

Abstract
Recombinant adeno-associated virus (rAAV) vectors have the unique property of being able to perform genomic targeted integration (TI) without inducing a double-strand break (DSB). In order to improve our understanding of the mechanism behind TI mediated by AAV and improve its efficiency, we performed an unbiased genetic screen in human cells using a promoterless AAV-homologous recombination (AAV-HR) vector system. We identified that the inhibition of the Fanconi anemia complementation group M (FANCM) protein enhanced AAV-HR-mediated TI efficiencies in different cultured human cells by ∼6- to 9-fold. The combined knockdown of the FANCM and two proteins also associated with the FANCM complex, RecQ-mediated genome instability 1 (RMI1) and Bloom DNA helicase (BLM) from the BLM-topoisomerase IIIα (TOP3A)-RMI (BTR) dissolvase complex (RMI1, having also been identified in our screen), led to the enhancement of AAV-HR-mediated TI up to ∼17 times. AAV-HR-mediated TI in the presence of a nuclease (CRISPR-Cas9) was also increased by ∼1.5- to 2-fold in FANCM and RMI1 knockout cells, respectively. Furthermore, knockdown of FANCM in human CD34+ hematopoietic stem and progenitor cells (HSPCs) increased AAV-HR-mediated TI by ∼3.5-fold. This study expands our knowledge on the mechanisms related to AAV-mediated TI, and it highlights new pathways that might be manipulated for future improvements in AAV-HR-mediated TI.
Keywords: AAV, genome editing, homologous recombination, targeted integration, AAV-HR, GeneRide, CRISPR/Cas9, nuclease-free
Graphical Abstract
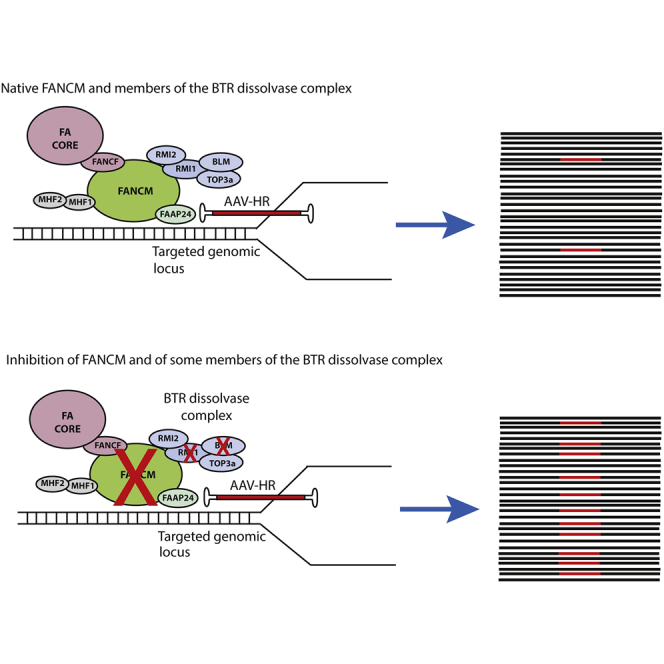
Genome editing using AAV vectors has been used to site-specifically modify the host genome. In order to better understand and potentially improve genome-editing efficiencies, de Alencastro et al. identified that the inhibition of FANCM and members of the BTR dissolvase complex increased the efficiency of AAV-mediated genome editing.
Introduction
Classical recombinant adeno-associated virus (rAAV) vectors have shown promise in a number of recent clinical trials.1, 2, 3, 4 However, the episomal nature of the AAV genome results in a significant decline of AAV-mediated gene expression due to cell division during organ regeneration and development,5,6 representing a significant limitation for the treatment of many pediatric diseases and stem cell-based therapies. The inherent ability of AAV to integrate into genomic loci has been well characterized in the past, and the inverted terminal repeats (ITRs) have been shown to drive this mechanism since they possess recombinogenic properties by activation of DNA damage response pathways.7, 8, 9 With the aim of achieving targeted integration (TI) at specific loci, various groups have modified AAV cassettes, introducing homologous sequences that flank the gene of interest, taking advantage of an upstream endogenous promoter for driving transgene expression. This strategy, also referred to as AAV-homologous recombination (HR) (AAV-HR), has been exploited for various in vivo genome-editing approaches, alone or in combination with nucleases.10, 11, 12 Several laboratories, including ours, have described TI efficiencies of up to 1% when using AAV-HR vectors without nucleases in liver in vivo.11,13 Nevertheless, the efficiency of TI using AAV-HR vectors can change depending on the cell type, the AAV dose, and the cell cycle. Even though the TI rates are higher when a nuclease is used to induce a DNA break at a specified genomic site, there are additional challenges associated with their use, such as potential off-targeting events14, 15, 16, 17 and the need to utilize multiple vectors to deliver the nuclease machinery to the cells in certain applications.18, 19, 20
Results
In order to identify cellular factors that influence the efficiency of AAV-HR-mediated TI, we performed an unbiased genome-wide screen in a library of near haploid human cells (HAP1) mutagenized by retroviral insertions.21,22 This library, which carries knockouts in nearly all non-essential genes due to the retrovirus insertions, has been successfully used in several different genome-wide screens such as in the identification of the AAVR receptor.22 Building upon a previously described vector design,13 we developed a promoterless AAV vector containing homology arms targeting the highly expressed human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) locus in HAP1 cells and expressing the blasticidin resistance gene (BLAS) (termed AAV-GAPDH(Hap1)-2A-BLAS) (Figure 1A). In cells undergoing TI, the BLAS gene is expressed from a chimeric mRNA that produces both GAPDH and BLAS using a P2A ribosomal skipping sequence. We rationalized that HAP1 cells containing knockouts in genes involved in repressing AAV-HR-mediated TI would be enriched after selection with blasticidin after infection with the GAPDH(Hap1)-2A-BLAS vector. Importantly, random integration in a non-targeted portion of the genome would unlikely result in activation of neighboring sequences due to the lack of a vector-encoded promoter. We chose the AAV-DJ capsid for packaging the AAV-GAPDH(Hap1)-2A-BLAS construct given its high efficiency in transducing cells in vitro23 (>90% of the HAP1 cells transduced when a multiplicity of infection (MOI) >10,000 was used). The functionality of the AAV-GAPDH(Hap1)-2A-BLAS vector was confirmed by establishing precise targeting of the GAPDH locus in blasticidin-resistant wild-type HAP1 cells, as determined by PCR analysis on clonal cell populations and Southern blot analysis on pooled cells (Figures S1A–S1C).
Figure 1.

Experimental Design for Targeting the GAPDH Locus Using a Promoterless AAV Vector and Its Use for the Identification of Factors Associated with AAV-HR-Mediated TI in the HAP1 Mutagenesis Screen
(A) Scheme of AAV-GAPDH(Hap1)-2A-BLAS targeting construct. The resulting recombination event results in expression of a single mRNA that is translated into two proteins, the one encoded by the endogenous GAPDH genomic locus and the transgene encoded by the vector (blasticidin resistance gene). Expression of blasticidin resistance is driven by the endogenous GAPDH promoter. (B) Scheme of the HAP1 mutagenesis screen for factors associated with AAV-mediated TI. (C) The network containing 21 identified proteins in the HAP1 mutagenesis screen (indicated with a star) as well as known NHEJ and HR factors were mapped using the STRING software (https://string-db.org/) where the links between proteins represent possible interactions. Different line colors represent the types of evidence for the interactions. Known interactions are indicated by the light blue line (curated database) and the pink line (experimentally determined). Predicted interactions are indicated by a red line (gene fusion evidence), green line (neighborhood evidence), dark blue line (co-occurrence evidence), yellow line (text-mining evidence), black line (co-expression evidence), and purple line (protein homology evidence).
The HAP1 screen was performed using a MOI of 16,000 and two different blasticidin concentrations, one moderate and another more stringent, in order to detect the most hits independent of the degree of selective pressure used. After antibiotic selection to eliminate clones that had not undergone TI, linear amplification-mediated (LAM)-PCR followed by deep-sequencing analysis of insertion sites was performed to identify the enriched knockouts (Figure 1B). Several genes associated with DNA maintenance were identified. Those genes that were identified in both blasticidin selection conditions, not present in previous HAP1 screens, and had a known possible biological function associated with TI and AAV biology were selected to be analyzed. The full dataset is presented in Table S1. The candidate genes and their interactions with the main factors associated with DNA repair, from both the HR and non-homologous end joining (NHEJ) pathways (Table S1), were determined using the STRING software for protein interaction analysis (Figure 1C).
One factor that was enriched in both screens and known to interact with several proteins of the DNA repair machinery was FANCM (Fanconi anemia [FA] complementation group M). In order to assess whether FANCM was involved in repression of AAV-HR-mediated TI, FANCM knockout HAP1 cell lines were generated using the CRISPR-Cas9 system,24 and RNAi was used to further validate this hypothesis (Figure 2A). Two clones containing frameshift insertions or deletions (indels) in exon 1 were generated (FANCM#B12, 32-bp deletion; FANCM#C1, 77-bp insertion) (Figure S2A), and their knockout was confirmed by western blot analysis (Figure 2B). Next, both HAP1 FANCM knockout cell clones as well as their wild-type counterparts were transduced with an AAV-HR vector containing the GFP coding sequence (AAV-GAPDH(Hap1)-2A-GFP) (Figure S2B), so that positive TI events could be quantified by flow cytometry analysis (Figure S2C). As in the previous experiment, precise targeting of the GAPDH locus was confirmed by Southern blot analysis on GFP-sorted cells (Figure S2D). Forty-eight hours after AAV-HR transduction we observed an ∼6- to 8-fold increase in GFP-expressing cells in the two knockout clones compared to the wild-type cells (Figure 2C; Figure S3). Next, in order to demonstrate that the increase in the TI efficiency was not restricted to HAP1 cells, we introduced a variety of indels using the CRISPR-Cas9 system in the FANCM gene in HeLa cells after transfection with two different single guide RNAs (sgRNA) targeting exons 1 and 4 (indel frequency was found to be approximately 78% [exon 1] and 36% [exon 4] for each guide by TIDE [tracking of indels by decomposition]25) (Figures S4A and S4B). Due to a single-nucleotide polymorphism (SNP) in the targeted GAPDH sequence between HeLa and HAP1 cells, a new AAV-HR vector with GAPDH homology arm sequences based on the gene sequence in HeLa cells was generated (termed AAV-GAPDH(HeLa)-2A-GFP) and packaged using the AAV-DJ capsid. Similar to what we had observed in HAP1 clonal knockout cells, TI efficiency was increased ∼9-fold in the HeLa cell pool that had been treated with the FANCM sgRNA as compared to the pool that had been treated with a scramble sgRNA control (Figures 2D; Figure S4C). In addition, we detected an increase of ∼7-fold in the TI levels after reducing the FANCM mRNA in HeLa cells to ∼38% of the wild-type level using a small interfering RNA (siRNA) knockdown strategy (Figure 2E; Figure S4D). This independent verification supported our observation that the increase in the TI efficiency was not a consequence of potential off-target events from the CRISPR-Cas9 system and suggested that a full knockout of FANCM is not required to achieve a substantial increase in TI using AAV-HR vectors.
Figure 2.

Characterization of the FANCM Knockout Cells and Transduction Analysis
(A) Schematic representation of experimental design used for specific knockout/knockdown of FANCM. (B) Western blot analysis of wild-type HAP1 cells and both FANCM knockout clones (FANCM molecular weight, 238 kDa). The loading control is a non-specific band recognized by the anti-FANCM antibody. (C) GFP expression in wild-type and the FANCM knockout HAP1 clones after transduction with AAV-GAPDH(Hap1)-2A-GFP (MOI of 16,000). Cells were plated in triplicate. (D) GFP expression in FANCM knockout HeLa cells after transduction with AAV-GAPDH(HeLa)-2A-GFP (MOI of 50,000). Cells were plated in triplicate. (E) GFP expression in HeLa cells initially transfected with siRNA-FANCM for 72 h followed by AAV-GAPDH(HeLa)-2A-GFP transduction (MOI of 50,000). Cells were plated in quadruplicate. For (C)–(E), GFP expression was analyzed 48 h post-transduction by flow cytometry analysis. Statistical analysis: (C) one-way ANOVA with Dunnet’s post hoc test; (D and E) unpaired t test. Error bars represent the standard deviation of the mean. ∗∗∗∗p < 0.0001, for differences between groups.
The FANCM protein is known to interact with several different proteins to execute its various cellular functions.26 In order to evaluate whether the inhibition of other proteins that interact with FANCM results in increased AAV-HR-mediated TI levels, we first analyzed whether proteins known to interact with FANCM (Figure S5A) were also enriched in the HAP1 screen. We identified the RecQ-mediated genome instability 1 (RMI1) and FANCF genes (Table S1). The former gene product is part of the Bloom DNA helicase (BLM)-topoisomerase IIIα (TOP3A)-RMI (BTR) dissolvase complex and was more enriched in the screen than the FANCF gene (Table S1). Based on this observation, we used the same workflow as described for FANCM (Figure 2A). We generated RMI1 HAP1 knockout cells and selected two clones for further analysis (RMI1#D10, 2-bp deletion; RMI1#E5, 61-bp insertion) (Figure S5B). Their full knockout was confirmed by western blot (Figure 3A). After transduction with the AAV-GAPDH(Hap1)-2A-GFP vector, an ∼3-fold increase in TI compared to wild-type cells was detected (Figure 3B; Figure S5C). We verified the enhanced RM1-mediated TI in HeLa cells by generating a pool of knockouts using the CRISPR-Cas9 system with two different sgRNAs for exon 1 of RMI1 (indel frequency was found to be approximately 46% and 49% for each guide by TIDE;25 Figures S6A and S6B) followed by AAV-GAPDH(HeLa)-2A-GFP vector transduction. Similar to the data obtained with the FANCM protein knockout, AAV-HR-mediated TI was increased ∼7-fold compared to sgRNA-scramble control-treated cells (Figure 3C; Figure S6C).
Figure 3.

Characterization of the Association of BTR Dissolvase Complex with FANCM and the Effect on AAV-HR-Mediated TI
(A) Western blot analysis of HAP1 wild-type and both RMI1 knockout clones (RMI1 molecular weight, 75 kDa). GAPDH was used as the loading control. (B) GFP expression in wild-type and the RMI1 knockout HAP1 clones after transduction with AAV-GAPDH(Hap1)-2A-GFP (MOI of 16,000). Cells were plated in triplicate. (C) GFP expression in RMI1 knockout HeLa cells after transduction with AAV-GAPDH(HeLa)-2A-GFP (MOI of 50,000). Cells were plated in triplicate. (D) GFP expression in HeLa cells initially transfected with siRNA for FANCM, RMI1, BLM, and scramble for 72 h followed by AAV-GAPDH(HeLa)-2A-GFP transduction (MOI of 50,000). Two biological replicates starting with separate transfection experiments were performed (n = 2 and n = 3). For (B)–(D), GFP expression was analyzed 48 h post-transduction by flow cytometry analysis. Statistical analysis: (B and D) one-way ANOVA with Dunnett’s post hoc test; (C) unpaired t test. Error bars represent the standard deviation of the mean. ∗∗p < 0.01, ∗∗∗∗p < 0.0001, for differences between groups.
In order to address whether the simultaneous inhibition of both FANCM and RMI1 would result in even higher levels of AAV-HR-mediated TI, we transfected HeLa cells with different combinations of siRNAs directed against FANCM, RMI1, and BLM. We decided to include a siRNA against BLM, as inhibition of this BTR dissolvase complex protein has been previously described to be associated with an increase in the level of gene targeting.27, 28, 29 Seventy-two hours after siRNA transfection, a portion of the cells were harvested for mRNA analysis while the remainder were transduced with AAV-GAPDH(HeLa)-2A-GFP. At the time of AAV-GAPDH(HeLa)-2A-GFP transduction, knockdown efficiency of FANCM, RMI1, and BLM was shown to be approximately 35%, 33%, and 23% relative to wild-type levels, respectively. As expected, the TI frequencies were ∼5-, ∼1.5-, and ∼2-fold higher in cells transfected with siRNAs for FANCM, RMI1, and BLM, respectively, compared to cells transfected with the siRNA-scramble control (Figure 3D; Figure S7A). Knockdown of RMI1 in combination with BLM had a small additive effect, supporting the fact that these proteins are associated with the same pathway. Remarkably, when the FANCM knockdown was combined with either RMI1 or BLM, the AAV-HR-mediated TI frequency was further enhanced to ∼7- and ∼14-fold, respectively. With simultaneous knockdown of all three factors, a ∼17-fold enhancement of AAV-HR-mediated TI compared to controls was obtained (Figure 3D; Figure S7A).
Even though FANCM and RMI1 were identified in the screen in the absence of nucleases, we wanted to address whether their inhibition would also enhance AAV-HR-mediated TI when used in combination with a nuclease. Thus, we designed five sgRNAs that target the genomic GAPDH (sgRNA-GAPDH) but not the AAV-HR vector sequence. Each sgRNA was individually tested by transfection into HeLa cells, and the sgRNA capable of inducing the highest levels of indels was chosen for the experiment (∼32% for guide#13 analyzed by TIDE25). Both sgRNA and AAV-GAPDH(Hap1)-2A-GFP were delivered to the HAP1 knockout clones (FANCM#B12 and RMI#D10) as well as wild-type HAP1 cells. TI levels were quantified 48 h later by flow cytometry (Figure 4A). As shown in Figures 4B and S7B, knockout of both FANCM and RMI1 increased the TI efficiency compared to wild-type cells by ∼1.5- and 2-fold, respectively.
Figure 4.

FANCM, RMI1, and TP53BP1 Knockout and Their Consequence on AAV-HR-Mediated TI
(A) Scheme of the nuclease-mediated AAV-HR assay using HAP1 cells. (B) GFP expression in wild-type, FANCM, and RMI1 knockout HAP1 clones (FANCM#B12 and RMI1#D10) initially electroporated with a plasmid expressing both Cas9 and sgRNA-GAPDH followed by AAV-GAPDH(Hap1)-2A-GFP transduction (MOI of 800). Two biological replicates using separate electroporations were performed for wild-type and FANCM samples (n = 2 in each experiment) and only one for the RMI1 sample (n = 2). The indel frequency was estimated using TIDE software to be approximately 10% in each cell clone. (C) GFP expression in wild-type, TP53BP1, and FANCM#B12 knockout HAP1 clones transduced with AAV-GAPDH(Hap1)-2A-GFP (MOI of 16,000). Cells were plated in triplicate. (D) Same experiment as in (B) but using the TP53BP1 knockout clone instead of RMI1. The indel frequency was estimated using TIDE software to be approximately 10% in each cell clone. For (B)–(D), GFP expression was analyzed 48 h post-transduction by flow cytometry analysis. Statistical analysis: (B–D) one-way ANOVA with Dunnett’s post hoc test. Error bars represent the standard deviation of the mean. ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, for differences between groups. ns, not significant.
Inhibition of NHEJ factors has been shown to improve the TI efficiency in both nuclease-mediated and nuclease-free approaches.30, 31, 32, 33, 34 However, some of the most well-studied NHEJ factors (LIG4, PRKDC, TP53BP1, XRCC5 [Ku86], and XRCC6 [Ku70]) were not enriched in our HAP1 screen (Table S1) in spite of the fact that all are expressed in HAP1 cells (https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-5867/samples/). In addition, because only XRCC6 (Ku70) and XRCC5 (Ku86) are known to be essential in human somatic cells,31,35 the knockout of the other proteins should not be lethal in HAP1 cells. Thus, we decided to test whether the inhibition of TP53BP1, a central protein in the NHEJ pathway that inhibition has recently been shown to increase nuclease-mediated TI levels,30,36,37 could also increase nuclease-free AAV-HR-mediated TI. We treated the TP53BP1 knockout HAP1 cell line (Horizon) (Figure S8A), wild-type HAP1 cells, and the FANCM knockout HAP1 clone (#B12) with the AAV-GAPDH(Hap1)-2A-GFP vector, and found that TI was similar in both the control and TP53BP1 knockout cell lines but significantly enhanced in the FANCM knockout line (Figure 4C; Figure S8B). In contrast, when the CRISPR-Cas9 system was used to induce DNA breaks in the GAPDH locus, AAV-HR-mediated TI was similarly enhanced in both the knockout cell lines (∼1.5- to 1.7-fold compared to wild-type) (Figure 4D; Figure S8C). Therefore, the abrogation of TP53BP1 enhances nuclease-dependent but not nuclease-free AAV-HR mediated TI.
In order to test our approach in a primary human stem cell context to assure that the findings were not specific to transformed cancer cell lines, we used human CD34+ hematopoietic stem and progenitor cells (HSPCs), which are extensively utilized for ex vivo cell therapies.38 We first tested siRNA transient knockdown of FANCM in CD34+ HSPCs. We used nucleofection as the transfection method since it has been successfully applied to deliver the ribonucleoprotein (RNP) complex Cas9/sgRNA in these types of cells.39,40 Upon siRNA-FANCM nucleofection, we were able to detect an ∼70% reduction in FANCM gene expression in CD34+ HSPCs after 48 h (Figure 5A). However, the knockdown of the target genes was not as efficient when we used a triple siRNA treatment to simultaneously knock down FANCM, BLM, and RMI1 in HSPCs (Figure 5A). Next, we designed an experiment in which we combined the siRNA-FANCM nucleofection with an AAV-HR vector using a MOI of 500,000 (Figure 5B). For this experiment, we used a promoterless AAV6 vector containing the IL2RG cDNA (AAV6-IL2RG), which had been previously characterized in combination with CRISPR-Cas9 nuclease transfection.40 Anti-FANCM or scrambled control siRNAs were nucleofected into CD34+ HSPCs, and after 48 h the cells were transduced with the AAV6-IL2RG vector. Forty-eight hours after transduction, cells were analyzed for IL2RG cDNA integration by Droplet Digital PCR (ddPCR). CD34+ HSPCs from two different human umbilical cord blood male donors (donors 3 and 4) treated with siRNA against FANCM showed an ∼2.7- and ∼4.4-fold increase in the AAV-HR-mediated TI levels, respectively, compared to the siRNA-scramble control (Figure 5C). Importantly, the combinatorial treatment with the three different siRNAs targeting FANCM, BLM, and RMI1 did not result in a further increase of TI (∼2.4-fold in the cells from donor 4) when compared to the cells treated with a siRNA against FANCM only (Figure 5C, donor 4), perhaps due to less efficient siRNA knockdown using three siRNAs in the same transfection.
Figure 5.

FANCM Knockdown Increased AAV-HR-Mediated TI in CD34+ HSPCs
(A) mRNA levels in CD34+ HSPCs from donor 1 upon siRNA-FANCM, siRNA scramble, and the triple combination siRNA-FANCM+BLM+RMI1 nucleofection treatment. RNA analysis was performed 48 h after siRNA nucleofection. Data are shown as fold change over the siRNA scramble group. Cells were plated in duplicate. (B) Schematic representation of experimental design used to test nuclease-free AAV6-IL2RG in CD34+ HSPCs and the ddPCR design. (C) Quantification of AAV6-IL2RG integration upon siRNA-FANCM and siRNA scramble nucleofection (donor 3) and siRNA-FANCM, siRNA scramble and the triple combination siRNA-FANCM+BLM+RMI1 (donor 4) nucleofection followed by AAV6-IL2RG transduction (MOI of 500,000). AAV transduction without siRNA treatment was used as a positive control. The two graphs represent two different CD34+ HSPC donors. Cells were plated in duplicates, treated with siRNA, and infected with AAV6-IL2RG. Data are shown as mean values of ddPCR measurements. Statistical analysis: (A and C) one-way ANOVA with Tukey’s post hoc test. Error bars represent the standard deviation of the mean. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, for differences between groups. ns, not significant.
Discussion
There is a need to increase the efficiency of TI in different gene-targeting approaches in order to expand their application. In this study, we performed an unbiased genome-wide screen in human cells that resulted in the identification of proteins that repress the AAV-HR-mediated TI process possibly through different mechanisms. To our knowledge, this is the first time that inhibition of either FANCM or RMI1 protein levels has been shown to increase AAV-HR-mediated TI in mammalian cells.
FANCM is a large protein (2,048 aa), with different isoforms and domains, and the most highly conserved protein of the FA pathway. FANCM interacts with a number of different proteins and plays an important role in genome maintenance, such as repair of DNA crosslinks, prevention of sister chromatid exchanges, disruption of D-loops, prevention of chromosomal breakage, and repair of stalled DNA replication forks.41,42 In order to perform specific functions, such as binding to inter-strand crosslink DNA, FANCM recruits and interacts with two distinct complexes of the DNA repair machinery: the FA core and the BTR dissolvase complexes.43 BTR is a large protein complex that consists of BLM, TOP3α, and the RMI sub-complex (RMI1 and RMI2).44 This complex is also associated with genome maintenance, mainly operating on DNA structures resembling replication and recombination intermediates such as double-Holliday junctions, replication forks, and G-quadruplexes.45 It is known that RMI1 and RMI2 physically anchor the BTR complex to FANCM, and impairment of this interaction has been shown to result in genome instability by compromising the complex’s ability to dissociate mobile D-loops and thus increasing sister chromatid exchange events.43,46, 47, 48, 49 The HR process is complex, and different pathways and proteins can be exploited for DNA repair, such as break-induced replication, double-Holliday junction, and synthesis-dependent strand annealing. As shown in this study, the inhibition of FANCM in association with two proteins of the BTR complex further enhanced the levels of AAV-HR-mediated TI, suggesting that they are involved in two different pathways. In addition, the observation that cells deficient in these proteins have distinct phenotypes supports the hypothesis that these proteins are not redundant and may act in different cellular processes. Importantly, we speculate that higher levels of TI may be obtained in primary cells by optimizing siRNA and vector delivery in order to achieve more efficient knockdown and template availability, respectively.
Although mutations in many of the FA complementation groups have been associated with the FA recessive disorder, no monogenic disease has been linked to mutations in the FANCM gene, making its function less clear. Moreover, the Fancm knockout mouse model has been shown to present a normal lifespan with no major pathological findings with the exception of increased likelihood of developing cancer at a late stage of their life.50 Nevertheless, mutations in FANCM have previously been reported to be associated with cancer predisposition in some patients, mainly in those with breast cancer.51, 52, 53 Mutations in BLM, on the other hand, are known to cause Bloom syndrome, a disease characterized by genome instability.54 To our knowledge, no diseases have been linked to mutations in the RMI1 gene, although a genetic variant in the RMI1 gene appears to be associated with acute myeloid leukemia and myelodysplastic syndromes.55 Importantly, knockout of RMI1 is embryonic lethal in mice,56,57 demonstrating an important role of RMI1 during development. Strategies to transiently inhibit these proteins for a very limited time and in a specific tissue using small molecule drugs or siRNAs prior to AAV-HR vectors exposure are unlikely to result in long-term negative consequences, but additional studies are required to confirm this premise.
Inhibition/blockage of the NHEJ pathway has been successfully used to increase the efficiency of nuclease-mediated TI30,32 and nuclease-free TI.31,33 However, we did not detect any of the most important factors of the NHEJ pathway (described above) enriched in our nuclease-free AAV-HR HAP1 screen (Table S1). In addition, we did not detect any increase in the nuclease-free AAV-HR-mediated TI in TP53BP1 knockout HAP1 cells, even though we showed that its absence indeed enhanced the TI levels when the CRISPR-Cas9 system was used to induce DNA breaks. Although we cannot exclude the possibility of the HAP1 cells with knockouts in genes associated with the NHEJ pathway having a selective disadvantage, our findings suggest that in the absence of inducing DNA breaks, inhibition of the NHEJ pathway may have little or no effect on the AAV-HR-mediated TI levels in nuclease-free systems. Possibly, this is due to the fact that the NHEJ machinery does not compete with the main pathway associated with nuclease-free AAV-HR-mediated TI. Further studies are needed to address whether this is true for the inhibition of other NHEJ factors as well as in different nuclease-free TI approaches.
The absence of FANCM and RMI1 was also shown to improve AAV-HR-mediated TI when the CRISPR-Cas9 system was used. However, the level of TI enhancement was not as prominent as what had been observed in the absence of nucleases. One of the reasons may be that nuclease-mediated TI occurs through a different pathway in which FANCM and RMI1 may not have a major role. In addition, nuclease-mediated TI may be approaching saturation in HAP1 cells, which is directly correlated with the levels of breaks induced with the sgRNA. Nevertheless, a 2-fold increase in nuclease-mediated TI is substantial and may provide benefit for this type of approach where less template and nuclease may be required to reduce potential toxicity.
Finally, we also demonstrated a significant increase of nuclease-free AAV-HR-mediated TI in the clinically relevant human CD34+ HSPCs upon FANCM knockdown. Although the increase in TI was slightly lower than those achieved in HAP1 and HeLa cells, the discrepancy among the different cells might be due to the differences in the mechanism of TI utilized by the cells relative to their status (immortalized versus primary), their cell cycle (dividing versus non-dividing), intrinsic donor variations, and different levels of FANCM, RMI1, and BLM expression, or it might in part be due to technical differences resulting in different knockdown efficiencies in the cell systems used. Further investigation of those differences may allow us to improve the AAV-HR-mediated TI levels in human CD34+ HSPCs up to the levels we have achieved in HeLa and HAP1 cells, especially if combined with cell cycle induction prior to AAV transduction or enhancing AAV delivery by electroporation-aided transduction (EAT)58. Several clinical trials using modified CD34+ HSPCs through the use of nucleases or lentiviral vectors are already underway.59,60 However, reducing the risk of genotoxicity using a safer nuclease-free approach to engineering CD34+ HSPCs might be envisioned for future clinical trials through a better understanding of the AAV-HR-mediated TI mechanism that will lead to improved nuclease-free gene-editing technologies.
Materials and Methods
Construction of Plasmids
A human genomic GAPDH segment was PCR amplified from DNA obtained from HAP1 cells using GAPDH-F1 (5′-GACTGTACAGGGCTGCTCACATATTCTGG-3′) and GAPDH-R4 (5′-CTGTGTACAGAGTGTATGTGGCTGTGGCCC-3′) (both containing BsRG1 sites for cloning) (3,481 bp) and inserted between AAV2 ITRs into BsrGI restriction sites in a modified pTRUF backbone.61 The genomic segment spans approximately 1,700 bp upstream and 1,700 bp downstream to the GAPDH stop codon. We then synthesized a 1,359-bp fragment spanning the region at the end of the GAPDH locus between the two SexA1 sites to be cloned in the vector. In this fragment, the GAPDH stop codon was removed, and it was inserted in an optimized P2A coding sequence preceded by a linker coding sequence (glycine-serine-glycine) and followed by the BLAS or GFP cDNA (without the start codon) to get pAB269-GAPDH-2A-BLAS and pAB269-GAPDH-2A-GFP that served in the construction of the rAAV-DJ targeting vector. Restriction enzymes were obtained from New England Biolabs. The plasmids for rAAV production were generated using an EndoFree plasmid megaprep kit (QIAGEN).
rAAV Vector Production and Titration
rAAV-DJ vectors were produced as previously described using a Ca3(PO4)2 transfection protocol followed by CsCl gradient purification.61 AAV vectors containing BLAS and GFP were titered on GAPDH and GFP, respectively, by TaqMan qPCR with the following primer/probe sets: GAPDH, forward, 5′-GCACCACTACTTCAGAGAAC-3′, reverse, 5′-CCCACAGAATAGCTTCTTCC-3′, probe, 5′-FAM-TTTCCTCTCCTCGCTCCAGTCCTA-BHQ-1-3′; GFP, forward, 5′-GACGTAAACGGCCACAAGTT-3′, reverse, 5′-GAACTTCAGGGTCAGCTTGC-3′, probe, 5′-FAM-CGAGGGCGATGCCACCTACG-BHQ-1-3′. For CD34+ HSPC experiments, the AAV6-IL2RG virus was used40 (Figure 1B in Pavel-Dinu et al.,40 NGFR construct). AAV6-IL2RG vector production and characterization have been described previously.40
Cell Lines
All cells were grown in media supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, and 100 IU/mL penicillin/streptomycin in a humidified incubator at 37°C with 5% CO2. HAP1 cells21 were cultured in complete Iscove’s modified Dulbecco’s medium (IMDM) media. HeLa cells (ATCC catalog no. CCL-2) were cultured in complete DMEM media supplemented with 1 mM sodium pyruvate.
Virus Transductions
HAP1 cells were seeded at 1.5 × 105 cells/well (24-well plate) and infected with AAV at an MOI of 16,000 viral genomes/cell (unless otherwise specified) in complete HAP1 medium. HeLa cells were seeded at 0.5 × 105 cells/well (24-well plate) and infected with AAV at an MOI of 50,000 viral genomes/cell (unless otherwise specified) in complete HeLa medium. Virus infectivity was determined 48 h post-infection by measuring GFP using flow cytometry.
Southern Blot
Genomic DNA (10 μg) from HAP1 cells was digested overnight with SpeI (cuts backbone) and XhoI (cuts genomic DNA) (both from New England Biolabs). Digested DNA was run in a 0.8% Tris-borate-EDTA (TBE) gel (80 V [10 min] and 20 V [overnight]). DNA was transferred to an Amersham Hybond-XL membrane. Probes for BLAS (400 bp) and GFP (574 bp) were generated by amplification from the AAV-HR vectors using the following primers and internally labeled with [α-32P]deoxycytidine triphosphate (dCTP) using the BcaBEST labeling kit (Takara): BLAS probe, forward, 5′-CTAGCGCCAAGCCTTTGTCT-3′, reverse, 5′-TAGCCCTCCCACACATAACC-3′; GFP probe, forward, 5′-GACGTAAACGGCCACAAGTT-3′, reverse, 5′-TGCTCAGGTAGTGGTTGTCG-3′.
Haploid Genetic Screen
The haploid genetic screen was performed similar to the protocol described21 with minor changes. Briefly, gene-trap virus was used to create a mutagenized HAP1 library. Of this mutagenized library, 300 million cells were infected with AAV-GAPDH(Hap1)-2A-BLAS at an MOI of 16,000. After 48 h, culture media of half of the cells were replaced with 20 μg/mL (stringent selection) and the other half with 5 μg/mL (moderate selection) of blasticidin. Blasticidin selection was maintained for 7 days before harvesting the cells for DNA analysis. Thirty million cells of the resistant population were used for genomic DNA isolation. Sequence analysis of gene-trap insertion sites was performed using the MiSeq platform (Illumina), and the significance of enrichment for each gene in the screen was calculated by comparing how often that gene was mutated and how often the gene carried an insertion in the control dataset (due to random integration). We found that the LRRC8 gene, which is related to blasticidin import62 and therefore served as a positive control, was enriched in both screens (positions 1 and 7), demonstrating a high degree of reproducibility of the experiment as well as its biological relevance.
Generation of Isogenic HAP1 Knockout Cell Lines
CRISPR-Cas9 gene-editing technology was used to generate isogenic knockout alleles by targeting exonic sequences of FANCM and RMI1. CRISPR sequence targeting oligonucleotides were designed using the Zhang Lab CRISPR design tool (https://zlab.bio/guide-design-resources) as follows: FANCM-1B, guide, 5′-TCAAGTATCTCCCGATCATC-3′; RMI1-6, guide, 5′- GATCTTGTAGTTTCAGTCAT3′. Oligonucleotides corresponding to the guide RNA were synthesized (Integrated DNA Technologies) and directly cloned into the Cas9-expressing plasmid px458 (Addgene #48138). HAP1 cells were transiently transfected with the plasmid encoding the guide RNA-encoding plasmid using Lipofectamine 3000 reagent (Thermo Fisher Scientific). After 48 h, GFP-expressing cells were subcloned using the BD FACSJazz cell sorter or the BD Influx cell sorter at the Stanford Shared FACS facility. Cells were expanded during 2 weeks and screened genotypically for the mutated allele after extracting genomic DNA from subclones (using the quick DNA universal 96-kit from Zymo Research, CA, USA). A 400- to 800-bp region that encompasses the guide RNA-targeted site was amplified and amplicons were sequenced (Sequetech, CA, USA) to identify subclones with KO mutations. The TP53BP1 knockout HAP1 cell line was obtained from Horizon Discovery (HZGHC000413c010) and its deletion in exon 2 (40 bp) was verified by Sanger sequencing (Sequetech, CA, USA).
Generation of HeLa Knockout Cells
CRISPR-Cas9 gene-editing technology was used to generate mixed knockout alleles by targeting exonic sequences of FANCM and RMI1. CRISPR sequence-targeting oligonucleotides were designed using the CRISPR design tool provided online by the Zhang Lab (https://zlab.bio/guide-design-resources) as follows: FANCM-1A, guide, 5′-GTTGCTTGTCGCGGCGTACG-3′; FANCM-ex4, guide, 5′-GAAAAGCTTATTGTTCCGCT-3′; RMI1-3, guide, 5′-TACTGATCTGAGGGATTTGG-3′; RMI1-6, guide, 5′-GATCTTGTAGTTTCAGTCAT-3′, non-targeting guide, 5′-GAAAACGTGAAAGTGTTAGG-3′. Oligonucleotides corresponding to the guide RNA were synthesized (Integrated DNA Technologies) and directly cloned into the Cas9-expressing plasmid px459 (Addgene #62988). HeLa cells were transiently transfected with guide RNA-encoding plasmid using Lipofectamine 2000 reagent (Thermo Fisher Scientific). After 48 h, transfected cells were re-plated and on the next day re-transfected with a different guide RNA-encoding plasmid targeting a different genomic sequence. 48 h after the second round of transfection, cells were either re-plated for AAV transduction (see Virus Transductions for details) or harvested for indel analysis. Indel frequency was calculated using TIDE software25 after amplifying and sequencing the targeted locus.
siRNA Knockdown
All siRNAs were purchased from Dharmacon and resuspended with nuclease-free water to 100 μM prior use, including siRNA-FANCM (individual: ON-TARGETplus human FANCM siRNA [J-021955-08-0002]), siRNA-RMI1 (SMARTpool: ON-TARGETplus RMI1 siRNA [L-014527-01-0005]), siRNA-BLM (SMARTpool: ON-TARGETplus BLM siRNA [L-007287-00-0005]), and siRNA-scramble (ON-TARGETplus non-targeting siRNA #1 [D-001810-01-50]). On the day before siRNA transfections, 0.4 × 105 HeLa cells were plated per well (24-well format). On the next day, transfection complexes were prepared with siRNA (a total of 750 ng/reaction [rxn]) and Lipofectamine 3000 reagent (2 μL/rxn) (Thermo Fisher Scientific). For experiments using three different siRNAs, 250 ng of each siRNA was mixed. For experiments using one siRNA for either FANCM, RMI1, or BLM, 250 ng of each siRNA was combined with 500 ng of siRNA-scramble. After a 15-min incubation at room temperature, complexes were added to the cells in complete culture medium and incubated for 72 h.
CD34+ HSPCs
Fresh umbilical cord purified CD34+ HSPCs were obtained from four different healthy male donors (donors 1, 3, and 4) with informed consent through Binn’s Family Cord Blood Research Program at Stanford University. CD34+ HSPCs were cultured for 24 h at 37°C, 5% CO2 and 5% O2 before the siRNA nucleofection.40
siRNAs (750 ng/well) were nucleofected using P3 nucleofection solution (Lonza) and Lonza Nucleofector 4d (program DZ-100).40 For triple treatment experiments using three different siRNAs, 250 ng of each siRNA was mixed. CD34+ HSPCs were seeded at 1 × 106 cells/well at the time of siRNA nucleofection in 48-well plates. On the same day 1 μg of siRNA was nucleofected.
For the AAV transduction experiment, we used 2 × 105 CD34+ HSPCs per well nucleofecting 750 ng of siRNA in each well. After 48 h, CD34+ HSPCs were nucleofected and, within 15 min from nucleofection, transduced with 5 × 105 vector genomes (vg)/cell of AAV6-IL2RG.40 48 h after AAV infection, ddPCR analyses were performed (described below).
mRNA Expression Analysis
RNA was extracted from HeLa cells using the RNeasy mini kit (QIAGEN, animal cells spin protocol). RNA was treated with DNase I (QIAGEN) for 15 min, according to the manufacturer’s instructions. cDNA was synthesized using the SuperScript III reverse transcriptase kit (Invitrogen) according to the manufacturer’s instructions. qPCR was performed to analyze gene expression in HeLa cells transfected with siRNAs using 2× Brilliant III master mix (Stratagene). qPCR was performed in duplicate using the Rotor-Gene 6000 thermal cycler (Corbett/QIAGEN), and results were analyzed with the Rotor-Gene 6000 application software. For CD34+ HSPC experiments the RNA was extracted with an RNA micro kit (QIAGEN) and cDNA was prepared as described above. qPCR was performed using the CFX384 Touch real-time PCR detection system (Bio-Rad). Gene expression was normalized against β-actin using the ΔΔCt method and was calculated relative to untransfected cells or cells transfected with siRNA-scramble control. The threshold was set up as 0.1 for all samples.
Primers are described below.
Primers for Human mRNA
The primers for human mRNA were as follows: β-actin, forward, 5′-GTCACCAACTGGGACGACAT-3′, reverse, 5′-GTACATGGCTGGGGTGTTGA-3′; FANCM, forward, 5′-CAATCCCACATGGCCGAAATG-3′, reverse, 5′-TCCGAGAGCTTTATGAGCTTC-3′; BLM, forward, 5′-GCTCAGGAAAGTGACTCTCTG-3′, reverse, 5′-TTCTTCTGGAGAAGGTGGAAC-3′; RMI1, forward, 5′-GAGCAGTGGAGGTTTCAAGTA-3′, reverse, 5′-CATGCCATGCAGCTAAAAGCC-3′.
Flow Cytometry
The number of GFP-expressing cells was evaluated using the BD FACSCalibur instrument and data were analyzed using the FlowJo software package. For sorting of GFP-expressing HAP1 cells, the BD FACSJazz cell sorter or the BD Influx cell sorter at the Stanford Shared FACS Facility were used.
Immunoblot Analysis
After a wash with cold PBS, cells were treated with radioimmunoprecipitation assay (RIPA) buffer (Cell Signaling Technology, #9806) supplemented with 1% protease inhibitor cocktail (Sigma, P8340). Total cell lysates were obtained by centrifugation at 12,000 rpm for 15 min at 4°C, and 30 μg of lysate was boiled for 10 min in the sample buffer and analyzed by SDS-PAGE (NuPAGE, 4%–12% Bis-Tris protein gels) and immunoblotting. The following antibodies were used: anti-RMI1 (rabbit polyclonal [Proteintech, 14630-1-AP], 1:1,000 dilution) and anti-FANCM (rabbit polyclonal,63 1:2,000 dilution). Anti-GAPDH (mouse monoclonal [Life Technologies, AM4300], 1:1,000 dilution) was used as loading control for the RMI1 analysis. For the FANCM analysis, the loading control is a non-specific band recognized by the anti-FANCM antibody.
CRISPR-Cas9-Mediated TI in the GAPDH Locus
CRISPR-Cas9 gene-editing technology was used to induce nuclease-mediated TI in the GAPDH locus. Five specific CRISPR sequence-targeting oligonucleotides were designed to target only the GAPDH locus and not the targeting vector using the Zhang Lab CRISPR design tool (https://zlab.bio/guide-design-resources): GAPDH guide #11, 5′-GGTCCAGGGGTCTTACTCCT-3′; GAPDH guide #12, 5′- TTACTCCTTGGAGGCCATGT-3′; GAPDH guide #13, 5′-CCAGGGGTCTTACTCCTTGG-3′; GAPDH guide #18, 5′-CCTCCAAGGAGTAAGACCCC-3′; GAPDH guide #19, 5′-CTTACTCCTTGGAGGCCATG-3′; non-targeting, 5′-GAAAACGTGAAAGTGTTAGG-3′. Oligonucleotides corresponding to the guide RNA were synthesized and cloned into px459, as described above. The indel frequency of each guide was assessed as described above. HAP1 cells were electroporated (program X-005, Lonza Nucleofector 2b) with guide RNA-encoding plasmid (5 μg of DNA for 1 × 106 cells) using 1 M buffer reagent.64 After electroporation, 3 × 105 cells were plated (24-well plate) and infected with AAV (MOI of 800). After 24 h, medium containing AAV was removed and replaced with fresh medium. Cells were harvested 48 h after transduction and analyzed by flow cytometry. Indel frequency was analyzed using a small aliquot of cells 24 h after electroporation.
ddPCR Quantification of TI Events in CD34+ HSPCs
Absolute quantification of the levels of genomic TI was carried out using ddPCR (Bio-Rad) as described.40
Statistical Analysis
GraphPad Prism was used for statistical analysis. Groups of two were analyzed by a non-parametric test (unpaired t test). More than two groups were compared by parametric test, α = 0.05 (one-way ANOVA). Statistical significance was assumed with p value <0.05 (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). Data are shown as mean ± SD.
Data Availability
The dataset generated and/or analyzed during the current study has been deposited in the ArrayExpress database at EMBL-EBI (https://www.ebi.ac.uk/arrayexpress/): E-MTAB-9247.
Acknowledgments
We would like to thank the Stanford Shared FACS Facility and its staff, Xuhuai Ji (Stanford Functional Genomics Facility), and members of the Kay laboratory for intellectual discussions and support. This work was supported by grants from the NIH to M.A.K. (R01 HL064274) and J.E.C (R01 AI130123). G.d.A. was supported by a fellowship from the Child Health Research Institute of Stanford University. The funding organizations played no role in experimental design, data analysis, or manuscript preparation.
Author Contributions
G.d.A. and M.A.K. directed the study. G.d.A., K.P., F.Z., S.P., K.M., J.E.C., M.H.P., and M.A.K. generated reagents and protocols, performed experiments, and analyzed data. M.T. performed Southern blots. H.J. and R.M. performed western blots. M.K.C. performed electroporations. M.P.-D. and F.P. designed and performed the experiments in CD34+ HSPCs and analyzed the data. A.S. purified CD34+ HSPC cells from healthy donors. G.d.A., K.P., F.P., and M.A.K. wrote the manuscript and generated the figures. All authors reviewed, edited, and commented on the manuscript.
Conflicts of Interest
G.d.A. and M.A.K. are inventors on a patent containing information presented in this paper. M.A.K. has commercial affiliations. M.A.K. has stock and/or equity in companies with technology broadly related to this paper. M.H.P. serves on the Scientific Advisory Boards for CRISPR Therapeutics and Allogene Therapeutics. Neither company had input into the design, execution, data analysis, or publication of the work presented in this manuscript. The remaining authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.10.020.
Supplemental Information
References
- 1.Nathwani A.C., Reiss U.M., Tuddenham E.G.D., Rosales C., Chowdary P., McIntosh J., Della Peruta M., Lheriteau E., Patel N., Raj D. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 2014;371:1994–2004. doi: 10.1056/NEJMoa1407309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mendell J.R., Al-Zaidy S., Shell R., Arnold W.D., Rodino-Klapac L.R., Prior T.W., Lowes L., Alfano L., Berry K., Church K. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 2017;377:1713–1722. doi: 10.1056/NEJMoa1706198. [DOI] [PubMed] [Google Scholar]
- 3.George L.A., Sullivan S.K., Giermasz A., Rasko J.E.J., Samelson-Jones B.J., Ducore J., Cuker A., Sullivan L.M., Majumdar S., Teitel J. Hemophilia B gene therapy with a high-specific-activity factor IX variant. N. Engl. J. Med. 2017;377:2215–2227. doi: 10.1056/NEJMoa1708538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bennett J., Wellman J., Marshall K.A., McCague S., Ashtari M., DiStefano-Pappas J., Elci O.U., Chung D.C., Sun J., Wright J.F. Safety and durability of effect of contralateral-eye administration of AAV2 gene therapy in patients with childhood-onset blindness caused by RPE65 mutations: a follow-on phase 1 trial. Lancet. 2016;388:661–672. doi: 10.1016/S0140-6736(16)30371-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang L., Bell P., Lin J., Calcedo R., Tarantal A.F., Wilson J.M. AAV8-mediated hepatic gene transfer in infant rhesus monkeys (Macaca mulatta) Mol. Ther. 2011;19:2012–2020. doi: 10.1038/mt.2011.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang L., Wang H., Bell P., McMenamin D., Wilson J.M. Hepatic gene transfer in neonatal mice by adeno-associated virus serotype 8 vector. Hum. Gene Ther. 2012;23:533–539. doi: 10.1089/hum.2011.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller D.G., Petek L.M., Russell D.W. Adeno-associated virus vectors integrate at chromosome breakage sites. Nat. Genet. 2004;36:767–773. doi: 10.1038/ng1380. [DOI] [PubMed] [Google Scholar]
- 8.Hirsch M.L. Adeno-associated virus inverted terminal repeats stimulate gene editing. Gene Ther. 2015;22:190–195. doi: 10.1038/gt.2014.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vasileva A., Linden R.M., Jessberger R. Homologous recombination is required for AAV-mediated gene targeting. Nucleic Acids Res. 2006;34:3345–3360. doi: 10.1093/nar/gkl455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller D.G., Wang P.-R., Petek L.M., Hirata R.K., Sands M.S., Russell D.W. Gene targeting in vivo by adeno-associated virus vectors. Nat. Biotechnol. 2006;24:1022–1026. doi: 10.1038/nbt1231. [DOI] [PubMed] [Google Scholar]
- 11.Russell D.W., Hirata R.K. Human gene targeting by viral vectors. Nat. Genet. 1998;18:325–330. doi: 10.1038/ng0498-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharma R., Anguela X.M., Doyon Y., Wechsler T., DeKelver R.C., Sproul S., Paschon D.E., Miller J.C., Davidson R.J., Shivak D. In vivo genome editing of the albumin locus as a platform for protein replacement therapy. Blood. 2015;126:1777–1784. doi: 10.1182/blood-2014-12-615492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barzel A., Paulk N.K., Shi Y., Huang Y., Chu K., Zhang F., Valdmanis P.N., Spector L.P., Porteus M.H., Gaensler K.M., Kay M.A. Promoterless gene targeting without nucleases ameliorates haemophilia B in mice. Nature. 2015;517:360–364. doi: 10.1038/nature13864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiao A., Wang Z., Hu Y., Wu Y., Luo Z., Yang Z., Zu Y., Li W., Huang P., Tong X. Chromosomal deletions and inversions mediated by TALENs and CRISPR/Cas in zebrafish. Nucleic Acids Res. 2013;41:e141. doi: 10.1093/nar/gkt464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adikusuma F., Piltz S., Corbett M.A., Turvey M., McColl S.R., Helbig K.J., Beard M.R., Hughes J., Pomerantz R.T., Thomas P.Q. Large deletions induced by Cas9 cleavage. Nature. 2018;560:E8–E9. doi: 10.1038/s41586-018-0380-z. [DOI] [PubMed] [Google Scholar]
- 16.Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kosicki M., Tomberg K., Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018;36:765–771. doi: 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmelas C., Grimm D. Split Cas9, not hairs—advancing the therapeutic index of CRISPR technology. Biotechnol. J. 2018;13:e1700432. doi: 10.1002/biot.201700432. [DOI] [PubMed] [Google Scholar]
- 19.Laoharawee K., DeKelver R.C., Podetz-Pedersen K.M., Rohde M., Sproul S., Nguyen H.-O., Nguyen T., St Martin S.J., Ou L., Tom S. Dose-dependent prevention of metabolic and neurologic disease in murine MPS II by ZFN-mediated in vivo genome editing. Mol. Ther. 2018;26:1127–1136. doi: 10.1016/j.ymthe.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang Y., Wang L., Bell P., McMenamin D., He Z., White J., Yu H., Xu C., Morizono H., Musunuru K. A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat. Biotechnol. 2016;34:334–338. doi: 10.1038/nbt.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carette J.E., Raaben M., Wong A.C., Herbert A.S., Obernosterer G., Mulherkar N., Kuehne A.I., Kranzusch P.J., Griffin A.M., Ruthel G. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature. 2011;477:340–343. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pillay S., Meyer N.L., Puschnik A.S., Davulcu O., Diep J., Ishikawa Y., Jae L.T., Wosen J.E., Nagamine C.M., Chapman M.S., Carette J.E. An essential receptor for adeno-associated virus infection. Nature. 2016;530:108–112. doi: 10.1038/nature16465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grimm D., Lee J.S., Wang L., Desai T., Akache B., Storm T.A., Kay M.A. In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J. Virol. 2008;82:5887–5911. doi: 10.1128/JVI.00254-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brinkman E.K., Chen T., Amendola M., van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42:e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ling C., Huang J., Yan Z., Li Y., Ohzeki M., Ishiai M., Xu D., Takata M., Seidman M., Wang W. Bloom syndrome complex promotes FANCM recruitment to stalled replication forks and facilitates both repair and traverse of DNA interstrand crosslinks. Cell Discov. 2016;2:16047. doi: 10.1038/celldisc.2016.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo G., Santoro I.M., McDaniel L.D., Nishijima I., Mills M., Youssoufian H., Vogel H., Schultz R.A., Bradley A. Cancer predisposition caused by elevated mitotic recombination in Bloom mice. Nat. Genet. 2000;26:424–429. doi: 10.1038/82548. [DOI] [PubMed] [Google Scholar]
- 28.So S., Nomura Y., Adachi N., Kobayashi Y., Hori T., Kurihara Y., Koyama H. Enhanced gene targeting efficiency by siRNA that silences the expression of the Bloom syndrome gene in human cells. Genes Cells. 2006;11:363–371. doi: 10.1111/j.1365-2443.2006.00944.x. [DOI] [PubMed] [Google Scholar]
- 29.Wang W., Seki M., Narita Y., Sonoda E., Takeda S., Yamada K., Masuko T., Katada T., Enomoto T. Possible association of BLM in decreasing DNA double strand breaks during DNA replication. EMBO J. 2000;19:3428–3435. doi: 10.1093/emboj/19.13.3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Canny M.D., Moatti N., Wan L.C.K., Fradet-Turcotte A., Krasner D., Mateos-Gomez P.A., Zimmermann M., Orthwein A., Juang Y.C., Zhang W. Inhibition of 53BP1 favors homology-dependent DNA repair and increases CRISPR-Cas9 genome-editing efficiency. Nat. Biotechnol. 2018;36:95–102. doi: 10.1038/nbt.4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fattah F.J., Lichter N.F., Fattah K.R., Oh S., Hendrickson E.A. Ku70, an essential gene, modulates the frequency of rAAV-mediated gene targeting in human somatic cells. Proc. Natl. Acad. Sci. USA. 2008;105:8703–8708. doi: 10.1073/pnas.0712060105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maruyama T., Dougan S.K., Truttmann M.C., Bilate A.M., Ingram J.R., Ploegh H.L. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2015;33:538–542. doi: 10.1038/nbt.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paulk N.K., Loza L.M., Finegold M.J., Grompe M. AAV-mediated gene targeting is significantly enhanced by transient inhibition of nonhomologous end joining or the proteasome in vivo. Hum. Gene Ther. 2012;23:658–665. doi: 10.1089/hum.2012.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robert F., Barbeau M., Éthier S., Dostie J., Pelletier J. Pharmacological inhibition of DNA-PK stimulates Cas9-mediated genome editing. Genome Med. 2015;7:93. doi: 10.1186/s13073-015-0215-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li G., Nelsen C., Hendrickson E.A. Ku86 is essential in human somatic cells. Proc. Natl. Acad. Sci. USA. 2002;99:832–837. doi: 10.1073/pnas.022649699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jayavaradhan R., Pillis D.M., Goodman M., Zhang F., Zhang Y., Andreassen P.R., Malik P. CRISPR-Cas9 fusion to dominant-negative 53BP1 enhances HDR and inhibits NHEJ specifically at Cas9 target sites. Nat. Commun. 2019;10:2866. doi: 10.1038/s41467-019-10735-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haapaniemi E., Botla S., Persson J., Schmierer B., Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018;24:927–930. doi: 10.1038/s41591-018-0049-z. [DOI] [PubMed] [Google Scholar]
- 38.High K.A., Roncarolo M.G. Gene therapy. N. Engl. J. Med. 2019;381:455–464. doi: 10.1056/NEJMra1706910. [DOI] [PubMed] [Google Scholar]
- 39.Bak R.O., Dever D.P., Porteus M.H. CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nat. Protoc. 2018;13:358–376. doi: 10.1038/nprot.2017.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pavel-Dinu M., Wiebking V., Dejene B.T., Srifa W., Mantri S., Nicolas C.E., Lee C., Bao G., Kildebeck E.J., Punjya N. Gene correction for SCID-X1 in long-term hematopoietic stem cells. Nat. Commun. 2019;10:1634. doi: 10.1038/s41467-019-09614-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gari K., Décaillet C., Delannoy M., Wu L., Constantinou A. Remodeling of DNA replication structures by the branch point translocase FANCM. Proc. Natl. Acad. Sci. USA. 2008;105:16107–16112. doi: 10.1073/pnas.0804777105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yan Z., Delannoy M., Ling C., Daee D., Osman F., Muniandy P.A., Shen X., Oostra A.B., Du H., Steltenpool J. A histone-fold complex and FANCM form a conserved DNA-remodeling complex to maintain genome stability. Mol. Cell. 2010;37:865–878. doi: 10.1016/j.molcel.2010.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deans A.J., West S.C. FANCM connects the genome instability disorders Bloom’s syndrome and Fanconi anemia. Mol. Cell. 2009;36:943–953. doi: 10.1016/j.molcel.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 44.Singh T.R., Ali A.M., Busygina V., Raynard S., Fan Q., Du C.H., Andreassen P.R., Sung P., Meetei A.R. BLAP18/RMI2, a novel OB-fold-containing protein, is an essential component of the Bloom helicase-double Holliday junction dissolvasome. Genes Dev. 2008;22:2856–2868. doi: 10.1101/gad.1725108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ouyang K.J., Woo L.L., Ellis N.A. Homologous recombination and maintenance of genome integrity: cancer and aging through the prism of human RecQ helicases. Mech. Ageing Dev. 2008;129:425–440. doi: 10.1016/j.mad.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 46.Hoadley K.A., Xue Y., Ling C., Takata M., Wang W., Keck J.L. Defining the molecular interface that connects the Fanconi anemia protein FANCM to the Bloom syndrome dissolvasome. Proc. Natl. Acad. Sci. USA. 2012;109:4437–4442. doi: 10.1073/pnas.1117279109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Manthei K.A., Keck J.L. The BLM dissolvasome in DNA replication and repair. Cell. Mol. Life Sci. 2013;70:4067–4084. doi: 10.1007/s00018-013-1325-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seki M., Nakagawa T., Seki T., Kato G., Tada S., Takahashi Y., Yoshimura A., Kobayashi T., Aoki A., Otsuki M. Bloom helicase and DNA topoisomerase IIIα are involved in the dissolution of sister chromatids. Mol. Cell. Biol. 2006;26:6299–6307. doi: 10.1128/MCB.00702-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu D., Guo R., Sobeck A., Bachrati C.Z., Yang J., Enomoto T., Brown G.W., Hoatlin M.E., Hickson I.D., Wang W. RMI, a new OB-fold complex essential for Bloom syndrome protein to maintain genome stability. Genes Dev. 2008;22:2843–2855. doi: 10.1101/gad.1708608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bakker S.T., van de Vrugt H.J., Rooimans M.A., Oostra A.B., Steltenpool J., Delzenne-Goette E., van der Wal A., van der Valk M., Joenje H., te Riele H., de Winter J.P. Fancm-deficient mice reveal unique features of Fanconi anemia complementation group M. Hum. Mol. Genet. 2009;18:3484–3495. doi: 10.1093/hmg/ddp297. [DOI] [PubMed] [Google Scholar]
- 51.Bogliolo M., Bluteau D., Lespinasse J., Pujol R., Vasquez N., d’Enghien C.D., Stoppa-Lyonnet D., Leblanc T., Soulier J., Surrallés J. Biallelic truncating FANCM mutations cause early-onset cancer but not Fanconi anemia. Genet. Med. 2018;20:458–463. doi: 10.1038/gim.2017.124. [DOI] [PubMed] [Google Scholar]
- 52.Catucci I., Osorio A., Arver B., Neidhardt G., Bogliolo M., Zanardi F., Riboni M., Minardi S., Pujol R., Azzollini J. Individuals with FANCM biallelic mutations do not develop Fanconi anemia, but show risk for breast cancer, chemotherapy toxicity and may display chromosome fragility. Genet. Med. 2018;20:452–457. doi: 10.1038/gim.2017.123. [DOI] [PubMed] [Google Scholar]
- 53.Neidhardt G., Hauke J., Ramser J., Groß E., Gehrig A., Müller C.R., Kahlert A.K., Hackmann K., Honisch E., Niederacher D. Association between loss-of-function mutations within the FANCM gene and early-onset familial breast cancer. JAMA Oncol. 2017;3:1245–1248. doi: 10.1001/jamaoncol.2016.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ellis N.A., Groden J., Ye T.-Z., Straughen J., Lennon D.J., Ciocci S., Proytcheva M., German J. The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell. 1995;83:655–666. doi: 10.1016/0092-8674(95)90105-1. [DOI] [PubMed] [Google Scholar]
- 55.Broberg K., Höglund M., Gustafsson C., Björk J., Ingvar C., Albin M., Olsson H. Genetic variant of the human homologous recombination-associated gene RMI1 (S455N) impacts the risk of AML/MDS and malignant melanoma. Cancer Lett. 2007;258:38–44. doi: 10.1016/j.canlet.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 56.Chen H., You M.J., Jiang Y., Wang W., Li L. RMI1 attenuates tumor development and is essential for early embryonic survival. Mol. Carcinog. 2011;50:80–88. doi: 10.1002/mc.20694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guiraldelli M.F., Eyster C., Pezza R.J. Genome instability and embryonic developmental defects in RMI1 deficient mice. DNA Repair (Amst.) 2013;12:835–843. doi: 10.1016/j.dnarep.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Charlesworth C.T., Camarena J., Cromer M.K., Vaidyanathan S., Bak R.O., Carte J.M., Potter J., Dever D.P., Porteus M.H. Priming human repopulating hematopoietic stem and progenitor cells for Cas9/sgRNA gene targeting. Mol. Ther. Nucleic Acids. 2018;12:89–104. doi: 10.1016/j.omtn.2018.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Naldini L. Genetic engineering of hematopoiesis: current stage of clinical translation and future perspectives. EMBO Mol. Med. 2019;11:e9958. doi: 10.15252/emmm.201809958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dever D.P., Porteus M.H. The changing landscape of gene editing in hematopoietic stem cells: a step towards Cas9 clinical translation. Curr. Opin. Hematol. 2017;24:481–488. doi: 10.1097/MOH.0000000000000385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grimm D., Pandey K., Nakai H., Storm T.A., Kay M.A. Liver transduction with recombinant adeno-associated virus is primarily restricted by capsid serotype not vector genotype. J. Virol. 2006;80:426–439. doi: 10.1128/JVI.80.1.426-439.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee C.C., Freinkman E., Sabatini D.M., Ploegh H.L. The protein synthesis inhibitor blasticidin S enters mammalian cells via leucine-rich repeat-containing protein 8D. J. Biol. Chem. 2014;289:17124–17131. doi: 10.1074/jbc.M114.571257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Meetei A.R., Medhurst A.L., Ling C., Xue Y., Singh T.R., Bier P., Steltenpool J., Stone S., Dokal I., Mathew C.G. A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat. Genet. 2005;37:958–963. doi: 10.1038/ng1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chicaybam L., Sodre A.L., Curzio B.A., Bonamino M.H. An efficient low cost method for gene transfer to T lymphocytes. PLoS One. 2013;8:e60298. doi: 10.1371/journal.pone.0060298. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The dataset generated and/or analyzed during the current study has been deposited in the ArrayExpress database at EMBL-EBI (https://www.ebi.ac.uk/arrayexpress/): E-MTAB-9247.