

Abstract
Recombinant adeno-associated virus (rAAV) vectors have the unique ability to promote targeted integration of transgenes via homologous recombination at specified genomic sites, reaching frequencies of 0.1%–1%. We studied genomic parameters that influence targeting efficiencies on a large scale. To do this, we generated more than 1,000 engineered, doxycycline-inducible target sites in the human HAP1 cell line and infected this polyclonal population with a library of AAV-DJ targeting vectors, with each carrying a unique barcode. The heterogeneity of barcode integration at each target site provided an assessment of targeting efficiency at that locus. We compared targeting efficiency with and without target site transcription for identical chromosomal positions. Targeting efficiency was enhanced by target site transcription, while chromatin accessibility was associated with an increased likelihood of targeting. ChromHMM chromatin states characterizing transcription and enhancers in wild-type K562 cells were also associated with increased AAV-HR efficiency with and without target site transcription, respectively. Furthermore, the amenability of a site to targeting was influenced by the endogenous transcriptional level of intersecting genes. These results define important parameters that may not only assist in designing optimal targeting vectors for genome editing, but also provide new insights into the mechanism of AAV-mediated homologous recombination.
Keywords: rAAV, homologous recombination, chromatin, genomic states affecting HR
Graphical Abstract
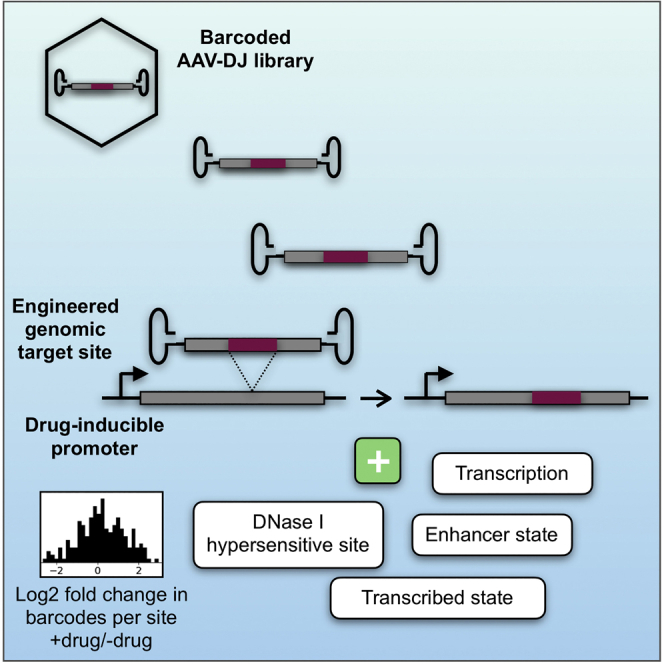
AAV vectors promote targeted integration of transgenes via homologous recombination at frequencies of 0.1%–1%. Spector et al. studied genomic parameters that influence targeting efficiencies in a large scale by targeting barcoded AAV-DJ vectors to engineered genomic sites, identifying transcription and enhancers as features of interest in targeting vector strategies.
Introduction
Site-specific gene targeting is a burgeoning field in gene therapy and genome engineering, providing the ability to readily generate models of gene disruption and gene introduction. While most recombinant adeno-associated virus (rAAV) transduction events are episomal, we have previously used the vector’s ability to induce homologous recombination (HR) for targeted integration of transgenes into the host genome downstream of an endogenous promoter. Targeted integration is achieved in the absence of a site-specific break induced by a nuclease such as transcription activator-like effector nucleases (TALENs) or CRISPR-Cas9 using expression-incompetent vectors (AAV-HR).1 Not only do nuclease-free targeting systems reduce the potential for toxicity associated with induced DNA breaks,2, 3, 4 but a vector lacking a promoter reduces the chance for oncogene activation from off-target vector integration.5,6 This simple system requires chromosomal homology arms as short as 750 bp7 flanking a coding sequence, and it easily meets the limited 4.7-kb packaging capacity of rAAV for many coding sequences. AAV-HR has previously been used both for targeted integration of whole transgenes as well as targeted correction of insertions and deletions, achieving a targeting rate up to 1% of cells.1,7, 8, 9, 10, 11, 12, 13, 14, 15, 16
Exactly what processes govern AAV-HR are still under investigation. Gene targeting with rAAV vectors occurs at rates several orders of magnitude higher than with plasmid DNA, and there is evidence to suggest that the AAV inverted terminal repeats (ITRs) are recombinogenic.17, 18, 19, 20 Studies to elucidate host factors that mediate AAV-HR demonstrate that it occurs via the HR pathway. For example, there is an improvement in AAV-HR after knocking down the non-homologous end joining (NHEJ) protein KU70,16,21 which competes with HR proteins for repair.20, 21, 22 Additionally, knocking down the HR proteins RAD54L, RAD54B, and XRCC3 reduces or abolishes stable integration via AAV-HR,18 evidence that AAV-HR requires the Rad51/Rad54 pathway of HR.
Transcription has long been known to increase HR23 and gene targeting24 in mammalian cells.
Studies in S. cerevisiae have made the connection between transcription and recombination, called “transcription-associated recombination” (TAR)25,26 and enumerated among its causes the formation of co-transcriptional RNA:DNA hybrids, or R loops,25,27 that can both stall movement of replication forks and expose single-stranded DNA to damage and recombination proteins. Consistent with these findings, we recently showed that knocking down the FANCM gene, which helps to maintain genome stability by reconciling transcription-replication conflicts,28,29 increased rates of AAV-HR up to 9-fold.30 A previous study of AAV-HR genome wide at engineered target sites demonstrated a preference for targeting at sites of convergent transcription and replication,31 where stalling and fork collapse can stimulate TAR.26,32
Recent literature has suggested that a “histone code” may be responsible for determining which repair proteins (HR or NHEJ) are recruited at DNA damage sites. H3K36me3, a mark of transcription elongation, is involved in recruiting CtIP and RAD51 to transcriptionally active loci,33 while histone H4 acetylation in cis to H4K20me1/2 diminishes recruitment of 53BP1, a protein important for promoting NHEJ.34 HR is also the preferred repair method within heterochromatin during the G2 phase.35, 36, 37 H3K6me3 is a feature of facultative and constitutive heterochromatin,38,39 not precluding a similar mechanism of recruitment. However, HR in heterochromatin is slower and requires unique factors compared to euchromatin,40,41 presumably due to the degree of chromatin compaction.36,41 H3K36me3 and H4K20me1 marks are both features of a transcribed chromatin state, while other combinations of marks such as H3K4me1, H3K27ac, and H3K9ac, as well as DNase I hypersensitivity, typically mark more accessible chromatin such as regulatory elements.42 Homologous recombination is also a characteristic feature of transposable elements such as long interspersed nuclear elements (LINEs) and short interspersed nuclear elements (SINEs).43 Dispersed repetitive elements can undergo allelic and non-allelic HR, resulting in deletions or duplications that contribute to genetic instability.43, 44, 45
Previous studies of AAV-HR have asked whether it is also subject to position effects. The first studies investigating the role of position effects on AAV-HR from nearby or intersecting chromosomal features did not find a significant correlation between transcription and targeting rate.10,12 A subsequent large-scale, genome-wide investigation reported an enrichment for targeting transcription units, although it could not solely be attributed to transcription due to the fact that the engineered target site contained its own promoter and expressed at a level sufficient for selective drug resistance.12,31 What these systems lacked was the ability to understand the impact of position effects on AAV-HR while controlling for transcription at the target site itself. In this study, we describe a high-throughput system to map and quantify precision AAV-HR genome wide that allows for decomposition of the complex role of transcription on AAV-HR by exploiting an engineered, drug-inducible locus and rAAV vectors that are designed to integrate unique barcodes. By associating multiple barcodes with a single locus as a measure of the AAV-HR efficiency at that site and controlling for sequence variation at the target site, our system adds a novel dimension that could not be tested by the design of earlier studies.
Results
In order to investigate genomic AAV-HR events, we used a dual Tet-On lentiviral vector system to introduce target sites into the near-haploid human HAP146 cell line (Figure 1A). HAP1 cells infected with lentivirus particles were selected as a blasticidin-resistant, EGFP-positive polyclonal population (Figure 1B; Figures S1 and S4). The experiment was divided into two treatment groups, with half of the population exposed to doxycycline upon transduction with the rAAV-DJ barcoded library. Doxycycline, a tetracycline-family antibiotic, has been shown to affect mitochondrial biogenesis and downregulate DNA-dependent protein kinase (DNA-PK) at concentrations of 25 μM or higher.47 Inhibition of DNA-PK can lead to upregulation of the HR repair pathway.48 However, at the low concentrations of doxycycline used in inducible expression systems, typically 1 μg/mL (1.95 μM) or lower, there is evidence to suggest that doxycycline does not affect DNA-PK expression (Table S149), and our study used a doxycycline concentration 10-fold lower. We chose to use the AAV-DJ capsid for packaging the pAAV-Luc-P2A-mScarlet-GFP construct given its high efficiency in transducing cells in vitro.50 The population was subsequently enriched for correctly targeted cells (mScarlet+/EGFP−) over two rounds of fluorescence-activated cell sorting (FACS) (Figures S2–S4).
Figure 1.

Vector Design and Experimental Scheme
(A) The rAAV-DJ vector (rAAV) encodes an mScarlet coding sequence followed by a stop codon, barcode of 12 degenerate nucleotides (BC), and an additional 38 bp to introduce a frameshift after HR. These are flanked by 1.6-kb homology arms comprising a (5′) partial firefly luciferase (F Luc) coding sequence followed by 2A-peptide sequence and (3′) EGFP coding sequence and partial mouse albumin 3′ UTR (mAlb 3′ UTR). The target site (provirus), generated by integration of a lentivirus vector, encodes firefly luciferase and EGFP coding sequences linked by a 2A-peptide and followed by the mouse albumin 3′ UTR, under the control of a TRE3Gs tetracycline-responsive promoter (pTRE). It also encodes a Tet-On 3G transactivator (Tet3G)-IRES-blasticidin resistance gene (BlastR) cassette under control of the human ubiquitin C (hUbC) promoter. Stop codons are excluded from coding sequences that immediately precede 2A-peptides, and start codons are excluded from coding sequences that immediately follow 2A-peptides. After integration by HR, firefly luciferase and mScarlet+barcode are fused at the DNA and RNA levels, but two separate proteins are produced as the result of ribosomal skipping. The stop codon and frameshift introduced by HR abolish EGFP expression. (B) A polyclonal population of >1,000 clones was generated by infecting wild-type HAP1 cells with the lentiviral vector at an MOI of <0.1. Clones were selected by blasticidin resistance and FACS. The polyclonal population was plated in two biological replicates for each experimental arm (+doxycycline and −doxycycline), then transduced with the barcoded rAAV-DJ library under the indicated doxycycline exposure. After several weeks, both experimental arms were exposed to doxycycline for sorting targeted cells (mScarlet+/EGFP−). Similarly, DNA and RNA were harvested after doxycycline exposure. (C) (1) Lentiviral provirus integration sites were sequenced from the 3′ LTR by LM-PCR, including a locked nucleic acid (blocking LNA) to inhibit PCR amplification into the provirus sequence.51 (2) For mapping barcodes, DNA was digested with the complementary overhang restriction enzymes AseI (cleaving just downstream of the barcode) and NdeI and then self-circularized. Fragments from circularized DNA were PCR amplified across the barcode and ligated adjacent genomic DNA, as well as into genomic DNA from the 5′ LTR. Genomic loci that overlapped in LM-PCR and iPCR were considered “targeted sites.” (3) Number of unique barcodes mapped to each genomic locus is referred to as “barcode heterogeneity.” Barcodes were amplified from cDNA, and counts were normalized to corresponding barcode counts from genomic DNA to measure targeted site expression.
In order to map lentiviral provirus sites in the blasticidin-selected/EGFP-sorted polyclonal population, genomic DNA was subjected to ligation-mediated PCR followed by next-generation sequencing (NGS)51 (Figure 1C). 1,474 unique sites were recovered in passaging phase samples (Table S2), with approximately 80%–90% of those sites intersecting between any two biological replicates, regardless of doxycycline treatment. There was a strong correlation between the abundance of a given site when compared between all pairwise combinations of biological replicates (R2 = 0.85, Figure S5A), giving confidence that clones were consistently represented between the different samples. Furthermore, the pairwise abundance of clones recovered from the polyclonal population just prior to rAAV transduction and during the passaging phase was well correlated (+doxycycline sites R2 = 0.67, −doxycycline sites R2 = 0.72, Figures S5B and S5C), suggesting that clones were minimally influenced by uneven clonal expansion.
Lentivirus/HIV-1 preferentially integrates within transcription units.52,53 Schröder et al.54 previously identified an enrichment for HIV-1 integration sites at transcription units as well as at Alu elements, which are known to be enriched in gene-rich regions,52,55 as well as an underrepresentation at long terminal repeat (LTR) and mammalian-wide interspersed repeat (MIR) elements.54 Consistent with this paradigm, we identified a significant enrichment for our provirus sites relative to a set of random sites at GENCODE genes (p < 0.001) and at Alu elements (p < 0.001), and an underrepresentation at LTR (p < 0.001) and MIR elements (p < 0.05) (Figure S6A). Provirus sites were also underrepresented at LINE-1 elements (p < 0.001), which may be consistent with their enrichment in gene-poor regions.56 Furthermore, using a permutation test, we found that a previously identified set of recurrent integration genes (RIGs) that are known lentivirus/HIV-1 hotspots55 were enriched at our provirus sites (Figure S6B).
In order to enumerate AAV-HR events at each provirus site, we quantified the number of unique barcodes integrated at each site, where each unique barcode represents a distinct rAAV vector that targeted the site. To estimate the influence of transcription on AAV-HR while controlling for position effects, we compared barcode heterogeneity from the populations of cells transduced in the presence or absence of doxycycline. The details of this analysis are described below.
In order to map integrated barcodes to each provirus site, genomic DNA from the enriched population of cells was harvested and subjected to inverse PCR (iPCR) followed by NGS57 (Figure 1C). To quantify barcode expression as a proxy for the transcriptional activity of the provirus site promoter, we prepared DNA and cDNA derived from RNA transcripts from the enriched population of cells for NGS of just the barcodes57 (Figure 1C). We refer to the number of unique barcodes mapped to each provirus site (Table S2) as the barcode heterogeneity, which is a measure of the AAV-HR efficiency at a given site.
In order to investigate the effect of transcription alone on AAV-HR, we controlled for position effects by comparing barcode heterogeneity between paired (shared) sites from different doxycycline/non-doxycycline treatment groups. Notably, barcode heterogeneity was significantly greater at paired sites in doxycycline- versus non-doxycycline-treated samples (p = 0.0021, Figure 2A), which is also shown by the right shift of the frequency histogram of log2 fold change between doxycycline- and non-doxycycline-treated samples in Figure 2C, left panel.
Figure 2.

Effect of Target Site Transcriptional Induction on AAV-HR Efficiency
Following FACS enrichment of cells with AAV-HR events, the number of unique barcodes mapped to each site (barcode heterogeneity) and average expression were quantified at targeted provirus sites, using the set of barcodes recovered in both iPCR and DNA barcode sequencing samples (see Materials and Methods). Here, measurements are compared only for those provirus sites targeted in both doxycycline- and non-doxycycline-treated samples. (A and B) Barcode heterogeneity (A) and expression (B) at targeted sites, measured at all pairwise targeted sites between treatment groups (concatenated biological replicates). Spearman’s rank correlation coefficient ρ is shown. p values were determined by a one-sided Wilcoxon signed-rank test (H0 = +doxycycline is not greater than −doxycycline) for barcode heterogeneity and a two-sided Wilcoxon signed-rank test for expression (n = 194). (C) Frequency histogram of log2 fold change in barcode heterogeneity and site expression for each pair of sites plotted in (A) and (B), respectively. For expression, RNA was extracted after administering doxycycline to both groups regardless of doxycycline exposure at the time of rAAV transduction.
There was no significant difference in the distribution of targeted site expression between paired sites from different doxycycline treatment groups when the RNA was extracted after administering doxycycline to both groups (p = 0.12, Figures 2B and 2C, right panel), confirming that we get similar expression from a site once it is targeted, regardless of the condition (+doxycycline or −doxycycline) in which it was transduced with the rAAV library.
Notably, the likelihood of AAV-HR with doxycycline exposure was 1.24-fold that without doxycycline exposure (relative risk ratio, 95% confidence interval 1.002–1.533, Figures 3A and 3B), indicating that a broader range of sites was amenable to targeting when target sites were transcriptionally induced. Thus, there are two main effects of target site transcription: increased AAV-HR efficiency as well as an increase in the number of unique sites targeted. We reasoned that provirus sites targeted in only one treatment group while still being present in the polyclonal population transduced under both conditions would represent sites at which targeting was truly dependent on target site transcription. Among these sites, the likelihood of AAV-HR with doxycycline exposure was 2.54-fold that without doxycycline exposure (relative risk ratio, 95% confidence interval 1.707–3.783, Figure 3A), confirming our observation that target site transcription makes a broader range of sites amenable to AAV-HR.
Figure 3.

Effect of Target Site Transcriptional Induction on Number of Unique Sites Targeted
(A) Relative risk ratio of a site having at least one integrated barcode in a doxycycline-treated sample compared to a non-doxycycline-treated sample. Sites from biological duplicates were assembled into a 2 × 2 contingency table for which the exposure is +doxycycline/−doxycycline and the outcome is targeted/not targeted. 95% confidence intervals are shown. We consider the relative risk ratio statistically significant when the 95% confidence interval does not overlap 1, shown by white circles. All sites, all targeted sites (considered out of n = 3,901 provirus sites across all samples); Group-specific, sites targeted in one treatment group and not the other. For group-specific sites, the set of provirus sites was first filtered to sites present in the polyclonal population transduced in both treatment groups but targeted exclusively in one treatment group or the other (n = 277). There were no zero values in the tables. (B) Ideogram of all provirus sites considered for targeting (1,246 sites; black bars, overlay), showing targeted sites below each chromosome in +doxycycline samples (gray bars, first row) and targeted sites in −doxycycline samples (black bars, second row), generated using the NCBI Genome Decoration Page (https://www.ncbi.nlm.nih.gov/genome/tools/gdp/).
To identify other factors such as nearby genes, repeat elements, or marks of chromatin accessibility that associate with AAV-HR when the polyclonal population is transduced in the presence or absence of doxycycline, we sought to identify genomic features correlated with either the number of unique barcodes integrated at each site or the amenability of a site to targeting. We hypothesized that proximal chromosomal features may be responsible for making some provirus sites more or less amenable to targeting in the context of target site transcription. First, we focused on features similar to those examined by other groups studying AAV-HR: genes; LTR, Alu, MIR, and LINE-1 elements; low-complexity and simple repeats; and DNase I hypersensitive sites (Figure S7). We calculated the relative risk ratio that a provirus site was targeted, given that it intersected the given feature (Figure 4A). Importantly, note that, given our data, we were unable to determine whether the provirus site itself disrupted the activity of any of these features.
Figure 4.

Association of Targeted Sites with Chromosomal Features
(A) Relative risk ratio of a site having at least one integrated barcode, given that the site intersects the feature indicated above the plot. Sites were assembled into a 2 × 2 contingency table for which the exposure is intersection/no intersection and the outcome is targeted/not targeted. There were no 0 values in the tables. +doxycycline, n = 2,013; −doxycycline, n = 1,888. 95% confidence intervals are shown. We consider the relative risk ratio statistically significant when the 95% confidence interval does not overlap 1, shown by white circles. DNase I hypersensitive sites were obtained by intersecting provirus sites with DNase I-seq called peaks (see Materials and Methods). Low-complexity repeats were excluded due to large confidence intervals. (B and C) Relative risk ratio of a targeted site intersecting the feature indicated, given that it was targeted in a doxycycline-treated sample, for (B) all sites (+doxycycline, n = 181; −doxycycline, n = 137) or (C) sites targeted in only one treatment group (+doxycycline, n = 64; −doxycycline, n = 25). For (C), provirus sites were initially filtered to sites present in both treatment groups and targeted exclusively in one treatment group, as in Figure 3 (Group-specific). For (B) and (C), only targeted sites were assembled into a 2 × 2 contingency table for which the exposure is +doxycycline/−doxycycline and the outcome is intersection/no intersection. 0.5 was added to all cells for tables with a 0 value, as was the case for DNase I hypersensitive sites in (C). DHS, DNase I hypersensitive site.
GENCODE genes were associated with a non-significant increase in the likelihood of AAV-HR (+doxycycline relative risk ratio = 1.30 and 95% confidence interval 0.80–2.10, −doxycycline relative risk ratio = 1.50 and 95% confidence interval 0.84–2.67, Figure 4A). However, DNase I hypersensitive sites were associated with a significant increase in the likelihood of AAV-HR in both treatment groups (+doxycycline relative risk ratio = 2.82 and 95% confidence interval 1.19–6.67, −doxycycline relative risk ratio = 4.17 and 95% confidence interval 1.96–8.87). DNase I hypersensitive sites are a widely recognized surrogate marker of accessible chromatin that are useful for mapping regulatory elements such as promoters and enhancers.58 We might expect that, in non-doxycycline-treated samples, the accessible chromatin state may have a greater effect on AAV-HR in the absence of the stimulatory effect of target site transcription. Consistent with studies of lentivirus integration,53 it is noteworthy that only a few percent of our provirus sites intersect DNase I hypersensitive sites, while more than 80% of provirus sites intersect genes (Figure S6A). Interestingly, in non-doxycycline-treated samples, Alu elements were associated with a decreased likelihood of AAV-HR (relative risk ratio = 0.50, 95% confidence interval 0.30–0.83), and LINE-1 elements were associated with an increased likelihood of AAV-HR (relative risk ratio = 1.64, 95% confidence interval 1.11–2.42) (Figure 4A). However, there was no significant difference in the likelihood of a targeted site intersecting one of these features with doxycycline exposure than without (Figure 4B), indicating no significant difference between the treatment groups. The reason for significance in the one treatment group is unclear. However, when filtering to sites that were targeted exclusively in one treatment group or the other, the likelihood of intersecting a LINE-1 element with doxycycline exposure was only 0.22-fold that without doxycycline exposure (relative risk ratio, 95% confidence interval 0.07–0.70, Figure 4C). It remains to be further explored how target site transcription influences the relative preference of targeting provirus sites that intersect LINE-1 elements. Our results also indicated that the relative risk of targeting in doxycycline- and non-doxycycline-treated samples was not influenced by whether a provirus was integrated into an LTR, MIR element, or simple repeat (Figure 4A).
To further examine the relationship of targeted provirus sites with nearby chromosomal features, we checked whether there were spatial correlations between targeted sites and these features using the distribution of relative distances59 rather than the intersections between them (Figure S8). This analysis showed that targeted provirus sites tend to be found closer to genes and farther from Alu elements, regardless of doxycycline treatment, a pattern that seems to carry over from the findings for direct intersections (Figure 4A). Importantly, no significant difference in spatial correlation between the two treatment groups was observed for any of the tested features (paired sample Kolmogorov-Smirnov test), suggesting that any differences we saw were independent of target site transcription.
Although we did not identify an increased likelihood for AAV-HR at provirus sites intersecting genes, we reasoned that we might see differences in targeting preference if we stratified the genes by expression level, as was previously observed.31 We took advantage of publicly available, transcriptome-wide RNA sequencing in wild-type HAP1 cells.60 We found that targeted sites in both treatment groups were enriched at genes with higher expression levels and more scarce at genes with lower expression levels (Figure 5, left, +doxycycline p = 2.6e−02, −doxycycline p = 3.6e−04), and there was no significant difference between treatment groups (Cochran-Armitage trend test, p = 0.19). Additionally, for non-doxycycline-treated samples, barcode heterogeneity was consistently greater at sites in the “high” expression bin compared to the “low” or “medium” bins (one-sided Mann-Whitney U test, p = 0.02 and p = 0.01, respectively; for +doxycycline p = 0.06 and p = 0.004), suggesting that AAV-HR target site preference is influenced by the expression level of intersecting genes and, in the absence of target site transcription, AAV-HR efficiency is as well.
Figure 5.

Association of Targeted Sites with GENCODE Genes by Expression Level
Genes intersecting provirus sites were split into equal-sized bins after ranking mean FPKM values for these genes from lowest to highest. Left y axis (bar chart) indicates the number of targeted sites in each bin. Right y axis (boxplot) indicates the barcode heterogeneity for targeted sites in each bin. Boxplot whiskers extend the first and third quartiles by 1.5 × interquartile range (IQR) with outlying data points shown as circles. All genes intersecting provirus sites (targeted gene counts +doxycycline 46, 47, 71 and −doxycycline 27, 36, 62), genes transcribed in the opposite direction relative to the doxycycline-inducible promoter (targeted gene counts +doxycycline 31, 23, 32 and −doxycycline 21, 15, 28), and genes transcribed in the same direction relative to the doxycycline-inducible promoter (targeted gene counts +doxycycline 21, 26, 40 and −doxycycline 12, 23, 35) are shown. p values shown for binned targeted sites were determined by a one-way chi-square test against the uniform distribution. A Cochran-Armitage trend test was used to compare between treatment groups but no significant differences were detected. A chi-square test of independence was used to compare gene counts at genes transcribed in the opposite versus same direction within each treatment group but no significant differences were detected. Median FPKM and interquartile range of transcripts in each bin for all transcripts in the source study and the genes intersecting provirus sites are provided in Table S3.
It was previously shown that a preference to target highly transcribed genes could largely be explained by the orientation of target site transcription relative to the gene in which it was embedded, with a preference for genes transcribed in the opposite direction to the target site.31 We also investigated whether this was the case for our data, keeping in mind that the target site cassette is not expressed in non-doxycycline-treated samples. Surprisingly, in our system, the preference for the most highly expressed genes held for genes transcribed in the same direction as the target site (Figure 5, right, +doxycycline p = 3.5e−02, −doxycycline p = 3.4e−03), but not for those transcribed in the opposite direction (Figure 5, center), regardless of doxycycline treatment. The discrepancy with the previous study could be due to our smaller relative sample size and the distribution of where the target sites are positioned within the genes, an absence of 3′-to-5′ transcriptional read-through of the provirus, or the collision of convergent RNA polymerase IIs (RNA Pol IIs) at the Tet-On transactivator/blasticidin resistance cassette downstream of the target site rather than at the target site itself. The Tet-On transactivator/blasticidin resistance cassette is ubiquitously expressed downstream of the target site (Figure 1A). The transient accumulation of negatively supercoiled DNA behind RNA Pol II on either proviral expression cassette could generate a recombinogenic block to RNA Pol II procession from a co-directionally transcribed gene, possibly explaining the preference in our study for co-directionally transcribed genes.
Without binning by expression level, there was no significant difference in the number of targeted sites that intersected genes transcribed in the opposite rather than same direction for either treatment group (+doxycycline 87 versus 86, −doxycycline 60 versus 64, respectively). Likewise, barcode heterogeneity at genes transcribed in the same direction was not higher than genes transcribed in the opposite direction for either treatment group, without binning (Mann-Whitney U test).
As mentioned previously, we have not evaluated how the provirus site itself influences endogenous transcription, nor have we explored other factors that could contribute to the observed preferences, such as replication fork direction.
We also hypothesized that certain chromatin states would be more or less conducive to AAV-HR. For example, without target site transcription to stimulate AAV-HR, the degree of accessibility of the surrounding chromatin or presence of proximal endogenous regulatory elements may be a more potent predictor of AAV-HR efficiency. In order to assess whether certain epigenetic features might influence target site preference or AAV-HR efficiency, we used genomic annotations derived from ChromHMM states learned across 127 reference epigenomes for 25 states,42 sourced from the Roadmap Epigenomics Project61 and ENCODE project,62 focusing specifically on the K562 chronic myeloid leukemia-derived cell line. Chromatin states are predicted by the combinatorial presence or absence of multiple types of epigenetic marks, such as histone modifications, histone variants, and regions of open chromatin.63
In both treatment groups, chromatin states characterizing transcription were associated with an increased likelihood of AAV-HR (+doxycycline, transcribed and 3′ preferential [Tx3′], relative risk ratio = 1.416, 95% confidence interval 1.02–1.966; -doxycycline, transcribed and 5′ preferential [Tx5′,], relative risk ratio = 1.735, 95% confidence interval 1.177–2.558) (Figure 6A). Furthermore, enhancer states were also associated with an increased likelihood of AAV-HR (+doxycycline, active enhancer 2 [EnhA2], relative risk ratio = 3.227, 95% confidence interval 1.393–7.475; −doxycycline, weak enhancer 2 [EnhW2], relative risk ratio = 2.796, 95% confidence interval 1.003–7.793). The 25-state ChromHMM model defines transcribed chromatin states as being enriched in the histone marks H3K36me3, H4K20me1, and H3K79me2, while enhancer states are enriched in the marks H3K4me1, H3K27ac, H3K9ac, and H3K4me2/3, as well as DNase I hypersensitivity and the histone variant H2A.Z. In order to assess the relationship between AAV-HR efficiency and certain epigenetic features, we fit independent models to predict the presence of an overlapping feature in both K562 ChromHMM states and ENCODE annotations,64 using as our predictor the barcode heterogeneity at each targeted site. In doxycycline-treated samples, the odds of overlapping a transcribed chromatin state (Tx, strong transcription) increased with higher AAV-HR efficiency (p = 0.008), while in non-doxycycline-treated samples, the odds of overlapping an active enhancer state (EnhA2), as well as the active enhancer mark H3K27ac, increased with higher AAV-HR efficiency (p = 0.046 and p = 0.023, respectively) (Figure 6B).
Figure 6.

Chromatin States and Epigenetic Measures Associated with AAV-HR
(A) Relative risk ratio of a site having at least one integrated barcode, given that it overlaps the indicated ChromHMM chromatin state segment, using chromatin state predictions in K562 cells. Sites were assembled into a 2 × 2 contingency table for which the exposure is intersection/no intersection and the outcome is targeted/not targeted. 95% confidence intervals are shown. We consider the relative risk ratio statistically significant when the 95% confidence interval does not overlap 1, shown by white circles. States with large confidence intervals were excluded but are given in Tables S4 and S5. Where incidence for either group is 0, 0.5 was added to all cells prior to computing relative risk ratio. (B) Predicting the presence of an overlapping ChromHMM or ENCODE feature peak in K562 cells from barcode heterogeneity at targeted sites using independent logistic regression models, filtering out features with high standard deviation. Exp(log odds ratio) represents the change in odds of overlapping a given feature for every unit increase in barcode heterogeneity. (C) Predicting barcode heterogeneity at targeted sites from the proportion of cell types assigned to a given ChromHMM state or ENCODE feature peak in the region over that site using independent linear regression models, filtering out features with high standard deviation. The cell types are a subset of seven cell types shared by both the Roadmap and ENCODE annotations (GM12878, H1-hESC, HSMM, HUVEC, K562, NHEK, and NHLF). Beta represents the mean change in barcode heterogeneity given a one-unit change in the proportion of assigned cell types. For (B) and (C), data were centered and scaled to a mean of 0 and standard deviation of 1 prior to model fitting. 95% confidence intervals are shown. A feature is considered predictive of targeting when the 95% confidence interval does not overlap 0, shown by white circles. Estimates and standard errors for all states are provided in Tables S6 and S7. For regression analyses, sites with barcode heterogeneity greater than the third quartile+3 × IQR of their respective treatment group were excluded.
In order to assess whether certain epigenetic features might influence AAV-HR efficiency, we then fit independent models to predict AAV-HR efficiency for each treatment group. In this study, we used as input the proportion of cell types annotated as a given ChromHMM state or ENCODE epigenetic measure at each targeted provirus site, for the subset of cell types shared between the two annotation sets (GM12878, H1-hESC, HSMM, HUVEC, K562, NHEK, NHLF). A chromatin state characterizing transcribed chromatin (Tx, strong transcription) was associated with increased AAV-HR efficiency for doxycycline-treated samples (p = 0.008), while the H3K4me1 enhancer-associated mark was associated with decreased AAV-HR efficiency (p = 0.024) (Figure 6C). Chromatin states characterizing weak enhancers (weak enhancer 1 [EnhW1] and EnhW2) were associated with increased AAV-HR efficiency for non-doxycycline-treated samples (p = 0.016 and p = 0.025, respectively). We also fit similar linear regression models using as input the proportion of all 127 reference epigenomes annotated as a given ChromHMM state. Consistent with the seven cell type model, state Tx was associated with increased AAV-HR efficiency for doxycycline-treated samples and state EnhW1 was associated with increased AAV-HR efficiency for non-doxycycline-treated samples (Table S8).
These findings suggest that, based on features representing a single related cell type or a composite of multiple diverse cell types, transcribed chromatin and the potential for greater chromatin accessibility at or activation in the vicinity of endogenous enhancers are important for AAV-HR. No significant associations were observed for promoter, repressed polycomb, or quiescent chromatin states. While the annotations used in these models were not from HAP1 cells, our findings are informative in establishing a direction for further investigation, suggesting that transcription and specific chromatin environments both may influence target site preference and the efficiency of AAV-HR.
Discussion
There is a need to improve the efficiency of AAV-HR in order to make the use of this safe and permanent gene-targeting technology viable for a larger number of diseases and applications. Herein, we describe a novel system for detecting and quantifying AAV-HR events comprised of more than 1,000 engineered, drug-inducible provirus sites that are infected by a library of barcoded rAAV vectors. This system uniquely allows for decomposition of the effects of target site transcription and other factors on AAV-HR. By modulating target site transcription at the time of rAAV transduction, we were able to evaluate its effect on AAV-HR efficiency and target site preference while controlling for position effects, a heretofore unexplored dimension of large-scale rAAV-mediated gene targeting. Moreover, by controlling for target site transcription, we began to identify chromosomal features that improve AAV-HR efficiency. While it is difficult to compare AAV-HR rates between different studies due to differences in serotype, multiplicity of infection, homology arm length, and target sequence,7,12,14 our system controlled for these variables over multiple genomic loci by using an engineered target site and rAAV vectors that differ only in a central barcode sequence.
Using this system, we found that AAV-HR efficiency was improved by target site transcription and there was a positive effect on the amenability of a site to targeting due to other factors such as the level of endogenous transcription from intersecting genes and accessible chromatin. The number of provirus sites amenable to AAV-HR was reproducibly higher when the target site was transcribed, suggesting that target site transcription might be able to compensate for sites that are less prone to HR. Whether this effect is due to transcription alone or to the interplay of transcription and other factors remains unclear.
Consistent with our results and those of others supporting a model in which transcription through the target site stimulates rAAV-mediated targeting, we identified that a ChromHMM transcribed chromatin state was associated with higher AAV-HR efficiency (Figure 6B). Further investigation is needed to understand why this is the case for doxycycline-treated samples only. We also found evidence that a ChromHMM enhancer state was associated with higher AAV-HR efficiency in the absence of target site transcriptional activation (Figure 6B). A recent study of HIV proviruses in Jurkat cells showed that the expression of a provirus inserted in a gene showed little correlation with the expression of the host gene, while the expression level of proviruses associated with proximal endogenous enhancers were significantly higher than average.65 This finding could explain the association of AAV-HR efficiency with an enhancer state in non-doxycycline-treated samples, as the enhancer may have a compensatory effect on the target site promoter in the absence of doxycycline and thereby influence AAV-HR efficiency. Importantly, note that the use of these features assumes that provirus sites adopt a chromatin conformation comparable to the average of the region in which they integrate. Furthermore, if a particular chromatin state or epigenetic measure is already favored for lentivirus integration, such as the H3K36me3 histone modification,66 that feature might not be as useful a predictor in our regression models.
Notably, the efficiency of AAV-HR at one provirus site in particular, which intersects the co-directionally transcribed gene ENAH, was reproducibly more than two orders of magnitude greater than the median targeting rate (Figure 2A). Despite the comparatively high targeting rate, the clonal abundance of this site varied only minimally between the pre-transduction time point and passaging phase of the experiment (+doxycycline pre-transduction = 30, post-transduction by replicate = 21 and 19; −doxycycline pre-transduction = 29, post-transduction by replicate = 23 and 21). Interestingly, this site is only 68 bp from the nearest restriction site with the potential for more efficient amplification during library preparation. However, the number of unique, accepted barcodes integrated at all sites and the distance to the nearest cut site show little correlation (Spearman ρ = −0.07), and the depth of sequencing used here plus the use of unique barcodes to mark individual AAV-HR events rather than read count alone suggest that this phenomenon is not fully explained by the short distance to the cut site. Future studies will provide an opportunity to explore the propensity of this site to AAV-HR.
TAR is a phenomenon that has been associated in eukaryotic cells with transcription in general,25,67 convergent transcription,69, 70, 71 and convergent transcription and replication.71 Target site transcription alone or combined with transcriptional read-through from genes intersecting the target site, or an opposing replication fork, could be responsible for the enhanced AAV-HR efficiency we observe when the polyclonal population is transduced in the presence of doxycycline. However, we cannot exclude the broader effects that the provirus has on the surrounding chromatin, including any effects of the ubiquitous promoter driving expression of the Tet-On transactivator/blasticidin resistance cassette. In characterizing the epigenetic consequences of rAAV-mediated gene targeting, Li et al.13 showed that after insertion of an ubiquitous promoter at the target site, the target site and surrounding chromatin up to 8.4 kb away were marked by an increase in the H3K27Ac histone mark, and the target site was marked by a reduction in the H3K27me3 histone mark.
We also observed that a provirus site was less likely to be targeted if it intersected Alu elements, and for non-doxycycline-treated samples this difference was significant. Alu elements are mobile elements belonging to the class of SINEs that make up 11% of the human genome and are present in more than 1 million copies, associating with more than three-quarters of genes.56,72 It has been suggested that the high level of sequence divergence between Alu elements can shift the reliance for repair of induced double-strand breaks at Alu elements from Alu/Alu recombination to variable-length NHEJ, which results in deletions between Alu elements.44 Allelic and non-allelic HR events are disruptive to genome integrity,45,73 and stimulation of AAV-HR at an Alu element has the potential to disrupt the drug-inducible expression cassette (Figure 1A).
Alternatively, a provirus site was more likely to be targeted when it intersected LINE-1 elements, and for non-doxycycline-treated samples this difference was significant. LINE-1 elements are autonomous transposable elements that make up 17% of genome mass and are generally enriched in gene-poor regions.56 Full-length LINE-1s are known to be silenced via CpG methylation, histone deacetylation, and H3K9me3 deposition as a way to constrain LINE-1 endonuclease expression,75, 76, 77, 78 so it was surprising that provirus sites intersecting these elements were more likely to be targeted. It is possible that the ubiquitous promoter of the Tet-On transactivator/blasticidin resistance cassette (Figure 1A) could reverse transcriptional silencing in the surrounding region and make the LINE-1 elements subject to transcriptional activation, or integration of the lentivirus itself disrupts the repressive modifications. It has also been demonstrated that transcriptional silencing of LINE-1 elements often occurs within introns of transcriptionally active genes, leading to downregulation of host gene expression.74 It is possible that reversal of this silencing by provirus integration upregulates surrounding gene expression to the effect of stimulating AAV-HR. The observed importance of endogenous transcriptional activity to stimulate AAV-HR in the absence of target site transcription may explain why these LINE-1-intersecting provirus sites are more likely to be targeted in this treatment group.
Taken together with previous studies of AAV-HR, the data from this study provide future directions of research to optimize the genomic target site in AAV-HR applications. Cell type-specific RNA sequencing and chromatin immunoprecipitation to identify epigenetic measures of transcriptional activation over the target site may provide more specific evidence of an environment capable of promoting AAV-HR in our cell line. Mapping of replication origins and/or endogenous transcripts as well as the timing of these processes may provide the second level of evidence that processes shown to enhance AAV-HR are occurring in concert. Since R loop formation has been associated with transcription-associated recombination and is proposed to be a mechanism by which convergent transcription and replication enhance AAV-HR,31 the cell type-specific mapping of R loops by pulldown of DNA:RNA hybrids, identification of R loop-prone sequences, or modulation of these structures presents a potential avenue for future investigation to pinpoint the mechanism of AAV-HR and optimize genomic target sites.
Materials and Methods
Plasmid Construction
All PCR, restriction, and ligation enzymes and buffers were purchased from New England Biolabs (NEB, Ipswich, MA, USA) except when following unmodified published protocols, where other products are indicated. Except where indicated, amplified and digested DNA was purified using either the QIAEX II gel extraction kit (QIAGEN, Germantown, MD, USA) or the Monarch PCR & DNA cleanup kit (NEB, Ipswich, MA, USA). All final plasmid preparations were generated using EndoFree kits from QIAGEN (Germantown, MD, USA). All constructed plasmid sequences were confirmed by Sanger sequencing (Sequetech, Mountain View, CA, USA and MCLAB, South San Francisco, CA, USA) and restriction digest plus agarose gel electrophoresis. The presence of ITRs in rAAV transfer plasmids was confirmed by restriction digest with AhdI and XmaI. All primers used in cloning were ordered from Integrated DNA Technologies (IDT, Coralville, IA, USA). Primers containing P5 and P7 adapters were high-performance liquid chromatography (HPLC) purified. Plasmid sequences are available upon request.
The original third-generation lentivirus transfer plasmid pCW22-Nkx2-1 was provided as a gift from the Winslow Lab at Stanford University School of Medicine (Stanford, CA, USA). To generate pCW22-Luc-P2A-GFP, the existing promoter containing the tetracycline-responsive element was replaced with pTRE3Gs by joining multiple regions from other plasmids containing pTRE3G, the cytomegalovirus (CMV) promoter, and a synthesized gBlock (IDT, Coralville, IA, USA) by restriction cloning into a subcloning vector. The existing Tet-Advanced reverse tetracycline transactivator (rtTA) sequence was directly replaced by the Tet-On 3G rtTA,78 which was amplified from a synthesized gBlock (IDT, Coralville, IA) and inserted by restriction cloning using EcoRI and XmaI. The full target site containing firefly luciferase, P2A-EGFP, and murine albumin 3′ UTR sequence was generated by PCR amplification from other Kay Lab plasmids and Gibson cloning79 downstream of the pTRE3Gs promoter in the subcloning vector. The entire subcloned expression cassette was restriction cloned into pCW22-Nkx2-1 using PacI and ClaI. A mock-targeted control transfer plasmid for FACS compensation was generated by cloning the mScarlet sequence (Michael Lin Lab, Stanford University School of Medicine, Stanford, CA, USA) into pCW22-Luc-P2A-GFP using BlpI. The third-generation lentivirus packaging and envelope plasmids pRSV-Rev, pMDLg/pRRE, and pMD2.G were purchased from Addgene (Watertown, MA, USA; IDs 12253, 12251, and 12259).
To generate the rAAV transfer plasmid pAAV-Luc-P2A-mScarlet-GFP, homology arm regions were generated by PCR amplification and Gibson cloning similarly to their homologous regions in the lentivirus plasmid. The restriction cloning sites BmgBI and NheI were added to the mScarlet sequence by PCR for cloning between the P2A and EGFP sequences. A cloning site for the barcode was included downstream of the mScarlet sequence, followed by an AseI restriction site used for iPCR. mScarlet plus two 1,600-bp flanking homology regions were inserted into a single-stranded rAAV-DJ ITR-containing plasmid from our laboratory.
Double-stranded barcoded inserts were generated by annealing and extension of the following single-stranded oligonucleotides (IDT, Coralville, IA, USA) where Ns indicate degenerate random nucleotides, as previously described:80,81 NheI-AAVbc, forward, 5′-GGTATGGATGAACTCTATGCTAGCACGGAAATACGATGTCGGGA-3′; XhoI-AAVb, reverse, 5′-ATTAATCTCGAGNNNNNNNNNNNNTCCCGACATCGTATTTCCGT-3′. Briefly, two 100-μL reactions each containing 1 μM each oligonucleotide and 1× NEB 2.0 buffer were incubated as follows: 10 min at 70°C, decrease 0.1°C/s to 30°C, hold at 4°C. Annealed oligonucleotides were extended by addition of 0.03 mM 2′-deoxynucleoside 5′-triphosphates (dNTPs) and 5 U Klenow fragment (3′ ® 5′ exo-) for 1 h at 37°C. The reactions were pooled and purified using 5PRIME Phase Lock Gel heavy tubes (Thermo Fisher Scientific). Double-stranded molecules were inserted downstream of mScarlet in pAAV-Luc-P2A-mScarlet-GFP by restriction cloning with NheI-HF and XhoI, using a backbone/insert ratio of 1:1.43. Plasmid DNA for the barcoded rAAV library was generated as described.81,82 The resulting yield from plasmid preparation was 3.2 mg.
Library diversity was estimated at 3.6 million clones by plating serial dilutions of the initial inoculation. Since we subsequently sorted about 50,000 cells per replicate, simulating choosing 50,000 barcodes from a pool of 3.6 million results in each barcode being selected on average 0.014 times, suggesting a low possibility of getting duplicate barcodes. A subset of colonies was sent for Sanger sequencing to assess the frequency of more than one barcode being ligated into the plasmid backbone. In 4 out of 100 clones, two backbones with single barcodes attached at one end had ligated to each other. One clone of the 100 did not match the plasmid backbone. Barcodes were amplified from plasmid DNA using the following site-specific primers containing Illumina P5 and P7 adaptor sequences and internal multiplexing barcodes: AAVbc-plasmid read 1, 5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTTAACGCGCCCTTGCTCACATTAAT-3′; AAVbc-plasmid read 2, 5′-CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCTGGATTATGGACGAGCTGTACAAGTAAG-3′. PCR products were purified from a 2% 1× TAE agarose gel with 1× SYBR Safe DNA gel stain (Thermo Fisher Scientific) using a QIAquick gel extraction kit (QIAGEN), performing all reactions at room temperature to avoid denaturing the short PCR products, then sequenced using a MiSeq reagent kit v3 (Illumina, San Diego, CA, USA) with 2 × 75-bp paired-end reads to check library diversity and integrity of the restriction sites surrounding the barcodes. In 484,966 reads, there were 400,427 different barcode sequences. This number did not change after clustering with Starcode,83 allowing for an edit distance of 2 at a ratio of 5.0.
Cell Culture
All cells were grown in media supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific, Waltham, MA, USA unless noted), 100 IU/mL penicillin-streptomycin (Thermo Fisher Scientific, Waltham, MA, USA), and 1 mM sodium pyruvate (Thermo Fisher Scientific, Waltham, MA, USA) in a humidified incubator at 37°C with 5% CO2. HAP1 cells46 were obtained from the Carette Lab at Stanford University School of Medicine (Stanford, CA, USA) and cultured in complete Iscove’s modified Dulbecco’s medium (IMDM) (4 mM l-glutamine, 25 mM HEPES) (Thermo Fisher Scientific, Waltham, MA, USA) using tetracycline-negative FBS (Gemini Bio-Products, West Sacramento, CA, USA). HEK293T cells for lentivirus and rAAV production were cultured in complete DMEM (with glucose, without l-glutamine and sodium pyruvate) supplemented with 2 mM l-glutamine and 20 mM HEPES (Thermo Fisher Scientific, Waltham, MA, USA).
Prior to all fluorescence-activated sorting steps, cells were rinsed twice in Dulbecco’s PBS (DPBS), resuspended in DPBS + 2% FBS, and strained through a 35-μm cell strainer (Thermo Fisher Scientific). Cells were maintained on ice and 1 μM SYTOX blue dead cell stain was added just prior to sorting. Sorting was performed on a BD FACSAria Fusion or BD FACSAria II sorter (BD Biosciences, San Jose, CA, USA) by Stanford Shared FACS Facility staff.
Viral Vector Stocks
Lentivirus vectors were produced by co-transfection of pCW22-Luc-P2A-GFP, pRSV-Rev, pMDLg/pRRE, and pMD2.G into HEK293T cells seeded on 0.001% poly-l-lysine-coated dishes at amounts previously described for 12 × 15-cm dishes.84 5e6 cells were seeded per dish and the following day transfected with plasmids diluted in Opti-MEM (2 mL per dish) (Thermo Fisher Scientific) and combined with 1 mg/mL polyethylenimine (PEI; linear, molecular weight [MW] 25,000 Da) (Polysciences, Warrington, PA, USA) at a ratio of 1:4 (μg of DNA/μL of PEI). Medium was replaced after 16 h and collected 48 h later. Viral particles were harvested and concentrated from the medium using Lenti-X concentrator (Takara Bio USA, Mountain View, CA, USA) and then resuspended in complete IMDM for use on HAP1 cells, and stored at −80°C. Vector stocks were titered for infectious particles by serial dilution on HAP1 cells and blasticidin selection.
rAAV vectors were packaged in serotype DJ capsids by co-transfection of pAAV-Luc-P2A-mScarlet-GFP, pAd5 adenoviral helper plasmid, and AAV-DJ rep/cap packaging plasmid using the calcium phosphate method, harvested from cell lysate, and concentrated by cesium chloride gradient centrifugation as previously described.81 Single-stranded vector genomes were extracted using the NucleoSpin virus kit (Takara Bio USA, Mountain View, CA, USA) and titered by SYBR Green qPCR using Apex qPCR GREEN master mix without ROX (Genesee Scientific, San Diego, CA, USA) on a Bio-Rad CFX384 real-time system (Bio-Rad, Hercules, CA, USA). Serial dilutions were run in quadruplicate using a gBlock (IDT) standard and primers homologous to the firefly luciferase sequence as follows: forward, 5′-TAAGGTGGTGGACTTGGACA-3′; reverse, 5′-GTTGTTAACGTAGCCGCTCA-3′. Packaging of full-length vector genomes (4.3 kb) was confirmed by alkaline denaturing Southern blot using 3e9 vector genomes.
Generating Lentivirus-Infected Cell Line
On day 0, 3.25e6 HAP1 cells per dish were seeded in two 10-cm dishes. On day 1, lentiviral particles were added in 4 mL of medium with 8 μg/mL Polybrene at an MOI of <0.1, keeping one dish uninfected as a control for drug selection. Plates were gently swirled regularly for 6 h, then left to sit overnight. On day 2, medium was replaced with fresh complete IMDM. On day 3, blasticidin S hydrochloride was added at 7 μg/mL to select for infected clones. Medium was replaced with fresh complete IMDM+blasticidin every other day. On day 11, approximately 1,000 clones were pooled and expanded into three T-225 flasks. With approximately 1,000 clones per dish, the effective MOI was on the order of 3e−4 viral particles per cell (1e3 clones/3.25e6 cells), at which point there is a negligible likelihood of having more than one insertion per cell according to a Poisson distribution (see table 3 in Horizon Discovery85). Cells were passaged regularly in 7 μg/mL blasticidin S hydrochloride. On day 15, 100 ng/mL doxycycline hyclate (Sigma-Aldrich, St. Louis, MO, USA) was added to the medium. On day 18, 4.5e6 GFP+/SYTOX blue− cells were sorted on a BD FACSAria Fusion sorter (BD Biosciences, San Jose, CA, USA) by Stanford Shared FACS Facility staff to establish a polyclonal population in which all expression cassettes are doxycycline responsive. Sorted cells were expanded and stored in liquid nitrogen.
Gene Targeting
On day 0, 100 ng/mL doxycycline or an equal volume of DPBS was added to half the lentivirus-infected polyclonal population. On day 1, both doxycycline-treated and untreated cells were seeded at 5e6 cells per dish in 4 × 15-cm dishes (two dishes per biological replicate). Medium was replaced every other day with fresh complete IMDM with or without doxycycline, consistent with the starting condition. On day 2, cells were transduced with the barcoded rAAV library at an MOI of 1e5 vector genomes/cell. On day 3, each dish was expanded into four T-225 flasks in the absence of any doxycycline. Cells were passaged regularly in order to dilute episomal rAAV genomes,86 retaining at least 1,000-fold representation of targeted cells, assuming a 0.1%–0.2% targeting rate.
FACS Enrichment of Targeted Cells
On day 7, 100 ng/mL doxycycline was added to all cells to prepare for the first round of sorting. On day 10, 36e6 cells per replicate were put through a sorter, producing 40,000–70,000 mScarlet+/GFP+ cells per replicate. HAP1 cells infected with single-positive lentivirus controls were used for compensation control. Cells were allowed to recover in medium absent doxycycline. On day 18, doxycycline was added to all cells to prepare for the second round of sorting to eliminate cells that were mScarlet+ due to off-target integrations and to conclusively identify mScarlet+ cells once episomal vector genomes are fully diluted. On day 21, 6e6 cells per replicate were put through a sorter, retaining only mScarlet+/GFP− cells. On day 26, 100 ng/mL doxycycline was added to all cells to prepare for nucleic acid extraction. For DNA extraction, one T-225 flask was seeded with 10e6 cells. For RNA extraction, six-well plates were seeded with 0.4e6 cells per well. On day 28, cells for DNA extraction were pelleted and frozen at −20°C in aliquots of 12e6 cells. Cells for RNA extraction were harvested in 1× Monarch RNA protection reagent (NEB, Ipswich, MA, USA) (750 μL/well) and stored at −80°C.
NGS Library Preparation
All primers used in NGS library preparation that contain P5 and P7 adapters were ordered from IDT (Coralville, IA, USA) as Ultramer DNA oligonucleotides. All PCR master mixes were made inside a PCR laminar flow cabinet with a designated set of pipettes. DNA or RNA extraction from frozen cells or cell lysate and library preparation for NGS were performed in two independent technical replicates for every biological replicate. Genomic DNA was extracted from frozen cell pellets of 12e6 cells using the GeneJET genomic DNA purification kit (Thermo Fisher Scientific). Samples were quantified using a Qubit double-stranded DNA (dsDNA) high-sensitivity (HS) assay kit (Thermo Fisher Scientific) and quality checked on a 0.8% 1× TAE agarose gel.
Total RNA was extracted using the Monarch total RNA miniprep kit (NEB) from samples frozen in 1× protection buffer, including in-column DNase I treatment. Samples were quantified and quality checked by NanoDrop and by Bioanalyzer, using the RNA 6000 Nano kit (Agilent Technologies, Santa Clara, CA, USA). For each technical replicate, 2 μg of RNA was reverse transcribed with RNase H treatment in a 21-μL reaction using a SuperScript III first-strand cDNA synthesis kit (Thermo Fisher Scientific) with oligo(dT) primers and including a no-reverse transcriptase negative control.
Ligation-Mediated PCR
Ligation-mediated PCR (LM-PCR) is designed to amplify genomic DNA downstream of the lentivirus 3′ LTR. Cells were harvested for LM-PCR just prior to rAAV transduction (one replicate per treatment group) and during the passaging phase, just before FACS to isolate targeted cells (corresponding to the two biological replicates per treatment group). LM-PCR was performed as previously described for HIV-151 with the following modifications. PCR2 primer sequences were adapted for use with standard Illumina primer sequences. The following modified format was used for PCR2 linker primer sequences: 5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT(Z)20–21-3′, which consists of a P5 adaptor sequence and primer landing site followed by a 20- to 21-nt site-specific primer for each linker (Z). The following modified format was used for PCR2 HIV LTR primer sequences: 5′-CAAGCAGAAGACGGCATACGAGATXXXXXXXXGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT(Z)0,2,4,6AGACCCTTTTAGTCAGTGTGGAAAATC-3′, which consists of a P7 adaptor sequence, 8-bp i7 index (Xs), P7 primer landing site, a variable length spacer (Z) associated with each i7 index to offset the common sequence for improved diversity in sequencing, and a site-specific primer for PCR. A combination of four different linker-specific sequences (20- to 21-nt landing site of PCR2 linker primer) and four different 8-bp i7 indexes were used to multiplex technical replicates. To generate linkers, single-stranded linker oligonucleotides corresponding to linkers iSL-1, iSL-2, iSL-3, and iSL-451 were annealed in a 50-μL reaction with 20 μM each oligonucleotide and 1× T4 DNA ligase buffer (NEB) under the following conditions: 30 min at 37°C, 5 min at 95°C, cool to room temperature for 1 h. For the second round of PCR, five cycles of linear amplification plus seven cycles of exponential amplification were performed. Negative controls of uninfected wild-type HAP1 cells and a no-DNA reaction were prepared in parallel. After the second round of PCR, samples were allowed to sit at 4°C for 24 h to encourage loss of A overhangs left by the DNA polymerase. Samples were quantified using the NEBNext library quant kit for Illumina (NEB) and confirmed by Bioanalyzer using the HS DNA kit (Agilent Technologies). Technical replicates were diluted to 20 nM, pooled in equimolar amounts, and sequenced by the Stanford Functional Genomics Facility using the MiSeq reagent kit v3 (Illumina) with 2 × 300-bp reads. Initial demultiplexing was done using i7 indexes, producing 12 million 2 × 300-bp paired-end reads.
iPCR
iPCR is designed to amplify genomic DNA upstream of the lentivirus 5′ LTR as well as the integrated barcode. iPCR was performed similarly as described57 with some modifications. For each technical replicate, 4 μg of genomic DNA was digested in a 200-μL reaction with 1× NEB buffer 3.1, 134 U of AseI, and 134 U of NdeI, which generate compatible overhangs. There is an AseI site just downstream of the barcode cloning site in pAAV-Luc-P2A-mScarlet-GFP. AseI (“ATTAAT’) cuts on average every 1,966 bp in the genome, while NdeI (“CATATG”) cuts on average every 3,189 bp (http://tools.neb.com/∼posfai/TheoFrag/TheoreticalDigest.human.html). Digests were purified through a Monarch PCR & DNA cleanup kit column (NEB). For each technical replicate, 3 × 600 ng of independent ligations were prepared in 400 μL with 1× T4 DNA ligase buffer and 2,800 U of high concentration T4 DNA ligase and incubated at 4°C overnight. Final PCR template was obtained by phenol/chloroform/isoamyl alcohol extraction and ethanol precipitation as described.57
iPCR was performed in two rounds with the following primer design: iPCR-1 read 1, 5′-AATGATACGGCGACCACCGAGATCTACACXXXXXXXXACACTCTTTCCCTACACGACGCTCTTCCGATCT(Z)0,2,4,6NNNNNNNNNNTGTACAAGTAAGCTAGCACGGAA-3′; iPCR-1 read 2, 5′-GGTTTCCCTTTCGCTTTCAAGTCCCTG-3′; iPCR-2 read 1, 5′-AATGATACGGCGACCACCGAGATCTACAC-3′; iPCR-2 read 2, 5′-CAAGCAGAAGACGGCATACGAGATXXXXXXXXGTGACTGGAGTTCAGACGTGTGCTCTTCCGATC(Z)0,2,4GCTCAGATCTGGTCTAACCAGAGAG-3′. iPCR-1 read 1 binds just upstream of the barcode, iPCR-1 read 2 binds just internal to the 5′ LTR and is designed to enrich for PCR products from the 5′ LTR since iPCR-2 read 2 anneals to both 5′ and 3′ LTRs, iPCR-2 read 1 binds to the tail of iPCR-1 read 1, and iPCR-2 read 2 binds to the 5′ end of the LTR. iPCR-1 read 1 and iPCR-2 read 2 consist of (in order): P5 and P7 adapters, Xs (i5 and i7 indexes), Illumina P5 and P7 primer binding sites, Zs (variable length spacers), Ns (random nucleotides) (unique molecular identifier [UMI]; not used in analysis), site-specific primer.
Negative controls were prepared in parallel and included non-rAAV-transduced, lentivirus-infected cells that lack a read 1 primer binding site, rAAV-transduced wild-type HAP1 cells that lack a lentivirus provirus site (and read 2 primer binding site), a no-T4 DNA ligase reaction (non-circularized DNA), and a no-DNA reaction. In the first round of iPCR, 4 × 25-μL reactions were prepared for each independent ligation (12 independent reactions total per technical replicate that were not pooled until after the second round of iPCR), using the standard protocol for Q5 hot start high-fidelity DNA polymerase but with 0.1 μM primers and 5 μL of DNA template. Cycling conditions were as follows: 30 s at 98°C, 10 s at 98°C, 30 s at 64°C, 5 min at 72°C, repeat from step 2 (four times), 2 min at 72°C, hold at 4°C. Primers and dNTPs were removed from first round reactions by addition of 2 U/μL ExoI and 0.2 U/μL shrimp alkaline phosphatase (rSAP) under the following conditions: 1 h at 37°C, 15 min at 80°C. In the second round of iPCR, 5 μL of each first round template was put in a new 50-μL reaction using the standard protocol for Q5 hot start high-fidelity DNA polymerase but with 0.3 μM primers. Cycling conditions were as follows: 30 s at 98°C, 10 s at 98°C, 20 s at 67°C, 4 min at 72°C, repeat from step 2 (35 times), 2 min at 72°C, hold at 4°C. Smears from positive reactions only were confirmed by running 10 μL of second round iPCR on a 1.2% TAE gel. Equal volumes of the 12 independent reactions per technical replicate were pooled and cleaned up using 0.8× Agencourt AMPure XP beads (Beckman Coulter, Pasadena, CA, USA). Samples were quantified on a Bioanalyzer using a DNA 12000 kit (Agilent Technologies). Technical replicates were diluted to 10 nM, pooled in equimolar amounts, and sequenced by the Stanford Functional Genomics Facility using the NextSeq mid-output kit (Illumina) with 2 × 150-bp reads. Reads were demultiplexed using i5 and i7 indexes, producing 108 million 2 × 150-bp paired end reads.
Barcode PCR
Barcode PCR was designed to amplify just the barcodes from either DNA (“normalization sample”) or cDNA (“expression sample”). Negative controls were prepared in parallel and included non-rAAV-transduced, lentivirus-infected cells, rAAV-transduced wild-type HAP1 cells that lack a lentivirus provirus site, a no-DNA/cDNA reaction, and a no-reverse transcriptase control for expression samples. Notably, expression barcodes could not be detectably amplified from cDNA without addition of doxycycline to the tissue culture medium, which is why all samples were doxycycline induced prior to RNA extraction regardless of doxycycline treatment at the time of transduction.
Barcode PCR was performed in two rounds with the following primer designs.
Normalization Primers
Normalization primers were as follows: DNA BC-1 read 1, 5′-AATGATACGGCGACCACCGAGATCTACACXXXXXXXXACACTCTTTCCCTACACGACGCTCTTCCGATCT(Z)0,2,4,6NNNNNNNNNNTGTACAAGTAAGCTAGCACGGAA-3′; DNA BC-1 read 2, 5′-ACCAACAGAAAAGATGAGTCCTGA-3′; DNA BC-2 read 1, 5′-GGTTTCCCTTTCGCTTTCAAGTCCCTG-3′; DNA BC-2 read 2.1, 5′ CAAGCAGAAGACGGCATACGAGATXXXXXXXXGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT(Z)2,4,6,8,10CCTCGCCCTTGCTCACATT-3′; and DNA BC-2 read 2.2, 5′-CAAGCAGAAGACGGCATACGAGAT-3′.
Expression Primers
Expression primers were as follows: RNA BC-1 read 1, 5′-AATGATACGGCGACCACCGAGATCTACACXXXXXXXXACACTCTTTCCCTACACGACGCTCTTCCGATCT(Z)0,2,4,6NNNNNNNNNNTGTACAAGTAAGCTAGCACGGAA-3′; RNA BC-1 read 2, 5′-CAAGCAGAAGACGGCATACGAGATXXXXXXXXGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT(Z)2,4,6,8,10CCTCGCCCTTGCTCACATT-3′; RNA BC-2 read 1, 5′-ACCAACAGAAAAGATGAGTCCTGA-3′; and RNA BC-2 read 2, 5′-CAAGCAGAAGACGGCATACGAGAT-3′.
Read 1 primers are identical to iPCR read 1 primers. DNA BC-1 read 2 binds within the final exon of the murine albumin 3′ UTR, outside of the rAAV homology region to promote amplification only of integrated barcodes.87 DNA BC-2 read 2.1 binds just downstream of the barcode. DNA BC-2 read 2.2 binds to the tail of DNA BC-2 read 2.1 to enrich for the correct product.87 RNA BC-1 read 2 is identical to DNA BC-2 read 2.1, and RNA BC-2 read 2 is identical to DNA BC-2 read 2.2. RNA BC-2 read 2 binds to the tail of RNA BC-1 read 2.
PCR of normalization and expression samples was performed similarly as described57 with some modifications. In the first round of PCR, 10 × 25-μL independent reactions were prepared for each technical replicate (to be pooled only after the second round of PCR) containing 100 ng of DNA or 1.68 μL of cDNA using the standard protocol for Q5 hot start high-fidelity DNA polymerase but with 0.1 μM primers. Cycling conditions were as follows: 30 s at 98°C, 10 s at 98°C, 30 s at 64°C, 15 s (expression) or 1 min (normalization) at 72°C, repeat from step 2 (four times), 2 min at 72°C, hold at 4°C. Primers and dNTPs were removed from first-round reactions to prevent amplification from episomal or off-target integrated rAAV by ExoI-rSap addition as described for iPCR. In the second round of PCR, 5 μL of each first round template was put in a new 50-μL reaction using the standard protocol for Q5 hot start high-fidelity DNA polymerase but with 0.3 μM primers. Cycling conditions were as follows: 30 s at 98°C, 10 s at 98°C, 20 s at 68°C (normalization) or 69°C (expression), 15 s at 72°C, repeat from step 2 (30 times for normalization or 25 times for expression, 2 min at 72°C, hold at 4°C. Bands from positive reactions only and library size were confirmed by running 5 μL on a 2% 1× TAE agarose gel. Equal volumes of the 10 independent reactions per technical replicate were pooled and cleaned up with 1.2× (normalization) or 1× (expression) Agencourt AMPure XP beads (Beckman Coulter). Samples were quantified on a Bioanalyzer using a high-sensitivity DNA kit (Agilent Technologies). Technical replicates were diluted to 10 nM, pooled in equimolar amounts, and sequenced by the Stanford Functional Genomics Facility using the HiSeq 4000 kit (Illumina) with 2 × 100-bp reads. Reads were demultiplexed using i5 and i7 indexes, producing 283 million 2 × 100-bp paired end reads.
Read Trimming and Alignment
Ligation-Mediated PCR
BBTools v38.60 (https://sourceforge.net/projects/bbmap/) BBDuk was used to quality trim read 3′ ends and filter out low-quality reads while error-correcting overlapping regions. UMI-tools88 v1.0.0 extract was used to trim reads of primer sequence and extract UMIs. In read 1, linker-unique sequences and UMIs were extracted while discarding all remaining sequence up to the linker-genomic DNA “breakpoint” using –extract-method = regex with the Python regex
‘(?P<cell_1>(linker-unique-sequences)){s<=3}(?P<umi_1>.{12})(?P<discard_1> CTCCGCTTAAGGGACT){s<=1}.*’.
In Read 2, primer and LTR sequences were discarded using the Python regex
‘(?P<discard_1> variable length spacer){s<=1}(? P<discard_2>AGACCCTTTTAGTCAGTGTGGAAAATC){s<=3}(?P<umi_1>TCTAGCA).*’.
A perfect match was required for the 7 bp between the end of the PCR primer and the end of the LTR. BBTools BBDuk was then used to filter out reads for which there was amplification into the provirus from the 5′ LTR, rather than into the genome from the 3′ LTR as desired. BBDuk was also used to trim overlapping reads of primer read-through at their 3′ ends, which was possible once the primer sequence had been removed from the read 5′ ends during the UMI extraction step.
BBTools demuxbyname was used to complete demultiplexing of technical replicates using the read 1 linker-unique sequences, allowing for a hamming distance of 3. All distinct linker-unique sequences have an edit distance of >10 from one another. Genome index generation and read alignment were performed using the STAR aligner v2.7.1a.89 Genome indexes for LM-PCR were generated with –sjdbOverhang 300 using GENCODE release 31 (GRCh38.p12).90 Paired reads were aligned to the genome using STAR with the non-default parameters –alignIntronMax 1 –alignMatesGapMax 2500 to prohibit spliced alignments. We ultimately filtered for uniquely mapped and properly matched read pairs.
UMI-tools dedup was used to deduplicate aligned reads using the –ignore-umi flag in order to deduplicate only on mapping coordinates. We ultimately chose not to use the UMIs for deduplication because clonal abundance is more accurately determined by the number of unique linker-genome breakpoints in read 1 for each fixed LTR junction in read 2.51 For each read pair, we required that they maintain the correct predicted orientation: the LTR junction read is “upstream” of the breakpoint read if aligned to the + strand, or "downstream” if aligned to the − strand. Read 2 LTR junction positions were clustered within 20 bp to generate a set of consensus junctions. Provirus “sites” were defined as the 20-bp genomic interval starting from the consensus LTR junction directed toward the breakpoint. We then computed clonal abundance as the depth-normalized number of unique breakpoints for each junction after clustering breakpoint positions within 5 bp, scaled to the maximum number of breakpoints in any sample. We required that consensus sites be recovered in both technical replicates associated with a biological replicate and have at least two unique breakpoints to be accepted (Table S2). Per biological replicate, per site clonal abundance is the average between technical replicates. A set of population-wide consensus sites was generated by repeating the junction clustering process across all technical replicates, which is reasonable because every clone originated from the same polyclonal population, even though not every clone was recovered in each biological replicate. For comparing targeted and untargeted provirus sites, a BED file of AseI/NdeI double-cut restriction fragments in hg38 was generated using the HiC-Pro91 script digest_genome.py, and bedtools92 closest was used to eliminate provirus sites not within a reasonable distance to the nearest cut site upstream of the 5′ LTR.
iPCR
BBTools v38.60 (https://sourceforge.net/projects/bbmap/) BBDuk was used to quality trim read 3′ ends and filter out low-quality reads while error-correcting overlapping regions. UMI-tools88 v1.0.0 extract was used to trim reads of primer sequence and extract barcodes. In read 1, barcodes were extracted while discarding all remaining sequence up to the reconstituted AseI/NdeI restriction site (excluding the last two bases, which differ between the two restriction sites) using –extract-method = regex with the Python regex
‘(?P<discard_1>variable length spacer){s<=1}(?P<umi_1>.{10})(?P<discard_2>TGTA)(?P<discard_3>.{13})(?P<discard_4>ACGGAAATACGATGTCGGGA){s<=2}(?P<cell_1>.{12})(?P<discard_5>CTCGAG)(?P<discard_6>ATTA){s<=1}.’
In read 2, primer and LTR sequences (including the U3 region) were discarded using the Python regex ‘(?P<discard_1>variable length spacer’){s<=1}(?P<discard_2>.{25})(?P<umi_1>ACCC)(?P<discard_3>AGTACAAGCA{5}GCAGATCTTGTCTTCGTTGGGAGTGAATTAGC){s<=5}(?P<discard_4>CCTTCCA).∗’
BBTools BBDuk was then used to filter out reads for which there was amplification into the provirus from the 3′ LTR rather than into the genome from the 5′ LTR as desired. BBDuk was also used to trim overlapping reads of primer read-through at their 3′ ends, which was possible once the primer sequence had been removed from the read 5′ ends during the barcode extraction step. Genome index generation and read alignment were performed using the STAR aligner v2.7.1a.89 Genome indexes for iPCR were generated with -sjdbOverhang 150 using GENCODE release 31 (GRCh38.p12).90 Paired reads were aligned to the genome using STAR with the non-default parameters –alignIntronMax 1 –alignMatesGapMax 3850 to prohibit spliced alignments. We ultimately filtered for uniquely mapped and properly matched read pairs.
Consensus sites were generated by clustering read 2 LTR junction positions within 20 bp, and technical replicates were subsequently merged. iPCR consensus sites were mapped back to provirus consensus sites. The junctions are expected to overlap by 5 bp due to a 5-bp duplication process at the site of lentivirus integration.48,50 To allow for some flexibility, we require that iPCR and LM-PCR LTR junctions be mapped to opposite strands and overlap at their LTR junction positions by not more than 10 bp (using bedtools92 v2.28.0 intersect).
Barcode PCR
Processing of normalization and expression reads was carried out in an identical manner. Forward and reverse reads were merged using BBTools v38.60 (http://sourceforge.net/projects/bbmap/) BBMerge with weak quality trimming at the 3′ end of reads while also filtering out low-quality reads and error-correcting overlapping regions. UMI-tools88 v1.0.0 extract was used to extract barcodes while discarding all remaining primer and vector sequence up to the XhoI barcode cloning site using the –extract-method = regex with the Python regex
‘(?P<discard_1>variable length spacer){s<=1}(?P<umi_1>.{10})(?P<discard_2>TGTA)(?P<discard_3>.{13})(?P<discard_4>ACGGAAATACGATGTCGGGA){s<=2}(?P<cell_1>.{12})CTCGAGATTA.∗’
Unique barcodes were collapsed and counted.
Mapping and Quantification of AAV-HR Events
Barcode processing and assignment were performed similarly as described with some modifications.57 Barcode processing started with the normalization barcodes. To produce a list of accepted barcodes, for each technical replicate, barcodes with at least five reads were retained, and the list of barcodes was filtered to those present in both technical replicates originating from the same biological replicate (Table S2). The counts of each barcode were well correlated between technical replicates (R2 ≥ 0.71 for normalization barcodes, R2 ≥ 0.91 for expression barcodes). A pseudocount of 1 was added to both normalization and expression read counts to account for barcodes with very low expression.93 Each normalization and expression technical replicate was then depth normalized and technical replicates were combined by averaging barcode counts. To adjust expression measurements by the representation of each barcode in the genomic DNA, expression counts were divided by normalization counts for each barcode. Notably, the counts of a barcode in the normalization sample and expression sample were well correlated (R2 ≥ 0.67). 83%–93% of normalization barcodes were recovered at detectable levels in the corresponding expression sample.
The set of accepted barcodes was filtered again by those barcodes recovered in iPCR. A barcode was assigned to a site when it mapped with at least two reads to the site and at least 90% of the total reads for the barcode mapped to the site, with fewer than 2.5% mapping to a secondary locus. As Akhtar et al.93 discussed, iPCR is non-saturating. Additionally, we were limited in our choice of restriction enzymes due to the large size of intervening sequence between the barcode and the 5′ LTR (4.3 kb). As a result, the distance between the provirus LTR and nearest restriction site can vary greatly, with shorter distances producing shorter PCR amplicons that are more efficiently amplified and shorter reads that are more efficiently sequenced. Even though some barcodes may be integrated, or provirus sites might be located, at places where the AseI/NdeI restriction site is not an optimal distance from the site of integration, we control for this by only comparing between samples with the same background (i.e., from the same polyclonal population). The number of unique barcodes mapping to a single provirus site is called the barcode heterogeneity and is normalized to the provirus abundance of that site (number of unique barcodes at LV site/relative abundance of LV site). A single site’s expression level was calculated as the mean of normalized expression for unique barcodes originating from that site. To compare targeted provirus sites between biological replicates within a single treatment group (+doxycycline or −doxycycline), barcodes from iPCR technical replicates were merged. To compare between the two treatment groups, values associated with the provirus sites recovered in independent biological replicates were concatenated. After concatenation, for both targeted and untargeted sites, independent replicates were kept as separate measurements for all analyses.
To check for enrichment of RIGs55 at provirus sites, gene sets of the same size as the RIG gene set were randomly selected with replacement from the set of all protein coding genes and intersected with the provirus sites using bedtools intersect.92 The size of the intersection is the number of RIGs overlapping one or more provirus sites. The p value is computed as the number of times the size of the intersection with random genes is at least as large as the size of the intersection with RIGs, divided by the number of gene set permutations. To identify intersecting or nearby genes and repeat elements for relative risk analyses and relative distance tests, genes were filtered from the GENCODE v31 basic annotation90 and repeats were taken from the RepeatMasker annotation for GRCh38, downloaded from the UCSC Table Browser.94 DNase I sequencing (DNase I-seq) called peaks in HAP1 cells were obtained from Gene Expression Omnibus (GEO: GSE90371).62 ChromHMM chromatin state segments for the K562 cell line (E123) were downloaded from https://egg2.wustl.edu/roadmap/data/byFileType/chromhmmSegmentations/ChmmModels/imputed12marks/jointModel/final/, using the hg38 liftover. For all analyses making comparisons to random sites, the set of random sites was generated using bedtools92 shuffle with the list of provirus sites as input and the option –chrom to permute the sites along the same chromosome, preserving per-chromosome site frequency. Sites were intersected using bedtools intersect. Relative distances were computed using bedtools reldist. For expression level binning of genes intersecting provirus sites, we used publicly available FPKM (fragments per kilobase per million mapped reads) values from RNA sequencing of wild-type HAP1 cells (GEO: GSE111272).60 Twenty-one intersected genes (10 transcribed in the opposite direction and 11 transcribed in the same direction as the provirus site) were excluded because they were not present in the RNA sequencing data.
To assess genomic features associated with the efficiency of targeting a site, we first filter to sites with barcode heterogeneity greater than 0 and exclude sites with barcode heterogeneity greater than the third quartile+3 × interquartile range (IQR) of their respective treatment group (12 targeted sites in doxycycline-treated samples and 7 targeted sites in non-doxycycline-treated samples). For the ChromHMM states, we downloaded state predictions for 25 states across 127 cell types from https://egg2.wustl.edu/roadmap/data/byFileType/chromhmmSegmentations/ChmmModels/imputed12marks/jointModel/final/ using the hg38 liftover, and intersected each with all targeted sites. If a site intersected more than one feature, it was assessed for both features. For ENCODE epigenetic measures, we downloaded encodeDCC broadPeak files across seven cell types (GM12878, H1-hESC, HSMM, HUVEC, K562, NHEK, and NHLF) from http://hgdownload.soe.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeBroadHistone/, then lifted over to hg38 coordinates using liftOver.64,95 In order to restrict the analysis to the same cell types across ChromHMM and ENCODE features, we used only the ChromHMM predictions for this subset of seven cell types; however, we also include an analysis with all 127 cell types in the Supplemental Information. We then use a linear regression model to assess whether any feature is associated with the barcode heterogeneity at a site, implemented using the lm function in R:96 N = β0 + β1P, where N is the barcode heterogeneity and P is the maximum value over a site for a given genomic feature, scaled to mean 0 and standard deviation of 1. If the 95% confidence interval around β1 does not overlap 0, we say that a feature is significantly associated with the degree of targeting. While we acknowledge that several of these features are correlated, as our goal is not to determine total predictive power but rather assess features associated with efficiency, we consider each feature independently here, expecting similar coefficient estimates for similar features. We assess all features but filter a subset of features with large confidence intervals due to the small number of sites assigned to those features.
K562 ChromHMM intersections were generated using chromatin state predictions for the K562 cell line (E123). K562 epigenetic measures were sourced from the following experiment IDs from https://www.encodeproject.org/experiments/ using replicated or pseudoreplicated peaks: ENCFF148POZ (H3K4me3), ENCFF031FSF (H3K27me3), ENCFF631VWP (H3K36me3), ENCFF159VKJ (H3K4me1), ENCFF038DDS (H3K27ac), ENCFF118PIE (H3K4me2), ENCFF212PQN (H3K79me2), ENCFF285EKW (H3K9me1), and ENCFF371GMJ (H3K9me3). We intersect these features with all targeted sites. We then use a logistic regression model to assess whether any feature is associated with the barcode heterogeneity at a site, implemented using the glm function in R96 with family binomial(link = ‘logit’), where the predictor variable is the barcode heterogeneity and the binary response variable is the presence or absence of an intersecting feature, scaled to mean 0 and a standard deviation of 1.
Statistical Analysis
Statistical tests were performed using Python 3.797 and the Python libraries SciPy98 v1.4.1, statsmodels99 v0.11.0, and Scikit-learn. R libraries96 were used where indicated.
Accession Numbers
Data are available from GEO: GSE151740.
Acknowledgments
We thank all Kay Lab members, Andrew Fire, Monte Winslow, and Joanna Wysocka at Stanford University School of Medicine (Stanford, CA, USA), and Nicolas Hengartner at the Los Alamos National Laboratory (LANL), Theoretical Biology and Biophysics group (Los Alamos, NM, USA) for helpful discussions. Next-generation sequencing for this project was performed by the Stanford Functional Genomics Facility. The Illumina HiSeq 4000 instrument was obtained using NIH S10 shared instrumentation grant S10OD018220. Cell sorting for this project was performed by the Stanford Shared FACS Facility, including on an instrument purchased by the Parker Institute for Cancer Immunotherapy. The data analysis was partially performed on the CTA cluster at the Center for Non-Linear Studies (CNLS) of LANL. This work was supported by National Institutes of Health grants R01 HL064274 (to M.A.K.) and U01 HG009431 (to S.B.M.). L.P.S. was supported by the Stanford Genome Training Program (NIH/NHGRI T32 HG000044). For work conducted at LANL, L.P.S. was funded by the CNLS sponsored by the LDRD program of LANL. N.M.F. was supported by the National Science Foundation graduate research fellowship DGE-1656518 and a graduate fellowship from the Stanford Center for Computational, Evolutionary and Human Genomics. N.S.A. was supported by a Stanford NIST/JIMB training grant.
Author Contributions
Conceptualization, L.P.S., M.T., and M.A.K.; Methodology, L.P.S. and M.T.; Investigation, L.P.S.; Formal Analysis, L.P.S., N.M.F., and N.S.A.; Visualization, L.P.S. and N.M.F.; Writing – Original Draft, L.P.S.; Writing – Review & Editing, L.P.S., M.T., N.M.F., N.S.A., S.B.M., and M.A.K.; Resources, S.B.M. and M.A.K.; Supervision, S.B.M. and M.A.K.
Declaration of Interests
M.A.K. is a co-founder, Board of Directors (BOD) member, advisor, and holds equity in LogicBio Therapeutics. While there is no intellectual property (IP) directly related to this study, LogicBio has licensed IP from Stanford University related to nuclease free AAV-mediated homologous recombination. S.B.M. is on the Scientific Advisory Board (SAB) of MyOme. The remaining authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.11.025.
Supplemental Information
References
- 1.Barzel A., Paulk N.K., Shi Y., Huang Y., Chu K., Zhang F., Valdmanis P.N., Spector L.P., Porteus M.H., Gaensler K.M., Kay M.A. Promoterless gene targeting without nucleases ameliorates haemophilia B in mice. Nature. 2015;517:360–364. doi: 10.1038/nature13864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xiao A., Wang Z., Hu Y., Wu Y., Luo Z., Yang Z., Zu Y., Li W., Huang P., Tong X. Chromosomal deletions and inversions mediated by TALENs and CRISPR/Cas in zebrafish. Nucleic Acids Res. 2013;41:e141. doi: 10.1093/nar/gkt464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adikusuma F., Piltz S., Corbett M.A., Turvey M., McColl S.R., Helbig K.J., Beard M.R., Hughes J., Pomerantz R.T., Thomas P.Q. Large deletions induced by Cas9 cleavage. Nature. 2018;560:E8–E9. doi: 10.1038/s41586-018-0380-z. [DOI] [PubMed] [Google Scholar]
- 4.Kosicki M., Tomberg K., Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018;36:765–771. doi: 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakai H., Wu X., Fuess S., Storm T.A., Munroe D., Montini E., Burgess S.M., Grompe M., Kay M.A. Large-scale molecular characterization of adeno-associated virus vector integration in mouse liver. J. Virol. 2005;79:3606–3614. doi: 10.1128/JVI.79.6.3606-3614.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chandler R.J., LaFave M.C., Varshney G.K., Trivedi N.S., Carrillo-Carrasco N., Senac J.S., Wu W., Hoffmann V., Elkahloun A.G., Burgess S.M., Venditti C.P. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J. Clin. Invest. 2015;125:870–880. doi: 10.1172/JCI79213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lisowski L., Lau A., Wang Z., Zhang Y., Zhang F., Grompe M., Kay M.A. Ribosomal DNA integrating rAAV-rDNA vectors allow for stable transgene expression. Mol. Ther. 2012;20:1912–1923. doi: 10.1038/mt.2012.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Russell D.W., Hirata R.K. Human gene targeting by viral vectors. Nat. Genet. 1998;18:325–330. doi: 10.1038/ng0498-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller D.G., Wang P.-R., Petek L.M., Hirata R.K., Sands M.S., Russell D.W. Gene targeting in vivo by adeno-associated virus vectors. Nat. Biotechnol. 2006;24:1022–1026. doi: 10.1038/nbt1231. [DOI] [PubMed] [Google Scholar]
- 10.Cornea A.M., Russell D.W. Chromosomal position effects on AAV-mediated gene targeting. Nucleic Acids Res. 2010;38:3582–3594. doi: 10.1093/nar/gkq095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Z., Lisowski L., Finegold M.J., Nakai H., Kay M.A., Grompe M. AAV vectors containing rDNA homology display increased chromosomal integration and transgene persistence. Mol. Ther. 2012;20:1902–1911. doi: 10.1038/mt.2012.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inoue N., Hirata R.K., Russell D.W. High-fidelity correction of mutations at multiple chromosomal positions by adeno-associated virus vectors. J. Virol. 1999;73:7376–7380. doi: 10.1128/jvi.73.9.7376-7380.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li L.B., Ma C., Awong G., Kennedy M., Gornalusse G., Keller G., Kaufman D.S., Russell D.W. Silent IL2RG gene editing in human pluripotent stem cells. Mol. Ther. 2016;24:582–591. doi: 10.1038/mt.2015.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Melo S.P., Lisowski L., Bashkirova E., Zhen H.H., Chu K., Keene D.R., Marinkovich M.P., Kay M.A., Oro A.E. Somatic correction of junctional epidermolysis bullosa by a highly recombinogenic AAV variant. Mol. Ther. 2014;22:725–733. doi: 10.1038/mt.2013.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sebastiano V., Zhen H.H., Haddad B., Bashkirova E., Melo S.P., Wang P., Leung T.L., Siprashvili Z., Tichy A., Li J. Human COL7A1-corrected induced pluripotent stem cells for the treatment of recessive dystrophic epidermolysis bullosa. Sci. Transl. Med. 2014;6:264ra163. doi: 10.1126/scitranslmed.3009540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paulk N.K., Wursthorn K., Wang Z., Finegold M.J., Kay M.A., Grompe M. Adeno-associated virus gene repair corrects a mouse model of hereditary tyrosinemia in vivo. Hepatology. 2010;51:1200–1208. doi: 10.1002/hep.23481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirsch M.L. Adeno-associated virus inverted terminal repeats stimulate gene editing. Gene Ther. 2015;22:190–195. doi: 10.1038/gt.2014.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vasileva A., Linden R.M., Jessberger R. Homologous recombination is required for AAV-mediated gene targeting. Nucleic Acids Res. 2006;34:3345–3360. doi: 10.1093/nar/gkl455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hirata R.K., Russell D.W. Design and packaging of adeno-associated virus gene targeting vectors. J. Virol. 2000;74:4612–4620. doi: 10.1128/jvi.74.10.4612-4620.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zentilin L., Marcello A., Giacca M. Involvement of cellular double-stranded DNA break binding proteins in processing of the recombinant adeno-associated virus genome. J. Virol. 2001;75:12279–12287. doi: 10.1128/JVI.75.24.12279-12287.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fattah F.J., Lichter N.F., Fattah K.R., Oh S., Hendrickson E.A. Ku70, an essential gene, modulates the frequency of rAAV-mediated gene targeting in human somatic cells. Proc. Natl. Acad. Sci. USA. 2008;105:8703–8708. doi: 10.1073/pnas.0712060105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Dyck E., Stasiak A.Z., Stasiak A., West S.C. Binding of double-strand breaks in DNA by human Rad52 protein. Nature. 1999;398:728–731. doi: 10.1038/19560. [DOI] [PubMed] [Google Scholar]
- 23.Nickoloff J.A., Reynolds R.J. Transcription stimulates homologous recombination in mammalian cells. Mol. Cell. Biol. 1990;10:4837–4845. doi: 10.1128/mcb.10.9.4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thyagarajan B., Johnson B.L., Campbell C. The effect of target site transcription on gene targeting in human cells in vitro. Nucleic Acids Res. 1995;23:2784–2790. doi: 10.1093/nar/23.14.2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aguilera A. The connection between transcription and genomic instability. EMBO J. 2002;21:195–201. doi: 10.1093/emboj/21.3.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gottipati P., Helleday T. Transcription-associated recombination in eukaryotes: link between transcription, replication and recombination. Mutagenesis. 2009;24:203–210. doi: 10.1093/mutage/gen072. [DOI] [PubMed] [Google Scholar]
- 27.Huertas P., Aguilera A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol. Cell. 2003;12:711–721. doi: 10.1016/j.molcel.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 28.Schwab R.A., Nieminuszczy J., Shah F., Langton J., Lopez Martinez D., Liang C.-C., Cohn M.A., Gibbons R.J., Deans A.J., Niedzwiedz W. The Fanconi anemia pathway maintains genome stability by coordinating replication and transcription. Mol. Cell. 2015;60:351–361. doi: 10.1016/j.molcel.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yan Z., Delannoy M., Ling C., Daee D., Osman F., Muniandy P.A., Shen X., Oostra A.B., Du H., Steltenpool J. A histone-fold complex and FANCM form a conserved DNA-remodeling complex to maintain genome stability. Mol. Cell. 2010;37:865–878. doi: 10.1016/j.molcel.2010.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De Alencastro G., Puzzo F., Pavel-Dinu M., Zhang F., Pillay S., Majzoub K., Tiffany M., Jang H., Sheikali A., Cromer M.K. Improved genome editing through inhibition of FANCM and members of the BTR dissolvase complex. Mol. Ther. 2020 doi: 10.1016/j.ymthe.2020.10.020. Published online October 22, 2020. doi.org/10.1016/j.ymthe.2020.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deyle D.R., Hansen R.S., Cornea A.M., Li L.B., Burt A.A., Alexander I.E., Sandstrom R.S., Stamatoyannopoulos J.A., Wei C.L., Russell D.W. A genome-wide map of adeno-associated virus-mediated human gene targeting. Nat. Struct. Mol. Biol. 2014;21:969–975. doi: 10.1038/nsmb.2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gottipati P., Cassel T.N., Savolainen L., Helleday T. Transcription-associated recombination is dependent on replication in mammalian cells. Mol. Cell. Biol. 2008;28:154–164. doi: 10.1128/MCB.00816-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aymard F., Bugler B., Schmidt C.K., Guillou E., Caron P., Briois S., Iacovoni J.S., Daburon V., Miller K.M., Jackson S.P., Legube G. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat. Struct. Mol. Biol. 2014;21:366–374. doi: 10.1038/nsmb.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang J., Cho N.W., Cui G., Manion E.M., Shanbhag N.M., Botuyan M.V., Mer G., Greenberg R.A. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat. Struct. Mol. Biol. 2013;20:317–325. doi: 10.1038/nsmb.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Riballo E., Kühne M., Rief N., Doherty A., Smith G.C.M., Recio M.-J., Reis C., Dahm K., Fricke A., Krempler A. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to γ-H2AX foci. Mol. Cell. 2004;16:715–724. doi: 10.1016/j.molcel.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 36.Beucher A., Birraux J., Tchouandong L., Barton O., Shibata A., Conrad S., Goodarzi A.A., Krempler A., Jeggo P.A., Löbrich M. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 2009;28:3413–3427. doi: 10.1038/emboj.2009.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Löbrich M., Shibata A., Beucher A., Fisher A., Ensminger M., Goodarzi A.A., Barton O., Jeggo P.A. γH2AX foci analysis for monitoring DNA double-strand break repair: strengths, limitations and optimization. Cell Cycle. 2010;9:662–669. doi: 10.4161/cc.9.4.10764. [DOI] [PubMed] [Google Scholar]
- 38.Chantalat S., Depaux A., Héry P., Barral S., Thuret J.-Y., Dimitrov S., Gérard M. Histone H3 trimethylation at lysine 36 is associated with constitutive and facultative heterochromatin. Genome Res. 2011;21:1426–1437. doi: 10.1101/gr.118091.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dhayalan A., Rajavelu A., Rathert P., Tamas R., Jurkowska R.Z., Ragozin S., Jeltsch A. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J. Biol. Chem. 2010;285:26114–26120. doi: 10.1074/jbc.M109.089433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kollárovič G., Topping C.E., Shaw E.P., Chambers A.L. The human HELLS chromatin remodelling protein promotes end resection to facilitate homologous recombination and contributes to DSB repair within heterochromatin. Nucleic Acids Res. 2020;48:1872–1885. doi: 10.1093/nar/gkz1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goodarzi A.A., Noon A.T., Deckbar D., Ziv Y., Shiloh Y., Löbrich M., Jeggo P.A. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell. 2008;31:167–177. doi: 10.1016/j.molcel.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 42.Ernst J., Kellis M. Large-scale imputation of epigenomic datasets for systematic annotation of diverse human tissues. Nat. Biotechnol. 2015;33:364–376. doi: 10.1038/nbt.3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Belancio V.P., Deininger P.L., Roy-Engel A.M. LINE dancing in the human genome: transposable elements and disease. Genome Med. 2009;1:97. doi: 10.1186/gm97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morales M.E., White T.B., Streva V.A., DeFreece C.B., Hedges D.J., Deininger P.L. The contribution of Alu elements to mutagenic DNA double-strand break repair. PLoS Genet. 2015;11:e1005016. doi: 10.1371/journal.pgen.1005016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hedges D.J., Deininger P.L. Inviting instability: transposable elements, double-strand breaks, and the maintenance of genome integrity. Mutat. Res. 2007;616:46–59. doi: 10.1016/j.mrfmmm.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carette J.E., Raaben M., Wong A.C., Herbert A.S., Obernosterer G., Mulherkar N., Kuehne A.I., Kranzusch P.J., Griffin A.M., Ruthel G. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature. 2011;477:340–343. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lamb R., Fiorillo M., Chadwick A., Ozsvari B., Reeves K.J., Smith D.L., Clarke R.B., Howell S.J., Cappello A.R., Martinez-Outschoorn U.E. Doxycycline down-regulates DNA-PK and radiosensitizes tumor initiating cells: implications for more effective radiation therapy. Oncotarget. 2015;6:14005–14025. doi: 10.18632/oncotarget.4159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Neal J.A., Dang V., Douglas P., Wold M.S., Lees-Miller S.P., Meek K. Inhibition of homologous recombination by DNA-dependent protein kinase requires kinase activity, is titratable, and is modulated by autophosphorylation. Mol. Cell. Biol. 2011;31:1719–1733. doi: 10.1128/MCB.01298-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ahler E., Sullivan W.J., Cass A., Braas D., York A.G., Bensinger S.J., Graeber T.G., Christofk H.R. Doxycycline alters metabolism and proliferation of human cell lines. PLoS ONE. 2013;8:e64561. doi: 10.1371/journal.pone.0064561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grimm D., Lee J.S., Wang L., Desai T., Akache B., Storm T.A., Kay M.A. In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J. Virol. 2008;82:5887–5911. doi: 10.1128/JVI.00254-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sherman E., Nobles C., Berry C.C., Six E., Wu Y., Dryga A., Malani N., Male F., Reddy S., Bailey A. INSPIIRED: a pipeline for quantitative analysis of sites of new DNA integration in cellular genomes. Mol. Ther. Methods Clin. Dev. 2016;4:39–49. doi: 10.1016/j.omtm.2016.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Desfarges S., Ciuffi A. Retroviral integration site selection. Viruses. 2010;2:111–130. doi: 10.3390/v2010111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lewinski M.K., Yamashita M., Emerman M., Ciuffi A., Marshall H., Crawford G., Collins F., Shinn P., Leipzig J., Hannenhalli S. Retroviral DNA integration: viral and cellular determinants of target-site selection. PLoS Pathog. 2006;2:e60. doi: 10.1371/journal.ppat.0020060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schröder A.R.W., Shinn P., Chen H., Berry C., Ecker J.R., Bushman F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110:521–529. doi: 10.1016/s0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- 55.Marini B., Kertesz-Farkas A., Ali H., Lucic B., Lisek K., Manganaro L., Pongor S., Luzzati R., Recchia A., Mavilio F. Nuclear architecture dictates HIV-1 integration site selection. Nature. 2015;521:227–231. doi: 10.1038/nature14226. [DOI] [PubMed] [Google Scholar]
- 56.Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 57.Akhtar W., Pindyurin A.V., de Jong J., Pagie L., Ten Hoeve J., Berns A., Wessels L.F.A., van Steensel B., van Lohuizen M. Using TRIP for genome-wide position effect analysis in cultured cells. Nat. Protoc. 2014;9:1255–1281. doi: 10.1038/nprot.2014.072. [DOI] [PubMed] [Google Scholar]
- 58.Song L., Crawford G.E. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb. Protoc. 2010;2010 doi: 10.1101/pdb.prot5384. pdb.prot5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Favorov A., Mularoni L., Cope L.M., Medvedeva Y., Mironov A.A., Makeev V.J., Wheelan S.J. Exploring massive, genome scale datasets with the GenometriCorr package. PLoS Comput. Biol. 2012;8:e1002529. doi: 10.1371/journal.pcbi.1002529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rodríguez-Castañeda F., Lemma R.B., Cuervo I., Bengtsen M., Moen L.M., Ledsaak M., Eskeland R., Gabrielsen O.S. The SUMO protease SENP1 and the chromatin remodeler CHD3 interact and jointly affect chromatin accessibility and gene expression. J. Biol. Chem. 2018;293:15439–15454. doi: 10.1074/jbc.RA118.002844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., Ziller M.J., Roadmap Epigenomics Consortium Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ernst J., Kellis M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol. 2010;28:817–825. doi: 10.1038/nbt.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rosenbloom K.R., Sloan C.A., Malladi V.S., Dreszer T.R., Learned K., Kirkup V.M., Wong M.C., Maddren M., Fang R., Heitner S.G. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res. 2013;41(Database issue):D56–D63. doi: 10.1093/nar/gks1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen H.-C., Martinez J.P., Zorita E., Meyerhans A., Filion G.J. Position effects influence HIV latency reversal. Nat. Struct. Mol. Biol. 2017;24:47–54. doi: 10.1038/nsmb.3328. [DOI] [PubMed] [Google Scholar]
- 66.Pradeepa M.M., Sutherland H.G., Ule J., Grimes G.R., Bickmore W.A. Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS Genet. 2012;8:e1002717. doi: 10.1371/journal.pgen.1002717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.García-Rubio M., Huertas P., González-Barrera S., Aguilera A. Recombinogenic effects of DNA-damaging agents are synergistically increased by transcription in Saccharomyces cerevisiae. New insights into transcription-associated recombination. Genetics. 2003;165:457–466. doi: 10.1093/genetics/165.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Prado F., Aguilera A. Impairment of replication fork progression mediates RNA polII transcription-associated recombination. EMBO J. 2005;24:1267–1276. doi: 10.1038/sj.emboj.7600602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Takeuchi Y., Horiuchi T., Kobayashi T. Transcription-dependent recombination and the role of fork collision in yeast rDNA. Genes Dev. 2003;17:1497–1506. doi: 10.1101/gad.1085403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.de la Loza M.C.D., Wellinger R.E., Aguilera A. Stimulation of direct-repeat recombination by RNA polymerase III transcription. DNA Repair (Amst.) 2009;8:620–626. doi: 10.1016/j.dnarep.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 71.Helmrich A., Ballarino M., Nudler E., Tora L. Transcription-replication encounters, consequences and genomic instability. Nat. Struct. Mol. Biol. 2013;20:412–418. doi: 10.1038/nsmb.2543. [DOI] [PubMed] [Google Scholar]
- 72.Polak P., Domany E. Alu elements contain many binding sites for transcription factors and may play a role in regulation of developmental processes. BMC Genomics. 2006;7:133. doi: 10.1186/1471-2164-7-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Callinan P.A., Batzer M.A. Retrotransposable elements and human disease. Genome Dyn. 2006;1:104–115. doi: 10.1159/000092503. [DOI] [PubMed] [Google Scholar]
- 74.Liu N., Lee C.H., Swigut T., Grow E., Gu B., Bassik M.C., Wysocka J. Selective silencing of euchromatic L1s revealed by genome-wide screens for L1 regulators. Nature. 2018;553:228–232. doi: 10.1038/nature25179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yu F., Zingler N., Schumann G., Strätling W.H. Methyl-CpG-binding protein 2 represses LINE-1 expression and retrotransposition but not Alu transcription. Nucleic Acids Res. 2001;29:4493–4501. doi: 10.1093/nar/29.21.4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hata K., Sakaki Y. Identification of critical CpG sites for repression of L1 transcription by DNA methylation. Gene. 1997;189:227–234. doi: 10.1016/s0378-1119(96)00856-6. [DOI] [PubMed] [Google Scholar]
- 77.Sultana T., van Essen D., Siol O., Bailly-Bechet M., Philippe C., Zine El Aabidine A., Pioger L., Nigumann P., Saccani S., Andrau J.-C. The landscape of L1 retrotransposons in the human genome is shaped by pre-insertion sequence biases and post-insertion selection. Mol. Cell. 2019;74:555–570.e7. doi: 10.1016/j.molcel.2019.02.036. [DOI] [PubMed] [Google Scholar]
- 78.Zhou X., Vink M., Klaver B., Berkhout B., Das A.T. Optimization of the Tet-On system for regulated gene expression through viral evolution. Gene Ther. 2006;13:1382–1390. doi: 10.1038/sj.gt.3302780. [DOI] [PubMed] [Google Scholar]
- 79.Gibson D.G. Enzymatic assembly of overlapping DNA fragments. Methods Enzymol. 2011;498:349–361. doi: 10.1016/B978-0-12-385120-8.00015-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Adachi K., Enoki T., Kawano Y., Veraz M., Nakai H. Drawing a high-resolution functional map of adeno-associated virus capsid by massively parallel sequencing. Nat. Commun. 2014;5:3075. doi: 10.1038/ncomms4075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pekrun K., Alencastro G.D., Luo Q.-J., Liu J., Kim Y., Nygaard S., Galivo F., Zhang F., Song R., Tiffany M.R. Using a barcoded AAV capsid library to select for clinically relevant gene therapy vectors. JCI Insight. 2019;4:e131610. doi: 10.1172/jci.insight.131610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kienle E., Senís E., Börner K., Niopek D., Wiedtke E., Grosse S., Grimm D. Engineering and evolution of synthetic adeno-associated virus (AAV) gene therapy vectors via DNA family shuffling. J. Vis. Exp. 2012;(62):3819. doi: 10.3791/3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zorita E., Cuscó P., Filion G.J. Starcode: sequence clustering based on all-pairs search. Bioinformatics. 2015;31:1913–1919. doi: 10.1093/bioinformatics/btv053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tiscornia G., Singer O., Verma I.M. Production and purification of lentiviral vectors. Nat. Protoc. 2006;1:241–245. doi: 10.1038/nprot.2006.37. [DOI] [PubMed] [Google Scholar]
- 85.Horizon Discovery. SMARTvector Lentiviral shRNA & shMIMIC Lentiviral MicroRNA Pooled Libraries: Technical Manual. https://horizondiscovery.com/-/media/Files/Horizon/resources/Technical-manuals/lentiviral-pooled-libraries-manual.pdf.
- 86.Porteus M.H., Cathomen T., Weitzman M.D., Baltimore D. Efficient gene targeting mediated by adeno-associated virus and DNA double-strand breaks. Mol. Cell. Biol. 2003;23:3558–3565. doi: 10.1128/MCB.23.10.3558-3565.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Winters I.P., Chiou S.-H., Paulk N.K., McFarland C.D., Lalgudi P.V., Ma R.K., Lisowski L., Connolly A.J., Petrov D.A., Kay M.A., Winslow M.M. Multiplexed in vivo homology-directed repair and tumor barcoding enables parallel quantification of Kras variant oncogenicity. Nat. Commun. 2017;8:2053. doi: 10.1038/s41467-017-01519-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Smith T.S., Heger A., Sudbery I. UMI-tools: modelling sequencing errors in unique molecular identifiers to improve quantification accuracy. Genome Res. 2017;27:491–499. doi: 10.1101/gr.209601.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Frankish A., Diekhans M., Ferreira A.-M., Johnson R., Jungreis I., Loveland J., Mudge J.M., Sisu C., Wright J., Armstrong J. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019;47:D766–D773. doi: 10.1093/nar/gky955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Servant N., Varoquaux N., Lajoie B.R., Viara E., Chen C.-J., Vert J.-P., Heard E., Dekker J., Barillot E. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015;16:259. doi: 10.1186/s13059-015-0831-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Akhtar W., de Jong J., Pindyurin A.V., Pagie L., Meuleman W., de Ridder J., Berns A., Wessels L.F.A., van Lohuizen M., van Steensel B. Chromatin position effects assayed by thousands of reporters integrated in parallel. Cell. 2013;154:914–927. doi: 10.1016/j.cell.2013.07.018. [DOI] [PubMed] [Google Scholar]
- 94.Karolchik D., Hinrichs A.S., Furey T.S., Roskin K.M., Sugnet C.W., Haussler D., Kent W.J. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32(Database issue):D493–D496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.R Core Team . R Foundation for Statistical Computing; 2018. R: A Language and Environment for Statistical Computing. [Google Scholar]
- 97.Python Core Team . Python Software Foundation; 2015. Python: A Dynamic, Open Source Programming Language. [Google Scholar]
- 98.Virtanen P., Gommers R., Oliphant T.E., Haberland M., Reddy T., Cournapeau D., Burovski E., Peterson P., Weckesser W., Bright J. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods. 2020;17:261–272. doi: 10.1038/s41592-019-0686-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Seabold S., Perktold J. Statsmodels: econometric and statistical modeling with Python. In: van der Walt S., Millman J., editors. Proceedings of the 9th Python in Science Conference. 2010. pp. 57–62. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.