

Abstract
Draft genomes of Penicillium roqueforti, Fusarium sororula, Chalaropsis populi, and Chrysoporthe puriensis are presented. Penicillium roqueforti is a model fungus for genetics, physiological and metabolic studies, as well as for biotechnological applications. Fusarium sororula and Chrysoporthe puriensis are important tree pathogens, and Chalaropsis populi is a soil-borne root-pathogen. The genome sequences presented here thus contribute towards a better understanding of both the pathogenicity and biotechnological potential of these species.
Keywords: Fusarium fujikuroi species complex (FFSC), Colombia, Pinus tecunumanii, Eucalyptus leaf pathogen
IMA GENOME – F 14A
Draft genome sequence of Penicillium roqueforti CECT 2905T
Introduction
Penicillium roqueforti is one of the economically most important fungal species within the genus Penicillium. This fungus is widely known in the food industry because it is responsible for the ripening of blue cheeses (Chávez et al. 2011). In addition, in recent years, P. roqueforti has acquired growing importance as a model fungus for genetics, physiological and metabolic studies, as well as for biotechnological applications (Coton et al. 2020).
Phylogenetically, P. roqueforti belongs to section Roquefortorum within the genus Penicillium (Houbraken et al. 2020; Fig. 1). P. roqueforti was originally described by Thom (1906), and the nomenclatural type of the species is the neotype IMI 024313 (Frisvad and Samson 2004). From this neotype strain, several ex-type strains have been obtained, which are stored in different culture collections around the world.
Fig. 1.

Phylogenetic tree obtained after Maximum Likelihood analysis of the genome sequence of Penicillium roqueforti CECT 2905T and related species. The analysis was done as detailed in the Materials and methods section. Bootstrap support values (> 50%) are shown at the nodes (bootstrap iterations = 1000). The tree was rooted using combined BenA, CaM and RPB2 regions from Aspergillus glaucus NRRL 116T. T = ex-type strain
Among the ex-type strains obtained from the neotype IMI 024313, P. roqueforti CECT 2905T is one of the most widely used. Many molecular studies have been carried out in this ex-type strain, including the analysis of regulatory genes of development and metabolism (García-Rico et al. 2009; Gil-Durán et al. 2015; Torrent et al. 2017; Rojas-Aedo et al. 2018), and importantly, P. roqueforti CECT 2905T has been used to demonstrate the functionality of all biosynthetic gene clusters (BGCs) for the production of secondary metabolites characterized so far in this fungal species (Hidalgo et al. 2014; Kosalková et al. 2015; Del-Cid et al. 2016; Fernández-Bodega et al. 2017; Rojas-Aedo et al. 2017).
Owing to the importance of P. roqueforti CECT 2905T as model for molecular studies and BGCs characterization, the availability of its genome would be very useful. Consequently, we report this genome resource here. The availability of the genome of P. roqueforti CECT 2905T will facilitate future studies on the functional characterization of further BGCs and genes related with the regulation of development and metabolism in this important fungal species.
Sequenced strain
USA: Connecticut: Storrs, isol. ex French Roquefort cheese, 1904, C. Thom (CECT 2905T, ex-type strain from the neotype IMI 024313).
Nucleotide sequence accession number
The genome sequence of Penicillium roqueforti CECT 2905T has been deposited in the DDBJ/ENA/ GenBank databases under the accession number MSQC00000000; Bio project PRJNA351232; Bio-sample SAMN05951162. The version described in this paper is version MSQC00000000.
Materials and methods
Penicillium roqueforti CECT 2905T was grown on CM broth as described before (Gil-Durán et al. 2015). Mycelium was harvested, washed with NaCl 0.9%, and high-molecular weight DNA was extracted exactly as was described by Bainbridge et al. (1990).
High-molecular weight DNA from P. roqueforti CECT 2905T was sequenced using the Illumina HiSeq 2000 platform at Macrogen (Seoul, Korea). A pair-end library with insert sizes of 550 bp was prepared using TruSeq DNA PCR Free kit, and used to generate 101 bp length reads. The quality of the data obtained was assessed using FastQC. Low quality data and adapters were removed with Trimmomatic v. 0.36 (Bolger et al. 2014). Genome assembly of high quality Illumina raw reads was performed with Bowtie2 v. 2.4.1 (Langmead and Salzberg 2012), using the genome of P. roqueforti FM164 (Cheeseman et al. 2014) as reference. The final assembly was subjected to completeness assessment using Benchmarking Universal Single-Copy Orthologs program (BUSCO v. 4.0.6; Seppey et al. 2019), utilizing Eurotiales odb10 dataset. Genes were predicted with AUGUSTUS v. 3.3.3 (Stanke et al. 2008) using the training dataset from Aspergillus nidulans. Finally, in order to identify BGCs, anti-SMASH fungal version v. 5 (Blin et al. 2019) was conducted with default parameters.
The phylogenetic analysis of P. roqueforti CECT 2905T and related species was done using combined β-tubulin (BenA), calmodulin (CaM) and RNA polymerase II second largest subunit (RPB2) regions (Guevara-Suarez et al. 2020). Sequences from related species were obtained from GenBank accessions reported by Houbraken et al. (2020), whereas those from P. roqueforti CECT 2905T were extracted from the sequence genome. Maximum Likelihood analysis was done in MegaX (Kumar et al. 2018) under GTR + G model.
Results and discussion
The phylogenetic tree based on the concatenated BenA, CaM and RPB2 regions confirmed that the sequenced genome belongs to the species P. roqueforti. P. roqueforti CECT 2905T clustered together with P. roqueforti CBS 221.30T with a bootstrap support of 100%. In addition, this P. roqueforti clade was clearly separated from the other five species within the Roquefortorum section (Fig. 1).
Table 1 summarizes the main metrics of the assembled genome sequence of P. roqueforti CECT 2905T. The assembled draft genome has a total length of 26.1 Mb corresponding to 1168 contigs with an N50 value of 70,366 bp, L50 value of 112, and an average GC content of 48.9%. AUGUSTUS predicted 9015 protein coding genes, with an average gene density of 345.4 genes per 1 Mb. BUSCO analysis reported a completeness score of 98.8% based on the identification of 4141 complete and 18 fragmented genes from a total of 4191 Eurotiales genes searched. The estimated genome size of P. roqueforti CECT 2905T is comparable to that of other P. roqueforti strains found in databases, namely JCM 22842 (27.1 Mb; GenBank accession number BCID00000000), UASWS P1 (27.9 Mb; GenBank accession number JNNS01000000) and FM164 (28 Mb; Cheeseman et al. 2014).
Table 1.
General characteristics of the genome of Penicillium roqueforti CECT 2905T
| Assembly metrics | |
| Total bases | 2,019,762,619 |
| Read count | 22,375,066 |
| GC (%) | 48.9 |
| Number of contigs | 1168 |
| Assembly size (Mb) | 26.1 |
| N50 (bp) | 70,366 |
| L50 | 112 |
| Predicted genes models | 9015 |
| Gene density (genes per Mb) | 345.4 |
| BUSCO completeness | 98.8% |
| Biosynthetic gene clusters (BGCs) | |
| PKS type I | 9 |
| PKS type I-NRPS | 3 |
| NRPS | 7 |
| NRPS-indole | 1 |
| NRPS-like fragments | 6 |
| NRPS-like fragment-indole | 1 |
| Terpene | 3 |
| Beta-lactone | 2 |
| Indole | 1 |
| Siderophore | 1 |
The BGCs prediction performed with anti-SMASH fungal version yielded a total of 34 regions associated with biosynthesis of secondary metabolites. The BGCs found correspond to type I polyketide synthases (PKS), non-ribosomal peptide synthetases (NRPS), NRPS-like fragments, PKS-NRPS, NRPS-indole, NRPS-like-indole, terpene, siderophore, and beta-lactone. The number of BGCs predicted in this work is in good agreement with previous estimation by Coton et al. (2020) who suggested that strains of P. roqueforti would contain between 34 and 37 BGCs. To date, only six BGCs have been functionally characterized in P. roqueforti CECT 2905T (Hidalgo et al. 2014; Kosalková et al. 2015; Del-Cid et al. 2016; Fernández-Bodega et al. 2017; Rojas-Aedo et al. 2017), so the availability of its draft genome, informed in this paper, will facilitate future studies of functional characterization of further BGCs in this important fungus.
Authors: Natalia Valdés, Mario Tello, Inmaculada Vaca, Carlos Gil-Durán, Gloria Levicán, and Renato Chávez*
*Contact: renato.chavez@usach.cl
IMA GENOME – F 14B
Draft genome assembly of Fusarium sororula
Introduction
Fusarium species within the Fusarium fuikuroi species complex (FFSC) are plant pathogens of various cultivated crops of economic importance (Kvas et al. 2009; Leslie and Summerell 2006). As such, numerous FFSC species have their genomes sequenced, with the first, F. verticillioides, published in 2010 (Ma et al. 2010). Currently, 51 FFSC species have genome sequences publicly available (www.ncbi.nlm.nih.gov).
In a study exploring the diversity of FFSC species associated with Pinus species in Colombia, five new species were described (Herron et al. 2015). One of these, Fusarium sororula, was isolated from diseased P. tecunumanii seedlings that displayed symptoms of wilt, shoot dieback and roots with lesions that were resin soaked (Steenkamp et al. 2012). These are all symptoms typical of infection by the pitch canker pathogen, F. circinatum. This new species was able to cause disease on susceptible P. patula, at similar levels as F. circinatum (Herron et al. 2015). Fusarium sororula is consequently a threat to global commercial forestry and the availability of its genome sequence will contribute to studies aimed at better understanding its biology and genetics.
Sequenced strain
Colombia: Angela Maria, Santa Rosa, 75°36′21″W 4°49′18″N, isolated from diseased Pinus tecunumanii seedlings, 2006, C.A. Rodas (CMW 25513; FCC 5425; PREM63211-dried culture) (Herron et al. 2015).
Nucleotide sequence accession number
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession JACWFA000000000. The version described in this paper is version JACWFA010000000.
Materials and methods
The Fusarium sororula isolate was grown on half strength potato dextrose agar (BD Difco™) and genomic DNA was extracted according to the protocol of Möller et al. 1992. One pair-end library (550 bp insert size, read length of 250 bp) was generated using the Illumina HiSeq 2500 platform at Macrogen (Seoul, Korea). Poor quality and duplicate reads were removed using Qiagen Genomics Workbench v 20.0.4 (CLCBio, Aarhus). Reads were assembled using SPAdes v 3.13.0 (Bankevich et al. 2012). Completeness of the genome assembly was evaluated with BUSCO v 4.0.6 (Seppey et al. 2019), using the hypocreales dataset. Annotation was done with the MAKER annotation pipeline (Cantarel et al. 2008) using Augustus (Stanke et al. 2006a, 2006b), Genemark ES (Ter-Hovhannisyan et al. 2008) and SNAP (Korf 2004). Gene model data from F. circinatum (Wingfield et al. 2012), F. fujikuroi (Wiemann et al. 2013), F. verticillioides and F. graminearum (Ma et al. 2010), as well as F. mangiferae and F. proliferatum (Niehaus et al. 2017), were included as additional evidence.
Results and discussion
The genome sequence of F. sororula was assembled into 328 scaffolds with a total genome size of 47,806,863 bp. The N50 value was 1,089,458 bp and the genome had a G + C content of 45.99%. BUSCO analyses showed that the assembly was 99.9% complete [4486 complete and single-copy BUSCOs, 7 complete and duplicated BUSCOs; 2 fragmented BUSCOs, 6 missing BUSCOs; n = 4494]. A total of 15,668 open reading frames (orfs) were predicted using the MAKER annotation pipeline, with a gene density of 327.74 orfs/Mb. Sequence comparisons indicated that all twelve known chromosomes typical for species in the FFSC are present. Phylogenetic analysis of sequences from the sequenced genome confirmed the taxonomic identity of the sequenced Fusarium strain as F. sororula (Fig. 2).
Fig. 2.

Maximum Likelihood tree based on the partial gene sequences of translation elongation factor 1-α and β-tubulin (Herron et al. 2015). Sequence alignments were assembled with MAFFT v 7.472 (Katoh et al. 2019). The program jModelTest v 2.1.10 (Darribo et al. 2012) was used to determine the best-fit substitution model (GTR + I + G substitution model) with gamma correction (Tavare 1986). A maximum Likelihood (ML) phylogenetic analysis was performed using PhyML v 3.1 (Guindon et al. 2010). Values at branch nodes are the bootstrapping confidence values with those ≥85% shown. The F. sororula isolate sequenced in this study is indicated in bold
This genome announcement is the third for a Fusarium species isolated from Pinus species in Colombia. The two previously sequenced and assembled genomes where for F. fracticaudum (Wingfield et al. 2018a) and F. pininemorale (Wingfield et al. 2017). This genome assembly for F. sorula is comparable to other American clade species from the FFSC isolated from Pinus species (Table 2; Wingfield et al. 2018a; Wingfield et al. 2017; Wingfield et al. 2018b), with genome size, G + C content and gene density all in a similar range.
Table 2.
Genome statistics for Fusarium sororula and its close relatives
| Genome size (Mb) | GC content (%) | Predicted orfsa | Gene density (orfs/Mb) | References | |
|---|---|---|---|---|---|
| F. sororula | 47.81 | 45.99 | 15,688 | 327.74 | This study |
| F. fracticaudum | 45.80 | 47.56 | 14,136 | 308.67 | Wingfield et al. (2018a) |
| F. pininemorale | 47.78 | 45.98 | 15,455 | 323.47 | Wingfield et al. (2017) |
| F. circinatum | 45.10 | 46.97 | 15,091 | 334.61 | Wingfield et al. (2018b) |
aDetermined as described in text
Authors: Lieschen De Vos*, Magriet A. van der Nest, Quentin C. Santana, Emma T. Steenkamp, Brenda D. Wingfield
*Contact: lieschen.bahlmann@fabi.up.ac.za
IMA GENOME – F 14C
Draft nuclear genome assembly of Chalaropsis populi, the second genome from the genus Chalaropsis
Introduction
Chalaropsis populi is a soil-borne root-pathogen in the family Ceratocystidaceae (Paulin-Mahady et al. 2002; de Beer et al. 2014). The first record of Ch. populi is from the early 1970’s where it was isolated from the bark of Populus and Salix spp. in Belgium (Veldeman 1971). This species was referred to as Chalaropsis populi, but no validly published description ever appeared (Kiffer and Delon 1983). Kiffer and Delon (1983) subsequently validated the name Chalaropsis populi, as Chalara populi, but in 2002 this species was once again redescribed and included in Thielaviopsis as T. populi (Paulin-Mahady et al. 2002). Most recently, the genus Chalaropsis was re-established and now includes three named species: Ch. ovoidea, Ch. populi, and Ch. thielavioides, although some evidence supports the recognition of a fourth taxon (de Beer et al. 2014).
Species of Chalaropsis are not considered of significant ecological or economic importance despite being predominantly isolated from diseased plant material (de Beer et al. 2014). The type species of the genus, Ch. thielavioides, is commonly associated with post-harvest moulding of carrot (Weber and Tribe 2004; Milosavljević et al. 2015; Xu et al. 2020), although the economic impact of the disease is negligible. Chalaropsis ovoidea is predominantly isolated from Fagus trees (and discoloured planks produced from the wood), but can occasionally be found in Quercus species as well (Nag Raj and Kendrick 1976; Kraj and Kowalski 2005). Chalaropsis populi was originally isolated from brown spots associated with trunk scab disease in Populus and Salix, prompting its description as a cambium killer (Kiffer and Delon 1983). A subsequent study also found Ch. populi in combination with other fungi from diseased roots of Populus and Euramericana trees, but the authors considered it a weak pathogen (Szabó and Harrington 2004).
The low level of importance of these pathogens has resulted in very little research effort focussed on Chalaropsis species. Nevertheless, the taxonomic positioning of Chalaropsis as a sister genus to many important pathogens in the genera Berkeleyomyces, Ceratocystis, and Endoconidiophora make this group of interest. A draft genome for Ch. thielavioides was generated by the RIKEN Center for Life Science Technologies, Division of Genomic Technologies, and is publicly available (GCA_001599435.1) (JCM-Riken 2016). In the current study, a draft genome sequence for Ch. populi is presented to accompany that of Ch. thielavioides. It is hoped that the availability of two genome sequences for Chalaropsis species will support future studies on comparative genomics, while also addressing the taxonomic complexities associated with asexual fungi.
Sequenced strain
Belgium: Gent: Moerzeke Populetum, isol. from necrosis in Populus gelrica, 1970, R. Veldeman (CMW 26388, CBS 486.71, CBS H-10141 - dried culture).
Nucleotide sequence accession number
This Whole Genome Shotgun project for Chalaropsis populi isolate CMW 26388 has been deposited at DDBJ/ENA/GenBank under the accession JADILG000000000. The version described in this paper is version JADILG010000000.
Materials and methods
Chalaropsis populi isolate CMW 26388 was obtained from the culture collection (CMW) of the Forestry and Agricultural Biotechnology Institute (FABI) based at the University of Pretoria and grown on 2% malt extract agar (MEA: 2% w/v, Biolab, South Africa) at 25 °C for the duration of the study. Genomic DNA was isolated from a 14-day old culture grown on a cellophane sheet using the DNeasy Plant Mini Kit (Qiagen, Germany). The isolated DNA was sent to the Agricultural Research Council Biotechnology Platform (ARC-BTP; Pretoria, South Africa) where it was used to prepare a pair-end library with an insert size of 500 bp. An Illumina HiSeq 2500 (Illumina, San Diego, CA) was used to generate 125 bp length reads from both ends of the insert.
The raw reads generated were imported and trimmed (using the Trim Sequences command and default values) in CLC Genomics Workbench v. 20.0.3 (CLCBio, Aarhus) before being used in a de novo assembly to generate a draft genome sequence. The untrimmed paired reads were also used for read-error correction and assembly with SPAdes v. 3.14.0 (Bankevich et al. 2012) using custom K-values (21, 33, 55, 77), applying the “careful” option to reduce mismatches and including the CLC-generated scaffolds as untrusted contigs. An estimation of the number of protein coding genes in the Ch. populi genome was made using the AUGUSTUS de novo prediction software with Fusarium graminearum gene models (Stanke et al. 2006a, 2006b; Keller et al. 2011). General genome statistics (genome length, GC content, N50 and L50 values) for the Ch. populi assembly were calculated using QUAST v. 5.0.1 (Gurevich et al. 2013), while both this genome and the Ch. thielavioides JCM 1933 assembly (JCM-Riken 2016) was assessed for completeness using the Benchmarking Universal Single Copy Orthologs tool (BUSCO v. 4.0.6) (Simão et al. 2015) using both the Fungi_odb10 and Ascomycota_odb10 datasets.
The publicly available genome sequence for Ch. thielavioides JCM 1933 was retrieved from the genome repository at the National Center for Biotechnology Information (NCBI) (JCM-Riken 2016). The beta-tubulin gene was extracted from the draft genome assemblies of both Ch. populi CMW 26388 and Ch. thielavioides JCM 1933 using CLC Genomics Workbench. These were used together with published sequences from Ch. ovoidea, Ch. thielavioides, Ch. populi, Ceratocystis adiposa and Berkeleyomyces basicola in a phylogenetic analysis to confirm the identity of the sequenced strains (Fig. 3). To do this, the one click mode phylogeny online tool (Dereeper et al. 2008, 2010) that included MUSCLE alignment (Edgar 2004), curation via Gblocks (Castresana 2000), PhyML steps (Guindon and Gascuel 2003) and a Maximum Likelihood test for branch support was used (Anisimova and Gascuel 2006). The tree was rooted using Ceratocystis adiposa and Berkeleyomyces basicola.
Fig. 3.

A Maximum Likelihood phylogeny based on the beta-tubulin gene from species of Chalaropsis. This analysis confirms the identity of the genome assembly presented here (shown in bold) as Chalaropsis populi. Interestingly, the publicly available Ch. thielavioides JCM 1933 genome (Genbank accession: GCA_001599435) grouped in the same clade as Ch. ovoidea. Ceratocystis adiposa and Berkeleyomyces basicola were used as outgroups, and the results for the approximate likelihood ratio test for branch support are shown as percentages
Results and discussion
The draft genome sequence of Ch. populi had a length of 23,877,278 bp present in 2158 contigs, of which 1398 were larger than 1000 bp. The genome had a GC content of 52.56%, an average coverage of 81x, a N50 value of 29,267 bp and a L50 value of 239. AUGUSTUS predicted 6654 protein coding genes, while BUSCO analysis reported a completeness score of 96.7 and 98.0% for Ch. populi for the respective Ascomycota and Fungi BUSCO datasets. This was based on the analysis of 1706 and 758 orthologs for the Ascomycota and Fungi datasets respectively, where 1650 and 743 were present and complete, while 48 and 13 copies were completely absent. The comparative BUSCO analysis for the Ch. thielavioides JCM 1933 genome assembly indicated a 84.1% (1435 complete and 266 missing BUSCOs) and 85.8% (650 complete and 108 missing BUSCOs) completeness for the Ascomycota and Fungi dataset.
Phylogenetic analysis using the beta-tubulin gene from the sequenced genome confirmed the identity of the isolate as Ch. populi (Fig. 3), although the publicly available genome sequence for Chalaropsis thielavioides JCM 1933 grouped closer to Ch. ovoidea than known Ch. thielavioides strains. When compared to the Ch. thielavioides genome, Ch. populi has a similar genome size (23,8 Mb for Ch. populi vs 23,3 Mb for Ch. thielavioides), although it is more fragmented (2158 vs 252 contigs) (JCM-Riken 2016). This is supported by the N50 values (29,267 bp for Ch. populi vs 161,617 bp for Ch. thielavioides). However, the Ch. populi genome was more complete based on the BUSCO assessments.
The Ch. populi draft assembly is the second genome sequence available for a Chalaropsis sp. This will support research efforts aimed at understanding the biology of these understudied fungal pathogens (Weber and Tribe 2004). For example, all three known species of Chalaropsis are considered asexual (de Beer et al. 2014), a stark contrast to the predominantly sexual species in the Ceratocystidaceae. Much work has been focussed on sexual reproduction in the family (e.g. Wilken et al. 2017; Nel et al. 2018; Simpson et al. 2018), and the availability of two genome sequences for putatively asexual members will be a valuable addition to this ongoing project. Together with the Ch. populi sequence, there are now 30 species residing in Ceratocystidaceae of which the genomes have been sequenced and these include representatives of ten genera (https://www.ncbi.nlm.nih.gov/datasets/genomes/?txid=1028423&term=Ceratocystidaceae&utm_source=assembly&utm_medium=referral&utm_campaign=:assemb). These sequences provide the opportunity to perform family-level analyses seeking to answer questions regarding speciation processes, host adaptations, and comparative genomics. They will also provide a basis for further functional studies (e.g. Sayari et al. 2019; Wilson et al. 2020) in Ceratocystidaceae.
Authors: Jostina R. Rakoma, Frances A. Lane, P. Markus Wilken*
* Contact: Markus.Wilken@fabi.up.ac.za
IMA GENOME– F 14D
Draft genome sequence of Chrysoporthe puriensis: the cause of a canket disease on Eucalyptus and Tibouchina
Introduction
The genus Chrysoporthe accommodates numerous economically important pathogens of plantation Eucalyptus species and other members of Myrtales (Gryzenhout et al. 2009). These fungi cause serious stem canker diseases, predominantly in tropical and subtropical parts of the world (Wingfield 2003). Chrysoporthe puriensis was first reported causing a stem canker disease on Tibouchina spp. in Brazil (Oliveira et al. 2021). Pathogenicity tests showed that C. puriensis is pathogenic on the hybrid Eucalyptus grandis × E. urophylla, suggesting that the fungus could threaten commercially-grown Eucalyptus plantations in South America (Oliveira et al. 2021). Similar to other Chrysoporthe spp. causing stem cankers on trees, C. puriensis is a potential threat to trees in Myrtales grown as non-natives for commercial purposes or where they are native. Countries such as Australia that has a mega-diverse Myrtales flora are especially vulnerable to these relatively wide host range pathogens (Burgess and Wingfield 2017); as has been seen for the globally spreading myrtle rust pathogen Austropuccinia psidii (Glen et al. 2007; Roux et al. 2016).
Genome sequences are available for three species of Chrysoporthe, which infect Eucalyptus. These include, C. austroafricana, C. cubensis, and C. deuterocubensis (Wingfield et al. 2015a, 2015b). The aim of this study was to sequence and assemble the genome of C. puriensis that will enable comparative genome studies focussed on further understanding the biology of Chrysoporthe species and to improve disease management strategies for them.
Sequenced strain
Brazil: Minas Gerais: Silveirânia, Tibouchina granulosa, 2018, M.E.S. Oliveira (CMW 54409).
Sequence accession numbers
The genome sequence of Chrysoporthe puriensis (isolate number CMW 54409) has been deposited in DDBJ/EMBL/GenBank databases under the accession numbers CP064894 - CP064907.
Materials and methods
Genomic DNA was extracted from freeze-dried mycelium of isolate CMW 54409 grown in malt yeast broth (2% malt extract, 0.5% yeast extract; Biolab, Midrand, South Africa) using the Qiagen® Genomic-tip DNA extraction protocol for plants and fungi. To verify the identification of the isolate, sequencing of the internal transcribed spacer (ITS) region and the partial β-tubulin gene (tub1 and tub2) was performed. The reference sequences were obtained from GenBank. Amphilogia gyrosa was used as an outgroup. Sequence datasets were aligned using an online version of MAFFT v.7 (Katoh and Standley 2013). A maximum likelihood analysis was performed using RAxML (Stamatakis 2014) using the GTR + G substitution model and branch support was calculated using 1000 bootstrap replicates.
Nanopore sequencing was conducted using the MinION sequencing device. The sequencing library was prepared using the Genomic DNA by Ligation (SQK-LSK109) protocol. The library was loaded on a MinION flow cell (R10.3) and sequencing run was carried out for 48 h. Base calling was conducted using ONT Guppy basecalling software v 4.0.14.
Nanopore reads were trimmed using Porechop (v0.2.1, https://github.com/rrwick/Porechop). The genome was assembled using Flye v 2.7 (Kolmogorov et al. 2019). The assembly was polished using Rebaler v0.2.0 (Wick et al. 2019), which runs multiple rounds of Racon v1.4.13 (Vaser et al. 2017) and followed by two rounds of polishing iterations with Medaka v1.0.3 (https://github.com/nanoporetech/medaka). Protein coding gene models were annotated using AUGUSTUS v.3.3 with Magnaporthe grisea as the model organism (Stanke and Morgenstern 2005). The assembled genome completeness was evaluated using the Benchmarking Universal Single-Copy Orthologs tool, BUSCO v. 4.1.3 by using the fungal lineage dataset (Simão et al. 2015).
Results and discussion
Phylogenetic analysis using three gene regions (ITS, tub1, and tub2) confirmed the taxonomic identity of isolate CMW 54409 as C. puriensis (Fig. 4). The assembly of C. puriensis consisted of 14 contigs, with the N50 of 4.78 Mb and L50 of 5. The calculated genome size was approximately 44.66 Mb and with a CG content of 53.91%. AUGUSTUS predicted 13,166 protein coding gene models in the assembled genome. BUSCO analysis using the sordariomycetes_odb10 dataset indicated the assembled genome to have a 98.3% completeness score. Of the 3817 BUSCO groups searches, 13 BUSCO orthologs were reported to be fragmented and 54 BUSCO groups were reported to be missing.
Fig. 4.
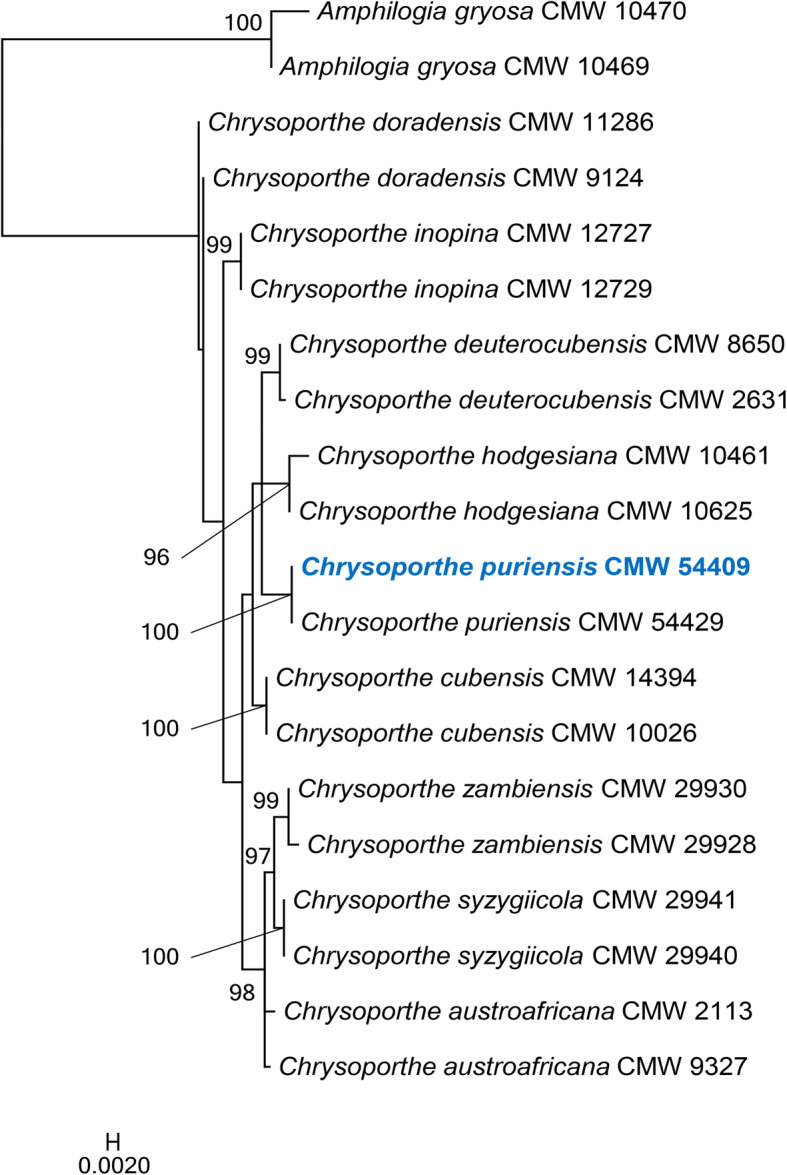
Maximum Likelihood tree based on ITS region and partial gene sequences of but1 and but2. Bootstrap values ≥65% are shown. The isolates used in this study are indicated in blue and bold
The estimated genome size and gene number for C. puriensis is similar to that of other Chrysoporthe species: Chrysoporthe ausroafricana (44.6 Mb, 13,484) (Wingfield et al. 2015a), C. cubensis (42.6 Mb, 13,121) (Wingfield et al. 2015b), C. deuterocubensis (43.9 Mb; 13,772) (Wingfield et al. 2015b). The draft genome sequence of C. puriensis generated here will be used for comparative genomics studies as well as to better understand its, biology and role as a tree pathogen.. Furthermore, the genome sequence will be useful for to develop molecular markers for population studies of the species and to determine its origin and pathways of movement in forests.
Authors: H. Suzuki, T.A. Duong*, M.A. Ferreira, M.J. Wingfield, B.D. Wingfield
*Contact: Tuan.Duong@fabi.up.ac.za
Acknowledgements
The authors of the contribution of Penicillium roqueforti CECT 2905T thank the support of DICYT-USACH. The authors of Penicillium roqueforti CECT 2905T study acknowledge the financial support of FONDECYT-Chile (projects numbers 1120833, 1150894 and 1170799) and CONICYT-Chile (fellowship CONICYT-PFCHA/Doctorado Nacional/2014-63140056). The authors thank the University of Pretoria, the Department of Science and Technology (DST)/National Research Foundation (NRF) Centre of Excellence in Tree Health Biotechnology (CTHB), South Africa, the Tree Protection Co-operative Programme (TPCP), the National Research Foundation and the DST-NRF SARChI chair in Fungal Genomics.
Adherence to national and international regulations
Not applicable.
Authors’ contributions
The authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Renato Chávez, Email: renato.chavez@usach.cl.
Lieschen De Vos, Email: lieschen.bahlmann@fabi.up.ac.za.
Tuan A. Duong, Email: Tuan.Duong@fabi.up.ac.za
P. Markus Wilken, Email: Markus.Wilken@fabi.up.ac.za.
References
- Anisimova M, Gascuel O. Approximate likelihood ratio test for branches: a fast, accurate and powerful alternative. Syst Biol. 2006;55:539–552. doi: 10.1080/10635150600755453. [DOI] [PubMed] [Google Scholar]
- Bainbridge BW, Spreadbury CL, Scalise FG, Cohen J. Improved methods for the preparation of high molecular weight DNA from large and small scale cultures of filamentous fungi. FEMS Microbiol Letters. 1990;54:113–117. doi: 10.1111/j.1574-6968.1990.tb03981.x. [DOI] [PubMed] [Google Scholar]
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blin K, Shaw S, Steinke K, Villebro R, Ziemert N, et al. antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Research. 2019;47:W81–W87. doi: 10.1093/nar/gkz310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess TI, Wingfield MJ. Pathogens on the move: a 100-year global experiment with planted eucalypts. Bioscience. 2017;67:14–25. doi: 10.1093/biosci/biw146. [DOI] [Google Scholar]
- Cantarel BL, Korf I, Robb SMC, Parra G, Ross E, et al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008;18:188–196. doi: 10.1101/gr.6743907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- Chávez R, Fierro F, García-Rico RO, Laich F (2011) Mold-fermented foods: Penicillium spp. as ripening agents in the elaboration of cheese and meat products. In: Leitao AL (ed) Mycofactories. Bentham Science Publisher Ltd, United Arab Emirates.
- Cheeseman K, Ropars J, Renault P, Dupont J, Gouzy J, et al. Multiple recent horizontal transfers of a large genomic region in cheese making fungi. Nature Communications. 2014;5:2876. doi: 10.1038/ncomms3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coton E, Coton M, Hymery N, Mounier J, Jany J-L. Penicillium roqueforti: an overview of its genetics, physiology, metabolism and biotechnological applications. Fungal Biol Rev. 2020;34:59–73. doi: 10.1016/j.fbr.2020.03.001. [DOI] [Google Scholar]
- Darribo D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9:772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Beer ZW, Duong TA, Barnes I, Wingfield BD, Wingfield MJ. Redefining Ceratocystis and allied genera. Stud Mycol. 2014;79:87–219. doi: 10.1016/j.simyco.2014.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del-Cid A, Gil-Durán C, Vaca I, Rojas-Aedo JF, García-Rico RO, et al. Identification and functional analysis of the mycophenolic acid gene cluster of Penicillium roqueforti. PLoS One. 2016;11:e0147047. doi: 10.1371/journal.pone.0147047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dereeper A, Audic S, Claverie JM, Blanc G. BLAST-EXPLORER helps you building datasets for phylogenetic analysis. BMC Evol Biol. 2010;10:8. doi: 10.1186/1471-2148-10-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, et al. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008;36:W465–W469. doi: 10.1093/nar/gkn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Bodega Á, Álvarez-Álvarez R, Liras P, Martín JF. Silencing of a second dimethylallyltryptophan synthase of Penicillium roqueforti reveals a novel clavine alkaloid gene cluster. Appl Microbiol Biotechnol. 2017;101:6111–6121. doi: 10.1007/s00253-017-8366-6. [DOI] [PubMed] [Google Scholar]
- Frisvad JC, Samson RA. Polyphasic taxonomy of Penicillium subgenus Penicillium - a guide to identification of food and air-borne terverticillate Penicillia and their mycotoxins. Stud Mycol. 2004;49:1–173. [Google Scholar]
- García-Rico RO, Chávez R, Fierro F, Martín JF. Effect of a heterotrimeric G protein α subunit on conidia germination, stress response, and roquefortine C production in Penicillium roqueforti. Int Microbiol. 2009;12:123–129. [PubMed] [Google Scholar]
- Gil-Durán C, Rojas-Aedo JF, Medina E, Vaca I, García-Rico RO, Villagrán S, et al. The pcz1 gene, which encodes a Zn (II)2Cys6 protein, is involved in the control of growth, conidiation, and conidial germination in the filamentous fungus Penicillium roqueforti. PLoS One. 2015;10:e0120740. doi: 10.1371/journal.pone.0120740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glen M, Aflenas AC, Zauza EAV, Wingfield MJ, Mohammed C. Puccinia psidii: a threat to the Australian environment and economy - a review. Australasian Plant Pathology. 2007;36:1–16. doi: 10.1071/AP06088. [DOI] [Google Scholar]
- Gryzenhout M, Wingfield BD, Wingfield MJ. Taxonomy, phylogeny, and ecology of bark-inhabiting and tree-pathogenic fungi in the Cryphonectriaceae. St. Paul, Minnesota U.S.A: The American Phytopathological Society; 2009. [Google Scholar]
- Guevara-Suarez M, García D, Cano-Lira JF, Guarro J, Gené J. Species diversity in Penicillium and Talaromyces from herbivore dung, and the proposal of two new genera of penicillium-like fungi in Aspergillaceae. Fungal Syst Evol. 2020;5:39–75. doi: 10.3114/fuse.2020.05.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W et al (2010) New algorithms and methods to estimate maximum- likelihood phylogenies: Assessing the performance of PhyML 3.0. Systematic Biology 59:307–21 [DOI] [PubMed]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herron DA, Wingfield MJ, Rodas CA, Marincowitz S, Steenkamp ET. Novel taxa in the Fusarium fujikuroi species complex from Pinus spp. Stud Mycol. 2015;80:131–150. doi: 10.1016/j.simyco.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo PI, Ullán RV, Albillos SM, Montero O, Fernández-Bodega MÁ, et al. Molecular characterization of the PR-toxin gene cluster in Penicillium roqueforti and Penicillium chrysogenum: cross talk of secondary metabolite pathways. Fungal Genet Biol. 2014;62:11–24. doi: 10.1016/j.fgb.2013.10.009. [DOI] [PubMed] [Google Scholar]
- Houbraken J, Kocsubé S, Visagie CM, Yilmaz N, Wang X-C, et al. Classification of Aspergillus, Penicillium, Talaromyces and related genera (Eurotiales): an overview of families, genera, subgenera, sections, series and species. Stud Mycol. 2020;95:5–169. doi: 10.1016/j.simyco.2020.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JCM-Riken BioResource Center . NBRP: Genome sequencing of Chalaropsis thielavioides JCM 1933. 2016. [Google Scholar]
- Katoh K, Rozewick J, Yamada KD. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2019;20:1160–1166. doi: 10.1093/bib/bbx108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usablility. Mol Biol Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller O, Kollmar M, Stanke M, Waack S. A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics. 2011;27:757–763. doi: 10.1093/bioinformatics/btr010. [DOI] [PubMed] [Google Scholar]
- Kiffer E, Delon R. Chalara elegans (Thielaviopsis basicola) and allied species. II - validation of two taxa. Mycotaxon. 1983;18:165–174. [Google Scholar]
- Kolmogorov M, Yuan J, Lin Y, Pevzner PA. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol. 2019;37:540–546. doi: 10.1038/s41587-019-0072-8. [DOI] [PubMed] [Google Scholar]
- Korf I. Gene finding in novel genomes. BMC Bioinformatics. 2004;5:59. doi: 10.1186/1471-2105-5-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosalková K, Domínguez-Santos R, Coton M, Coton E, García-Estrada C, et al. A natural short pathway synthesizes roquefortine C but not meleagrin in three different Penicillium roqueforti strains. Appl Microbiol Biotechnol. 2015;99:7601–7612. doi: 10.1007/s00253-015-6676-0. [DOI] [PubMed] [Google Scholar]
- Kraj W, Kowalski T. Identification of the polish strains of Chalara ovoidea using RAPD molecular markers. Acta Societatis Botanicorum Poloniae. 2005;74:35–42. [Google Scholar]
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvas M, Marasas WFO, Wingfield BD, Wingfield MJ, Steenkamp ET. Diversity and evolution of Fusarium species in the Gibberella fujikuroi complex. Fungal Diversity. 2009;34:1–21. [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie JF, Summerell BA. The Fusarium laboratory manual. Iowa: Blackwell; 2006. [DOI] [PubMed] [Google Scholar]
- Ma L-J, Van der Does HC, Borkovich KA, Coleman JJ, Daboussi M-J, et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature. 2010;464:367–373. doi: 10.1038/nature08850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milosavljević A, Trkulja N, Popović T, Ivanović Ž, Mitrović M, et al. First report of Thielaviopsis thielavioides, a causal agent of postharvest blackening on Daucus carota in Serbia. Plant Disease. 2015;99:1274–1275. doi: 10.1094/PDIS-12-14-1261-PDN. [DOI] [Google Scholar]
- Möller EM, Bahnweg G, Sandermann H, Geiger HH. A simple and efficient protocol for isolation of high molecular weight DNA from filamentous fungi, fruit bodies, and infected plant tissues. Nucleic Acids Res. 1992;20:6115–6116. doi: 10.1093/nar/20.22.6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nag Raj TR, Kendrick B. A monograph of Charala and allied genera. Waterloo: Wilfrid Laurier University Press; 1976. [Google Scholar]
- Nel WJ, Duong TA, Wingfield MJ, Wingfield BD, Hammerbacher A, et al. Heterothallism revealed in the root rot fungi Berkeleyomyces basicola and B. rouxiae. Fungal Biol. 2018;122:1031–1040. doi: 10.1016/j.funbio.2018.08.006. [DOI] [PubMed] [Google Scholar]
- Niehaus EM, Münsterkötter M, Proctor RH, Brown DW, Sharon A, et al. Comparative “omics” of the Fusarium fujikuroi species complex highlights differences in genetic potential and metabolite synthesis. Genome Biol Evol. 2017;8:3574–3599. doi: 10.1093/gbe/evw259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira MES, van der Merwe NA, Wingfield MJ, Wingfield BD, Soares TPF et al (2021) Chrysoporthe puriensis sp. nov. from Tibouchine spp. in Brazil: an emerging threat to Eucalyptus. Australasian Plant Pathol 50:29–40
- Paulin-Mahady AE, Harrington TC, McNew D. Phylogenetic and taxonomic evaluation of Chalara, Chalaropsis, and Thielaviopsis anamorphs associated with Ceratocystis. Mycologia. 2002;94:62–72. doi: 10.1080/15572536.2003.11833249. [DOI] [PubMed] [Google Scholar]
- Rojas-Aedo JF, Gil-Durán C, Del-Cid A, Valdés N, Álamos P, et al. The biosynthetic gene cluster for andrastin a in Penicillium roqueforti. Front Microbiol. 2017;8:813. doi: 10.3389/fmicb.2017.00813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas-Aedo JF, Gil-Durán C, Goity A, Vaca I, Levicán G, et al. The developmental regulator Pcz1 affects the production of secondary metabolites in the filamentous fungus Penicillium roqueforti. Microbiol Res. 2018;212:67–74. doi: 10.1016/j.micres.2018.05.005. [DOI] [PubMed] [Google Scholar]
- Roux J, Granados GM, Shuey L, Barnes I, Wingfield MJ, McTaggart AR. A unique genotype of the rust pathogen, Puccinia psidii, on Myrtaceae in South Africa. Australasian Plant Pathol. 2016;45:645–652. doi: 10.1007/s13313-016-0447-y. [DOI] [Google Scholar]
- Sayari M, van der Nest MA, Steenkamp ET, Adegeye OO, Marincowitz S, et al. Agrobacterium-mediated transformation of Ceratocystis albifundus. Microbiol Res. 2019;226:55–64. doi: 10.1016/j.micres.2019.05.004. [DOI] [PubMed] [Google Scholar]
- Seppey M, Manni M, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness. In: Kollmar M, editor. Gene prediction: methods and protocols, methods in molecular biology. New York: Humana; 2019. [DOI] [PubMed] [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- Simpson MC, Coetzee MPA, van der Nest MA, Wingfield MJ, Wingfield BD. Ceratocystidaceae exhibit high levels of recombination at the mating-type (MAT) locus. Fungal Biol. 2018;122:1184–1191. doi: 10.1016/j.funbio.2018.09.003. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Diekhans M, Baertsch R, Haussler D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics. 2008;24:637–644. doi: 10.1093/bioinformatics/btn013. [DOI] [PubMed] [Google Scholar]
- Stanke M, Morgenstern B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005;33:W465–W467. doi: 10.1093/nar/gki458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Schöffmann O, Morgenstern B, Waack S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics. 2006;7:62. doi: 10.1186/1471-2105-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Tzvetkova A, Morgenstern B. AUGUSTUS at EGASP: using EST, protein and genomic alignments for improved gene prediction in the human genome. Genome Biol. 2006;7:S11. doi: 10.1186/gb-2006-7-s1-s11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenkamp ET, Rodas CA, Kvas M, Wingfield BD. Fusarium circinatum and pitch canker of Pinus in Colombia. Australasian Plant Pathol. 2012;41:483–491. doi: 10.1007/s13313-012-0120-z. [DOI] [Google Scholar]
- Szabó I, Harrington TC. First report of Thielaviopsis populi on hybrid poplar in Hungary. Plant Pathol. 2004;53:249–249. doi: 10.1111/j.0032-0862.2004.00975.x. [DOI] [Google Scholar]
- Tavare S. Some probabilistic and statistical problems in the analysis of DNA sequences. In: Muira RM, editor. Some mathematical questions in biology: DNA sequence analysis. Providence, Rhode Island: The American Mathematical Society; 1986. [Google Scholar]
- Ter-Hovhannisyan V, Lomsadze A, Chernoff YO, Borodovsky M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Research. 2008;18:1979–1990. doi: 10.1101/gr.081612.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thom C. Fungi in cheese ripening: camembert and Roquefort. U.S.D.A. Bureau Anim Industry Bull. 1906;82:1–39. [Google Scholar]
- Torrent C, Gil-Durán C, Rojas-Aedo JF, Medina E, Vaca I, et al. Role of sfk1 gene in the filamentous fungus Penicillium roqueforti. Front Microbiol. 2017;8:2424. doi: 10.3389/fmicb.2017.02424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaser R, Sovic I, Nagarajan N, Sikic M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017;27:737–746. doi: 10.1101/gr.214270.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldeman R. ‘Chalaropsis sp.’ a new parasitic fungus on poplar, the cause of bark lesions. Mededelingen van de Facultiet Landbouwwetenschappen, Rijksuniversiteit Gent. 1971;36:1001–1005. [Google Scholar]
- Weber RW, Tribe HT. Thielaviopsis basicola and T. thielavioides, two ubiquitous moulds on carrots sold in shops. Mycologist. 2004;18:6–10. doi: 10.1017/S0269915X04001028. [DOI] [Google Scholar]
- Wick RR, Judd LM, Holt KE. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 2019;20:129–138. doi: 10.1186/s13059-019-1727-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiemann P, Sieber CMK, Von Bargen KW, Studt L, Niehaus E-M, et al. Unleashing the cryptic genome: genome-wide analyses of the rice pathogen Fusarium fujikuroi reveal complex regulation of secondary metabolism and novel metabolites. PLoS Pathogens. 2013;9:e1003475. doi: 10.1371/journal.ppat.1003475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilken PM, Steenkamp ET, van der Nest MA, Wingfield MJ, de Beer ZW, et al. Unexpected placement of the MAT1-1-2 gene in the MAT1-2 idiomorph of Thielaviopsis. Fungal Genet Biol. 2017;113:32–41. doi: 10.1016/j.fgb.2018.01.007. [DOI] [PubMed] [Google Scholar]
- Wilson AM, Wilken PM, van der Nest MA, Wingfield MJ, Wingfield BD. The novel Huntiella omanensis mating gene, MAT1-2-7, is essential for ascomatal maturation. Fungal Genet Biol. 2020;137:103335. doi: 10.1016/j.fgb.2020.103335. [DOI] [PubMed] [Google Scholar]
- Wingfield BD, Ades PK, Al-Naemi FA, Beirn LA, Bihon W, et al. Draft genome sequences of Chrysoporthe austroafricana, Diplodia scrobiculata, Fusarium nygamai, Leptographium lundbergii, Limonomyces culmigenus, Stagonosporopsis tanaceti, and Thielaviopsos punctulata. IMA Fungus. 2015;6:233–248. doi: 10.5598/imafungus.2015.06.01.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingfield BD, Bames I, De Beer ZW, De Vos L, Duong TA, et al. Draft genome sequences of Ceratocystis eucalypticola, Chrysoporthe cubensis, C. deuterocubensis, Davidsoniella irescens, Fusarium temperatum, Graphilbum fragrans, Penicillium nordicum, and Thielaviopsis musarum. IMA Fungus. 2015;6:493–506. doi: 10.5598/imafungus.2015.06.02.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingfield BD, Berger DK, Steenkamp ET, Lim H-J, Duong TA, et al. Draft genome of Cercospora zeina, Fusarium pininemorale, Hawksworthiomyces lignivorus, Huntiella decipiens and Ophiostoma ips. IMA Fungus. 2017;8:385–396. doi: 10.5598/imafungus.2017.08.02.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingfield BD, Bills GF, Dong Y, Huang W, Nel WJ, et al. Draft genome sequence of Annulohypoxylon stygium, Aspergillus mulundensis, Berkeleyomyces basicola (syn. Thielaviopsis basicola), Ceratocystis smalleyi, two Cercospora beticola strains, Coleophoma cylindrospora, Fusarium fracticaudum, Phialophora cf. hyalina, and Morchella septimelata. IMA Fungus. 2018;9:199–223. doi: 10.5598/imafungus.2018.09.01.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingfield BD, Liu M, Nguyen HDT, Lane FA, Morgan SW, et al. Nine draft genome sequences of Claviceps purpurea s. lat., including C. arundinis, C. humidiphila, and C. cf. spartinea, pseudomolecules for the pitch canker pathogen Fusarium circinatum, draft genome of Davidsoniella eucalypti, Grosmannia galeiformis, Quambalaria euclaypti, and Teratospahaeria destructans. IMA Fungus. 2018;9:401–418. doi: 10.5598/imafungus.2018.09.02.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingfield BD, Steenkamp ET, Santana QC, Coetzee MPA, Bam S, et al. First fungal genome sequence from Africa: a preliminary analysis. South African Journal of Science. 2012;108:1–9. [Google Scholar]
- Wingfield MJ. Daniel McAlpine memorial lecture increasing threat of diseases to exotic plantation forests in the southern hemisphere: lessons from Cryphonectria canker. Australasian Plant Pathol. 2003;32:133–139. doi: 10.1071/AP03024. [DOI] [Google Scholar]
- Xu K, Li J, Yang X, Zhang R, Li X, et al. Postharvest rot on carrot caused by Ceratocystis fimbriata and Chalaropsis thielavioides (≡ Thielaviopsis thielavioides) in China. J Gen Plant Pathol. 2020;86:322–325. doi: 10.1007/s10327-020-00919-1. [DOI] [Google Scholar]