

Abstract
Emerging evidence reveals that autophagy plays crucial roles in cardiac hypertrophy. Long noncoding RNAs (lncRNAs) are novel transcripts that function as gene regulators. However, it is unclear whether lncRNAs regulate autophagy in cardiac hypertrophy. Here, we identified a novel transcript named lncRNA Gm15834, which was upregulated in the transverse aortic constriction (TAC) model in vivo and the angiotensin-II (Ang-II)-induced cardiac hypertrophy model in vitro and was regulated by nuclear factor kappa B (NF-κB). Importantly, forced expression of lncRNA Gm15834 enhanced autophagic activity of cardiomyocytes and promoted myocardial hypertrophy, whereas silencing of lncRNA Gm15834 attenuated autophagy-induced myocardial hypertrophy. Mechanistically, we found that lncRNA Gm15834 could function as an endogenous sponge RNA of microRNA (miR)-30b-3p, which was downregulated in cardiac hypertrophy. Inhibition of miR-30b-3p enhanced cardiomyocyte autophagic activity and aggravated myocardial hypertrophy, whereas overexpression of miR-30b-3p suppressed autophagy-induced myocardial hypertrophy by targeting the downstream autophagy factor of unc-51-like kinase 1 (ULK1). Moreover, inhibition of lncRNA Gm15834 by adeno-associated virus carrying short hairpin RNA (shRNA) suppressed cardiomyocyte autophagic activity, improved cardiac function, and mitigated cardiac hypertrophy. Taken together, our study identified a novel regulatory axis encompassing lncRNA Gm15834/miR-30b-3p/ULK1/autophagy in cardiac hypertrophy, which may provide a potential therapy target for cardiac hypertrophy.
Keywords: cardiac hypertrophy, autophagy, NF-κB, lncRNA Gm15834, miR-30b-3p, ULK1
Graphical Abstract
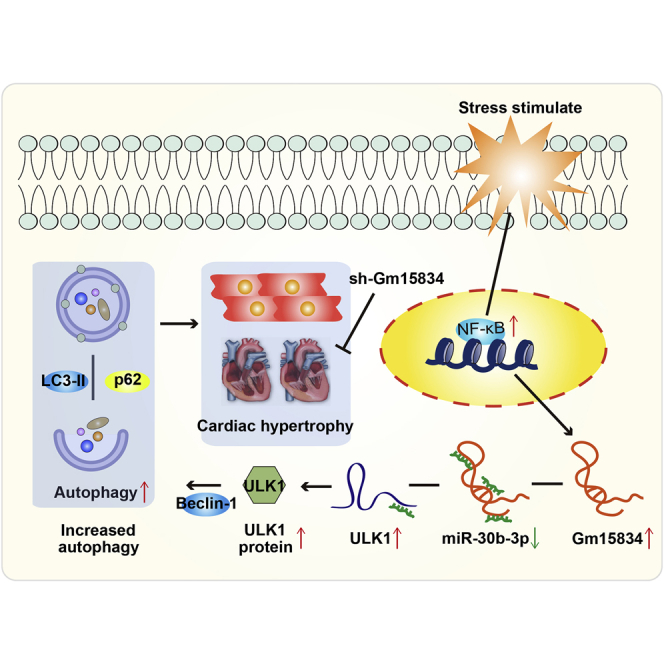
It is unclear whether long noncoding RNAs (lncRNAs) regulate autophagy in cardiac hypertrophy. Sun and colleagues identified a novel lncRNA Gm15834 and elucidated regulatory axis encompassing lncRNA Gm15834/miR-30b-3p/ULK1/autophagy in cardiac hypertrophy. In vivo silencing of lncRNA Gm15834 showed anti-hypertrophy effects, which may provide a potential therapy target for cardiac hypertrophy.
Introduction
Cardiac hypertrophy occurs in response to numerous pathologic stimuli. It is characterized by pathological myocardial hypertrophy, cardiomyocyte apoptosis, myocardial extracellular matrix (ECM) deposition, and fibrosis.1,2 Sustained cardiac hypertrophy can decrease myocardial contractility and compliance, which finally leads to heart failure and death. Thus, it is important to uncover the molecular mechanisms of cardiac hypertrophy.
Recent studies have found that noncoding RNAs (ncRNAs), such as microRNA (miR)-1, miR-133, and miR-23a, play pivotal roles in physiological and pathological processes of cardiac hypertrophy.3, 4, 5 MicroRNAs (miRNAs) exert functions by targeting the 3′ untranslated region (3′ UTR) of downstream genes. Moreover, a novel class of ncRNAs, long ncRNAs (lncRNAs), are considered as key regulators of complex biological processes. lncRNAs are more than 200 nucleotides long and without protein-coding potential; they participate in biological regulation, mainly via regulating translation, splicing, and gene expression at transcriptional and post-transcriptional levels.6, 7, 8 Importantly, lncRNAs can act as competitive endogenous RNAs (ceRNAs) of miRNAs, which can prevent the degradation of downstream mRNAs and induce multiple biological dysfunction. Some studies have found that lncRNAs regulate cardiac hypertrophy through miRNA-mediated regulatory axes.9,10 lncRNA CHRF can sponge endogenous miR-489 and regulate downstream target Myd88 in cardiac hypertrophy.11 Chen et al.12 have demonstrated that lncRNA CRRL functions as an endogenous sponge of miR-199a-3p and thereby increases the expression of downstream target Hopx, leading to increased cardiac injury. Liu et al.10 have revealed the H19-miR-675-CaMKIIδ axis as a negative regulator of cardiac hypertrophy.
Autophagy, an evolutionarily conserved and adaptive response of cells to cellular stress, has been demonstrated to play crucial roles in multiple regulatory processes of the cardiovascular system, such as cardiac homeostasis and pathology, including cardiac hypertrophy.13, 14, 15, 16, 17 The mechanism of autophagy in cardiac hypertrophy is still controversial. Suppression of autophagy activity has been found to attenuate cardiac hypertrophy.18,19 whereas other studies have suggested that increased autophagy can inhibit cardiac hypertrophy.14,20 Thus, the molecular mechanism of autophagy in cardiac hypertrophy should be investigated. Importantly, ncRNAs seem to participate in regulating cardiac hypertrophy by involvement in autophagy pathways. Cardiac-specific overexpression of the miR-199a leads to suppressed cardiomyocyte autophagy and induced cardiac hypertrophy via targeting glycogen synthase kinase 3β (GSK3β)/mammalian target of rapamycin (mTOR) complex signaling.15 Our previous studies found that endogenous miR-103 attenuated cardiomyocyte hypertrophy by reducing cardiac autophagy activity through inhibition of TRPV3 signaling in the pressure-overloaded hearts.21 Furthermore, few studies have demonstrated that lncRNAs regulate cardiac hypertrophy through the autophagy pathway. lncRNA Chast negatively regulates pleckstrin homology domain-containing protein family M member 1 (opposite strand of Chast), impeding cardiomyocyte autophagy and driving hypertrophy.22 Thus, it is essential to elucidate the molecular mechanism of lncRNA involvement in cardiac hypertrophy.
Here, we integrated two cardiac hypertrophy profiles and identified three upregulated lncRNAs as risk factors for cardiac hypertrophy. We focused on one such lncRNA (lncRNA Gm15834) and revealed that it was abundantly expressed in the heart and upregulated in cardiac hypertrophy. Our study showed that lncRNA Gm15834 was activated by transcription factor nuclear factor kappa B (NF-κB). Overexpression of lncRNA Gm15834 induced cardiac hypertrophy through promoting cardiomyocyte autophagy by targeting miR-30b-3p, which endogenously bound to the 3′ UTR of unc-51-like kinase (ULK1). Silencing of lncRNA Gm15834 by in vivo injection of adeno-associated virus 9 (AAV9) attenuated cardiac hypertrophy and improved cardiac function. Our study revealed a novel role of lncRNA in regulating cardiac hypertrophy via the autophagy pathway and the potential of the lncRNA Gm15834/miR-30b-3p/ULK1 axis as a therapeutic target for the treatment of cardiac hypertrophy.
Results
lncRNA Gm15834 Was a High-Risk Factor for Cardiac Hypertrophy
To identify the high-risk lncRNAs in cardiac hypertrophy, we integrated and processed two gene-expression datasets (RNA sequencing [RNA-seq] and gene microarray) to identify differentially expressed lncRNAs via bioinformatics protocols (see Materials and Methods; Figure 1A). As a result, 253 and 125 upregulated lncRNAs were considered as high-risk factors based on the RNA-seq dataset and microarray dataset, respectively. To reduce the false-positive results, we chose only the lncRNAs that were available in both databases. The results showed that three lncRNAs were upregulated in both datasets (lncRNA Gm15834, 4930473A02Rik, and Gm11508; Figure 1B). Furthermore, a cardiac hypertrophy lncRNA-mRNA coexpression network (CHLMCN) was constructed by integrating the high-correlated lncRNA-mRNA pairs of two datasets (Figure 1C). Previous studies reported that the random walk algorithm on the network could identify novel regulators.23,24 Thus, we performed random walk on CHLMCN to identify potential pathogenic lncRNAs by mapping known disease genes (Bdkrb1, Ttr, Pla2g5, Prkcb, Alox5ap, Il18bp, Klrb1a, Lta, Fkbp1b, and Hmox1) into CHLMCN. Interestingly, lncRNA Gm15834 was identified as a potential pathogenic risk factor of cardiac hypertrophy, according to the random walk algorithm (Table S1). The above results implied that lncRNA Gm15834 might be a high-risk factor in cardiac hypertrophy.
Figure 1.

lncRNA Gm15834 Was the High-Risk Factor of Cardiac Hypertrophy and Was Regulated by NF-κB
(A) The pipelines of identification of cardiac hypertrophy associated high-risk lncRNAs based on bioinformatics analysis. (B) The heatmaps of differentially expressed lncRNAs that identified from cardiac hypertrophy-related RNA-seq (left) and microarray (right) datasets. The common lncRNAs were highlighted in red. (C) The visualization of the lncRNA-mRNA coexpression network. lncRNA names were labeled in purple, and disease seed genes were labeled in blue. (D) The expression heatmap of lncRNA Gm15834 in multiple tissues. Rectangles represented the high-expressed tissues, rows represented data sources (Fantom5 project), and columns represented tissues. (E) UCSC genome browser tracks of lncRNA Gm15834 gene epigenomics signals in heart tissues. (F) The dynamic expression of lncRNA Gm15834 in different stages of heart development. (G) The effect of NF-κB on lncRNA Gm15834 promoter dual-luciferase activity. ∗p < 0.05 versus Gm15834-WT group; #p < 0.05 versus NF-κB + Gm15834-MUT group. n = 3.
lncRNA Gm15834 Was Abundantly Expressed in the Heart
To investigate the tissue-specific expression of lncRNA Gm15834, we searched the expression of lncRNA Gm15834 from the Mouse Genome Informatics (MGI) database (Figure 1D). lncRNA Gm15834 was highly expressed in six tissues, including the brain, thymus, testis, liver, lung, and heart. To validate the expression of lncRNA Gm15834 in the heart, we also downloaded the epigenetics signal from University of California, Santa Cruz (UCSC), genome browsers and found that the promoter of lncRNA Gm15834 was occupied by multiple activator signals (H3k27ac, H3k4me3, and Pol2), indicating that lncRNA Gm15834 had high transcriptional activity in the heart (Figure 1E). Furthermore, lncRNA Gm15834 showed a tendency to decreased expression during heart development (Figure 1F). Studies have demonstrated higher expression levels of the causative genes of cardiac hypertrophy in the early heart-development period than in the adult period.25 Furthermore, the BlastN result indicated that two fraction sequences of lncRNA Gm15834 were homologous to human sequences (Figure S1). We also identified the upstream transcription factors for lncRNA Gm15834, and the results showed that two NF-κB binding sites (transcription factor p65) were located in the promoter region of lncRNA Gm15834. Luciferase experiments validated that NF-κB regulated the expression of lncRNA Gm15834 directly (Figure 1G). Previous studies have shown that NF-κB is one of the key regulators of cardiac hypertrophy.26,27 Thus, all of these results implied that lncRNA Gm15834 was highly expressed in the heart and might play crucial roles in the pathological processes of cardiac hypertrophy.
lncRNA Gm15834 and Autophagy Were Both Activated in Cardiac Hypertrophy
To investigate the regulatory role of lncRNA Gm15834 in cardiac hypertrophy, we first established 3-week transverse aortic constriction (TAC) models in mice. 3 weeks after the surgery, the heart volume, heart weight/body weight ratio (HW/BW), left ventricle weight (LVW)/BW ratio, and LVW/tibial length ratio (TL) were significantly increased in TAC groups (Figures S2A and S2B). H&E staining marked an increased cardiomyocyte area in TAC groups (Figure S2C). Activated cardiac hypertrophy marker proteins (β-myosin, heavy polypeptide 7, cardiac muscle [β-MHC] and brain natriuretic peptide [BNP]) and impaired cardiac function were found in TAC groups (Figures S2D–S2F; Table S2). RNA-level changes of cardiac hypertrophy markers were also tested; the results showed that the levels significantly increased in TAC groups (Figure S2G). Second, we also used Ang-II to induce in vitro cardiomyocyte hypertrophy and detected the hypertrophy indices in HL-1 cell lines. Immunofluorescence staining showed that Ang-II significantly enlarged cardiomyocyte areas (Figure S3A). Hypertrophy marker proteins and RNAs (β-MHC and BNP) were both activated by Ang-II (Figures S3B–S3D). These results all indicated that in vivo and in vitro models were established.
We performed lncRNA sequencing for sham and TAC hearts, and the results showed that cardiac hypertrophy markers and lncRNA Gm15834 were upregulated in hypertrophic hearts (Figure 2A; with log2 [fold change (FC)] ≥ 1.5). Quantitative reverse transcriptase PCR (qRT-PCR) results verified that lncRNA Gm15834 was upregulated in the TAC model in vivo and in the Ang-II hypertrophy model in vitro (Figure 2B), implying that lncRNA Gm15834 might play a regulatory role in the pathogenesis of cardiac hypertrophy. Importantly, to explore the translational potential of lncRNA Gm15834, we also used qRT-PCR to test the expression of the human homologous region, and the results indicated that this conserved fraction was also upregulated in Ang-II-stimulated AC-16 cell lines (Figure 2B; bottom).
Figure 2.

lncRNA Gm15834 and Autophagy Were Activated in Cardiac Hypertrophy
(A) The expression heatmap of lncRNA sequencing data of cardiac hypertrophy models (left). Rows and columns represent samples and genes. The log2 (fold change) of each gene (right). (B) lncRNA Gm15834 was upregulated in cardiac hypertrophy in vivo and in vitro (top). Expression data were detected by qRT-PCR. ∗p < 0.05 versus sham/control group. n = 3. (Bottom) qRT-PCR results of lncRNA Gm15834 homologous region expression in human AC-16 cell lines. ∗p < 0.05 versus control group. n = 3. (C) Representative images of cardiac tissues that was captured by electron microscopy. Autophagosomes were hunted in the TAC group. Scale bar, 500 nm. (D) In vitro transfection of double-labeled LC3 autophagy adenovirus to count autolysosomes and autophagosomes from cardiomyocytes. ∗p < 0.05 versus control group. n = 3. Scale bar, 20 μm.
Meanwhile, previous studies demonstrated that abnormal autophagy activity could affect cardiac hypertrophy.28 Thus, we examined the autophagy level in our hypertrophy models. We observed autophagosomes on transmission electron microscope in the TAC hearts in vivo, and protein expression of autophagy markers (LC3-II and Beclin-1) was significantly increased, and p62 was decreased in TAC hearts (Figure 2C; Figures S4A–S4C). In vitro, LC3 adenovirus infection experiments monitored an increased autophagic activity after Ang-II stimulation (Figure 2D). Increased protein expression of LC3-II and Beclin-1 and decreased protein expression of p62 were observed simultaneously (Figures S4D–S4F). These results indicate increased autophagic activity in both in vitro and in vitro cardiac hypertrophy. However, autophagy has controversial roles in cardiac hypertrophy. To further demonstrate the promoting role of autophagy in our cardiac hypertrophy models, we used autophagy inhibitor 3-methyladenine (3MA) to block autophagy in vitro. The results showed that blocking autophagic activity delayed the progress of cardiomyocyte hypertrophy (Figures S5A and S5B). Previous studies suggested that some lncRNAs could regulate multiple pathological processes via autophagy, which inspired us to investigate whether lncRNA Gm15834 regulates cardiac hypertrophy through the autophagy signal axis.
lncRNA Gm15834 Was a Crucial Regulator of Cardiac Hypertrophy In Vivo and In Vitro
To explore the function of lncRNA Gm15834 in cardiac hypertrophy, we performed loss-of-function and gain-of-function experiments in vitro. First, we used a small interfering RNA (siRNA) to interfere with the expression of lncRNA Gm15834. The results showed that lncRNA Gm15834 was efficiently suppressed (Figure 3A). Knockdown of lncRNA Gm15834 attenuated hypertrophic cardiomyocyte size and suppressed the expression of cardiac hypertrophy marker proteins compared with the Ang-II group (Figures 3B–3D). Based on the results shown in Figure 2, we hypothesized that lncRNA Gm15834 was biologically involved in the development of cardiac hypertrophy via autophagy. Therefore, we examined the effect of lncRNA Gm15834 on autophagic activity in hypertrophy models. The results showed that knockdown of lncRNA Gm15834 also reduced the number of puncta of the monomeric red fluorescent protein (mRFP)-GFP-LC3 system and significantly decreased the expression levels of LC3-II and Beclin-1 but markedly activated the expression of p62 (Figures 3E–3H), indicating that high autophagy activity of cardiomyocytes induced by Ang-II was suppressed. Moreover, knockdown of lncRNA Gm15834 with short hairpin RNA (shRNA) #2 yielded the same results as siRNA interference (Figure S6). Second, we constructed the full-length plasmid of lncRNA Gm15834 and transfected into cardiomyocytes (Figure 4A). We found that overexpression of lncRNA Gm15834 significantly increased cardiomyocyte size (Figure 4B), marker protein expression (Figures 4C and 4D), and autophagy activity (Figures 4E–4H). Additionally, rescue experiments demonstrated that autophagy inhibitor 3MA counteracted the adverse effects of lncRNA Gm15834-induced cardiomyocyte hypertrophy and abnormal autophagy level (Figures S7A and S7B). These results indicated that lncRNA Gm15834 played an important role in cardiac hypertrophy via regulating cardiomyocyte autophagy levels. Further research is needed to uncover the regulatory pathways of lncRNA Gm15834.
Figure 3.

Knockdown of lncRNA Gm15834 Attenuated Cardiac Hypertrophy
(A) qRT-PCR results of lncRNA Gm15834 knockdown. ∗p < 0.05 versus control group; #p < 0.05 versus Ang-II group. n = 3. (B) Immunofluorescence results of cardiomyocyte areas from in vitro groups. ∗p < 0.05 versus control group; #p < 0.05 versus Ang-II group. n = 3. Scale bar, 50 μm. (C and D) Western blot results of β-MHC (C) and BNP (D) protein expression in lncRNA Gm15834 knockdown groups.. ∗p < 0.05 versus control group; #p < 0.05 versus Ang-II group. n = 6. (E) In vitro transfection of double-labeled LC3 autophagy adenovirus to count autolysosomes and autophagosomes from cardiomyocytes. ∗p < 0.05 versus control group; #p < 0.05 versus Ang-II group. n = 3. Scale bar, 20 μm. (F–H) Western blot results of Beclin-1 (F), LC3-II (G), and p62 (H) protein in lncRNA Gm15834 knockdown groups. ∗p < 0.05 versus control group; #p < 0.05 versus Ang-II group. n = 6.
Figure 4.

Overexpression of lncRNA Gm15834 Triggered Cardiac Hypertrophy
(A) qRT-PCR results of lncRNA Gm15834 overexpression. ∗p < 0.05 versus pcDNA group. n = 3. (B) Immunofluorescence results of cardiomyocyte areas from in vitro groups. ∗p < 0.05 versus control group; #p < 0.05 versus Gm15834 group. n = 3. Scale bar, 50 μm. (C and D) Western blot results of β-MHC (C) and BNP (D) protein expression in lncRNA Gm15834 overexpression groups. ∗p < 0.05 versus control group; #p < 0.05 versus Gm15834 group. n = 6. (E) In vitro transfection of double-labeled LC3 autophagy adenovirus to count autolysosomes and autophagosomes from cardiomyocytes. ∗p < 0.05 versus pcDNA group. n = 3. Scale bar, 20 μm. (F–H) Western blot results of LC3-II (F), Beclin-1 (G), and p62 (H) protein in lncRNA Gm15834 overexpression groups. ∗p < 0.05 versus control group; #p < 0.05 versus Gm15834 group. n = 6.
miR-30b-3p Was the Potential Target of lncRNA Gm15834
Many studies have reported that cytoplasm-located lncRNAs participate in physiologic and pathologic processes of cardiovascular diseases usually by acting as a sponge for decoy disease miRNAs. Fluorescence in situ hybridization results showed that lncRNA Gm15834 was predominantly localized in the cytoplasm of cardiomyocytes (Figure 5A). Thus, we speculated that lncRNA Gm15834 might exert regulatory function as a miRNA sponge. We performed bioinformatics analysis to identify the high-confidence decoy miRNAs of lncRNA Gm15834, via integrating miRanda algorithm and disease-relevant miRNA profiles. Briefly, three relevant miRNA profiles were reanalyzed; only miR-30b-3p was downregulated in the three datasets (Figure 5B), whereas it was predicted as a potential target of lncRNA Gm15834 by miRanda. Subsequently, we examined the expression level of miR-30b-3p in the cardiac hypertrophy model, where qRT-PCR showed that miR-30b-3p was downregulated both in vivo and in vitro (Figure 5C). Meanwhile, knockdown or overexpression of lncRNA Gm15834 had a severe impact on miR-30b-3p expression (Figure 5D). Interestingly, the results showed that two miRNA binding sites were located in the gene body of lncRNA Gm15834, and one of the binding sites was located in the conserved region (Figures 5E and S1). Thus, we hypothesized that lncRNA Gm15834 might regulate cardiac hypertrophy via targeting miR-30b-3p. Next, we validated the effect of miR-30b-3p on cardiac hypertrophy.
Figure 5.

miR-30b-3p Was the Downstream of lncRNA Gm15834 and Regulated Cardiac Hypertrophy via Regulating Autophagy
(A) Fluorescence in situ hybridization (FISH) was performed to detect the subcellular localization of lncRNA Gm15834. Scale bar, 20 μm. (B) Venn diagram of miR-30b-3p in 3 miRNA expression datasets of cardiac hypertrophy or heart failure. (C) qRT-PCR results of miR-30b-3p expression in TAC models and Ang-II induced models in vitro. ∗p < 0.05 versus sham/control group. n = 3. (D) qRT-PCR results of miR-30b-3p expression in lncRNA Gm15834 interference experiments. ∗p < 0.05 versus NC/pcDNA group. n = 3. (E) The binding sites between miR-30b-3p and lncRNA Gm15834, including the binding site of the conserved region. (F and G) Western blot results of β-MHC (F) and BNP (G) protein expression in miR-30b-3p overexpression and antagonism groups. ∗p < 0.05 versus control group; #p < 0.05 versus Ang-II group; @p < 0.05 versus Ang-II + miR-30b group. n = 6. (H) Immunofluorescence results of cardiomyocyte areas from in vitro groups. ∗p < 0.05 versus control group; #p < 0.05 versus Ang-II group; @p < 0.05 versus Ang-II + miR-30b group. n = 3. Scale bar, 50 μm. (I) In vitro transfection of double-labeled LC3 autophagy adenovirus to count autolysosomes and autophagosomes from cardiomyocytes. ∗p < 0.05 versus control group; #p < 0.05 versus Ang-II group; @p < 0.05 versus Ang-II + miR-30b group. n = 3. Scale bar, 20 μm. (J–L) Western blot results of LC3-II (J), Beclin-1 (K), and p62 (L) protein in miR-30b-3p overexpression and antagonism groups. ∗p < 0.05 versus control group; #p < 0.05 versus Ang-II group; @p < 0.05 versus Ang-II + miR-30b group. n = 6.
miR-30b-3p Inhibited the Hypertrophic Effect through Targeting ULK1
The miR-30 family was demonstrated to participate in cardiac hypertrophy by involvement in several pathways, including autophagy and p53 signaling pathways.29,30 To uncover the molecular mechanism of miR-30b-3p in cardiac hypertrophy, we first transfected the miR-30b-3p mimic and miR-30b-3p mimic + AMO-30b mixture into cardiomyocytes. Transfection of the miR-30b-3p mimic induced a dramatic increase in miR-30b-3p expression in Ang-II-treated cardiomyocytes, whereas transfection of additional AMO-30b abrogated this effect (Figure S8A). Functionally, transfection of miR-30b-3p decreased the protein levels of β-MHC and BNP, which were rescued by AMO-30b (Figures 5F and 5G). Congruously, overexpression of miR-30b-3p markedly decreased the Ang-II-induced hypertrophic cardiomyocyte size, whereas this effect was remarkably abolished by cotransfecting AMO-30b (Figure 5H). miR-30b-3p was considered as the target of lncRNA Gm15834, so we next investigated whether miR-30b-3p was involved in the regulation of autophagy. As expected, overexpression of miR-30b-3p significantly suppressed the autophagy activity, which could be elevated by AMO-30b (Figure 5I). We also validated the protein level of the autophagy markers. The results showed that LC3-II and Beclin-1 were suppressed, whereas p62 was elevated after miR-30b-3p transfection; transfection of additional AMO-30b yielded the reverse results (Figures 5J–5L). For strengthening the regulatory role of miR-30b-3p in cardiac hypertrophy and autophagy, we only transfected the antagonist AMO-30b into cardiomyocytes. The results of antagonizing miR-30b-3p showed autophagy activation and promotion of cardiomyocyte hypertrophy (Figures S8B and S8C). These results indicated that miR-30b-3p regulated cardiac hypertrophy via mediating cardiomyocyte autophagy.
Next, we used TargetScan software to identify the candidate downstream targets of miR-30b-3p. Interestingly, ULK1 3′ UTR had a binding site for miR-30b-3p (Figure 6A), which was reported as a crucial regulator in the autophagy pathway.31,32 Therefore, we speculated that ULK1 was the direct target of miR-30b-3p, which mediated the miR-30b-3p effect on autophagy. We transfected the miR-30b-3p mimic and AMO-30b into cardiomyocytes to detect the expression of ULK1 protein. The results showed that forced expression of miR-30b-3p inhibited the expression of ULK1, whereas knockdown of miR-30b-3p resulted in the opposite effect (Figure 6B). To validate the direct binding relationship between miR-30b-3p and ULK1, we constructed wild-type (WT) and mutated-type ULK1 plasmid with luciferase. By cotransfection of the luciferase plasmid and miR-30b-3p mimic/negative control (NC) into HEK293T cells, the miR-30b-3p mimic remarkably suppressed the luciferase activity of the WT ULK1 plasmid, but miR-30b-3p failed to affect the mutated-type ULK1 luciferase activity (Figure 6C). These results indicated miR-30b-3p-dependent regulation of cardiac hypertrophy via the ULK1/autophagy axis.
Figure 6.

lncRNA Gm15834 Regulated Cardiac Hypertrophy via the miR-30b-3p/ULK1 Axis
(A) The binding sites of miR-30b-3p and 3′ UTR of ULK1 in mouse and human. (B) Western blot results of ULK1 protein expression levels. ∗p < 0.05 versus control group; #p < 0.05 versus Ang-II group; @p < 0.05 versus Ang-II + miR-30b group. n = 6. (C) Luciferase assay results of direct interactions between miR-30b-3p and ULK1. ∗p < 0.05 versus ULK1-wt group. n = 3. (D) Luciferase assay results of direct interactions among lncRNA Gm15834, miR-30b-3p, and ULK1. ∗p < 0.05 versus ULK1-WT group; #p < 0.05 versus ULK1-WT + miR-30b-mimic group; @p < 0.05 versus ULK1-WT + miR-30b-mimic + Gm15834 group. n = 3. (E) AGO2 RNA immunoprecipitation assay was conducted to assess endogenous AGO2-binding RNAs in TAC hearts. IgG was used as the negative control (NC). qRT-PCR was used to detect the gene expression. ∗p < 0.05 versus IgG group. n = 3. (F) Western blot results of ULK1 protein expression levels in cotransfection experiments. ∗p < 0.05 versus control group; #p < 0.05 versus Gm15834 group. n = 6. (G and H) Western blot results of β-MHC (G) and BNP (H) protein expression in lncRNA Gm15834 and miR-30b-3p co-transfection groups. ∗p < 0.05 versus control group; #p < 0.05 versus Gm15834 group. n = 6. (I) Immunofluorescence results of cardiomyocyte areas from in vitro groups. ∗p < 0.05 versus control group; #p < 0.05 versus Gm15834 group. n = 3. Scale bar, 50 μm. (J) In vitro transfection of double-labeled LC3 autophagy adenovirus to count autolysosomes and autophagosomes from cardiomyocytes. ∗p < 0.05 versus control group; #p < 0.05 versus Gm15834 group. n = 3. Scale bar, 20 μm. (K–M) Western blot results of LC3-II (K), Beclin-1 (L), and p62 (M) protein in lncRNA Gm15834 and miR-30b-3p co-transfection groups. ∗p < 0.05 versus control group; #p < 0.05 versus Gm15834 group. n = 6.
lncRNA Gm15834 Promoted Myocardial Hypertrophy via the miR-30b-3p/ULK1/Autophagy Axis
Our results demonstrated that lncRNA Gm15834 promoted cardiomyocyte autophagy and hypertrophy; lncRNA Gm15834 bound to miR-30b-3p, and miR-30b-3p displayed an anti-hypertrophy role by suppressing ULK1-mediated autophagy activity. We hypothesized that lncRNA Gm15834-sponged miR-30b-3p might rescue the inhibition of ULK1 by miR-30b-3p and lead to high autophagy activity of cardiomyocytes, eventually causing myocardial hypertrophy. First, we used a luciferase reporter assay to investigate the effect of lncRNA Gm15834 on the expression of ULK1. Luciferase assay results showed that lncRNA Gm15834 rescued the inhibition effect of miR-30b-3p on ULK1 (Figure 6D). Second, we conducted AGO2 RNA immunoprecipitation (RIP) experiments on 3-week TAC hearts to extract the mixture of AGO2-enriched RNAs. Notably, qPCR testing showed that compared with NC of immunoglobulin G (IgG), lncRNA Gm15834 and miR-30b-3p were highly enriched (Figure 6E). These results implied that the lncRNA Gm15834/miR-30b-3p/ULK1 regulatory axis might exist. We subsequently performed lncRNA Gm15834 interference experiments to detect the effect of lncRNA Gm15834 on ULK1 expression. As a result, silenced lncRNA Gm15834 led to decreased ULK1 expression, whereas elevated lncRNA Gm15834 significantly increased ULK1 expression (Figures S9A–S9C).
To further investigate the effect of lncRNA Gm15834 on myocardial hypertrophy by mediating the miR-30b-3p/ULK1/autophagy axis, we cotransfected lncRNA Gm15834 and miR-30b-3p mimics into cardiomyocytes. The results showed that the induced expression of ULK1 by lncRNA Gm15834 was attenuated by miR-30b-3p mimics (Figure 6F). More importantly, miR-30b-3p mimics reversed the elevated cardiomyocyte size and β-MHC and BNP protein expression by lncRNA Gm15834 (Figures 6G–6I). Simultaneously, enhanced autophagic activity induced by lncRNA Gm15834 was suppressed by miR-30b-3p (Figures 6J–6M). Together, these results demonstrated that lncRNA Gm15834 regulated cardiomyocyte autophagy activity via the miR-30b-3p/ULK1 axis, and overexpression of miR-30b-3p attenuated the lncRNA Gm15834-induced cardiomyocyte hypertrophy.
Inhibition of lncRNA Gm15834 Attenuated Pressure Overload-Induced Cardiac Hypertrophy
The above-mentioned results suggested that lncRNA Gm15834 inhibited cardiac hypertrophy via the miR-30b-3p/ULK1/autophagy axis, and we next aimed to validate whether inhibition of lncRNA Gm15834 could attenuate cardiac hypertrophy in a mouse model. We packed the shRNAs and NCs in heart-specific AAV9 and injected it to the mouse models via tail-vein injection. After 3 weeks of modeling, we detected that AAV9 harboring shRNA was highly expressed in cardiac tissues (Figure 7A). The miR-30b-3p expression level was boosted by shRNA-mediated inhibition (Figure S10A). Then we examined the potential therapeutic effects of shRNA on cardiac hypertrophy in vivo. H&E staining results showed that knockdown for lncRNA Gm15834 significantly decreased cardiomyocyte size induced by TAC in vivo (Figure 7B). TAC-induced expression changes of cardiac hypertrophy marker proteins (β-MHC and BNP) were also reversed by AAV9 harboring shRNA injection (Figures 7C and 7D; Figures S10B and S10C). M-mode echocardiography showed attenuated left ventricle walls (left ventricle posterior wall end-diastolic diameter [LVPWd] and left ventricle anterior wall end-diastolic diameter [LVAWd]) after injection of AAV9 harboring shRNA to TAC model mice (Figure 7E; Table S3). The ejection fraction (EF) and fractional shortening (FS), two echocardiographic indices of the left ventricle contractile function, were enhanced by AAV9 harboring shRNA (Figure 7E; Table S3). Mechanistically, we also detected the expression of autophagy marker proteins (LC3-II, Beclin-1, and p62) and the target ULK1. Similarly, the expression changes of these proteins were reversed by shRNA (Figures 7F–7I; Figures S10B and S10C). These results suggested that cardiac-specific inhibition of lncRNA Gm15834 was able to delay the process of cardiac hypertrophy through blocking autophagy.
Figure 7.

Knockdown of lncRNA Gm15834 Attenuated Cardiac Hypertrophy In Vivo
(A) The protocol of AAV9-shRNA injection. GFP represents AAV9 harboring shRNA or sh-NC plasmid expression in situ. (B) H&E staining of cardiac tissues to assess the effects of AAV9 injection. Scale bar, 100 μm for 20× and 50 μm for 40×. (C and D) Western blot results of β-MHC (C) and BNP (D) protein expression in lncRNA Gm15834 AAV-9 knockdown groups. ∗p < 0.05 versus sham group; #p < 0.05 versus TAC group. n = 3. Results of TAC + sh_NC group were provided in Figure S10. (E) Typical images of mouse heart echocardiography and statistical graphs for left ventricle anterior wall end-diastolic diameter (LVAWd), left ventricle posterior wall end-diastolic diameter (LVPWd), left ventricle ejection fraction (LVEF), and shortening fraction (FS). ∗p < 0.05 versus sham group; #p < 0.05 versus TAC + NC group; @p < 0.05 versus TAC + sh_Gm15834 group. n = 3. (F–I) Western blot results of LC3-II (F), Beclin-1 (G), ULK1 (H), and p62 (I) protein in lncRNA Gm15834 AAV-9 knockdown groups. ∗p < 0.05 versus sham group; #p < 0.05 versus TAC group. n = 3. Results of TAC + sh_NC group were provided in Figure S10.
Discussion
In this study, we illustrated the molecular mechanism of the novel lncRNA Gm15834 effect on cardiac hypertrophy. First, we performed bioinformatics analysis and molecular biological experiments to validate that lncRNA Gm15834 was the best candidate risk factor for cardiac hypertrophy. Second, gain and loss experiments indicated that abnormal expression of lncRNA Gm15834 affected the level of myocardial hypertrophy. Third, miR-30b-3p was identified as a downstream target of lncRNA Gm15834 and was shown to have an anti-hypertrophy effect. Finally, we demonstrated that lncRNA Gm15834 regulated cardiac hypertrophy via rescuing ULK1 and mediating autophagy.
Importantly, lncRNA Gm15834 is the novel functional lncRNA in cardiac hypertrophy, which might regulate cardiac hypertrophy via autophagy. Autophagy is a highly dynamic cellular process involved in cardiac stress and cardiac injury.16,33,34 However, the role of autophagy in cardiac hypertrophy is controversial, depending on disease status and severity.35 Based on the time of onset, cardiac hypertrophy can be divided into compensatory stage (adaptive cardiac hypertrophy) and decompensation stage (maladaptive cardiac hypertrophy). On the positive side, adaptive autophagy provides compensatory energy for cardiomyocytes and plays a protective role in maintaining short-term homeostasis.28 Some pathways, such as AMPK/mTOR pathway and FoxO1, were demonstrated to be autophagy-related protective factors in cardiac hypertrophy.36, 37, 38 Notably, the above-mentioned protective roles were all conducted on 1-week mild hypertrophy models, which coincided with the physiological characters of adaptive cardiac hypertrophy. On the negative side, with the long-term stimulation or in severe stress, adaptive cardiac hypertrophy transforms to maladaptive hypertrophy. In this situation, previous studies demonstrated that excessive autophagy could induce cardiomyocyte death and finally lead to heart failure.34,39 Our previous study also demonstrated that inhibition of myocardial cell autophagy activity attenuated pressure overload-induced cardiac hypertrophy.21 In this study, our in vivo and in vitro cardiac hypertrophy models were both severe hypertrophy models (maladaptive stage), and we also validated that autophagy inhibitor 3MA could rescue Ang-II-induced cardiomyocyte hypertrophy. Despite the biological importance of lncRNAs, it is not yet clear whether lncRNAs are involved in the regulation of autophagy during cardiac hypertrophy. Thus, we also evaluated the effect of lncRNA Gm15834 on cellular autophagy activity. The results showed that overexpression of lncRNA Gm15834 activated cardiomyocyte autophagy and silencing of lncRNA Gm15834 could alleviate the cellular stress-induced autophagy activity. Additionally, autophagy inhibitor 3MA was able to block both the lncRNA Gm15834-induced cardiac hypertrophy and raised autophagy levels. These results suggested that lncRNA Gm15834 might regulate cardiac hypertrophy via autophagy.
We demonstrated that miR-30b-3p was the direct target of lncRNA Gm15834. We calculated and compared the expression of lncRNA Gm15834 and miR-30b-3p in the heart cells, including cardiomyocytes and fibroblasts. lncRNA Gm15834 was broadly expressed in both major cell types, whereas miR-30b-3p tended to be a cardiomyocyte-specific miRNA (Figures S11B–S11D). Therefore, here, we speculated that the lncRNA Gm15834/miR-30b-3p axis participated in regulatory processes of cardiac hypertrophy. Studies have revealed the members of the miR-30 family, such as miR-30a, miR-30b, miR-30c, and miR-30d, as crucial autophagy regulators in multiple diseases, including cardiac hypertrophy.29,30,40, 41, 42. miR-30b-3p is a novel transcript, and we confirmed its expression in the heart and especially in cardiomyocytes (Figure S11D). Our results demonstrated that exogenous overexpression of miR-30b-3p suppressed cardiac hypertrophy and myocardial cell autophagy. Furthermore, miR-30b-3p was demonstrated as the lncRNA Gm15834 downstream target.
To investigate the mechanism of the miR-30b-3p effect on cardiac hypertrophy and autophagy, we used TargetScan to predict the downstream targets. Intriguingly, we found that autophagy regulator ULK1 was listed in the miR-30b-3p target results. It has been reported that the AMPK/mTOR/ULK pathway is the master regulator of autophagy. Amino acid starvation or mTOR inhibition trigger ULK1 to phosphorylate Beclin-1 on Ser 14, and then activate the VPS34 complexes, inducing cell autophagy.32 Before autolysosome formation, autophagy signal is transduced via activating the ULK complex, which is composed of ULK1, ULK2, FIP200, and ATG13. Furthermore, ULK1 acts as a bridge between the upstream energy sensor AMPK/mTOR and the downstream autophagosome formation. However, the function of ULK1 in cardiac hypertrophy is unclear. In this study, we demonstrated that ULK1 served as the downstream target of miR-30b-3p, which mediated the inhibitory effect of miR-30b-3p on autophagy in cardiac hypertrophy. We plan to investigate the molecular mechanism of ULK1 on regulating autophagy in cardiac hypertrophy in our future work. Importantly, a luciferase experiment demonstrated that lncRNA Gm15834 rescued the degradation of miR-30b-3p to ULK1. Furthermore, cotransfection experiments showed that miR-30b-3p attenuated the effect of lncRNA Gm15834 on autophagy and cardiac hypertrophy via ULK1. These results suggested that lncRNA Gm15834 regulated myocardial cell autophagy and cardiac hypertrophy via miR-30b-3p/ULK1.
Our study also had some limitations. First, lncRNA Gm15834 was expressed both in cardiomyocytes and cardiac fibroblasts. Although we detected the expression changes of lncRNA Gm15834 and miR-30b-3p in primary cardiomyocytes, and the results were in lines with the experiments in HL-1 cell lines (Figures S11A–S11C), we plan to further explore and identify the novel mechanism of lncRNA Gm15834 in both cell types. Second, because of the lack of miRNA expression matrixes of cardiac hypertrophy in public data sources, we used three expression datasets from different situations, including cardiac aging, TAC, and the transgenic hypertrophy model. Nevertheless, these situations in original papers were all used to mimic severe hypertrophy or heart-failure models. Thus, we used these datasets to identify the potential miRNA targets of lncRNA Gm15834. The results would have been more precise if we had analyzed the datasets of the miRNA expression matrix in 3-week TAC hearts. Third, our data showed that only one miRNA binding site was located in the human homologous regions of lncRNA Gm15834, and conservation of ULK1 between mouse and human was not perfect. Although we found that the lncRNA Gm15834 homologous region was upregulated in human-stimulated AC-16 samples, further work is needed to investigate the regulatory axis of lncRNA Gm15834/miR-30b-3p/ULK1 in human cells. Finally, in this study, we only identified and focused on the mechanism of miR-30b-3p, and further studies will investigate the regulatory role of the reverse strand miR-30b-5p in autophagy and hypertrophy.
In summary, we identified the novel lncRNA Gm15834, a heart-abundant transcript that mediates autophagy to regulate cardiac hypertrophy. We demonstrated a novel regulatory axis encompassing lncRNA Gm15834/miR-30b-3p/ULK1/autophagy in cardiac hypertrophy, which may provide potential therapy targets for cardiac hypertrophy (Figure 8).
Figure 8.

The Mimic Mechanism of lncRNA Gm15834 Regulating Cardiac Hypertrophy via the miR-30b-3p/ULK1/Autophagy Axis
Inhibition of lncRNA Gm15834 attenuated cardiac hypertrophy in vivo.
Materials and Methods
Identification of High-Risk lncRNAs in Cardiac Hypertrophy
In this study, two sets of cardiac hypertrophy expression data were performed. All samples were sham- and TAC-operated. RNA-seq data were downloaded from the European Bioinformatics Institute (EBI: PRJEB2602). TopHat and Culfflinks software were performed to call gene expression (fragments per kilobase of transcript per million mapped reads [FPKM]). Gene microarray data were downloaded from the GEO database (GEO: GSE12337). For the microarray data, we only reserved the 8 samples of sham operation and TAC operation. The probe reannotation pipeline was used to identify lncRNA and mRNA expression. The Significance Analysis of Microarrays (SAM) test was used to identify differentially expressed genes for the two datasets (FC ≥ 2, p ≤ 0.05).
Optimization of High-Risk lncRNAs
For further optimization of high-risk lncRNAs, we integrated a network analysis and random walk algorithm based on our previous studies. Briefly, we performed the SAM test to identify differentially expressed mRNAs in two datasets and calculated Pearson correlation coefficients (PCC) between high-risk lncRNAs and mRNAs, respectively. lncRNA-mRNA interactions with PCC >0.8 were reserved. We merged all lncRNA-mRNA interactions into the CHLMCN. A random walk algorithm was performed to optimize high-risk lncRNAs. The significance of random walk was yielded from 3,000 times network shuffling.
lncRNA Sequencing for the Cardiac Hypertrophy Model In Vivo
To further validate the expression patterns of high-risk lncRNAs in our disease model, we also conducted two batches of ribosome-free and strand-specific lncRNA sequencing for sham and cardiac hypertrophy hearts with 3 replicates by the Illumina sequencing platform. Hisat2 and feature Counts software were used to call raw counts. DESeq2 was used for batch-effect removal and differential analysis.
Tissue-Specific Analysis
Three developmental stages of heart (embryo, postnatal, and 8-week adult) expression data were downloaded from the Encyclopedia of DNA Elements (ENCODE) database. All epigenetics data of the heart were downloaded from the UCSC genome browser, including H3k27ac, H3k4me3, p300, H3k27me3, and Pol2 signals. Reference genome was mm9. For the cardiomyocyte-specific analysis of lncRNA Gm15834 and miR-30b-3p, we downloaded a raw expression matrix from the GEO database with accession numbers GEO: GSE112316, GSE69924, and GSE58453 and performed a comparative analysis.
TAC Models
All experimental procedures conformed to the Guide for the Care and Use of Laboratory Animals published by the US NIH (publication, 8th edition, 2011). Our study conformed to regulations of the Ethic Committees of Harbin Medical University. 8-week-old male adult C57BL/6 mice were anesthetized by pentobarbital sodium (30 mg/kg intraperitoneally [i.p.]). The animals were then ventilated via tracheal intubation connected to a rodent ventilator (tidal volume 0.2 mL, respiratory rate 110 beats per minute [bpm]; Chengdu Taimeng, China). The left chest was opened at the second intercostals space, and the thymus glands were superiorly reflected. The transverse thoracic aorta between the innominate artery and left common carotid artery were dissected, and a 7-0 nylon suture was tied around the aorta against a 28-gauge needle. A sham group underwent thoracotomy and aortic dissection without constricting the aorta.
Cell Culture and Treatment
The cardiomyocyte-like HL-1 cell lines were cultured in recommended DMEM, which was supplemented with 10% fetal bovine serum (HyClone), 100 units/mL penicillin, and 100 mg/mL streptomycin (Beyotime). The cells were grown at 37°C in an atmosphere of 5% CO2 at a relative humidity of approximately 95%. Ang-II (#RAB0010; Sigma-Aldrich, St. Louis, MO, USA) 100 nM was treated to induce cardiomyocyte hypertrophy in vitro for 24 h. 3MA (#MB5063; Meilun Biotechnology, Dalian, China) 5 mM was used to block autophagy in vitro for 24 h. We also tested the expression of lncRNA Gm15834 and miR-30b-3p in neonatal mouse cardiomyocytes, which was cultured in 20% fetal bovine serum (HyClone), 100 units/mL penicillin, and 100 mg/mL streptomycin (Beyotime). Furthermore, AC-16, a type of human cardiomyocyte cell line, was used to investigate the translational potential of lncRNA Gm15834 in human disease. AC-16 cell lines were also supplemented with 10% fetal bovine serum, 100 units/mL penicillin, and 100 mg/mL streptomycin and were cultured in 5% CO2 at 37°C.
Western Blotting
Total protein samples were extracted from fresh tissues and cells based on the previous protocol.43 In brief, 50 μg protein samples was separated in 8%, 10%, or 15% SDS-PAGE gel and transferred onto a nitrocellulose membrane. After 5% nonfat milk blocking, the blots were incubated with primary antibodies, including BNP (1:200 dilution, sc-271185; Santa Cruz), Beclin-1 (1:2,000 dilution, #3495; Cell Signaling Technology), β-MHC (1:500 dilution, HPA001239; Sigma), ULK1 (1:2,000 dilution, #8054; Cell Signaling Technology), LC3-II (1:2,000 dilution, #2775; Cell Signaling Technology), p62 (1:2,000 dilution, #88588; Cell Signaling Technology), and β-actin (1:5,000 dilution, KC5A08; Kangcheng) at 4°C overnight. The bands were detected by an imaging system (LI-COR Biosciences) and quantified with ImageJ software by calculating intensity (area and density). β-actin was used as the internal control.
qRT-PCR
Total RNA from heart tissues and cells was extracted with Trizol (Invitrogen), according to the manufacturer’s protocol. cDNA was synthesized with RT (Takara, Japan). Relative gene expression was normalized to the Gapdh or U6 level in each sample. The primer sequences were as follows: miR-30b-3p RT primer: forward: 5′-GTCGTATCCAGTGCAGGGTCCGAGG-3′, reverse: 5′-TATTCGCACTGGATACGACAGCTGA-3′; miR-30b-3p: forward: 5′-CGGCGGTGTAAACATCCTACAC-3′, reverse: 5′-ATCCAGTGCAGGGTCCGAGG-3′; lncRNA Gm15834: forward: 5′-TGGAGGGCAAACTTAATCACAA-3′, reverse: 5′-AGGCTGGTAAGGGCATACTT-3′; U6: forward: 5′-GCTTCGGCAGCACATATACTAAAAT-3′, reverse: 5′-CGCTTCACGAATTTGCGTGTCAT-3′; Gapdh: forward: 5′-GTCGTGGAGTCTACTGGCGTCTTCA-3′, reverse: 5′-TCGTGGTTCACACCCATCACAAACA-3′; Myh7: forward: 5′-TACTTGCTACCCTCAGGTGGCT-3′, reverse: 5′-GCCTTGGATTCTCAAACGTGTC-3′; Nppb: forward: 5′-TATCTCAAGCTGCTTTGGGCA-3′, reverse: 5′-AACAACTTCAGTGCGTTACAGC-3′; homologous conserved region: forward: 5′-ACCTAGGACTAGTGACAACCT-3′, reverse: 5′-AGGATAATGGCTTACATGCACCA-3′.
siRNA and shRNA Treatment
siRNA and shRNA sequences that targeted to lncRNA Gm15834 were synthesized by GenePharma and ISBioTech, respectively. The siRNA sequence was forward: 5′-GCCUCUCUGUAGAAGAUAAGU-3′, reverse: 5′-UUAUCUUCUACAGAGAGGCAG-3′; NC sequence was forward: 5′-UUCUCCGAACGUGUCACGUTT-3′, reverse: 5′-ACGUGACACGUUCGGAGAATT-3′; shRNA sequence was 5′-CCAAATGCTCTGGAATCTA-3′; and shRNA NC sequence was 5′-TTCTCCGAACGTGTCACGT-3′. Plasmid or oligonucleotide vectors (1 μg/mL) and X-tremeGENE siRNA (Roche, Penzberg, Germany) were separately mixed with 300 μL of serum-free medium for 5 min. Then two mixtures were combined and incubated at room temperature for 18 min. The X-tremeGENE siRNA and plasmid mixture were added to the cells and incubated at 37°C for 24 h. Cells were divided into 4 groups: (1) control group; (2) Ang-II group; (3) Ang-II + si_Gm15834/Ang-II + sh_Gm15834 group: cells were treated with siRNA/shRNA for 24 h and then exposed to Ang-II for 24 h; and (4) Ang-II + NC group: cells were treated with siRNA NC/shRNA NC for 24 h and then exposed to Ang-II for 24 h.
Vector Construction and Transfection
The full length of lncRNA Gm15834 was synthesized by PCR and inserted into the pcDNA3.1 vector. The pcDNA3.1 empty vector was used as a NC. For transfection, HL-1 cell lines were washed with serum-free medium and then incubated in 5 mL of serum-free medium for 4–6 h. The transfection protocol was the same as vector transfection. Cells were divided into 4 groups: (1) control group; (2) Gm15834 group: cells were transfected with lncRNA Gm15834 plasmid for 24 h; (3) pcDNA group: cells were treated with pcDNA3.1 empty vector for 24 h; and (4) Ang-II group.
miRNA Mimics and Inhibitors Construction and Transfection
miR-30b-3p mimics and inhibitors were synthesized by GenePharma (Shanghai GenePharma, China). The miR-30b-3p mimics sequence was forward: 5′-CUGGGAUGUGGAUGUUUACGUC-3′, reverse: 5′-CGUAAACAUCCACAUCCCAGUU-3′; NC sequence was forward: 5′-UUCUCCGAACGUGUCACGUTT-3′, reverse: 5′-ACGUGACACGUUCGGAGAATT-3′; AMO-miR-30b-3p sequence was 5′-GACGUAAACAUCCACAUCCCAG-3′; and AMO NC sequence was 5′-GACGUAAACAUCCACAUCCCAG-3′. The transfection protocol was same as vector transfection. Cells were divided into 6 groups: (1) control group; (2) Ang-II group; (3) Ang-II + miR-30b group: cells were exposed to Ang-II for 24 h and then treated with miR-30b-3p mimics for 24 h; (4) Ang-II + miR-30b + AMO-30b group: cells were exposed to Ang-II for 24 h and then treated with miR-30b-3p mimics and AMO-miR-30b-3p for 24 h; (5) Ang-II + NC group: cells were exposed to Ang-II for 24 h and then treated with NC; (6) Ang-II + ANC group: cells were exposed to Ang-II for 24 h and then treated with AMO NC (ANC). In the Supplemental Information, we also conducted the knockdown experiments and divided cells into 4 groups: (1) control group; (2) AMO-30b group: control cells were treated with AMO-30b-3p for 24 h; (3) ANC group: cells were treated with AMO NC for 24 h; and (4) Ang-II group.
In Vivo AAV Infection
The second day after TAC surgery, the model mice were injected with AAV9-sh-lncRNA Gm15834 and AAV9-sh-NC via the tail vein (units: 1.2 × 1012 vg/mL, 100 μL; ISBioTech). Sham mice underwent the same protocol but were injected with 100 μL saline. Mice were divided into 4 groups: (1) sham group; (2) TAC group; (3) TAC + sh_Gm15834 group: TAC mice were injected with AAV9-sh-lncRNA Gm15834 for 3 weeks; and (4) TAC + NC group: TAC mice were injected with saline for 3 weeks. Additionally, in Figures 7 and S10, we also list the internal NC results of the TAC + sh_NC group: TAC mice were injected with AAV9-sh-NC for 3 weeks.
Immunofluorescence Staining
HL-1 cell lines cultured on glass coverslips were washed 3 times with cold PBS and then fixed with 4% paraformaldehyde for 15 min. The cell membrane was permeabilized with 0.4% Triton X-100 for 15 min. Glass coverslips were washed 3 times with PBS. The cells were incubated with α-smooth muscle actin (α-SMA) antibody (1:200 dilution, #19245; Cell Signaling Technology) at 4°C overnight. On the second day, cells were incubated with a fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit antibody for 1 h. 4′,6-diamidino-2-phenylindole (DAPI) staining (5 min at room temperature) was used to label nuclei. Immunofluorescence was analyzed under a fluorescence microscope (Leica AF6000, Germany).
Luciferase Reporter Assays
The ULK1 3′ UTR and lncRNA Gm15834 full-length sequences containing miR-30b-3p binding sites were amplified by PCR. The sequences were subcloned into the downstream luciferase gene (SacI and HindIII sites), respectively. For luciferase assay performed in HEK293T cells, 40 ng/well luciferase reporter vector, 10 pmol miR-30b-3p mimic, or mimic control was cotransfected into 48-well plates using Lipofectamine 2000 (Invitrogen). Cells were harvested at 24 h after transfection, and the luciferase activity was detected using the Dual Luciferase Reporter Assay kit (Promega), according to the manufacturer’s instructions. 100 μL luciferase substrate with 30 μL protein samples was analyzed in a luminometer. Firefly luciferase relative to Renilla luciferase was used as normalized luciferase activity.
AGO2-RNA Immunoprecipitation Assay
EZ-Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore, USA) was used based on the manufacturer’s protocol. 3 weeks TAC heart (hypertrophy heart) was harvested, grinded, and lysed in RIP lysis buffer. AGO2 and IgG antibodies were provided from Cell Signaling Technology (AGO2, #2897S) and the Magna RIP Kit. Magnetic beads were incubated with AGO2 or IgG antibodies for 30 min at room temperature. Cell lysates were immunoprecipitated with magnetic beads for 3 h at 4°C and overnight. Purified RNA was detected by qRT-PCR.
Echocardiographic Assessment
Sham and model mice were anesthetized and removed the chest hair. The Vevo 2100 Imaging System (Visual Sonics, Canada) was used to acquire ultrasound imaging. LVAWd, LVPWd, left ventricle EF (LVEF), and FS were measured and analyzed by Vevo 2100 software.
Electron Microscopy
Heart tissues were fixed with 2% glutaraldehyde containing 0.1 M sodium cacodylate. Samples were fixed using 1% osmium tetroxide, followed by dehydration with an increasing concentration gradient of ethanol and propylene oxide. Samples were then embedded, cut into 50 nm sections, and stained with 3% uranyl acetate and lead citrate. Images were acquired using electron microscope (Hitachi HT7650, Japan).
Autophagy Adenoviral Infection
HL-1 cell lines were plated in 6-well plates and allowed to infect adenovirus at 70% cell density. mRFP-GFP-LC3 adenoviral vectors were synthesized by Hanbio Tech (Shanghai, China). The principle of the assay was based on the different pH stability of GFPs and RFPs. The fluorescent signal of GFP could be quenched under the acidic condition (pH < 5) inside the lysosome, and the mRFP fluorescent signal had no significant change under the acidic condition. In green and red-merged images, autophagosomes were shown as yellow puncta (i.e., GFP+/RFP+), whereas autolysosomes were shown as red puncta (i.e., GFP−/RFP+). Autophagic flux was increased when both yellow and red puncta were increased in cells, whereas autophagic flux was blocked when only yellow puncta were increased without red puncta alteration or when both yellow and red puncta were decreased in cells.44 Adenoviral infection was performed according to the manufacturer’s instructions. After 24 h infection, LC3 puncta were measured with a fluorescence microscope (Leica AF6000, Germany).
Data Analysis
Expression data were represented as mean ± SEM. All data were analyzed statistically by one-way ANOVA implementing in R. If the ANOVA p value was significant, then paired t test was used to yield differences between experimental groups. Differences were considered as statistical significance with t test p <0.05.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (81872856); Key Project of Natural Science Foundation of Heilongjiang Province of China (ZD2017015); Postdoctoral Science Foundation of China (2019M661311); and Postdoctoral Science Foundation of Heilongjiang Province of China (LBH-Z19075).
Author Contributions
Conceived and Designed the Experiments, H.S.; Performed the Experiments, C.S., H.Q., Y.L., Y. Chen, P.S., S.Z., J.R., L.W., and Y. Cao; Analyzed the Data, C.S., H.Q., and Y.L.; Wrote the Paper, C.S. and H.S.
Conflicts of Interest
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.10.024.
Supplemental Information
References
- 1.Frey N., Olson E.N. Cardiac hypertrophy: the good, the bad, and the ugly. Annu. Rev. Physiol. 2003;65:45–79. doi: 10.1146/annurev.physiol.65.092101.142243. [DOI] [PubMed] [Google Scholar]
- 2.Heineke J., Molkentin J.D. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 3.Sayed D., Hong C., Chen I.Y., Lypowy J., Abdellatif M. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ. Res. 2007;100:416–424. doi: 10.1161/01.RES.0000257913.42552.23. [DOI] [PubMed] [Google Scholar]
- 4.Carè A., Catalucci D., Felicetti F., Bonci D., Addario A., Gallo P., Bang M.L., Segnalini P., Gu Y., Dalton N.D. MicroRNA-133 controls cardiac hypertrophy. Nat. Med. 2007;13:613–618. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- 5.Lin Z., Murtaza I., Wang K., Jiao J., Gao J., Li P.F. miR-23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proc. Natl. Acad. Sci. USA. 2009;106:12103–12108. doi: 10.1073/pnas.0811371106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mercer T.R., Dinger M.E., Mattick J.S. Long non-coding RNAs: insights into functions. Nat. Rev. Genet. 2009;10:155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- 7.Wang K.C., Chang H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell. 2011;43:904–914. doi: 10.1016/j.molcel.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hung T., Chang H.Y. Long noncoding RNA in genome regulation: prospects and mechanisms. RNA Biol. 2010;7:582–585. doi: 10.4161/rna.7.5.13216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu X.H., Yuan Y.X., Rao S.L., Wang P. LncRNA MIAT enhances cardiac hypertrophy partly through sponging miR-150. Eur. Rev. Med. Pharmacol. Sci. 2016;20:3653–3660. [PubMed] [Google Scholar]
- 10.Liu L., An X., Li Z., Song Y., Li L., Zuo S., Liu N., Yang G., Wang H., Cheng X. The H19 long noncoding RNA is a novel negative regulator of cardiomyocyte hypertrophy. Cardiovasc. Res. 2016;111:56–65. doi: 10.1093/cvr/cvw078. [DOI] [PubMed] [Google Scholar]
- 11.Wang K., Liu F., Zhou L.Y., Long B., Yuan S.M., Wang Y., Liu C.Y., Sun T., Zhang X.J., Li P.F. The long noncoding RNA CHRF regulates cardiac hypertrophy by targeting miR-489. Circ. Res. 2014;114:1377–1388. doi: 10.1161/CIRCRESAHA.114.302476. [DOI] [PubMed] [Google Scholar]
- 12.Chen G., Li H., Li X., Li B., Zhong L., Huang S., Zheng H., Li M., Jin G., Liao W. Loss of long non-coding RNA CRRL promotes cardiomyocyte regeneration and improves cardiac repair by functioning as a competing endogenous RNA. J. Mol. Cell. Cardiol. 2018;122:152–164. doi: 10.1016/j.yjmcc.2018.08.013. [DOI] [PubMed] [Google Scholar]
- 13.Proud C.G. Ras, PI3-kinase and mTOR signaling in cardiac hypertrophy. Cardiovasc. Res. 2004;63:403–413. doi: 10.1016/j.cardiores.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 14.Li Y., Chen C., Yao F., Su Q., Liu D., Xue R., Dai G., Fang R., Zeng J., Chen Y. AMPK inhibits cardiac hypertrophy by promoting autophagy via mTORC1. Arch. Biochem. Biophys. 2014;558:79–86. doi: 10.1016/j.abb.2014.06.023. [DOI] [PubMed] [Google Scholar]
- 15.Li Z., Song Y., Liu L., Hou N., An X., Zhan D., Li Y., Zhou L., Li P., Yu L. miR-199a impairs autophagy and induces cardiac hypertrophy through mTOR activation. Cell Death Differ. 2017;24:1205–1213. doi: 10.1038/cdd.2015.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levine B., Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 18.Cao D.J., Wang Z.V., Battiprolu P.K., Nan J., Morales C.R., Yongli K., Rothermel B.A., Gillette T.G., Hill J.A. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc. Natl. Acad. Sci. USA. 2011;108:4123–4128. doi: 10.1073/pnas.1015081108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pei H., Wang W., Zhao D., Su H., Su G., Zhao Z. G Protein-Coupled Estrogen Receptor 1 Inhibits Angiotensin II-Induced Cardiomyocyte Hypertrophy via the Regulation of PI3K-Akt-mTOR Signalling and Autophagy. Int. J. Biol. Sci. 2019;15:81–92. doi: 10.7150/ijbs.28304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie Y.P., Lai S., Lin Q.Y., Xie X., Liao J.W., Wang H.X., Tian C., Li H.H. CDC20 regulates cardiac hypertrophy via targeting LC3-dependent autophagy. Theranostics. 2018;8:5995–6007. doi: 10.7150/thno.27706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qi H., Ren J., e M., Zhang Q., Cao Y., Ba L., Song C., Shi P., Fu B., Sun H. MiR-103 inhibiting cardiac hypertrophy through inactivation of myocardial cell autophagy via targeting TRPV3 channel in rat hearts. J. Cell. Mol. Med. 2019;23:1926–1939. doi: 10.1111/jcmm.14095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Viereck J., Kumarswamy R., Foinquinos A., Xiao K., Avramopoulos P., Kunz M., Dittrich M., Maetzig T., Zimmer K., Remke J. Long noncoding RNA Chast promotes cardiac remodeling. Sci. Transl. Med. 2016;8:326ra22. doi: 10.1126/scitranslmed.aaf1475. [DOI] [PubMed] [Google Scholar]
- 23.Song C., Zhang J., Liu Y., Pan H., Qi H.P., Cao Y.G., Zhao J.M., Li S., Guo J., Sun H.L., Li C.Q. Construction and analysis of cardiac hypertrophy-associated lncRNA-mRNA network based on competitive endogenous RNA reveal functional lncRNAs in cardiac hypertrophy. Oncotarget. 2016;7:10827–10840. doi: 10.18632/oncotarget.7312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xuan P., Han K., Guo Y., Li J., Li X., Zhong Y., Zhang Z., Ding J. Prediction of potential disease-associated microRNAs based on random walk. Bioinformatics. 2015;31:1805–1815. doi: 10.1093/bioinformatics/btv039. [DOI] [PubMed] [Google Scholar]
- 25.Zeller R., Bloch K.D., Williams B.S., Arceci R.J., Seidman C.E. Localized expression of the atrial natriuretic factor gene during cardiac embryogenesis. Genes Dev. 1987;1:693–698. doi: 10.1101/gad.1.7.693. [DOI] [PubMed] [Google Scholar]
- 26.Brasier A.R., Jamaluddin M., Han Y., Patterson C., Runge M.S. Angiotensin II induces gene transcription through cell-type-dependent effects on the nuclear factor-kappaB (NF-kappaB) transcription factor. Mol. Cell. Biochem. 2000;212:155–169. [PubMed] [Google Scholar]
- 27.Young D., Popovic Z.B., Jones W.K., Gupta S. Blockade of NF-kappaB using IkappaB alpha dominant-negative mice ameliorates cardiac hypertrophy in myotrophin-overexpressed transgenic mice. J. Mol. Biol. 2008;381:559–568. doi: 10.1016/j.jmb.2008.05.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li L., Xu J., He L., Peng L., Zhong Q., Chen L., Jiang Z. The role of autophagy in cardiac hypertrophy. Acta Biochim. Biophys. Sin. (Shanghai) 2016;48:491–500. doi: 10.1093/abbs/gmw025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pan W., Zhong Y., Cheng C., Liu B., Wang L., Li A., Xiong L., Liu S. MiR-30-regulated autophagy mediates angiotensin II-induced myocardial hypertrophy. PLoS ONE. 2013;8:e53950. doi: 10.1371/journal.pone.0053950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raut S.K., Singh G.B., Rastogi B., Saikia U.N., Mittal A., Dogra N., Singh S., Prasad R., Khullar M. miR-30c and miR-181a synergistically modulate p53-p21 pathway in diabetes induced cardiac hypertrophy. Mol. Cell. Biochem. 2016;417:191–203. doi: 10.1007/s11010-016-2729-7. [DOI] [PubMed] [Google Scholar]
- 31.Lee J.W., Park S., Takahashi Y., Wang H.G. The association of AMPK with ULK1 regulates autophagy. PLoS ONE. 2010;5:e15394. doi: 10.1371/journal.pone.0015394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Russell R.C., Tian Y., Yuan H., Park H.W., Chang Y.Y., Kim J., Kim H., Neufeld T.P., Dillin A., Guan K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013;15:741–750. doi: 10.1038/ncb2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shintani T., Klionsky D.J. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu H., Tannous P., Johnstone J.L., Kong Y., Shelton J.M., Richardson J.A., Le V., Levine B., Rothermel B.A., Hill J.A. Cardiac autophagy is a maladaptive response to hemodynamic stress. J. Clin. Invest. 2007;117:1782–1793. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakai A., Yamaguchi O., Takeda T., Higuchi Y., Hikoso S., Taniike M., Omiya S., Mizote I., Matsumura Y., Asahi M. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 36.Xu X., Hua Y., Nair S., Bucala R., Ren J. Macrophage migration inhibitory factor deletion exacerbates pressure overload-induced cardiac hypertrophy through mitigating autophagy. Hypertension. 2014;63:490–499. doi: 10.1161/HYPERTENSIONAHA.113.02219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McMullen J.R., Sherwood M.C., Tarnavski O., Zhang L., Dorfman A.L., Shioi T., Izumo S. Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation. 2004;109:3050–3055. doi: 10.1161/01.CIR.0000130641.08705.45. [DOI] [PubMed] [Google Scholar]
- 38.Hariharan N., Ikeda Y., Hong C., Alcendor R.R., Usui S., Gao S., Maejima Y., Sadoshima J. Autophagy plays an essential role in mediating regression of hypertrophy during unloading of the heart. PLoS ONE. 2013;8:e51632. doi: 10.1371/journal.pone.0051632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Halapas A., Armakolas A., Koutsilieris M. Autophagy: a target for therapeutic interventions in myocardial pathophysiology. Expert Opin. Ther. Targets. 2008;12:1509–1522. doi: 10.1517/14728220802555554. [DOI] [PubMed] [Google Scholar]
- 40.Chen C., Yang S., Li H., Yin Z., Fan J., Zhao Y., Gong W., Yan M., Wang D.W. Mir30c Is Involved in Diabetic Cardiomyopathy through Regulation of Cardiac Autophagy via BECN1. Mol. Ther. Nucleic Acids. 2017;7:127–139. doi: 10.1016/j.omtn.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lerchenmüller C., Rabolli C.P., Yeri A.S., Kitchen R., Salvador A.M., Liu L.X., Ziegler O., Danielson K., Platt C., Shah R. CITED4 Protects Against Adverse Remodeling in Response to Physiological and Pathological Stress. Circ. Res. 2020;127:631–646. doi: 10.1161/CIRCRESAHA.119.315881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li X., Du N., Zhang Q., Li J., Chen X., Liu X., Hu Y., Qin W., Shen N., Xu C. MicroRNA-30d regulates cardiomyocyte pyroptosis by directly targeting foxo3a in diabetic cardiomyopathy. Cell Death Dis. 2014;5:e1479. doi: 10.1038/cddis.2014.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qi H., Liu Y., Li S., Chen Y., Li L., Cao Y., e M., Shi P., Song C., Li B., Sun H. Activation of AMPK Attenuated Cardiac Fibrosis by Inhibiting CDK2 via p21/p27 and miR-29 Family Pathways in Rats. Mol. Ther. Nucleic Acids. 2017;8:277–290. doi: 10.1016/j.omtn.2017.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu T., Guo F., Yu Y., Sun T., Ma D., Han J., Qian Y., Kryczek I., Sun D., Nagarsheth N. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell. 2017;170:548–563.e16. doi: 10.1016/j.cell.2017.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.