

Abstract
Gene editing is often touted as a permanent method for correcting mutations, but its long-term benefits in Duchenne muscular dystrophy (DMD) may depend on sufficiently high editing efficiencies to halt muscle degeneration. Here, we explored the persistence of dystrophin expression following recombinant adeno-associated virus serotype 6 (rAAV6):CRISPR-Cas9-mediated multi-exon deletion/reframing in systemically injected 2- and 11-week-old dystrophic mice and show that induction of low dystrophin levels persists for several months in cardiomyocytes but not in skeletal muscles, where myofibers remain susceptible to necrosis and regeneration. Whereas gene-correction efficiency in both muscle types was enhanced with increased ratios of guide RNA (gRNA)-to-nuclease vectors, obtaining high dystrophin levels in skeletal muscles via multi-exon deletion remained challenging. In contrast, when AAV-microdystrophin was codelivered with editing components, long-term gene-edited dystrophins persisted in both muscle types. These results suggest that the high rate of necrosis and regeneration in skeletal muscles, compared with the relative stability of dystrophic cardiomyocytes, caused the rapid loss of edited genomes. Consequently, stable dystrophin expression in DMD skeletal muscles will require either highly efficient gene editing or the use of cotreatments that decrease skeletal muscle degeneration.
Keywords: CRISPR/Cas9, Duchenne muscular dystrophy, gene editing, AAV, microdystrophin, dystrophin
Graphical Abstract
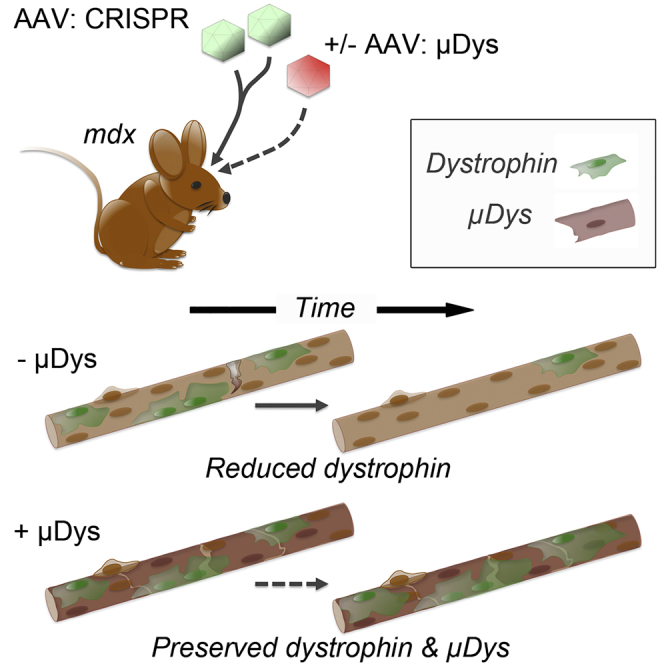
Bengtsson et al. show that the efficacy of dystrophin gene-editing in skeletal, but not cardiac, muscle depends on the rate of muscle necrosis/regeneration. The authors further show that muscle stabilization, here via codelivery of microdystrophin, significantly improves long-term outcomes of therapeutic gene editing in a mouse model of Duchenne muscular dystrophy.
Introduction
Duchenne muscular dystrophy (DMD) affects approximately 1 in 5,000 newborn males and is characterized by progressive muscle wasting, weakness, and premature death.1,2 DMD is caused by mutations in the gene-encoding dystrophin, which plays an essential role in maintaining muscle integrity by providing a structural link between the cytoskeleton and extracellular matrix. In the absence of dystrophin, multinucleated skeletal muscle cells (myofibers) undergo repeated bouts of necrosis and muscle stem cell (SC)-mediated regeneration (turnover), ultimately resulting in reduced regenerative capacity and the replacement of muscle mass with fibrotic and adipose tissue.3,4 Cardiomyocytes are also affected, but they display a delayed and very slow rate of necrosis and are not replaced by SCs. Our group has previously developed highly functional miniaturized versions of dystrophin (microdystrophin [μDys]), which are small enough, together with compact muscle-specific expression cassettes (MSECs), to be packaged into recombinant adeno-associated viral (rAAV) vectors for restoring expression of dystrophin following systemic infusion.5, 6, 7 AAV-mediated μDys gene-transfer therapies demonstrate tremendous therapeutic benefits in both mice and large-animal models of DMD7, 8, 9 and are currently being evaluated in three clinical trials.10 Potential limitations of μDys gene-transfer therapies are that μDys are not fully functional and that episomal vector genomes (vgs) and hence, μDys expression could be lost over time due to normal muscle turnover. Approaches that enable correction of endogenous dystrophin expression therefore represent attractive alternatives.11,12 We11,13 and others14, 15, 16, 17, 18 have demonstrated the use of CRISPR-Cas9-based gene editing for correcting dystrophin expression in cardiac and skeletal muscles of dystrophic mouse models following AAV-mediated delivery. Although dystrophin editing has been shown for multiple muscle groups following systemic gene delivery, optimizations to enhance efficiency and longevity of correction in dystrophic muscle and hence the long-term beneficial impact on pathophysiology are still being pursued. Whereas robust and persistent therapeutic benefits, including normalization of muscle function, are often reported following local intramuscular delivery of AAV:CRISPR-Cas9, translating local therapeutic responses to body-wide functional improvements following systemic administration is not straightforward. One challenge for systemic versus local gene editing is that higher effective vector doses (vgs per body weight [versus muscle weight]) are easier to achieve via local delivery. Equivalent vector doses needed to reach comparable body-wide therapeutic effects via systemic delivery present significant challenges for both the manufacturing and safe infusion of very high vector amounts needed to account for vector uptake in nontarget (nonmuscle) tissues, such as the liver, as well as the vector trapped in the interstitial space between blood microvasculature and muscle cells. Suboptimal systemic vector doses will likely fail to correct sufficient numbers of myonuclear genomes distributed along the entire length of myofibers. Since damage and necrosis can occur from tears at structurally compromised regions at any point along a myofiber, sufficient longitudinal distribution of dystrophin along the sarcolemma is crucial for preventing pathological myofiber turnover. Skeletal muscles of both DMD patients and animal models experience turnover throughout life, but dystrophic mdx mice display a uniquely accelerated pace of turnover during an early crisis period that occurs between 3 and 7 weeks postnatally.19 This provides an opportunity for conducting stringent tests of AAV-mediated dystrophin gene correction and of the resulting gene-edited dystrophins during a period of high skeletal muscle turnover.
Here, we present results describing dystrophin gene correction via deletion of exons 52 and 53 following AAV-mediated delivery of CRISPR-Cas9 to 2- and 11-week-old dystrophic (mdx4cv) mice. Our findings indicate that the long-lasting therapeutic impact in skeletal muscle is hampered by low levels of dystrophin gene correction that are insufficient to stabilize affected myofibers prior to their pathological turnover. This results in the loss of previously corrected myonuclei and therapeutic vgs. We also demonstrate that these limitations can be ameliorated by myofiber stabilization via μDys codelivery. The resulting preservation of transduced myofibers allows for enhanced genomic correction and continued expression of near-native dystrophin in skeletal muscles following in vivo gene editing. In contrast, stable editing and dystrophin expression are observed in cardiac muscles without codelivery of μDys, reflecting the paucity of cardiomyocyte loss. These results indicate that a functional long-term therapy for DMD using CRISPR-Cas9 will require highly efficient skeletal muscle gene editing and that such an approach could benefit greatly from complimentary approaches that stabilize myofibers to prevent loss of vector and corrected genomes. However, life-long therapies will also require genomic correction of muscle SCs (or satellite cells),20 since even normal myofibers exhibit gradual myonuclear turnover throughout life.
Results
CRISPR-Cas9 Correction of Dystrophin in mdx4cv Mice Persists in Cardiac but Not Skeletal Muscle
The mdx4cv mouse model of DMD carries a nonsense codon in exon 53, requiring deletion of both exons 52 and 53 (Δ5253) to generate a mRNA open reading frame.13 To improve the expression levels and duration of the Δ5253-corrected dystrophin (Δ5253-dys) following systemic delivery of rAAV serotype 6 (rAAV6):CRISPR-Cas9, we performed studies in which both the overall vector dose and the relative ratios of nuclease to single guide RNA (sgRNA) targeting vectors were varied. For this purpose, we utilized a dual AAV vector system that we previously validated for correcting the mdx4cv mouse mutation in vivo.13 Briefly, a nuclease vector was designed to express Cas9 derived from Staphylococcus aureus (SaCas9) using the creatine kinase (CK)8e MSEC, and a separate target vector contained two U6 promoter-driven sgRNA cassettes designed to target introns 51 and 53 so as to excise a 45-kb region, including exons 52-53 (Figure 1). To facilitate detection of successful transduction, a cytomegalovirus (CMV)-mCherry reporter gene was also included in the target vector (Figure 1). Four different doses and ratios of nuclease to sgRNA vgs were tested following systemic administration into young-adult (11-week-old) mdx4cv mice: 1 × 1013/2 × 1012 (n = 3), 2 × 1012/1 × 1013 (n = 3), 5 × 1012/5 × 1012 (n = 3), and 1 × 1013/1 × 1013 (n = 5). Immunofluorescence (IF) analysis at 12 weeks post-treatment demonstrated uniform dose-dependent expression of mCherry in hearts of treated mice but also highly mosaic expression in different skeletal muscle groups (Figure 2A), suggesting suboptimal vector transduction and/or retention of vgs. The presence of corrected Δ5253-dys-expressing fibers also varied greatly between treatment groups and muscle type with an enriched presence of Δ5253-dys-positive fibers in the hearts of all treatment groups compared to skeletal muscles (Figure 2B). Whereas the high dose of 1 × 1013/1 × 1013 vgs generated the greatest numbers of Δ5253-dys-expressing myofibers, they were nevertheless rare and highly variable among all muscle groups analyzed.
Figure 1.

AAV Vectors and Dystrophin Gene-Correction Strategy
(A) The dual-AAV6 vector CRISPR system comprised of a nuclease vector (N; left) expressing SaCas9 under control of the striated CK8e muscle-specific expression cassette (MSEC) and a target vector (T; right) containing two U6 promoter-driven single guide (sg)RNA expression cassettes and a mCherry reporter gene expression cassette under control of the CMV enhancer/promoter. (B) gRNAs produced from the target vector recruit SaCas9 to target sequences within introns 51 (i51) and 53 (i53) of the murine Dmd gene in striated muscle. Upon excision of the 45-kb intervening genomic region that contains the C to T nucleotide substitution in exon 53 of mdx4cv mice (encoding a TAA nonsense mutation), a slightly truncated but functional dystrophin protein lacking the amino acids encoded on exons 52 and 53 (Δ5253) is produced. (C) Schematic of AAV vector expressing microdystrophin 5 (μDys5)7 under control of the CK8e MSEC.
Figure 2.

Corrected Dystrophin Expression Persists in Cardiac but Not Skeletal Muscle following Systemic AAV-Mediated Dystrophin Gene Editing in Adult mdx4cv Mice
Immunofluorescence-based evaluation of muscle transduction (A) and dystrophin expression (B) at 12 weeks postsystemic delivery with varying doses of nuclease (rAAV6-CK8eSaCas9) and target (rAAV6-Δ5253CMVmCherry) vectors into young-adult (11-week-old mdx4cv) mice. The doses of the nuclease/target vectors: (a) 1 × 1013/2 × 1012 vector genomes (vgs), (b) 2 × 1012/1 × 1013 vgs, (c) 5 × 1012/5 × 1012 vgs, and (d) 1 × 1013/1 × 1013 vgs are shown above (A). (A) Varying levels of transduction based on direct visualization of mCherry expression in multiple muscle groups, including heart (He), diaphragm (Dia), gastrocnemius (Ga), soleus (Sol), extensor digitorum longus (Edl), quadriceps (Qu), and tibialis anterior (TA). Scale bar, 5 mm. Whereas mCherry expression is observed in hearts from all treatment groups, discernible patchy expression within skeletal muscle is only seen with the highest doses of the CMV-mCherry-containing target vector. (B) Widespread dystrophin expression is seen in cardiomyocytes of treated mice, with the greatest number of dystrophin-positive cardiomyocytes obtained with the highest dose of both nuclease and target vectors, as observed following immunofluorescent analysis of muscle cross-sections stained with antibodies against the C-terminal domain of dystrophin. Mosaic expression of dystrophin is seen within multiple skeletal muscle groups, with the highest frequency of dystrophin-positive myofibers being observed with the highest dose of both vectors. Few dystrophin-revertant fibers were observed in muscles of untreated mdx4cv control mice. Scale bar, 500 μm.
To facilitate accurate quantification, genomic DNA was harvested from hearts, diaphragms, gastrocnemius muscles, and livers of treated mice and analyzed for both vgs and edited (Δ5253)-dys genomes. Despite roughly comparable liver and cardiac muscle transduction, deletion of dystrophin exons 52-53 was not observed in the liver, underscoring the importance of tissue-specific gene regulatory cassettes (RCs) for AAV-mediated in vivo gene editing (Figures 3A and 3B; Table 1).13 Whereas all doses and ratios resulted in successful Δ5253 gene editing in the hearts, quantification via digital PCR (dPCR) showed that the high dose of 1 × 1013/1 × 1013 vg led to the highest Δ5253-correction efficiency (~1.9% of total genomes; Figure 3C, top; Table 1 [12-week data]). Since cardiomyocytes contribute only about 30%–35% of the total cellular genomes in rat cardiac muscle,21 this suggests that less than 6% of the cardiomyocytes exhibit corrected genomes 3 months after treatment (see Discussion). The highest dose also exhibited significantly elevated yet strikingly low overall levels of Δ5253 correction in skeletal muscles (~0.14% [diaphragm] and 0.22% [gastrocnemius] versus the other tested doses [Figure 3C, bottom; Table 1 (12-week data)]). Thus, even after correcting for nonmyonuclear genomes, less than 1% of myofiber dystrophin genes was corrected under these conditions.
Figure 3.

Systemic Dystrophin Correction Is Enhanced with Increased Vector Dose and Demonstrates Preferred Dependence on Target versus Nuclease Vector Availability
Comparison of dose and ratio of muscle-specific nuclease and target vectors (Figure 1A) at 12 weeks following systemic delivery into young-adult (11-week-old mdx4cv) mice. The dose and ratios are the same as in Figure 2. (Top two lines) Semiquantitative PCR depicting the presence of nuclease (367 bp product) and target (741 bp product) vgs (A) or the dystrophin exons 52-53 (Δ5253) genomic DNA-deletion product (Δ; 520 bp product) (B) in DNA isolated from hearts and livers of mdx4cv mice treated with varying doses of nuclease/targeting vectors: (a) 1 × 1013/2 × 1012 vgs, (b) 2 × 1012/1 × 1013 vgs, (c) 5 × 1012/5 × 1012 vgs, and (d) 1 × 1013/1 × 1013 vgs; (−), untreated. PCR amplicon size reference via GeneRuler 1 kb Plus DNA Ladder (Thermo Fisher Scientific). (C) Quantitative dPCR analysis of exons 52-53 (Δ5253)-deleted genomes versus total genomes for the various nuclease and target vector doses (a–d), as described above. (Top) Values for the heart; (bottom) diaphragm and gastrocnemius (Gastroc) values. Statistical significance was determined by two-way ANOVA with Tukey’s post hoc test. Values represent mean ± SEM (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). (D) Western analysis of cardiac versus liver lysates for dystrophin (detected with a C-terminal dystrophin antibody [Dys (CT)]) and SaCas9 expression. The results show dose-dependent and muscle-specific expression of near-full-length dystrophin in hearts but not livers of treated mice. Lower molecular weight bands observed in cardiac samples following C-terminal dystrophin antibody staining likely reflect partial dystrophin degradation and/or possibly shorter CRISPR-induced dystrophin isoforms of unknown therapeutic relevance. A wild-type (WT; C57BL/6) cardiac muscle control sample was loaded at 10%, 5%, and 1% of total protein loaded for treated and untreated mdx4cv mice.
Table 1.
Digital PCR (dPCR) Quantification of Δ5253-Corrected Dystrophin Genomes and Transcripts
| DNA |
Δ5253 (% Total Genomes) |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Treatment | Vector Dose N/T/μD (×1013) (vg/vg/vg) | Treat. Age (Weeks) | Post-treat. (Weeks) | Heart | Diaphragm | Gastroc | TA | Liver | |||||||
| CRISPR | (a): 1/0.2/– | 11 | 12 | 0.59 ± 0.07 | 0.043 ± 0.003 | 0.023 ± 0.007 | – | – | |||||||
| (b): 0.2/1/– | 11 | 12 | 1.42 ± 0.06 | 0.066 ± 0.008 | 0.060 ± 0.020 | – | – | ||||||||
| (c): 0.5/0.5/– | 11 | 12 | 1.23 ± 0.21 | 0.082 ± 0.022 | 0.085 ± 0.009 | – | – | ||||||||
| (d): 1/1/– | 11 | 12 | 1.88 ± 0.16 | 0.137 ± 0.018 | 0.220 ± 0.021 | – | 0.000a | ||||||||
| CRISPR | 0.5/0.6/– | 2 | 4 | 2.39 ± 0.51b | 0.180 ± 0.060b | 0.29 ± 0.150 | 0.153a | – | |||||||
| 0.5/0.6/– | 2 | 18 | 2.31 ± 0.14 | 0.015 ± 0.004 | 0.072 ± 0.042 | 0.032a | – | ||||||||
| CRISPR | 0.5/1/– | 2 | 18 | 2.57 ± 0.18 | 0.068 ± 0.016 | 0.077 ± 0.029 | – | – | |||||||
| CRISPR + μDys | 0.5/1/0.2 | 2 | 18 | 2.50 ± 0.11 | 0.623 ± 0.123 | 0.984 ± 0.101 | – | – | |||||||
| Target + μDys | –/1/0.2 | 2 | 18 | 0.004 ± 0.004b | 0.004 ± 0.004b | 0.005 ± 0.005b | – | – | |||||||
| mRNA – Δ5253 Transcripts (% Total Dystrophin Transcripts) | |||||||||||||||
| Mouse # | Heart | Diaphragm | Gastroc | ||||||||||||
| CRISPR | CRISPR + μDys | Target + μDys | CRISPR | CRISPR + μDys | Target + μDys | CRISPR | CRISPR + μDys | Target + μDys | |||||||
| 1 | 34.341 | 33.531 | 0.064 | 0.517 | 7.440 | 0.039 | 0.209 | 8.852 | 0.017 | ||||||
| 2 | 36.565 | 36.024 | 0.09 | 0.339 | 6.791 | 0.025 | 0.451 | 10.236 | 0.031 | ||||||
| 3 | 39.486 | 39.21 | 0.123 | 0.728 | 11.545 | 0.048 | 0.201 | 9.141 | 0.064 | ||||||
| Mean | 36.797 | 36.255 | 0.094 | 0.528 | 8.652 | 0.037 | 0.287 | 9.410 | 0.037 | ||||||
| SEM | 1.490 | 1.643 | 0.021 | 0.112 | 1.453 | 0.008 | 0.082 | 0.422 | 0.017 | ||||||
(Top) The percentage of genomes lacking exons 52-53 relative to total genomes for selected tissues of the different treatment groups, including heart, diaphragm, gastrocnemius (Gastroc), tibialis anterior (TA), and liver. Columns represent the following: nuclease (N), target (T), and μDys (μD) vector dose (×1013 vg); treatment age; time post-treatment; and percentage of genomes containing the Δ5253 deletion out of total genomes (encompassing genomes isolated from whole tissues, which include cells of myogenic, fibroblastic, adipocytic, and hematopoietic lineages, etc.). Vector dose values (vg/vg/vg) correspond to the delivered amount of nuclease, target, or μDys vectors (see Figure 1), respectively. Each value represents tissues obtained from three mice (n = 3) except where otherwise stated. (Bottom) Quantification of the percentage of dystrophin transcripts lacking exons 52-53 in heart, diaphragm, and gastrocnemius muscles of mice treated with nuclease and target vectors (CRISPR); nuclease, target, and microdystrophin vectors (CRISPR + μDys); or target and μDys vectors only (Target + μDys). RT-dPCR was performed on mRNA that was isolated from the same mice shown in the bottom three rows of the top (DNA) part of the table (n = 3). Values are represented as mean ± SEM. Dashes represent either that the μDys vector was not delivered or that samples were not analyzed. Decimal places are derived from each dPCR chip analysis, 25,000 genomes per sample, and most conditions were measured in triplicate.
n = 1.
n = 2.
The dPCR analyses of cardiac genomic DNA indicate that in our editing system, the relative availability of sgRNAs imparts a greater effect on editing efficiency in the heart than the availability of Cas9, as demonstrated by the observed Δ5253 deletion efficiencies for the four different vector doses (Table 1 [12-week data]). This trend was supported by results from western analysis of whole cardiac lysates, where all of the dose combinations tested resulted in the expression of Δ5253-dys protein but where higher doses of target vectors produced larger amounts of Δ5253-dys protein, regardless of nuclease vector dose (Figure 3D). Importantly, dose-dependent expression of SaCas9 was detected in cardiac but not liver samples of treated mice, again emphasizing the muscle specificity of our editing system (Figure 3D).13 Due to very low levels of Δ5253-dys gene correction in skeletal muscles, as determined by dPCR and IF analysis, Δ5253-dys expression in skeletal muscle could not be determined via western blotting. Further evidence of suboptimal Δ5253-dys correction in skeletal muscle was demonstrated by a lack of significant improvement in strength, based on measurements of specific force production for gastrocnemius, tibialis anterior (TA), and diaphragm muscles (Figure S1).
Temporal Dissipation of CRISPR-Mediated Dystrophin Gene Correction Due to Myofiber Turnover
Based on the noticeable differences in dystrophin correction between skeletal and cardiac muscle and from our previous observations of higher frequencies of Δ5253-dys-positive skeletal muscle myofibers at earlier time points (4 weeks post-treatment) following systemic treatment of young-adult mdx4cv mice (11 weeks old),13 we hypothesized that Δ5253-dys-corrected myofibers/myonuclei were being lost from skeletal but not cardiac muscles over time. To test this hypothesis, we transduced 2-week-old male mdx4cv mice with 5 × 1012/6 × 1012 vg of nuclease and target vectors, respectively (n = 7). Transduction at this early time point would force CRISPR-Cas9-mediated dystrophin correction to withstand the dramatic rate of skeletal muscle turnover that occurs during the “crisis” phase in mdx skeletal muscles, which starts around 3 weeks of age.19 A separate cohort of age-matched mice was injected with 5 × 1012 vg of rAAV6 vectors expressing μDys 5 (μDys5 or μDys) under control of the CK8e RC to serve as a reference, since robust μDys expression has been shown to persist through this crisis phase and much longer.7,22 Endpoint analyses at 4 (n = 2) or 18 (n = 5) weeks post-treatment demonstrated significant expression of the mCherry reporter (Figure 1) in the hearts of treated mice at both time points (Figure 4A). Conversely, whereas mCherry expression was weak but detectable in skeletal muscles at week 4 post-treatment, by week 18, the signal was greatly reduced (Figure 4A, top), suggesting an almost complete loss of skeletal muscle fibers that had been transduced by the mCherry-carrying vectors. A similar trend was observed while monitoring expression of Δ5253-dys in skeletal muscles via immunostaining, where a concomitant decrease in dystrophin-positive myofibers and increase in central nucleation were observed between 4 and 18 weeks post-treatment, suggesting that dystrophin-positive fibers are likely lost due to turnover (Figures 4A and S2). In contrast, strong and widespread expression was seen in the heart at both 4 and 18 weeks (Figure 4A). This loss of skeletal muscle Δ5252-dys expression is strikingly different from results using similar doses of AAV-μDys vectors5, 6, 7, 8 (see below).
Figure 4.

Dystrophin Correction Persists in the Heart but Is Lost over Time in Skeletal Muscles following Gene Editing of Exons 52-53 in 2-Week-Old mdx4cv Mice
(A) Muscle transduction levels, as detected by expression of mCherry from target vectors at 4 and 18 weeks post-transduction (top) is reduced in all muscle groups except the heart between 4 (left) and 18 (right) weeks post-CRISPR treatment. Whole heart cross-sections (assembled from multiple 100× magnification images) show widespread dystrophin expression in cardiac myocytes at both 4 and 18 weeks post-treatment (top rows, right). (Bottom) At either 4 or 18 weeks post-treatment, abundant dystrophin-expressing myocytes in hearts but only rare dystrophin-positive fibers in skeletal muscle groups, such as diaphragm, gastrocnemius, and TA. Scale bars, 1 mm. (B) Semiquantitative PCR of vgs (top) or edited genomic DNA (Δ5253; bottom) in mdx4cv mice systemically treated with AAV6-CK8e-μDys5 (μD) or AAV6-CRISPR-SaCas9 (CRISPR). Mice were infused with vector at 2 weeks of age and analyzed 14 weeks later (μD) or 4 and 18 weeks later (CRISPR). The PCR product sizes are the following: μDys vector (μD; 429 bp), target vector (741 bp), and nuclease (SaCas9) vector (367 bp) (top). The PCR product from exon 52-53-deleted genomes (Δ5253 or Δ) is 520 bp (bottom). PCR products from untreated mdx4cv muscle extracts are shown as a control. The GeneRuler 1 kb Plus DNA Ladder (Thermo Fisher Scientific) was included as a size standard. (C) Quantification via dPCR shows significantly enhanced Δ5253 correction in hearts versus diaphragm and gastrocnemius muscles at either 4 or 18 weeks post-transduction. Whereas cardiac Δ5253 correction remains stable between 4 and 18 weeks post-treatment, corrected genomes in diaphragm and gastrocnemius muscles appear to be drastically reduced during this same time period (also see Table 1). Statistical significance determined by two-way ANOVA with Tukey’s post hoc test. Values represent mean ± SEM (∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). Values that were too low to be visualized on the graphs are demarcated by # (see Table 1). (D) Western analysis of muscle lysates derived from heart, diaphragm, and TA muscles of CRISPR- or μDys5-treated mice showing expression of near-full-length dystrophin (Dys (CT)), μDys5 (μDys), SaCas9, and GAPDH. The results show a loss of edited dystrophin expression between 4 and 18 weeks in skeletal muscles but an increase in expression in the heart, despite lower SaCas9 expression at the later time point in all tissues examined. Conversely, μDys expression is clearly detected at 14 weeks post-transduction in all muscle groups. (E) Functional assessment of specific force generation in multiple skeletal muscle groups of CRISPR-treated (4 weeks [w], 18 w) versus age-matched untreated control (mdx) mice, including gastrocnemius, diaphragm, and TA muscles. Whereas only minor improvements were observed for diaphragm at 18 weeks post-treatment, there are no significant improvements in treated gastrocnemius or TA muscles at any time point when treatment is initiated prior to the rapid skeletal muscle turnover phase in mdx mice. The significantly reduced values in specific force for diaphragm at the 4-week time point (demarcated as ≠) are related to the immature state of the diaphragm at this early stage, making relevant comparisons challenging.23 Statistical significance determined by two-way ANOVA with Tukey’s post hoc test. Values represent mean ± SEM (∗p < 0.05).
Analysis of vg levels and Δ5253-genomic correction within cardiac and skeletal muscles was also performed to look for evidence of skeletal but not cardiac myocyte turnover. Nuclease and target vgs were detected by semiquantitative PCR in both cardiac and skeletal muscle at 4 weeks post-treatment (Figure 4B, top). Although cardiac vg levels appeared similar at both 4 and 18 weeks post-treatment, a drastic reduction in vgs was observed in diaphragm and TA muscles between 4 and 18 weeks (Figure 4B, top). In contrast, mice treated with μDys exhibited a robust presence of vgs in all muscle groups at 14 weeks post-treatment (Figure 4B, top). Similarly, hearts of treated mice exhibited robust and stable Δ5253 correction between weeks 4 and 18 post-treatment, whereas a clear reduction in Δ5253-corrected genomes was seen in diaphragm and TA muscles during the same time span (Figure 4B, bottom). Quantification via dPCR showed that whereas Δ5253 correction in the heart was significantly greater than in skeletal muscle at both weeks 4 and 18 post-treatment, the percentage of Δ5253-corrected genomes per total isolated genomes in the heart remained comparable between 4 (2.4%) and 18 weeks (2.3%) post-treatment (Figure 4C; Table 1 [4- and 18-week data]). Not surprisingly, an almost complete loss of edited genomes was observed in skeletal muscles over this same time period (Figure 4C; Table 1 [4- and 18-week data]). These findings were also reflected at the protein level, where Δ5253-dys expression was nearly undetectable in diaphragm and TA muscles at both weeks 4 and 18, whereas appearing to increase in hearts of gene-edited mice (Figure 4D). In line with the continued presence of vgs, cardiac SaCas9 expression persisted over time, whereas skeletal muscle expression declined between 4 and 18 weeks post-treatment. In contrast to the loss of expression in skeletal muscles of gene-edited mice, μDys expression was still detected in both heart and skeletal muscle at 14 weeks post-transduction (Figure 4D) and has been observed to remain at high levels for more than 2 years post-transduction in our lab.7 Unsurprisingly, physiological measurements of specific force did not reveal significant improvements in either gastrocnemius or TA muscles of gene-edited versus untreated mdx4cv mice, and only a slight increase was observed in the diaphragm at the 18-week time point (Figure 4E). Unfortunately, the immature state of the murine diaphragm at 6 weeks of age (4-week time point) renders comparisons of specific force to later time points challenging due to significantly reduced force values at this early time point.23
μDys Expression Abrogates Skeletal Muscle Turnover and Enhances the Persistence of CRISPR-Cas9-Mediated Δ5253-dys Correction
The considerable loss of vgs and Δ5253 expression in skeletal muscles but not the heart, coupled with the observation that high-level μDys expression is stable in mdx4cv skeletal muscles (Figures 4B and 4D),7 suggested that the Δ5253-dys levels were insufficient to halt myofiber necrosis and regeneration. Based on this, we hypothesized that codelivery of rAAV-μDys could serve to stabilize skeletal muscle and allow for longitudinal CRISPR-SaCas9-based dystrophin editing in the absence of myofiber turnover. To test this hypothesis, we again transduced 2-week-old mdx4cv male mice with rAAV6 nuclease (5 × 1012 vg) and target (1 × 1013 vg) vectors (that also contained an mCherry expression cassette), with or without codelivery of rAAV6-CK8e-μDys (2 × 1012 vg) (n = 3). As a control, we coinjected target vectors only (1 × 1013 vg) in combination with μDys vectors (1 × 1012 vg) (n = 3). Tissues from treated mice were harvested at 18 weeks post-transduction and analyzed for dystrophin gene editing and expression. The retention of vgs in muscles protected by μDys expression over the 18 weeks post-treatment was evident based on continued expression of the mCherry reporter in multiple muscle groups, including heart, diaphragm, gastrocnemius, TA, extensor digitorum longus, soleus, and quadriceps (Figure 5A). Encouragingly, myofibers expressing Δ5253-dys were far more prevalent in skeletal muscle when in vivo gene editing was carried out in conjunction with μDys gene transfer (Figure 5B). The retention of Δ5253-dys+ myofibers, dramatic reduction in central nuclei, and improved muscle morphology reinforce previous data showing that μDys effectively stabilizes skeletal myofibers, halting necrosis and regeneration (Figure S3).5, 6, 7, 8 In accordance with previous observations, similar frequencies of Δ5253-dys-expressing cardiomyocytes were detected following CRISPR treatment, with or without μDys codelivery (Figure 5B).
Figure 5.

Sarcolemmal μDys Enhances Persistence of Corrected Endogenous Dystrophin Expression in Skeletal Muscles of mdx4cv Mice following AAV-Mediated Dystrophin Gene Editing
Three different AAV6 vector combinations were infused into 2-week-old mdx4cv mice, followed by analysis of various muscles 18 weeks later. (Left) Infusion with nuclease and target vectors (CRISPR); (middle) infusion with the nuclease and target vectors and the μDys vector (CRISPR + μDys); (right) infusion with the target vector and the μDys vector but not the nuclease vector (Target + μDys). Note that only the target vector carries the mCherry reporter (Figure 1A). (A) Direct visualization of mCherry expression in multiple muscle groups of treated mice, including gastrocnemius, soleus (So), TA, extensor digitorum longus, heart, diaphragm, and quadriceps. Except for the heart, expression of mCherry is noticeably reduced in skeletal muscles of CRISPR-treated mice (left), whereas mice receiving μDys in conjunction with either CRISPR (middle) or with just target vectors (right) display strong expression in multiple muscle groups. Scale bar, 5 mm. (B) Expression of near-full-length dystrophin (Dys (C-term); left half of each), which recognizes endogenous dystrophin but not μDys and μDys (right half of each) in heart, diaphragm, and gastrocnemius muscles of mice treated with the same vector combinations, as shown in (A). The results show comparable frequencies of cardiomyocytes expressing near-full-length dystrophin following CRISPR treatment alone (left; CRISPR) or in combination with μDys (middle; CRISPR + μDys), whereas myofibers expressing near-full-length dystrophin were more prevalent in both diaphragm and gastrocnemius muscles following CRISPR and μDys codelivery (middle; CRISPR + μDys). Control mice receiving target vectors only in combination with μDys only exhibited weak background staining following C-terminal dystrophin antibody staining (right; Target + μDys). Scale bar, 250 μm.
Semiquantitative PCR revealed that vgs and the unique Δ5253 genomic deletion product were both detected at similar levels in the hearts of mice treated with either CRISPR or the combination of CRISPR and μDys (Figures 6A and 6B). In contrast, codelivery of μDys dramatically increased the amount of CRISPR vectors and the unique Δ5253 genomic deletion product retained in dystrophic skeletal muscles (Figures 6A and 6B). Quantification of Δ5253-deleted genomes via dPCR revealed comparable levels of dystrophin gene correction in the hearts of CRISPR-treated mice, with or without μDys (2.6% and 2.5%, respectively). In contrast, whereas the overall percent of Δ5253-corrected genomes in skeletal muscle is still low, it increased significantly in both diaphragm (0.07% versus 0.6%) and gastrocnemius (0.08% versus 1%) muscles following μDys codelivery (Figure 6C; Table 1). Quantification of indel formation at the individual target sites within introns 51 and 53 using inference of CRISPR edits (ICE) analysis also revealed an enhancement in the occurrence of target-site indel formation in DNA samples from skeletal muscles following codelivery of CRISPR with μDys (Figure 6D). Of note, detection of indels within PCR products generated across the individual target sites only represents editing events that did not result in the removal of the intervening genomic sequence (i.e., representing unsuccessful dystrophin correction).
Figure 6.

μDys Codelivery Prevents Loss of Edited Genomes, mRNA, and AAV Vectors during AAV-Mediated In Vivo Gene Editing
Dystrophic mdx4cv mice infused with the indicated vector combinations at 2 weeks of age were analyzed 18 weeks later. (A and B) Semiquantitative PCR demonstrating the presence of vgs (A) and the unique Δ5253 genomic deletion product (B) in heart, diaphragm, and gastrocnemius muscles of mdx4cv mice infused with nuclease and target vectors from Figure 1A (CRISPR), nuclease and target vectors and μDys (CRISPR + μDys), or μDys combined with the CRISPR target vector but not the nuclease vector (Target + μDys) (n = 3/group). Encouragingly, both vgs and the CRISPR-generated Δ5253-deletion product are retained in both hearts and skeletal muscles following μDys codelivery but were drastically reduced in skeletal muscles in the absence of μDys. Target vector PCR product (741 bp); μDys5 vector PCR product (555 bp); nuclease vector PCR product (367 bp); and Δ5253 deletion product PCR product (520 bp). Coinfusion with the μDys vector prevented the loss of nuclease/target vectors and Δ5253-deletion product in skeletal muscles. (C) Quantification of Δ5253-deletion product via dPCR showed comparable deletion efficiency in the heart with or without μDys but significantly enhanced deletion in both diaphragm and gastrocnemius following CRISPR + μDys codelivery. Statistical significance determined by two-way ANOVA with Tukey’s post hoc test. Values represent mean ± SEM (∗p < 0.05, ***p < 0.001 ∗∗∗∗p < 0.0001; # demarcates values too small to display on graph; see Table 1). (D) Quantification of indel formation at i51 and i53 target sites based on ICE analyses of PCR amplicons generated from DNA isolated from heart, diaphragm, and gastrocnemius muscles of mice treated with CRISPR (n = 3) or CRISPR + μDys (n = 3). Note that detected indels stem from genomic editing events where deletion of the 45-kb exon 52-53 region was unsuccessful (i.e., only one site was cut, or one site was cut and repaired before the other site was cut), as successful deletion would eliminate the possibility to generate a PCR amplicon across the individual target sites. Statistical significance determined by two-way ANOVA with Bonferroni post hoc test. Values represent mean ± SEM (∗p < 0.05). (E) Semiquantitative RT-PCR depicting the presence of unique exons 52-53 (Δ5253)-deleted dystrophin transcripts (Δ; 237 bp PCR product) in CRISPR-treated mouse hearts regardless of the presence of μDys. However, edited mRNAs were only detected at the 18-week time point in skeletal muscles (diaphragm and gastrocnemius) when combined with μDys treatment. U, unmodified native dystrophin transcripts (567 bp). (F) RT-dPCR quantification of corrected dystrophin transcripts (Δ5253) in heart, diaphragm, and gastrocnemius muscles reveals significant enrichment for Δ5253 transcripts in skeletal muscles following gene editing in the presence of μDys (# demarcates values too small to display on graph; see Table 1). Statistical significance determined by two-way ANOVA with Tukey’s post hoc test. Values represent mean ± SEM (*p < 0.01, ∗∗∗∗p < 0.0001).
As has been shown previously by us13 and others,15,17,24 differences in in vivo gene-correction efficiencies (with or without μDys) were significantly more pronounced at the transcript level compared with the genomic DNA level (Figure 6E). The proportion of edited versus nonedited dystrophin transcripts increased from 0.5% to 8.7% in the diaphragm and from 0.3% to 9.4% in the gastrocnemius muscle upon codelivery of μDys; in contrast, no μDys-related differences were observed in the heart (Figures 6E and 6F; Table 1). The greater effects at the transcript level have been attributed to protection of corrected transcripts from nonsense-mediated decay.13 The increases in transcript levels were reflected in increased dystrophin protein levels, as measured by western blot (Figure 7A). Coinfusion with the μDys vector led to approximately 2-fold higher expression levels in the diaphragm and more than a 7-fold increase in the gastrocnemius compared with mice receiving the CRISPR vector alone (Figure 7B). In contrast, a slight decrease in Δ5253-dys was observed in the heart, possibly due to competition with the μDys protein (Figure 7B).
Figure 7.

μDys Preserves Expression of CRISPR-Corrected Near-Full-Length Dystrophin and Improves Function in Skeletal Muscle
(A) Western analysis of lysates derived from heart, diaphragm, and gastrocnemius muscles of 20-week-old WT or AAV vector-treated mdx4cv mice (infused at 2 weeks of age with the same vector combinations as indicated in Figures 5 and 6). The WT lanes were loaded with 10% (3 μg) of the total protein loaded in the mdx4cv mouse sample lanes. Dys (CT), edited dystrophin expression, detected with a C-terminal dystrophin antibody; μDys, μDys expression detected with an N-terminal (hinge-1) dystrophin antibody (note that the μDys is ~1/3 the size of the edited, nearly full-length dystrophin [Dys (CT)], and so they do not comigrate on the gels); SaCas9, nuclease expression as detected using antibodies against a triple-HA epitope fused to the C-terminal end of SaCas9; GAPDH, expression as a loading control. CRISPR (nuclease + target vectors), n = 3; CRISPR + μDys, n = 3; target + μDys, n = 3. Note that CRISPR + μDys and Target + μDys sample loading order is different for gastrocnemius versus heart and diaphragm samples. (B) GAPDH normalized relative expression levels of near-full-length dystrophin (edited dystrophin) based on densitometry measurements of blots immunostained with Dys (CT) in heart (left), diaphragm (middle), and gastrocnemius (right) muscles. Significant preservation of dystrophin expression is observed in both diaphragm and gastrocnemius when gene editing is conducted in conjunction with μDys cotreatment. Statistical significance was determined via multiple Student’s t tests. Values represent mean ± SEM (∗p < 0.05, ∗∗p < 0.01). (C) Measurements of muscle-specific force in gastrocnemius and diaphragm muscles of treated versus untreated mdx4cv mice show significant improvements in muscle force generation upon μDys gene transfer, with or without concurrent gene editing. Statistical significance determined by two-way ANOVA with Tukey’s post hoc test. Values represent mean ± SEM (∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001).
Significant improvements in skeletal muscle-specific force generation were only observed for gastrocnemius and diaphragm muscles of mice receiving μDys gene transfer (Figure 7C). Previous studies show a clear increase in specific force due to μDys expression,7,23 but whether codelivery of μDys leads to persistent functional benefits attributable to Δ5253-dys was uncertain. It was clear, however, that μDys coexpression significantly increased retention of Δ5253-dys expression obtained via gene editing, and higher levels of near-full-length dystrophin should provide further amelioration of dystrophic muscle dysfunction over time.
Discussion
Our results demonstrate that myofiber turnover presents a significant challenge to the efficacy of CRISPR-Cas9-based dystrophin correction in skeletal muscle. In contrast, expression of edited dystrophin is maintained in the heart, which does not readily undergo necrosis and regeneration. The observed loss of dystrophin expression in skeletal muscle is likely related to current limitations in both the efficiency and rate of AAV-mediated gene-editing strategies. These issues are exacerbated in editing situations that require excision of one or more exons. For example, editing strategies using multiple sgRNAs to excise genomic regions (particularly larger regions) are particularly inefficient due to asynchronous DNA cleavage, where sequential target-site cleavage accompanied by indel-meditated destruction of recognized sequences causes failure to delete the intervening DNA region. Gene-edited dystrophins could display varying levels of functionality, possibly impacting the long-term stability of treated muscles. Whereas the structure and functionality of μDys have been thoroughly validated,7, 8, 9,22,25 novel CRISPR-generated dystrophins also need to be evaluated, as they could display reduced functionality and/or instability due to the removal of important protein binding sites or contain incompatible structural modifications, respectively.26 Encouragingly, online simulation (http://edystrophin.genouest.org) revealed the formation of a hybrid repeat 20/21 with possibly maintained filamentous architecture following deletion of exons 52 and 53 of dystrophin (Figure S4), resulting in a likely stable and functional protein containing all known protein binding sites. We have previously demonstrated continued expression of Δ5253-dys and improved muscle function for up to 18 weeks post-treatment following direct intramuscular delivery,13 where much higher relative doses and hence, editing efficiency can be more easily achieved compared to systemic delivery.
The loss of vgs and corrected myonuclei due to turnover may also be attributed to the lag time between vector delivery and the attainment of therapeutically beneficial dystrophin levels in myofibers. Unless dystrophin gene correction can be obtained in a sufficiently large percentage of myonuclei and lead to the recruitment of dystrophin along the entire myofiber before it experiences damage, delivered vgs as well as any successfully corrected myonuclei will be lost during the ensuing degeneration/regeneration process. Encouragingly, our results demonstrate that although overall correction compared to wild-type (WT) levels was still low, significant longer-term retention of dystrophin-corrected myonuclei was obtained in skeletal muscles (1% of total genomes) via concurrent gene-editing/μDys treatment. Furthermore, expression of μDys in skeletal muscle enhanced the presence of corrected dystrophin transcripts (up to ~30-fold) and dystrophin protein expression (up to ~7-fold) compared to gene editing in the absence of μDys. Whereas our observed levels of corrected versus total genomes may appear low, it is important to consider that “total genomes” also contain DNA derived from nonmyogenic nuclei, including fibroblasts, adipocytes, and infiltrating immune cells, all cell types that are highly abundant in dystrophic muscle. The heterogeneity of cell types coupled with the muscle-restricted expression of the CK8e RC limits Δ5253 correction to differentiated myonuclei and thus significantly reduces the percentage of “correctible” total genomes in muscle tissue. This results in an apparent reduction in correction efficiency (per total genomes) as compared to the use of a ubiquitously active RC (i.e., CMV).14, 15, 16,24,27,28 Although comparisons of genome correction efficiencies are affected by many parameters, the only examples of higher efficiencies than those observed in the present study have been obtained when a single site in the dystrophin gene has been targeted.17,29 Although these successes are important, this strategy is only narrowly applicable to patients where bypassing a single exon is sufficient for correcting dystrophin expression. For more complex and common mutations, where single exon targeting is not applicable or perhaps not nearly as permissible/efficient as for other exons, CRISPR-Cas9 and μDys codelivery shows clear promise for obtaining prolonged dystrophin correction. In such a scenario, one could envision the two treatments working synergistically. μDys expression from highly active RCs would quickly stabilize affected muscle groups by providing much improved function and halting further disease progression, whereas gene editing would provide continuing improvements over time due to the greater functional properties of near-full-length dystrophin relative to μDys. Although we chose to focus on μDys, additional methods that effectively stabilize dystrophic muscle could also prove beneficial,10 such as gene-replacement therapies using complementary genes other than dystrophin (for example, microutrophin or GALGT2)30, 31, 32, 33 or exon skipping using antisense oligonucleotides.34,35
One drawback to a dual-treatment method is the increased dose of AAV that may be required. Recent gene-therapy clinical trials that use high vector doses to achieve body-wide transduction have seen adverse events associated with complement activation, including thrombocytopenia, acute kidney injury, and cardiopulmonary insufficiency. Additionally, an AAV gene-therapy trial for myotubular myopathy (MTM) tragically had three patients develop sepsis and die following a sustained period of liver damage. Whereas the MTM patients all had prior liver disease due to the underlying MTM pathology, which is not relevant to DMD and which likely exacerbated an immune response that caused the liver damage, these cases illustrate that high AAV doses can cause severe adverse events in multiple organs that need to be modulated. The ongoing DMD clinical trials have worked to develop safety protocols to minimize risks associated with complement activation, such as the utilization of complement inhibitors and removal of empty AAV capsids, and with liver toxicity, such as with steroids protocols. Optimizations of AAV body-wide muscle delivery and CRISPR editing efficiencies could also allow for lower doses to be used and are being worked on by numerous groups across both fields. Additionally, a number of strategies are being developed to allow for safe AAV readministration, which could allow for our strategy to be broken into two different administrations—a μDys AAV to stabilize the muscle, followed by AAV-delivered CRISPR for gene editing—thus lowering each individual administration’s vector dose and improving the safety profile. Nonetheless, increased editing efficiencies and/or nonviral vector methods to prestabilize myofibers would not require the use of dual vectors.
Further improvements to dystrophin gene-editing efficiency should also result from an ability to target and edit genes in myogenic SCs.20 Efficient SC correction would prevent functional loss of edited myofiber nuclei upon necrosis/regeneration, as they would be replaced with edited nuclei from SCs. Currently, the efficiency of AAV-mediated gene delivery to SCs is controversial. Our previous studies in WT mice suggest that AAV vectors poorly target quiescent SCs.36 However, other studies in dystrophic mice suggest that SCs might be targeted, although the proportion of targeted quiescent SCs versus activated SCs and transit amplifying cells in muscles undergoing active regeneration is unclear.16,28 Additional reports have demonstrated persistence of low levels of dystrophin expression for up to 1 year following systemic delivery of CRISPR-Cas9 without the need for auxiliary (e.g., μDys) stabilization of muscle membranes.24,27 These studies utilized the ubiquitously active human CMV enhancer/promoter to drive expression of SaCas9 and conclude that this may have helped preserve dystrophin expression due to the possibility of edited SCs contributing to the regeneration of damaged myofibers. However, the greatest amount of long-term dystrophin correction was still observed in the heart, with significantly less correction in skeletal muscle. Since SCs only contribute to skeletal muscle regeneration, this would appear to indicate that corrected SCs contributed only minimally to muscle regeneration over time. The use of ubiquitously active gene RCs also complicates downstream genomic analyses by making it cumbersome to separate genomes corresponding to myogenic cells (many of which are postmitotic) from nonmyogenic fibroblasts, adipocytes, and infiltrating hematopoietic cells. In fact, AAV-mediated systemic gene editing using CMV-driven Cas9 expression has been shown to edit significant numbers of nonmyogenic cells within the skeletal muscle at equal or higher frequencies than SCs.28 Importantly, despite the potential for correcting SCs using ubiquitously active promoters, their use for controlling nuclease expression appears ill advised, as it drastically increases the risk for unwanted and potentially hazardous on- or off-target effects occurring in nonmyogenic and actively dividing cells.24,37 Ubiquitously active RCs also significantly increase the risk of eliciting cellular immune responses against vector-encoded transgenes.38, 39, 40
The fact that gene correction is preferentially retained in the heart indicates that potential immune responses against Cas9 in mice have comparatively little impact on treatment outcomes, as host immune responses raised against introduced vectors or gene products would almost assuredly affect cardiac and skeletal muscles alike.6,40 Although we did not investigate potential treatment-related immune responses directly, none were overtly apparent. This may have been partly due to our utilization of the CK8e RC to restrict expression of SaCas9 to postmitotic myonuclei, thereby reducing the likelihood of stimulating immune responses via antigen-presenting cells.38,41
Together, our data suggest that efficient and stable dystrophin gene editing in skeletal muscles could benefit greatly from combinatorial treatment approaches that rapidly halt myofiber necrosis and regeneration to enhance longitudinal dystrophin gene correction. Numerous challenges still lie ahead of clinical application of gene-editing strategies for treating DMD, including further optimization of editing efficiency, as well as additional investigations into the safety and efficacy of CRISPR-Cas-based genome-editing strategies in other DMD models. Higher and persisting skeletal muscle levels of gene correction than achieved in the present study need to be obtained, particularly when CRISPR strategies are applied in the therapeutic context of treating patients whose skeletal muscles are already undergoing extensive degeneration. Notwithstanding the hurdles that still need to be overcome before in vivo gene editing can be efficiently and safely applied in the clinic, this strategy shows promise for future treatments of DMD and other genetic neuromuscular disorders.
Materials and Methods
Cloning and Vector Production
Plasmids containing gene RCs for expression of Cas9 or sgRNAs flanked by AAV2 inverted terminal repeats (ITRs) were generated using standard cloning techniques.
A dual AAV6 vector system was tested, where a nuclease vector expressed SaCas942 under control of a synthetic muscle-specific CK (CK8e) gene RC was paired with a second vector carrying two U6-driven sgRNA expression cassettes targeting introns 51 and 53, plus a ubiquitously expressed CMV-mCherry reporter cassette. This dual vector system and the associated gRNA sequences were previously validated as a single vector system.13
AAV6 vectors expressing μDys5 under control of the CK8e gene RC were also produced and tested. Expression construct plasmids containing AAV2 ITRs were cotransfected with the pDGM6 packaging plasmid into subcultured HEK293 cells (American Type Culture Collection) using calcium phosphate-mediated transfection to generate AAV6 vectors that were harvested, purified via heparin-affinity chromatography, and concentrated using sucrose gradient centrifugation.43 Resulting titers were determined by Southern analyses using probes specific to CK8e or CMV for nuclease and targeting vectors, respectively.
Animals
All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Washington. Systemic vector delivery was accomplished via retroorbital injection of up to 200 μL Hank’s balanced salt solution (HBSS) containing nuclease and target vectors at doses varying from 2 to 10 × 1012 vg into 2- or 11-week-old male C57BL/6-mdx4cv (mdx4cv) mice. The mdx4cv mouse model of DMD harbors a nonsense C to T mutation in exon 53 leading to a loss of dystrophin expression.44 These mice exhibit ~10-fold lower frequencies of revertant dystrophin-expressing muscle fibers than the original mdxscsn mouse strain,45 which provides much greater assurance that dystrophin-corrected fibers resulted from gene targeting rather than spontaneous reversion.
Tissue Harvest and Processing
Muscles were harvested and analyzed at 4, 12, or 18 weeks post-transduction and compared to age-matched male noninjected mdx4cv mice. Cardiac and skeletal muscles were embedded in optimal cutting temperature (OCT) compound (VWR International) and fresh frozen in liquid nitrogen-cooled isopentane for subsequent cryosectioning and IF analysis. Additional muscle samples were snap frozen in liquid nitrogen and ground to a powder under liquid nitrogen in a mortar kept on dry ice for subsequent extraction of DNA, RNA, and protein.
Immunohistochemical Analyses
Muscle cross-sections (10 μm) were stained with hematoxylin & eosin (Sigma-Aldrich) or costained with antibodies raised against alpha 2-laminin (Sigma; rat monoclonal, 1:200) and the C-terminal domain of dystrophin (a kind gift from Dr. Stanley Froehner at the University of Washington, Department of Physiology and Biophysics; rabbit polyclonal, 1:500). Expression of μDys5 was detected using antibodies raised against the hinge-1 region of dystrophin (clone 1011b, mouse monoclonal immunoglobulin G2a [IgG2a]; Developmental Studies Hybridoma Bank [DSHB]), since μDys5 lacks the majority of the C-terminal domain and shows only limited immunoreactivity with few C-terminal antibodies that still share partial epitope homology. Slides were mounted using Permount mounting medium (Thermo Fisher Scientific) or ProLong Gold with 4′,6-diamidino-2-phenylindole (DAPI; Thermo Fisher Scientific) and imaged using an Olympus E1000 fluorescent microscope running SlideBook 6 acquisition software (3i, Denver, CO). Images were processed and assembled into figures using Photoshop CS5 (Adobe, San Jose, CA).
Nucleic Acid and Protein Analyses
Nucleic acids were isolated from ground muscle tissue using Trizol reagent (Invitrogen), and RNA was extracted according to the manufacturer’s recommendations. DNA was extracted from the Trizol suspension using a modified protocol described by Shirley Zhu at Stanford University, relying on back extraction with 4 M guanidine thiocyanate, 50 mM sodium citrate, and 1 M Tris base. Analysis of dystrophin transcripts by RT-PCR was performed on cDNA produced from muscle lysates using the Superscript IV VILO cDNA Synthesis Kit (Invitrogen). Semiquantitative PCR and RT-PCR amplicons across the targeted region were generated using Phire Hot Start II polymerase (New England Biolabs [NEB]) and separated via gel electrophoresis on 2% agarose gels. PCR amplicons generated across the individual cut sites in introns 51 and 53 using Phusion proofreading polymerase (NEB) were submitted for Sanger sequencing.
The resulting reads were subjected to ICE analysis (https://www.synthego.com/products/bioinformatics/crispr-analysis), where the Sanger sequencing data from treated samples were compared to an untreated control sample to establish editing frequency at each target site. Deletion of exons 52-53 at the genome and transcript levels was quantified using a QuantStudio 3D dPCR system (Thermo Fisher Scientific) using the manufacturer’s proprietary reagents in combination with primer/probe sets, detecting either native unmodified dystrophin alleles/transcripts or alleles/transcripts lacking the sequence spanning the two target sites (Δ5253). Data analysis was performed using the dPCR AnalysisSuite software at Thermo Fisher Scientific’s Cloud-based Connect platform. Quantification of VIC/FAM calls was performed using the dPCR AnalysisSuite’s built-in target/total algorithm. Representative scatterplots depicting positive and negative calls are presented in Figure S5. Sequences of sgRNA spacers, as well as for PCR primers and probes, are listed in the Supplemental Information.
Muscle proteins were extracted in radioimmunoprecipitation analysis (RIPA) buffer, supplemented with 5 mM EDTA and 4% protease inhibitor cocktail (Sigma; catalog number [Cat #]P8340) for 1 h on ice with gentle agitation every 15 min. Total protein concentration was determined using the Pierce bicinchoninic acid (BCA) Assay Kit (Thermo Fisher Scientific). Muscle lysates from WT (3, 1.5, or 0.3 μg) and untreated mdx4cv (30 μg) and treated mdx4cv (30 μg) mice were denatured at 100°C for 10 min, quenched on ice, and separated via gel electrophoresis after loading onto Bolt 4%–12% Bis-Tris polyacrylamide gels (Invitrogen). Protein transfer to 0.45 μm polyvinylidene fluoride (PVDF) membranes was performed overnight at constant 46 V at 4°C in Towbin’s buffer containing 20% methanol. Blots were blocked for 1 h at room temperature in 5% nonfat dry milk (NFDM) before overnight incubation with antibodies raised against the C-terminal domain of dystrophin (only detects Δ5253-dys; Froehner Lab; rabbit polyclonal, 1:15,000), 1011b anti-hinge-1 of dystrophin (detects μDys and Δ5253-dys; DSHB, University of Iowa, Iowa City, IA), anti-hemagglutinin (HA; Roche; rat monoclonal horseradish peroxidase [HRP] conjugated; 1:2,000) for detection of HA-tagged SaCas9, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Sigma; rabbit polyclonal, 1:100,000). HRP-conjugated secondary antibody staining (1:50,000) was performed for 1 h at room temperature prior to signal development using Clarity Western Enhanced Chemiluminescent (ECL) substrate (Bio-Rad) and visualization using a ChemiDoc MP imaging system (Bio-Rad). Gel- and blot-band densitometry measurements were performed on unsaturated images using ImageJ software (National Institutes of Health).
Muscle Physiology
Treated and age-matched control mdx4cv mice were anesthetized via inhalation of 1% Isoflurane in oxygen and assayed in situ (gastrocnemius and TA) and ex vivo (diaphragm) for force generation.46 For in situ measurements, a 4-0 silk suture was tied around the distal TA/gastrocnemius tendon and to a lever attached to a force transducer. For ex vivo measurements, a 6-0 silk suture was tied between the 1st and 2nd rib of a thin strip of diaphragm muscle on one side and attached to a small hook in a chamber filled with oxygenated (95% O2/5% CO2) physiological solution comprised of 131 mM NaCl, 5 mM KCl, 1.8 mM CaCl2 × 2 H2O, 0.5 mM MgCl2 × 6 H2O, 0.4 mM NaH2PO4, 24 mM NaHCO3, and 5.5 mM glucose that was maintained at 37°C.23,47 At the opposite side of the diaphragm strip, another 6-0 suture was tied through to a small portion of the central diaphragm tendon, still attached to the muscle strip, and tied to a force transducer. After determination of optimal muscle fiber length (L0) the maximum isometric tetanic force was measured during electrical stimulation using Dynamic Muscle Control version (v.)5.420 software (Aurora Scientific). Muscle cross-sectional area (CSA) was calculated by dividing muscle mass (milligram) by fiber length (millimeter) and 1.06 mg/mm3 (density of mammalian skeletal muscle). Specific force values were obtained by normalizing maximum isometric tetanic force production to CSA.
Statistical Analyses
Data values are represented as mean ± SEM and were analyzed using Prism8 (GraphPad). Measurements were analyzed for statistical significance using two-way analysis of variance (ANOVA) multiple comparison tests with Turkey’s post hoc tests unless otherwise stated. Statistical significance was set to p < 0.05.
Acknowledgments
We thank the Viral Vector Core of the Seattle Wellstone Muscular Dystrophy Specialized Research Center for generating rAAV vectors and James Allen, Christine Halbert, Darren Bissett, James Moore, Quynh Nguyen, and Xiolan Chen for advice and assistance. This work was supported by NIH grants R01AR44533 and P50AR065139 (to S.D.H. and J.S.C.) and grant RRG 715234 from the Muscular Dystrophy Association (to J.S.C.).
Author Contributions
Conceptualization, N.E.B., S.D.H., and J.S.C.; Methodology, N.E.B., H.T., S.D.H., and J.S.C.; Investigation, N.E.B. and H.T.; Formal Analysis, N.E.B., S.D.H., H.T., and J.S.C.; Writing – Original Draft, N.E.B.; Writing – Review & Editing, S.D.H. and J.S.C.; Supervision, S.D.H. and J.S.C.
Declaration of Interests
N.E.B., S.D.H., and J.S.C. are inventors on patents covering muscle-specific gene editing. S.D.H. and J.S.C. are inventors on patents covering muscle-specific gene regulatory cassettes and microdystrophins. J.S.C. holds equity in and is a member of the Scientific Advisory Board of Solid Biosciences.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.11.003.
Supplemental Information
References
- 1.Emery A.E.H., Muntoni F. Third Edition. Oxford University Press; 2003. Duchenne Muscular Dystrophy. [Google Scholar]
- 2.Mendell J.R., Shilling C., Leslie N.D., Flanigan K.M., al-Dahhak R., Gastier-Foster J., Kneile K., Dunn D.M., Duval B., Aoyagi A. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann. Neurol. 2012;71:304–313. doi: 10.1002/ana.23528. [DOI] [PubMed] [Google Scholar]
- 3.Batchelor C.L., Winder S.J. Sparks, signals and shock absorbers: how dystrophin loss causes muscular dystrophy. Trends Cell Biol. 2006;16:198–205. doi: 10.1016/j.tcb.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 4.Ervasti J.M., Ohlendieck K., Kahl S.D., Gaver M.G., Campbell K.P. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature. 1990;345:315–319. doi: 10.1038/345315a0. [DOI] [PubMed] [Google Scholar]
- 5.Harper S.Q., Hauser M.A., DelloRusso C., Duan D., Crawford R.W., Phelps S.F., Harper H.A., Robinson A.S., Engelhardt J.F., Brooks S.V., Chamberlain J.S. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat. Med. 2002;8:253–261. doi: 10.1038/nm0302-253. [DOI] [PubMed] [Google Scholar]
- 6.Gregorevic P., Blankinship M.J., Allen J.M., Crawford R.W., Meuse L., Miller D.G., Russell D.W., Chamberlain J.S. Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat. Med. 2004;10:828–834. doi: 10.1038/nm1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramos J.N., Hollinger K., Bengtsson N.E., Allen J.M., Hauschka S.D., Chamberlain J.S. Development of Novel Micro-dystrophins with Enhanced Functionality. Mol. Ther. 2019;27:623–635. doi: 10.1016/j.ymthe.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gregorevic P., Allen J.M., Minami E., Blankinship M.J., Haraguchi M., Meuse L., Finn E., Adams M.E., Froehner S.C., Murry C.E., Chamberlain J.S. rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat. Med. 2006;12:787–789. doi: 10.1038/nm1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Le Guiner C., Servais L., Montus M., Larcher T., Fraysse B., Moullec S., Allais M., François V., Dutilleul M., Malerba A. Long-term microdystrophin gene therapy is effective in a canine model of Duchenne muscular dystrophy. Nat. Commun. 2017;8:16105. doi: 10.1038/ncomms16105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crudele J.M., Chamberlain J.S. AAV-based gene therapies for the muscular dystrophies. Hum. Mol. Genet. 2019;28(R1):R102–R107. doi: 10.1093/hmg/ddz128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bengtsson N.E., Seto J.T., Hall J.K., Chamberlain J.S., Odom G.L. Progress and prospects of gene therapy clinical trials for the muscular dystrophies. Hum. Mol. Genet. 2016;25(R1):R9–R17. doi: 10.1093/hmg/ddv420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Young C.S., Pyle A.D., Spencer M.J. CRISPR for Neuromuscular Disorders: Gene Editing and Beyond. Physiology (Bethesda) 2019;34:341–353. doi: 10.1152/physiol.00012.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bengtsson N.E., Hall J.K., Odom G.L., Phelps M.P., Andrus C.R., Hawkins R.D., Hauschka S.D., Chamberlain J.R., Chamberlain J.S. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun. 2017;8:14454. doi: 10.1038/ncomms14454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Long C., Amoasii L., Mireault A.A., McAnally J.R., Li H., Sanchez-Ortiz E., Bhattacharyya S., Shelton J.M., Bassel-Duby R., Olson E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 2016;351:400–403. doi: 10.1126/science.aad5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nelson C.E., Hakim C.H., Ousterout D.G., Thakore P.I., Moreb E.A., Castellanos Rivera R.M., Madhavan S., Pan X., Ran F.A., Yan W.X. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2016;351:403–407. doi: 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tabebordbar M., Zhu K., Cheng J.K.W., Chew W.L., Widrick J.J., Yan W.X., Maesner C., Wu E.Y., Xiao R., Ran F.A. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 2016;351:407–411. doi: 10.1126/science.aad5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Amoasii L., Long C., Li H., Mireault A.A., Shelton J.M., Sanchez-Ortiz E., McAnally J.R., Bhattacharyya S., Schmidt F., Grimm D. Single-cut genome editing restores dystrophin expression in a new mouse model of muscular dystrophy. Sci. Transl. Med. 2017;9:eaan8081. doi: 10.1126/scitranslmed.aan8081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duchêne B.L., Cherif K., Iyombe-Engembe J.P., Guyon A., Rousseau J., Ouellet D.L., Barbeau X., Lague P., Tremblay J.P. CRISPR-Induced Deletion with SaCas9 Restores Dystrophin Expression in Dystrophic Models In Vitro and In Vivo. Mol. Ther. 2018;26:2604–2616. doi: 10.1016/j.ymthe.2018.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duddy W., Duguez S., Johnston H., Cohen T.V., Phadke A., Gordish-Dressman H., Nagaraju K., Gnocchi V., Low S., Partridge T. Muscular dystrophy in the mdx mouse is a severe myopathy compounded by hypotrophy, hypertrophy and hyperplasia. Skelet. Muscle. 2015;5:16. doi: 10.1186/s13395-015-0041-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mauro A. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 1961;9:493–495. doi: 10.1083/jcb.9.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nag A.C. Study of non-muscle cells of the adult mammalian heart: a fine structural analysis and distribution. Cytobios. 1980;28:41–61. [PubMed] [Google Scholar]
- 22.Banks G.B., Judge L.M., Allen J.M., Chamberlain J.S. The polyproline site in hinge 2 influences the functional capacity of truncated dystrophins. PLoS Genet. 2010;6:e1000958. doi: 10.1371/journal.pgen.1000958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Faulkner J.A., Ng R., Davis C.S., Li S., Chamberlain J.S. Diaphragm muscle strip preparation for evaluation of gene therapies in mdx mice. Clin. Exp. Pharmacol. Physiol. 2008;35:725–729. doi: 10.1111/j.1440-1681.2007.04865.x. [DOI] [PubMed] [Google Scholar]
- 24.Nelson C.E., Wu Y., Gemberling M.P., Oliver M.L., Waller M.A., Bohning J.D., Robinson-Hamm J.N., Bulaklak K., Castellanos Rivera R.M., Collier J.H. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019;25:427–432. doi: 10.1038/s41591-019-0344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gregorevic P., Blankinship M.J., Allen J.M., Chamberlain J.S. Systemic microdystrophin gene delivery improves skeletal muscle structure and function in old dystrophic mdx mice. Mol. Ther. 2008;16:657–664. doi: 10.1038/mt.2008.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iyombe-Engembe J.P., Ouellet D.L., Barbeau X., Rousseau J., Chapdelaine P., Lagüe P., Tremblay J.P. Efficient Restoration of the Dystrophin Gene Reading Frame and Protein Structure in DMD Myoblasts Using the CinDel Method. Mol. Ther. Nucleic Acids. 2016;5:e283. doi: 10.1038/mtna.2015.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hakim C.H., Wasala N.B., Nelson C.E., Wasala L.P., Yue Y., Louderman J.A., Lessa T.B., Dai A., Zhang K., Jenkins G.J. AAV CRISPR editing rescues cardiac and muscle function for 18 months in dystrophic mice. JCI Insight. 2018;3:124297. doi: 10.1172/jci.insight.124297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goldstein J.M., Tabebordbar M., Zhu K., Wang L.D., Messemer K.A., Peacker B., Kakhki S.A., Gonzalez-Celeiro M., Shwartz Y., Cheng J.K.W. In Situ Modification of Tissue Stem and Progenitor Cell Genomes. Cell Rep. 2019;27:1254–1264.e7. doi: 10.1016/j.celrep.2019.03.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Amoasii L., Hildyard J.C.W., Li H., Sanchez-Ortiz E., Mireault A., Caballero D., Harron R., Stathopoulou T.R., Massey C., Shelton J.M. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science. 2018;362:86–91. doi: 10.1126/science.aau1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Odom G.L., Gregorevic P., Allen J.M., Finn E., Chamberlain J.S. Microutrophin delivery through rAAV6 increases lifespan and improves muscle function in dystrophic dystrophin/utrophin-deficient mice. Mol. Ther. 2008;16:1539–1545. doi: 10.1038/mt.2008.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kennedy T.L., Guiraud S., Edwards B., Squire S., Moir L., Babbs A., Odom G., Golebiowski D., Schneider J., Chamberlain J.S., Davies K.E. Micro-utrophin Improves Cardiac and Skeletal Muscle Function of Severely Affected D2/mdx Mice. Mol. Ther. Methods Clin. Dev. 2018;11:92–105. doi: 10.1016/j.omtm.2018.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nguyen H.H., Jayasinha V., Xia B., Hoyte K., Martin P.T. Overexpression of the cytotoxic T cell GalNAc transferase in skeletal muscle inhibits muscular dystrophy in mdx mice. Proc. Natl. Acad. Sci. USA. 2002;99:5616–5621. doi: 10.1073/pnas.082613599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu R., Jia Y., Zygmunt D.A., Martin P.T. rAAVrh74.MCK.GALGT2 Protects against Loss of Hemodynamic Function in the Aging mdx Mouse Heart. Mol. Ther. 2019;27:636–649. doi: 10.1016/j.ymthe.2019.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peccate C., Mollard A., Le Hir M., Julien L., McClorey G., Jarmin S., Le Heron A., Dickson G., Benkhelifa-Ziyyat S., Piétri-Rouxel F. Antisense pre-treatment increases gene therapy efficacy in dystrophic muscles. Hum. Mol. Genet. 2016;25:3555–3563. doi: 10.1093/hmg/ddw201. [DOI] [PubMed] [Google Scholar]
- 35.Dzierlega K., Yokota T. Optimization of antisense-mediated exon skipping for Duchenne muscular dystrophy. Gene Ther. 2020;27:407–416. doi: 10.1038/s41434-020-0156-6. [DOI] [PubMed] [Google Scholar]
- 36.Arnett A.L.H., Konieczny P., Ramos J.N., Hall J., Odom G., Yablonka-Reuveni Z., Chamberlain J.R., Chamberlain J.S. Adeno-associated viral (AAV) vectors do not efficiently target muscle satellite cells. Mol. Ther. Methods Clin. Dev. 2014;1:14038. doi: 10.1038/mtm.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hanlon K.S., Kleinstiver B.P., Garcia S.P., Zaborowski M.P., Volak A., Spirig S.E., Muller A., Sousa A.A., Tsai S.Q., Bengtsson N.E. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun. 2019;10:4439. doi: 10.1038/s41467-019-12449-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hartigan-O’Connor D., Kirk C.J., Crawford R., Mulé J.J., Chamberlain J.S. Immune evasion by muscle-specific gene expression in dystrophic muscle. Mol. Ther. 2001;4:525–533. doi: 10.1006/mthe.2001.0496. [DOI] [PubMed] [Google Scholar]
- 39.Mendell J.R., Campbell K., Rodino-Klapac L., Sahenk Z., Shilling C., Lewis S., Bowles D., Gray S., Li C., Galloway G. Dystrophin immunity in Duchenne’s muscular dystrophy. N. Engl. J. Med. 2010;363:1429–1437. doi: 10.1056/NEJMoa1000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crudele J.M., Chamberlain J.S. Cas9 immunity creates challenges for CRISPR gene editing therapies. Nat. Commun. 2018;9:3497. doi: 10.1038/s41467-018-05843-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salva M.Z., Himeda C.L., Tai P.W., Nishiuchi E., Gregorevic P., Allen J.M., Finn E.E., Nguyen Q.G., Blankinship M.J., Meuse L. Design of tissue-specific regulatory cassettes for high-level rAAV-mediated expression in skeletal and cardiac muscle. Mol. Ther. 2007;15:320–329. doi: 10.1038/sj.mt.6300027. [DOI] [PubMed] [Google Scholar]
- 42.Ran F.A., Cong L., Yan W.X., Scott D.A., Gootenberg J.S., Kriz A.J., Zetsche B., Shalem O., Wu X., Makarova K.S. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blankinship M.J., Gregorevic P., Allen J.M., Harper S.Q., Harper H., Halbert C.L., Miller A.D., Chamberlain J.S. Efficient transduction of skeletal muscle using vectors based on adeno-associated virus serotype 6. Mol. Ther. 2004;10:671–678. doi: 10.1016/j.ymthe.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 44.Im W.B., Phelps S.F., Copen E.H., Adams E.G., Slightom J.L., Chamberlain J.S. Differential expression of dystrophin isoforms in strains of mdx mice with different mutations. Hum. Mol. Genet. 1996;5:1149–1153. doi: 10.1093/hmg/5.8.1149. [DOI] [PubMed] [Google Scholar]
- 45.Danko I., Chapman V., Wolff J.A. The frequency of revertants in mdx mouse genetic models for Duchenne muscular dystrophy. Pediatr. Res. 1992;32:128–131. doi: 10.1203/00006450-199207000-00025. [DOI] [PubMed] [Google Scholar]
- 46.Dellorusso C., Crawford R.W., Chamberlain J.S., Brooks S.V. Tibialis anterior muscles in mdx mice are highly susceptible to contraction-induced injury. J. Muscle Res. Cell Motil. 2001;22:467–475. doi: 10.1023/a:1014587918367. [DOI] [PubMed] [Google Scholar]
- 47.Lynch G.S., Rafael J.A., Hinkle R.T., Cole N.M., Chamberlain J.S., Faulkner J.A. Contractile properties of diaphragm muscle segments from old mdx and old transgenic mdx mice. Am. J. Physiol. 1997;272:C2063–C2068. doi: 10.1152/ajpcell.1997.272.6.C2063. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.