985). Genetic mapping and DNA sequence analysis of mutations in the polA gene of Escherichia coli. Journal of Molecular Biology, 186(2), 283–293. [DOI] [PubMed] [Google Scholar]
- Julou T, Mora T, Guillon L, Croquette V, Schalk IJ, Bensimon D, & Desprat N (2013). Cell-cell contacts confine public goods diffusion inside Pseudomonas aeruginosa clonal microcolonies. Proceedings of the National Academy of Sciences of the United States of America, 110(31), 12577–12582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamarthapu V, Epshtein V, Benjamin B, Proshkin S, Mironov A, Cashel M, & Nudler E (2016). ppGpp couples transcription to DNA repair in E. Coli. Science, 352(6288), 993–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitani T, Yoda K, Ogawa T, & Okazaki T (1985). Evidence that discontinuous DNA replication in Escherichia coli is primed by approximately 10 to 12 residues of RNA starting with a purine. Journal of Molecular Biology, 184(1), 45–52. [DOI] [PubMed] [Google Scholar]
- Kogoma T (1994). Escherichia coli RNA polymerase mutants that enhance or diminish the SOS response constitutively expressed in the absence of RNase HI activity. Journal of Bacteriology, Vol. 176, pp. 1521–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogoma T (1997a). Stable DNA replication: interplay between DNA replication, homologous recombination, and transcription. Microbiology and Molecular Biology Reviews : MMBR, 61(2), 212–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogoma T (1997b). Stable DNA replication: interplay between DNA replication, homologous recombination, and transcription. Microbiol Mol Biol Rev, 61(2), 212–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogoma T, Barnard KG, & Hong X (1994). RecA, Tus protein and constitutive stable DNA replication in Escherichia coli rnhA mutants. Molecular & General Genetics : MGG, Vol. 244, pp. 557–562. [DOI] [PubMed] [Google Scholar]
- Kogoma Tokio. (1978). A Novel Escherichia coli Mutant Capable of DNA Replication in the Absence of Protein Synthesis. Journal of Molecular Biology, 121(1), 55–69. [DOI] [PubMed] [Google Scholar]
- Kogoma Tokio, Hong X, Cadwell GW, Barnard KG, & Asai T (1993). Requirement of homologous recombination functions for viability of the Escherichia coli cell that lacks RNase HI and exonuclease V activities. Biochimie, 75, 89–99. [DOI] [PubMed] [Google Scholar]
- Kogoma Tokio, & von Meyenburg K (1983). The origin of replication, oriC, and the dnaA protein are dispensable in stable DNA replication (sdrA) mutants of Escherichia coli K-12. The EMBO Journal, 2(3), 463–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg A, & Baker TA (1992). DNA Replication (2nd ed.). New York: W. H. Freeman. [Google Scholar]
- Kuzminov A (2001). Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proceedings of the National Academy of Sciences of the United States of America. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecointe F, Serena C, Velten M, Costes A, McGovern S, Meile JC, Polard P (2007). Anticipating chromosomal replication fork arrest: SSB targets repair DNA helicases to active forks. Embo J, 26(19), 4239–4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, & Manley JL (2005). Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell, 122(3), 365–378. [DOI] [PubMed] [Google Scholar]
- Lindahl G, & Lindahl T (1984). Initiation of DNA replication in Escherichia coli: RNase H-deficient mutants do not require the dnaA function. Molecular & General Genetics : MGG, 196(2), 283–289. [DOI] [PubMed] [Google Scholar]
- Liu X, Wang X, Reyes-Lamothe R, & Sherratt D (2010). Replication-directed sister chromosome alignment in Escherichia coli. Mol Microbiol, 75(5), 1090–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JE, Massoni SC, & Sandler SJ (2010). RecA4142 causes SOS constitutive expression by loading onto reversed replication forks in Escherichia coli K-12. Journal of Bacteriology, 192(10), 2575–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marceau AH (2012). Functions of single-strand DNA-binding proteins in DNA replication, recombination, and repair. Methods in Molecular Biology, 922, 1–21. [DOI] [PubMed] [Google Scholar]
- Marceau AH, Soon B, Massoni SC, George NP, Sandler SJ, Marians KJ, & Keck JL (2011). Structure of the SSB-DNA polymerase III interface and its role in DNA replication. EMBO Journal, 30, 4236–4247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlynn P, Savery NJ, & Dillingham MS (2012). The conflict between DNA replication and transcription. Mol Microbiol, 85(1), 12–20. [DOI] [PubMed] [Google Scholar]
- Merrikh H, Zhang Y, Grossman AD, & Wang JD (2012). Replication-transcription conflicts in bacteria. Nature Reviews. Microbiology, 10(7), 449–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer RR, & Laine PS (1990). The single-stranded DNA-binding protein of Escherichia coli. Microbiol Rev, 54(4), 342–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel B (2005). After 30 years of study, the bacterial SOS response still surprises us. PLoS Biology, 3(7), 1174–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller H, Zhou Z, Wollman AJ, & Leake MC (2015) Superresolution imaging of single DNA molecules using stochastic photoblinking of minor groove and intercalating dyes. Methods 88:81–88. [DOI] [PubMed] [Google Scholar]
- Mirkin EV, & Mirkin SM (2007). Replication fork stalling at natural impediments. Microbiology and Molecular Biology Reviews : MMBR, 71(1), 13–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk M, & Kinross J (1972). Conditional lethality of recA and recB derivatives of a strain of Escherichia coli K-12 with a temperature-sensitive deoxyribonucleic acid polymerase I. J. Bacteriol, 109234, 971–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moolman MC, Krishnan S. T. iruvad., Kerssemakers JWJ, van den Berg A, Tulinski P, Depken M, … Dekker NH (2014). Slow unloading leads to DNA-bound β2-sliding clamp accumulation in live Escherichia coli cells. Nature Communications, 5, 5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HD, Yadav T, Giri S, Saez B, Graubert TA, & Zou L (2017). Functions of Replication Protein A as a Sensor of R Loops and a Regulator of RNaseH1. Molecular Cell, 65(5), 832–847.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell M, Langston L, & Stillman B (2013). Principles and concepts of DNA replication in bacteria, archaea, and eukarya. Cold Spring Harbor Perspectives in Biology, 5(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa T, & Okazaki T (1980). Discontinuous DNA replication. Annu Rev Biochem, 49, 421–457. [DOI] [PubMed] [Google Scholar]
- Ogawa T, & Okazaki T (1984). Function of RNase H in DNA replication revealed by RNase H defective mutants of Escherichia coli. Molecular & General Genetics : MGG, 193(2), 231–237. [DOI] [PubMed] [Google Scholar]
- Ogawa T, Pickett GG, Kogoma T, & Kornberg A (1984). RNase H confers specificity in the dnaA-dependent initiation of replication at the unique origin of the Escherichia coli chromosome in vivo and in vitro. Proceedings of the National Academy of Sciences of the United States of America, 81(4), 1040–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parajuli S, Teasley DC, Murali B, Jackson J, Vindigni A, & Stewart SA (2017). Human ribonuclease H1 resolves R-loops and thereby enables progression of the DNA replication fork. Journal of Biological Chemistry, 292(37), 15216–15224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petzold C, Marceau AH, Miller KH, Marqusee S, & Keck JL (2015). Interaction with single-stranded DNA binding protein stimulates Escherichia coli ribonuclease HI enzymatic activity. Journal of Biological Chemistry, jbc.M115.655134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz RT, & O’Donnell M (2010). What happens when replication and transcription complexes collide? Cell Cycle (Georgetown, Tex.), 9(13), 2537–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado F, & Aguilera A (2005). Impairment of replication fork progression mediates RNA polII transcription-associated recombination. Embo J, 24(6), 1267–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proshkin S, Rahmouni AR, Mironov A, & Nudler E (2010). Cooperation between translating ribosomes and RNA polymerase in transcription elongation. Science, 328(5977), 504–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Lamothe R (2012). Use of fluorescently tagged SSB proteins in in vivo localization experiments. Methods in Molecular Biology, 922, 245–253. [DOI] [PubMed] [Google Scholar]
- Reyes-Lamothe R, Nicolas E, & Sherratt DJ (2012). Chromosome replication and segregation in bacteria. Annual Review of Genetics, 46, 121–143. [DOI] [PubMed] [Google Scholar]
- Reyes-Lamothe R, Possoz C, Danilova O, & Sherratt DJ (2008). Independent positioning and action of Escherichia coli replisomes in live cells. Cell, 133(1), 90–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Lamothe R, Sherratt DJ, & Leake MC (2010). Stoichiometry and architecture of active DNA replication machinery in Escherichia coli. Science, 328(5977), 498–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronayne EA, Wan YCS, Boudreau BA, Landick R, & Cox MM (2016). P1 Ref Endonuclease: A Molecular Mechanism for Phage-Enhanced Antibiotic Lethality. PLoS Genetics, 12(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostas K, Morton SJ, Picksley SM, & Lloyd RG (1987). Nucleotide sequence and LexA regulation of the Escherichia coli recN gene. Nucleic Acids Research, 15(13), 5041–5049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders K, Lin CL, Smith AJ, Cronin N, Fisher G, Eftychidis V, … Dillingham MS (2017). The structure and function of an RNA polymerase interaction domain in the PcrA/UvrD helicase. Nucleic Acids Research, 45(7), 3875–3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler SJ (2005). Requirements for replication restart proteins during constitutive stable DNA replication in Escherichia coli K-12. Genetics, 169(4), 1799–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler SJ, Samra HS, & Clark AJ (1996). Differential suppression of priA2::kan phenotypes in Escherichia coli K-12 by mutations in priA, lexA, and dnaC. Genetics, 143(1), 5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler Steven J., McCool JD, Do TT, & Johansen RU (2001). PriA mutations that affect PriA-PriC function during replication restart. Molecular Microbiology, 41(3), 697–704. [DOI] [PubMed] [Google Scholar]
- Shaner NC, et al. (2013) A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat Methods 10(5):407–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shereda RD, Kozlov AG, Lohman TM, Cox MM, & Keck JL (2008). SSB as an organizer/mobilizer of genome maintenance complexes. Crit Rev Biochem Mol Biol, 43(5), 289–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sliusarenko O, Heinritz J, Emonet T, & Jacobs-Wagner C (2011). High-throughput, subpixel precision analysis of bacterial morphogenesis and intracellular spatio-temporal dynamics. Mol Microbiol, 80(3), 612–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubry N, Wang A, & Reyes-Lamothe R (2019). Replisome activity slowdown after exposure to ultraviolet light in Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America, 116(24), 11747–11753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivatsan A, Tehranchi A, MacAlpine DM, & Wang JD (2010). Co-orientation of replication and transcription preserves genome integrity. PLoS Genetics, 6(1), e1000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stylianidou S, Brennan C, Nissen SB, Kuwada NJ, & Wiggins PA (2016). SuperSegger: robust image segmentation, analysis and lineage tracking of bacterial cells. Molecular Microbiology, 102(4), 690–700. [DOI] [PubMed] [Google Scholar]
- Sun W, & Godson GN (1996). Interaction of Escherichia coli primase with a phage G4ori(c)-E. coli SSB complex. J Bacteriol, 178(23), 6701–6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syeda AH, Wollman AJM, Hargreaves AL, Howard JAL, Brüning J-G, McGlynn P, & Leake MC (2019). Single-molecule live cell imaging of Rep reveals the dynamic interplay between an accessory replicative helicase and the replisome. Nucleic Acids Research, 47(12), 6287–6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tehranchi AK, Blankschien MD, Zhang Y, Halliday JA, Srivatsan A, Peng J, … Wang JD (2010). The transcription factor DksA prevents conflicts between DNA replication and transcription machinery. Cell, 141(4), 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrall ES, Kath JE, Chang S, & Loparo JJ (2017). Single-molecule imaging reveals multiple pathways for the recruitment of translesion polymerases after DNA damage. Nature Communications, 8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga M, Imamoto N, & Sakata-Sogawa K (2008) Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat Methods 5(2):159–161. [DOI] [PubMed] [Google Scholar]
- Trautinger BW, Jaktaji RP, Rusakova E, & Lloyd RG (2005). RNA polymerase modulators and DNA repair activities resolve conflicts between DNA replication and transcription. Molecular Cell, 19(2), 247–258. [DOI] [PubMed] [Google Scholar]
- Tuduri S, Crabbé L, Conti C, Tourrière H, Holtgreve-Grez H, Jauch A, Pasero P (2009). Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nature Cell Biology, 11(11), 1315–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uphoff S, Reyes-Lamothe R, De Leon FG, Sherratt DJ, & Kapanidis AN (2013). Single-molecule DNA repair in live bacteria. Proceedings of the National Academy of Sciences of the United States of America, 110(20), 8063–8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyemura D, & Lehman I (1976). Biochemical characterization of mutant forms of DNA polymerase I from Escherichia coli. I. The polA12 mutation. The Journal of Biological Chemistry, 251(13), 4078–4084. [PubMed] [Google Scholar]
- Vilette D, Ehrlich DS, & Michel B (1996). Transcription-induced deletions in plasmid vectors: M13 DNA replication as a source of instability. Molecular and General Genetics, 252(4), 398–403. [DOI] [PubMed] [Google Scholar]
- von Meyenburg K, Boye E, Skarstad K, Koppes L, & Kogoma T (1987). Mode of initiation of constitutive stable DNA replication in RNase H-defective mutants of Escherichia coli K-12. J Bacteriol, 169(6), 2650–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahba L, Amon JD, Koshland D, & Vuica-Ross M (2011). RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Mol Cell, 44(6), 978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker GC, Smith BT, and Sutton MD (2000). The SOS response to DNA damage. In Storz R, Hengge-Aronis G (Ed.), Bacterial Stress Responses (pp. 131–144). American Society of Microbiology Press. [Google Scholar]
- Wang JD, Berkmen MB, & Grossman AD (2007). Genome-wide coorientation of replication and transcription reduces adverse effects on replication in Bacillus subtilis. Proc Natl Acad Sci U S A, 104(13), 5608–5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn RS, & Gottesman ME (2011). Transcription termination maintains chromosome integrity. Proc Natl Acad Sci U S A, 108(2), 792–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessel SR, Marceau AH, Massoni SC, Zhou R, Ha T, Sandler SJ, & Keck JL (2013). PriC-mediated DNA replication restart requires PriC complex formation with the single-stranded DNA-binding protein. J Biol Chem, 288, 17569–17578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westover KD, Bushnell D. a, & Kornberg RD (2004). Structural basis of transcription: separation of RNA from DNA by RNA polymerase II. Science (New York, N.Y.), 303(5660), 1014–1016. [DOI] [PubMed] [Google Scholar]
- Willetts NS, Clark a. J., & Low B (1969). Genetic location of certain mutations conferring recombination deficiency in Escherichia coli. Journal of Bacteriology, 97(1), 244–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawadzki P, Stracy M, Ginda K, Zawadzka K, Lesterlin C, Kapanidis AN, & Sherratt DJ (2015). The Localization and Action of Topoisomerase IV in Escherichia coli Chromosome Segregation Is Coordinated by the SMC Complex, MukBEF. Cell Reports, 13(11), 2587–2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.