

SUMMARY
There is increasing evidence that gut microbiome perturbations, also known as dysbiosis, can influence colorectal cancer development. To understand the mechanisms by which the gut microbiome modulates cancer susceptibility, we examine two wild-type mouse colonies with distinct gut microbial communities that develop significantly different tumor numbers using a mouse model of inflammation-associated tumorigenesis. We demonstrate that adaptive immune cells contribute to the different tumor susceptibilities associated with the two microbial communities. Mice that develop more tumors have increased colon lamina propria CD8+ IFNγ+ T cells before tumorigenesis but reduced CD8+ IFNγ+ T cells in tumors and adjacent tissues compared with mice that develop fewer tumors. Notably, intratumoral T cells in mice that develop more tumors exhibit increased exhaustion. Thus, these studies suggest that microbial dysbiosis can contribute to colon tumor susceptibility by hyperstimulating CD8 T cells to promote chronic inflammation and early T cell exhaustion, which can reduce anti-tumor immunity.
In Brief
The gut microbiome is capable of modulating intestinal inflammation and tumorigenesis. Yu et al. demonstrate that dysbiosis can lead to increased susceptibility to inflammation-associated colon tumorigenesis via the induction of pro-inflammatory CD8+ IFNγ+ T cells, which can lead to increased T cell exhaustion within the tumor microenvironment.
Graphical Abstract
INTRODUCTION
Colorectal cancer (CRC) is the third most common cause of cancer and fourth leading cause of cancer-related deaths worldwide (Arnold et al., 2017). In addition to the presence of specific gene mutations, host immune responses significantly contribute to the development and progression of CRC (Markman and Shiao, 2015). For example, chronic inflammation results in the production of DNA-damaging reactive oxygen species and pro-inflammatory mediators that promote cellular survival and growth. Consequently, patients with inflammatory bowel disease (IBD) are at increased risk for developing CRC (Eaden et al., 2001). Host immunity also contributes to immune surveillance and anti-tumor activity (Markman and Shiao, 2015; Pernot et al., 2014). Consistently, tumors with higher densities of effector CD8 T cells and a Th1 gene signature are associated with better prognosis (Galon et al., 2006). Thus, factors that influence the balance of these processes likely affect CRC risk.
The gut microbiome is recognized as having a role in modulating both host immunity and susceptibility to colon tumorigenesis. For example, the gut microbiota can influence frequencies of intestinal lamina propria (LP) immune cells, notably regulatory T cells (Tregs) and T helper 17 (Th17) cells (Atarashi et al., 2011; Furusawa et al., 2013; Geva-Zatorsky et al., 2017) which can affect inflammatory responses and tumor susceptibility (Műzes et al., 2012; Razi et al., 2019; Wu et al., 2009). Certain human gut bacteria can promote IFNγ production by CD8 T cells and anti-tumor immunity in mice (Tanoue et al., 2019). How these immunomodulatory activities ultimately contribute to CRC susceptibility remains to be fully elucidated.
CRC patients exhibit altered gut microbiomes typically characterized by decreased Firmicutes and increased Bacteroidetes bacteria compared with healthy controls (Ahn et al., 2013; Baxter et al., 2014; Wang et al., 2012; Zackular et al., 2014). Specific bacteria have consistently been enriched in colon tumors, such as Escherichia coli positive for the polyketide synthase (pks) pathogenicity island, enterotoxigenic Bacteroides fragilis (ETBF), and Fusobacterium nucleatum. Some proposed mechanisms, largely based on mice studies, include the promotion of DNA damage (Arthur et al., 2012; Nougayrède et al., 2006), the induction of pro-inflammatory Th17 responses (Wu et al., 2009), and the inhibition of tumor-infiltrating lymphocytes (Gur et al., 2015). However, these bacteria are not associated with all CRC cases, and therefore, other bacterial populations likely contribute to tumor susceptibility via other mechanisms. Nonetheless, additional studies are needed to understand the mechanisms by which perturbations in the gut microbiome, also known as dysbiosis, affect colon tumorigenesis.
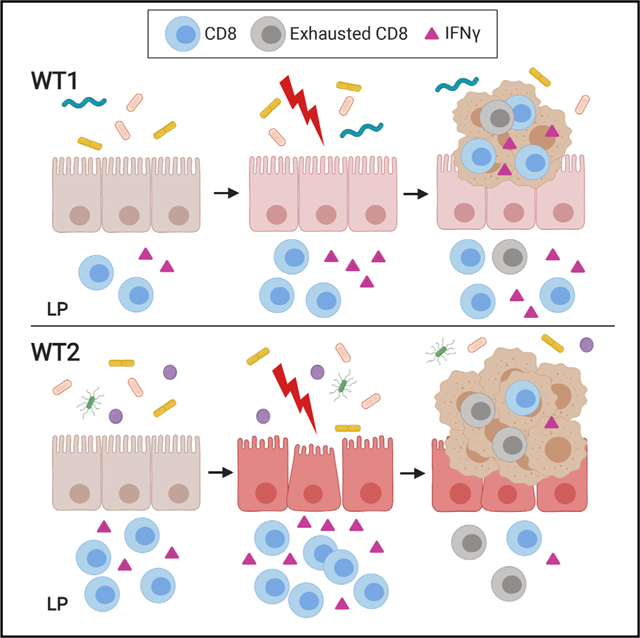
In this study, we take advantage of the different susceptibilities of two wild-type(WT) C57BL/6Jmouse colonies in our facility anduse a mouse model of inflammation-associated tumorigenesis to identify factors that contribute to increased inflammation and colon tumorigenesis. Specifically, mice from our “WT1” colony developed an average of 5 tumors, and mice from our “WT2” colony developed an average of 15 tumors. Sequencing of fecal bacteria from WT1 and WT2 mice indicate that the two colonies have distinct microbiomes, which when transferred into germ-free (GF) WT mice recapitulated their respective tumor susceptibilities. Interestingly, cohousing and cross-fostering studies resulted in intermediate tumor phenotypes with hybrid microbiomes. By cross referencing microbiome compositions after microbiome transfer with tumor outcomes, we identified candidate bacteria consistently associated with low or high tumor burdens. Additionally, naive and acutely inflamed WT2 mice have increased colon LP CD8+ IFNγ+ T cells compared with WT1 mice. However, after tumor development, WT2 mice exhibited decreased intratumoral CD8+ IFNγ+ T cell numbers and increased CD8 T cell exhaustion. Both GF Rag1−/− and specific pathogen-free (SPF) Cd8−/− mice colonized with WT2 microbiota resulted in fewer tumor numbers than SPF WT2 mice. These results strongly suggest that CD8 T cells may play a pro-inflammatory and pro-tumorigenic role in the context of a dysbiotic microbiome and that dysbiosis can contribute to increased tumor susceptibility by increasing T cell exhaustion.
RESULTS
Differences in Gut Microbiome Composition Directly Contribute to Inflammation-Associated Tumor Susceptibility
We have previously demonstrated that alterations in the gut microbiome can directly contribute to tumorigenesis (Zackular et al., 2013). In this study, we used the azoxymethane (AOM)/dextran sulfate sodium (DSS) model of inflammation-associated colon tumorigenesis (De Robertis et al., 2011; Tanaka et al., 2003), in which mice are given an intraperitoneal injection of the carcinogen AOM followed by three rounds of water containing 2% DSS, which compromises the intestinal barrier, resulting in increased epithelial permeabilization and inflammation (Chassaing et al., 2014). In this model, tumors develop by weeks 8–10 (Neufert et al., 2007; Zackular et al., 2013) (Figure 1A). Although this model is driven by epithelial injury and inflammation and has been used to understand the pathogenesis of IBD-associated CRC, there are features of this model that also recapitulate sporadic CRC, including the predominance of tumor mutations that dysregulate Wnt signaling (Greten et al., 2004; Zhan et al., 2013), which occurs in the majority of human CRC, as well as the progression from adenomas to adenocarcinomas (Suzuki et al., 2004). AOM/DSS treatment also results in microbiome alterations similar to that observed in human CRC patients such as reduced species richness and alpha diversity as well as significant shifts in beta diversity (Zackular et al., 2013).
Figure 1. Two Colonies of C57BL/6J WT Mice Have Different Colon Tumor Burdens in the AOM/DSS Model of Inflammation-Associated Colon Tumorigenesis.

(A) Schema of AOM/DSS model.
(B) Representative photo of WT1 and WT2 tumors. Scale bar, 1 cm.
(C) Representative micrographs of H&E stains of colon tissue at the tumor endpoint at 40× magnification. Tumors are denoted by black arrows. Scale bar, 100 μm.
(D and E) Tumor numbers (D) and sizes (E) are shown. WT1, n = 42; WT2, n = 36.
(F) Representative graph of percentage weight change during AOM/DSS treatment compared with day 0. n = 5 mice/group.
Data are mean ± SEM and are representative of or pooled from at least three experiments. *p < 0.05 by Mann-Whitney test.
See also Figure S1.
In our previous study (Zackular et al., 2013), we observed C57BL/6 (B6) WT mice with higher tumor numbers after AOM/DSS treatment than what we have typically observed (Chen et al., 2008; Zhan et al., 2016). Specifically, when we compared the two mouse colonies, “WT1” mice developed 5 tumors on average, whereas “WT2” mice developed 15 tumors on average (Figures 1B–1D). WT2 mice not only developed larger tumors but also exhibited increased weight loss after DSS treatment compared with WT1 mice (Figures 1E and 1F). Additionally, during the acute inflammatory response to the first round of DSS (day 12), WT2 mice have significantly more colonic inflammation on the basis of levels of stool lipocalin-2, a surrogate marker for intestinal inflammation (Chassaing et al., 2012), and histologic scoring (Figures S1A and S1B).
Although both WT1 and WT2 colonies originate from Jackson B6 mice and are housed in the same mouse room, WT2 mice were generated during backcrossing of WT1 mice with a transgenic knockout (KO) mouse strain that exhibited increased tumor susceptibility after AOM/DSS treatment (Chen et al., 2008) and were also housed on the same rack as the KO strain. We therefore hypothesized that dysbiotic microbiota from the KO strain were transferred to WT2 mice, resulting in similarly increased susceptibility to colon tumorigenesis. To examine microbiome differences between the two colonies, we performed 16S rRNA sequencing on stool-extracted DNA from untreated WT1 and WT2 mice. Although no difference in species richness was measured between the gut microbiomes of the two colonies, WT2 microbiomes had increased alpha diversity compared with WT1 microbiomes (Figures 2A and 2B). Beta diversity was visualized by nonmetric multidimensional scaling (NMDS) ordination, which showed two separate clusters representing the microbiomes of WT1 and WT2 mice, suggesting that WT1 and WT2 colonies have distinct microbiome compositions (Figure 2C). Consistently, the gut microbial community structures of WT1 and WT2 mice were significantly dissimilar, as determined by θYC (thetaYC) distances and analysis of molecular variance (AMOVA) (Figure 2D).
Figure 2. Gut Microbiome Compositions in WT1 and WT2 Mice Are Significantly Different.

(A–C) 16S rRNA sequencing of naive WT1 and WT2 fecal microbiota was performed, and richness (A) and alpha diversity (B) were measured. Beta diversity (C) is shown as a nonmetric dimensional scaling (NMDS) plot. *p < 0.05 by Mann-Whitney test.
(D) Microbiome composition dissimilarity based on thetaYC distances. *p < 0.05 by AMOVA.
(E) Relative bacteria family abundances of naive WT1 and WT2 mice. n = 15/group
(F) The most differentially abundant OTUs between WT1 (blue) or WT2 (red) microbiomes were determined using LEfSe pairwise analysis. OTUs with LDA scores greater than 4 are shown.
Data are pooled from at least five experiments; WT1, n = 51; WT2, n = 45, unless otherwise noted.
WT1 and WT2 microbiomes are differentially abundant in certain bacteria families. WT1 mice have increased Anaeroplasmataceae, Erysipelotrichaceae, Clostridiales, and Sutterellaceae bacteria, while WT2 mice have increased Prevotellaceae and Helicobacteraceae bacteria (Figure 2E). To identify specific differences between the two microbiomes, we used linear discriminant analysis (LDA) effect size (LEfSe) analysis (Segata et al., 2011) to identify operational taxonomic units (OTUs), which are bacterial sequences that are at least 97% identical, significantly associated with WT1 or WT2 microbiomes (Figure 2F). Consistent with the relative abundance data, among the most differentially significant OTUs, WT1 mice are enriched in a member of the Erysipelotrichaceae family, and WT2 mice are enriched in a member of the Prevotellaceae family, which has also been observed in human CRC patients (Sobhani et al., 2011; Wang et al., 2012).
To determine whether the microbiome differences between WT1 and WT2 colonies are directly responsible for the differences in tumor phenotype, we gavaged anaerobically prepared stool and cecal homogenates of SPF WT1 or WT2 mice into GF WT mice. Additionally, to determine whether cultivable bacteria from WT1 or WT2 microbiomes also contribute to tumor susceptibility, stool and cecal homogenates were plated onto specific media plates and anaerobically cultured. Bacterial colonies were resuspended in anaerobic PBS and gavaged into GF WT mice. After 4 weeks of colonization, GF WT mice had stool bacteria levels comparable with those of SPF mice as measured by qPCR using universal bacteria primers (data not shown), and their microbiomes also recapitulated the donor or input composition by thetaYC measurement (Figures S2A and S2B). After AOM/DSS treatment, GF WT mice with WT1 bacteria (SPF WT1 > GF) developed significantly fewer and smaller tumors compared with GF WT mice with WT2 bacteria (SPF WT2 > GF), suggesting that microbiome differences directly promote tumor burden differences (Figures 3A–3C). Interestingly, GF WT mice gavaged with cultivable WT2 bacteria (cult WT2 > GF) also developed more tumors compared with GF WT mice gavaged with cultivable WT1 bacteria (cult WT1 > GF) but developed significantly fewer and smaller tumors compared with SPF WT2 > GF (Figures 3A–3C). These data suggest that the different tumor susceptibilities of WT1 and WT2 mice can be attributed to gut microbiome differences and that the absence or presence of specific bacterial populations, some of which may be cultivable, can determine tumor outcomes.
Figure 3. The Gut Microbiomes of WT1 and WT2 Mice Directly Contribute to Tumor Susceptibility.

(A and B) Tumor number (A) and sizes (B) of AOM/DSS-treated GF WT mice that were colonized with SPF WT1 or WT2 whole-stool and cecal homogenates or anaerobically cultivable WT1 or WT2 stool and cecal bacteria for 4 weeks. Tumors were counted 10 days after the last DSS round. Data are mean ± SEM. n = 4 or 5/group. *p < 0.05 by Mann-Whitney test.
(C) Representative micrographs of H&E stains of colon tissue at the tumor endpoint at 40× magnification. Tumors are denoted by black arrows. Scale bar, 1 mm.
See also Figure S2.
Cohousing and Cross-Fostering Are Insufficient to Completely Transfer Microbiomes and Are Associated with Intermediate Phenotype Transmission
We next wanted to examine if the tumor burden phenotype could be transferred between WT1 and WT2 mice via cohousing, which has been commonly used to transmit microbiome-dependent phenotypes. Age-matched female WT1 and WT2 mice were first mostly cohoused in a 2:2 ratio for 4 weeks before AOM/DSS treatment (Figure 4A, “coh”). Cohoused WT1 and WT2 mice exhibited significant tumor burden differences similar to non-cohoused control mice (Figure 4B, “coh”). Although the microbiota of cohoused WT1 and WT2 mice were not significantly different from each other after 4 weeks of cohousing on the basis of thetaYC distances, cohoused WT1 microbiome compositions remained significantly different from control WT2 microbiomes (Figure S2C). Cohoused WT2 microbiomes also remained distinct from control WT1 microbiomes on the basis of thetaYC measurements, suggesting incomplete microbiome transfer (Figure S2C). Additionally, by NMDS ordination, cohoused WT1 and cohoused WT2 mice cluster separately (Figure S2D).
Figure 4. Specific Bacterial Populations Correlate with High or Low Tumor Susceptibilities.

(A) WT1 and WT2 mice were cohoused in a 2:2 ratio(coh) for 4 weeks or a 1:1 ratio (oto) for 6 weeks or cross-fostered (CF). Mice were treated with AOM/DSS and sacrificed for tumor counting on days 60–70 depending on the experiment.
(B) Number of tumors in indicated mice after AOM/DSS treatment. The control group includes pooled control mice from all cohousing and cross-fostering experiments. Data are mean ± SEM; controls: WT1, n = 27; WT2, n = 20; coh: n = 9/group; oto: n = 10/group; CF: WT1(WT2), n = 18; WT2(WT1), n = 14. *p < 0.05 by Mann-Whitney test.
(C) Phylogenetic tree of nine bacterial candidates associated with low (blue) or high (red) tumor burdens on the basis of sequence similarity.
(D and E) Relative bacterial abundances in mice at the time of AOM injection that were associated with low (D) or high (E) tumor burdens. Black lines denote mean relative abundances. Poisson regression p values are shown. LDA scores were determined using LEfSe analyses. Low, n = 85; high, n = 38.
Data shown are pooled from at least two independent experiments per microbiome transfer method.
See also Figure S2.
Although cohousing did not result in complete transmission of tumor phenotype from either WT1 or WT2 mice to the other, two cohoused mice developed tumors similar in number to mice from the other colony (Figure 4B, black boxes). These mice were cohoused in a 1:1 ratio (one WT1 and one WT2 mouse), which may have resulted in more efficient microbiome transfer by coprophagy. We therefore performed one-to-one (oto) cohousing experiments and also extended the cohousing time to 6 weeks to improve microbiome and phenotype transmission. However, significant tumor burden differences were still observed between oto cohoused WT1 and WT2 mice (Figure 4B, “oto”). Furthermore, oto cohoused WT2 mice developed an intermediate tumor burden phenotype, and no differences in tumor number between oto cohoused and control WT1 mice were measured (Figure 4B, “oto”). With regard to microbiome composition, oto cohoused WT2 mice were significantly different from WT1 controls, again suggesting incomplete microbiome transfer despite oto cohousing (Figure S2E). By NMDS ordination, microbiome compositions of oto cohoused WT1 and WT2 mice clustered between control WT1 and WT2 mice, suggesting that oto cohoused mice have a hybrid microbiome, which may explain the intermediate tumor phenotype (Figure S2F).
As the gut microbiome of an adult mouse is typically determined by maternal transmission, cross-fostering is another microbiome transfer method and may be more effective than cohousing (Daft et al., 2015; Moore and Stanley, 2016). We performed cross-fostering experiments in which WT1 or WT2 pups were switched within 48 h of birth to a WT2 or WT1 nursing mother, respectively. Cross-fostered mice were treated with AOM/DSS after reaching 6 weeks of age. Unlike with cohousing, cross-fostered WT1 and WT2 mice no longer developed significantly different tumor burdens; however, average tumor numbers were again intermediate between control WT1 and WT2 mice (Figure 4B, “CF”). When we examined gut microbiome compositions of cross-fostered mice, we determined that the microbial community of WT1 mice cross-fostered by WT2 nursing mothers (WT1[WT2]) and WT2 mice cross-fostered by WT1 nursing mothers (WT2[WT1]) remained distinct (Figure S2G). Although microbiomes of WT2(WT1) and non-cross-fostered WT1 mice were overall similar, the microbiome compositions of WT1(WT2) and non-cross-fostered WT2 mice were still significantly different despite improved microbiome transfer compared with cohousing on the basis of thetaYC and NMDS ordination (Figures S2G and S2H). There were also significant differences in specific bacterial populations between WT1(WT2) or WT2(WT1) mice and WT2 or WT1 mice by LEfSe (data not shown), respectively, which may explain why the tumor phenotype was not fully transmissible.
Specific Bacteria Are Associated with Low or High Tumor Burdens
As we observed multiple tumor phenotypes associated with various microbial communities that were generated from different microbiome transfer methods, we reasoned that certain bacterial populations may consistently be associated with either low or high tumor burdens regardless of WT colony origin. We therefore examined 16S rRNA sequences from the stool of cohoused and cross-fostered WT1 and WT2, control WT1 and WT2 mice, and GF WT mice conventionalized with WT1 or WT2 microbiota that was collected just prior to AOM/DSS treatment to identify OTUs that directly correlated with tumor outcomes after AOM/DSS treatment. On the basis of relative abundances alone, we observed, for example, increased abundance of Prevotellaceae in day 0 mice that developed high tumor numbers and increased abundance of Anaeroplasmataceae in mice that developed low tumor burdens (Figure S2I). OTUs that were significantly associated with low or high tumor numbers were determined by the combination of the following analyses: Metastats (White et al., 2009), LEfSe (Segata et al., 2011), random forest (Zackular et al., 2015), and linear and Poisson regression. In particular, we identified nine OTUs belonging to at least five different bacterial families that were significantly associated with either low or high tumor numbers (Figure 4C). Many of these bacteria have not been previously classified, but phylogenetic tree analysis on the basis of 16S sequence similarity suggests that some of the bacteria within a family may be closely related (Figure 4C) (Clarridge, 2004; Edgar, 2018).
Specifically, two OTUs from the Lachnospiraceae family are significantly associated and predictive of low tumor burdens. These OTUs are also more abundant in mice that eventually developed low tumor numbers (Figure 4D). On the other hand, seven OTUs, including bacteria in the Prevotellaceae family, are predictive of high tumor burdens and are more abundant in mice that develop high tumor numbers (Figures 2E, 4E, and S2I). Of note, although Helicobacteraceae appeared to be more abundant in WT2 mice (Figure 2E), it was not significantly associated with tumor numbers by linear regression or Metastats analyses. Altogether, these data reveal nine bacterial populations that may have tumor-suppressive or tumor-promoting activities.
Dysbiotic Microbiome from WT2 Mice Promotes Inflammation-Associated Tumorigenesis via Adaptive Immune Cells
An important characteristic of the gut microbiota is its ability to modulate host immune responses (Belkaid and Hand, 2014; Geva-Zatorsky et al., 2017). To determine whether the difference in tumor susceptibilities between WT1 and WT2 mice were associated with altered baseline immune responses, LP immune cell compositions from the entire colon of untreated WT1 and WT2 mice were analyzed using flow cytometry. No differences in colon LP monocytes, neutrophils, dendritic cells (DCs), or natural killer (NK) cells were observed; however, WT2 mice have increased B and T cell populations compared with WT1 mice, suggesting that the WT2 microbiome may exert its tumor-promoting effect via adaptive immune cells (Figure 5A).
Figure 5. Adaptive Immune Cells Contribute to Increased Tumor Susceptibility of WT2 Mice.

(A) Colon LP immune cells from untreated WT1 andWT2 mice were analyzed using flow cytometry. Data are pooled from at least two independent experiments. n = 5–7/group.
(B) GF Rag1−/− mice were gavaged with SPF WT1 or WT2 stool and cecal contents followed by AOM/DSS treatment after 4 weeks of colonization. Mice were sacrificed on day 61 for tumor counting.
(C and D) Tumor number (C) and sizes (D) are shown. n = 4–6/group.
Data are mean ± SEM. *p < 0.05 by Mann-Whitney test.
See also Figure S3.
To examine this possibility, GF Rag1−/− mice were colonized with SPF WT1 or WT2 stool and cecal homogenates, which resulted in successful colonization of WT1 and WT2 microbiomes after 4 weeks (Figures S3A and S3B). After induction of tumors by AOM/DSS, GF Rag1−/− mice with WT2 microbiota developed fewer and smaller tumors compared with SPF WT2 controls (Figures 5C and 5D), suggesting that the dysbiotic WT2 microbiome promotes tumor susceptibility by acting on adaptive immune cells. GF Rag1−/− mice with WT1 microbiota developed slightly increased tumor numbers compared with SPF WT1 control mice (Figure 5C), which may indicate a protective role for adaptive immune cells in the context of the WT1 microbiome. Of note, GF Rag1−/− had significantly higher tumor numbers compared with that of SPF Rag1−/−, which is consistent with previous reports demonstrating increased susceptibility of GF mice to DSS-induced injury and AOM/DSS-induced tumorigenesis (Maslowski et al., 2009; Zhan et al., 2013)
Naive WT2 Mice that Have Increased Tumor Susceptibility Have Increased IFNγ-Producing CD8 T Cells in the Colon LP
We and others have previously shown a role of T cells in affecting tumor susceptibility in the AOM/DSS model (Becker et al., 2004; Zhan et al., 2016). To determine if T cell activity was altered between WT1 and WT2 mice, we examined LP T cell cytokine production after ex vivo stimulation by phorbol myristate cetate (PMA) and ionomycin. Specifically, colon LP CD8+, but not CD4+, IFNγ+ T cells were increased in naive WT2 mice (Figures 6A and 6B), which was not observed in the mesenteric lymph nodes (Figures S4A–S4C). In contrast, there were no differences in colon LP CD4+ IL-17+ or CD4+ Foxp3+ cells (Figure 6B), suggesting that Th17 and Treg cells do not contribute to the increased inflammation and tumor susceptibility of WT2 mice. Furthermore, we measured increased activated CD8+ CD69+ cells, resident memory CD8+ CD69+ CD103+ cells, and effector memory CD8+ CD44+ CD62L− cells in the colon LP of WT2 mice, whereas there were no differences in activated, resident memory, or effector memory CD4 T cells in the colon LP between WT1 and WT2 mice (Figures S4D–S4F).
Figure 6. Naive WT2 Mice Exhibit Increased CD8+ IFNγ T Cells in LP, which Partly Mediates Increased Tumor Susceptibility.

(A) Numbers of CD4 and CD8 T cells in colon LP T cells of WT1 and WT2 mice as determined using flow cytometry. Data are pooled from at least two independent experiments. n = 8/group.
(B) Colon LP immune cells were ex vivo-stimulated with PMA and ionomycin with monensin for 4 h, and IFNγ, FoxP3, and IL-17 were measured using flow cytometry. Representative flow plots are shown. Data are pooled from at least two independent experiments. n = 8 or 9/group.
(C) SPF Cd8−/− mice were treated with antibiotics for 1 week prior to three consecutive gavages of SPF WT1 or WT2 microbiota followed by AOM and three rounds of 2% DSS after 4 weeks of bacteria colonization.
(D and E) Tumor number (D) and sizes (E) were measured on day 62 of AOM/DSS. n = 4–6/group.
Data are mean ± SEM. *p < 0.05 by Mann-Whitney test.
See also Figures S3–S7.
We next analyzed the colon LP immune cell composition during the acute inflammatory response to DSS, specifically on day 12, when immune cell infiltration typically peaks (data not shown). As in naive mice, there were significantly increased CD3 and CD8, but not CD4, T cells in WT2 mice (Figures S4G and S4H). Additionally, colon LP CD8+ IFNγ+ T cells were increased in WT2 mice, but no differences in CD4+ IFNγ+, CD4+ IL-17+, or CD4+ Foxp3+ subsets were measured (Figure S4I). Colon LP monocytes and neutrophils were also elevated to a greater extent than in WT1 mice (Figure S4G), likely reflecting greater levels of inflammation in WT2 mice due to DSS treatment (Figure S1).
To determine whether CD8 T cells specifically contributed to increased tumorigenesis in WT2 mice, SPF Cd8−/− mice were treated with an antibiotic cocktail and antifungal water before gavage of SPF WT1 or WT2 stool and cecal contents for 3 consecutive days. Four weeks after the last gavage, mice were treated with AOM/DSS to induce tumors (Figure 6C). By NMDS ordination, Cd8−/− mice colonized with WT1 microbiota (WT1 > CD8 KO) clustered with WT1 control mice, and Cd8−/− mice colonized with WT2 microbiota (WT2 > CD8 KO) clustered with WT2 control mice, suggesting near complete microbiome transfer (Figures S3C and S3D). Importantly, WT1 > CD8 KO mice developed similar tumor numbers as WT1 control mice, whereas WT2 > CD8 KO mice developed significantly fewer and smaller tumors compared with WT2 control mice (Figures 6D and 6E), strongly suggesting that the dysbiotic WT2 microbiome mediates its tumor-promoting effects in part via pro-inflammatory CD8 T cells, although CD8-independent mechanisms likely also contribute.
To determine if WT2 microbiota directly contributed to the increased CD8+ IFNγ+ T cell numbers in naive WT2 mice, we gavaged WT2 mice stool and cecal homogenates into SPF WT1 mice after antibiotic and antifungal water treatment, as described in Figure 6C. After 9 weeks of recolonization, WT1 mice reconstituted with WT2 microbiota (WT1+WT2bac) had microbiomes closely resembling WT2 donor input as well as antibiotic-treated WT2 mice reconstituted with WT2 microbiota (WT2+WT2bac) by NMDS ordination, although there were still significant differences between groups on the basis of thetaYC distances (Figures S5A and S5B). Importantly, WT1+WT2bac mice had increased colon LP CD3 and CD8, but not CD4, T cells, as well as increased CD8+ IFNγ+ T cells compared with the control WT1 mice that were recolonized with WT1 bacteria (WT1+WT1bac) (Figures S5C and S5D). WT1+WT2bac mice also had increased CD4+ IFNγ+ T cells, but no differences in CD4+ IL-17+ or CD4+ Foxp3+ subsets, compared with WT1+WT1bac mice (Figure S5D).
We also gavaged GF WT mice with SPF WT1 or WT2 microbiota as in Figure 3. After 8 weeks of colonization to allow microbial and immune cell reconstitution, colon LP immune cell populations were analyzed using flow cytometry. Although no differences in CD3, CD8, or CD4 T cells were observed (Figure S6A), GF WT mice colonized with WT2 microbiota had increased colon LP CD8+ IFNγ+ T cells, but no differences in CD4+ IFNγ+, CD4+ IL-17+ or CD4+ Foxp3+ subsets, similar to what is observed in naive SPF WT2 mice (Figure S6B).
To explore potential mechanisms of how the WT2 microbiome may alter CD8 T cell responses in the colons of WT2 mice, we examined the expression of the T cell chemoattractants CXCL9 and CXCL10 in the colon epithelium of naive WT1 and WT2 mice by qPCR, and observed no differences, suggesting that the increase in CD8 T cells in the colon LP of WT2 mice is not necessarily due to increased recruitment via epithelial chemokine production (Figure S7A). There were also no differences in the expression of IL-6 or the antimicrobial peptides Reg3γ and β-defensin2, which may predispose WT2 mice to inflammation (Figure S7A). Interestingly, in WT2 epithelium cells, there was increased IL-1β expression, which has been shown to promote effector T cell responses, including the production of IFNγ (Ben-Sasson et al., 2013), as well as occludin, although the significance of this is unclear. Production of IL-12 by DCs can also promote CD8 T cell activation and IFNγ production (Brzoza et al., 2004; Curtsinger et al., 1999; Trinchieri, 1998). To determine if WT2 bacteria promote increased IL-12 production by DCs, we cultured WT1 and WT2 bone marrow-derived DCs (BMDCs) with heat-killed (HK) stool microbiota from either WT1 or WT2 mice and measured IL-12 in the supernatant. HK WT2 microbiota promoted increased IL-12 production by both WT1 and WT2 BMDCs compared with HK WT1 microbiota (Figure S7B). Altogether, these data strongly support a role for the WT2 microbiota in upregulating colon LP IFNγ CD8 T cell responses.
Intratumoral T Cells in WT2 Mice Exhibit an Exhausted Phenotype with Decreased IFNγ Production
CD8 T cells and IFNγ production are important for anti-tumor immunity (Castro et al., 2018; Galon et al., 2006; Mager et al., 2016; Pagès et al., 2005); consistently, increased CD8 T cells and a Th1 gene signature within the tumor microenvironment correlate with improved CRC patient outcomes (Galon et al., 2006). Yet our data suggest that increased homeostatic levels of CD8+ IFNγ+ T cells, associated with dysbiosis, can promote inflammation and tumorigenesis. To determine the activity of CD8 T cells within tumors of WT1 and WT2 mice, we analyzed the immune composition of tumor tissue (“tum”) and non-tumor tissue adjacent to tumors (“adj”) using flow cytometry. No differences in total T cells or CD4 T cells were measured between WT1 and WT2 mice in both tumor and adjacent tissues (Figures 7A and 7B). However, contrary to naive and acute inflammatory conditions, WT2 CD8 T cells are decreased in both tumor and adjacent tissue compared with WT1 tissues (Figure 7B). Furthermore, both intratumoral WT2 CD8 and CD4 T cells have reduced IFNγ responses compared with WT1 CD8 and CD4 T cells upon ex vivo stimulation (Figures 7C and 7D), whereas no differences in intratumoral CD4+ IL-17+ or CD4+ Foxp3+ cells were observed (Figures 7E and 7F).
Figure 7. Intratumoral WT2 T Cells Display Decreased IFNγ Activity and Increased Exhaustion.

(A and B) Numbers of T cells (A) and CD4 and CD8 T cells (B) in tumor (“tum”) and adjacent (“adj”) tissue from AOM/DSS-treated WT1 and WT2 mice as analyzed using flow cytometry on day 60. n = 6–10/group.
(C–F) Representative flow plots after ex vivo stimulation of cells for 4 h and analysis of IFNγ (N = 8–10/group), FoxP3, and IL-17 (n = 4–6/group).
(G and H) Representative flow plots and quantification of PD-1, Tim-3, and Lag-3 on CD8 (G) or CD4 T (H) cells. n = 5–7/group. n = 5–7/group.
Data are mean ± SEM and are pooled from at least two independent experiments. *p < 0.05 by Mann-Whitney test.
On the basis of the pattern of increased CD8+ IFNγ+ T cells at baseline and during chronic inflammation followed by reduced CD8+ IFNγ+ T cells within the tumor microenvironment and adjacent normal tissue in WT2 mice, we hypothesized that the dysbiotic WT2 microbiome promotes T cell exhaustion. T cell exhaustion can occur as a result of prolonged antigen exposure and chronic inflammation (Wherry and Ahmed, 2004; Wherry et al., 2003) and is also observed with tumor-infiltrating lymphocytes, which can promote tumor progression (Baitsch et al., 2011; Fourcade et al., 2010; Grosso et al., 2009; Lee et al., 1999; Matsuzaki et al., 2010; Sakuishi et al., 2010). To determine if intratumoral WT2 T cells exhibit an exhausted phenotype, we measured inhibitory receptors typically expressed by exhausted T cells using flow cytometry. Indeed, there were greater numbers of PD-1+ Lag-3+ and PD-1+ Tim-3+ CD8 T cells within WT2 tumors compared with WT1 tumors (Figure 7G). Additionally, tumor-infiltrating WT2 CD4 T cells also exhibited increased PD-1 and Lag-3 expression compared with WT1 CD4 T cells (Figure 7H). Altogether, these data suggest that dysbiosis can lead to T cell exhaustion by chronically inducing IFNγ production in CD8 T cells, which may result in impaired immune surveillance and increased susceptibility to tumorigenesis.
DISCUSSION
In this study, we took advantage of a unique system in which two distinct microbial communities in WT B6 mice are associated with different disease outcomes to identify host-microbial interactions that underlie susceptibility to inflammation-associated tumorigenesis. WT2 mice, which develop significantly more tumors after AOM/DSS treatment than WT1 mice, have an altered microbiome that is associated with increased LP CD8+ IFNγ+ T cells at baseline and during acute inflammatory responses in the colon. Although CD8 T cells are known for their cytotoxic, anti-tumor activity, our results suggest that in the presence of dysbiosis, they can also have a pathogenic role by promoting damaging, chronic inflammation and consequently tumor development. Consistently, Rag1−/− and Cd8−/− mice transplanted with microbiomes from WT2 donor mice developed significantly fewer tumors. However, the absence of CD8 T cells in the presence of a WT2 microbiome does not completely suppress tumorigenesis, suggesting the possibility that WT2 microbiota can also promote tumorigenesis via CD8-independent mechanisms and/or CD8 T cells can also contribute to tumor suppression. Although there is significant evidence supporting a role of commensal-specific CD4 T cells in mediating inflammatory Th1 and Th17 responses (Cong et al., 1998; Sorini et al., 2018), less is known about the contribution of CD8 T cells in driving colon inflammation. CD8 T cells have been implicated in IBD, whereby increased colon LP activated cytotoxic CD8 T cells are associated with active disease (Muller et al., 1998). In certain mouse models, host antigen-reactive CD8 T cells have been shown to be major drivers of colitis (Nancey et al., 2006; Punit et al., 2015; Westendorf et al., 2006). However, whether WT2 microbiota promotes the generation of commensal-specific CD8 T cells to potentiate colitis-associated tumorigenesis remains to be determined.
How dysbiosis promotes the accumulation of activated CD8+ IFNγ+ T cells in naive WT2 mice is unclear and may be multifactorial in nature. It has been previously shown that Bifidobacterium is capable of priming DCs to enhance CD8 T cell proliferation and IFNγ production (Sivan et al., 2015). Similarly, we observed increased IL-12 production by BMDCs stimulated with HK WT2 microbiota. Therefore, it is possible that the presence of certain bacterial populations in the gut of WT2 mice selectively expands and primes the activation of colon LP CD8 T cells via DC activation. Gnotobiotic mouse studies also suggest that the accumulation of CD8+ IFNγ+ T cells is a result of chemokine induction by specific bacteria proximal to the intestinal epithelium (Tanoue et al., 2019); however, we did not observe any differences in epithelial production of CXCL9 or CXCL10 in WT1 and WT2 mice. Given that bacteria-produced metabolites can have immunomodulatory effects (Kim, 2018; Levy et al., 2016; Smith et al., 2013), it is also possible that WT2 microbiota-specific metabolites may contribute to increased colon LP CD8+ IFNγ+ T cells. For example, the gut microbiota can modulate levels of all-trans-retinoic acid levels that can promote CD8 T cell responses in CRC (Bhattacharya et al., 2016).
Importantly, our results suggest a role of dysbiosis in promoting T cell exhaustion, which, within the tumor microenvironment, can reduce anti-tumor immunity. Specifically, in contrast to increased colon LP CD8+ IFNγ+ T cells in naive and acutely inflamed WT2 mice, there were significantly decreased tumor-infiltrating CD8+ IFNγ+ cells as well as in normal adjacent tissue. A potential explanation for the observed reduction in CD8+ IFNγ+ cells is that chronic stimulation and activation of intestinal LP CD8 T cells by WT2 gut microbiota lead to their exhaustion and result in reduced immune surveillance and ultimately increased tumor burden. Conversely, it is also possible that WT1 microbiota limits T cell exhaustion during the development of inflammation and tumors in the colon. Thus, it may be important to identify bacterial populations that not only induce protective CD8+ IFNγ+ responses within the tumor microenvironment but also attenuate T cell exhaustion to maximize anti-tumor immunity.
In this study, we identified several OTUs that were predictive of tumor susceptibility by analyzing the gut microbiomes of WT1 and WT2 mice as well as of mice from multiple fecal transfer experiments with hybrid microbiomes. We and others have used similar approaches and statistical tools to identify differential OTUs between groups that correlate with disease outcomes (Caruso et al., 2019; Seregin et al., 2017; Surana and Kasper, 2017; Zackular et al., 2015). Here, the low or high tumor burdens of AOM/DSS-treated mice cannot be attributed to just a single bacteria species or family. For example, the bacteria family Prevotellaceae is increased in naive WT2 mice and strongly predictive of high tumor burdens, while Lachnospiraceae is significantly associated with decreased inflammation-associated tumorigenesis. Decreased Lachnospiraceae have been found in IBD and CRC patients (Morgan et al., 2012; Wang et al., 2012), suggesting that they have anti-inflammatory function, possibly via butyrate production (Wang et al., 2012). In contrast, Prevotellaceae are enriched in IBD and CRC patients and are also associated with increased susceptibility to DSS-induced colitis in mice, which suggests that they may be pro-inflammatory (Elinav et al., 2011; Flemer et al., 2017; Sobhani et al., 2011; Wang et al., 2012). Similarly, a member of the Ruminococcaceae was associated with high tumor burdens, and Ruminococcaceae have also been found to be enriched in IBD and CRC patients (Joossens et al., 2011; Wang et al., 2012). Furthermore, a phylogenetic tree based on 16S rRNA sequence similarity suggests that the OTU candidates associated with different tumor burdens but from the same family are evolutionarily distinct from one another (Figure 4C) (Clarridge, 2004; Woese, 1987). Regardless, as we have only identified a correlation between the identified OTUs and tumor outcomes, it still remains to be determined whether these bacteria regulate CD8 T cell responses and tumor susceptibility alone or via interactions with other bacterial populations.
There has been significant interest in optimizing microbiome transfer to control for or determine microbiome-specific effects on phenotype. The gold standard is fecal transplantation of donor microbiota into recipient GF mice. In this study, the transfer of WT1 or WT2 microbiomes and their corresponding tumor phenotype was optimal using this system. Cohousing is a widely used method of microbiome transfer between mice of dissimilar microbiome compositions in host-microbiome association studies, but the optimal length of time of cohousing and ratio of mice harboring two different communities in a cage remain unclear. Here, we show that 4 weeks of 2:2 cohousing resulted in poor microbiome and phenotype transfer. Transfer was significantly improved with 6 weeks of 1:1 cohousing; however, microbiome analyses demonstrated that the transfer was still incomplete, with significant differences in OTU membership between cohoused mice by LEfSe (data not shown), likely explaining the intermediate tumor phenotype. Generation of hybrid microbiomes and incomplete transfer by cohousing were also observed by others (Caruso et al., 2019; Robertson et al., 2019; Surana and Kasper, 2017). In this study, cross-fostering provided better microbiome transfer from WT1 and WT2 mothers to WT2 and WT1 pups, respectively, than cohousing, demonstrating the strong influence of maternal transmission of the microbiome to offspring, although as with 1:1 cohousing, significant differences in microbiome composition remained. Despite the improvement in microbiome transfer, the tumor phenotype of cross-fostered mice remained intermediate between control WT1 and WT2 mice. It is possible that the transfer of WT1 and WT2 microbiomes was incomplete with these methods given the difficulty of transferring mucosa-associated adherent bacteria (Robertson et al., 2019). Alternatively, the lack of complete phenotype transmissibility with either cohousing or cross-fostering may also reflect the poor survivability of obligate anaerobes outside the colon. Interestingly, on the basis of thetaYC dissimilarity, we observed effective microbiome transfer in mice that were gavaged with cecal and fecal contents from donor mice after antibiotic depletion, although not necessarily as complete as in GF mice. Nonetheless, this approach may still be a reasonable alternative to GF mice in determining the effects of specific microbial communities on phenotype. Other groups also observed similar transfer results by this method (Le Roy et al., 2018; Shen et al., 2015; Staley et al., 2017). Nevertheless, further studies are needed to determine the antibiotic combination, length of treatment, and number of gavages for optimal transfer of microbiota and phenotype.
In summary, our data demonstrate a role of the gut microbiome in altering CD8 T cell activity in the colon LP to affect tumorigenesis through early CD8 T cell activation, which can have long-term negative consequences by increasing susceptibility to inflammation and exhaustion, which can result in colon tumorigenesis. Future studies are needed to determine the mechanism by which the microbiota activates CD8 T cells and promotes their exhaustion.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Grace Chen (gchenry@umich.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
SPF WT1 and Cd8−/− (B6.129S2-Cd8atm1Mak) mice (all C57BL/6 background) were originally purchased from Jackson Laboratory and bred in-house at the University of Michigan (UM). WT2 mice were generated from WT littermates (Nod1+/+) of backcrosses between B6/J mice originally purchased from Jackson Laboratory and transgenic Nod1−/− mice (> F8) and subsequently established as a separate wild-type colony on a different rack from WT1 to minimize cross-contamination of WT1 and WT2 microbiomes. Adult male or female 6–10 week-old mice were used except in antibiotic experiments where mice began antibiotic treatment at 4–5 weeks old and cohousing experiments which were done with adult females. Cross-fostered WT1 and WT2 pups were switched to a nursing dam of the opposite colony within 48 hours of birth. GF WT and GF Rag1−/− mice (all C57BL/6 background, derived from Jackson Laboratory mice) were bred and housed at the UM Germ-free mouse facility. Sterility was regularly verified by aerobic and anaerobic cultures, Gram stains and qPCR. GF mice were conventionalized via oral gavage of cecal and stool contents from either SPF WT1 or WT2 mice. Animal studies were conducted under protocols approved by the University of Michigan Committee on the Use and Care of Animals.
METHOD DETAILS
Inflammation-Induced Colon Tumorigenesis
Age- and sex-matched 6–10 week-old mice were injected intraperitoneally with 10mg azoxymethane (Sigma) per kg mouse weight. After five days, mice were treated with three cycles of 2% dextran sulfate sodium (MP Bio, m.w. = 36,000–50,000) given for five days in the drinking water followed by 16 days of regular drinking water. Depending on the particular experiment, mice were sacrificed between days 60–70 after AOM injection as specified in the Figure. This time range does not significantly affect tumor numbers between mice of the same group (data not shown). For tumor counting, colons were flushed of stool with PBS, longitudinally cut open and grossly counted using a magnifier and measured with calipers. For experiments involving GF and conventionalized GF mice, mice were treated with either 1.5% or 2% depending on the experiment. For example, in Figure 3, all groups were treated with two rounds of 2% and a final round of 1.5% DSS water due to increased mortality of GF+WT2 microbiota after two DSS rounds. In Figures 5B–5D, all groups received three rounds of 1.5% DSS water to ensure 100% survival.
Bacteria Preparation for Oral Gavage
Whole SPF stool and cecal contents were isolated from WT1 and WT2 mice. Specifically, stool and cecal contents were collected under strictly anaerobic conditions (85% N2, 10% H2, 5% CO2) in a Coy anaerobic chamber immediately after euthanasia and homogenized in anaerobic PBS. Homogenates were also plated onto either brain-heart infusion (BHI, BD) agar supplemented with 10% horse blood (Quad Five), chopped-meat carbohydrate broth (CMCB) agar (Hehemann et al., 2012), or YCFA agar (Ze et al., 2012). Plates were cultured at 37°C anaerobically for 48 hours. Cultivable bacteria were resuspended from plates using anaerobic PBS and gavaged into GF WT mice.
Antibiotic and Antifungal Treatment of Mice
4–5 week old mice were treated with antibiotic and antifungal water for one week to target Gram-positive, Gram-negative and anaerobic bacteria and to prevent fungal blooms (Kennedy et al., 2018). Mice were treated with an antibiotic cocktail consisting of 0.5g/L vancomycin (Pfizer), 2g/L streptomycin (Sigma), 0.75 g/L metronidazole (Sigma), and 0.5g/L fluconazole (Sigma) that was sterilefiltered prior to administration. 2% sucralose (Apriva, Kroger®) was added to increase the antibiotic solution palatability (Abt et al., 2012). Mice then received regular water for 24 hours, gavaged with WT1 or WT2 stool and cecal homogenates for three consecutive days, then AOM/DSS treatment four weeks later.
Isolation of Lamina Propria Cells
Colon LP cells were isolated as previously described (Chen et al., 2008; Zhan et al., 2016). Briefly, whole colons were isolated from naive 6–10 week old WT1 and WT2 mice and cut into small pieces and washed with HBSS (GIBCO) supplemented with 2.5% heat-inactivated FBS (Sigma Aldrich) and penicillin/streptomycin (HBSS+). After two washes with magnetic stirring, colon pieces were incubated in HBSS+ with 1mM DTT (Invitrogen) at 37°C. Colon pieces were washed once and incubated twice in HBSS+ with 1mM EDTA (Lonza) at 37°C with magnetic stirring for 30 minutes. After washing twice, colon tissue was digested in HBSS+ with 400 I.U./mL type III collagenase (Worthington) and 10 μg/mL DNase I (Worthington) at 37°C with magnetic stirring. The single cell suspension was filtered through a 70-micron filter and colon LP immune cells were collected at the interface of a 40%/75% Percoll gradient after centrifugation. For tumor endpoint analyses, tumor or adjacent tissue were cut from the colons and processed as described above but with a single EDTA step.
Flow Cytometry and Intracellular Cytokine Staining
LP cells were isolated from mice at day 0, day 12 of AOM/DSS or at the tumor endpoint (typically day 60). For intracellular staining, cells were incubated for 4 hours at 37°C with 1) GolgiStop (monensin, BD), 100 ng/ml PMA (Sigma) and 1000 ng/ml ionomycin, or 2) with GolgiStop alone (“no stim”). Cells were surface stained, fixed and permeabilized using the Foxp3/Transcription Factor Staining Buffer Set (eBioscience), and then incubated with fluorochrome-conjugated antibodies against PE-Cy7 IFNγ (Biolegend), FITC IL17A (Biolegend), and PE Foxp3 (eBioscience). Samples were analyzed by a BD LSRFortessa, FACSCanto II, or FACSAria II flow cytometer. Foxp3 staining was done on unstimulated cells.
Preparation of Heat-Killed Bacteria
Stool pellets from SPF WT1 and WT2 mice were homogenized in PBS, strained through a 40um cell strainer, and centrifuged at 1000rpm for 10 s to pellet debris. The supernatant was centrifuged at 5000rpm for 10 minutes. The white bacteria layer of the pellet was diluted in PBS and incubated at 95°C for one hour. Heat-killed bacteria suspensions were stored at −20°C until use.
BMDC Preparation and Stimulation
BMDCs were prepared from WT1 and WT2 mice as previously described (Toubai et al., 2019). Briefly, bone marrow cells were cultured for seven days in RPMI (GIBCO) supplemented with 10% FBS (Sigma Aldrich), 1% penicillin/streptomycin, 1% L-glutamine, and 20 ng/ml GM-CSF (Peprotech) (BMDC-media). BMDCs were isolated using the CD11c Microbeads Ultrapure (Miltenyi Biotec) kit. 4×105 BMDCs were cultured with 4×106 heat-killed bacteria for four hours, washed once in PBS, and were resuspended in fresh BMDC-media for another 20 hours.
ELISA (lipocalin-2 and IL-12p70)
Stool was collected on day 0, 12, 26 or 60 of AOM/DSS and homogenized in PBS at 100 mg/ml. Homogenates were diluted to a range of 1:100 to 1:100000 and lcn-2 was measured using the Lcn-2/NGAL ELISA kit (R&D Systems). IL-12p70 was measured in 24 hour supernatants of heat-killed bacteria-stimulated BMDCs (described below) using the IL-12p70 ELISA kit (R&D Systems).
Histologic Scoring
Histological assessment of H&E sections was performed in a blinded fashion by a pathologist (KAE) using a previously described scoring system with modifications (Chen et al., 2008). A point scale was used to denote the severity of inflammation (0 = none, 1 = mild/mucosa only, 2 = moderate infiltration of mucosa/extension to submucosa, 3 = severe in mucosa and submucosa, and 4 = transmural), severity of epithelial loss (0 = none, 1 = mild/basal 1/3 of glands, 2 = moderate/basal 2/3 crypts, 3 = severe where only surface epithelium remains, 4 = ulceration/erosion), and quantification of adenoma/carcinoma (0 = none, 1 = single focus, 2 = multiple foci). Each parameter was multiplied by a factor reflecting the percentage of colon involvement (0%, 25% or less, 26%–50%, 51%–75%, or 76%–100%) and then summed to obtain the overall score.
Isolation of Bacterial DNA and 16S rRNA Sequences Analyses
16S rRNA analysis was performed with fecal samples that had been collected from 6–12 week old mice on the day of AOM injection and were frozen at −20 or −80°C. Bacterial DNA was isolated using the PowerSoil-htp 96 Well Soil DNA isolation kit (QIAGEN) with the epMotion 5075 or manually with the DNeasy PowerSoil Kit (QIAGEN). The V4 region of the 16S rRNA gene was amplified using custom barcoded primers, sequenced with the Illumina MiSeq Personal Sequencing platform, and processed using the mothur software package to reduce sequencing errors and remove chimeras as previously described (Kozich et al., 2013; Schloss et al., 2009). Sequences were aligned to the SILVA 16S rRNA sequence database (Pruesse et al., 2007). Sequences were clustered into operational taxonomic units (OTUs) using a 97% similarity cutoff. Parallel sequencing and processing of a mock community allowed us to determine a sequencing error rate of 1.38%. Dissimilarity in community structure between samples was calculated using the θYC (thetaYC) distance metric. ThetaYC distances between samples were used for two-dimensional ordination analysis by non-metric dimensional scaling (NMDS). Microbial alpha diversity was calculated using the inverse Simpson index and the observed number of OTUs (richness) (Zackular et al., 2013). Analysis of molecular variance (AMOVA) was used to determine significance between community structure differences of different groups of samples. A random forest regression model (see Statistical Analysis) was used to identify OTUs that are associated with the number of tumors on the day of AOM injection (Zackular et al., 2015). Linear and Poisson regression analyses were used to correlate OTUs to tumor burdens. OTUs identified to be predictive of tumor outcomes based on random forest (see Statistical Analysis) and regression models were cross-referenced with LEfSe and Metastats methods, which were used to identify OTUs associated with low (0–5 tumors) or high (> 15) tumor burdens based on abundance, using an LDA score cutoff of 3 and an abundance difference cutoff of 0.003, respectively. OTUs that were significant in all five statistical models were chosen as candidates. A phylogenetic tree of the candidates was generated based on the V4 region sequence of the 16S rRNA gene using the Clustal Omega program by the EMBL-EBI (Madeira et al., 2019).
RNA Isolation and qPCR
Intraepithelial cells were isolated from untreated WT1 and WT2 mice from the EDTA steps of LP immune cell isolation. Total RNA was isolated using the Nucleospin RNA kit (Macherey-Nagel). cDNA synthesis was performed using the iScript cDNA synthesis kit (BioRad) and used for quantitative PCR using SYBR Green on the ABI 7900HT. Gene transcript levels were normalized to actin. Primer sequences can be found in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD3 APC-Cy7 clone 145-2C11 | BioLegend | Cat#100330; AB_1877170 |
| CD3 PE-Cy7 clone 17A2 | BioLegend | Cat#100220; AB_1732057 |
| CD3 FITC clone 145-2C11 | BioLegend | Cat#100204; AB_312661 |
| CD45.2 BV421 clone 104 | BioLegend | Cat#109832; AB_2565511 |
| CD4 APC clone GK1.5 | eBioscience | Cat#17-004-82; AB_469320 |
| CDS PerCP-Cy5.5 clone 53-6.7 | BioLegend | Cat#100732; AB_893423 |
| CDS APC-Cy7 clone 53-6.7 | BioLegend | Cat#100714; AB_312753 |
| CD69 APC clone H1.2F3 | BioLegend | Cat#104514; AB_492S43 |
| CD44 PE clone IM7 | BioLegend | Cat#103007; AB_312958 |
| CD62L APC-Cy7 clone MEL-14 | BioLegend | Cat#104427; AB_S3079S |
| Tim-3 PerCP-Cy5.5 clone BS.2C12 | BioLegend | Cat#134011; AB_2632735 |
| PD-1 PE-Cy7 clone RMP1-30 | BioLegend | Cat#109109; AB_572016 |
| Lag-3 PE clone C9B7W | BioLegend | Cat#125207; AB_2133344 |
| IFNγ PE-Cy7 clone XMG1.2 | BioLegend | Cat#505826; AB_2295770 |
| IL-17 FITC clone TC11-1SH10.1 | BioLegend | Cat#506907; AB_536009 |
| Foxp3 PE clone FJK-16 s | eBioscience | Cat#12-5773-S0; AB_465936 |
| NK1.1 PerCP-Cy5.5 clone PK136 | BD PharMingen | Cat#561111 |
| Ly6C FITC clone HK1.4 | BioLegend | Cat#12S005; AB_11S6134 |
| Ly6G PE-Cy7 clone 1AS | BioLegend | Cat#127617; AB_1S77262 |
| CD11b PE clone M1/70 | BD PharMingen | Cat#5616S9; AB_394775 |
| CD11c APC clone N41S | BioLegend | Cat#117310; AB_313779 |
| B220 Pacific Blue clone RA3-6B2 | BioLegend | Cat#103227; AB_492S76 |
| CD103 PE clone 2E7 | BioLegend | Cat#121405; AB_53594S |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Azoxymethane (AOM) | Sigma Aldrich | Cat#A5486 |
| Dithiothreitol (DTT) | Invitrogen | Cat#ILT1550S013 |
| AccuGENe 0.5 M EDTA Solution | Lonza | Cat#BMA51201 |
| Deoxyribonuclease I | Worthington | Cat#LS002007 |
| Collagenase, Type 3 | Worthington | Cat#LS0041S3 |
| Recombinant Murine GM-CSF | Peprotech | Cat#315-03 |
| Percoll | GE Healthcare Life Sciences | Cat#170S9101 |
| Phorbol 12-myristate 13-acetate | Sigma Aldrich | Cat#PS139-5MG |
| lonomycin | Sigma Aldrich | Cat#I9657 |
| BD GolgiStop Protein Transport Inhibitor | BD | Cat#554724 |
| Fluconazole | Sigma Aldrich | Cat#PHR1160 |
| Vancomycin Hydrochloride, USP | Pfizer | N/A |
| Streptomycin sulfate salt | Sigma Aldrich | Cat#S6501 |
| Metronidazole | Sigma Aldrich | Cat#M1547 |
| BHI | BD | Cat#237500 |
| Defibrinated Horse Blood | Quad Five | Cat#210 |
| Granulated Agar | Thermo Fisher Scientific | Cat#BP9744-500 |
| Bacto Agar | BD | Cat#DF0140-01-0 |
| Beef extract | Sigma-Aldrich | Cat#B4SSS |
| Pancreatic digest of casein | BioWorld | Cat#30620060-1 |
| Yeast extract | Fluka Analytical | Cat#70161 |
| Dipotassium phosphate | Thermo Fisher Scientific | Cat#BP363-500 |
| Cysteine | Sigma-Aldrich | Cat#C7352 |
| Glucose | Sigma-Aldrich | Cat#158968 |
| Galactose | Sigma-Aldrich | Cat#G0625 |
| Fructose | Sigma-Aldrich | Cat#F0127 |
| Mannose | Acros Organics | Cat#150600 |
| N-acetyl glucosamine | Sigma-Aldrich | Cat#A3286 |
| Xylose | Sigma-Aldrich | Cat#X3877 |
| Hematin | Sigma-Aldrich | Cat#H3281 |
| Histidine | Sigma-Aldrich | Cat#H8000 |
| Menadione | Sigma-Aldrich | Cat#M5625 |
| Cyanocobalamin | Sigma-Aldrich | Cat#V2876 |
| p-Aminobenzoic acid | Sigma-Aldrich | Cat#A9878 |
| Folic acid | Sigma-Aldrich | Cat#F7876 |
| Biotin | Sigma-Aldrich | Cat#B4501 |
| Nicotinic acid | Sigma-Aldrich | Cat#N4126 |
| Calcium pantothenate | Sigma-Aldrich | Cat#P2250 |
| Riboflavin | Sigma-Aldrich | Cat#R7649 |
| Thiamine HCl | Sigma-Aldrich | Cat#T4625 |
| Pyridoxine HCl | Sigma-Aldrich | Cat#P9755 |
| Thioctic acid | Sigma-Aldrich | Cat#T5625 |
| Adenine | Sigma-Aldrich | Cat#A2786 |
| Guanine | Sigma-Aldrich | Cat#G11950 |
| Thymine | Sigma-Aldrich | Cat#T0895 |
| Cytosine | Sigma-Aldrich | Cat#C3506 |
| Uracil | Sigma-Aldrich | Cat#U1128 |
| Magnesium sulfate anhydrous | Sigma-Aldrich | Cat#M7506 |
| Manganese(II) sulfate monohydrate | Mallinkrodt | Cat#6192 |
| Sodium chloride | Sigma-Aldrich | Cat#S7653 |
| Iron(II) sulfate heptahydrate | Sigma-Aldrich | Cat#215422 |
| Calcium chloride | Sigma-Aldrich | Cat#C1016 |
| Zinc sulfate heptahydrate | JT Baker Chemical Company | Cat#4382 |
| Copper(II) sulfate pentahydrate | Sigma-Aldrich | Cat#C7631 |
| Boric acid | Sigma-Aldrich | Cat#B6768 |
| Sodium molybdate dehydrate | JT Baker Chemical Company | Cat#3764 |
| Nickel(II) chloride | Sigma-Aldrich | Cat#339350 |
| Alanine | Sigma-Aldrich | Cat#A7469 |
| Arginine | Sigma-Aldrich | Cat#A8094 |
| Asparagine | Sigma-Aldrich | Cat#A4159 |
| Aspartic Acid | Sigma-Aldrich | Cat#A93100 |
| Glutamic Acid | Sigma-Aldrich | Cat#G1501 |
| Glutamine | Sigma-Aldrich | Cat#G8540 |
| Glycine | Thermo Fisher Scientific | Cat#BP381-5 |
| Isoleucine | Sigma-Aldrich | Cat#I2752 |
| Leucine | Sigma-Aldrich | Cat#L8000 |
| Lysine | Sigma-Aldrich | Cat#L5501 |
| Methionine | Sigma-Aldrich | Cat#M9625 |
| Phenylalanine | Sigma-Aldrich | Cat#P2126 |
| Proline | Sigma-Aldrich | Cat#P5607 |
| Serine | Sigma-Aldrich | Cat#84959 |
| Threonine | Sigma-Aldrich | Cat#T8625 |
| Tyrosine | Sigma-Aldrich | Cat#T3754 |
| Valine | Sigma-Aldrich | Cat#V0500 |
| Tryptone | Thermo Fisher Scientific | Cat#BP1421 |
| Sodium bicarbonate | Sigma-Aldrich | Cat#S5761 |
| Acetic acid | Acros Organics | Cat#222140010 |
| Propionic acid | Acros Organics | Cat#149300010 |
| Isobutyric acid | Alfa Aesar | Cat#79-31 -2 |
| Isovaleric acid | Alfa Aesar | Cat#503-74-2 |
| Valeric acid | Alfa Aesar | Cat#109-52-4 |
| Potassium dihydrogen phosphate | Thermo Fisher Scientific | Cat#P284 |
| Dextran sulfate sodium salt (colitis grade, m.w. = 36,000-50,000) | MP Bio | Cat#160110 |
| Critical Commercial Assays | ||
| DNeasy PowerSoil Kit | QIAGEN (formerly Mo Bio) | Cat#12888-100 |
| Nucleospin RNA kit | Macherey-Nagel | Cat#740955.50 |
| SYBR Green PCR Master Mix | Applied Biosystems | Cat#4309155 |
| iScript cDNA Synthesis Kit | 3io-Rad | Cat#1708890 |
| Foxp3 / Transcription Factor Staining Buffer Set | eBioscience | Cat#00-5523-00 |
| Mouse Lipocalin-2/NGAL DuoSet ELISA kit | R&D Systems | Cat#DY1857 |
| Mouse IL-12 p70 DuoSet ELISA kit | R&D Systems | Cat#DY419 |
| Deposited Data | ||
| 16S rRNA Sequencing Data | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| WT1 mice | Purchased from Jackson Laboratory and bred in the Cancer Center mouse room | N/A |
| WT2 mice | Derived from mice purchased from Jackson _aboratory and bred in the Cancer Center mouse room | N/A |
| CD8−/− mice | Purchased from Jackson Laboratory and bred n the Cancer Center mouse room | N/A |
| GF Rag1−/− mice | Germfree facility at UM | N/A |
| GF WT mice | Germfree facility at UM | N/A |
| Oligonucleotides | ||
| qPCR primer: Eub F | 5′-AGAGTTTGATCCTGGCTC-3′ | N/A |
| qPCR primer: Eub R | 5′-TGCTGCCTCCCGTAGGAGT-3′ | N/A |
| qPCR primer: IL-1β F | 5′-GATCCACACTCTCCAGCTGCA-3′ | N/A |
| qPCR primer: IL-1β R | 5′-CAACCAACAAGTGATATTCTCCATG-3′ | N/A |
| qPCR primer: CXCL9 F | 5′-GGAACCCTAGTGATAAGGAATGCA-3′ | N/A |
| qPCR primer: CXCL9 R | 3′-TGAGGTCTTTGAGGGATTTGTAGTG-5′ | N/A |
| qPCR primer: CXCL10 F | 5′-GACGGTCCGCTGCAACTG-3′ | N/A |
| qPCR primer: CXCL10 R | 3′-CTTCCCTATGGCCCTCATTCT-5′ | N/A |
| qPCR primer: IL-6 F | 5′-CACAT GTTCTCTGGGAAATCG-3′ | N/A |
| qPCR primer: IL-6 R | 5′-TTTCTGCAAGTGCATCATCG-3′ | N/A |
| qPCR primer: occludin F | 5′-GGGAATGTCCAGAACGAGAAGA-3′ | N/A |
| qPCR primer: occludin R | 5′-CGTGGCAATGAACACCATGA-3′ | N/A |
| qPCR primer:β-defensin2 F | 5′-AAGTATTGGATACGAAGCAG-3′ | N/A |
| qPCR primer:β-defensin2 R | 5′-TGGCAGAAGGAGGACAAATG-3′ | N/A |
| qPCR primer: Reg3g F | 5′-TCAGGTGCAAGGTGAAGTTG-3′ | N/A |
| qPCR primer: Reg3g R | 5′-GGCCACTGTTACCACTGCTT-3′ | N/A |
| qPCR primer: actin F | 5′-CAACTTGATGTATGAAGGCTTTGGT-3′ | N/A |
| qPCR primer: actin R | 5′-ACTTTTATTGGTCTCAAGTCAGTGTACAG-3′ | N/A |
| Software and Algorithms | ||
| GraphPad Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| FlowJo | FlowJo | https://www.flowjo.com/ |
| mothur | Mothur | https://www.mothur.org/ |
| Microsoft Excel | Microsoft | N/A |
| R | https://www.r-project.org/ | N/A |
| BioRender | https://biorender.com/ | N/A |
Graphics
Graphical Abstract was created with BioRender.com
QUANTIFICATION AND STATISTICAL ANALYSIS
Continuous data are shown as mean ± SEM. Sample sizes can be found in the Figure Legends. Statistical analyses were performed using GraphPad Prism7, R and mothur software. Statistically significant differences were determined by Mann-Whitney when only two groups are compared (e.g., tumor numbers, fecal Lcn-2 levels). Differences in bacterial community structure were analyzed using AMOVA in mothur (Excoffier et al., 1992). P values below 0.05 were considered statistically significant and are represented as follows: * p < 0.05. A random forest (RF) regression model was used to identify OTUs that are associated with the number of tumors that developed. RF is a decision tree-based approach that allows for nonlinear relationship between the OTU and tumor count data and interactions between OTUs. The RF model included all 521 OTUs with a total normalized count over all mice > 0.005. It was fit using the randomForest package in R with default parameters except that we increased the number of trees in RF to 1,000 trees (Breiman, 2001). Finally, OTUs were ranked by importance in the RF model as determined by the percent reduction in the mean square error (MSE) when an OTU was removed from the model. As an alternative to the machine learning method, we also used parametric regression models to study the effect of each OTU on the tumor count data. Specifically, we considered the simple linear regression (lm function in R) and Poisson regression (glm function in R). The resulting p values were adjusted to control the false discovery rate (Benjamini and Yekutieli, 2001).
DATA AND CODE AVAILABILITY
The accession number for the FASTQ sequences reported in this paper is Sequence Read Archive: PRJNA557261.
Supplementary Material
Highlights.
Dysbiosis can lead to increased colon tumor susceptibility and CD8+ IFNγ+ T cells
Increased colon CD8+ IFNγ+ T cells are associated with more inflammation and tumors
Dysbiosis and increased tumorigenesis are associated with greater T cell exhaustion
ACKNOWLEDGMENTS
We thank Joseph Zackular, Nielsen Baxter, Nicholas Pudlo, Gabriel Nuñez, Asma Nusrat, Yatrik Shah, and Sergey Seregin for their advice and critical manuscript reading, and the University of Michigan (UM) Flow Cytometry, Germ-Free and Gnotobiotic, Cancer Center Immunology, DNA Sequencing, Histology, and Microscopy Cores for technical support. This work was supported by a UM Rackham Research Grant and NIH grant T32 AI007413 (A.I.Y.) and NIH grant R01 CA166879, an American Cancer Society Research Scholar Grant, and the Tom Liu Memorial Golf Tournament Fund from the UM Rogel Cancer Center (G.Y.C.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.03.035.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abt MC, Osborne LC, Monticelli LA, Doering TA, Alenghat T, Sonnenberg GF, Paley MA, Antenus M, Williams KL, Erikson J, et al. (2012). Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity 37, 158–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Sinha R, Pei Z, Dominianni C, Wu J, Shi J, Goedert JJ, Hayes RB, and Yang L (2013). Human gut microbiome and risk for colorectal cancer. J. Natl. Cancer Inst. 105, 1907–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, and Bray F (2017). Global patterns and trends in colorectal cancer incidence and mortality. Gut 66, 683–691. [DOI] [PubMed] [Google Scholar]
- Arthur JC, Perez-Chanona E, Mühlbauer M, Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B, Rogers AB, et al. (2012). Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 338, 120–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, et al. (2011). Induction of colonic regulatory T cells by indigenous Clostridium species. Science 331, 337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baitsch L, Baumgaertner P, Devêvre E, Raghav SK, Legat A, Barba L, Wieckowski S, Bouzourene H, Deplancke B, Romero P, et al. (2011). Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J. Clin. Invest. 121, 2350–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter NT, Zackular JP, Chen GY, and Schloss PD (2014). Structure of the gut microbiome following colonization with human feces determines colonic tumor burden. Microbiome 2, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker C, Fantini MC, Schramm C, Lehr HA, Wirtz S, Nikolaev A, Burg J, Strand S, Kiesslich R, Huber S, et al. (2004). TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity 21, 491–501. [DOI] [PubMed] [Google Scholar]
- Belkaid Y, and Hand TW (2014). Role of the microbiota in immunity and inflammation. Cell 157, 121–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Sasson SZ, Wang K, Cohen J, and Paul WE (2013). IL-1β strikingly enhances antigen-driven CD4 and CD8 T-cell responses. Cold Spring Harb. Symp. Quant. Biol. 78, 117–124. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, and Yekutieli D (2001). The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 29, 1165–1188. [Google Scholar]
- Bhattacharya N, Yuan R, Prestwood TR, Penny HL, DiMaio MA, Reticker-Flynn NE, Krois CR, Kenkel JA, Pham TD, Carmi Y, et al. (2016). Normalizing microbiota-induced retinoic acid deficiency stimulates protective CD8(+) T cell-mediated immunity in colorectal cancer. Immunity 45, 641–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman L (2001). Random forests. Mach. Learn. 45, 5–32. [Google Scholar]
- Brzoza KL, Rockel AB, and Hiltbold EM (2004). Cytoplasmic entry of Listeria monocytogenes enhances dendritic cell maturation and T cell differentiation and function. J. Immunol. 173, 2641–2651. [DOI] [PubMed] [Google Scholar]
- Caruso R, Ono M, Bunker ME, Nunez G, and Inohara N (2019). Dynamic and asymmetric changes of the microbial communities after cohousing in laboratory mice. Cell Rep. 27, 3401–3412.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro F, Cardoso AP, Goncalves RM., Serre K, and Oliveira MJ (2018). Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Front. Immunol. 9, 847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassaing B, Srinivasan G, Delgado MA, Young AN, Gewirtz AT, and Vijay-Kumar M (2012). Fecal lipocalin 2, a sensitive and broadly dynamic noninvasive biomarker for intestinal inflammation. PLoS ONE 7, e44328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassaing B, Aitken JD, Malleshappa M, and Vijay-Kumar M (2014). Dextran sulfate sodium (DSS)-induced colitis in mice. Curr. Protoc. Immunol. 104, 15.25.1–15.25.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GY, Shaw MH, Redondo G, and Núñez G (2008). The innate immune receptor Nod1 protects the intestine from inflammation-induced tumorigenesis. Cancer Res. 68, 10060–10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarridge JE 3rd. (2004). Impact of 16S rRNA gene sequence analysis for identification of bacteria on clinical microbiology and infectious diseases. Clin. Microbiol. Rev. 17, 840–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong Y, Brandwein SL, McCabe RP, Lazenby A, Birkenmeier EH, Sundberg JP, and Elson CO (1998). CD4+ T cells reactive to enteric bacterial antigens in spontaneously colitic C3H/HeJBir mice: increased T helper cell type 1 response and ability to transfer disease. J. Exp. Med. 187, 855–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtsinger JM, Schmidt CS, Mondino A, Lins DC, Kedl RM, Jenkins MK, and Mescher MF (1999). Inflammatory cytokines provide a third signal for activation of naive CD4+ and CD8+ T cells. J. Immunol. 162, 3256–3262. [PubMed] [Google Scholar]
- Daft JG, Ptacek T, Kumar R, Morrow C, and Lorenz RG (2015). Cross-fostering immediately after birth induces a permanent microbiota shift that is shaped by the nursing mother. Microbiome 3, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Robertis M, Massi E, Poeta ML, Carotti S, Morini S, Cecchetelli L, Signori E, and Fazio VM (2011). The AOM/DSS murine model for the study of colon carcinogenesis: From pathways to diagnosis and therapy studies. J. Carcinog. 10, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaden JA, Abrams KR, and Mayberry JF (2001). The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut 48, 526–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R (2018). Taxonomy annotation and guide tree errors in 16S rRNA databases. PeerJ 6, e5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI, and Flavell RA (2011). NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier L, Smouse PE, and Quattro JM (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131, 479–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flemer B, Lynch DB, Brown JMR, Jeffery IB, Ryan FJ, Claesson MJ, O’Riordain M, Shanahan F, and O’Toole PW (2017). Tumour-associated and non-tumour-associated microbiota in colorectal cancer. Gut 66, 633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, Kirkwood JM, Kuchroo V, and Zarour HM (2010). Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 207, 2175–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, et al. (2013). Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450. [DOI] [PubMed] [Google Scholar]
- Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, Tosolini M, Camus M, Berger A, Wind P, et al. (2006). Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313, 1960–1964. [DOI] [PubMed] [Google Scholar]
- Geva-Zatorsky N, Sefik E, Kua L, Pasman L, Tan TG, Ortiz-Lopez A, Yanortsang TB, Yang L, Jupp R, Mathis D, et al. (2017). Mining the human gut microbiota for immunomodulatory organisms. Cell 168, 928–943.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, and Karin M (2004). IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 118, 285–296. [DOI] [PubMed] [Google Scholar]
- Grosso JF, Goldberg MV, Getnet D, Bruno TC, Yen H-R, Pyle KJ, Hipkiss E, Vignali DAA, Pardoll DM, and Drake CG (2009). Functionally distinct LAG-3 and PD-1 subsets on activated and chronically stimulated CD8 T cells. J. Immunol. 182, 6659–6669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M, Enk J, Bar-On Y, Stanietsky-Kaynan N, Coppenhagen-Glazer S, et al. (2015). Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity 42, 344–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hehemann JH, Kelly AG, Pudlo NA, Martens EC, and Boraston AB (2012). Bacteria of the human gut microbiome catabolize red seaweed glycans with carbohydrate-active enzyme updates from extrinsic microbes. Proc. Natl. Acad. Sci. U S A 109, 19786–19791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joossens M, Huys G, Cnockaert M, Preter VD, Verbeke K, Rutgeerts P, Vandamme P, and Vermeire S (2011). Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut 60, 631–637. [DOI] [PubMed] [Google Scholar]
- Kennedy EA, King KY, and Baldridge MT (2018). Mouse microbiota models: comparing germ-free mice and antibiotics treatment as tools for modifying gut bacteria. Front. Physiol. 9, 1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH (2018). Immune regulation by microbiome metabolites. Immunology 154, 220–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozich JJ, Westcott SL, Baxter NT, Highlander SK, and Schloss PD (2013). Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Roy T, Debédat J, Marquet F, Da-Cunha C, Ichou F, Guerre-Millo M, Kapel N, Aron-Wisnewsky J, and Clément K (2018). Comparative evaluation of microbiota engraftment following fecal microbiota transfer in mice models: age, kinetic and microbial status matter. Front. Microbiol. 9, 3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PP, Yee C, Savage PA, Fong L, Brockstedt D, Weber JS, Johnson D, Swetter S, Thompson J, Greenberg PD, et al. (1999). Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat. Med. 5, 677–685. [DOI] [PubMed] [Google Scholar]
- Levy M, Thaiss CA, and Elinav E (2016). Metabolites: messengers between the microbiota and the immune system. Genes Dev. 30, 1589–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeira F, Park YM, Lee J, Buso N, Gur T, Madhusoodanan N, Basutkar P, Tivey ARN, Potter SC, Finn RD, et al. (2019). The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47, W636–W641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mager LF, Wasmer MH, Rau TT, and Krebs P (2016). Cytokine-induced modulation of colorectal cancer. Front. Oncol. 6, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markman JL, and Shiao SL (2015). Impact of the immune system and immunotherapy in colorectal cancer. J. Gastrointest. Oncol. 6, 208–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, Schilter HC, Rolph MS, Mackay F, Artis D, et al. (2009). Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461, 1282–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki J, Gnjatic S, Mhawech-Fauceglia P, Beck A, Miller A, Tsuji T, Eppolito C, Qian F, Lele S, Shrikant P, et al. (2010). Tumor-infiltrating NYESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl. Acad. Sci. U S A 107, 7875–7880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RJ, and Stanley D (2016). Experimental design considerations in microbiota/inflammation studies. Clin. Transl. Immunol. 5, e92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, Reyes JA, Shah SA, LeLeiko N, Snapper SB, et al. (2012). Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 13, R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller S, Lory J, Corazza N, Griffiths GM, Z’graggen K, Mazzucchelli L, Kappeler A, and Mueller C (1998). Activated CD4+ and CD8+ cytotoxic cells are present in increased numbers in the intestinal mucosa from patients with active inflammatory bowel disease. Am. J. Pathol. 152, 261–268. [PMC free article] [PubMed] [Google Scholar]
- Műzes G, Molnár B, and Sipos F (2012). Regulatory T cells in inflammatory bowel diseases and colorectal cancer. World J. Gastroenterol. 18, 5688–5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nancey S, Holvöet S, Graber I, Joubert G, Philippe D, Martin S, Nicolas JF, Desreumaux P, Flourié B, and Kaiserlian D (2006). CD8+ cytotoxic T cells induce relapsing colitis in normal mice. Gastroenterology 131, 485–496. [DOI] [PubMed] [Google Scholar]
- Neufert C, Becker C, and Neurath MF (2007). An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation-driven tumor progression. Nat. Protoc. 2, 1998–2004. [DOI] [PubMed] [Google Scholar]
- Nougayrède JP, Homburg S, Taieb F, Boury M, Brzuszkiewicz E, Gottschalk G, Buchrieser C, Hacker J, Dobrindt U, and Oswald E (2006). Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science 313, 848–851. [DOI] [PubMed] [Google Scholar]
- Pagès F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, Mlecnik B, Kirilovsky A, Nilsson M, Damotte D, et al. (2005). Effector memory T cells, early metastasis, and survival in colorectal cancer. N. Engl. J. Med. 353, 2654–2666. [DOI] [PubMed] [Google Scholar]
- Pernot S, Terme M, Voron T, Colussi O, Marcheteau E, Tartour E, and Taieb J (2014). Colorectal cancer and immunity: what we know and perspectives. World J. Gastroenterol. 20, 3738–3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, and Glöckner FO (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punit S, Dube PE, Liu CY, Girish N, Washington MK, and Polk DB (2015). Tumor necrosis factor receptor 2 restricts the pathogenicity of CD8(+) T cells in mice with colitis. Gastroenterology 149, 993–1005.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razi S, Baradaran Noveiry B, Keshavarz-Fathi M, and Rezaei N (2019). IL-17 and colorectal cancer: From carcinogenesis to treatment. Cytokine 116, 7–12. [DOI] [PubMed] [Google Scholar]
- Robertson SJ, Lemire P, Maughan H, Goethel A, Turpin W, Bedrani L, Guttman DS, Croitoru K, Girardin SE, and Philpott DJ (2019). Comparison of co-housing and littermate methods for microbiota standardization in mouse models. Cell Rep. 27, 1910–1919.e2. [DOI] [PubMed] [Google Scholar]
- Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, and Anderson AC (2010). Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 207, 2187–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, and Huttenhower C (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seregin SS, Golovchenko N, Schaf B, Chen J, Pudlo NA, Mitchell J, Baxter NT, Zhao L, Schloss PD, Martens EC, et al. (2017). NLRP6 protects IL10−/− mice from colitis by limiting colonization of Akkermansia muciniphila. Cell Rep. 19, 733–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen TC, Albenberg L, Bittinger K, Chehoud C, Chen YY, Judge CA, Chau L, Ni J, Sheng M, Lin A, et al. (2015). Engineering the gut microbiota to treat hyperammonemia. J. Clin. Invest. 125, 2841–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Lei YM, Jabri B, Alegre ML, et al. (2015). Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350, 1084–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, Glickman JN, and Garrett WS (2013). The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobhani I, Tap J, Roudot-Thoraval F, Roperch JP, Letulle S, Langella P, Corthier G, Tran Van Nhieu J, and Furet JP (2011). Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS ONE 6, e16393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorini C, Cardoso RF, Gagliani N, and Villablanca EJ (2018). Commensal bacteria-specific CD4+ T cell responses in health and disease. Front. Immunol. 9, 2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley C, Kaiser T, Beura LK, Hamilton MJ, Weingarden AR, Bobr A, Kang J, Masopust D, Sadowsky MJ, and Khoruts A (2017). Stable engraftment of human microbiota into mice with a single oral gavage following antibiotic conditioning. Microbiome 5, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surana NK, and Kasper DL (2017). Moving beyond microbiome-wide associations to causal microbe identification. Nature 552, 244–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki R, Kohno H, Sugie S, and Tanaka T (2004). Sequential observations on the occurrence of preneoplastic and neoplastic lesions in mouse colon treated with azoxymethane and dextran sodium sulfate. Cancer Sci. 95, 721–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Kohno H, Suzuki R, Yamada Y, Sugie S, and Mori H (2003). A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci. 94, 965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanoue T, Morita S, Plichta DR, Skelly AN, Suda W, Sugiura Y, Narushima S, Vlamakis H, Motoo I, Sugita K, et al. (2019). A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature 565, 600–605. [DOI] [PubMed] [Google Scholar]
- Toubai T, Fujiwara H, Rossi C, Riwes M, Tamaki H, Zajac C, Liu C, Mathew AV, Byun J, Oravecz-Wilson K, et al. (2019). Host NLRP6 exacerbates graft-versus-host disease independent of gut microbial composition. Nat. Microbiol. 4, 800–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinchieri G (1998). Interleukin-12: a cytokine at the interface of inflammation and immunity. Adv. Immunol. 70, 83–243. [DOI] [PubMed] [Google Scholar]
- Wang T, Cai G, Qiu Y, Fei N, Zhang M, Pang X, Jia W, Cai S, and Zhao L (2012). Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 6, 320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westendorf AM, Fleissner D, Deppenmeier S, Gruber AD, Bruder D, Hansen W, Liblau R, and Buer J (2006). Autoimmune-mediated intestinal inflammation-impact and regulation of antigen-specific CD8+ T cells. Gastroenterology 131, 510–524. [DOI] [PubMed] [Google Scholar]
- Wherry EJ, and Ahmed R (2004). Memory CD8 T-cell differentiation during viral infection. J. Virol. 78, 5535–5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, and Ahmed R (2003). Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 77, 4911–4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JR, Nagarajan N, and Pop M (2009). Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput. Biol. 5, e1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woese CR (1987). Bacterial evolution. Microbiol. Rev. 51, 221–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, Huso DL, Brancati FL, Wick E, McAllister F, et al. (2009). A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 15, 1016–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zackular JP, Baxter NT, Iverson KD, Sadler WD, Petrosino JF, Chen GY, and Schloss PD (2013). The gut microbiome modulates colon tumorigenesis. MBio 4, e00692–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zackular JP, Rogers MA, Ruffin MT 4th, and Schloss PD (2014). The human gut microbiome as a screening tool for colorectal cancer. Cancer Prev. Res. (Phila.) 7, 1112–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zackular JP, Baxter NT, Chen GY, and Schloss PD (2015). Manipulation of the gut microbiota reveals role in colon tumorigenesis. MSphere 1, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ze X, Duncan SH, Louis P, and Flint HJ (2012). Ruminococcus bromii is a keystone species for the degradation of resistant starch in the human colon. ISME J. 6, 1535–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Y, Chen PJ, Sadler WD, Wang F, Poe S, Núñez G, Eaton KA, and Chen GY (2013). Gut microbiota protects against gastrointestinal tumorigenesis caused by epithelial injury. Cancer Res. 73, 7199–7210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Y, Seregin SS, Chen J, and Chen GY (2016). Nod1 limits colitis-associated tumorigenesis by regulating IFN-γ production. J. Immunol. 196, 5121–5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the FASTQ sequences reported in this paper is Sequence Read Archive: PRJNA557261.